
Siccanin-related drimane meroterpenoids: biological activities and synthesis†‡
Shengxin
Sun§
,
Xia
Wang
,
Nvdan
Hu
,
Shiqi
Fu
and
Shengkun
Li
 *
*
State Key Laboratory of Green Pesticide, Center for R&D of Fine Chemicals of Guizhou University, Guiyang, 550025, China. E-mail: SKL505@outlook.com
First published on 4th November 2024
Abstract
Covering: 1962 to 2023
Drimane (hydro)quinones biosynthetically arise from the combination of drimane-type terpenoids with phenols or equivalents. Since the isolation of siccanin in 1962 (structure identified in 1967), over 200 natural drimane (hydro)quinones have been reported. They are widespread with remarkably diverse architectures and biological functions, which are achieved by varying either the drimane subunit, hydroquinone segment, or the fusion types of drimane and hydroquinone segment both of them. This type of natural products has drawn increasing attention in the discovery of novel pharmaceutical leads. Enormous efforts have been devoted to developing efficient and divergent synthesis approaches to facilitate the SAR study of drimane (hydro)quinones, aiming for more promising functional leads. This review is arranged mainly in terms of scaffold types of drimane (hydro)quinones and further categorized on the basis of biological functions. The mechanisms of action are also briefly discussed. Synthetic methods are categorized according to the strategies forging the Csp2–Csp3 linker between drimane segments and (hydro)quinone subunits.
 Shengxin Sun | Shengxin Sun obtained his master's degree from Nanjing Agricultural University and is now conducting his doctoral studies at Guizhou University under the supervision of Prof. Shengkun Li. His research focuses on the efficient and modular synthesis and function-oriented optimization of drimane (hydro)quinones. |
 Shengkun Li | Shengkun Li earned his PhD from Northwest A & F University in 2013, during which he spent two years at Rutgers University. He later started independent research as an associate professor at Nanjing Agricultural University and moved to Guizhou University as a professor and “Cheung Kong Young Scholar” in 2020. His research focuses on ligands discovery inspired by quinones (e.g. CoQ and PQQ). |
1. Introduction
Natural products continue to serve as models or inspirations for lead discovery because they have been selected through evolutionary pressure for high potency and specificity against biological targets. With diverse and intriguing scaffolds of attractive functions and bioactivities, natural products have widely affected the discovery of drugs and functional materials.1 Biogenetically originating from polyketide–terpenoid, drimane meroterpenoids are an important family of natural products that have attracted intensive attention from both synthetic and biological communities. One of the most eminent examples is the five-ring-fused antifungal natural product siccanin (via infra), which was isolated in 1962 as an antifungal antibiotic. Its high-order complexity, natural scarcity, and limited availability have largely impeded its further investigation in the pharmaceutical industry. Since its isolation, over 200 framework-related but structurally simplified natural drimane meroterpenoids have been reported. Structurally assembling an sp3-hybridized drimane skeleton with sp2-hybridized aromatic units,2 these natural products together with rearranged analogs show a wealth of bioactivities (via infra), including antifungal, antibacterial, antiviral, insecticidal, cytotoxic, antioxidant, algicidal, anti-inflammatory, antifeedant, and immunomodulatory activities. Although increasing attention has been drawn from both chemical biology and chemistry communities, there is no comprehensive insight specifically dealing with scaffold-based activities and synthetic strategies (Fig. 1). This review is arranged mainly in terms of scaffold-dependent functions and synthetic efforts. The first section will highlight siccanin-related scaffolds of drimane meroterpenoids accompanied with important biological characteristics. Synthetic strategies are categorized by accentuating how to forge the linker between the drimane segments and (hydro)quinone subunits. The original building blocks (natural feedstocks or commercial synthons) have also been provided in every section to help researchers choose appropriate strategies and starting materials. The mechanisms of action are also briefly mentioned, and function-oriented optimization is succinctly discussed for the discovery of pharmaceutical leads or probes. We have emphasized the key characteristics and major synthetic strategies by providing typical applications, rather than attempting comprehensive listings. We anticipate that the current review may encourage analogous efforts on different complex natural products that have been limited by biosynthetic accessibility and synthetic challenges. | ||
| Fig. 1 Siccanin-related meroterpenoids: bioactivity and synthesis. | ||
2. Characteristics of siccanin-related drimane meroterpenoids
2.1 Siccanin and its biological importance
As a preeminent meroterpenoind with a drimane sesquiterpene combined with orcinol, (−)-siccanin (1) was isolated in 1962 by Ishibashi et al. from the BH-34 fungi strain of Helminthosporium siccans as a prototypical complicated antibiotic,3,4 whose absolute configuration was confirmed by X-ray (Cu Kα irradiation) diffraction of its p-bromobenzenesulfonate in 1967 (Fig. 2).5 (−)-Siccanin (1) showed excellent antifungal activities against Trichophyton interdigitale and Trichophyton asteroids, with the corresponding MIC value of 0.1 μg mL−1.3,4 It was demonstrated to possess inhibitory effects against up to 51 fungal strains.6 Alongside its potential in the treatment of superficial dermatophyte infections,7 it also showed moderate antibacterial activity, of which the MIC against Escherichia coli is 12.5 μg mL−1.3,4 In 2016, this compound was discovered as a potent broad-spectrum anti-trypanosomatid lead.8 Its pharmaceutical importance has drawn considerable attention in the investigation of the model of action. As early as 1971, it was reported to disrupt the mitochondria of Trichophyton mentagrophytes by influencing succinate dehydrogenase.9 In 2009, Mogi et al. demonstrated that siccanin (1) acts as a species-selective succinate dehydrogenase inhibitor.10 Recently, siccanin (1) was rediscovered as a dual-target inhibitor of mitochondrial complex II and complex III.11 Considering the unique species-selective inhibition by siccanin (1) against the biologically important mitochondria, it could serve as a promising model for new chemotherapeutics. | ||
| Fig. 2 Structure and biological profiles of siccanin. | ||
The limited natural supply and the stagnant progress in practical total synthesis (vide infra) make it difficult to investigate siccanin (1) in detail, especially in applied pharmaceuticals. The structural analysis showed that the key framework of siccanin (1) is drimane (hydro)quinone, which is embedded in a large number of meroterpenoids. Inspired by the principles12 in the function-oriented synthesis, one can envisage that the comprehensive investigation of siccanin (1) and the scaffold-related distinct drimane meroterpenoids may significantly contribute to the discovery and practical acquirement of simplified natural models or mimics to facilitate lead innovation. Some typical drimane meroterpenoids with distinct scaffolds and biological importance will be discussed in the following parts, whose subgroups were arranged by the unsaturated core drimane.
2.2 Drimane substituted (hydro)quinones and analogues
Drimane-substituted (hydro)quinone is the basic scaffold of siccanin, with a drimane sesquiterpene combined with an aromatic segment through a single Csp2–Csp3 linkage. Featuring Δ8,12-drimane (hydro)quinone (Fig. 3), the biogenetically unique (−)-tauranin (2) with monohydroxy-p-benzoquinone was first isolated from the mycelia of Oospora aurantia (Cooke) by Kawashima et al. in 1964.13–15 The absolute configuration of (−)-tauranin was determined by comparison of its degradation homodrimanic acid with the known enantiospecific acid from ambrein and sclareol. It was further verified by enantioselective synthesis.15 Structural determination of (−)-3-ketotauranin (3-oxotauranin), together with the revision of the originally proposed 3α-hydroxytauranin to be 3β-hydroxytauranin, were also accomplished by enantioselective total synthesis.16 (−)-Tauranin was also reported by Gunatilaka et al. from Phyllosticta spinarum, together with three natural analogues: (−)-3-ketotauranin (3), (+)-3α-hydroxytauranin (4) and (−)-12-hydroxytauranin (5). Among them, (−)-tauranin (2) showed antiproliferative activity against NCI-H460, MCF-7, SF-268, PC-3M, and MIA Pa Ca-2, with the IC50 values of 4.3, 1.5, 1.8, 3.5 and 2.8 μM, respectively. It could induce apoptosis in the PC-3M and NIH 3T3 cell lines.17 | ||
| Fig. 3 Siccanin-related Δ8,12 drimane (hydro)quinones. | ||
A typical fungitoxic drimane hydroquinone, (+)-zonarol (6), was first isolated in 1973 from the brown seaweed Dictyopteris zonarioides.18 Monomethylated zonarol (7) was firstly isolated from the marine brown alga Dictyopteris undulata by Bipin C.19 The zonarol analogues, (+)-zonaroic acid (8) and (+)-zonarone (9), were firstly isolated from the brown seaweed Dictyopteris undulata by Fenical and Kakisawa, respectively.20,21 The absolute configurations of these natural meroterpenoids were established by enantioselective total synthesis.22–24 Zonarol (6) showed remarkable inhibition against four plant pathogenic fungi, including Phytophthora cinnamomi, Rhizoctonia solani, Sclerotinia sclerotiorum, and Sclerotium rolfsii.18 Zonarol (6), zonaroic acid (8), and zonarone (9) showed toxicity to fish, with the corresponding MIC values of 17, 13, and 7 ppm, respectively.21 Zonarol (6) and zonarone (9) also possessed antifeedant activity against the young abalone at a concentration of 75 μg of each sample, with the electivity index (Ei) values of 0.85 and 0.92, respectively.25 They were detected to have cytotoxicity against the tumor cell lines L-929 (murine fibroblasts), K-562 (human leukemia), and HeLa (human cervix carcinoma).26 Promising anti-inflammatory activity of zonarol (3) was detected against 3α-hydroxysteroid dehydrogenase (3α-HSD), by inhibiting ROS production in SOZ-stimulated granulocytes.27
(+)-Hyatellaquinone (10) was first isolated in 1993 from a southern Australian marine sponge, Spongia sp.,28 and then from the alga Peyssonnelia sp. together with an antiviral analog peyssonol A (11) with an EC50 value of 1 μM against HIV.29,30 Samadi et al. assigned the absolute configuration of the naturally occurring (+)-hyatellaquinone (10)31 through the comparison of optical rotations with the synthetic (−)-enantiomer. The relative configuration of peyssonol A was revised as 11 by racemic total synthesis by Snyder in 2010.32 The artificial one showed cytotoxicity against KB cells with an IC50 value of 14 μM. The (±)-hyatellaquinone (10) showed antitumor activity against cell lines HM02, HepG2, and MCF7, with corresponding GI50 values of 5.3, 6.0 and 2.4 μg mL−1, respectively.26 (+)-Hyatellaquinone showed excellent cytotoxicity against breast cancer (IC50 = 4.45 μg mL−1), small cell lung cancer (IC50 = 10.9 μg mL−1), L-929 (GI50 = 20.9 μM), K-562 (GI50 = 8.4 μM) and HeLa (CC50 = 72.1 μM) cell lines.33 It also provided good anti-inflammatory activity against 3α-hydroxysteroid dehydrogenase (3α-HSD).27
In 2000, Kohama et al. isolated (−)-F-12509A (12) from Trichopezizella barbata SANK 25395,34 whose absolute configuration was confirmed by enantiospecific synthesis.35 It was demonstrated to permeate the plasma membrane and effectively inhibit rat liver SPH kinase, with an IC50 value of 18 μM.34 Two antitumor drimenyl cyclohexenone derivatives,36,37 (+)-purpurogemutantin (13) and (−)-purpurogemutantidin (14), were isolated from Penicillium purpurogenum G59. The absolute configurations were determined by extensive spectroscopic methods, especially 2D NMR and ECD analysis. Noteworthily, (−)-purpurogemutantidin (14) showed excellent cytotoxicity against K562 and HL-60, with corresponding IC50 values of 0.93 and 2.48 μM, respectively.36 The cytotoxic drimane quinone (−)-penicillium A (15) was isolated from Penicillium sp. F00120, and its relative configuration was confirmed by NOESY spectra. Its GI50 values against mouse melanoma (B16), human melanoma (A375), and human cervical carcinoma (HeLa) were 27.37, 22.88, and 44.05 μg mL−1, respectively. The median toxic concentrations (TC50) against the host cells HeLa, African green monkey kidney (Vero), and Madin–Darby canine kidney (MDCK) cells were 40.72, 133.52, and 43.00 μg mL−1, respectively.38
Three drimane (hydro)quinones, (+)-macrophorins A–C (16–18), featuring a special ethylene oxide moiety on the benzene ring were obtained from the fungus causing macrophoma fruit rot of apple. The absolute stereochemistry of (+)-macrophorins A–C was elucidated by CD spectra and a comparison of its optical rotation with that of biformene. (+)-Macrophorin A (16) was detected to have more promising antibacterial activity and cytotoxicity than the other two analogs, highlighting the crucial role of 3-substituents.39 The analog (+)-macrophorin D (19) was reported as a self-growth inhibitor,40 and the absolute configuration was revised through chemical correlation by Fujimoto et al.40,41 Macrophorin A (16) and 4′-oxomacrophorin A (24) showed significant in vitro cytotoxicity against the K562, MCF-7, DU145, U937, H1975, SGC-7901, A549, MOLT-4, HL60 and HeLa cell lines, with IC50 values ranging from 0.19 to 35.4 μM.37
Three malonyl analogs macrophorins E, F, and G (20–22) were isolated from the Botryosphaeria berengeriana, and they possessed antifungal activities against two phytopathogenic fungi (B. berengeriana and G. fujikuroi), with the IC50 values ranging from 3 to 25 μg per disc.42 (+)-4′-Oxomacrophorin D (23) and (+)-4′-oxomacrophorin A (24) were acquired from Ascomycete Eupenicillium crustaceum, and their absolute configurations were determined via chemical correlation. They showed immunosuppressive effects against Con A-induced (IC50 = 4.5 and 0.5 μg mL−1) and LPS-induced (IC50 = 2.0 and 0.25 μg mL−1) proliferations of mouse splenic lymphocytes.41 The oxidized macrophorin analogue (+)-2′,3′-epoxy-13-hydroxy-4′-oxomacrophorin A (25) was produced by Hymenopsis sp. (MYC-1703; NRRL 37638), whose structure was confirmed by X-ray crystallographic analysis. It showed good antibacterial activity against Staphylococcus aureus (ATCC 25923) and Bacillus subtilis (ATCC 6051), and antifungal activity against Aspergillus flavus and Fusarium verticillioides.43 In 2014, three macrophorin-related metabolites called (−)-neomacrophorin I, II, and III (26, 27, 28) were isolated from the culture broth of Trichoderma sp. 1212-03. The absolute configurations were established by comparing their ECD spectra with those of related compounds. These compounds showed antifungal activity against the phytopathogenic fungi Cochliobolus miyabeanus. Hashimoto et al. showed that only (−)-neomacrophorin I (26) could inhibit human colon adenocarcinoma (COLO 201) cell proliferation (IC50 = 46 μg mL−1), demonstrating the indispensable role of the quinone unit.44 In 2019, Kimura et al. presented another three members of this family, neomacrophorins IV (29), V (30), and VI (31), from the same source. The cytotoxicity and proteasomal activities were investigated, in which neomacrophorins IV (29) and VI (31) demonstrated the most potent activity against HL60, with IC50 values of 1.3 and 0.3 μM, respectively. Neomacrophorin IV (29) could inhibit the chymotrypsin-like, trypsin-like, and caspase-like sites of proteasomes, with IC50 values of 5.3, 22.5, and 3.9 μM, respectively.45 (+)-Myrothecol A, (−)-myrothecol B, and (+)-myrothecols C-F (32–37) characterized by an ethylene oxide or polyhydroxy decorated quinone were isolated from a Myrothecium sp. SC0265. The absolute configurations were assigned by CD/TDDFT calculations.46 (+)-Myrothecols G and H (38 and 39) from the mycelia solid cultures of Myrothecium sp. SC0265, were elucidated by spectroscopic data combined with theoretical conformational analysis.47 (+)-Myrothecol A (32) stood out in both the antibacterial test and cytotoxic screening. Its MIC values against Staphylococcus aureus and Bacillus cereus are 12.5 and 25.0 μg mL−1, respectively. The IC50 values against human carcinoma A549, HeLa, and HepG2 cells are 8.0, 7.9, and 15.2 μM, respectively. The epoxide analog nitrosporeunol G (40) was isolated by Lin et al. from the marine microorganisms of a mutated strain of Arctic Streptomyces nitrosporeus YBH10-5. The configuration was elucidated through the comparison of the NOE interaction and CD effects with that of macrophorin A.48
Similar to that of (+)-zonarol (6), the structure of the antifungal natural Δ7,8-isomer (+)-isozonarol (41) was confirmed by enantioselective total synthesis (Fig. 4).18,22 (+)-Isozonarone (42) and (+)-zonarone (9) were isolated from the brown alga Dictyopteris undulate, whose absolute configuration was also confirmed by enantioselective total synthesis.21,23 Isozonarone (42, MIC = 3 ppm) showed >5-fold enhanced toxicity to fish than isozonarol (41),21 while they demonstrated paralleled antifeedant activity against the young abalone Haliotis discus hannai, with electivity index (Ei) values of 0.78 and 0.85, respectively.25 The antioxidant activity of isozonarol (41, EC50 = 71 μM) is much better than that of isozonarone (42, EC50 = 145 μM) in the DPPH radical scavenging test.24
 | ||
| Fig. 4 Typical Δ7,8 drimane (hydro)quinones. | ||
Both of them showed cytotoxicity against the tumor cell lines L-929 (murine fibroblasts), K-562 (human leukemia), and HeLa (human cervix carcinoma).26 Isozonarone (42) also showed good anti-inflammatory activity against 3α-hydroxysteroid dehydrogenase (3α-HSD) by inhibiting ROS production.27 Isozonarol (41) showed algicidal activity toward Heterosigma akashiwo, Chattonella marina, Chattonella antiqua, and Heterocapsa circularisquama, with average mortalities of 77%, 15%, 57% and 100% at 1 μg mL−1 after 4 h.49 The enantiomers ent-isozonarol and ent-isozonarone showed significant antitumor activity against cell lines P-338, A-549, HT-29, and MEL-28, with IC50 values ranging from 0.16 to 3.2 μM, respectively.50 (−)-20-O-acetyl-21-hydroxy-ent-isozonarol (43) was isolated from the genus Dysidea, and its absolute configuration was verified by chemical derivatization. It showed excellent cytotoxicity against the human tumor cell lines MDA-MB-231, A-549, and HT-29, with LC50 values of 11.8, 11.8, and 14.0 μM, respectively.51 (−)-21-Hydroxy-ent-isozonarone (44) was isolated from the New Zealand sponge Dysidea cf. cristagalli, and its absolute configuration was assumed to be the same as that of meroterpenoid 43. It and compound 43 showed anti-inflammatory activity by inhibiting superoxide production. They also showed antiproliferative activity against HL60 cells, with IC50 values of 0.37 and 0.34 μM, respectively.52
(−)-Siphonodictyal C (45) was first isolated from Siphonodictyon coralliphagum,53 and its structure was later revised by Schmitz et al., who also isolated siphonodictyol I (46) from a Micronesian sponge, Aka sp.54 The relative configurations (excluding the 9-positions) were reported. Siphonodictyal C (45) possessed a stronger antibacterial activity against S. aureus and B. subtilis than siphonodictyal B. Beside the inhibition against the marine bacterium Vibrio anguillarum, siphonodictyal C (45) could inhibit CDK4/cyclin D1 complexation, with the IC50 value of 9 μg mL−1.53,54
Peyssonoic acid A (47) with a range of bioactivities was isolated from the crustose red alga Peyssonnelia sp., whose relative configuration was confirmed by NOESY spectra, and further confirmed by total synthesis.32 It has a modest antibacterial effect and also possesses a certain inhibitory effect against the human ovarian cancer cell line (IC50 = 34.5 μM).55 The cytotoxic drimane quinone (+)-isohyatellaquinone (48) was separated from the sponge Dactylospongia elegans, and the absolute configuration was confirmed by enantiospecific synthesis.
The IC50 values of (+)-isohyatellaquinone against breast cancer (BC) and small cell lung cancer (NCI-H187) cells were 6.69 and 11.52 μg mL−1, respectively.33 (−)-Epoxyphomalins A (49), B (50), C (51), D (52), and E (53) were isolated from the marine-derived fungus Phoma sp. and Paraconiothyrium sp. The absolute configurations of (−)-epoxyphomalins A and B were established, employing a combination of CD measurements and NMR analysis. The relative configurations of (−)-epoxyphomalins C, D and E were confirmed by NOESY spectra.56,57 Of a panel of 36 human tumor cell lines, compounds 49, 50, 52 displayed excellent cytotoxicity against 12 cell lines, with the IC50 values between 0.010 and 18.46 μg mL−1.56,57 (−)-Epoxyphomalin A (49) and B (50) were proposed to exert their cytotoxic effect through inhibiting the 20S proteasome.56 The structurally rare mero sesquiterpenoids called (−)-craterellins A–B, and (+)-craterellins C–D (54–57) were isolated from the cultures of basidiomycete Craterellus odoratus.58,59 Their relative configurations were confirmed by ROESY spectrum. The absolute configuration of (+)-craterellin A was determined by the modified Mosher's method and single-crystal X-ray diffraction analysis using Cu Kα.60 Among them, (−)-craterellin A (54) showed significant inhibitory activity against human 11β-HSD2 with an IC50 value of 1.5 μg mL−1.58,59 The compound (−)-smenodiol (58) was firstly isolated from the Seychelles sponge Smenospongia sp. by Faulkner et al., and the configuration was determined by NOEDS measurements.61 (−)-Dactylosponol (59) and (−)-dactylospontriol (60) were isolated from the Dactylospongia elegans, and the absolute configurations were assumed by comparison with the reported compounds related to isozonarol.62
(−)-Yahazunol (61), as a typical example of 8-hydroxy drimane hydroquinones, was first isolated from the brown seaweed Dictyopteris undulata in 1979 (Fig. 5).63 The absolute configuration was confirmed by enantioselective total synthesis.64 It exhibits cytotoxic activity against tumor cell lines HM02, HepG2, and MCF7, with GI50 values of 4.2, 7.1, and 6.0 μg mL−1, respectively.26 It also exerts cell lysis activity against the red tide microalgal species, especially against H. akashiwo, C. marina, C. antiqua, and H. circularisquama at 1 μg mL−1, with the average mortality of 67%, 88%, 97% and 98% at 4 h, respectively.49 Interestingly, the natural enantiomer, (+)-ent-yahazunol (62), was isolated from the genus Dysidea collected in the Gulf of California. It exhibits cytotoxicity against MDA-MB-231 and A-549, with LC50 values of 27.7 and 17.4 μM, respectively.51
 | ||
| Fig. 5 8-Hydroxy, Δ8,9 and Δ9,11 drimane (hydro)quinones. | ||
The drimane hydroquinone containing a phenolic aldehyde fragment, (+)-siphonodictyal A (63), was isolated from the burrowing sponge Siphonodicryon corulliphugum. It was reported to show antibacterial activity against Staphylococcus aureus (S. aureus) and Bacillus subtilis (B. subtilis).65 Its formylated analog (+)-albaconol (64) was isolated from the Basidiomycetes Albatrellus confluens.66 The absolute configuration was elaborated by enantioselective total synthesis in 2008.67 This compound was synthesized and detected to have a certain antifungal activity against S. sclerotiorum, with an EC50 value of 24.35 μM. Meanwhile, it also showed antitumor activity against HepG2 and MCF-7, with EC50 values of 10.44 and 10.08 μM.68 The anticancer regioisomer (+)-neoalbaconol (65) was isolated from the fruiting body of Albatrellus confluens, which could mediate the phosphoinositide-3 kinase (PI3-K)/Akt-hexokinase 2 (HK2) pathway, resulting in energy depletion.69
Li et al. reported a concise synthesis and a brief antifungal SAR exploration of yahazunol-related meroterpenoids, starting from the enantiopure (−)-sclareol, in which (+)-neoalbaconol (65) possesses antifungal activities against S. sclerotiorum and R. solani with EC50 values of 40.38 and 35.16 μM, respectively.68
The drimane-substituted benzoic acid, (+)-dictyvaric acid (66), was isolated from the brown alga Dictyopteris divaricata Okam.70 In 2012, Baran et al. disclosed an enantioselective total synthesis to confirm its absolute configuration.71 Li et al. reported that it could serve as an antifungal hit against S. sclerotiorum and R. solani, with EC50 values of 22.16 and 22.54 μM, respectively.68 A sulfated siphonodictyal-related derivative called siphonodictyal A sulfate (67) was isolated from the marine sponge Aka coralliphaga, which possessed the same relative configuration as siphonodictyal A and displayed 25% inhibition in the DPPH assay at 200 μM.72 (−)-Dysidphenol C (68), with weak antibacterial activity, was isolated from the South China Sea Sponge Dysidea sp. Its absolute configuration was assigned mainly by calculated ECD spectra.73 The cytotoxic (−)-hippomeroterpene B (69) was achieved from the Vietnamese sponge Hippospongia fistulosa, and the relative configuration was confirmed by analysis of the NOESY spectrum. It demonstrated a modest cytotoxic activity against Hep-G2, MCF-7, SK-LI-1 and SK-Mel-2 (10–22% inhibition at 100 μM).74
Recently, the antibacterial drimane hydroquinone (−)-xishaeleganin B (70) was isolated from the Xisha marine sponge Dactylospongia elegans. It was presumed to have the same absolute configuration as ent-yahazunol. It showed significant inhibition against Staphylococcus aureus (MIC = 1.5 μg mL−1), Streptococcus pyogenes (MIC = 1.5 μg mL−1) and Enterococcus faecium (MIC = 3 μg mL−1).75 Drimane hydroquinone (+)-71, with its relative configuration determined by NOESY, was isolated from the marine macroalga Gracilaria salicornia as a potent anti-inflammatory agent against pro-inflammatory enzymes (COX-1, COX-2, and 5-LOX), with corresponding IC50 values of 1.72, 1.56 and 1.90 mM, respectively. It also showed potent antioxidant activities in scavenging DPPH and ABTS, with IC50 values of 1.51 and 1.88 mM, respectively.76
Siphonodictyol H is a member of the natural siphonodictyal family and featured Δ8,9-drimane substituted hydroquinones, whose relative configuration is depicted in 72. The antibacterial activity against S. aureus and B. subtilis is not so significant compared with its analog siphonodictyal B.53 (+)-Wiedendiol A (73) was acquired from the marine sponge Xestoepongia wiedenmayeri as an antiatherosclerosis ingredient, with the IC50 value of 5 μM against cholesteryl ester transfer protein (CETP).77 Its absolute configuration was elucidated through enantioselective total synthesis.78 The analogous metabolite, (−)-zonarenone (74), without assigned stereochemistry was isolated from the brown alga, Dictyopteris undulata by Hirao et al., and exhibited algicidal activity against H. akashiwo, C. marina, C. antiqua, and H. circularisquama, with the average mortality of 58%, 32%, 48% and 100% after 4 h at 1 μg mL−1, respectively.49
Another distinct drimane (hydro)quinone called spongiaquinone (75), featuring a Csp2–Csp2 linker between drimane and quinone segment, was isolated from the marine sponge Stelospongia corzulata.79 The absolute configuration was confirmed by chemical degradation and comparison with a previously assigned structure.28 It holds cytotoxicity against cell lines HM02, HepG2, and MCF7, with GI50 values of 3.1, 3.6, and 2.6 μg mL−1, respectively.26 The GI50 or CC50 values against L-929, K-562, and HeLa cell lines are over 10 μg mL−1. It also showed good anti-inflammatory activity against 3α-hydroxysteroid dehydrogenase (3α-HSD), by inhibiting ROS production in SOZ-stimulated granulocytes.27 (−)-Wiedendiol B (76), isolated from the marine sponge Xestoepongia wiedenmayeri, showed promising antiatherosclerosis activity, with the IC50 value of 5 μM against cholesteryl ester transfer protein (CETP).77 It exhibits cytotoxicity against L-929 (GI50 = 63.1 μM), K-562 (GI50 = 35.8 μM) and HeLa (CC50 = 38.1 μM) cell lines.27 In 1997, Barrero reported on its enantioselective total synthesis and confirmed its absolute configuration.80 (+)-Deoxyspongiaquinol (77) and (+)-deoxyspongiaquinone (78), together with their chloro-analogs (+)-(E)-chlorodeoxyspongiaquinol (79) and (+)-(E)-chlorodeoxyspongiaquinone (80), respectively, were isolated from a southern Australian marine sponge Euryspongia sp.81 The shown structures were further consolidated by the enantioselective total synthesis.82 The antibacterial phenolic aldehyde (−)-siphonodictyal B (81) was isolated from the burrowing sponge Siphonodicryon corulliphugum. Its stereochemistry was revised in 2015 by George et al.65,83 It was demonstrated to be antifungal against Aspergillus fumigatus. Siphonodictyal B (81), siphonodictyal B1 (82), and siphonodictyal B2 (83) were detected to show cytotoxic activity against L929 mouse fibroblasts.84 Siphonodictyal B and 8-epi-siphonodictyal B could inhibit PI3Kα, with the IC50 values of 2.6 and 3.3 μM, respectively.85 The sulfated hydroquinones (−)-siphonodictyals B1 (82), B2 (83), and B3 (84) were isolated from the Caribbean sponge Aka coralliphagum by Köck et al. Their relative configuration were elucidated on the basis of ROESY spectra. Siphonodictyals B1 and B2 were observed to show antibacterial activity against Staphylococcus aureus and antifungal performance against Botrytis cinerea. Siphonodictyal B2 showed more promising DPPH radical-scavenging activity, with a scavenging rate of 88.8% at 200 μM.72,84
2.3 Benzopyran-fused-naphthane-type drimane (hydro)quinones
(−)-Chromazonarol (85) was isolated from the brown seaweed Dictyopteris undulata (zonarioides) and structurally featured a benzopyran-fused-naphthane scaffold (Fig. 6).86 Its enantiomer (+)-ent-chromazonarol (86) was isolated from the sponge Disidea pallescens as a non-crystalline gum.87 Interestingly, another natural isomeric chroman-sesquiterpenoid called (−)-8-epi-chromazonarol (87) was also isolated.88 The absolute configurations of the pair of enantiomers were confirmed by enantioselective total synthesis.50,89 These stereoisomers also differentiate from each other in the biological profiles. Chromazonarol was reported to be toxic to fish with a MIC value of 17 ppm.21 It exhibits potent antifeedant activity against the young abalone Haliotis discus hannai with an electivity index (Ei) value of 0.8.25 Promising algicidal activity at 1 μg mL−1 was also observed against Heterosigma akashiwo, Chattonella antiqua and Heterocapsa circularisquama, with an average mortality of 78%, 42%, 93% after 4 h.49 Meanwhile, moderate antioxidant activity was detected in DPPH radical scavenging activity, with the corresponding EC50 value of 121 μM.24 (+)-ent-Chromazonarol (86) was detected to show antitumor activity against cell lines P-338, A-549, HT-29, and MEL-28, with the IC50 values of 15.91, 15.91, 15.91 and 15.91 μM, respectively.50 The IC50 values against BAEC, A549, H116, PSN1, and SKBR3 are 45, 14, 14, 15, and 13 μM, respectively.90 Anti-inflammatory (+)-cyclospongiaquinone-1 (88) and (−)-dehydrocyclospongiaquinone-1 (89) were isolated from the marine sponge Stelospongia conulata, which have distinct appearances due to an additional double bond conjugated with the quinone ring. The common characteristic is the presence of the methoxylated quinone skeleton.79,91 The absolute configurations were confirmed in 2017 through enantioselective total synthesis.92 The oxidized chromazonarol derivative, (+)-puupehenone (90), has drawn increasing attention in research ever since its isolation as a sponge metabolite in 1979. The absolute configuration was confirmed by X-ray data. It exerts a wide range of bioactivities,93–98 including antibacterial, antifungal, insecticidal, antitumor, antioxidant, and anti-feedant activities. For example, the antifungal MIC values against Candida albicans and Trichophyton mentagrophytes are 3.1 and 1.6 μg mL−1, respectively.93 It exhibited potent antifungal activity against C. neoformans ATCC 90113, and C. krusei ATCC 6258, with the IC50 values of 0.38 and 1.49 μg mL−1, respectively.99 The MIC values against Trichomonas vaginalis and M. tuberculosis (H37Rv) are 3.1 μg mL−1 and 12.5 μg mL−1, respectively.93,100 The antitumor effect against A549, MCF-7, HT-29, and P338 cell lines is noteworthy, with IC50 values of 0.1–1.3 μg mL−1.94,95 The potent mechanisms of action of puupehenone (90) was either experimentally or virtually investigated. The antitumor activities were realized through the interaction or modulation with HIF-2α in the context of renal cell carcinoma.101 Glycogen synthase kinase-3 beta (GSK-3β) was speculated to be a possible kinase target by virtual screening molecular docking.102 The antifungal ability was executed by disrupting the Hsp90 activity and the cell wall integrity pathway.103 Puupehenone (90) exhibited a much better inhibitory effect against 15-LOX (IC50 = 0.76 μM) over that of 12-LOX (IC50 = 8.3 μM). It was selected as a potent inhibitor of NOX104 and 15-lipoxygenase,105 with the IC50 values of 1.3 μM and 0.68 μM, respectively. It could serve as the most potent inhibitor of NADH oxidase.106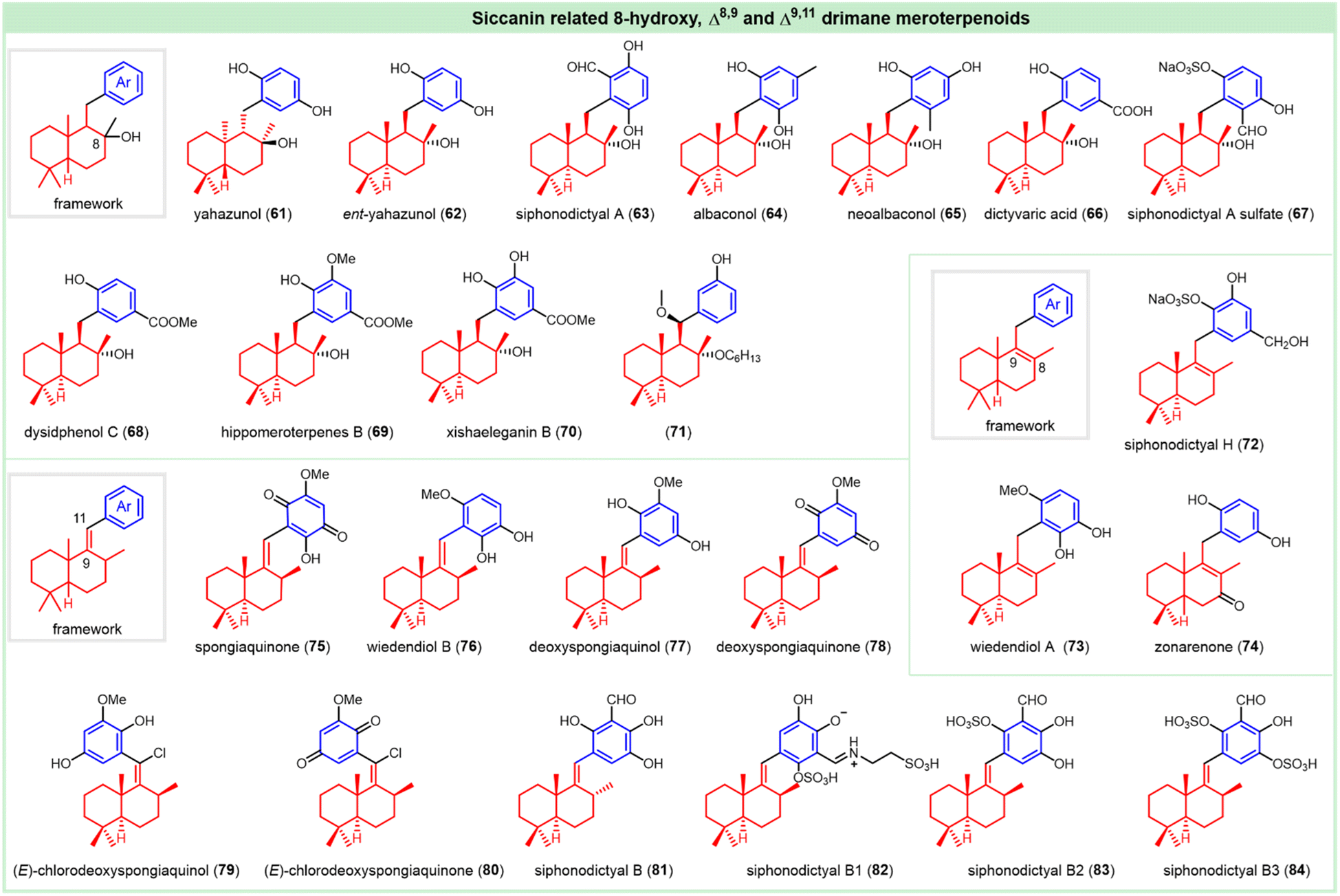 | ||
| Fig. 6 Benzopyran-fused-naphthane-type drimane (hydro)quinones. | ||
Besides the innate versatile biological potentials of puupehenone itself, structurally-related compounds could facilitate the discovery science of pharmaceutically important entities due to a wide series of bioactivities, including antiangiogenic, antitumoral, antioxidant, antimicrobial, and immunomodulatory profiles.104 The semisynthetic puupehediol, 8-epi-puupehedione, 8-epi-puupehediol, 8-epi-9,11-dihydro-puupehedione, and 8-epi-9,11-dehydro-puupehediol showed anti-endothelial activities in vitro against the BAEC, A549, H116, PSN1, SKBR3 cell lines.90
It was demonstrated that puupehenone (90) and its related analogs could effectively inhibit capillary tube formation.90 The synthetic epimer of puupehedione, 8-epipuupehedione, could be an attractive model for treating leukemia through modulating proliferation, survival, and extra-cellular matrix re-modeling. It was demonstrated to strongly inhibit matrix metalloproteinase-2 and urokinase.107 Three other puupehenone analogs, called (+)-cyanopuupehenone (91), (+)-21-chloropuupehenone (92), and (+)-puupehedione (93), were isolated from the Verongid sponge. Their relative configurations were confirmed by HMBC and NOE experiments. The absolute configurations of compounds 92 and 93 were further confirmed via enantioselective synthesis.108,109 They showed cytotoxicity, antiviral, immunomodulatory and antifungal activities.99,109,110 Noteworthily, 21-chloropuupehenone could significantly inhibit 12-LOX (IC50 = 0.7 μM)104 and outperformed puupehenone (90) in antiatherosclerosis, with an IC50 value of 0.3 μM against cholesteryl ester transfer protein (CETP).77 15α-Cyanopuupehenol (94) was isolated from the marine sponge order Verongida and possessed a wide range of bioactivities, including antiviral potential and antifungal activity, together with cytotoxicity. Noteworthily, it led to 98% reduction of Herpes simplex II virus at 5 μg mL−1 (ref. 111) and effectively inhibited M. tuberculosis (H37Rv) at 12.5 μg mL−1. (+)-21-Chloropuupehenol (95) and (−)-15-oxopuupehenol (96) were isolated from a species of O'ahu sponges. The absolute configurations were elaborated by enantioselective total synthesis starting from enantiopure drimane equivalents.108,112,113 Compound 96 showed antimalarial activity against Plasmodium falciparum clones D6W2, with the IC50 values of 2.0 and 1.3 μg mL−1, respectively.112 (−)-15α-Methoxypuupehenol (97), with its relative configuration confirmed by NOESY spectra, was isolated from the marine sponge Hyrtios species. It was reported to show excellent antimicrobial and antifungal activities, with a much lower cytotoxicity than puupehenone (90) against KB cells. It was also effective against both chloroquine-susceptible and resistant P. falciparum strains.114 Its antitumor effects were associated with the modulation of Stat3, CyclinB1, Alk, ezrin, merlin, and Erk1/2 functions.115 Puupehenone analogs, 98–100, were isolated from an Indonesian sponge extract of a Hyrtios sp., which were demonstrated to show LO inhibition activity.
Their structures were consolidated by both complete sets of spectra and the comparison with previous properties.116
The sesquiterpene–dihydroquinone, (−)-puupehanol (101), was isolated from the marine sponge Hyrtios sp. The absolute configuration was addressed by experimentally and theoretically calculated ECD.99 (+)-Puupehenol (102) was isolated as an antioxidant from a Hawaiian deep-water Dactylospongia sp. sponge, and it also showed antimicrobial properties against both Staphylococcus aureus and Bacillus cereus. The relative configuration of (+)-puupehenol was confirmed by gNOESY.117 A puupehenol derivative called (+)–(5S, 8S, 10S)-19-methoxy-9,15-ene-puupehenol (103) was acquired from the Australian sponge Hyrtios digitatus, whose absolute configuration was supported by the ECD spectrum. This compound is active against scavenger receptor-class B Type 1 HepG2 (SR-B1 HepG2) with the EC50 value of 1.78 μM.118 (−)-BE-40644 (104), from Actinoplanes sp. A40644, acts as a strong selective inhibitor against thioredoxin (TRX) systems (IC50 0.12 μg mL−1 for E. coli, IC50 0.8 μg mL−1 for human). Its absolute configuration was confirmed by enantioselective total synthesis in 2009.15 It could be considered not only as a tool to investigate the biological significance of the TRX system, but also as a candidate to be developed as an anticancer agent.119,120
Kampanol C (relative configuration shown as 105) was isolated as an inhibitor of farnesyl-protein transferase (FPT) from a fungal culture of Stachybotrys kampalensis Hans.121 (+)-Hongoquercins A and B (106, 107) were acquired from the Staphylococcus aureus, showing moderate to significant antibacterial activities against a wide range of bacteria with MIC values of 2–16 μg mL−1. Their relative stereochemistries were supported by NOESY spectra,122 and the absolute configuration of compound 106 was confirmed by enantioselective total synthesis.123 (+)-UPA0043 (108) and (+)-UPA0044 (109),124 with the absolute configurations determined by enantioselective total synthesis, exhibited significant cytotoxic and antifungal activities. The tetracyclic analog (−)-phomoarcherin C (110) was isolated from Phomopsis archeri,125 and its absolute configuration was confirmed through enantioselective total synthesis in 2019.126
Meroterpenoids of the austalide family were characterized by linking the drimane subunit with the isobenzofuran-1-one skeleton (Fig. 7). Austalides K and L (111, 112) were isolated from Aspergillus ustus.127–129 Austalides V and W (113–114) were obtained from the culture of the marine fungus Penicillium rudallense. (−)-17S-Dihydroaustalide K (115) was achieved from the alga-derived fungi Penicillium thomii Maire and Penicillium lividum Westling. The absolute configurations were established by ECD spectra, together with other spectra comparison.130,131 Compounds 112–115 showed potent osteoclast differentiation inhibitory activity with ED50 values ranging from 2.0 μM to 2.5 μM.130 Farnesyl-protein transferase (FPTase) is a critical enzyme that participates in the post-translational modification of the Ras protein.
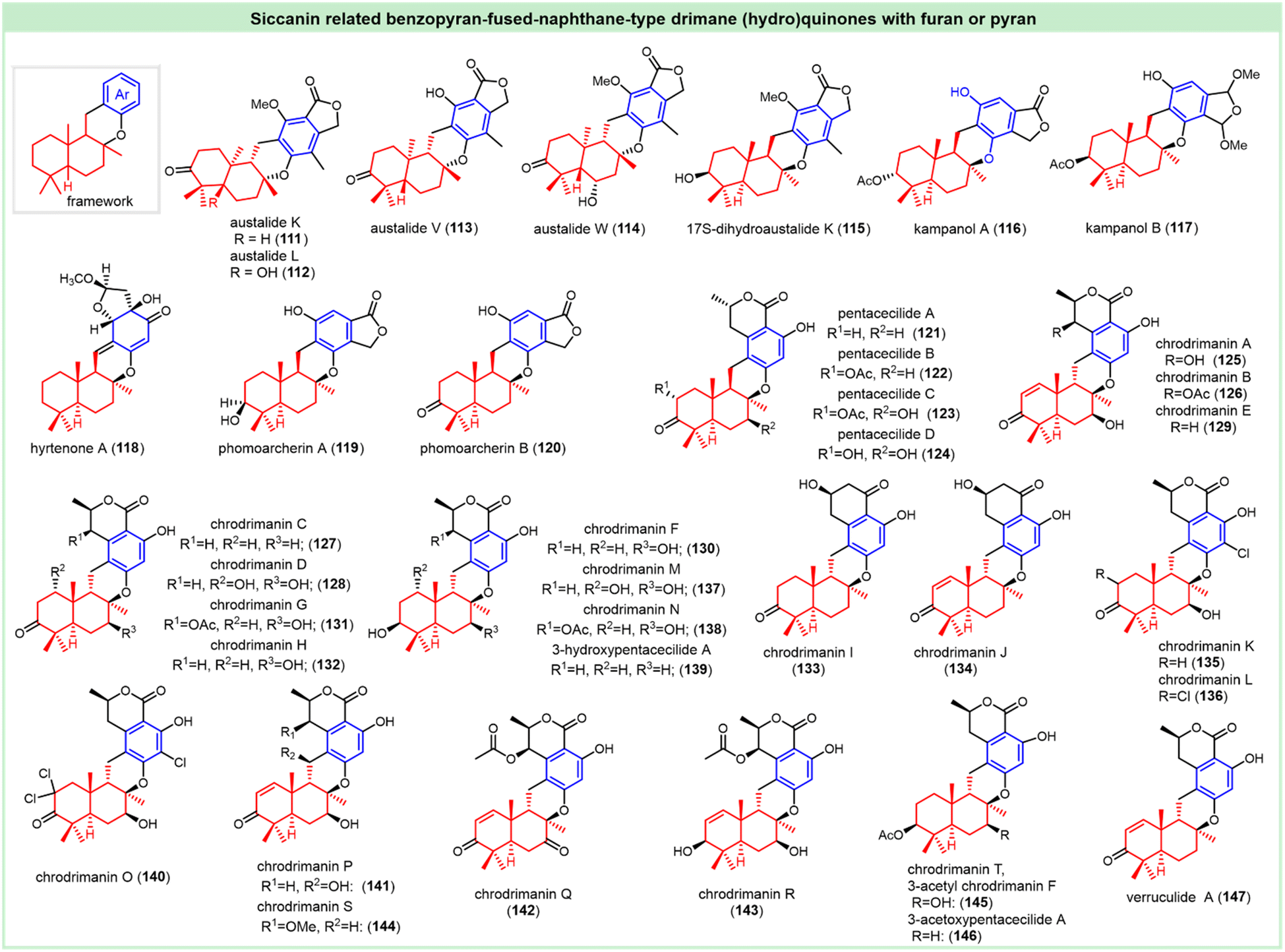 | ||
| Fig. 7 Benzopyran-fused-naphthane drimane (hydro)quinones with furan or pyran. | ||
Kampanols A–B (116 and 117), from a fungal culture Stachybotrys kampalensis Hans, performed as FPT inhibitors for the discovery of anticancer leads. The relative configurations were deduced from NOEDS experiments.121 The IC50 values against Ras rHFPTase of kampanols A and B were 13 and 7 μM, respectively, demonstrating much better potentials than the analog kampanol C. Kampanol A was also cytotoxic against KKU-M213, with an IC50 value of 19.6 μg mL−1.125 (−)-Hyrtenone A (118) was isolated from a marine sponge and its absolute configuration was confirmed by X-ray. It could inhibit 15-HLO, 15-SLO, and 12-HLO, with IC50 values of 59, 39 and 31 μM, respectively.132
The pentacyclic meroterpenoids (−)-phomoarcherins A–B (119–120) were isolated from Phomopsis archeri. The absolute configurations were determined by X-ray crystallographic analysis of the p-bromobenzoate derivative of compound 119. Despite their similar structural features, these two meroterpenoids showed distinct inhibitory effects against five cholangiocarcinoma cell lines (KKU-100, KKU-M139, KKU-M156, KKU-M213 and KKU-M214). Phomoarcherin A (119) could selectively inhibit KKU-M213 (IC50 = 16.6 μg mL−1), while the oxidative phomoarcherin B (120) demonstrated promising effects against the other four cell lines with the IC50 values ranging from 0.1 to 8 μg mL−1. Phomoarcherin B (120) also showed excellent antimalarial activity against Plasmodium falciparum, with an IC50 value of 0.79 μg mL−1.125
Pentacecilides A to D (121–124) were obtained from the fermentation broth of Penicillium cecidicola FKI-3765-1,133,134 and their absolute configurations were elucidated via the modified Mosher method.134 Compounds 121 and 122 could inhibit the synthesis of cholesteryl ester in different cell lines (macrophages, and ACAT1- and ACAT2-CHO cells) with IC50 values of 0.69 to 10.8 μM without any cytotoxic effect, which may be due to the inhibition of acyl-CoA: cholesterol acyltransferase.133,134
The chrodrimanins family of natural meroterpenoids (125–132) was isolated from the strain YO-2 of Talaromyces sp.135,136 The absolute configuration of chrodrimanin B (126) was confirmed by Mosher's method and the configurations of other chrodrimanins congers were deduced. Chrodrimanin B (126) outperformed the other analogs in the insecticidal evaluation against the third instar larva of the silkworm, with a LD50 value of 10 μg g−1.135 Matsuda et al. found that the insecticidal mechanism of chrodrimanin B (126) is a potent, non-open-channel-blocking antagonist on B. mori RDL, with an IC50 of 1.13 nM. It exhibited competitive actions at low concentrations and non-competitive actions at higher concentrations. The much weaker blocking action on human α1β2γ2 GABAR compared to RDL was used to rationalize the design of safer insecticides based on chrodrimanin B (126).137 Chrodrimanin A (125), E (129), and F (130) showed inhibitory activities against influenza virus A (H1N1), with IC50 values of 21, 55, and 57 μM, respectively.138 Chrodrimanin A (125) and H (132) could inhibit the activity of protein tyrosine phosphatase 1B (PTP1B) with IC50 values of 8.5 and 14.9 μM.139
(−)-Chrodrimanins I and J (133 and 134), with a unique cyclohexanone instead of the δ-lactone ring of chrodrimanin-related metabolites, were isolated from the Antarctic moss-derived fungus Penicillium funiculosum GWT2-24. The absolute configurations were unambiguously determined by X-ray crystallographic analysis using Cu Kα radiation.138 (−)-Chrodrimanins K–N (135–138) and a previously semisynthetic 3-hydroxypentacecilide A (139) were isolated from the fungus Penicillium sp. SCS-KFD09. The absolute configurations were determined by a single-crystal X-ray diffraction or synthesis from the assigned structure. Compounds 135, 138, and 139 displayed anti-H1N1 activity with IC50 values of 74 and 58 and 34 μM, respectively.140 Chrodrimanins O–T (140–145) were obtained from marine worms. Recently, some ester derivatives of chrodrimanin, including 3-acetyl (−)-chrodrimanin F (145, chrodrimanin T) and (−)-3-acetoxypentacecilide A (146) were separated from the DES mutant of the marine-derived fungus Penicillium chrysogenum S-3–25.141 The absolute configurations were elucidated by spectroscopic data and ECD analysis.141–143 Chrodrimanin O (140) represents the first example of an unusual trichlorinated meroterpenoid with a unique dichlorine functionality. Chrodrimanins G (131), O (140), R (143), and S (144) inhibited protein tyrosine phosphatase 1B (PTP1B) with IC50 values of 39.6, 71.6, 62.5, and 63.1 μM, respectively.142 Compounds 130 and 145 showed a stronger inhibitory effect on HL-60 cells with IC50 values of 8.7 and 8.1 μM, respectively.141 (−)-Verruculide A (147), as the dehydroxyl chrodrimanin E, was isolated from the culture broth of the Indonesian ascidian-derived Penicillium verruculosum TPU1311. Its structure was confirmed by CD spectra and NOE difference experiments. It was reported to inhibit the activity of protein tyrosine phosphatase 1B (PTP1B) with an IC50 value of 8.4 μM.139
2.4 Benzofuran-spiro-naphthane-type drimane (hydro)quinones
This subclass of drimane (hydro)quinones features a benzofuran-spiro-naphthane framework (Fig. 8). A typical example is (+)-isochromazonarol (148), which was isolated from the sponge Dictyopteris undulata. Though it is related to zonarol and chromazonarol, the stereochemistry of this compound was undefined. It showed algicidal activity against H. akashiwo, C. antiqua, and H. circularisquama at 1 μg mL−1, with an average mortality of 83%, 37% and 100%, respectively.49 (+)-Cyclospongiaquinone-2, as the cyclized derivative of spongiaquinone, was isolated from the marine sponge Stelospongia conulata. The configuration described as compound 149 was confirmed by X-ray data.79 Another three analogs called (+)-7,8-dehydrocyclospongiaquinone-2 (150), (−)-9-epi-7,8-dehydrocyclospongiaquinone-2 (151) and (+)-cyclospongiacatechol (152) were isolated from the sponge Dactylospongia elegans, and their relative configurations were mainly confirmed by NOE correlations.33,144 Compounds 150 and 151 did not exhibit any obvious cytotoxic activities against breast cancer and small lung cancer (NCI-H187) cells.33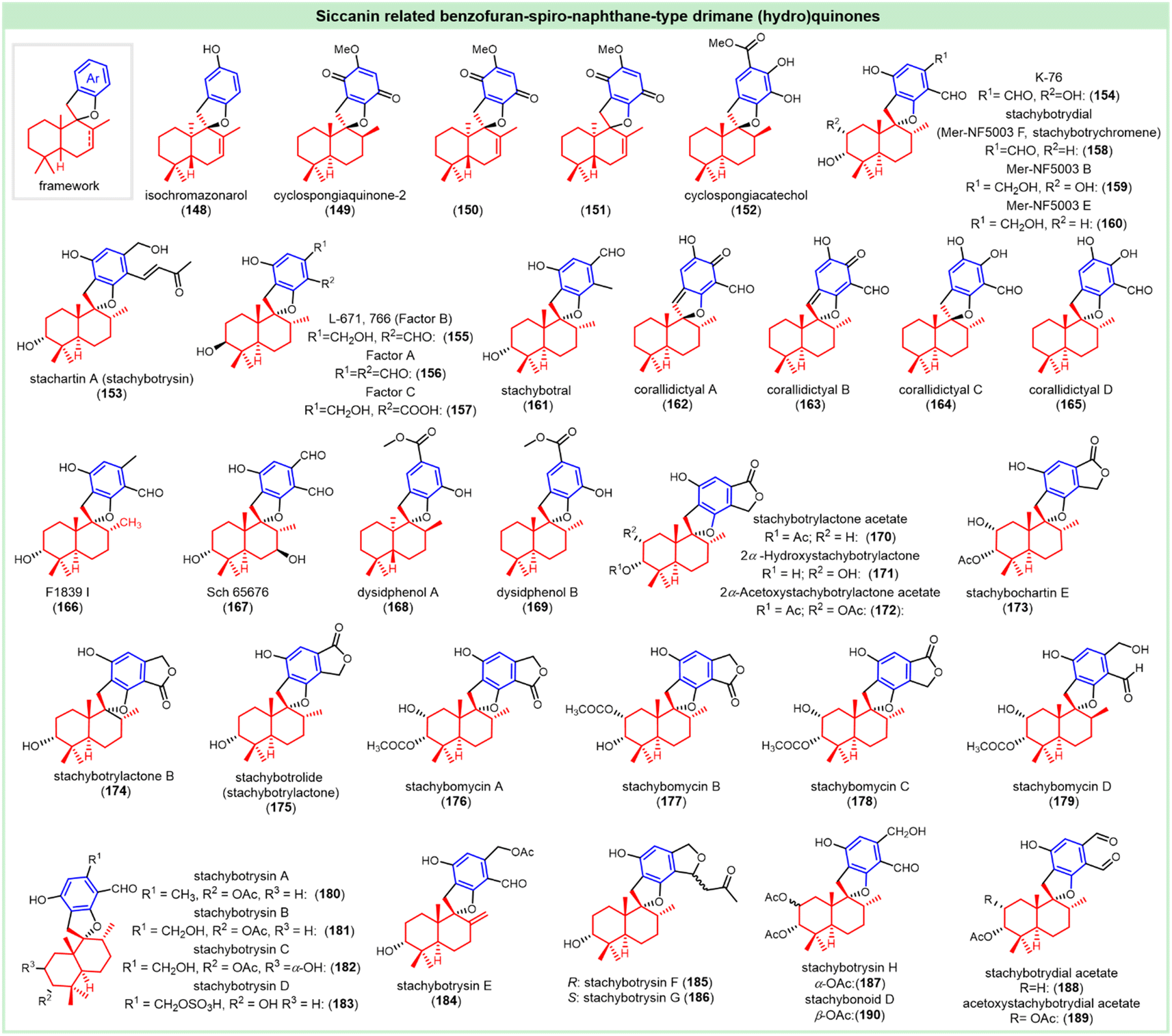 | ||
| Fig. 8 Benzofuran-spiro-naphthane-type drimane (hydro)quinones. | ||
A phenylspirodrimane-type analog named (−)-stachartin A (153) was isolated from the culture of Stachybotrys chartarum.145 It holds the same structure as stachybotrysin (153), whose absolute structure was proposed by single-crystal X-ray structure analysis. It exhibited an inhibitory effect on osteoclast differentiation via suppressing the RANKL-induced activation of p-ERK, p-JNK, p-p38, c-Fos, and NFATc1.146 The complement inhibitor (−)-K-76 (154) was obtained from the Stachybotrys complementi nov. sp. The absolute configuration was established by CD spectra of its monobenzoate and dibenzoate derivatives. It has been shown to improve the symptoms of experimental glomerulonephritis,147,148 and inhibit pancreatic cholesterol esterase, with an IC50 value of 0.2 mM.149 An inositol mono-phosphatase inhibitor L-671, 776 (155, factor B), together with two other analogs factors A and C (156, 157), were isolated from the culture of the hyphomycete, Memnoniella echinata ATCC 20928. The structures were elucidated by comparison with that of K-76. The IC50 values of L-671, 776 (155) against Myo-inositol mono-phosphatase, Myo-inositol 1,4,5-triphosphatase 3-kinase and IMPase, were 0.4 mM, 3 mM and 0.46 mM, respectively.150,151 Factors A and C (156, 157) could inhibit IMPase with IC50 values of 70 and 200 μM, respectively.151 A K-76 related fungal metabolite (from Stachybotrys cylindrospora), stachybotrydial, with the relative configuration established as compound 158,152,153 was detected to show antibacterial activity against Staphylococcus aureus, with a MIC value of 32 μg mL−1.154 It turned out to be potent inhibitors of pancreatic cholesterol esterase and human protein kinase CK2, with IC50 values of 60 μM and 4.43 μM, respectively.149,155 The analogs, Mer-NF5003 B, E (159, 160)156 and stachybotral (161)157 were isolated from the culture broth of Stachybotrys fungi. The relative configurations of Mer-NF5003 B, E were confirmed by NOE correlation,156 and compound 160 showed weak cytotoxicity against three human cancer cell lines K562, Hela and HL60.158
Corallidictyal A (162) and corallidictyal B (163) were obtained as a mixture with the IC50 value of 28 μM against protein kinase C. These spirosesquiterpene aldehydes were shown to inhibit cultured vero cells (IC50 = 1 μM),159 and showed antifungal activity against Hansenula anomala, Aspergillus fumigatus, Botrytis cinerea, and Pythium debaryanum.84 The sulfated products corallidictyal C (164) and corallidictyal D (165) were isolated from the Caribbean sponge Aka coralliphagum, which exhibited antibacterial, antifungal, and cytotoxic activities. Besides the detailed elucidation of relative configurations (162–165),84 the absolute configurations of (−)-corallidictyals B and D were elucidated via enantiospecific synthesis.160 F1839-I (166) was isolated from cultures of Stachybotrys sp. F-1839, which showed a certain inhibitory effect against pancreatic cholesterol esterase with an IC50 value of 0.27 mM.149 Its absolute configuration was confirmed by enantiospecific total synthesis.160 Besides the anti-HIV activity, it was also active against the NCI-H460, BGC823, Daoy, and HepG2 cell lines, with IC50 values of 15.8, 21.9, 41.5, and 18.4 μM, respectively.161 (−)-Sch 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 676, with the relative configuration shown in compound 167, represented the first CMV protease inhibitor in this class, with an IC50 value of 9.8 μg mL−1.162 The sesquiterpenoid phenols called (+)-dysidphenol A (168) and (−)-dysidphenol B (169) were isolated from Dysidea sp., showing antibacterial activity against Escherichia coli, Bacillus subtilis, and Staphylococcus aureus. The absolute configurations were determined by ECD spectra.73 The stachybotrylactone-related metabolites, including (−)-stachybotrylactone acetate (170), (−)-2α-hydroxystachybotrylactone (171) and (−)-2α-acetoxystachybotrylactone acetate (172), were obtained from the fungus Stachybotrys chartarum.163 Compound 171 was determined by X-ray, and the others isolated from the same source were assigned by spectral comparison. This endophytic fungus obtained from Pinellia ternate also produced (−)-stachybochartin E (173),164 whose absolute configuration was elucidated through ECD calculations and the modified Mosher's method. Stachybotrylactone (175),165 with the same scaffold of previously reported stachybotrolide,166 was isolated from the crinoid-derived fungus Stachybotrys chartarum and confirmed by single crystal X-ray diffraction (Cu Kα radiation).66 Stachybotrylactone B (174) was acquired from the culture of Stachybotrys sp. KCB13F013, and its configuration was determined mainly by the modified Mosher's method.146 Recently, (−)-stachybomycins A–C (176–178) together with the reduced analog (−)-stachybomycin D (179) were also acquired from Stachybotrys sp. SCSIO 40434. The absolute configurations were determined by X-ray diffraction (compound 176), ECD spectrum (177 and 178), or spectral comparison (179) with a known compound.167 Compound 170 showed antiviral activity against the influenza A virus with an IC50 value of 18.9 μM.161 Compound 175 showed antibacterial activity against Staphylococcus aureus DHFR, Staphylococcus aureus and Methicillin-resistant Staphylococcus aureus CCARM3167, with corresponding IC50 values of 41.5 μM, 32 μg mL−1 and 32 μg mL−1, respectively.154
676, with the relative configuration shown in compound 167, represented the first CMV protease inhibitor in this class, with an IC50 value of 9.8 μg mL−1.162 The sesquiterpenoid phenols called (+)-dysidphenol A (168) and (−)-dysidphenol B (169) were isolated from Dysidea sp., showing antibacterial activity against Escherichia coli, Bacillus subtilis, and Staphylococcus aureus. The absolute configurations were determined by ECD spectra.73 The stachybotrylactone-related metabolites, including (−)-stachybotrylactone acetate (170), (−)-2α-hydroxystachybotrylactone (171) and (−)-2α-acetoxystachybotrylactone acetate (172), were obtained from the fungus Stachybotrys chartarum.163 Compound 171 was determined by X-ray, and the others isolated from the same source were assigned by spectral comparison. This endophytic fungus obtained from Pinellia ternate also produced (−)-stachybochartin E (173),164 whose absolute configuration was elucidated through ECD calculations and the modified Mosher's method. Stachybotrylactone (175),165 with the same scaffold of previously reported stachybotrolide,166 was isolated from the crinoid-derived fungus Stachybotrys chartarum and confirmed by single crystal X-ray diffraction (Cu Kα radiation).66 Stachybotrylactone B (174) was acquired from the culture of Stachybotrys sp. KCB13F013, and its configuration was determined mainly by the modified Mosher's method.146 Recently, (−)-stachybomycins A–C (176–178) together with the reduced analog (−)-stachybomycin D (179) were also acquired from Stachybotrys sp. SCSIO 40434. The absolute configurations were determined by X-ray diffraction (compound 176), ECD spectrum (177 and 178), or spectral comparison (179) with a known compound.167 Compound 170 showed antiviral activity against the influenza A virus with an IC50 value of 18.9 μM.161 Compound 175 showed antibacterial activity against Staphylococcus aureus DHFR, Staphylococcus aureus and Methicillin-resistant Staphylococcus aureus CCARM3167, with corresponding IC50 values of 41.5 μM, 32 μg mL−1 and 32 μg mL−1, respectively.154
Phenylspirodrimane derivatives called stachybotrysins A–G (180–186) were isolated from the fungus Stachybotrys chartarum, showing antiviral activity against the HIV-1 virus and influenza A virus (IAV). The absolute configurations were established on the basis of ECD, the modified Mosher's method, or single-crystal X-ray analysis.161,165 Stachybotrysins A–C, and G were weakly active against the cell lines HepG2, K562, HeLa, and HL60.158,161 Stachybotrysin B turned out to be a potent inhibitor of human protein kinase CK2, with an IC50 value of 13.42 μM.155 Both stachybotrysin C and stachybotrylactone exhibited moderate anti-inflammatory activity by inhibiting the production of nitric oxide (NO) in RAW264.7 cells, with the IC50 value of 27.2 μM.165 (−)-Stachybotrysin H, with the relative configuration shown in compound 187, was reported to show weak cytotoxicity against three human cancer cell lines K562, Hela and HL60.158 The acetate derivatives including stachybotrydial acetate (188) and acetoxystachybotrydial acetate (189) turned out to be potent inhibitors of human protein kinase CK2, with IC50 values of 0.69 μM and 1.86 μM, respectively.155 The compound (−)-stachybonoid D (190) was isolated from the crinoid-derived fungus Stachybotrys chartarum 952, whose absolute configuration was mainly confirmed by NOE correlation and ECD spectrum.165
2.5 Indane-fused-naphthane-type drimane (hydro)quinones
(−)-Pelorol (191)168 is an exemplar natural product belonging to the indane-fused-naphthane-type drimane meroterpenoids, which was isolated from the sponge Petrosaspongia metachromia in 2000 (Fig. 9). The absolute configuration was confirmed by enantioselective total synthesis. It is toxic to brine shrimp with LC50 values of between 5 and 10 μg mL−1. It was detected to be anti-inflammatory via modulating the inositol-5-phosphatase SHIP.169 The possibility as a promising anti-melanoma model was consolidated by exerting cytotoxicity and inducing changes in miRNA expression and their downstream effectors.170 It was also reported to act as an inhibitor against PI3Kβ with an IC50 value of 38.17 μM.171 The methylated pelorol (pelorol A, 192) was isolated from the marine sponge Spongia pertusa Esper, and the absolute configuration was proposed to be the same as that of pelorol by spectral comparison.172 Two reduced analogs, (−)-akaol A (193) and (−)-akaol B (194), were acquired from a marine sponge of the genus Aka sp. After the elaboration of relative configurations,54 the absolute configuration of (−)-akaol A was determined by enantioselective total synthesis.173 (−)-19-O-Methylpelorol, with the absolute configuration shown in compound 195 by ECD spectra, was obtained from Dactylospongia sp. as an anti-inflammatory and cytotoxic agent with the IC50 values of between 5.1 and 9.2 μM against the given models.174 | ||
| Fig. 9 Indane-fused-naphthane-type drimane (hydro)quinones. | ||
Tetracyclic meroterpenoids (−)-dasyscyphins A–C (196–198) were isolated from the fermentation broth of the ascomycete Dasyscyphus niveus, and the relative configurations were depicted by NOESY spectra. The attached cytotoxicity demonstrated the important role of the unsaturated segment of (hydro)quinone, in which dasyscyphins B and C are active against several human cell lines and display moderate antimicrobial activities.175 (+)-Dasyscyphins D and E (199, 200) were isolated from Dasyscyphus niveus, and both inhibited the germination of conidia of Magnaporthe grisea at 25 μg mL−1. The relative configurations were confirmed by NOESY spectra, and the relative configuration of dasyscyphin D was also determined by X-ray crystallography.176 (+)-Dasyscyphins F and G (201 and 202) were obtained from Stictidaceae (Ostropales, Ascomycota). The absolute configurations were established by ECD spectroscopy.177 It is speculated that the quinol in dasyscyphin B (197) might account for the promising activity.175 Dasyscyphin C (198) showed antibacterial activity against MRSA, Pseudomonas aeruginosa, and Bacillus anthracis, with IC50 values of 16, 63, and 2 μg mL−1, respectively. It also exhibited anticancer activities against human cancer cell lines MDA-MB-435, MDA-MB-231, and OVCAR3, with IC50 values of 14.4, 12.2, and 10.4 μM, respectively.177 Dasyscyphins C (198) and F (201) showed anticancer activities against lines MDA-MB-435, MDA-MB-231, and OVCAR3 with IC50 values ranging from 4 to 16 μM. Dasyscyphin C is also promising in the discovery of antimicrobial leads, with the MIC value of 2 μg mL−1 against B. anthracis.177 The sulfated analogue, (−)-akadisulfate A, with the relative configuration shown in compound 203, was isolated from Aka coralliphaga, showing modest antioxidative activity.72
Characterized by an appendant alkyne unit, (+)-walsucochin A (204) and (−)-walsucochin B (205) were isolated from Walsura cochinchinensis. The absolute configurations were assigned by ROESY experiment and CD spectra. They were demonstrated to show significant cell-protecting activities against H2O2-induced cell damage.178 Walsucochinoids C–N (206–217)179 were isolated from the twigs and leaves of Walsura cochinchinensis. The relative configurations were confirmed by ROESY data, and walsucochinoid C was assigned absolute configuration (206) by X-ray crystallography. Walsucochinoids D and E were mild inhibitors of mouse and human 11β-HSD1 with the IC50 values of 13.4 μM and 8.25 μM, respectively.179
2.6 Other drimane (hydro)quinones
(−)-Cyclozonarone (218) was isolated from the alga Dictyopteris undulata as a typical example of the naphthane-fused-naphthane-type drimane (hydro)quinones (Fig. 10). The absolute configuration was established by comparison of its spectroscopic data and optical rotation with those of (+)-cyclozonarone.17 It revealed potent antifeedant activity against the young abalone Haliotis discus hannai with an electivity index (Ei) value of 0.93.17,25 (±)-Cyclozonarone showed certain cytotoxicity.26 Two cyclozonarone analogs, (+)-(5S,10S)-4′-hydroxymethylcyclozonarone (219) and phyllospinarone (220), were obtained from Phyllosticta spinarum. The absolute configuration of compound 219 was confirmed by comparison with that of (+)-cyclozonarone, and compound 220 with an ambiguous tertiary alcohol was assumed as an intermediate in the biosynthesis of compound 219.17 Isolated from the deep-water sponge Neopetrosia cf. proxima, (+)-neopetrosiquinone A (221) outperformed (−)-neopetrosiquinone B (222) in inhibiting both the DLD-1 human colorectal adenocarcinoma cell line (IC50 3.7 vs. 9.8 μM) and the PANC-1 human pancreatic carcinoma cell line (IC50 6.1 vs. 13.8 μM). The IC50 value of neopetrosiquinone A against the AsPC-1 human pancreatic carcinoma cell line is 6.1 μM.180 The absolute configurations were then confirmed by enantioselective total synthesis. Neopetrosiquinone B was reported to be antiproliferative against A-549, MCF-7, and T-48 cell lines, with IC50 values of 8.3, 7.7, and 11.5 μM, respectively.181 | ||
| Fig. 10 Other siccanin-related meroterpenoids. | ||
Characterized by an oxygenated 7-membered oxepane functionality between drimane and benzene ring, (+)-bis(sulfato)-cyclosiphonodictyol A (223) and (+)-cyclosiphonodictyol A (224) were isolated from the sponge Sipbonodictyon coralliphagum and Aka coralliphagum, respectively.182 The absolute configurations were elucidated through enantioselective total synthesis in 2020.183 The former one possessed otherwise seldom two aromatic sulfate functionalities, and could inhibit the binding of [3H]-LTB4 to intact human neutrophils with an IC50 value of 44 μM.184 Cyclosiphonodictyol A (224) showed moderate activity against Gram-positive bacteria, including Staphylococcus aureus (MRSA), Staphylococcus aureus (MSSA), and Micrococcus luteus, with the corresponding IC50 values of 117, 117 and 58 μM, respectively.182 (−)-Dictyoceratidaquinone, presumed as compound 225 without the stereochemistry disclosed, and featuring a cyclopropane fragment fused to drimane, was isolated from a dictyoceratidan marine sponge Spongia sp.185
2.7 Categorized activities of siccanin-related drimane meroterpenoids
Natural products have provided remarkable opportunities for lead innovation since ancient times. The structural diversity of siccanin-related drimane meroterpenoids contributes to their wide range of activities, which have proved to play versatile and important roles in the discovery of new chemotypes of pharmaceutical importance. An eminent example is the promising antifungal ingredient siccanin. The aforementioned sections were arranged according to the key scaffold of the natural products (chemical structure oriented), and this section will be briefly focused on the biological importance (bioactivity oriented), which will be valuable for scientists who are interested in achieving or modulating specific activities or targets with distinct chemotypes. Only the natural products with judgable or (semi)quantitative data were provided in the following sections (See ESI‡ for more details).| Compounds | Activity index | Biological species | Ref. |
|---|---|---|---|
| Siccanin (1) | MIC = 0.1 μg mL−1 | T. interdigitale, T. asteroids | 3 |
| MIC = 5.0 μg mL−1 | G. fujikuroi, M. bataticola | 3 | |
| Macrophorin A (16) | IC50 = 6 μg per disc | B. berengeriana, G. fujikuroi | 42 |
| Macrophorin E (20) | IC50 > 10 μg per disc | B. berengeriana, G. fujikuroi | 42 |
| Macrophorin F (21) | IC50 = 3 μg per disc | G. fujikuroi | 42 |
| IC50 = 6 μg per disc | B. berengeriana | 42 | |
| Macrophorin G (22) | IC50 > 10 μg per disc | B. berengeriana, G. fujikuroi | 42 |
| Albaconol (64) | EC50 = 24.35 μM | S. sclerotiorum | 68 |
| Neoalbaconol (65) | EC50 > 30 μM | S. sclerotiorum, R. solani | 68 |
| Dictyvaric acid (66) | EC50 = 22.16 μM | S. sclerotiorum | 68 |
| EC50 = 22.54 μM | R. solani | 68 | |
| Puupehenone (90) | IC50 = 0.38 μg mL−1 | C. neoformans ATCC 90113 | 99 |
| IC50 = 1.49 μg mL−1 | C. krusei ATCC 6258 | 99 | |
| MIC = 1.6 μg mL−1 | T. mentagrophytes | 93 and 186 | |
| IC50 = 2.67–5.63 μg mL−1 | C. albicans ATCC 90028, C. glabrata ATCC 90030, A. fumigatus ATCC 90906 | 99 | |
| 21-Chloropuupehenone (92) | IC50 = 5.73 μg mL−1 | C. neoformans ATCC 90113 | 99 |
This potential was attenuated sharply by the introduction of a chloro-substituent to the quinone segment, as exemplified by 21-chloropuupehenone with an IC50 value of 5.73 μg mL−1. Both macrophorin A (16) and macrophorin F (21) act as promising models in the discovery of antifungal entities for controlling Botryosphaeria berengeriana and Gibberella fujikuroi with the IC50 values ranging from 3 μg per disc to 6 μg per disc. It is worth mentioning that the drimane-substituted (hydro)quinones, albaconol (64), and dictyvaric acid (66), exhibited promising inhibitory effects against agriculturally important pathogens Rhizoctonia solani and Sclerotinia sclerotiorum with the EC50 values lower than 25 μM.
| Compounds | Activity index | Biological species | Ref. |
|---|---|---|---|
| Tauranin (2) | IC50 = 1.5–4.3 μM | NCI-H460, MCF-7, SF-268, PC-3M, MIA Pa Ca-2 | 17 |
| Purpurogemutantidin (14) | IC50 = 0.93–31 μM | K562, HL-60, HeLa, BGC-823, MCF-7 | 36 |
| Macrophorin A (16) | MIC = 0.3 ppm | L-5178Y | 39 |
| IC50 = 1.17–5.28 μM | K562, MCF-7, Hela, DU145, U937, H1975, SGC-7901, A549, MOLT-4, HL60 | 37 | |
| 4′-Oxomacrophorin A (24) | IC50 = 0.19–6.74 μM | K562, MCF-7, U937, H1975, SGC-7901, A549, MOLT-4, HL60 | 37 |
| Neomacrophorin IV (29) | IC50 = 1.3–22.5 μM | HL60, chymotrypsin-like, trypsin-like, caspase-like | 45 |
| Neomacrophorin VI (31) | IC50 = 0.3–76.1 μM | HL60, chymotrypsin-like, trypsin-like, caspase-like | 45 |
| 20-O-Acetyl-21-hydroxy-ent-isozonarol (43) | IC50 = 0.37 μM | HL60 | 52 |
| 21-Hydroxy-ent-isozonarone (44) | IC50 = 0.37 μM | HL60 | 52 |
| Epoxyphomalin A (49) | IC50 = 0.017–11.420 μg mL−1 | 12 of a panel of 36 human tumor cell lines 20S proteasome | 56 and 57 |
| Epoxyphomalin B (50) | IC50 = 0.017–11.420 μg mL−1 | 12 of a panel of 36 human tumor cell lines 20S proteasome | 56 and 57 |
| Epoxyphomalin D (52) | IC50 = 0.72–1.43 μM | Prostate PC3M, bladder BXF 1218 L | 56 |
| Puupehenone (90) | IC50 = 0.1–1.3 μg mL−1 | P388, A549, MCF-7, HT-29, CV-1, PROT, DNA, RNA, KB | 9,94,95,110,114 and 187 |
| IC50 = 0.68–15 μM | BAEC, A549, H116, PSN1, SKBR3, CETP, 12-, and 15-lipoxygenases | 77,90,105 and 187 | |
| MIC = 1 μg mL−1 | LOVO | 110 | |
| 21-Chloropuupehenone (92) | MIC = 1 μg mL−1 | LOVO | 110 |
| IC50 = 0.3 μM | CETP | 77 | |
| IC50 = 0.2–6 μg mL−1 | P-388, A-549, HT-29, CV-1, KB, GR, DHFR, TS, PROT, DNA, Topo II | 110 | |
| 20-Methoxy-9, 15-ene-puupehenol (99) | IC50 = 1.78 μM | HepG2 | 118 |
| BE-40644 (104) | IC50 = 0.08–22 μg mL−1 | E. coli TRX, human TRX, yeast GSSG reductase | 119 |
| Phomoarcherin B (120) | IC50 = 0.1–9.4 μg mL−1 | KKU-100, KKU-M139, KKU-M156, KKU-M214, KB | 125 |
| Pentacecilide A (121) | IC50 = 0.69–3.65 μM | ACAT1, ACAT2-CHO | 134 |
| Stachybotrydial acetate (188) | IC50 = 0.69 μM | Human protein kinase CK2 | 155 |
| Acetoxystachybotrydial acetate (189) | IC50 = 1.86 μM | Human protein kinase CK2 | 155 |
| Compounds | Activity index | Biological species | Ref. |
|---|---|---|---|
| Siccanin (1) | MIC = 12.5–50 μg mL−1 | E. coli, M. tuberculosis 607, B. subtilis, S. aureus 209P, S. lutea, P. aeruginosa | 3 |
| Zonarol (6) | Average mortality = 17–98% at 1 μg mL−1 | H. akashiwo, C. antiqua, H. circularisquama | 49 |
| Zonaroic acid (8) | Average mortality = 15–45% at 1 μg mL−1 | H. akashiwo, C. antiqua, H. circularisquama | 49 |
| Zonarone (9) | Average mortality = 32–100% at 1 μg mL−1 | H. akashiwo, C. marina, C. antiqua, H. circularisquama | 49 |
| Macrophorin A (16) | MIC = 6.2–25 ppm | S. aureus, Trichophyton spp. | 39 |
| Myrothecol A (32) | MIC = 12.5, 25.0 μg mL−1 | S. aureus, B. cereus | 46 |
| Myrothecol B (33) | MIC = 50.0, 100.0 μg mL−1 | S. aureus, B. cereus | 46 |
| Myrothecol C (34) | MIC = 50.0, 100.0 μg mL−1 | S. aureus, B. cereus | 46 |
| Myrothecol D (35) | MIC = 100.0 μg mL−1 | S. aureus | 46 |
| Myrothecol E (36) | MIC = 50.0 μg mL−1 | S. aureus | 46 |
| Isozonarol (41) | Average mortality = 15–100% at 1 μg mL−1 | H. akashiwo, C. marina, C. antiqua, H. circularisquama | 49 |
| Yahazunol (61) | Average mortality = 67–98% at 1 μg mL−1 | H. akashiwo, C. marina, C. antiqua, H. circularisquama | 49 |
| Dysidphenol C (68) | MIC = 50.0 μg mL−1 |
E. coli (25922), B. subtilis (6633), S. aureus (25923) |
73 |
| Xishaeleganin B (70) | MIC = 1.5–3.0 μg mL−1 | S. aureus, S. pyogenes, E. faecium | 75 |
| Chromazonarol (85) | Average mortality = 42–93% after 4 h at 1 μg mL−1 | H. akashiwo, C. antiqua, H. circularisquama | 49 |
| Puupehenone (90) | MIC = 12.5 μg mL−1, IC50 = 2.0 μg mL−1 | M. tuberculosis (H37Rv) | 100 |
| Hongoquercin A (106) | MIC = 2–8 μg mL−1 | S. aureus, S. haemolyticus GC 4546, E. faecalis, E. faecium, B. cereus GC 4561, S. lutea GC 4562 | 122 |
| Isochromazonarol (148) | Average mortality = 37–100% after 4 h at 1 μg mL−1 | H. akashiwo, C. antiqua, H. circularisquama | 49 |
| Dysidphenol A (168) | MIC = 100 μg mL−1 |
E. coli (25922), B. subtilis (6633), S. aureus |
73 |
| Stachybotrolide stachybotrylactone (175) | IC50 = 32 μg mL−1 | S. aureus | 154 |
| Dasyscyphin C (198) | MIC = 2–63 μg mL−1 | MRSA, P. aeruginosa, B. anthracis | 177 |
| Dasyscyphin F (201) | MIC = 31–125 μg mL−1 | MRSA, P. aeruginosa, B. anthracis | 177 |
| Compounds | Activity index | Biological species | Ref. |
|---|---|---|---|
| Peyssonol A (11) | IC50 = 1 μM, EC50 = 1 μM | HIV, HIV | 29 and 30 |
| Penicilliumin A (15) | TC50 = 40.72–133.52 μg mL−1 | Coxsackievirus B3, herpes simplex virus type I (HSV-1), influenza A virus subtype H5N3 (A/H5N3) | 38 |
| Chrodrimanin A (125) | IC50 = 21 μM | Influenza A virus (H1N1) | 138 |
| Chrodrimanin E (129) | IC50 = 55 μM | Influenza A virus (H1N1) | 138 |
| Chrodrimanin F (130) | IC50 = 57 μM | Influenza A virus (H1N1) | 138 |
| Chrodrimanin K (135) | IC50 = 74 μM | Influenza A virus (H1N1) | 140 |
| Chrodrimanin N (138) | IC50 = 58 μM | Influenza A virus (H1N1) | 140 |
| 3-Hydroxypentacecilide A (139) | IC50 = 34 μM | Influenza A virus (H1N1) | 140 |
| F1839-I (166) | IC50 = 15.6 μM | HIV | 161 |
| Stachybotrylactone acetate (170) | IC50 = 18.9 μM | IAV | 161 |
| Stachybotrysin A (180) | IC50 = 19.6 μM, IC50 = 12.4 μM | HIV, IAV | 161 |
| Stachybotrysin B (181) | IC50 = 19.2 μM | HIV | 161 |
| Stachybotrysin E (184) | IC50 = 20.5 μM, IC50 = 45.6 μM | HIV, IAV | 161 |
| Stachybotrysin F (185) | IC50 = 35.7 μM, IC50 = 14.6 μM | HIV, IAV | 161 |
| Stachybotrysin G (186) | IC50 = 18.1 μM, IC50 = 23.4 μM | HIV, IAV | 161 |
The tetracyclic meroterpenoids (including benzopyran-fused-naphthene and benzofuran-spiro-naphthene meroterpenoids) demonstrated significant advantages in antiviral discovery. Among them, peyssonol A showed significant anti-HIV activity, with the EC50 value of 1 μM.
| Compounds | Activity index | Biological species | Ref. |
|---|---|---|---|
| Zonarol (6) | E i = 0.85 | Haliotis discus hannai | 25 |
| Zonarone (9) | E i = 0.92 | Haliotis discus hannai | 25 |
| Isozonarol (41) | E i = 0.78 | Haliotis discus hannai | 25 |
| Isozonarone (42) | E i = 0.85 | Haliotis discus hannai | 25 |
| Chromazonarol (85) | E i = 0.8 | Haliotis discus hannai | 25 |
| Puupehenone (90) | MIC = 3.1 μg mL−1 | T. vaginalis | 93 |
| IC50 = 0.6–2.1 μg mL−1 | P. falciparum | 114 | |
| 15-Oxopuupehenol (96) | IC50 = 1.3 μg mL−1 | P. falciparum (W2 clone) | 112 |
| IC50 = 2.0 μg mL−1 | P. falciparum (D6 clone) | 112 | |
| 15α-Methoxypuupehenol (97) | IC50 = 0.4–1.4 μg mL−1 | P. falciparum | 114 |
| Phomoarcherin B (120) | IC50 = 0.79 μg mL−1 | P. falciparum | 125 |
| Chrodrimanin B (126) | LD50 = 10 μg g−1 | Silkworm | 135 |
| Chrodrimanin D (128) | LD50 = 20 μg g−1 | Silkworm | 136 |
| Chrodrimanin E (129) | LD50 = 10 μg g−1 | Silkworm | 136 |
| Chrodrimanin F (130) | LD50 = 50 μg g−1 | Silkworm | 136 |
| Pelorol (191) | LC50 = 5–10 μg mL−1 | Brine shrimp | 168 |
| Compound | Activity index | Biological species | Ref. |
|---|---|---|---|
| Isozonarol (41) | EC50 = 71 μM | DPPH scavenging | 24 |
| Isozonarone (42) | EC50 = 145 μM | DPPH scavenging | 24 |
| 20-O-Acetyl-21-hydroxy-ent-isozonarol (43) | IC50 = 3.0 μM | Superoxide production by human neutrophils | 52 |
| 21-Hydroxy-ent-isozonarone (44) | IC50 = 11.0 μM | Superoxide production by human neutrophils | 52 |
| 13-[[2-(Hexyloxy)-2,5,5,8a-tetramethyldecahydro-1-naphthalenyl] (methoxy) methyl]benzenol (71) | IC50 = 1.51 mM | DPPH scavenging | 76 |
| Chromazonarol (85) | EC50 = 71 μM | DPPH scavenging | 24 |
| Puupehenone (90) | IC50 = 32 μM | DPPH scavenging | 97 |
| IC50 = 0.76 μM | 15-HLO, 15-LOX | 104 and 132 | |
| IC50 = 1.3–8.3 μM | 15-SLLO, 12-HLO, 12-LOX, NOX | 104 and 132 | |
| 21-Chloropuupehenone (92) | IC50 = 0.7–2.4 μM | 15-HLO, 15-SLLO, 12-HLO, 12-LOX | 104 and 132 |
| Stachybotrolide (stachybotrylactone) (175) | IC50 = 17.9 μM | NO in RAW264.7 | 165 |
| Stachybotrysin C (182) | IC50 = 27.2 μM | NO in RAW264.7 | 165 |
| 19-O-Methylpelorol (195) | IC50 = 5.1–9.2 μM | Inflammatory cytokines IL-6, IL-1β, IL-8, PEG2 | 174 |
3. Synthesis of siccanin and related drimane (hydro)quinones
Siccanin and its structurally-related meroterpenoids display a wealth of activities (via infra), including antifungal, antibacterial, antiviral, insecticidal, cytotoxicity, antioxidant, algicidal, anti-inflammatory, antifeedant, and immunomodulatory activities. The biological importance assures this kind of natural product to be an ideal model for the discovery of new chemotypes of a given activity, or probing the important biological pathways of interest. The realization of efficient synthesis is a pivotal objective in facilitating the chemistry and chemical biology of this kind of natural product. This section deals with the synthetic strategies used to access siccanin and the related structurally simplified meroterpenoids. The synthetic approaches are categorized based on how the Csp3–Csp2 linker is forged between the drimane segments and (hydro)quinone subunits, and other synthetic efforts are also briefly discussed.3.1 Advances in the synthesis of siccanin
The antifungal antibiotic siccanin features a unique scaffold involving the unusual cis-, syn-, and cis-fused A/B/C ring system, which is regarded as a drimane sesquiterpene combined with orcinol. The important characteristic of lead innovation stems from not only its remarkable antifungal activity against a variety of fungi (vide supra), but also from no obvious deleterious effects. So, it has garnered considerable attention from the synthetic communities. The first total synthesis of racemic siccanin was reported in 1981 by Yoshikoshi and coworkers, starting from the readily available keto ester (S1) in 19 steps (Scheme 1).188,189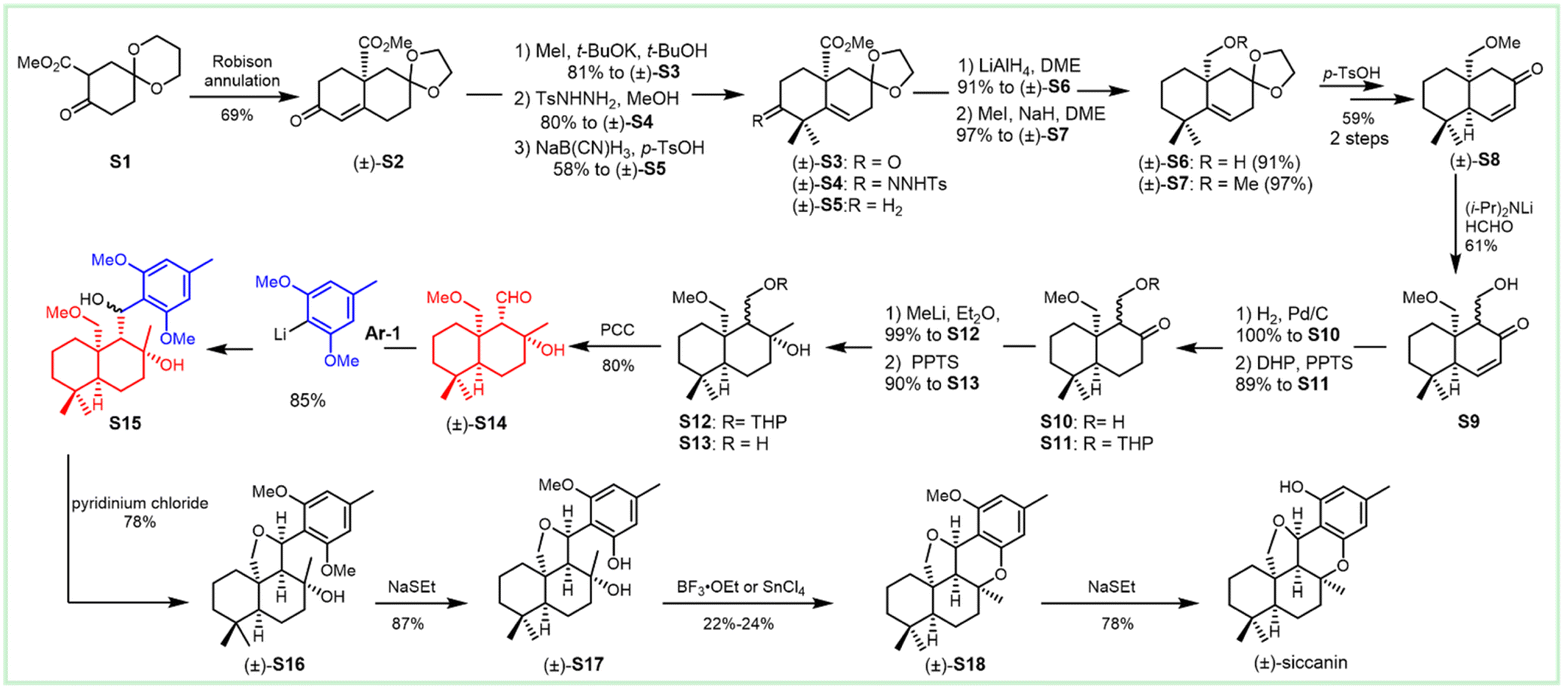 | ||
| Scheme 1 Synthesis of racemic siccanin from keto ester S1. | ||
Robinson annulation of the intermediate S1 with methyl vinyl ketone yielded octalone S2, which was smoothly methylated to give dimethyl ketone S3. Transformation of the hindered ketone to methylene was achieved by reduction of its tosylhydrazone S4. The subsequent reduction of octalin S5 with LiAlH4 afforded alcohol S6, followed by methylation to give methyl ether S7. A concomitant deacetalization and isomerization through the treatment of p-TsOH afforded the conjugated enone S8. Regioselective hydroxymethylation with gaseous formaldehyde provided the hydroxy ketone intermediate S9 with an unclarified configuration of the newly formed chiral center. Hydrogenation, followed by the protection of the hydroxy group, gave tetrahydropyranyl ether S11, which was then methylated (excess MeLi, Et2O) to produce carbinol S12. Subsequent hydrolysis and oxidation led to the drimane synthon S14, whose stereochemistry was concluded by comparison with that of the unambiguously assigned siccanin. The introduction of the aromatic ring was carried out by coupling the aldehyde S14 with a lithiated orcinol dimethyl ether Ar-1. Noteworthily, the synthetic strategy by the addition of aryl-metallic reagents to carbonyl acts as the key step to finish the sp2–sp3 linker between the drimane synthon and aromatic segments, which is detailed in the following section. Tetrahydrofuran ring formation (S16) by intramolecular attack of the methoxymethyl group to a benzylic carbonium ion was effectively performed by the treatment with pyridinium chloride. Partial demethylation with NaSEt, followed by Lewis acid-catalyzed cyclization, proceeded to give siccanin methyl ether S18, which produced racemic siccanin after the demethylation.
In 1998, Liu et al. used a different approach to acquire the drimane segment S24 for the concomitant attack by Grignard reagent Ar-2 (Scheme 2).190 The ferric chloride-catalyzed Diels–Alder cycloaddition of dienone ester S19 with diene S20 occurred readily to give the adduct S21, possessing the complete sesquiterpenoid framework of siccanin with the correct oxidation level and relative stereochemistry. Barton deoxygenation of the derivative of compound S22 afforded the intermediate S23, which took 2-functional manipulations (including deprotection and oxidation) to afford the drimane synthon (S24). The subsequent nucleophilic attack by 2,6-dimethoxy-4-methylmagnesium bromide produced the stereoisomer S25 with the full carbon-framework of siccanin. The following transformations to siccanin require consecutive closures of two ether rings. The tetrahydrofuran ring was readily formed in almost quantitative yield, on exposure to pyridinium chloride in refluxing dichloromethane. The single crystal X-ray analysis revealed that the intermediate S26 is an epimer of that required by siccanin, which was expected to be thermodynamically stable. The sequential demethylation and cyclization gave isosiccanin methyl ether.
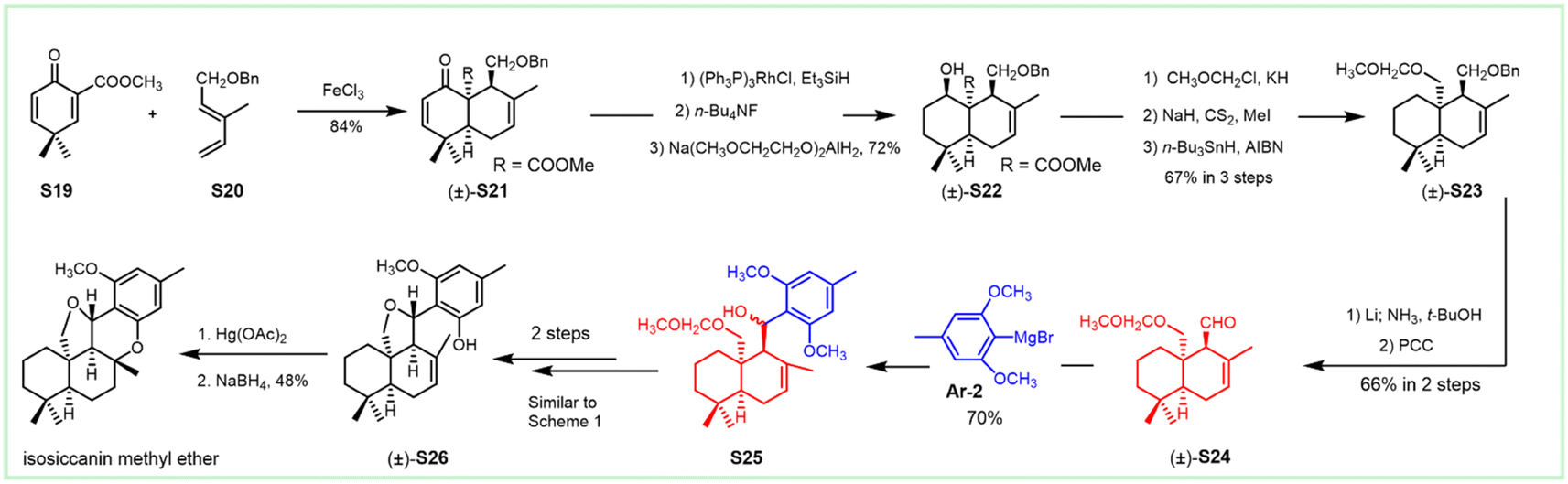 | ||
| Scheme 2 Synthesis of isosiccanin methyl ether from dienone ester S19 and diene S20. | ||
The Trost group reported on the Pd-catalyzed reductive cyclization of diynes as the key step for the construction of the drimane meroterpenoids (Scheme 3).191 Differentiation of the hydroxy groups of the diol S28 was realized by monsilylation to deliver S29, which was treated with phosphonates for acquiring the desired cis-diyne S30. The Pd-catalyzed reductive cyclization was attempted for the readily available cis-diynes (S30, S31 and S32), in which compound S32 provided promising results with diene S33 as a geometrically homogeneous crystalline in good yield. This intermediate underwent cyclization through treatment with protonic acid (TsOH), and deprotection with mercaptoethanol normally led to the tetrahydrofuran S35. The BF3 etherate satisfactorily mediated the formation of pyran, and this cyclization afforded siccanin methyl ether S36, which underwent O-demethylation to provide (±)-siccanin with the desired relative configuration of the natural specimen. Alternatively, the simultaneous formation of tetrahydrofuran and pyran could be accomplished in 1 step through the Lewis acid (BF3-etherate)-assisted cyclization of meroterpenoid S34, which was accessible through partial demethylation of the precursor S33.
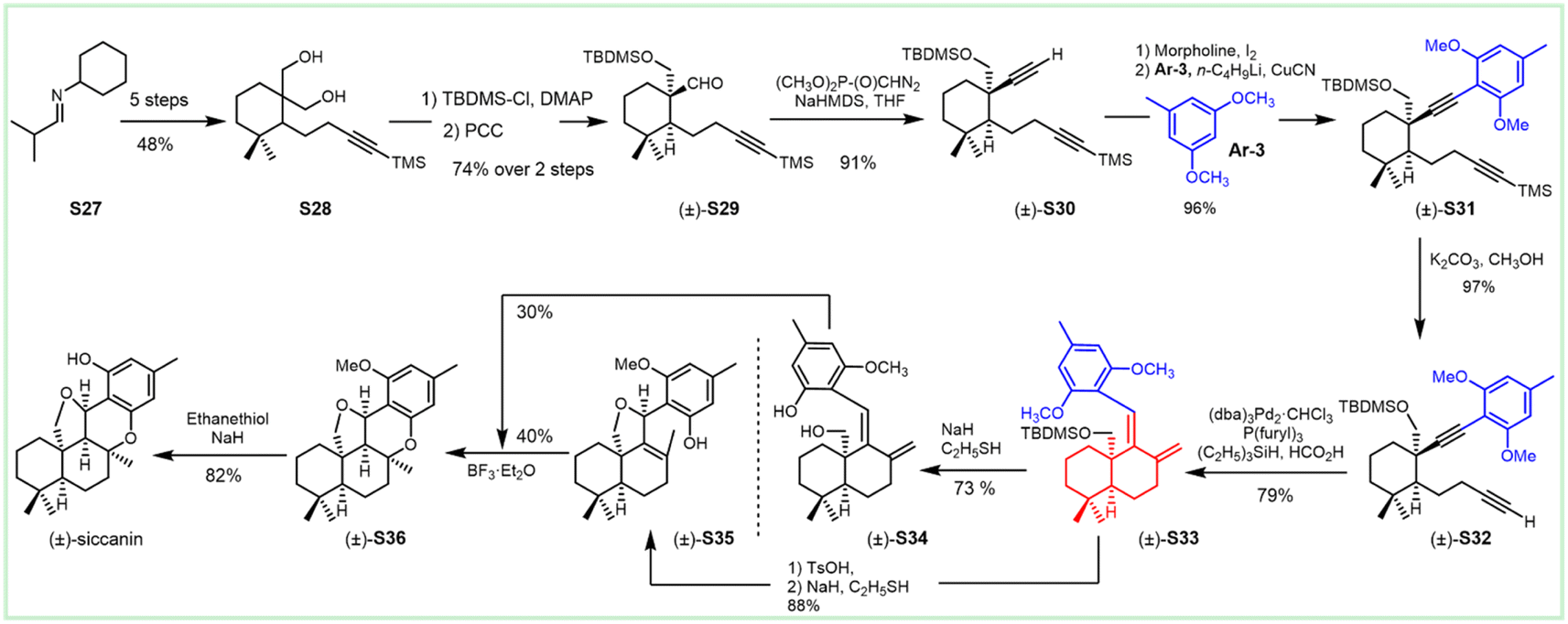 | ||
| Scheme 3 Synthesis of racemic siccanin from S27. | ||
The first enantioselective biomimetic total synthesis of (−)-siccanin was documented by Trost et al. in 2003, and featured Pd-catalyzed asymmetric allylic alkylation and epoxyolefin radical cyclization (Scheme 4). They finished the total synthesis of (−)-siccanin, starting from the readily available allyl carbonates S38 and chiral alcohol S37 in 13 linear steps.192 Both E- and Z-isomers of the allylic carbonate S38 could be employed in allylic alkylation for the enantioselective cyclization to obtain pyran S39, which underwent dihydroxylation, followed by oxidative cleavage, to afford chiral aldehyde S40. Chiral alcohol S37 was utilized to synthesize sulfone S41via the Mitsunobu reaction, followed by oxidation. The sulfone underwent Julia olefination to give diene S42. Enantioselective sharpless dihydroxylation (d.r. 10:1), followed by hydrogenation, smoothly afforded chroman diol S43. DDQ oxidation afforded chromene diol S44, which was readily converted into epoxide S45 in 2 steps. The Lewis acids-catalyzed cationic cyclization was utilized, while no productive results were observed. A radical cyclization in the presence of Cp2TiIIICl was deployed for a 6-exo-trig cyclization to form the precursor S46 with the full carbon framework and right stereochemistry of natural (−)-siccanin. The following remote iodination and cyclization under Suarez conditions led to the pentacyclic compound S36, which was smoothly demethylated to afford (−)-siccanin (1) in good yield.
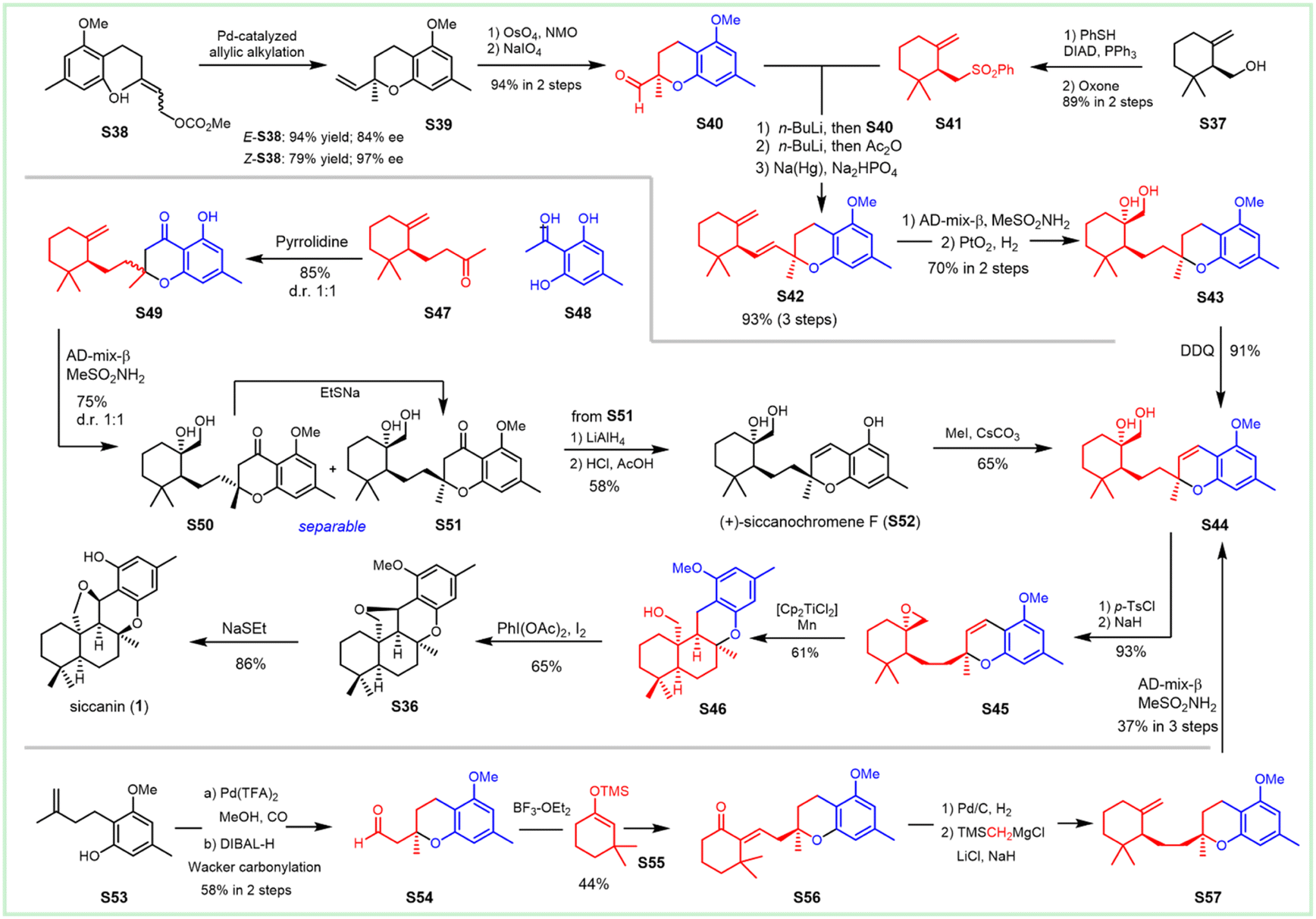 | ||
| Scheme 4 Asymmetric synthesis of siccanin from S37, S38 or S47, S48 or S53. | ||
A readily available natural product possessing appropriate stereochemistry and functionalities could serve as a “chiral pool”, and find wide applications in the enantioselective synthesis of related complicated natural products.
Moral and Barrero et al. reported a formal synthesis of (−)-siccanin from (+)-3, 4-dihydro-γ-ionone, which could be obtained easily from the natural source Bellardia trixago.193 The key step comprises the pyrrolidine-catalyzed cascade condensation of dihydroionone S47 with commercially available 2,6-dihydroxy-4-methylacetophenone S48 for the construction of chromanone S49 as an epimeric mixture (1:1). The sharpless asymmetric dihydroxylation with AD-mix β provided the separable mixture of diols S50 and S51.
The undesired chromanone epimer S50 could be epimerized to S51, possessing the right stereochemistry determined by the natural (−)-siccanin. Reduction with LAH, followed by acid treatment, afforded (+)-siccanochromene F (S52), which underwent chemoselective methylation of the aromatic hydroxy group to produce the precursor S44, required by the enantioselective synthesis of (−)-siccanin reported by Trost et al.192
Another formal synthesis of (−)-siccanin was achieved by Tietze through an enantioselective domino Wacker/carbonylation/methoxylation to form the chroman ring with a quaternary stereogenic center.194 The cascade manipulations of alkenyl phenol S53 in the presence of the Pd/(R, R)-Bn-BOXAX chiral catalyst, and p-benzoquinone as an oxidant under an atmosphere of carbon monoxide in methanol at room temperature, enantioselectively delivered the benzopyran acetate. The subsequent reduction delivered chiral aldehyde S54, which smoothly underwent the scale-up aldol reaction with silyl enol ether S55. Martin's sulfurane was introduced instead of the Burgess reagent in the dehydration step to improve the productivity of S56. The chemoselective hydrogenation of the α,β-unsaturated ketone was obtained by using palladium on charcoal (10 mol%) in CH2Cl2 at room temperature. Peterson olefination provided a good approach in the introduction of a methylene group for the preparation of S57, which could be employed for the synthesis of the desired precursor S44, en route to the synthesis of (−)-siccanin.
3.2 Addition of aryl-metallic reagents to carbonyl
The addition of aryl-metallic reagents to the drimane carbonyl group was widely recruited for the efficient construction of the full carbon framework of siccanin and its related drimane meroterpenoids. A variety of drimane intermediates (Ka-1–Ka-10) were successfully synthesized from structurally distinct terpenoids (Fig. 11). Noteworthily, many complex but easily available natural products with definite absolute configuration were employed as chiral pools for the efficient asymmetric synthesis of the desired drimane synthons. Synthesis of albicanal (Ka-2) and its isomers (Ka-1, Ka-5) was widely explored because of the synthetic values of the downstream transformation to drimane meroterpenoids, and their biological importance as natural products. | ||
| Fig. 11 Synthesis of divergent drimane α-aldehyde or drimanyl (pseudo)halide synthons. | ||
The commercially available and inexpensive diterpenoid sclareol and sclareolide were widely utilized for the synthesis of the drimane synthons through degradation of the side chain. Typically, (+)-sclareolide (SM-1) with unambiguous absolute configuration could be recruited for the synthesis of chiral (+)-albicanol, which was transformed to the stereochemically definite albicanal (Ka-2),31,195 its enal isomers Ka-1,196 crolein aldehyde Ka-5,92 together with hydroxy aldehyde (Ka-3).197 The hydrogenated chiral drimanic synthon Ka-4 could be synthesized from the bulky chiral feedstock (−)-sclareol (SM-2).80,198 Targeting the synthesis of albicanal (Ka-2), (1) direct approaches by employing the chiral pools sclareolide (SM-1)31,195 and manool (SM-4),199 together with (2) the indirect ways (racemic synthesis followed by resolution) utilizing racemic albicanol (SM-12)15 and farnesyl acetate (SM-10)200 could be successfully employed. Meanwhile, the synthesis of the enantiomer epi-Ka-2 was realized starting from β-ionone (SM-11).64 The other starting materials typically included monoterpenoids nerol (SM-7) and SM-8,35,201,202 sesquiterpenoids farnesol and its acetate, ionone and racemic albicanol. Complicated terpenoids, including trans-communic acid (SM-5) and 18β-glycyrrhetinic acid (SM-6), were also employed in the synthesis of drimanic carbonyl synthons. With these drimane intermediates available (Fig. 11), a plethora of meroterpenoids (vide infra) can achieved by the choice of appropriate aryl-metallic reagents. The enantioselective syntheses of natural products and its analogues with definite configuration are discussed in the following sections.
(+)-Wiedendiol-A was successfully synthesized enantioselectively by utilizing the drimane aldehyde (Ka-1) with unambiguous absolute configuration (Scheme 5), which could be acquired starting from (+)-sclareolide (SM-1). The following addition of the lithiated Ar-4 to Ka-1 led to the drimane meroterpenoid KM-1.196 Aiming for the enantiospecific synthesis of wiedendiol A, Barrero recruited (−)-sclareol (SM-2) and (+)-cis-abienol (SM-3) as the chiral pool starting materials for the preparation of drimane aldehyde Ka-1, which was further reacted with aryllithium derived from Ar-5 to get drimane meroterpenoids KM-2.78 Either of the hydroxylated precursors, KM-1 or KM-2, could enantiospecifically provide (+)-wiedendiol-A after 2 functional manipulations. Through these synthetically similar approaches, the meroterpenoid intermediate KM-3 prepared from (−)-sclareol in 13 steps, was submitted in the synthesis of (+)-puupehedione via another 7 steps.109 With the adduct KM-4 from coupling lithiated Ar-7 and ka-1 as an intermediate, a 9-step synthesis of (+)-ent-chromazonarol in 19% overall yield was released starting from (+)-sclareolide (SM-1). The configuration of 8-β-methyl group was confirmed by NOESY experiment and compared with known ones.203 Synthesis of isozonarone and isozonarol in 9 and 10 steps from (+)-manool (SM-4), respectively, also relied on the same chiral drimane synthon Ka-1, by choosing the lithium reagent of Ar-8 in the coupling step.204
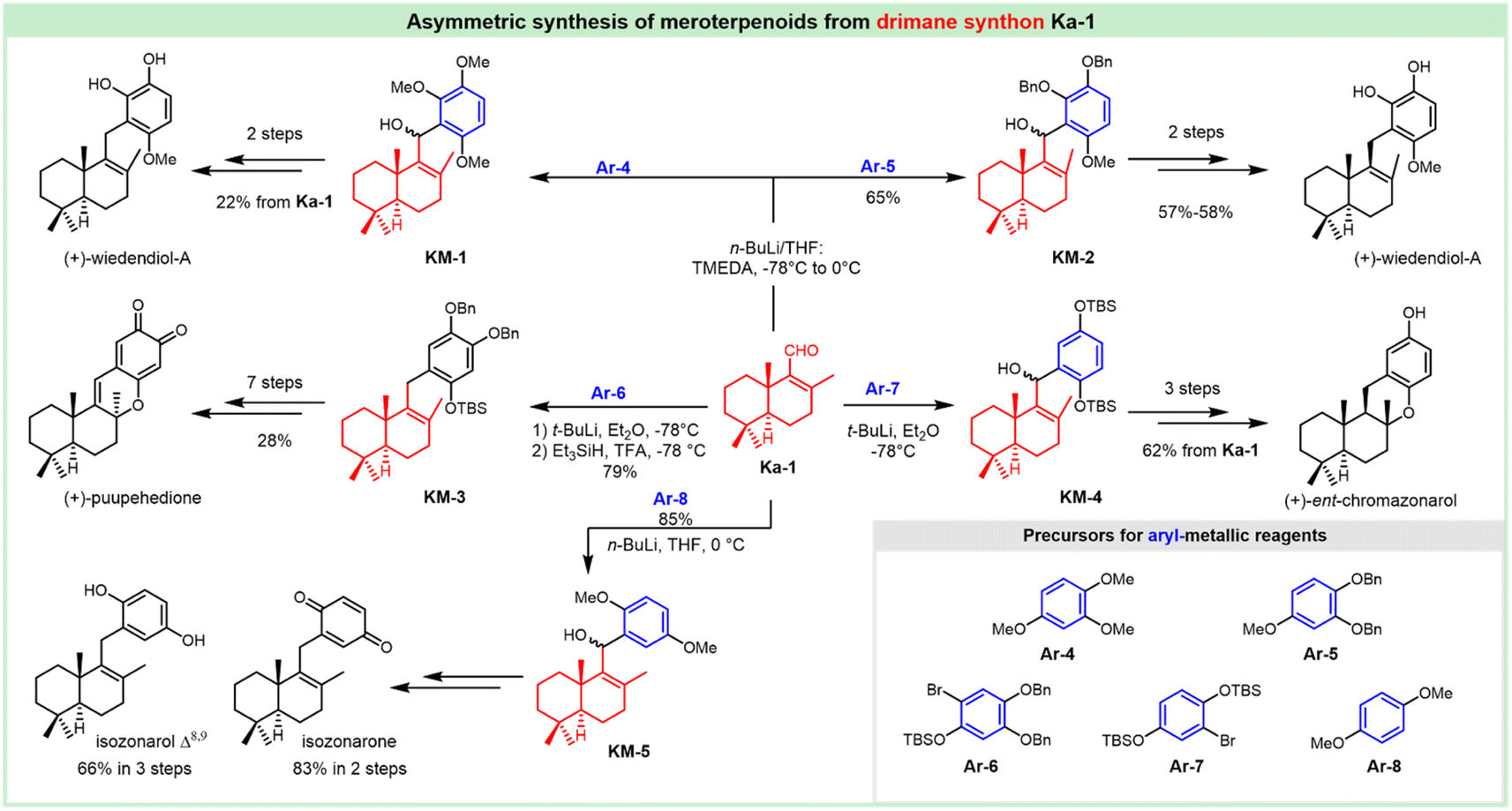 | ||
| Scheme 5 Asymmetric synthesis of meroterpenoids from drimane synthon Ka-1. | ||
Both the drimane synthon Ka-2 and its enantiomer as key intermediates have facilitated the synthesis of a large number of drimane meroterpenoids (Scheme 6). The enantioselective synthesis of this couple of enantiomers is substrate-dependent. The chiral pool approaches were also widely found in other sections for the efficient construction of the desired stereochemistry. (−)-Hyatellaquinone was prepared in 10 steps from (+)-sclareolide (SM-1) with 13% overall yield.195 Addition of the lithium reagent of 1,2,4,5-tetramethoxybenzene (Ar-9) to Ka-2 afforded the drimane meroterpenoid enantiomer of KM-6, which was assigned as ent-KM-6 and was transformed to (−)-hyatellaquinone in 3 steps.31 This total synthesis assisted the revision of the absolute configuration of the naturally occurring one, which was depicted as (+)-hyatellaquinone.31 The enantiospecific synthesis of (+)-ent-zonarol and (+)-ent-zonarone, with (+)-manool (SM-4) as the starting material, also went through the utilization of aldehyde segment (+)-Ka-2 in coupling with the organolithium reagent Ar-10via the intermediate ent-KM-7.199 Its enantiomer ent-Ka-2 was reported to be synthesized from β-ionone (SM-11) in 10 steps, which was then reacted with lithiated aromatics Ar-11 or Ar-12 to get the natural products yahazunol, zonarone, and zonarol isolated from Dictyopteris undulata Okamura.64 Based on the two readily available enantiomers of albicanal, as a tailored key synthetic hub, hyatellaquinone and spongiaquinone,205 (−)-albaconol,67,206 (−)-tauranin and (−)-BE-40644,15 and (−)-F-12509A35 were successfully prepared by choosing different aryl organolithium reagents for nucleophilic addition to the carbonyl group (Scheme 6). Interestingly, the key drimane intermediate Ka-2 could be synthesized in 4 steps from farnesyl acetate (SM-10), which could also efficiently facilitate the racemic synthesis of zonarone, macrophorin A and 4′-macrophorin A.200
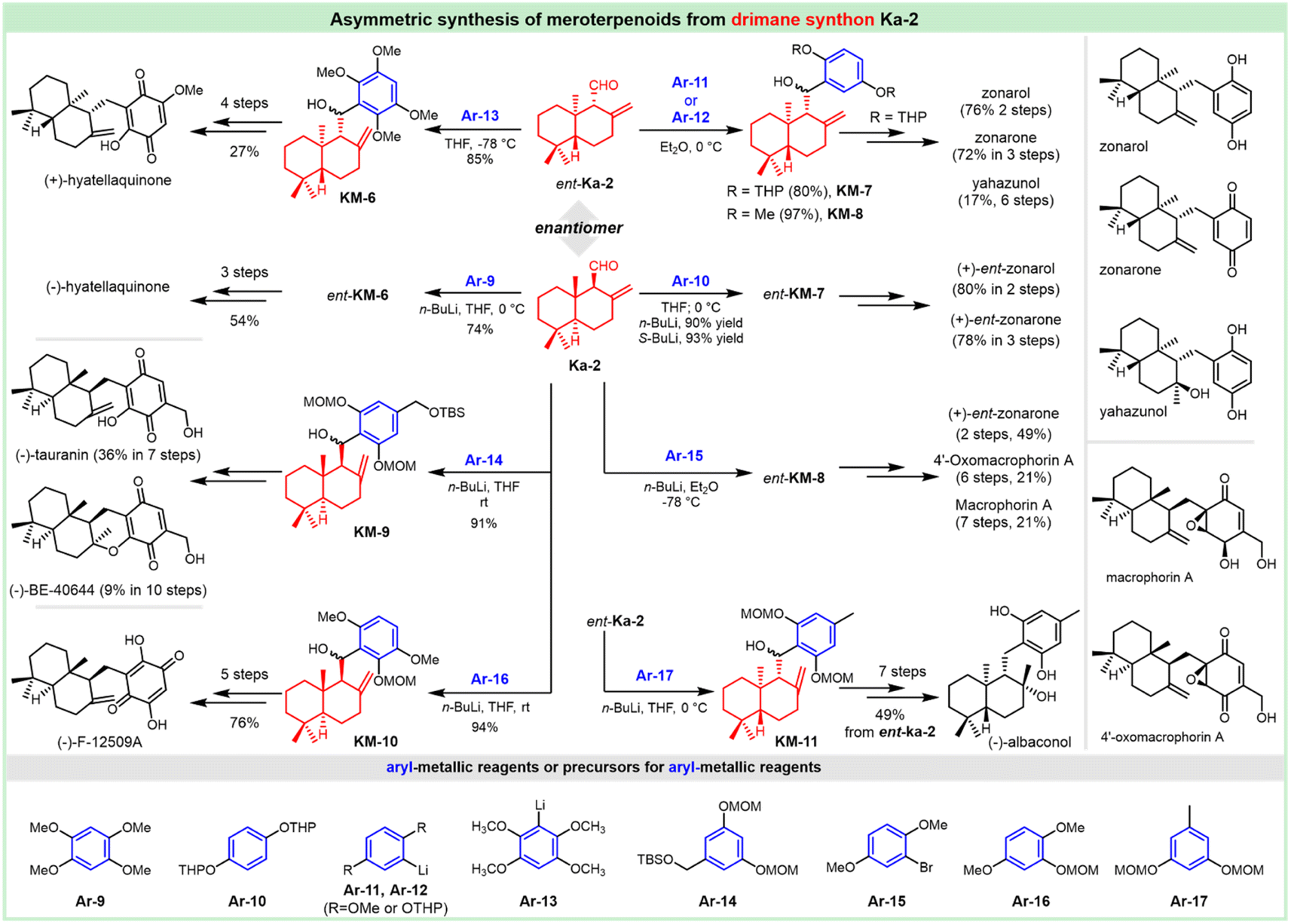 | ||
| Scheme 6 Asymmetric synthesis of meroterpenoids from enantiomers of drimane synthon Ka-2. | ||
The key intermediates Ka-3, including 8-α-OH-Ka-3 and its epimer 8-β-OH-Ka-3, were accessible from sclareolide (Scheme 7). Through the coupling of 8-β-OH-Ka-3 and the in situ produced lithium reagent of Ar-18, total synthesis of (+)-puupehenone was realized in 8 steps starting from (+)-sclareolide in 3% overall yield.197 Deprotonation of ketone Ar-20 LDA, followed by coupling with aldehyde 8-β-OH-Ka-3, efficiently gave the precursor KM-14 for the synthesis of puupehenone after dehydration and oxidation in 2 or 3 steps.
 | ||
| Scheme 7 Asymmetric synthesis of meroterpenoids from drimane synthon Ka-3. | ||
The concomitant reduction delivered puupehenol in almost quantitative yield, which could be oxidized to puupehedione.207 Drimane segment 8-α-OH-Ka-3 was coupled with the lithiated reagent from Ar-19 to get the epimeric benzyl alcohols KM-13 in 68% yield. This enabled the synthesis of (−)-pelorol from (+)-sclareolide with 6% overall yield in 11 steps.169
Two isomers of drimane synthon Ka-4, differentiating in the configuration of the methyl group at 8-position (8-β-CH3 and 8-α-CH3), were prepared from (−)-sclareol (SM-1) in 6 steps and from (+)-cis-abienol (SM-3) in 4 steps, respectively (Scheme 8).78,80,198 Utilization of the aryllithium reagent from Ar-23 in the nucleophilic attack to the carbonyl group of 8-β-CH3-Ka-4 helps in the first construction of wiedendiol B starting from (−)-sclareol via the epimeric benzyl alcohols KM-17 as a key intermediate.78,80 The drimanic aldehyde 8-α-Ka-4 and its epimer 8-β- CH3-Ka-4 successfully found their application in obtaining (−)-siphonodictyal B and (+)-8-epi-siphonodictyal B, respectively.78,83,85 The synthetic value was strengthened by not only the structural revision of siphonodictyal B, but also the further conversion into liphagal in a one-pot cascade reaction. In 2019, George et al. established a divergent total synthesis of siphonodictyal B and corallidictyals A–D, starting from the natural feedstock sclareolide with drimane aldehyde (8-α-CH3-Ka-4) as the key intermediate.208
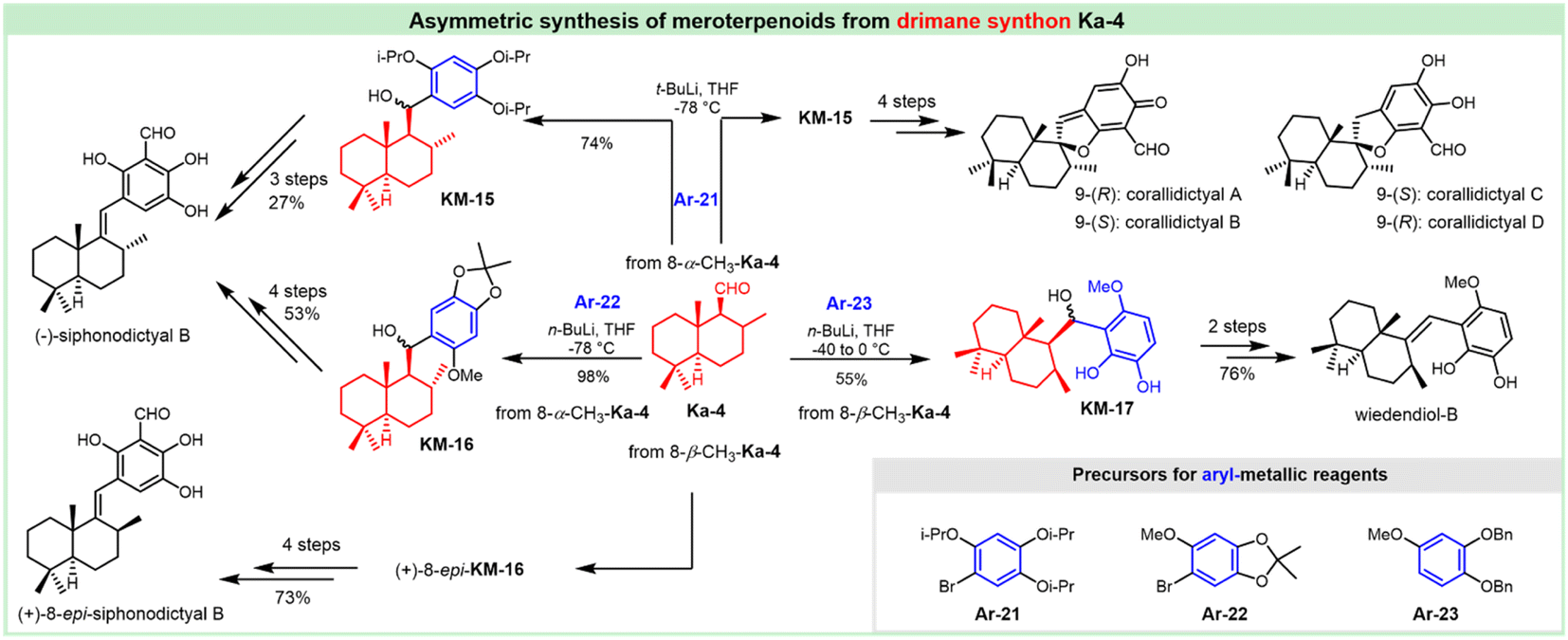 | ||
| Scheme 8 Asymmetric synthesis of meroterpenoids from drimane synthon Ka-4. | ||
Drimane intermediate Ka-5 and its enantiomer are valuable in the acquiring of drimane meroterpenoids (Scheme 9). The drimane aldehyde ent-Ka-5 was synthesized from β-ionone (SM-11) in 11 steps,64 while the enantiomer Ka-5 was prepared starting from commercially inexpensive chiral pool natural products (+)-sclareolide (SM-1) in 5 steps92 or from (−)-sclareol (SM-2) in 7 steps.209 The tailored enantiomer was reacted with 2-lithiohydroquinone di-THP ether (Ar-12) to deliver the natural products isozonarone and isozonarol isolated from Dictyopteris undulata Okamura.64 One could envision that the enantiomers of natural isozonarone and isozonarol (ent-isozonarone) could be prepared by choosing the enantiopure drimane synthon Ka-5 in coupling with appropriate lithiated derivatives via the enantiomer or equivalent of the intermediate KM-20. The first enantiospecific synthesis of (−)-15-oxopuupehenol, together with an improved synthesis of (+)-puupehenone, (+)-puupehedione, and (+)-15-cyanopuupehenone, was accomplished by the utilization of Ka-5 in coupling with an organolithium reagent from Ar-25.209 By altering the aromatic metallic reagents (e.g., lithiated Ar-24 or Ar-26), this intermediate (Ka-5) was also deployed in the efficient asymmetric preparation of marine sesquiterpene quinones (+)-cyclospongiaquinone-1 and (−)-dehydrocyclospongiaquinone-1,92 together with the synthesis of (−)-akaol A.210 A 4-step synthesis of the racemic intermediate (±)-Ka-5 was reported by Mehta et al. from farnesyl acetate (SM-10), which was leveraged in coupling with the lithiated reagent derived from Ar-15 to acquire racemic 1′-epi-craterellin A.200
 | ||
| Scheme 9 Asymmetric synthesis of meroterpenoids from drimane synthon Ka-5. | ||
Starting from the natural product (−)-sclareol, the enantiomers of the naturally occurring bioactive meroterpenoids isozonarol, isozonarone and chromazonarol could be prepared via the coupling of the drimane aldehyde Ka-6 with the lithium reagent derivatized from Ar-7 in 3–5 steps (Scheme 10).50 Noteworthily, ent-chromazonarol was also a natural metabolite from sponge Disidea pallescens. This coupling partner is synthetically available from (−)-sclareol (SM-2) in 4 steps.211 The key intermediate, racemic drimanoyl chloride Ka-7, was synthesized with either β-ionone or farnesol as a starting material. It was employed in the synthesis of (±)-wiedendiol B by the coupling with the lithiated Ar-27, followed by another 3 manipulations (Scheme 10).212 Replacing the hydroquinone precursor by the Ar-29-derived aryllithium in the coupling step enabled a 14-step racemic synthesis of deoxyspongiaquinone from farnesol (SM-9).82 The drimane aldehyde intermediate Ka-8 is achievable from nerol (SM-7) in 10 steps or farnesol (SM-9) in 5 steps, which was employed in the coupling with the Ar-28-related aryllithium for the synthesis of the proposed peyssonol A in 2 steps (Scheme 10).30,32 It is noteworthy that Snyder and coworkers accomplished the racemic total synthesis through polyene cyclizations, and revised the structure of this brominated natural product.32 This facilitated the anti-HIV SAR investigation of peyssonol A-related diastereoisomers and truncated analogues. The first enantiospecific synthesis of the cytotoxic UPA0043 and UPA0044 was reported successfully starting from commercially available 18β-glycyrrhetinic acid (SM-6), which served as the precursor of the drimane precursor Ka-9 (Scheme 10). It was reacted with lithiated Ar-30 to get the drimane meroterpenoid KM-27, which was transformed to UPA0043 and UPA0044 in 5 and 6 steps, respectively.124 This enantioselective synthesis helped the establishment of the absolute stereochemistries of the isolated natural specimens, as described in Scheme 10. The drimane aldehyde intermediate Ka-10 could be obtained from communic acid (SM-5) in 9 steps, and it enabled the first synthesis of dasyscyphins E, which featured the efficient coupling of the enantiopure sesquiterpene synthon Ka-10 with the lithiated Ar-31 as the key constructive step (Scheme 10).213
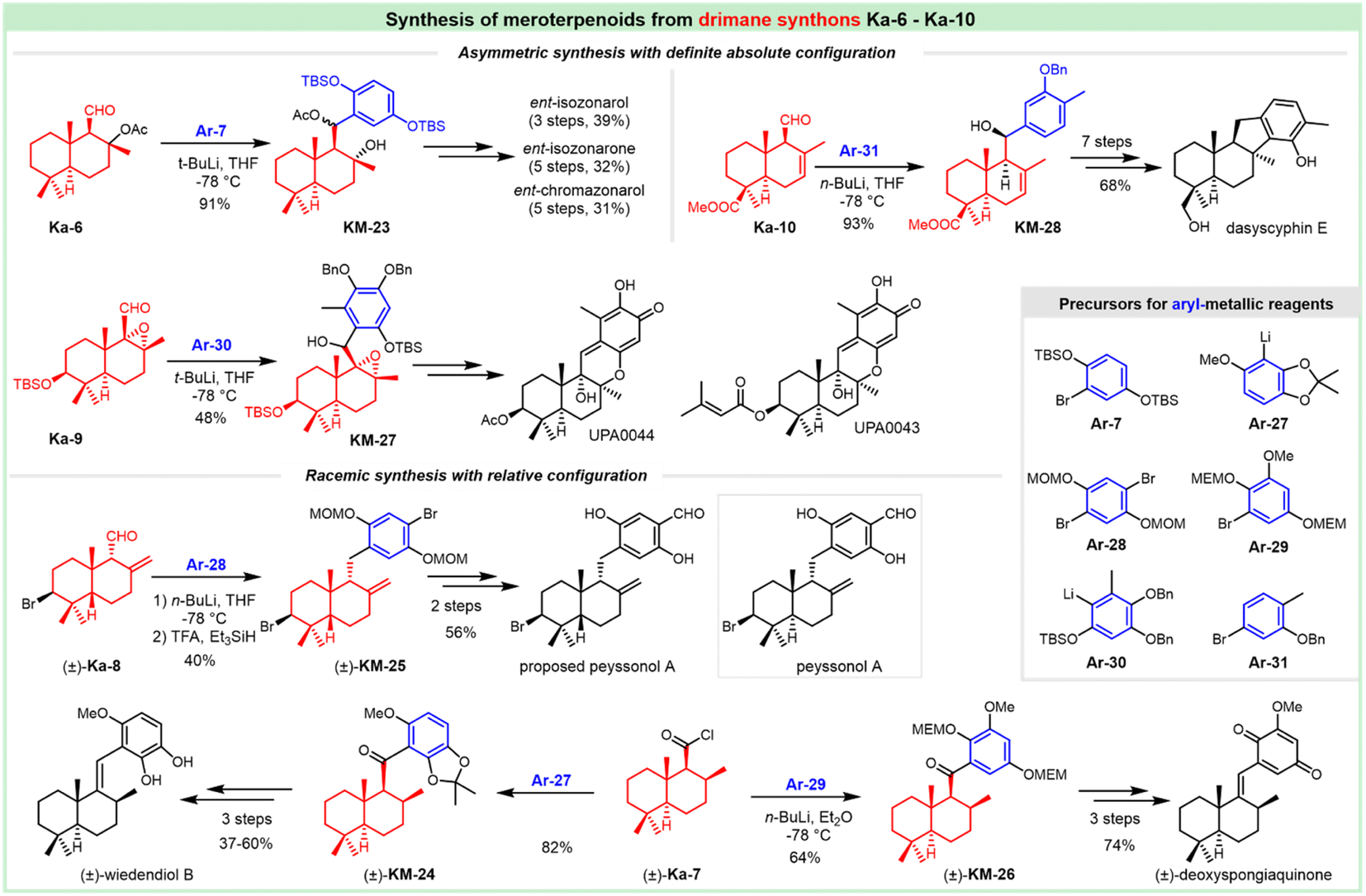 | ||
| Scheme 10 Synthesis of meroterpenoids from drimane synthon Ka-6–Ka-10. | ||
3.3 Coupling of aryl-metallics and drimanyl (pseudo)halides
The readily available and diverse nucleophilic aryl-metallics can be coupled with a couple of drimanyl (pseudo)halides for the construction of the framework of meroterpenoids (Scheme 11). A racemic total synthesis of the complement inhibitor K-76 was established from 6-acetoxy-4-methyl-4-hexenal (SM-13), featuring the allylic bromide Kb-1 as the key coupling partner. Noteworthily, synthesis of this drimanic synthon was realized through Hg(OTFA)2-assisted electrophile-induced polyene cyclizations. Attempts in the coupling of bromide Kb-1 with the lithiated derivative of Ar-33 under previously established conditions were not successful. However, the in situ transmetalation via the addition of CuCN prepared the corresponding aryl copper derivative of Ar-33 and smoothly led to the coupled product KM-31, which took another 4 functional manipulations to achieve the desired K-76.214 The enantiospecific total synthesis of the tetracyclic terpenoid L-671,776 was successfully reported from the chiral pool 18-β-glycyrrhetinic acid (SM-6) as the starting material, during which the structure of the natural product was revised. The key step is the coupling of the higher order organocuprate in situ synthesized from Ar-34 with the allylic bromide Kb-2, which was synthesized from 18β-glycyrrhetinic acid according to the reported method.215,216 Similarly, the key intermediate Kb-3 synthesized from (−)-sclareol in 7 steps reacted smoothly with the aryl copper derivative from Ar-5 to get the dibenzyl-protected drimane meroterpenoid KM-30, which underwent deprotection to deliver wiedendiol-A.78,217 (±)-BE-40644 was prepared through a similar approach. The drimanic building block (±)-Kb-3 was synthesized starting from geranylacetone in 6 steps with 15% overall yield. The lithiation of the aromatic intermediate Ar-32 with n-BuLi and concomitant transmetalation with CuCN afforded the desired organocopper reagent, which was smoothly coupled with the drimanic synthon (±)-Kb-3 to give the meroterpenoid derivative (±)-KM-29. This key constructive step facilitated the racemic synthesis of (±)-BE-40644.120 | ||
| Scheme 11 Synthesis of meroterpenoids via coupling of aryl-metallics and drimanyl halides. | ||
A distinct synthetic logic featuring the late-stage construction of the drimane units via polyene cyclization was also conducted for preparing a number of drimane meroterpenoids (Scheme 12). The key constructive step is the coupling of the farnesol or its scaffold equivalents with aromatic equivalents, preceding the construction of the drimane units. Taking the synthesis of drimane meroterpenoids from epoxyfarnesyl acetate as an example, Li2CuCl4-catalyzed allylic substitution reactions of the aryl Grignard reagent Ar-35 and Ar-36 by epoxyfarnesyl acetate afforded the epoxyfarnesyl aromatic structures Kc-1 or Kc-2. The titanocene-catalyzed epoxypolyene cyclization of these intermediates efficiently built the precursors KM-33 and KM-34 for drimane meroterpenoids zonarone, zonarol, puupehedione, and 8-epi-puupehedione.218 The first enantioselective total synthesis of (−)-walsucochin B was accomplished from geraniol in 15 steps, which also featured the coupling of the enantioenriched allylic bromide Kc-3 with metallic reagent derived from Ar-37 as the crucial step for the scaffold. The desired stereochemistry of the epoxide was constructed efficiently by Shi-asymmetric epoxidation. The following cationic polyolefin cyclization of the chiral epoxide, which was initiated by the presence of Et2AlCl, afforded the integral framework of the desired natural product (−)-walsucochin B.219 The total synthesis of austalide-related natural products was accomplished with some biomimetic transformations as key constructive steps. Polyketide aromatization of a trans, trans-farnesol (SM-10)-derived β,δ-diketodioxinone Kc-6 was realized through Pd2(dba)3-catalyzed decarboxylative allylic rearrangement and base-mediated cyclization.
 | ||
| Scheme 12 Synthesis of meroterpenoids via coupling of aryl-metallics with farnesyl ester and polyolefin cyclization. | ||
Noteworthily, this approach established the aromatic segment of meroterpenoids first, producing the corresponding farnesyl resorcylate Kc-7 for polyene cyclization. The subsequent titanium(III)-mediated reductive radical cyclization of its epoxide furnished the racemic meroterpenoid KM-38. The concomitant phenylselenonium ion-induced diastereoselective cyclization obtained the essential carbon framework of the austalide-related natural products, which could access (±)-17S-dihydroaustalide K and (±)-austalide K via sequential functional group-interconversions.220
3.4 Michael addition of aryl-metallics
The aforementioned organometallic reagents could serve as carbanions for the 1,4-conjugate addition to the activated olefins, which were embedded in or could serve as the desired drimane units of the meroterpenoids (Scheme 13). The racemic bicyclic exo-α,β-unsaturated ketone (±)-Kd-1 was synthesized and utilized in the synthesis of (±)-zonarol and (±)-isozonarol.221 The enantioenriched Michael acceptors (−)-Kd-1 and (+)-Kd-1 were synthetically available from geranylacetone through diastereoselective differentiation of the corresponding β-hydroxyl esters by the enantiopure (R)-L-(naphthyl)ethyl isocyanate. These two diastereomers of carbamates were readily separable by SiO2 chromatography.22 Utilization of 2,5-dimethoxyphenylmagnesium bromide (Ar-15) in the nucleophilic attack of these Michael acceptors efficiently provided enantiospecific chiral (+)-zonarol and (−)-zonarol.221 Facile oxidation afforded (+)-zonarone and (−)-zonarone in good yields.22 As showcased in Scheme 13, the cuprate catalyzed conjugated 1,4-addition of 2,4-dibenzyloxyphenylmagnesium bromide from Ar-39 to nordrim-9-en-8-one (+) Kd-1 was employed successfully as a key constructive step in the asymmetric synthesis of (−)-yahazunol.26 | ||
| Scheme 13 Asymmetric synthesis of meroterpenoids via Michael addition of aryl-metallics. | ||
3.5 Friedel–Crafts alkylation
As can be seen in the characteristics of siccanin and related natural products, many drimane meroterpenoids hold electron-rich aromatic segments caused by the decoration with one or more electro-donating groups (e.g., OH or MeO). By virtue of the innate features, the Lewis acid-catalyzed electrophilic alkylation of this kind of aromatic units with drimanyl alcohols provides a powerful tool that allows the preparation of drimane meroterpenoids. The α-drimanic alcohols (including synthons Ke-1, Ke-2, and Ke-3) were already available, which could be accessed by utilizing the starting materials α-ionone, Wieland–Miescher ketone and sclareolide (SM-1), respectively (Scheme 14). The hydroxyketone Ke-1 was prepared starting from α-ionone as a drimane synthon, and was directly arylated by the cationic resin AmberLyst-assisted Friedel–Crafts reaction with benzyl ether Ar-40 easily prepared from commercial sesamol. This afforded ketone KM-41 in good yield, which underwent methenylation and isomerization to get the O-allyl phenol framework necessary for NIS-PPh3-catalyzed spiroannulation. The regioselective and stereoselective achievement of spirodihydrobenzofuran facilitated the first total synthesis of corallidictyal D starting from α-ionone.222 | ||
| Scheme 14 Asymmetric synthesis of meroterpenoids via Friedel–Crafts alkylation. | ||
Starting from the enantiospecific Wieland–Miescher ketone, the chirality of the substrate-oriented asymmetric total synthesis of meroterpenoids (−)-F1839-I and (−)-corallidictyals B and D was established with overall yields of 13.7% (13 steps), 14.9% (11 steps) and 18.6% (10 steps), respectively. The catalytic Friedel–Crafts alkylation of the electron-rich arenes Ar-41 or Ar-42 with allylic alcohols Ke-2 or Ke-3 formed the complete carbon framework of the desired meroterpenoid natural products.160 This strategy was also employed by the same group in the enantiospecific total synthesis of chromazonarol, hongoquercins A and B, 8-epi-puupehediol and (+)-8-epi-puupehedione. The key is the careful variation of the electron-rich aromatic segments in the Friedel–Crafts reaction with allylic alcohol Ke-3.198,223
This approach is also applicable in the alkylation with farnesol, which featured a postponed drimane construction through polyolefin cyclization. The BF3-Et2O-assisted Friedel–Crafts alkylation of 4-methoxyphenol Ar-45 and sesamol Ar-46 with farnesol provided the (E,E)-2-farnesyl-4-methoxyphenol and (E,E)-2-farnesyl-4,5-methylenedioxyphenol. These two farnesyl aromatics were subjected to the Lewis acid (SnCl4)-assisted chiral Brønsted acid (axial phenols)-induced biomimetic cyclization, which elaborated the essential carbon scaffold of (−)-chromazonarol and (+)-8-epi-puupehedione.89
3.6 Diels–Alder cycloaddition
With a broad scope and operational simplicity, the [4 + 2]-electrocycloaddition of a conjugated diene and a dienophile is synthetically powerful in establishing unsaturated six-membered rings, especially for the naphthane-fused-naphthane-type drimane (hydro)quinones, e.g., cyclozonarone. Enantioselective synthesis of the drimane-related diene intermediates (+)-Kf-1, (−)-Kf-1, and Kf-2 could be accomplished through the chiral pool natural products (+)-albicanol, (−)-polygodial and trans-communic acid, respectively (Scheme 15).224–226 Meanwhile, the synthesis of racemic Kf-1 could be achieved through the polyene cyclization of epoxy-farnesol acetate.227 With the electron-poor quinone as the dienophile, cyclozonarones with different steric configurations could be accessible from cycloaddition with the diene intermediate in 1 or 2 steps.224,225,227 Replacing the conjugated diene species by carboxylate intermediate Kf-2 in cycloaddition with quinone led to the enantiospecific synthesis of the marine meroterpenoids neopetrosiquinone A and B.181 | ||
| Scheme 15 Enantiospecific substrate-oriented asymmetric synthesis of meroterpenoids via Diels–Alder cycloaddition. | ||
The Diels–Alder cycloaddition was also widely used in the synthesis of indane-fused-naphthane-type drimane (hydro)quinones, featuring a late-stage formation of the appendant (hydro)quinones units (Scheme 15). The enantiospecific diene intermediates Kf-3 and Kf-4 were accessible from (−)-sclareol and abietic acid or cupressic acid (SM-35), respectively. The first enantiospecific synthesis of akaol A was reported from (−)-sclareol, in which the cycloaddition of the key silyl dienol ether Kf-3 with methyl propiolate Ag-1 as the dienophile served as a key constructive step. This furnished the construction of the full carbon framework, and the following further oxidation with DDQ afforded the tetracyclic compound required by akaol A.173 The diene intermediate Kf-4 was used to react with methyl propiolate Ag-1 in 2 steps to get the drimane meroterpenoid KM-50, which was adopted in the synthesis of dasyscyphin B228 and dasyscyphin E.229 The Diels–Alder cycloaddition also finds wide applications in the synthesis of benzopyran-fused-naphthane-type and oxy-doped seven-membered ring-type drimane meroterpenoids. The masked diene, tricyclic α,β-enone kf-5 was obtained from (−)-sclareol in 2 steps, consisting of the ozone-lead(IV) degradation and base-mediated cyclization. The acid-catalyzed cyclization of α,β-enone kf-5 with E-1,2-bis(phenylsulphonyl)ethylene Ag-2 and isopropenyl acetate produced an isomer-mixture of the tetracyclic bis-sulphone KM-51.
The successive functional manipulations on this established scaffold led to hongoquercin A and cyclospongiaquinone-1, with each completed in 7 steps.230 The dienophile Ag-2 was also recruited in the efficient asymmetric construction of the carbon framework of cyclosiphonodictyol A and bis(sulfate)-cyclosiphonodictyol A. The methyl vinyl ketone Kf-6 could be achieved from (+)-sclareolide in 6 steps, which was subjected to the synthesis of dienol ester required in the Diels–Alder cycloaddition. This was further investigated for obtaining cyclosiphonodictyol A and its disulfate derivative.183 The enal intermediate Kf-7 was prepared from the enantioenriched Wieland–Miescher ketone in 11 steps. It can be employed in a phenyl boronic acid-mediated 6π-electrocyclization with orcinol Ar-48, delivering the inseparable epimers KM-53. The following Pd-C hydrogenation protocol for reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and deprotection of the benzyl group in one pot provided the desired isomer, which was required for getting (−)-phomoarcherin C.126
C double bond and deprotection of the benzyl group in one pot provided the desired isomer, which was required for getting (−)-phomoarcherin C.126
3.7 Suzuki–Miyaura coupling
The Suzuki–Miyaura coupling is one of the most powerful reactions for C–C bond formation, combining an organohalide or pseudo-halide electrophile and an organoboron nucleophile. The predominance stems not only from the mild conditions and broad functional tolerance, but also from the bench stability of the organoboron species.Bench-stable drimanyl organoborons, including borono-sclareolide (Kg-1) and drimanyl Bpin (Kg-2), have already been successfully employed in the modular synthesis of natural drimane meroterpenoids and a wide array of un-natural mimics, which will contribute greatly to lead innovation (Scheme 16). Borono-sclareolide (Kg-1) was synthesized from (+)-sclareolide in 5 steps, which was recruited for the Suzuki–Miyaura coupling with the readily available aryl halides (Ar-49, Ar-47, Ar-19, Ar-50, Ar-51). This approach has found many applications in obtaining (−)-zonarol, (−)-isozonarol, (+)-yahazunone, (+)-chromazonarol, 8-epi-puupehedione, (+)-yahazunol, (−)-zonarone, (−)-isozonarone, (+)-8-epi-puupehedione, puupehenol and (−)-pelorol.71,171,231 The other drimane synthon, drimanyl Bpin (Kg-2), could be synthesized from the commercially inexpensive (−)-sclareol (SM-2) in 3 steps by employing decarboxylative borylation. Then, it was submitted in Suzuki–Miyaura coupling as a powerful platform that enabled the expedient and enantioselective synthesis of drimane meroterpenoids, including (+)-yahazunol, (+)-chromazonarol, (+)-8-epi-puupehedione, (+)-dictyvaric acid, (+)-neoalbaconol and (+)-albaconol. This method also led to a large number of natural analogs of meroterpenoids.2,68 Noteworthily, the role of the pseudo-halide electrophile and organoboron nucleophile can be interchanged for the construction of drimane meroterpenoids. A nickel-catalyzed decarboxylative cross-coupling paradigm between drimane redox-active esters and phenylboronic acid was established to access a wide array of drimane meroterpenoids.
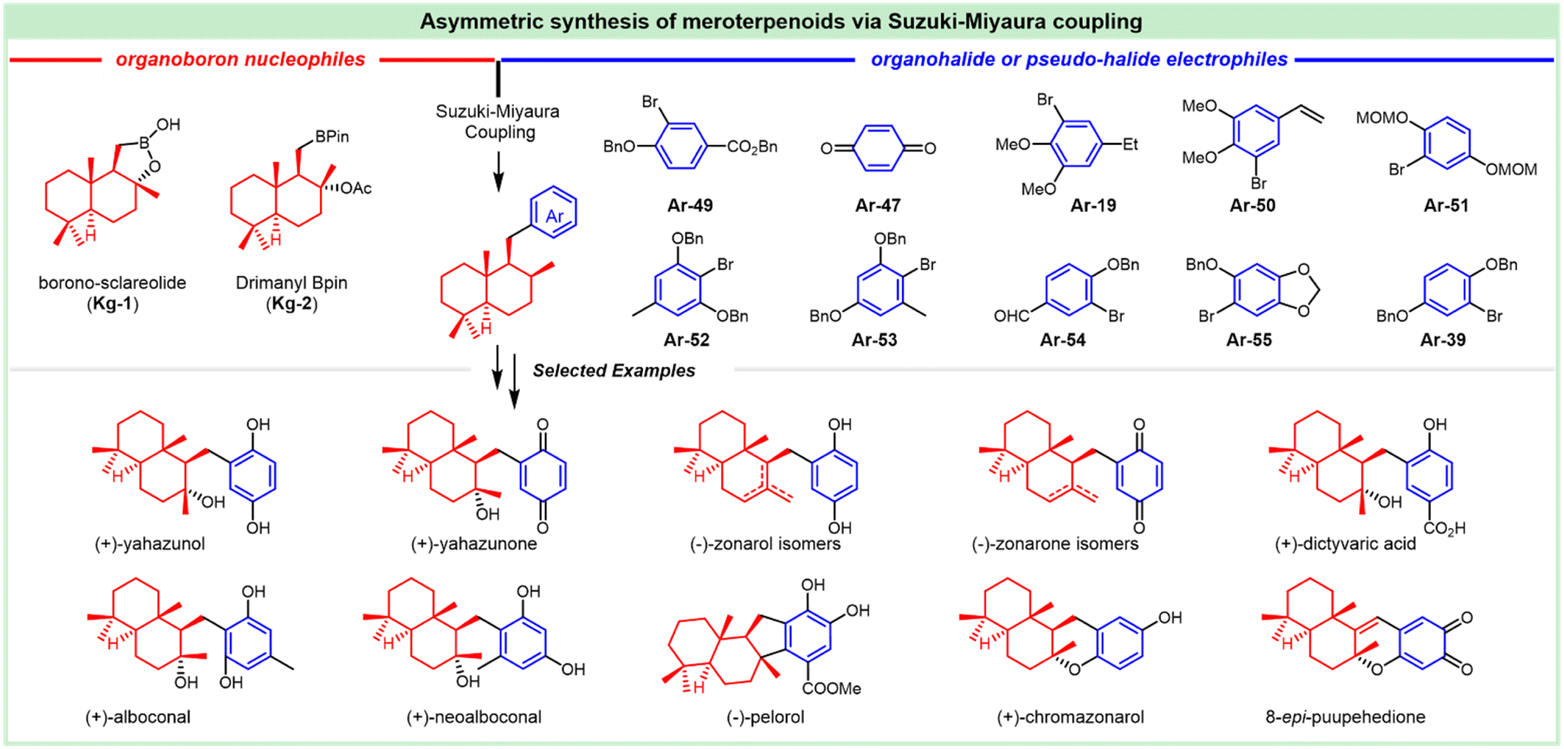 | ||
| Scheme 16 Asymmetric synthesis of meroterpenoids via Suzuki–Miyaura coupling. | ||
The bench-stable redox-active drimane ester drimanyl TCNHPI is easily available from the inexpensive feedstock sclareol.232
3.8 Carbonylation coupling
Carbonylation usually refers to the installation of carbon monoxide (CO) or an equivalent into the parent compound. Herein, this C1-synthon could be used for linking the nor-drimane (as electrophile) and the aryl nucleophiles through transition metal catalysis, under conditions similar to that of Suzuki–Miyaura coupling or Stille coupling.An enantioselective total synthesis of (−)-8-epi-chromazonarol was established from the commercially available R-carvone as the starting material (Scheme 17). The two essential coupling segments were bicyclic triflate Kh-1 and the aryl stannane derived from Ar-40. A typical lithium–halogen exchange/tributyltin chloride capture cascade was employed for converting the aryl bromide to aryl stannane. The requisite bicyclic triflate Kh-1 was easily enantiospecifically accomplished from (R)-carvone in 5 steps. With a practical synthesis of these two coupling components, Pd-catalyzed Stille carbonylative cross-coupling was performed successfully for constructing the desired α,β-unsaturated aryl ketone KM-54. This intermediate experienced an intramolecular oxa-Michael cyclization with the assistance of hydrazine hydrate, and then led to 8-epi-chromazonarol in another 7 steps.233 A similar approach was also employed in a concise total synthesis of (−)-15-oxopuupehenol in 8 steps (the longest linear route) from R-(−)-carvone in an overall yield of 18%. The key distinct feature was that accessibility to the entailed oxo-meroterpenoid KM-55 was accomplished by Suzuki carbonylative coupling of bicyclic vinyl triflate Kh-1 and aryl boronic acid Ar-41. The resulting compact tetra-substituted α,β-unsaturated aryl ketone KM-55 was exploited successfully to build the unique chromanone core via KOH-promoted intramolecular cyclization. With these key transformations, the synthesis of (−)-15-oxopuupehenol and (+)-puupehenone and formal syntheses of (−)-puupehenol and (+)-puupehedione were finished.234
 | ||
| Scheme 17 Asymmetric synthesis of meroterpenoids via carbonylation coupling. | ||
3.9 Carbene migratory insertion
Carbene migratory insertion was also employed in the enantiospecific construction of the essential carbon framework of drimane meroterpenoids (Scheme 18). The carbene precursors including drimanal hydrazones, Ki-1 and Ki-2, were prepared starting from (+)-sclerolide. The palladium-catalyzed tandem carbene migratory insertion was developed for coupling with various substituted aryl iodides (Ar-42, Ar-43, Ar-44, Ar-45, Ar-46, Ar-47). The epimer (8-epi-Ki-1) of drimanal hydrazones Ki-1 was also prepared successfully. Specifically, the key drimanal trimethoxystyrene 8-epi-KM-56 was constructed by the Pd-catalyzed cross-coupling of the aryl iodine Ar-42 and a drimanal hydrazone 8-epi-Ki-1. One could envision that the 8-epimers of the selected meroterpenoids depicted in Scheme 18 and scaffold-related natural products will be efficiently constructed in a modular approach. The benzopyran-fused ring system of puupehedione was installed efficiently by the in situ CAN-oxidation/intramolecular oxa-Stork-Danheiser transposition tandem reaction, which facilitated the enantioselective semisynthesis of puupehenone and puupehenol.235 With sclerolide as an easily available material, the applicability of this approach was also showcased by the divergent synthesis of (+)-8-epi-puupehedione, (+)-chromazonarol, (+)-yahazunol, (+)-yahazunone.236 siphonodictyal B, corallidictyals C/D,237 and (−)-15-oxopuupehenol.238 The synthetic applications were also demonstrated by the formal synthesis of (−)-pelorol and 9-epi- pelorol.239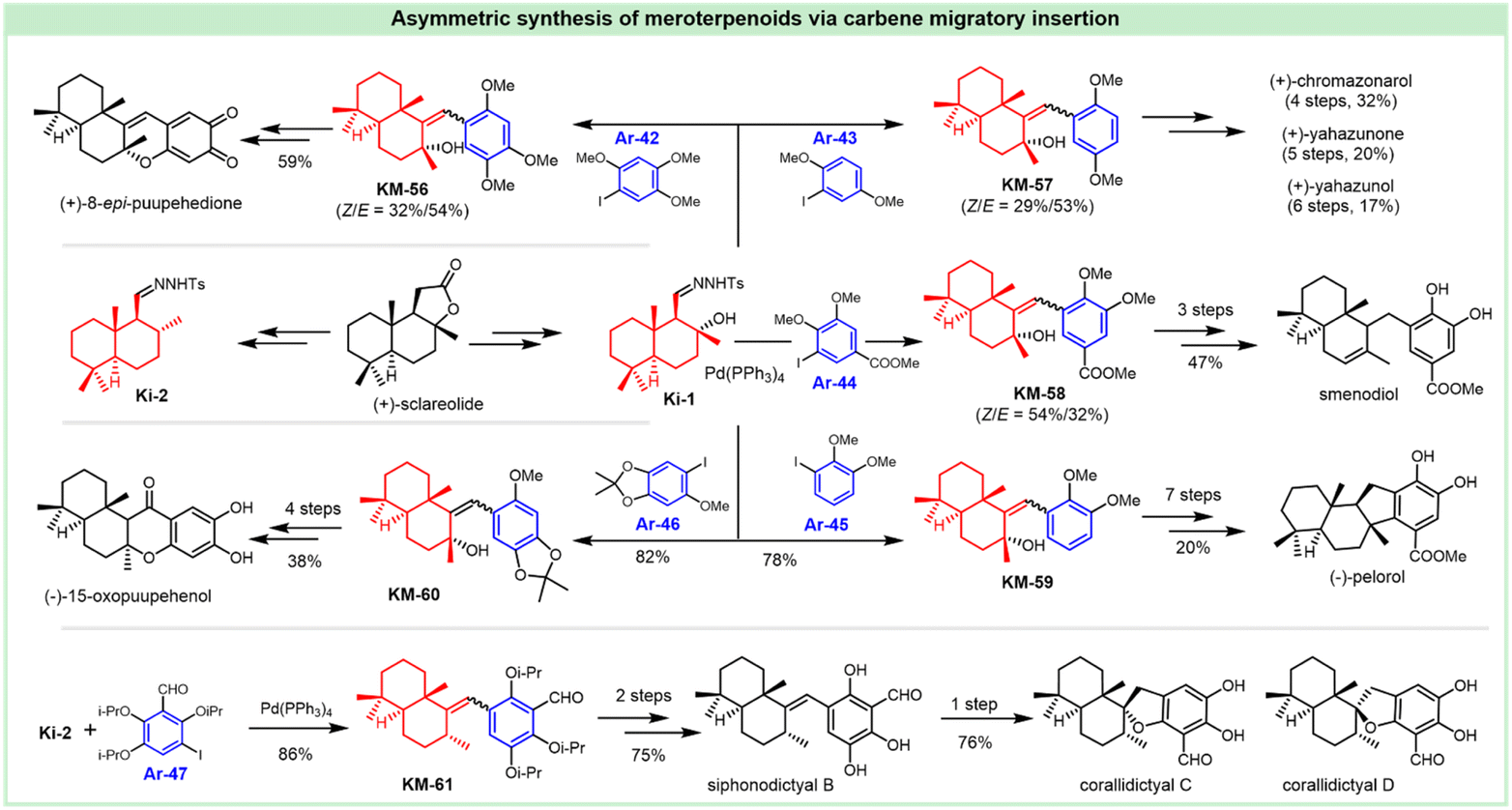 | ||
| Scheme 18 Asymmetric synthesis of meroterpenoids via carbene migratory insertion. | ||
3.10 Barton decarboxylative coupling
Considering the easy availability of homodrimanic acid from the commercially inexpensive (−)-sclareol, the decarboxylative arylation was envisaged for the synthesis of drimane meroterpenoids (Scheme 19). The commonly occurring quinone subunits can be introduced easily to the drimane segment. The cascade comprising radical decarboxylation, followed by quinone addition, was designed and conducted for the scalable synthesis of (+)-yahazunol in 5 longest linear sequences (LLS) starting from (−)-sclareol. The divergent translational potentials of (+)-yahazunol were demonstrated by the expedient preparation of (−)-zonarone, (−)-isozonarone, (−)-zonarol, (−)-isozonarol, (+)-chromazonarol, and (+)-yahazunone. This approach also enables the formal synthesis of puupehenol, puupehedione, and hongoquercin A.240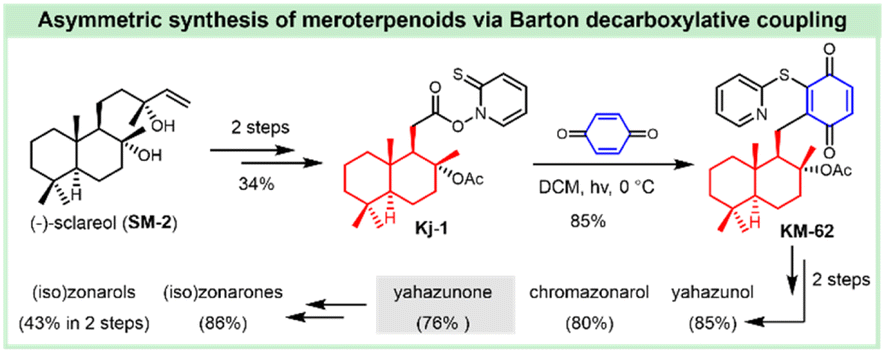 | ||
| Scheme 19 Asymmetric synthesis of meroterpenoids via Barton decarboxylative coupling. | ||
4. Summary and outlook
With diverse and intriguing scaffolds of attractive functions and bioactivities, natural products continue to serve as inspirations for lead innovation. The five-ring-fused antifungal natural product siccanin and related meroterpenoids are an important family of natural products that has attracted intensive attention from both synthetic and biological communities. While these natural products themselves are not well-organized for direct application as drugs or agrochemicals, the pre-validated bioactivities and distinct scaffolds have demonstrated that they can serve as good models for the design of new leads. For example, the polycyclic natural product siccanin could serve as a prototypical complicated antifungal agent against a large range of fungal strains without obvious toxicity. The polycyclic natural product siccanin has clearly demonstrated its capability in treating superficial dermatophyte infections. The benzopyran-fused-naphthane-type drimane (hydro)quinone, puupehenone, is well known for its diverse biological potentials, ranging from antifungal, anticancer, antibacterial, antiviral, antioxidative, and anti-inflammatory to insecticidal potentials. In contrast to the burgeoning progress in the discovery of medicinally important chemical entities, the efforts in the agrochemical discovery are still in its infancy. Although some modes of action or potential targets were discussed in the previous reports, e.g., siccanin was demonstrated to be an inhibitor of both mitochondrial complex II and complex III, more insights are necessary in the investigation of the specific targets and selectivity. Structural biology may be introduced and serve as a powerful tool for obtaining more detailed information about the interactions, which will be helpful in molecular design. In addition, other siccanin-related pharmaceutically important but structurally simplified and synthetically tractable ones (e.g., zonarol, puupehenone, pelorol, chrodrimanin and stachybotrysin) will show great value.Although various activities were described for siccanin and related meroterpenoids, many natural products did not produce judgable or (semi)quantitative data, especially for the complicated ones whose pharmacophores are challenging to verify. The function-oriented discovery and the concomitant optimization may facilitate the investigation of biologically important factors. Although many efforts were showcased for the construction of siccanin and some biologically important complicated meroterpenoids, the practicality has limited their further application in acquiring enough amount samples. For this point, more robust and modular synthetic strategies are necessary, as the developed classic methodologies frequently include the usage of unstable and flammable organometallic reagents. The recently developed Suzuki coupling or synthetic congeners avoided several requirements, including cryogenic conditions, inert atmosphere, and pyrophoric reagents. Synergistic effects with this approach could be envisaged by choosing the easily available and inexpensive chiral pool natural products, which will feature chemical fidelity and a good functional tolerance. The introduction of nonprecious metals or light-mediated synthesis will improve the synthetic practicality, which contributes greatly to the high throughput experimentation for library generation in routine medicinal applications in the identification of pharmaceutically promising and structurally novel scaffolds. In addition, establishing an asymmetric synthesis through easily available chiral catalysts with achiral and inexpensive raw starting materials will be desirable. The concomitant and late-stage functionalizations may accelerate the discovery of simplified drimane meroterpenoids as starting points for biological exploration. Valuable and practical starting points will be selected for the creation of a multitude of biologically important chemotypes for lead innovation.
5. Author contributions
This manuscript was written and revised by Shengxin Sun, Shengkun Li, and checked by Xia Wang, Nvdan Hu, and Shiqi Fu. Shengkun Li conceived and designed this work.6. Conflicts of interest
The authors declare no competing financial interest.7. Acknowledgments
We thank the reviewer for the nice comments. This work was financially supported by National Key R&D Program of China (2022YFD1700300), National Natural Science Foundation of China (22477019), and Guizhou Provincial Science and Technology Projects (ZK[2023]Key Program 008).8. Notes and references
- G. T. M. Bitchagno, V. A. Nchiozem-Ngnitedem, D. Melchert and S. A. Fobofou, Nat. Rev. Chem, 2022, 6, 806–822 CrossRef.
- X. Wang, S. Zhang, P. Cui and S. Li, Org. Lett., 2020, 22, 8702–8707 Search PubMed.
- K. Ishibashi, J. Antibiot., Ser. A, 1963, 15, 161–167 Search PubMed.
- B. Wu, V. Oesker, J. Wiese, S. Malien, R. Schmaljohann and J. F. Imhoff, Mar. Drugs, 2014, 12, 1924–1938 CrossRef.
- K. Ishibashi and M. Shirasaka, Tetrahedron Lett., 1967, 23, 2177–2179 Search PubMed.
- M. G. Bellotti and L. Riviera, Chemioterapia, 1985, 4, 431–433 CAS.
- F. Nemlioǧlu, J. Int. Med. Res., 1979, 7, 332–334 CrossRef.
- N. Nihashi, D. K. Inaoka, C. Tsuge, E. O. Balogun, Y. Osada, Y. Goto, Y. Matsumoto, T. Nara, T. Mogi, S. Harada and K. Kiyoshi, in Kala Azar in South Asia: Current Status and Sustainable Challenges, ed. E. Noiri and T. K. Jha, Springer International Publishing, Cham, 2016, pp. 101–122 Search PubMed.
- K. Nose and A. Endo, J. Bacteriol., 1971, 105, 176–184 Search PubMed.
- T. Mogi, T. Kawakami, H. Arai, Y. Igarashi, K. Matsushita, M. Mori, K. Shiomi, S. Ōmura, S. Harada and K. Kita, J. Biol. Chem., 2009, 146, 383–387 CAS.
- K. Komatsuya, T. Sakura, K. Shiomi, S. Ōmura, K. Hikosaka, T. Nozaki, K. Kita and D. K. Inaoka, Pharmaceuticals, 2022, 15, 903 CrossRef CAS PubMed.
- P. A. Wender, R. V. Quiroz and M. C. Stevens, Acc. Chem. Res., 2015, 48, 752–760 CrossRef CAS.
- K. Kawashima, K. Nakanishi, M. Tada and H. Nishikawa, Tetrahedron Lett., 1964, 5, 1227–1231 CrossRef.
- K. Kawashima, K. Nakanishi and H. Nishikawa, Chem. Pharm. Bull., 1964, 12, 796–803 CrossRef CAS PubMed.
- S. Ishii, M. Fujii and H. Akita, Chem. Pharm. Bull., 2009, 57(10), 1103–1106 CrossRef CAS PubMed.
- M. Goehl and K. Seifert, Eur. J. Org Chem., 2015, 2015, 6249–6258 CrossRef CAS.
- E. M. Wijeratne, P. A. Paranagama, M. T. Marron, M. K. Gunatilaka, A. E. Arnold and A. A. Gunatilaka, J. Nat. Prod., 2008, 71, 218–222 CrossRef CAS.
- W. Fenical, J. J. Sims, D. Squatrito, R. M. Wing and P. Radlick, J. Org. Chem., 1973, 38, 2383–2386 CrossRef CAS PubMed.
- C. Joshi Bipin, M. Kazaoka and A. Trischman Jacqueline, Res. J. Chem. Sci., 2012, 2, 9–13 CAS.
- G. Cimino, S. de Stefano, W. Fenical, L. Minale and J. J. Sims, Experientia, 1975, 31, 1250–1251 CrossRef CAS.
- H. Kakisawa, M. N. Dave, T. Kusumi, M. O. Ishitsuka and T. Iwashita, Heterocycles, 1984, 22, 2301–2307 CrossRef.
- K. Mori and M. Komatsu, Bull. Soc. Chim. Belg., 1986, 95, 771–781 CrossRef CAS.
- J. Schroder, C. Magg and K. Seifert, Tetrahedron Lett., 2000, 41, 5469–5473 CrossRef CAS.
- M. Kumagai, K. Nishikawa, H. Matsuura, T. Umezawa, F. Matsuda and T. Okino, Molecules, 2018, 23, 1211–1218 CrossRef PubMed.
- K. Kurata, K. Taniguchi and M. Suzuki, Phytochemistry, 1996, 41, 749–752 CrossRef CAS.
- T. Laube, W. Beil and K. Seifert, Tetrahedron, 2005, 61, 1141–1148 CrossRef CAS.
- T. Laube, A. Bernet, H. M. Dahse, I. D. Jacobsen and K. Seifert, Bioorg. Med. Chem., 2009, 17, 1422–1427 CrossRef CAS.
- R. J. Capon, D. R. Groves, S. Urban and R. G. Watson, Aust. J. Chem., 1993, 46, 1245–1253 CrossRef CAS.
- R. Talpir, A. Rudi, Y. Kashman, Y. Loya and A. Hizi, Tetrahedron, 1994, 50, 4179–4184 CrossRef CAS.
- D. S. Treitler, Z. Li, M. Krystal, N. A. Meanwell and S. A. Snyder, Bioorg. Med. Chem. Lett., 2013, 23, 2192–2196 CrossRef CAS.
- S. Poigny, T. Huor, M. Guyot and M. Samadi, J. Org. Chem., 1999, 64, 9318–9320 CrossRef CAS.
- S. A. Snyder, D. S. Treitler and A. P. Brucks, J. Am. Chem. Soc., 2010, 132, 14303–14314 CrossRef CAS PubMed.
- K. W. L. Yong, A. Jankam, J. N. A. Hooper, A. Suksamrarn and M. J. Garson, Tetrahedron, 2008, 64, 6341–6348 CrossRef CAS.
- K. Kono, M. Tanaka, T. Ogita, T. Hosoya and T. Kohama, J. Antibiot., 2000, 53, 459–466 CrossRef CAS PubMed.
- N. Maezawa, N. Furuichi, H. Tsuchikawa and S. Katsumura, Tetrahedron Lett., 2007, 48, 4865–4867 CrossRef CAS.
- S. M. Fang, C. B. Cui, C. W. Li, C. J. Wu, Z. J. Zhang, L. Li, X. J. Huang and W. C. Ye, Mar. Drugs, 2012, 10, 1266–1287 CrossRef CAS.
- W. J. He, X. J. Zhou, X. C. Qin, Y.-X. Mai, X.-P. Lin, S.-R. Liao, B. Yang, T. Zhang, Z.-C. Tu, J.-F. Wang and Y. Liu, Nat. Prod. Res., 2017, 31, 604–609 CrossRef CAS PubMed.
- X. Lin, X. Zhou, F. Wang, K. Liu, B. Yang, X. Yang, Y. Peng, J. Liu, Z. Ren and Y. Liu, Mar. Drugs, 2012, 10, 106–115 CrossRef CAS.
- T. Sassa and H. Yoshikoshi, Agric. Biol. Chem., 1983, 47, 187–189 CAS.
- T. Sassa and M. Nukina, Agric. Biol. Chem., 1984, 48, 1923–1925 CAS.
- H. Fujimoto, E. Nakamura, Y. P. Kim, E. Okuyama, M. Ishibashi and T. Sassa, J. Nat. Prod., 2001, 64, 1234–1237 CrossRef CAS.
- T. Sassa, A. Ishizaki, M. Nukina, M. Ikeda and T. Sugiyama, Biosci., Biotechnol., Biochem., 1998, 62, 2260–2262 CrossRef CAS PubMed.
- L. E. Schmidt, S. T. Deyrup, J. Baltrusaitis, D. C. Swenson, D. T. Wicklow and J. B. Gloer, J. Nat. Prod., 2010, 73, 404–408 CrossRef CAS PubMed.
- A. Hirose, H. Maeda, A. Tonouchi, T. Nehira and M. Hashimoto, Tetrahedron, 2014, 70, 1458–1463 CrossRef CAS.
- S. Uesugi, Y. Honmura, M. Nishiyama, K. Kusakabe, A. Tonouchi, T. Yamashita, M. Hashimoto and K. I. Kimura, Bioorg. Med. Chem., 2019, 27, 115161 CrossRef CAS PubMed.
- Y. Fu, P. Wu, J. Xue and X. Wei, J. Nat. Prod., 2014, 77, 1791–1799 CrossRef CAS.
- Y. Fu, P. Wu, J. Xue, H. Li and X. Wei, Mar. Drugs, 2015, 13, 3360–3367 CrossRef CAS PubMed.
- D. Liu, A. Yang, C. Wu, P. Guo, P. Proksch and W. Lin, Bioorg. Med. Chem. Lett., 2014, 24, 5288–5293 CrossRef CAS PubMed.
- F. Ishibashi, S. Sato, K. Sakai, S. Hirao and K. Kuwano, Biosci., Biotechnol., Biochem., 2013, 77, 1120–1122 CrossRef CAS.
- A. F. Barrero, E. J. Alvarez-Manzaneda, M. M. Herrador, R. Chahboun and P. Galera, Bioorg. Med. Chem. Lett., 1999, 9, 2325–2328 CrossRef CAS PubMed.
- E. Perez-Garcia, E. Zubia, M. J. Ortega and J. L. Carballo, J. Nat. Prod., 2005, 68, 653–658 CrossRef CAS.
- C. E. McNamara, L. Larsen, N. B. Perry, J. L. Harper, M. V. Berridge, E. W. Chia, M. Kelly and V. L. Webb, J. Nat. Prod., 2005, 68, 1431–1433 CrossRef CAS PubMed.
- B. W. Sullivan, D. J. Faulkner, G. K. Matsumoto, C. H. He and J. Clardy, J. Org. Chem., 1986, 51, 4568–4573 CrossRef CAS.
- V. Mukku, R. A. Edrada, F. J. Schmitz, M. K. Shanks, B. Chaudhuri and D. Fabbro, J. Nat. Prod., 2003, 66, 686–689 CrossRef CAS.
- A. L. Lane, L. Mular, E. J. Drenkard, T. L. Shearer, S. Engel, S. Fredericq, C. R. Fairchild, J. Prudhomme, K. Le Roch, M. E. Hay, W. Aalbersberg and J. Kubanek, Tetrahedron, 2010, 66, 455–461 CrossRef CAS PubMed.
- I. E. Mohamed, S. Kehraus, A. Krick, G. M. König, G. Kelter, A. Maier, H.-H. Fiebig, M. Kalesse, N. P. Malek and H. Gross, J. Nat. Prod., 2010, 73, 2053–2056 CrossRef CAS.
- I. E. Mohamed, H. Gross, A. Pontius, S. Kehraus, A. Krick, G. Kelter, A. Maier, H.-H. Fiebig and G. M. König, Org. Lett., 2009, 11, 5014–5017 CrossRef CAS.
- L. Zhang, Y. Shen, F. Wang, Y. Leng and J.-K. Liu, Phytochemistry, 2010, 71, 100–103 CrossRef CAS.
- H. Guo, T. Feng, Z.-H. Li and J.-K. Liu, J. Asian Nat. Prod. Res., 2012, 14, 950–955 CrossRef CAS PubMed.
- C. J. Zheng, C.-L. Shao, M. Chen, Z.-G. Niu, D.-L. Zhao and C. Y. Wang, Chem. Biodiversity, 2015, 12, 1407–1414 CrossRef CAS.
- Y. Venkateswarlu, D. J. Faulkner, J. L. R. Steiner, E. Corcoran and J. Clardy, J. Asian Nat. Prod. Res., 1991, 56, 6271–6274 CAS.
- J. Rodríguez, E. Quiñoá, R. Riguera, B. M. Peters, L. M. Abrell and P. Crews, Tetrahedron, 1992, 48, 6667–6680 CrossRef.
- M. Ochi, H. Kotsuki, K. Muraoka and T. Tokoroyama, Bull. Chem. Soc. Jpn., 1979, 52, 629–630 CrossRef CAS.
- T. Laube, J. Schroder, R. Stehle and K. Seifert, Tetrahedron, 2002, 58, 4299–4309 CrossRef CAS.
- B. Sullivan, P. Djura, D. E. McIntyre and D. J. Faulkner, Tetrahedron, 1981, 37, 979–982 CrossRef CAS.
- D. Zhi-Hui, D. Ze-Jun and L. Ji-Kai, Helv. Chim. Acta, 2001, 84, 259–262 CrossRef.
- M. Fujii, S. Ishii, R. Saito and H. Akita, Tetrahedron, 2008, 64, 5147–5149 CrossRef CAS.
- X. Wang, N. Hu, W. Kong, B. Song and S. Li, Eur. J. Med. Chem., 2022, 227, 113912 CrossRef CAS PubMed.
- Q. Deng, X. Yu, L. Xiao, Z. Hu, X. Luo, Y. Tao, L. Yang, X. Liu, H. Chen, Z. Ding, T. Feng, Y. Tang, X. Weng, J. Gao, W. Yi, A. M. Bode, Z. Dong, J. Liu and Y. Cao, Cell Death Dis., 2013, 4, e804 CrossRef CAS PubMed.
- F. Song, X. Fan, X. Lu, J. Zhao, L. Han and J. Shi, Chin. Chem. Lett., 2004, 15, 316–318 Search PubMed.
- D. D. Dixon, J. W. Lockner, Q. Zhou and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 8432–8435 CAS.
- L. Shubina, A. Kalinovsky, T. Makarieva, S. Fedorov, S. Dyshlovoy, P. Dmitrenok, I. Kapustina, E. Mollo, N. Utkina, V. Krasokhin, V. Denisenko and V. Stonik, Nat. Prod. Commun., 2012, 7, 487–490 CAS.
- X. Zhang, H. Y. Xu, A. M. Huang, L. Wang, Q. Wang, P. Y. Cao and P. M. Yang, Chem. Pharm. Bull., 2016, 64, 1036–1042 CrossRef CAS PubMed.
- V. K. Thu, D. T. Trang, D. T. T. Hang, N. X. Nhiem, T. H. Quang, P. H. Yen, B. H. Tai and P. V. Kiem, Phytochem. Lett., 2021, 44, 115–119 CrossRef CAS.
- B. Chen, Q. Zhao, Y. C. Gu, L. Lan, C. Y. Wang and Y. W. Guo, Mar. Drugs, 2022, 20, 118 Search PubMed.
- K. Chakraborty, T. Antony and M. Joy, Algal Res., 2019, 40, 101472 CrossRef.
- S. J. Coval, M. A. Conover, R. Mierzwa, A. King, M. S. Puar, D. W. Phife, J.-K. Pai, R. E. Burrier, H.-S. Ahn, G. C. Boykow, M. Patel and S. A. Pomponi, Bioorg. Med. Chem. Lett., 1995, 5, 605–610 CrossRef CAS.
- A. F. Barrero, E. J. Alvarez-Manzaneda and R. Chahboun, Tetrahedron, 1998, 54, 5635–5650 CrossRef CAS.
- R. Kazlauskas, P. T. Murphy, R. G. Warren, R. J. Wells and B. John F, Aust. J. Chem., 1978, 31, 2685–2697 CrossRef CAS.
- A. F. Barrero, E. J. AlvarezManzaneda and R. Chahboun, Bioorg. Med. Chem. Lett., 1997, 38, 8101–8104 CAS.
- S. Urban and R. Capon, Aust. J. Chem., 1996, 49, 611–615 CrossRef CAS.
- M. Goehl and K. Seifert, Eur. J. Org Chem., 2014, 2014, 6975–6982 CrossRef CAS.
- A. W. Markwell-Heys, K. K. W. Kuan and J. H. George, Org. Lett., 2015, 17, 4228–4231 CrossRef CAS.
- A. Grube, M. Assmann, E. Lichte, F. Sasse, J. R. Pawlik and M. Köck, J. Nat. Prod., 2007, 70, 504–509 CrossRef CAS PubMed.
- T. Kikuchi, K. Narita, K. Saijo, C. Ishioka and T. Katoh, Eur. J. Org Chem., 2016, 2016, 5659–5666 CrossRef CAS.
- W. Fenical and O. Mcconnell, Specialia, 1975, 31, 1004–1005 CAS.
- G. Cimino, S. D. Stefano and L. Minale, Specialia, 1975, 31, 1117–1118 CAS.
- P. Djura, D. B. Stierle, B. Sullivan and D. J. Faulkner, J. Org. Chem., 1980, 45, 1435–1441 CrossRef CAS.
- H. Ishibashi, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 11122–11123 CrossRef CAS.
- M. E. Castro, M. González-Iriarte, A. F. Barrero, N. Salvador-Tormo, R. Muñoz-Chápuli, M. A. Medina and A. R. Quesada, Int. J. Cancer, 2004, 110, 31–38 CrossRef CAS.
- D.-S. Lee, I. H. Hwang, N.-K. Im, G.-S. Jeong and M. K. Na, Nat. Prod. Sci., 2015, 21, 268–272 CrossRef CAS.
- Y. Takeda, K. Narita and T. Katoh, Eur. J. Org Chem., 2017, 2017, 901–907 CrossRef CAS.
- B. N. Ravi, H. P. Perzanowski, R. A. Ross, T. R. Erdman and P. J. Scheuer, Pure Appl. Chem., 1979, 51, 1893–1900 CAS.
- S. Kohmoto, O. J. McConnell, A. Wright, F. Koehn, W. Thompson, M. Lui and K. M. Snader, J. Nat. Prod., 1987, 50, 336 CrossRef CAS.
- R. E. Longley, O. J. McConnell, E. Essich and D. Harmody, J. Nat. Prod., 1993, 56, 915–920 CrossRef CAS.
- S. Takamatsu, T. W. Hodges, I. Rajbhandari, W. H. Gerwick, M. T. Hamann and D. G. Nagle, J. Nat. Prod., 2003, 66, 605–608 CrossRef CAS PubMed.
- N. K. Utkina, V. A. Denisenko and V. B. Krasokhin, Tetrahedron Lett., 2011, 52, 3765–3768 CrossRef CAS.
- K. L. Cheney, A. White, I. W. Mudianta, A. E. Winters, M. Quezada, R. J. Capon, E. Mollo and M. J. Garson, PLoS One, 2016, 11, e0145134 CrossRef PubMed.
- W.-H. Xu, Y. Ding, M. R. Jacob, A. K. Agarwal, A. M. Clark, D. Ferreira, Z.-S. Liang and X.-C. Li, Bioorg. Med. Chem. Lett., 2009, 19, 6140–6143 CrossRef CAS PubMed.
- K. A. El Sayed, P. Bartyzel, X. Shen, T. L. Perry, J. K. Zjawiony and M. T. Hamann, Tetrahedron, 2000, 56, 949–953 CrossRef CAS.
- T. C. McKee, D. Rabe, H. R. Bokesch, T. Grkovic, E. L. Whitson, T. Diyabalanage, A. W. W. Van Wyk, S. R. Marcum, R. S. Gardella, K. R. Gustafson, W. M. Linehan, J. B. McMahon and D. P. Bottaro, J. Nat. Prod., 2012, 75, 1632–1636 CrossRef CAS.
- H. Y. Ebrahim and K. A. El Sayed, Mar. Drugs, 2016, 14, 57 CrossRef PubMed.
- S. K. Tripathi, Q. Feng, L. Liu, D. E. Levin, K. K. Roy, R. J. Doerksen, S. R. Baerson, X. Shi, X. Pan, W.-H. Xu, X.-C. Li, A. M. Clark and A. K. Agarwal, mSphere, 2020, 5, e00818–e00819 CrossRef CAS PubMed.
- B. Martinez-Poveda, A. R. Quesada, M. A. Medina, A. R. Quesada and M. A. Medina, Mar. Drugs, 2017, 15, 325 CrossRef.
- S. J. Robinson, E. K. Hoobler, M. Riener, S. T. Loveridge, K. Tenney, F. A. Valeriote, T. R. Holman and P. Crews, J. Nat. Prod., 2009, 72, 1857–1863 CrossRef CAS PubMed.
- M. L. Ciavatta, M. P. Lopez Gresa, M. Gavagnin, V. Romero, D. Melck, E. Manzo, Y.-W. Guo, R. van Soest and G. Cimino, Tetrahedron, 2007, 63, 1380–1384 CrossRef CAS.
- B. Martínez-Poveda, A. R. Quesada and M. Á. Medina, J. Cell. Mol. Med., 2008, 12, 701–706 CrossRef PubMed.
- D. H. Hua, X. Huang, Y. Chen, S. K. Battina, M. Tamura, S. K. Noh, S. I. Koo, I. Namatame, H. Tomoda, E. M. Perchellet and J.-P. Perchellet, J. Org. Chem., 2004, 69, 6065–6078 CrossRef CAS PubMed.
- A. F. Barrero, E. J. Alvarez-Manzaneda, R. Chahboun, M. Cortés and V. Armstrong, Tetrahedron, 1999, 55, 15181–15208 CrossRef CAS.
- M. T. Hamann, P. J. Scheuer and M. Kelly-Borges, J. Org. Chem., 1993, 58, 6565–6569 CrossRef CAS.
- M. T. Hamann and P. J. Scheuer, Tetrahedron Lett., 1991, 32, 5671–5672 CrossRef CAS.
- S. S. Nasu, B. K. S. Yeung, M. T. Hamann and P. J. Scheuer, J. Org. Chem., 1995, 60, 7290–7292 CrossRef CAS.
- O. Arjona, M. Garranzo, J. Mahugo, E. Maroto, J. Plumet and B. Sáez, Tetrahedron Lett., 1997, 38, 7249–7252 CrossRef CAS.
- M.-L. Bourguet-Kondracki, F. Lacombe and M. Guyot, J. Nat. Prod., 1999, 62, 1304–1305 CrossRef CAS PubMed.
- T. S. Hilliard, G. Miklossy, C. Chock, P. Yue, P. Williams and J. Turkson, Mol. Cancer Ther., 2017, 16, 601–613 CrossRef CAS PubMed.
- I. C. Pina, M. L. Sanders and P. Crews, J. Nat. Prod., 2003, 66, 2–6 CrossRef CAS.
- K. Hagiwara, J. E. Garcia Hernandez, M. K. Harper, A. Carroll, C. A. Motti, J. Awaya, H.-Y. Nguyen and A. D. Wright, J. Nat. Prod., 2015, 78, 325–329 CrossRef CAS PubMed.
- H. A. Wahab, N. B. Pham, J. N. A. Hooper, R. J. Quinn, H. A. Wahab, T. S. T. Muhammad, T. S. T. Muhammad and J. N. A. Hooper, Mar. Drugs, 2016, 15, 6 CrossRef PubMed.
- K. Torigoe, N. Wakasugi, N. Sakaizumi, T. Ikejima, H. Suzuki, K. Kojiri and H. Suda, J. Antibiot., 1996, 49, 314–317 CrossRef CAS.
- H. Tsujimori and K. Mori, Biosci. Biotechnol. Biochem., 2001, 65, 167–171 CrossRef CAS PubMed.
- S. B. Singh, D. L. Zink, M. Williams, J. D. Polishook, M. Sanchez, K. C. Silverman and R. B. Lingham, Bioorg. Med. Chem. Lett., 1998, 8, 2071–2076 CrossRef CAS.
- D. M. Roll, J. K. Manning and G. T. Carter, J. Antibiot., 1998, 51, 635–639 CrossRef CAS PubMed.
- H. Tsujimori, M. Bando and K. Mori, Eur. J. Org Chem., 2000, 2000, 297–302 CrossRef.
- K. I. Takao, T. Sasaki, T. Kozaki, Y. Yanagisawa, K. I. Tadano, A. Kawashima and H. Shinonaga, Org. Lett., 2001, 3, 4291–4294 CrossRef CAS.
- C. Hemtasin, S. Kanokmedhakul, K. Kanokmedhakul, C. Hahnvajanawong, K. Soytong, S. Prabpai and P. Kongsaeree, J. Nat. Prod., 2011, 74, 609–613 CrossRef CAS.
- D. H. Dethe and B. VijayKumar, J. Org. Chem., 2019, 84, 14053–14060 CrossRef CAS PubMed.
- R. M. Horak, P. S. Steyn and R. Vleggaar, J. Chem. Soc., Perkin Trans. 1, 1985, 1, 357–361 RSC.
- R. M. Horak, P. S. Steyn, R. Vleggaar and C. J. Rabie, J. Chem. Soc., Perkin Trans. 1, 1985, 1, 345–356 RSC.
- R. M. Horak, P. S. Steyn, R. Vleggaar and C. J. Rabie, J. Chem. Soc., Perkin Trans. 1, 1985, 1, 363–367 RSC.
- W. Wang, J. Lee, K.-J. Kim, Y. Sung, K.-H. Park, E. Oh, C. Park, Y. J. Son and H. Kang, J. Nat. Prod., 2019, 82, 3083–3088 CrossRef CAS PubMed.
- O. I. Zhuravleva, M. P. Sobolevskaya, E. V. Leshchenko, N. N. Kirichuk, V. A. Denisenko, P. S. Dmitrenok, S. A. Dyshlovoy, A. M. Zakharenko, N. Y. Kim and S. S. Afiyatullov, J. Nat. Prod., 2014, 77, 1390–1395 CrossRef CAS.
- T. Amagata, S. Whitman, T. A. Johnson, C. C. Stessman, C. P. Loo, E. Lobkovsky, J. Clardy, P. Crews and T. R. Holman, J. Nat. Prod., 2003, 66, 230–235 CrossRef CAS.
- H. Yamazaki, K. Kobayashi, D. Matsuda, K. Nonaka, R. Masuma, S. Ōmura and H. Tomoda, J. Antibiot., 2009, 62, 195–200 CrossRef CAS.
- H. Yamazaki, N. Ugaki, D. Matsuda and H. Tomoda, J. Antibiot., 2010, 63, 315–318 CrossRef CAS.
- H. Hayashi, Y. Oka, K. Kai and K. Akiyama, Biosci., Biotechnol., Biochem., 2012, 76, 745–748 CrossRef CAS PubMed.
- H. Hayashi, Y. Oka, K. Kai and K. Akiyama, Biosci., Biotechnol., Biochem., 2012, 76, 1765–1768 CrossRef CAS.
- Y. Xu, S. Furutani, M. Ihara, Y. Ling, X. Yang, K. Kai, H. Hayashi and K. Matsuda, PLoS One, 2015, 10, e0122629 CrossRef.
- H. Zhou, L. Li, W. Wang, Q. Che, D. Li, Q. Gu and T. Zhu, J. Nat. Prod., 2015, 78, 1442–1445 CrossRef CAS PubMed.
- H. Yamazaki, W. Nakayama, O. Takahashi, R. Kirikoshi, Y. Izumikawa, K. Iwasaki, K. Toraiwa, K. Ukai, H. Rotinsulu, D. S. Wewengkang, D. A. Sumilat, R. E. P. Mangindaan and M. Namikoshi, Bioorg. Med. Chem. Lett., 2015, 25, 3087–3090 CrossRef CAS PubMed.
- F.-D. Kong, Q.-Y. Ma, S.-Z. Huang, P. Wang, J.-F. Wang, L.-M. Zhou, J.-Z. Yuan, H.-F. Dai and Y.-X. Zhao, J. Nat. Prod., 2017, 80, 1039–1047 CrossRef CAS PubMed.
- H. Qiao, S.-H. Zhang, Y. Dong, Y. Yang, R. Xu, B. Chen, Y. Wang, T.-J. Zhu, C.-B. Cui, G.-G. Zhang and C.-W. Li, Nat. Prod. Res., 2022, 36, 1834–1841 CrossRef CAS.
- F.-D. Kong, R.-S. Zhang, Q.-Y. Ma, Q.-Y. Xie, P. Wang, P.-W. Chen, L.-M. Zhou, H.-F. Dai, D.-Q. Luo and Y.-X. Zhao, Fitoterapia, 2017, 122, 1–6 CrossRef CAS.
- Y. Fu, C. Li, J. Zhu, L. Zhang, Y. Wang, Q. Chen, L. Xu, S. Zhang, Y. Fang and T. Liu, Biochem. Syst. Ecol., 2020, 93, 104186 CrossRef CAS.
- L. Du, Y.-D. Zhou and D. G. Nagle, J. Nat. Prod., 2013, 76, 1175–1181 CrossRef CAS.
- W. X. Chunyu, Z. G. Ding, M. G. Li, J. Y. Zhao, S. J. Gu, Y. Gao, F. Wang, J. H. Ding and M. L. Wen, Helv. Chim. Acta, 2016, 99, 583–587 CrossRef CAS.
- J. W. Kim, S. K. Ko, H. M. Kim, G. H. Kim, S. Son, G. S. Kim, G. J. Hwang, E. S. Jeon, K.-S. Shin, I. J. Ryoo, Y. S. Hong, H. Oh, K. H. Lee, N. K. Soung, D. Hashizume, T. Nogawa, S. Takahashi, B. Y. Kim, H. Osada, J.-H. Jang and J. S. Ahn, J. Nat. Prod., 2016, 79, 2703–2708 CrossRef CAS.
- K. Hong, T. Kinoshita, W. Miyazaki, T. Izawa and K. Inoue, J. Immunol., 1979, 122, 2418–2423 CrossRef CAS.
- H. Kaise, M. Shinohara, W. Miyazaki, T. Izawa, Y. Nakano, M. Sugawara, K. Sugiura and K. Sasaki, J. Chem. Soc., Chem. Commun., 1979, 726–727 RSC.
- K. Sakai, K. Watanabe, K. Masuda, M. Tsuji, K. Hasumi and A. Endo, J. Antibiot., 1995, 48, 447–456 CrossRef CAS PubMed.
- Y. K. Lam, C. F. Wichmann, M. S. Meinz, L. Guariglia, R. A. Giacobbe, S. Mochales, L. Kong, S. S. Honeycutt, D. Zink and G. F. Bills, et al. , J. Antibiot., 1992, 45, 1397–1403 CrossRef CAS.
- S. Stefanelli, F. Sponga, P. Ferrari, C. Sottani, E. Corti, C. Brunati and K. Islam, J. Antibiot., 1996, 49, 611–616 CrossRef CAS PubMed.
- W. A. Ayer and S. Miao, Can. J. Chem., 1993, 71, 487–493 CrossRef CAS.
- A. Jagels, Y. Hövelmann, A. Zielinski, M. Esselen, J. Köhler, F. Hübner and H. U. Humpf, Mycotoxin Res., 2018, 34, 179–185 CrossRef CAS PubMed.
- Y.-J. Kwon, M.-J. Sohn, H.-J. Kim and W.-G. Kim, Biol. Pharm. Bull., 2014, 37, 1406–1410 CrossRef CAS.
- S. Haidar, F. M. Juergens, D. Aichele, A. Jagels, H. U. Humpf and J. Jose, Molecules, 2021, 26, 4453 CrossRef CAS.
- R. Kaneto, K. Dobashi, I. Kojima, K. Sakai, N. Shibamoto, T. Yoshioka, H. Nishida, R. Okamoto, H. Akagawa and S. Mizuno, J. Antibiot., 1994, 47, 727–730 CrossRef CAS PubMed.
- L. S. Kamalov, S. F. Aripova and M. I. Isaev, Chem. Nat. Compd., 1999, 35, 82–85 CrossRef CAS.
- X. H. Ma, W. M. Zheng, K. H. Sun, X. F. Gu, X. M. Zeng, H. T. Zhang, T. H. Zhong, Z. Z. Shao and Y. H. Zhang, Nat. Prod. Res., 2019, 33, 386–392 CrossRef CAS.
- J. A. Chan, A. J. Freyer, B. K. Carté, M. E. Hemling, G. A. Hofmann, M. R. Mattern, M. A. Mentzer and J. W. Westley, J. Nat. Prod., 1994, 57, 1543–1548 CrossRef CAS PubMed.
- D. H. Dethe, B. D. Dherange, S. Ali and M. M. Parsutkar, Org. Biomol. Chem., 2016, 15, 65–68 RSC.
- J. Zhao, J. Feng, Z. Tan, J. Liu, J. Zhao, R. Chen, K. Xie, D. Zhang, Y. Li, L. Yu, X. Chen and J. Dai, J. Nat. Prod., 2017, 80, 1819–1826 CrossRef CAS PubMed.
- M. Chu, R. Mierzwa, I. Truumees, A. King, M. Patel, J. Pichardo, A. Hart, B. Dasmahapatra, P. R. Das and M. S. Puar, Tetrahedron Lett., 1996, 37, 3943–3946 CrossRef CAS.
- B. B. Jarvis, J. Salemme and A. Morais, Nat. Toxins, 1995, 3, 10–16 CrossRef CAS.
- H. Zhang, M. H. Yang, F. F. Zhuo, N. Gao, X. B. Cheng, X. B. Wang, Y. H. Pei and L. Y. Kong, RSC Adv., 2019, 9, 3520–3531 RSC.
- P. Zhang, Y. Li, C. Jia, J. Lang, S. I. Niaz, J. Li, J. Yuan, J. Yu, S. Chen and L. Liu, RSC Adv., 2017, 7, 49910–49916 RSC.
- L. S. Kamalov, S. F. Aripova and M. I. Isaev, Chem. Nat. Compd., 1998, 34, 616–619 CrossRef CAS.
- P. Mou, Q. Zhang, J. Peng, X. Jiang, L. Zhang, Z. Zhou, C. Zhang and Y. Zhu, Fitoterapia, 2021, 152, 104937 CrossRef CAS PubMed.
- J. H. Kwak, F. J. Schmitz and M. Kelly, J. Nat. Prod., 2000, 63, 1153–1156 CrossRef CAS PubMed.
- L. Yang, D. E. Williams, A. Mui, C. Ong, G. Krystal, R. van Soest and R. J. Andersen, Org. Lett., 2005, 7, 1073–1076 CrossRef CAS.
- S. Carpi, S. Carpi, B. Polini, S. Brogi, V. Calderone, P. Nieri, E. Scoditti, B. Polini, S. Brogi, V. Calderone, P. Nieri, P. Proksch and S. S. Ebada, Mar. Drugs, 2022, 20, 427 CrossRef CAS.
- Y. Luo, H. Chen, J. Weng and G. Lu, Mar. Drugs, 2016, 14, 118 CrossRef.
- W. Z. Tang, H. M. Zhao, Y. Tian, S. W. Dai, A. Zhang, H. W. Lin, C. X. Zhang and F. Yang, Tetrahedron Lett., 2022, 93, 153690 CrossRef CAS.
- E. Alvarez-Manzaneda, R. Chahboun, E. Alvarez, A. Fernandez, R. Alvarez-Manzaneda, A. Haidour, J. M. Ramos and A. Akhaouzan, Chem. Commun., 2012, 48, 606–608 RSC.
- J. Li, F. Yang, Z. Wang, W. Wu, L. Liu, S.-P. Wang, B.-X. Zhao, W.-H. Jiao, S.-H. Xu and H.-W. Lin, Org. Biomol. Chem., 2018, 16, 6773–6782 RSC.
- V. Rojas de la Parra, V. Mierau, T. Anke and O. Sterner, Tetrahedron, 2006, 62, 1828–1832 CrossRef CAS.
- J. C. Liermann, H. Kolshorn, H. Anke, E. Thines and T. Opatz, J. Nat. Prod., 2008, 71, 1654–1656 CrossRef CAS.
- L. Flores-Bocanegra, M. Augustinovic, H. A. Raja, S. J. Kurina, A. C. Maldonado, J. E. Burdette, J. O. Falkinham, C. J. Pearce and N. H. Oberlies, Tetrahedron Lett., 2021, 68, 152896 CrossRef CAS PubMed.
- Z. W. Zhou, S. Yin, H. Y. Zhang, Y. Fu, S. P. Yang, X. N. Wang, Y. Wu, X. C. Tang and J. M. Yue, Org. Lett., 2008, 10, 465–468 CrossRef CAS PubMed.
- M. L. Han, Y. Shen, Y. Leng, H. Zhang and J. M. Yue, RSC Adv., 2014, 4, 19150–19158 RSC.
- P. L. Winder, H. L. Baker, P. Linley, E. A. Guzmán, S. A. Pomponi, M. C. Diaz, J. K. Reed and A. E. Wright, Bioorg. Med. Chem., 2011, 19, 6599–6603 CrossRef CAS PubMed.
- I. Chayboun, E. Boulifa, A. I. Mansour, F. Rodriguez-Serrano, E. Carrasco, P. J. Alvarez, R. Chahboun and E. Alvarez-Manzaneda, J. Nat. Prod., 2015, 78, 1026–1036 CrossRef CAS.
- Q. Göthel and M. Köck, Beilstein J. Org. Chem., 2014, 10, 613–621 CrossRef PubMed.
- J. A. Gil, F. Arias, R. Chahboun and E. Alvarez-Manzaneda, J. Org. Chem., 2020, 85, 3799–3805 CrossRef CAS PubMed.
- K. B. Killday, A. E. Wright, R. H. Jackson and M. A. Sills, J. Nat. Prod., 1995, 58, 958–960 CrossRef CAS.
- N. K. Utkina and M. V. Veselova, Chem. Nat. Compd., 1990, 26, 37–40 CrossRef.
- M. T. Hamann and P. J. Scheuer, J. Org. Chem., 1993, 58, 6565–6569 CrossRef CAS.
- M. Gordaliza, Mar. Drugs, 2010, 8, 2849–2870 CrossRef CAS PubMed.
- R. M. Lukes, G. I. Poos and L. H. Sarett, J. Am. Chem. Soc., 1952, 74, 1401–1405 CrossRef CAS.
- M. Kato, K. Heima, Y. Matsumura and A. Yoshikoshi, J. Am. Chem. Soc., 1981, 103, 2434–2435 CrossRef CAS.
- H. J. Liu and B. Ramani, Tetrahedron Lett., 1988, 29, 6721–6724 CrossRef CAS.
- B. M. Trost, F. J. Fleitz and W. J. Watkins, J. Am. Chem. Soc., 1996, 118, 5146–5147 CrossRef CAS.
- B. M. Trost, H. C. Shen and J.-P. Surivet, Angew. Chem., Int. Ed., 2003, 42, 3943–3947 CrossRef CAS.
- A. Castillo, L. Silva, D. Briones, J. F. Quilez del Moral and A. F. Barrero, Eur. J. Org Chem., 2015, 2015, 3266–3273 CrossRef CAS.
- L. F. Tietze, S. Jackenkroll, D. Ganapathy and J. R. Reiner, Synlett, 2016, 27, 96–100 CrossRef CAS.
- K. Kuchkova, Synthesis, 1997, 1997, 1045–1048 CrossRef.
- S. Chackalamannil, Y. Wang, Y. Xia and M. Czarniecki, Tetrahedron Lett., 1995, 36, 5315–5318 CrossRef CAS.
- S. Quideau, M. Lebon and A. M. Lamidey, Org. Lett., 2002, 4, 3975–3978 CrossRef CAS.
- J. H. George, J. E. Baldwin and R. M. Adlington, Org. Lett., 2010, 12, 2394–2397 CrossRef CAS PubMed.
- J. Villamizar, A. L. Orcajo, J. Fuentes, E. Tropper and R. Alonso, J. Chem. Res., Synop., 2002, 395–397 CrossRef CAS.
- S. Garai and G. Mehta, Tetrahedron Lett., 2014, 55, 6252–6256 CrossRef CAS.
- T. Hata, K. Tanaka and S. Katsumura, Tetrahedron Lett., 1999, 40, 1731–1734 CrossRef CAS.
- N. Furuichi, T. Hata, H. Soetjipto, M. Kato and S. Katsumura, Tetrahedron, 2001, 57, 8425–8442 CrossRef CAS.
- J. H. Huang and X. G. Lei, Sci. China: Chem., 2013, 56, 349–353 CrossRef CAS.
- J. Villamizar, J. Fuentes, E. Tropper, A. L. Orcajo and R. Alonso, Synth. Commun., 2003, 33, 1121–1129 CrossRef CAS.
- A. Bernet, J. Schröder and K. Seifert, Helv. Chim. Acta, 2003, 86, 2009–2020 CrossRef CAS.
- H. Akita, M. Nozawa, A. Mitsuda and H. Ohsawa, Tetrahedron: Asymmetry, 2000, 11, 1375–1388 CrossRef CAS.
- H. S. Wang, H. J. Li, J. L. Wang and Y. C. Wu, Green Chem., 2017, 19, 2140–2144 RSC.
- K. K. W. Kuan, A. W. Markwell-Heys, M. C. Cruickshank, D. P. Tran, R. M. Adlington, J. E. Baldwin and J. H. George, Bioorg. Med. Chem., 2019, 27, 2449–2465 CrossRef CAS PubMed.
- E. J. Alvarez-Manzaneda, R. Chahboun, I. B. Perez, E. Cabrera, E. Alvarez and R. Alvarez-Manzaneda, Org. Lett., 2005, 7, 1477–1480 CrossRef CAS PubMed.
- H. M. Liu, D. Chen, W. D. Xu, L. Z. Dang and H. B. Qin, Org. Chem. Front., 2018, 5, 1886–1889 RSC.
- A. F. Barrero, E. A. Manzaneda, J. Altarejos, S. Salido, J. M. Ramos, M. S. J. Simmonds and W. M. Blaney, Tetrahedron, 1995, 51, 7435–7450 CrossRef CAS.
- A. Bernet and K. Seifert, Helv. Chim. Acta, 2006, 89, 784–796 CrossRef CAS.
- F. Jiménez, A. Fernández, E. Boulifa, A. I. Mansour, R. Alvarez-Manzaneda, R. Chahboun and E. Alvarez-Manzaneda, J. Org. Chem., 2017, 82, 9550–9559 CrossRef.
- J. E. McMurry and M. D. Erion, J. Am. Chem. Soc., 1985, 107, 2712–2720 CrossRef CAS.
- J. R. Falck, S. Manna, S. Chandrasekhar, L. Alcaraz and C. Mioskowski, Tetrahedron Lett., 1994, 35, 2013–2016 CrossRef CAS.
- J. R. Falck, K. K. Reddy and S. Chandrasekhar, Tetrahedron Lett., 1997, 38, 5245–5248 CrossRef CAS.
- Y. Arima, M. Kinoshita and H. Akita, Tetrahedron: Asymmetry, 2007, 18, 1701–1711 CrossRef CAS.
- A. Gansäuer, J. Justicia, A. Rosales, D. Worgull, B. Rinker, J. M. Cuerva and J. E. Oltra, Eur. J. Org Chem., 2006, 2006, 4115–4127 CrossRef.
- S. Xu, J. Gu, H. Li, D. Ma, X. Xie and X. She, Org. Lett., 2014, 16, 1996–1999 CrossRef CAS.
- T.-K. Ma, P. J. Parsons and A. G. M. Barrett, J. Org. Chem., 2019, 84, 4961–4970 CrossRef CAS.
- S. C. Welch and A. S. C. P. Rao, Tetrahedron Lett., 1977, 18, 505–508 CrossRef.
- M. J. Cano, H. Bouanou, R. Tapia, E. Alvarez, R. Alvarez-Manzaneda, R. Chahboun and E. Alvarez-Manzaneda, J. Org. Chem., 2013, 78, 9196–9204 CrossRef CAS PubMed.
- D. H. Dethe, G. M. Murhade, B. D. Dherange and S. K. Sau, Eur. J. Org Chem., 2017, 2017, 1143–1150 CrossRef CAS.
- J. Schroder, B. Matthes and K. Seifert, Tetrahedron Lett., 2001, 42, 8151–8152 CrossRef CAS.
- M. Cortés, J. A. Valderrama, M. Cuellar, V. Armstrong and M. Preite, J. Nat. Prod., 2001, 64, 348–349 CrossRef.
- W. Peñta, J. T. López and M. Cortés, Synth. Commun., 1989, 19, 2841–2850 CrossRef.
- A. Rosales Martínez, L. Pozo Morales and E. Díaz Ojeda, Synth. Commun., 2019, 49, 2554–2560 CrossRef.
- A. Akhaouzan, A. Fernandez, A. I. Mansour, E. Alvarez, A. Haidoeur, R. Alvarez-Manzaneda, R. Chahboun and E. Alvarez-Manzaneda, Org. Biomol. Chem., 2013, 11, 6176–6185 RSC.
- A. Fernandez, E. Boulifa, F. Jimenez, S. Mahdjour, A. I. Mansour, R. Chahboun and E. Alvarez-Manzaneda, Tetrahedron, 2017, 73, 6549–6557 CrossRef CAS.
- A. Fernandez, E. Alvarez, R. Alvarez-Manzaneda, R. Chahboun and E. Alvarez-Manzaneda, Chem. Commun., 2014, 50, 13100–13102 RSC.
- A. Si, A. D. Landgraf, S. Geden, S. J. Sucheck and K. H. Rohde, ACS Omega, 2022, 7, 23487–23496 CrossRef CAS.
- S. Sun, X. Wang, R. Csuk and S. Li, J. Nat. Prod., 2023, 86, 1420–1427 CrossRef CAS.
- L. Liu, H. Song, P. Chen, Z. Yuan, S. Feng, W. Zhang, B. Fang, X. Xie and X. She, Org. Chem. Front., 2018, 5, 3013–3017 RSC.
- H. Song, L. Liu, M. Yang, G. Wu, P. Chen, X. Xie and X. She, Org. Chem. Front., 2020, 7, 35–42 RSC.
- H. S. Wang, H. J. Li, X. Nan, Y. Y. Luo and Y. C. Wu, J. Org. Chem., 2017, 82, 12914–12919 CrossRef CAS PubMed.
- H. S. Wang, H. J. Li, Z. G. Zhang and Y. C. Wu, Eur. J. Org Chem., 2018, 2018, 915–925 CrossRef CAS.
- J. L. Wang, H. J. Li and Y. C. Wu, J. Org. Chem., 2018, 83, 8716–8723 CrossRef CAS PubMed.
- Y. F. Cheng, H. J. Li, Y. L. Zhang and Y. C. Wu, Nat. Prod. Res., 2021, 37, 1265–1270 CrossRef PubMed.
- H. S. Wang, X. Nan, H. J. Li, Z. Y. Cao and Y. C. Wu, Org. Biomol. Chem., 2021, 19, 9439–9447 RSC.
- S. Zhang, X. Wang, J. Hao, D. Li, R. Csuk and S. Li, J. Nat. Prod., 2018, 81, 2010–2017 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Wenjun Wu on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4np00025k |
| § First author. |
| This journal is © The Royal Society of Chemistry 2025 |