Mamoalosi A.
Selepe
*a,
Siyanda T.
Mthembu
b and
Molahlehi S.
Sonopo
c aDepartment of Chemistry, Faculty of Natural and Agricultural Sciences, University of Pretoria, Lynnwood Road, Hatfield, Pretoria 0002, South Africa. E-mail: mamoalosi.selepe@up.ac.za bDepartment of Physical and Earth Sciences, Faculty of Natural and Applied Sciences, Sol Plaatje University, P/Bag x 5008, Kimberley, 8300, South Africa cApplied Radiation Department, South African Nuclear Energy Corporation Ltd, Pelindaba, Brits 0240, South Africa
Received
31st October 2024
First published on 11th February 2025
Abstract
Covering: 2012 to 2024
Isoflavonoids are phenolic compounds with wide structural diversity and a plethora of biological activities. Owing to their structural variation and potential health-promoting and other benefits, they have been targeted for synthesis. Herein, we review the synthesis of natural isoflavonoids belonging to different classes that include isoflavones, isoflavanones, isoflavans, isoflavenes, pterocarpans, rotenoids, coumaronochromones, and coumestans. The synthetic methodologies employed and advancements in synthetic strategies are highlighted.
Mamoalosi A. Selepe
Mamoalosi Selepe completed her undergraduate study at the National University of Lesotho. She received BSc (honours) and PhD degrees from the University of KwaZulu-Natal (South Africa). Her PhD study was on analysis and synthesis of isoflavonoids from Eriosema, under the supervision of Prof. Fanie van Heerden and Prof. Siegfried Drewes. Upon completion of PhD study, she joined iThemba Pharmaceuticals, and subsequently the University of Pretoria. Her research interests are on isolation of compounds from natural sources and synthesis of natural compounds and derivatives.
Siyanda T. Mthembu
Siyanda Mthembu is lecturer at Sol Plaatje University teaching undergraduate and postgraduate chemistry in Faculty of Natural and Applied Sciences. He holds a PhD, MSc and BSc honours in Chemistry from the University of the Witwatersrand (South Africa). Siyanda developed a keen interest in the field of chemistry early in life, curious about how the universe works. His key areas of research focus on synthetic organic chemistry and medicinal chemistry. He is presently a recipient of University Capacity Development Programme, Junior Research Fellows Programme grant investigating synthesis of pyrrole derivatives via enaminone intermediate.
Molahlehi S. Sonopo
Molahlehi Sonopo obtained his MSc degree in Chemistry from the University of the Free State and PhD in Chemistry from the University of KwaZulu-Natal (South Africa). He was awarded a joint post-doctoral research position by the South African Nuclear Energy Corporation (Necsa) and iThemba Pharmaceuticals after completing PhD study. Currently he is the research leader for the Radiolabelling section within the Applied Radiation department of research and innovation division (R&I) at Necsa. His current research focuses on radiolabelling of bioactive compounds, natural products isolation, synthetic method development for specific natural products and total synthesis.
1. Introduction
Isoflavonoids are a class of flavonoids that possess a 3-phenylchroman skeleton, which is derived from a 1,2-aryl migration during the biosynthesis of flavonoids.1 Isoflavonoids occur mainly in the Leguminosae family,1–4 however, there is a limited number of isoflavonoids that have been identified from non-leguminous plants.5,6 Despite their limited distribution, isoflavonoids have large structural variation and are divided into subclasses that include isoflavones, isoflavanones, isoflavans, pterocarpans, rotenoids, 3-arylcoumarins, coumaronochromones, coumestans, and others depending on the oxidation state of the chroman ring and the presence of additional heterocyclic rings.1,7,8 Structural diversity also arises from the substitution patterns and the structural modifications that include hydroxylation,2,3 methylation,2,3 prenylation,3,9 glycosylation,3,4,10,11 and oligomerisation.3,4,12–15
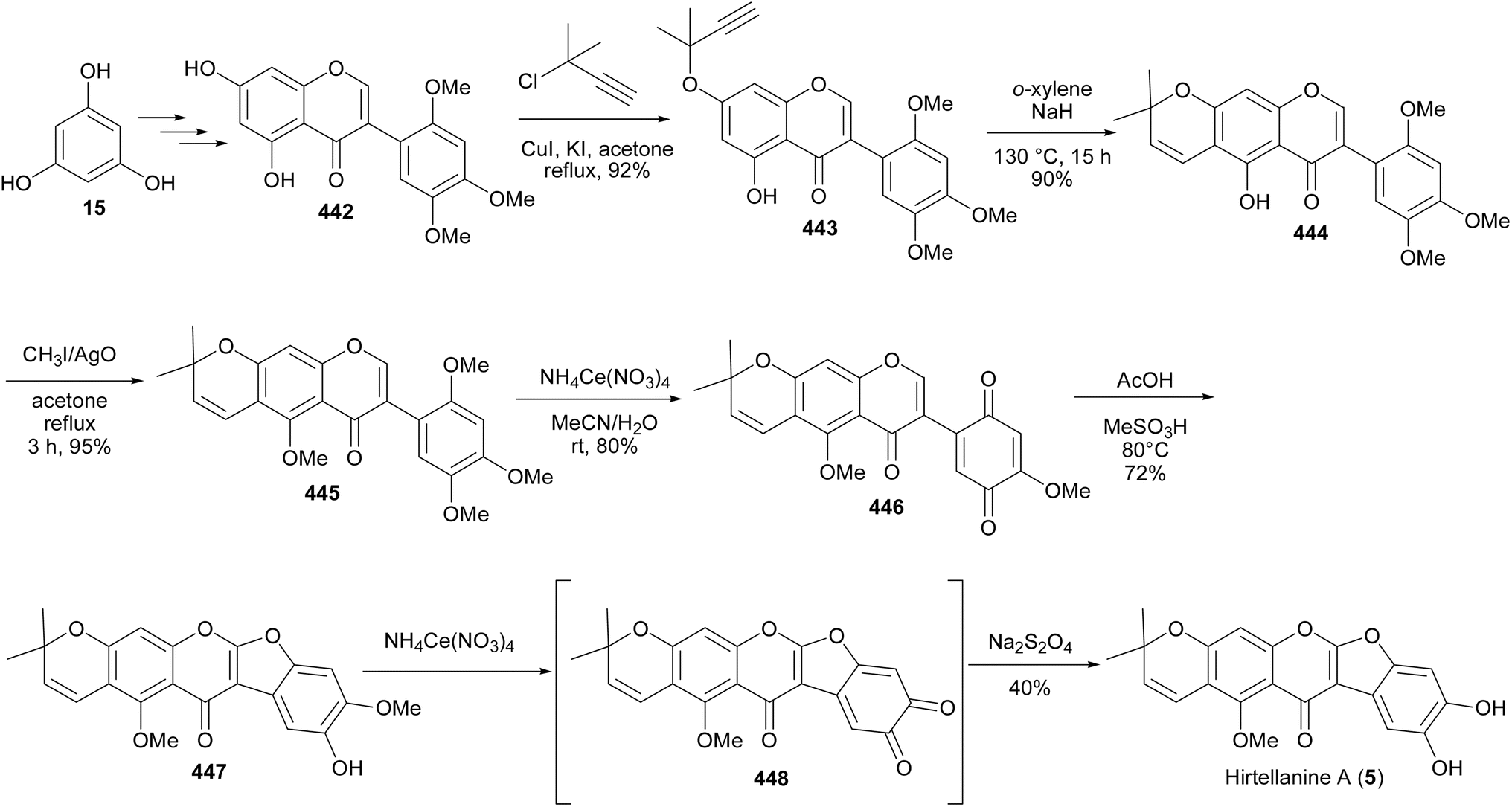
In the plant kingdom, isoflavonoids act as phytoalexins that offer protection to plants against microorganisms.5,16 Apart from interaction with microorganisms, isoflavonoids have been reported to exhibit biological activities with potential health-promoting benefits to humans.8,17–21 These include oestrogenic,22,23 anti-inflammatory,24 anti-microbial,17,24–26 vasorelaxation,27 antiarthritic,28 cardioprotective,29 neuroprotective,20 antiviral,30 immunosuppressive,31,32 osteoprotective,33 chemoprotective and antiproliferative activities.34–36 Examples of bioactive isoflavonoids are shown in Fig. 1. They comprise of a C-glucosylated isoflavone, puerarin (1);18,20,37 prenylated isoflavans, eryzerin C (2);38,39 a pterocarpan, (+)-(6aS,11aS)-2,3,9-trimethoxypterocarpan [(+)-3];40,41 a rotenoid, amorphispironone B (4);34 a pyranocoumarochromone, hirtellanine A (5);31,32 and a coumestan, psoralidin (6).33,42,43
Fig. 1 Examples of bioactive isoflavonoids.
Owing to their important biological activities, the synthesis of isoflavonoids and their derivatives has been of interest to researchers.32,37,41,44–47 Several synthetic strategies have been developed for the different classes and applied to the synthesis of natural isoflavonoids. The methods range from classical condensations and rearrangements,43,48–54 to metal-catalysed cross-coupling reactions of pre-functionalised precursors55–61 and transformations facilitated by direct C–H activation amongst others.62–64 In this review, the synthesis of natural isoflavonoids belonging to different subclasses is discussed. The main focus is on reports published from 2012 to 2024. The sections are divided based on the subclasses of isoflavonoids and further (where permissible) based on methodologies that include conventional and newly developed methods. For chiral isoflavonoids that include isoflavanones, isoflavans, pterocarpans and rotenoids, recent studies on syntheses of racemic mixtures, as well as stereoselective syntheses encompassing the use of chiral pool building blocks,65–67 metal-catalysed reactions in the presence of chiral ligands,68,69 organocatalysed stereoselective syntheses,70–75 and stereoselective hydrogenation and hydrogen transfer are discussed.76–79
2. Synthesis of isoflavones
Traditionally, isoflavones are synthesised from 2-hydroxyphenyl benzyl ketone (2-hydroxydeoxybenzoin) and chalcone precursors.49,53 The construction of isoflavones from 2-hydroxydeoxybenzoins requires the incorporation of an additional carbon unit to afford a C6C3C6 skeleton. This is achieved by formylation or acylation at the benzylic position and consequent O-cyclisation.49,50,53 The synthesis of isoflavones from chalcones follows a biomimetic sequence that involves oxidative rearrangement of the B-ring aryl group to the 3 position.53 Depending on the reaction conditions, this often leads to the formation of α-formyldeoxybenzoins or α-acetaldeoxybenzoins as intermediates, which undergo ring-closure with the hydroxy group on the phenyl A-ring to afford isoflavones.48,51,52 The syntheses of isoflavones from the deoxybenzoin and chalcone precursors were carried out as early as the 1920s and the 1950s and the methods continue to be modified and applied to the synthesis of natural isoflavones.53,80–84 Modified deoxybenzoin-based methodologies developed lately involve Pd(II)-catalysed oxidative cyclisation of α-methylenedeoxybenzoins,85 and CuI-catalysed intramolecular cyclisation of 2-bromo-α-formyldeoxybenzoins.60,61 Other recent synthetic strategies for isoflavones include oxidation of isoflavenes obtained by ring-closing metathesis,86 tandem demethylation and ring/opening cyclisation of methoxybenzoylbenzofurans,87 and metal-catalysed cross-coupling reactions of functionalised chromones that encompass the Suzuki–Miyaura,55,56,88 the Negishi cross-coupling,57,58 and the Stille cross-coupling reactions.59,89 Very recently, new methods that involve direct arylation of 2-hydroxyenaminoketones for the synthesis of isoflavones have been developed.90–93
2.1. Deoxybenzoin route
2.1.1 Simple isoflavones. Several natural isoflavones have been synthesised by the deoxybenzoin route. The deoxybenzoin intermediates 11 can be accessed by reactions of phenols 8 with phenylacetonitriles 7 followed by hydrolysis.94,95 Alternatively, the benzyl ketones can be synthesised by Friedel–Crafts acylation of phenols 8 with phenylacetic acids 9 or acyl chlorides 10.80,96–98 Deoxybenzoins 11 can be converted into isoflavones 14 by treatment with reagents containing activated units that include ethyl formate, ethyl orthoformate,53N,N-dimethylformamide (DMF),80,96N,N-dimethylformamide dimethyl acetal (DMF–DMA)98,99 and bis(dimethylamino)-t-butoxymethane [HC[N(CH3)2]2O-tBu].97 Using ethyl formate in the presence of sodium metal first converts the deoxybenzoins 11 into 2-hydroxyisoflavanone intermediates 12, which upon dehydration form the requisite isoflavones 14.53 Treatment of 11 with ethyl oxalyl chloride in pyridine affords the intermediates 13 and subsequently isoflavones 14 upon hydrolysis and decarboxylation.53 A depiction of the synthesis of isoflavones by the deoxybenzoin route is shown in Scheme 1.
Scheme 1 Synthesis of isoflavones from deoxybenzoin intermediates.
6,8-Dimethylgenistein (19), an isoflavone with a fully substituted A-ring was isolated from Henriettella fascicularis.100 It showed a stronger binding affinity to estrogen receptor β than estrogen receptor α and moderate antiestrogenic activity with cultured Ishikawa cells.100 This compound was synthesised by two independent synthetic routes (the deoxybenzoin route and the Suzuki–Miyaura coupling reaction).80 Following the deoxybenzoin route, the synthesis was initiated by formylation of phloroglucinol (15) to give compound 16, which was reduced to give the dimethylated phloroglucinol 17. BF3·Et2O-mediated coupling of 17 with 4-hydroxyphenylacetic acid (18) followed by treatment of the resulting benzyl ketone with PCl5/DMF gave 6,8-dimethylgenistein (19) in a 14% yield (Scheme 2).80
Scheme 2 Synthesis of 6,8-dimethylgenistein (19).
2.1.2 Glucosylated isoflavones. O-Glucosylated isoflavones 27 and 30 together with their aglycone derivatives from Apios americana were synthesised for the first time by Asebi and co-workers.98,101 The synthesis of the compounds featured acylation of trimethoxybenzene 21 with dimethoxyphenylacetic acid 20 to give a mono-demethylated deoxybenzoin 22 in a 79% yield, Scheme 3. Condensation of the benzyl ketone 22 with DMF–DMA gave an isoflavone 23,99 which was demethylated with BBr3 to give aglycone precursor 24. Selective phase-transfer O-glucosylation of 24 using 2,3,4,6-tetra-O-pivaloyl-α-D-glucopyranosyl bromide [(Pv)4GlcBr] in the presence of aliquat 336 and K2CO3 gave compound 26,102 from which 7-β-D-glucopyranosyloxy-2′-hydroxygenistein (27) was accessed by deprotection of the pivaloyl groups, Scheme 3. The synthesis of 7-β-D-glucopyranosyloxy-2′-hydroxy-5-O-methylgenistein (30) was accomplished by the pivaloyl protection of the 2′- and 4′-hydroxy groups of 26 followed by methylation of the 5-hydroxy group of 28 to afford isoflavone 29, and finally cleavage of all the pivaloyl protecting groups under basic conditions (Scheme 4).98
Scheme 3 Synthesis of isoflavone glucoside 27.
Scheme 4 Synthesis of isoflavone glucoside 30.
Puerarin (1) is a C-glucosylated isoflavone isolated from Pueraria lobata (Willd.).103 It has many important biological activities that include antioxidant, antihyperglycemic, anti-inflammatory and neuroprotective effects.20,104–106 The first synthesis of puerarin (1) was reported by Lee and co-workers.37 The synthesis involved the coupling of aryllithium species with pyranolactone for the C-glucosylation and oxidative rearrangement of a chalcone for isoflavone formation.37 In 2018, Zhou and colleagues synthesised puerarin (1) by coupling alkyl-substituted resorcinol 31 with glucosyl trifluoroacetimidate 32 to afford C-glucoside 34 in a 46.2% yield. Removal of the tert-butyl group followed by oxidation of the resulting diarylethene 35 gave deoxybenzoin 36.107 Modified Vilsmeier–Haack formylation and debenzylation gave the isoflavone 37 and the targeted puerarin (1), respectively (Scheme 5).107,108
Scheme 5 Synthesis of puerarin (1).
Although the deoxybenzoin route avoids the use of toxic TTN(III) that was utilised by Lee and colleagues for the synthesis of puerarin (1),37,107 the synthesis in Scheme 4 gave comparatively low yields of the C-glucosylated precursor 34. Also, the t-butyl deprotection and oxidation steps leading to the deoxybenzoin intermediate 36 were low yielding.107
Although synthesis of isoflavones by the deoxybenzoin route was first reported almost 100 years ago,109 the method is still utilised for the synthesis of natural isoflavones that include simple isoflavones, glucosylated isoflavones and prenylated isoflavones.80,94,97,98,107 The main advantage of the method is that the deoxybenzoin precursors can be synthesised from non-protected phenylacetic acids and phenols, thereby eliminating the protection and deprotection steps.80,94 However, the formation of deoxybenzoin and their conversion into isoflavones provide relatively low yields of the products depending on the reaction conditions and substituents present.80,97 Moreover, the harsh acidic conditions employed necessitate the installation of sensitive substituents such as prenyl groups upon the complete construction of the isoflavone core structure and in several steps.94
2.2. Oxidative rearrangement of chalcones
Unlike deoxybenzoins, chalcones can be readily prepared by a Claisen–Schmidt condensation of readily available benzylaldehydes and acetophenones. Earlier protocols utilised thallium nitrate [TTN(III)] and thallium acetate [Tl(OAc)3] to facilitate oxidative rearrangement of chalcones.52,53 Hypervalent iodine reagents that include phenyliodine(III) bis(trifluoroacetate), (diacetoxyiodo)benzene/p-toluenesulfonic acid, [hydroxy(tosyloxy)iodo]benzene and [bis(trifluoroacetoxy)iodo]benzene are other reagents that have been developed to facilitate the aryl migration.53,82,110
2.2.1 Glycosylated isoflavones. 8-β-D-Glucopyranosylgenistein 46 was identified as a major constituent of the extract of Genista tenera with anti-hyperglycemic activity.111 Jesus and colleagues synthesised this compound and conducted further antidiabetic activity studies.81 The synthesis is shown in Schemes 6. The first step entailed preparation of the C-glucosylated acetophenone 42. This was accomplished in 49% yield by Sc(OTf)3-catalysed reaction of 2,4-dibenzyloxy-6-hydroxyacetophenone (39) with perbenzylglucosyl acetate 40. Alternatively, compound 42 could be synthesised by TMSOTf-catalysed reaction of phloroacetophenone (38) with glucosylating agent 41 followed by dibenzylation. Aldol condensation of 42 and benzyladehyde 43 gave a chalcone 44, which underwent rearrangement into isoflavone 45 upon oxidation with TTN(III). Finally, debenzylation of 44 gave 8-β-D-glucopyranosylgenistein (46).81
Scheme 6 Synthesis of 8-β-D-glucopyranosylgenistein (46).
2.2.2 Prenylated isoflavones. Several prenylated isoflavones and pyranoisoflavones have been synthesised by oxidative rearrangement of chalcones.82–84,112,113 Owing to the sensitivity of the prenyl side chain to highly oxidative and acidic conditions employed, the complete installation of the prenyl group has been conducted in the late stages of the synthesis, upon complete construction of the isoflavone core structure.82–84 Some examples of prenylisoflavones and dimethylpyranoisoflavones that were synthesised by oxidative rearrangement of chalcones are wighteone,82 derrubone,82 lupisoflavone,82,83 erysubin F, other Erythrina isoflavones84 and barbigerone.113
Lupisoflavone (57), the antifungal prenylisoflavone from Lupinus albus114 was synthesised by Hossain and colleagues together with other prenylisoflavones.82,83 Initially they utilised microwave irradiation in several key steps, which led to higher yields and shorter reaction times.82 The synthesis of lupisoflavone (57) under conventional conditions was reported in 2012.83 As shown in Scheme 7, benzoylchalcone 49, obtained in a sequence of steps from iodoacetophenone 47 and benzaldehyde 48, underwent oxidative rearrangement upon treatment with a mixture of [hydroxyl(toxyloxy)iodo]benzene (HTIB) and [bis(trifluoroacetoxy)iodo]benzene (BTIB) to afford acetal intermediate 50. O-Cyclisation of acetal 50 under basic conditions gave 6-iodoisoflavone 51. Sonogashira coupling of 51 with 2-methyl-3-butyn-2-ol followed by reduction of the alkynylisoflavone 52 gave 53. Sequential protection of phenolic groups with benzoyl and tosyl groups and subsequent dehydration of the 2-hydroxybutyl group gave a mixture of 6-prenylisoflavone 56 and the regioisomer, 6-(3-methyl-3-butenyl) isoflavone. Finally, lupisoflavone (57) was obtained by successive deprotection of tosyl and benzoyl groups.83
Scheme 7 Synthesis of lupisoflavone (57).
Several prenylated isoflavones from the genus Erythrina have been synthesised by Kwesiga and colleagues by 2,3-oxidative aryl migration of flavanones obtained from chalcones.84,112 These included a diprenylated isoflavone erysubin F (66). The synthesis of erysubin F (66) was initiated by MOM protection of allylbenzaldehyde 58 followed by Claisen–Schmidt condensation of the resulting 59 with O-allylacetophenone 60 to afford chalcone 61 in 74% yield. Claisen rearrangement and O-cyclisation gave flavanone 62 in 50% yield, which upon treatment with BTI and trimethylorthoformate gave isoflavone 63 in 20% yield, together with flavone 64 in 18% yield. Double cross-coupling metathesis of bisallylisoflavone 63 with 2-methyl-2-butene using the second-generation Grubbs catalyst A rendered bisprenylated isoflavone 65, which was deprotected to give erysubin F (66), Scheme 8.84
Scheme 8 Synthesis of erysubin F (66).
Barbigerone (70) is a pyranoisoflavone that was first isolated from Tephrosia barbigera.115 It has also been identified from other sources, especially from the Millettia genus.116,117 It has been determined to exhibit biological activities that include antiparasitic activity,118 cytotoxicity against several cancer cells including MDR cells119,120 and anti-inflammatory activities.113 The synthesis of barbigerone has been reported by several groups.113,121 The synthesis by Wei and colleagues proceeded via condensation of pyranoacetophenone 67 with benzaldehyde 68 to give chalcone 69. Oxidative migration of the trimethoxybenzene ring in the presence of TTN(III) gave an acetal intermediate, which underwent oxycyclisation to afford barbigerone 70, Scheme 9.113
Scheme 9 Synthesis of barbigerone (70).
Despite the continued application of the oxidative rearrangement of chalcones for the synthesis of isoflavones, the high cost, toxicity and adverse environmental effects of thallium reagents make the method unattractive.82 Environmentally friendly and less toxic hypervalent iodine reagents have been developed to facilitate the aryl shift.53,110 The challenge with these reagents is inconsistencies in product yields. Some studies reported moderate to high yields of isoflavones obtained by oxidative rearrangement of hypervalent iodine reagents, while other studies reported low to moderate yields. The low yields result from the formation of other untargeted products including benzofurans and flavones.53,84,110 Moreover, like the deoxybenzoin route, the incorporation of the prenyl groups is restricted to the late-stages of the synthesis due to the oxidative reaction conditions that are incompatible with such substituents. This can pose challenges in controlling the regioselectivity and therefore, necessitate the attachment of such groups in multiple steps.84,112
2.3. Modified deoxybenzoin and chalcone routes
A modified deoxybenzoin route that involved CuI-catalysed intramolecular cyclisation of α-formylated 2-bromodeoxybenzoin was reported by Zhu's group in 2011.60 Later (2016) Semenov and colleagues synthesised a potential anti-cancer compound, glaziovianin A (77) and several derivatives from 3-(2-bromophenyl)-3-oxopropanal intermediates using Zhu's conditions.60,61 However, the α-formyldeoxybenzoin precursors in Semenov's synthesis were prepared by rearrangement of epoxychalcones.48,122 As illustrated in Scheme 10, the synthesis of glaziovianin A (77) commenced with the preparation of bromoacetophenone 72 and benzaldehyde 73, from plant metabolites, methyleugenol and apiol, respectively. Condensation of 72 and 73 gave chalcone 74, which was converted into epoxide 75. Treatment of epoxide 75 with BF3·OEt2 and subsequent CuI-mediated cyclisation of the resulting bromodiarylketoaldehyde 76 gave glaziovianin A (77).61
Scheme 10 Synthesis of glaziovianin A (77) by Semenov and colleagues.61
2.4. Transformation of methoxybenzoylbenzofurans into isoflavones
Kunyane and colleagues reported the unexpected conversion of methoxybenzoylbenzofurans into isoflavones through a one-pot sequence that involved demethylation and oxycyclisation, resulting in furan ring-opening and chromone ring-formation.87 A series of isoflavone derivatives were prepared by demethylation of 2′-methoxybenzoylbenzofurans. The method was further applied to the synthesis of glaziovianin A (77), Scheme 11. The synthesis was initiated by the preparation of benzoquinone 81 from trimethoxybenzaldehyde 80. Condensation of acetophenone 78 with DMF–DMA and subsequent coupling of the resulting enaminone intermediate 79 with benzoquinone 81 under acidic conditions gave 2′-methoxybenzoylbenzofuran 82 in 45% yield. Demethylation with TMSI gave a separable mixture of isoflavone 84 and isoflavone quinone 83. Methylation of the isoflavone 84 gave the targeted compound 77, Scheme 11.
Scheme 11 Synthesis of glaziovianin A (77).
2.5. Functionalised chromones
Metal-catalysed cross-coupling reactions of C-3 functionalised chromones have been employed successfully for the synthesis of isoflavones. Access to functionalised chromone coupling partners could be attained from enaminone precursors or by direct C-3 metalation or halogenation of chromones.57,123,124 Metal-catalysed cross-coupling reactions such as the Suzuki–Miyaura reaction,55,56,88 the Negishi and Stille cross-coupling reactions utilise the 3-halogenated or metallated chromone precursors for the synthesis of isoflavones.55–59,89 In other instances, triarylbismuths have also been employed as coupling partners for chromonetriflates.125 In comparison to the Suzuki–Miyaura reaction, studies on the synthesis of isoflavones by the Negishi and Stille reactions are still limited.
2.5.1 Negishi cross-coupling reaction. Klier and colleagues reported regioselective metalation of chromones and quinolones leading to C-2 zincated chromones upon treatment with TMP2Zn·2MgCl2·2LiCl, whereas chromone-3-zinc chlorides were obtained with TMPZnCl·LiCl in the absence of MgCl2.57 Isoflavone derivatives could be synthesised by Pd-catalysed Negishi cross-coupling of aryl iodides or aryl bromides with the C-3 zincated chromones. As shown in Scheme 12, the natural compound biochanin A (88) was synthesised by treatment of chromone 85 with TMPZnCl·LiCl followed by Pd-catalysed coupling with p-methoxyphenyl iodide 86 and debenzylation of 87. The isoflavones 87 and biochanin A (88) were obtained in 95% and 88% yields, respectively (Scheme 12).57
Scheme 12 Synthesis of biochanin A (88) by Negishi cross-coupling reaction.
On the other hand, Zhang and colleagues prepared a series of isoflavones at room temperature by nickel-catalysed Negishi coupling of halochromones with arylzinc bromides in the presence of LiCl, as an additive.58 Under these conditions, the natural compound 7,4′-dimethoxyisoflavone (92)126 was synthesised in 76–82% yield by coupling of 3-iodochromone 89 or 3-bromochromone 90 with 4-methoxyphenylzinc bromide (91), Scheme 13.58
Scheme 13 Synthesis of 7,4′-dimethoxyisoflavone (92) by Negishi cross-coupling reaction.
2.5.2 Stille cross-coupling reactions. Chang and co-workers synthesised isoflavones by Pd-catalysed Stille coupling of 3-bromochromones with arylbutylstannanes. The employed conditions were tolerant to air and water and several isoflavone derivatives could be synthesised in good yields including daidzein (94). Coupling of 3-bromo-7-methoxychromone 90 with arylstannane 93 in the presence of PdCl2(NH3)2/2,2′-ammonium bipyridyl catalytic system and TBAF gave dimethoxyisoflavone 92, which was demethylated with pyridine hydrochloride to afford daidzein (94) in 63% yield, Scheme 14.59
Scheme 14 Synthesis of daidzein (94) by Stille cross-coupling reactions.
2.5.3 Suzuki–Miyaura coupling reaction. 2.5.3.1 Simple isoflavones. Alternative to the deoxybenzoin route, Jung and colleagues also synthesised 6,8-dimethylgenistein (19) and isosideroxylin (101) by the Suzuki–Miyaura reaction, Scheme 15.80 This necessitated further functionalisation of the dimethylphloroglucinol 17 into 3-iodochromone 98. To achieve this, compound 17 was converted into trimethyl ether 95, which was acylated to render acetophenone 96. The reaction of acetophenone 96 with DMF–DMA followed by iodine-mediated cyclisation of the resulting enaminoketone 97 gave 3-iodochromone 98. The Suzuki–Miyaura cross-coupling reaction of the 3-iodochromone 98 with 4-hydroxyphenylboronic acid (99) and subsequent demethylation with BBr3 gave dimethylgenistein (19). Selective demethylation of 100 with BCl3 rendered isosideroxylin (101), Scheme 15.80
Scheme 15 Synthesis of 6,8-dimethylgenistein (19) and isosideroxylin (101).
2.5.3.2 Prenylated isoflavones. Isoflavones bearing prenyl substituents or their long-chain derivatives such as geranylated analogues, as well as those with dimethylpyran and furan ring systems that originated from O-cyclisation of prenyl groups have been conveniently synthesised by the Suzuki–Miyaura reaction.44,88,127,128 Several strategies were employed for the introduction of the prenyl side chains, which include direct C-prenylation using prenylbromide,129O-prenylation followed by Claisen rearrangement129,130 and allylation followed by cross-coupling metathesis.128,129 The dimethylpyran scaffold is often assembled by condensation of phenolic rings of the isoflavones with prenal or by O-propargylation followed by sigmatropic rearrangements.44,88,131
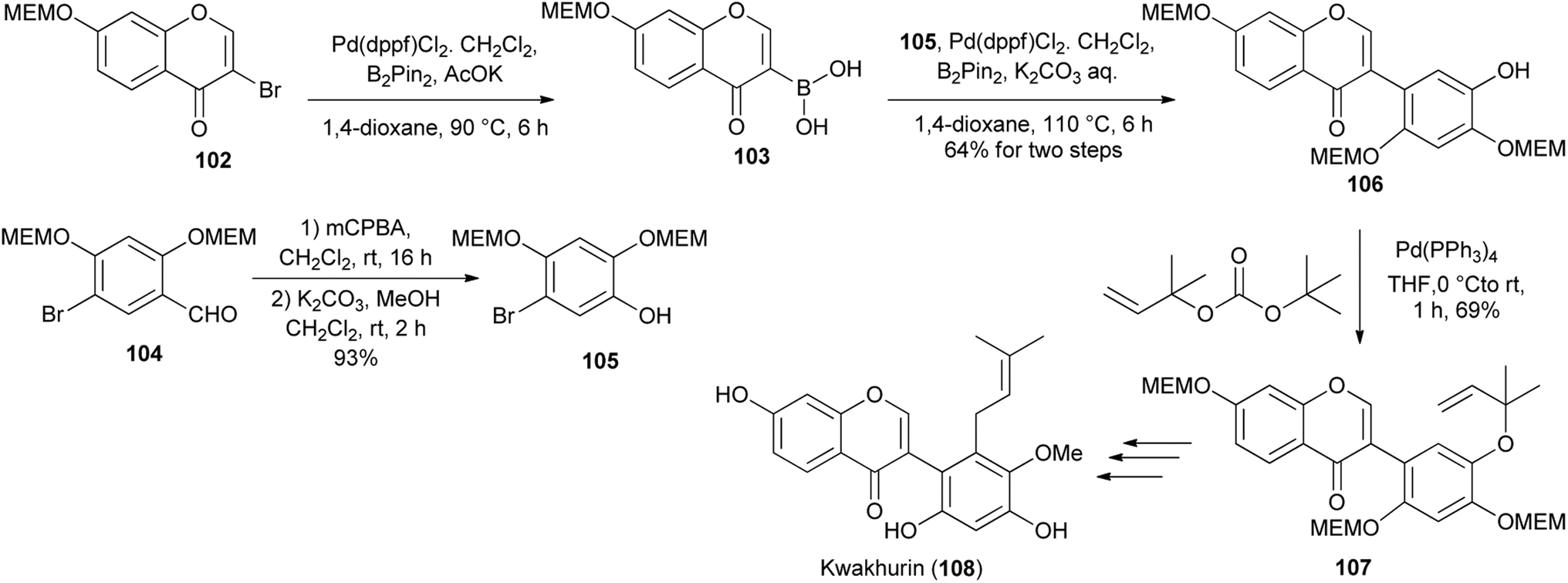
Kwakhurin (108), a B-ring prenylated isoflavone isolated from Pueraria mirifica was determined to exhibit moderate estrogenic activity.132 It was first synthesised by Ito and colleagues in 2005133 and a modified synthesis route was disclosed by Tsuji and co-workers in 2020.130 As illustrated in Scheme 16, the synthesis entailed coupling of bromophenol 105 (synthesised from aldehyde 104) with boronic acid 103 that was obtained from 3-bromochromone 102 to give isoflavone 106. Pd-catalysed O-allylation followed by Claisen rearrangement of the resulting 1,1-dimethylallyl ether 107, methylation and desilylation gave kwakhurin (108), Scheme 16.130,133
Scheme 16 Synthesis of kwakhurin (108) by Tsuji and co-workers.130
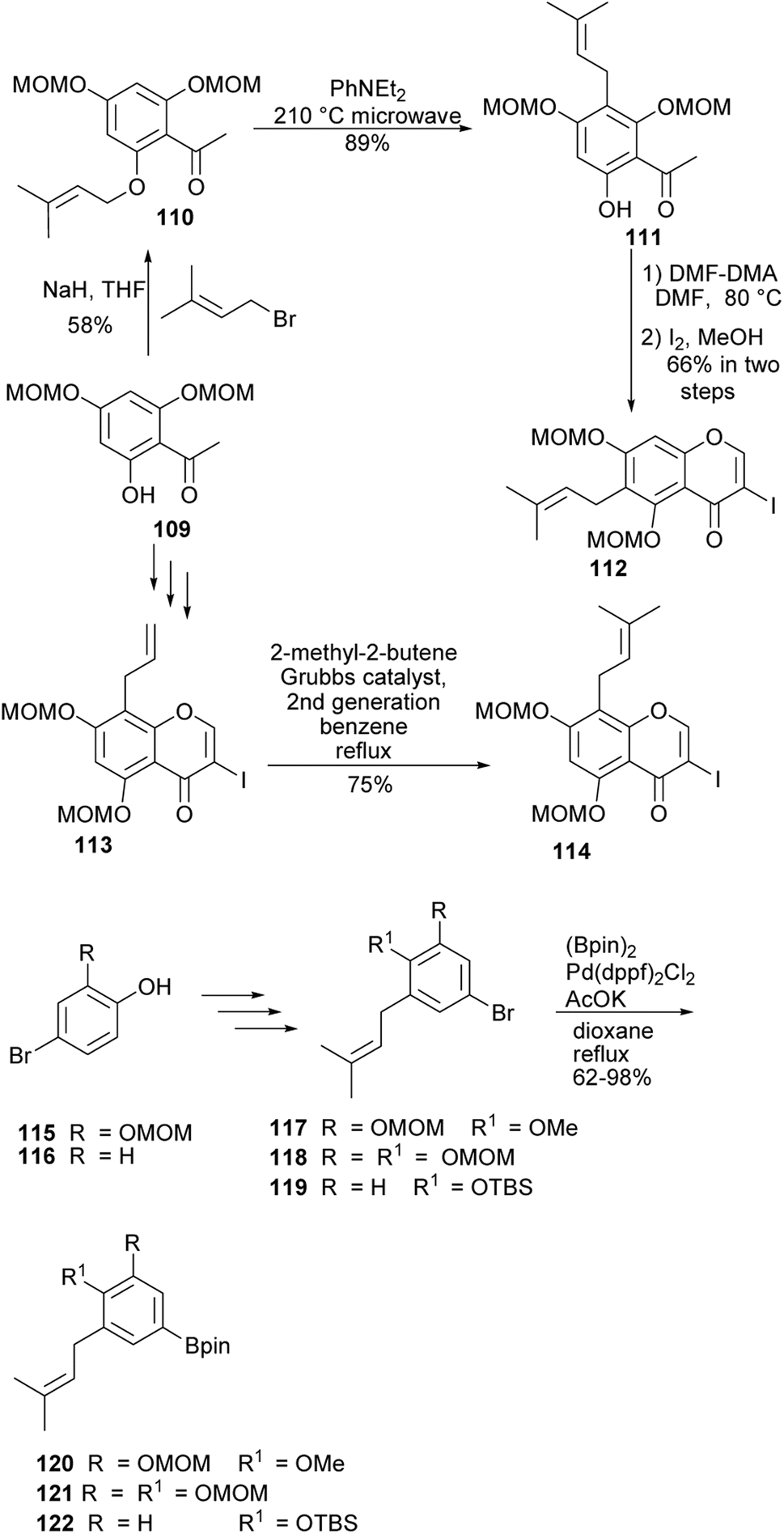
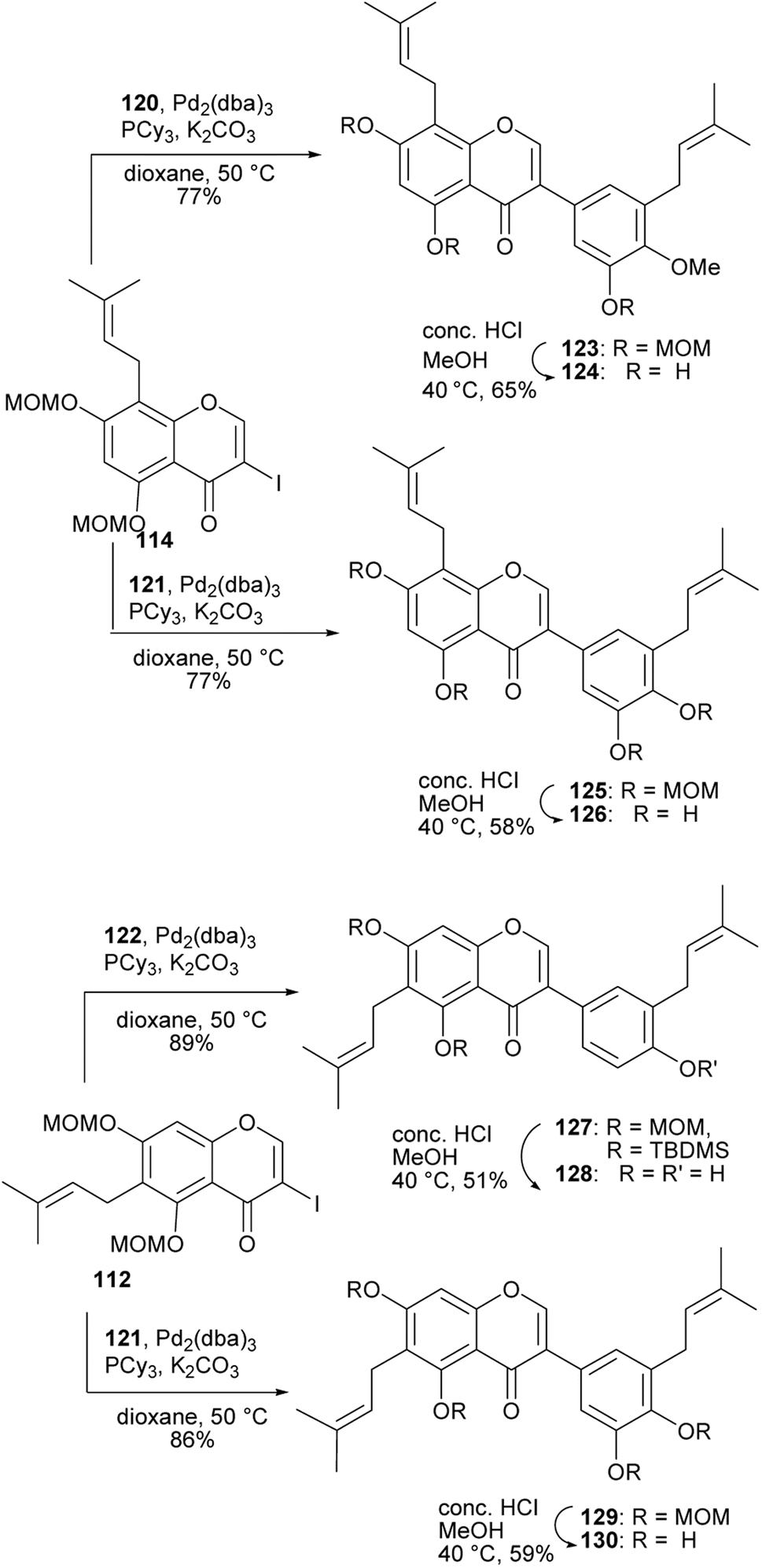
4′-O-Methylgrynullarin (124) was isolated from Derris scandens flowers together with other structurally related compounds,134 glyurallin B (126),135 isoangustone A (128)136 and lupalbigenin (130).134,137 The compounds 124, 128, and 130 were determined to exhibit preferential cytotoxicity against pancreatic cancer cell lines.134 The synthesis of 4′-O-methylgrynullarin (124) and related compounds 126, 128, and 130 was reported by Okada and colleagues.129 The key step involved the Suzuki–Miyaura coupling of the prenylboronate esters 120, 121, and 122 with prenyl-3-iodochromones 112 and 114 (Schemes 17 and 18). Three strategies were followed for early-stage C-prenylation, viz. cross-coupling metathesis, O-prenylation followed by Claisen rearrangement and direct C-prenylation using prenylbromide.129 As shown in Scheme 17, the C-6 dimethylallyliodochromone precursor 112 was prepared by O-prenylation of acetophenone 109 followed by Claisen rearrangement of 110 and condensation of the resulting prenylacetophenone 111 with DMF–DMA and iodine-mediated cyclisation. Alternatively, the C-8 prenyl group on the iodochromone 114 was introduced by cross-coupling metathesis of 2-methyl-2-butene with allylchromone 113 prepared from acetophenone 109. The boronate precursors 120, 121, and 122 were synthesised by direct C-prenylation of bromophenols 115 and 116, followed by alkylation or silylation of the hydroxy groups to afford bromobenzenes 117, 118, and 119 and finally Pd-catalysed cross coupling with bis(pinacolato)diboron (Bpin)2, Scheme 17.129 The Suzuki–Miyaura coupling reaction of 3-iodochromone 114 with boronate esters 120 and 121, followed by MOM deprotection gave methylgrynullarin (124) and glyurallin B (126), respectively. Isoangustone A (128) and lupalbigenin (130) were synthesised by the Pd-cross coupling reaction of iodochromone 112 with boronate esters 122 and 121, respectively, followed by removal of the MOM and silyl protecting groups, Scheme 18.129
Scheme 17 Syntheses of 3-iodochromones and boronate ester precursors.
Scheme 18 Total synthesis of isoflavones 124, 126, 128, and 130.
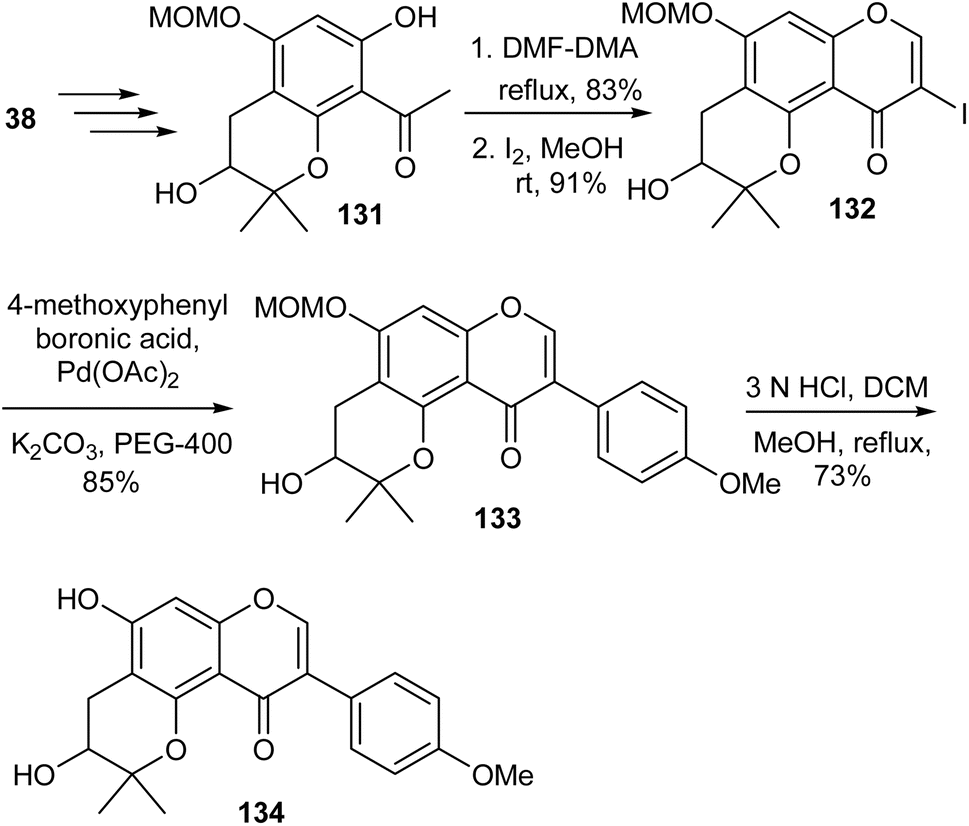
Cudraisoflavone J (134), a neuroprotective isoflavone bearing a 3-hydroxy-2,2-dimethyldihydropyran scaffold was isolated together with other structurally related compounds from the fruits of Cudrania tricuspidata (Carr.) Bur. (Moraceae).138 The synthesis of rac-cudraisoflavone J (134) was reported by Lee's group.139 As illustrated in Scheme 19, the synthesis was initiated by the preparation of dimethyldihydropyranoacetophenone 131 obtained from phloroacetophenone (38) by primary steps that involved MOM protection, O-prenylation, Claisen rearrangement, and oxidative cyclisation. Compound 131 was converted into 3-iodochromone 132 by a modified Gammill procedure.123 The Suzuki coupling of 132 with p-methoxyphenylboronic acid followed by MOM deprotection of the resulting isoflavone 133 gave rac-cudraisoflavone J (134).139 In addition to synthesis of the racemate, the group conducted stereoselective synthesis of R and S cudraisoflavone J enantiomers using Sharpless asymmetric dihydroxylation for induction of stereochemistry. Moreover, kinetic resolution was also employed to afford the R and S enantiomers, and Mosher's ester method was used to confirm the absolute configuration of the compounds.139
Scheme 19 Total synthesis of (±)-cudraisoflavone J (134).
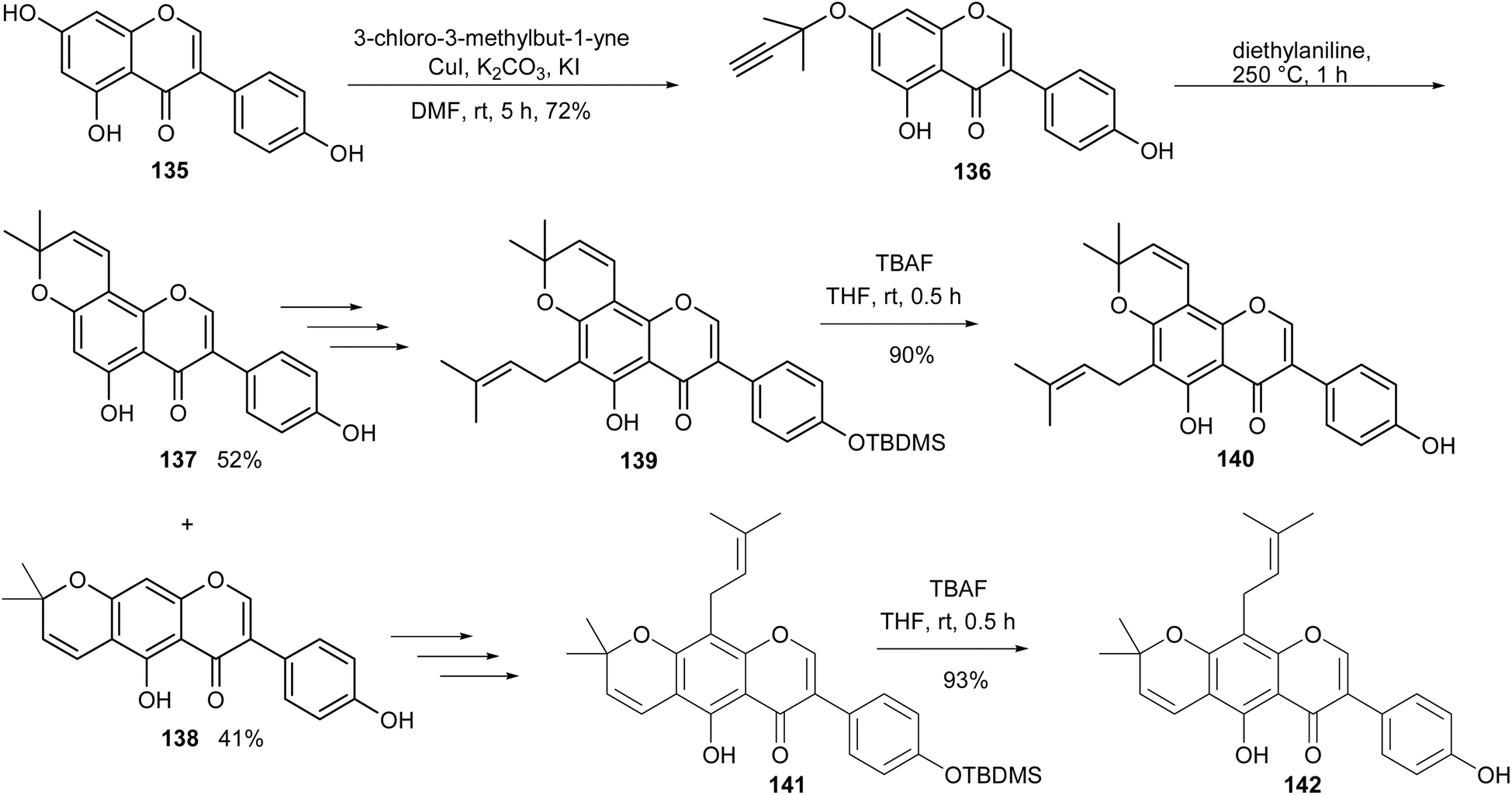
Scandenone (142) and osajin (140) are isoflavones that possess a dimethylpyran scaffold and a 3,3-dimethylallyl side chain in the A-ring. The compounds have been isolated from different sources that include Derris, Flemingia, Millettia, and Tephrosia genera and from non-leguminous families.5,140–144 They have been reported to exhibit cytotoxicity against different cancer cells,35,142 and nitric oxide production inhibition.145 The synthesis of scandenone (142) and osajin (140) involved the Suzuki–Miyaura coupling reaction as the key step for the construction of genistein (135), followed by O-propargylation and Harfenist–Thom rearrangement of 136 to afford the dimethylpyranoisoflavones 137 and 138.146 TBDMS protection of 137 and 138 followed by O-prenylation and sigmatropic rearrangement gave isoflavones 139 and 141, respectively, from which osajin (140) and scandenone (142) were obtained upon TBDMS deprotection, Scheme 20.131 Several derivatives were also synthesised from compounds 140 and 142 and evaluated for anti-inflammatory activity.131
Scheme 20 Total synthesis of scandenone (142) and osajin (140).
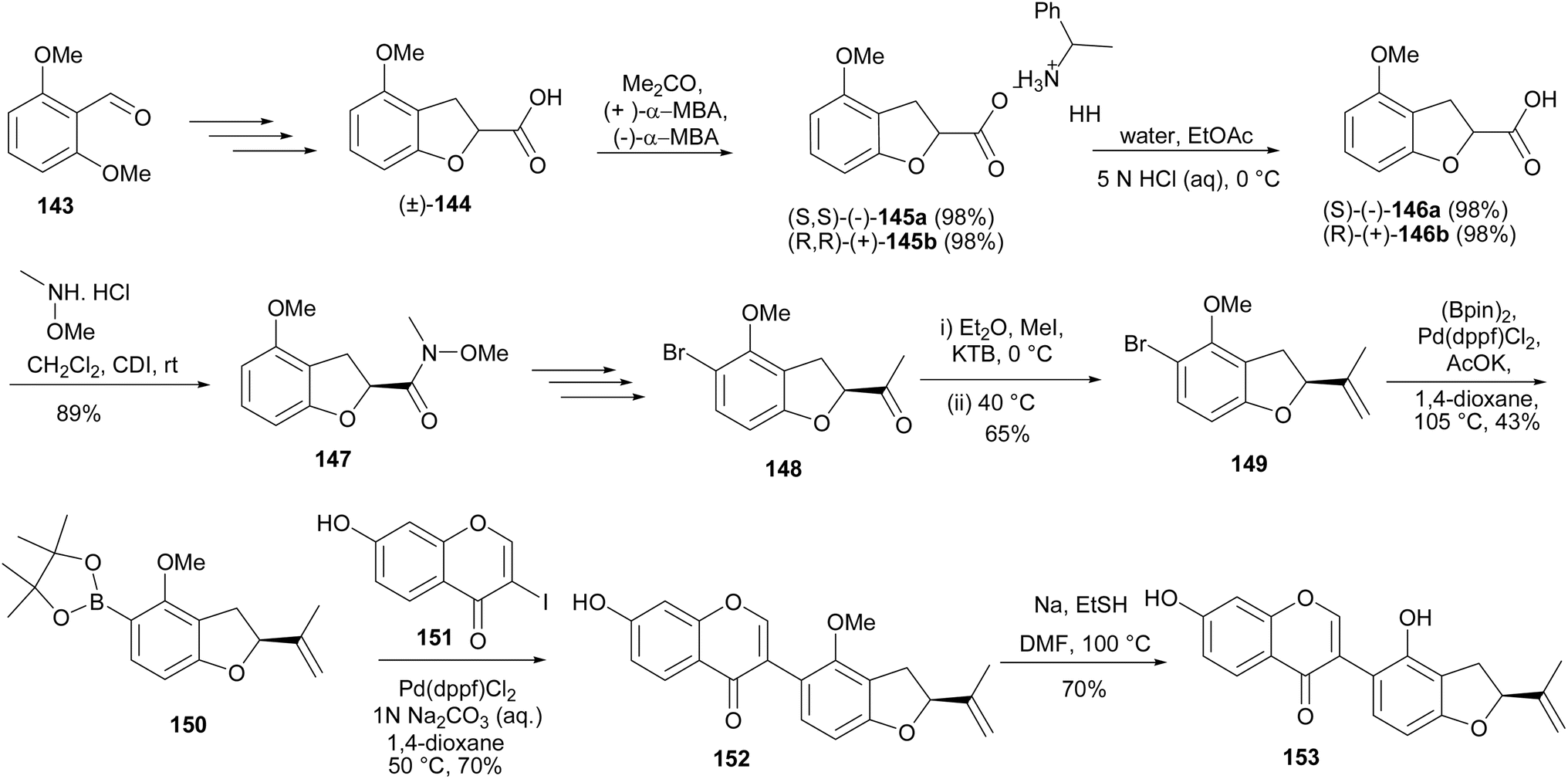
A furanoisoflavone with potential anti-obesity activity from Crotalaria albida, crotadihydrofuran C (153), was synthesised by Sun and coworkers.147,148 As shown in Scheme 21, the synthesis was accomplished by coupling of 3-iodochromone 151 and furanylboronate ester 150 to afford the isoflavone 152, which was demethylated to give crotadihydrofuran C (153). The 5-boronate ester 150 was in turn prepared in a sequence of steps that entailed the conversion of a dimethoxybenzaldehyde 143 into rac-2,3-dihydro-2-carboxy-4-methoxybenzofuran (144). The rac-144 was resolved into R and S enantiomers by coordination with enantiomers of α-methylbenzylamine (α-MBA), followed by hydrolysis. Amide coupling of the (S)-146a gave 147, which was selectively brominated to afford 148 in a sequence of steps that involved blocking the para-position by iodination, followed by bromination, methylation with methylmagnesium bromide, and concurrent deiodination. Finally, Wittig reaction of 148 and Pd-catalysed coupling of the resulting 149 with bis(pinacolato)diboron gave the boronate ester 150, Scheme 21.148
Scheme 21 Total synthesis of crotadihydrofuran C (153).
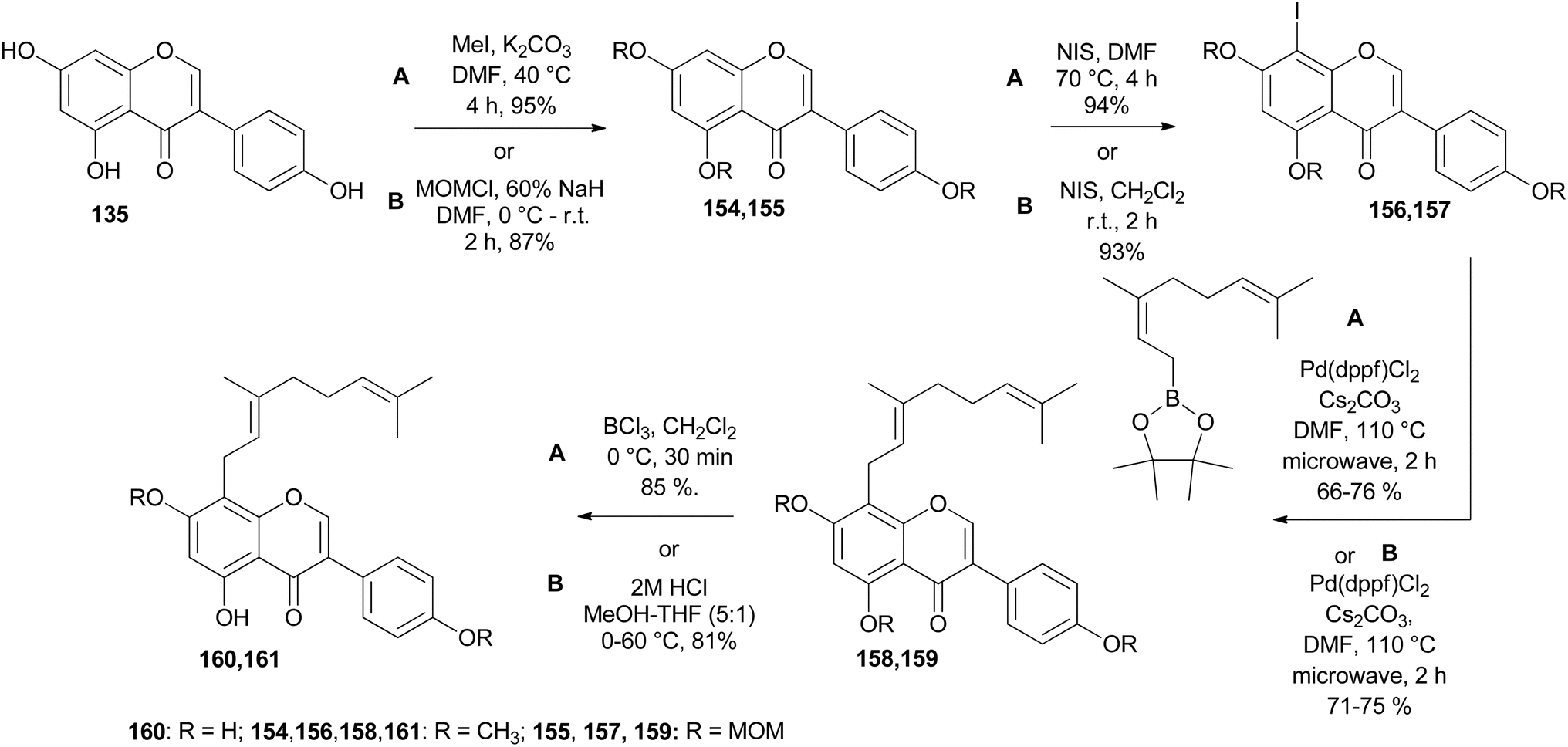
In other instances, the Suzuki–Miyaura reaction was used for the installation of substituents to the isoflavones nuclei.149 Geranylated isoflavones from Lespedeza homoloba and Dalbergia paniculata, lespedezols E1 (160) and 8-geranyl-7-O-methylbiochanin A (161), respectively,150,151 were synthesised from genistein 135, Scheme 22. Alkylation with methyl iodide or MOMCl followed by iodination gave iodinated isoflavones 156 and 157. The Suzuki–Miyaura coupling of 8-iodoisoflavones 156 and 157 with geranyl boronate ester and subsequent MOM deprotection or selective demethylation of 158 and 159 gave the compounds 160 and 161.149
Scheme 22 Total synthesis of lespedezol E1 (160) and 8-geranyl-7-O-methylbiochanin A (161).
2.6. Direct arylation of 2-hydroxyenaminoketones
More recently, different research groups have developed methods for the synthesis of isoflavones by direct arylation of 2-hydroxyenaminoketones.90–93 These provide an advantage of eliminating the pre-functionalisation steps required in metal-catalysed C–C forming reactions that utilise functionalised chromones as precursors. However, the applications of most of the newly developed methodologies involving direct transformation of enaminones are yet to be demonstrated in the synthesis of natural isoflavones.
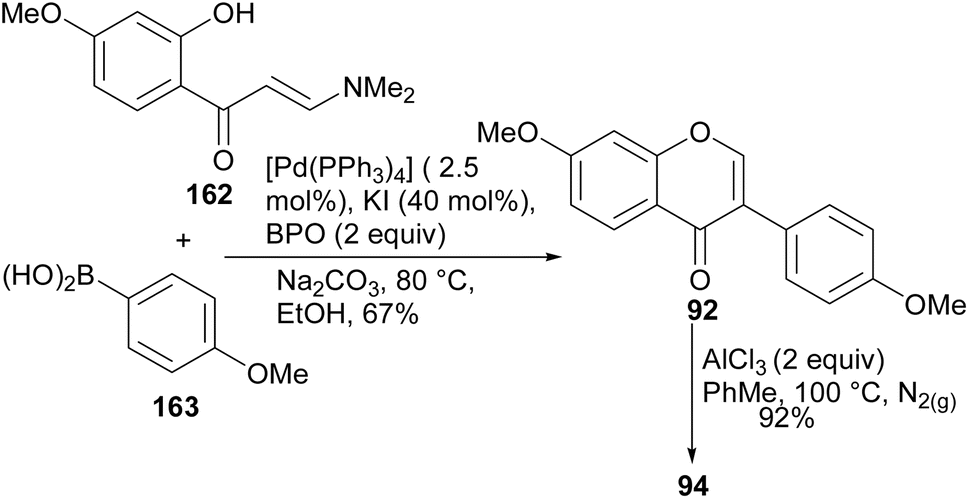
Wan and colleagues developed a method for direct arylation of 2-hydroxyenaminomes with boronic acids in the presence of Pd catalyst, benzoyl peroxide (BPO) and catalytic KI.90 The reaction proceeded through the momentary formation of 3-iodochromone and in situ arylation. Several C-3 arylated derivatives were synthesised including the natural isoflavone daidzein (94). Coupling of enaminone 162 with boronic acid 163 under established conditions gave the isoflavone 92, which was demethylated to afford daidzein (94), Scheme 23.90
Scheme 23 Synthesis of daidzein (94) by direct arylation of 2-hydroxyenaminone.
Although many new synthetic strategies have been developed for isoflavones, their application has been demonstrated mainly in the synthesis of simple isoflavones. Isoflavones with additional substituents such as glycosyl and prenyl units continue to be synthesised by well-established protocols that include the deoxybenzoin route, oxidative rearrangement of chalcones, and the Suzuki–Miyaura reaction. As already mentioned, both the chalcone and deoxybenzoin routes use harsh conditions that necessitate the installation of sensitive groups in the late stages of the synthesis. The Suzuki–Miyauara reaction on the other hand makes use of benign reaction conditions that enable incorporation of such substituents in the early stages of the synthesis. Moreover, a variety of boronic acids are available commercially that enable the ease of synthesis of simple isoflavones and derivatives. The main disadvantage of the Suzuki–Miyaura reaction and other chromone-based metal-catalysed reactions is that they require pre-derivatisation of the chromones and the coupling partners. These increase the number of synthetic steps. Moreover, since the added atoms are eliminated during the cross-coupling reactions, these methodologies are not atom-economic. The methods also utilise transition metals to catalyse the reactions, which are often expensive. In a recent study, isoflavones were synthesised by demethylation and concomitant cyclisation of methoxybenzoylbenzofuran intermediates, prepared by conjugate addition of 2-methoxyenaminoketones with benzoquinones. Although there was no pre-functionalisation of the coupling partners, the method could be improved by optimising the conditions for direct synthesis of isoflavones from 2-hydroxyenaminoketones and benzoquinones. This will eliminate the demethylation step and avoid the use of the toxic BBr3 or volatile TMSI. The synthesis of isoflavones by direct arylation of 2-hydroxyenaminoketones presents an advancement in the synthesis of isoflavones. Therefore, it is encouraged that the scope of these reactions be extended to the synthesis of natural isoflavones.
3. Synthesis of isoflavanones and isoflavans
3.1. Synthesis of racemates
Isoflavanones and isoflavans have been synthesised in racemic form and stereoselectively. Different synthetic strategies were followed for the synthesis of racemates. These included hydrogenation of isoflavones or isoflavenes,152–154 reaction of deoxybenzoin with paraformaldehyde and diethylamine,155,156 [3 + 2]-annulation of chromenes with 1,4-benzoquinones followed by reduction of the resulting pterocarpans to afford isoflavans, and finally oxidation of the isoflavans to isoflavanones.157
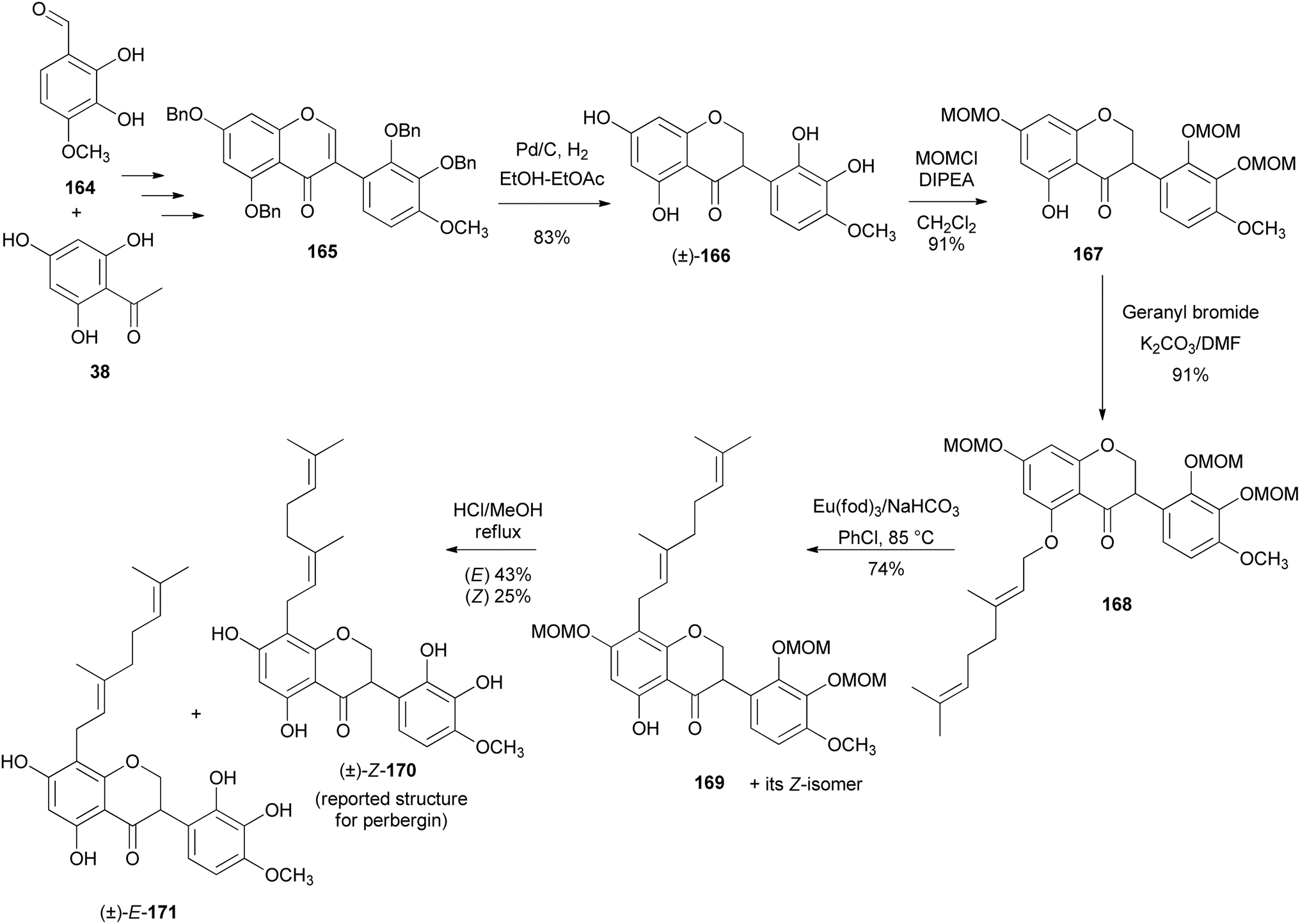
3.1.1 Isoflavanones. Almabruk et al. synthesised proposed [(±)-Z-171] and its isomer [(±)-E-170] over nine steps.152 The key steps involved the synthesis of isoflavanone (±)-166 and C-8 geranylation. The isoflavanone (±)-166 was prepared by reduction of the isoflavone 165, which was synthesised starting from commercially available 2,3-dihydroxy-4-methoxybenzylaldehyde (164) and trihydroxyacetophenone 38.152 Selective MOM protection of the isoflavanone (±)-166 gave compound 167 (91%), which upon O-geranylation under mild conditions afforded compound 168 in good yield (91%) (Scheme 24). The first attempted p-Claisen–Cope rearrangement of the O-geranyl group leading to C-geranylated products proved impossible, but instead resulted in cleavage of the geranyl side chain. This was remedied by employing modified conditions reported by Zhang's group utilising the europium catalyst Eu(fod)3 in the presence of NaHCO3,158 resulting in an E- and a Z-geranylated isoflavanone 169 in 74% yield. Following failure to separate the isoflavanone 169 isomers by HPLC using a chiral-phase column, deprotection of 169 in MeOH under reflux in the presence of 3 N aqueous HCl followed by separation using chiral-phase HPLC (Chiralcel OD-R column) afforded pure (±)-E-171 (43%) and (±)-Z-170 (25%), as well as a mono-MOM-protected intermediate (20%) (Scheme 24).152 Analysis and comparison of the NMR data of the synthesised perbergin [(±)-Z-170] with those of the isolated compound159 prompted structural revision of the isolated compound to that of a C-6 geranylated isoflavanone.152 The synthesised isoperbergins were evaluated for antibacterial activity. They showed good activity against Rhodococcus fascians, Mycobacterium smegmatis, and Staphylococcus aureus, but not against the Gram-negative bacteria Pseudomonas aeruginosa and Escherichia coli.152
Scheme 24 Synthesis of proposed perbergin [(±)-Z-170] and its isomer [(±)-E-171].
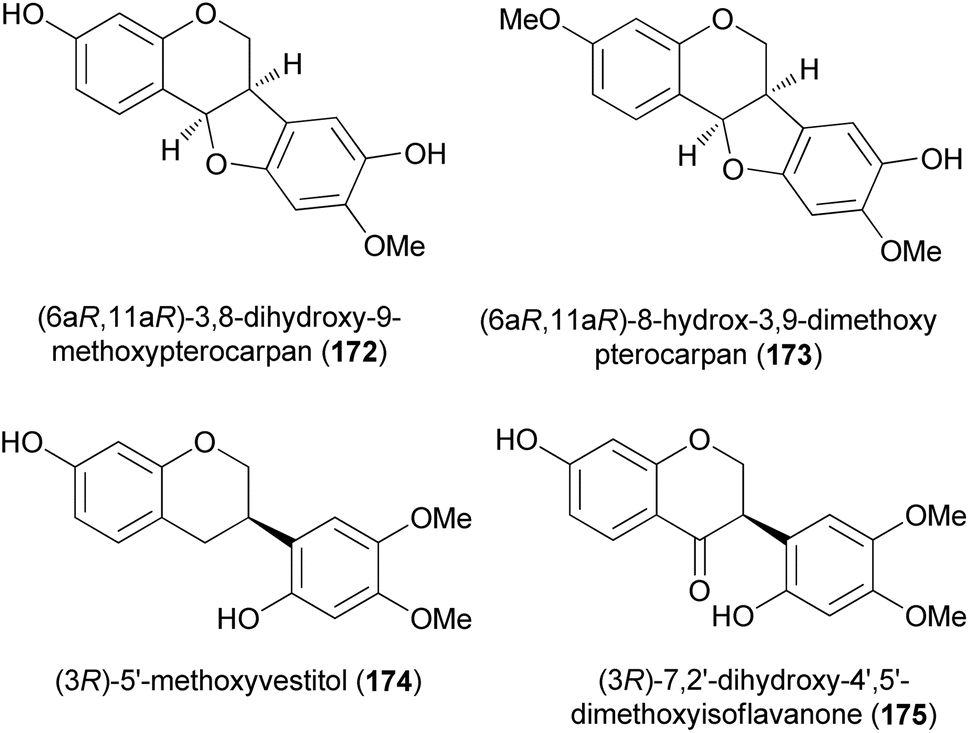
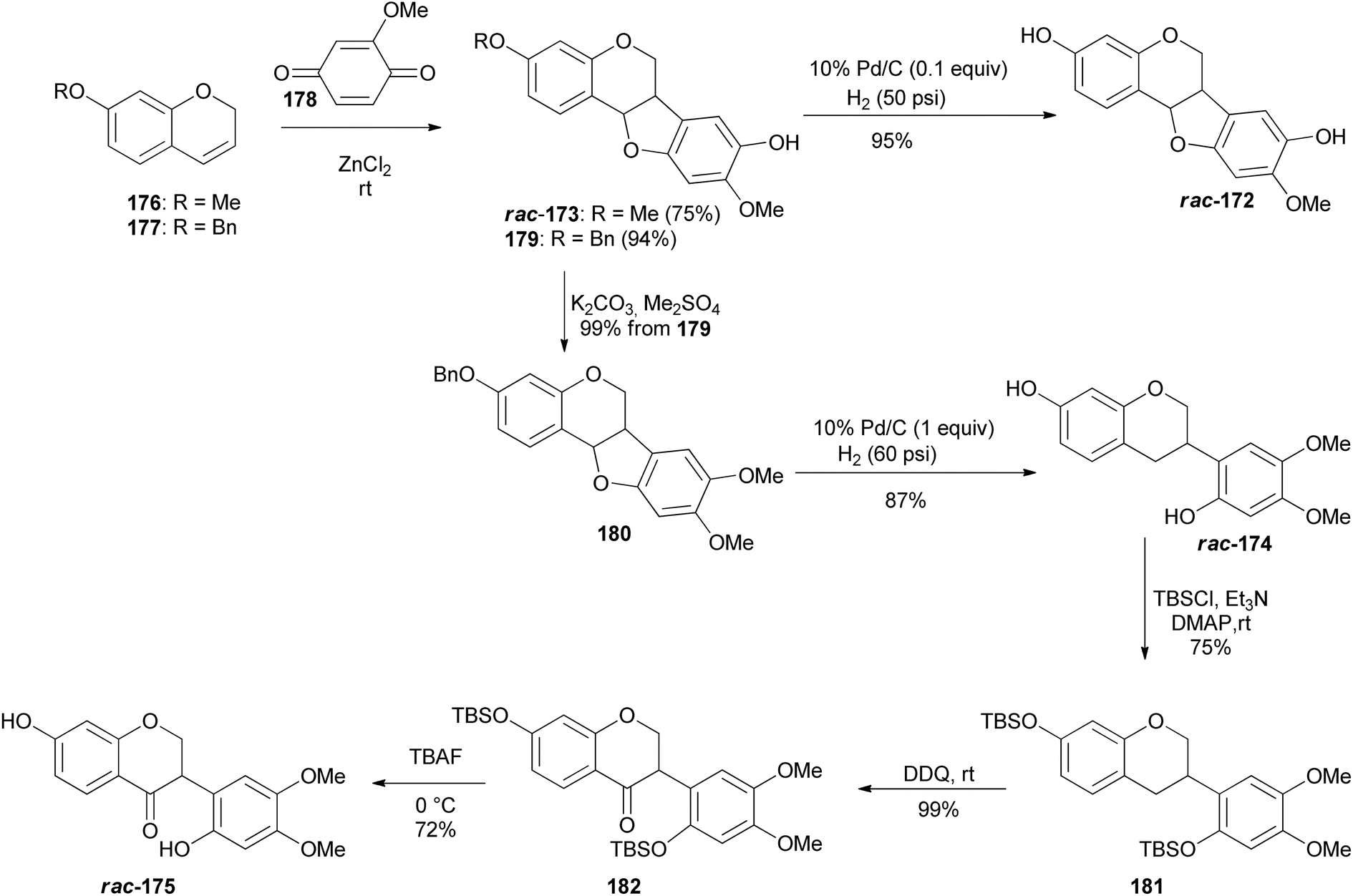
D. K. Singh et al. recently synthesised racemic pterocarpans and isoflavanones isolated from Dalbergia oliveri using a ZnCl2-mediated [3 + 2]-annulation and reductive ring cleavage/reoxidation as key steps. This marked the first total synthesis of 172, 174, and 175 which would pave the way for more biological studies among these compounds (Fig. 2).157 Chromenes 176 and 177 were reacted with 2-methoxy-1,4-benzoquinone (178) in the presence of ZnCl2 at room temperature to afford the corresponding pterocarpans 173 and 179 in good yields (Scheme 25). The ZnCl2 catalyst was preferred over known catalysts like TiCl4 and Ti(O-i-Pr)4 by Engler et al.160 for the same transformation because of ease of manipulation. Pterocarpan 179 was debenzylated using Pd/C catalyst in the presence of hydrogen gas to give rac-172 in a 95% yield. The racemic isoflavan 174 was then synthesised from 179 by methylation of the phenolic group first using Me2SO4 to give 180 in 99% yield, followed by reductive cleavage of the benzylic position of 180 with a large amount of Pd/C to render isoflavan 174 in 87% yield (Scheme 25). The benzylic position of isoflavan 174 could not be oxidised under several oxidation conditions. Protection of the two hydroxy groups of 174 with TBSCl furnished 181 in a 75% yield, followed by exposure of 181 to DDQ to afford 182, and finally desillylation of 182 with TBAF gave rac-175 as a pale yellow amorphous solid in a 72% yield.157
Fig. 2 Naturally occurring pterocarpans, isoflavan, and isoflavanone.
Scheme 25 ZnCl2-mediated [3 + 2]-annulation.
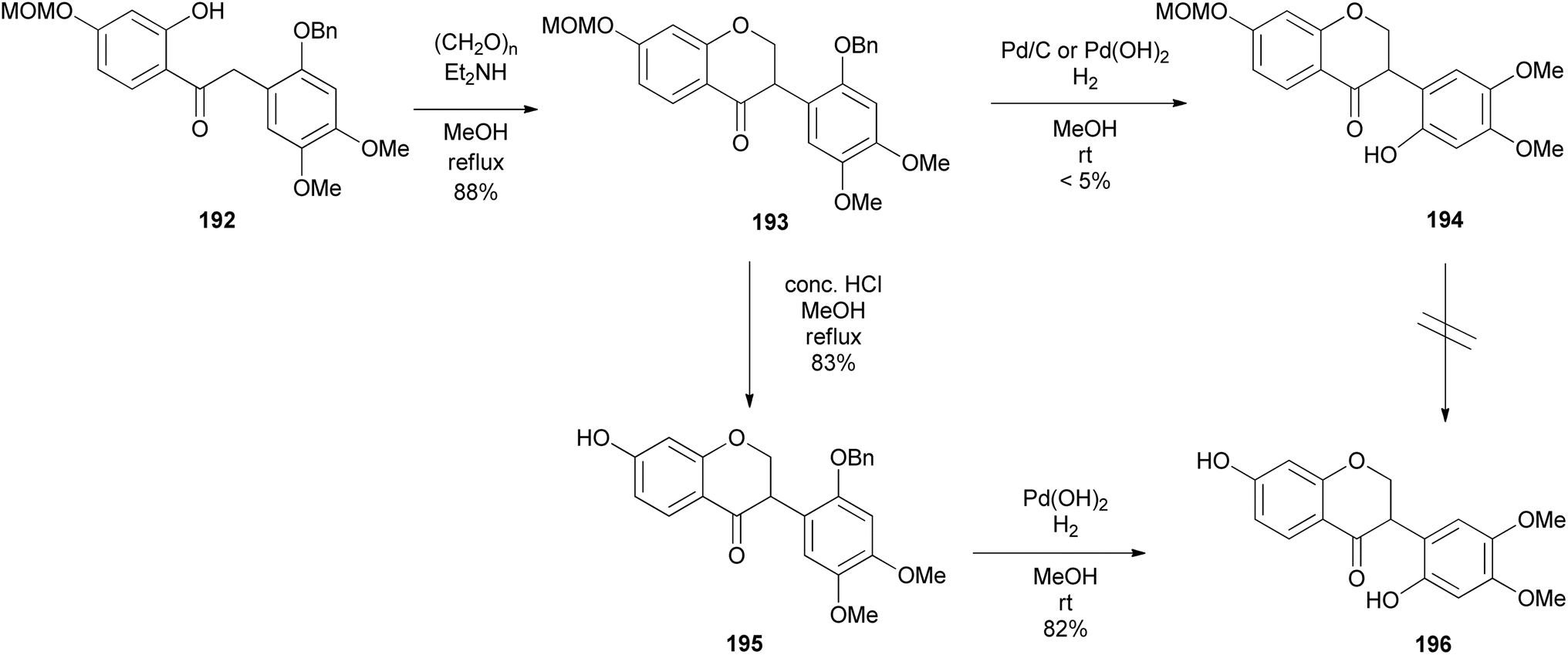
T. Kim et al. (2022) reported the synthesis of a phytoestrogen, 7,2′-dihydroxy-4′,5′-dimethoxyisoflavanone (196) in a gram scale over 11 steps.156 Large quantities of isoflavone 196 made it possible to do further biological studies, although the route was longer as compared to the sophisticated synthesis of isoflavanone 196 by Singh et al. (2018) with low yields.157 The synthesis followed the deoxybenzoin route. However, the deoxybenzoin intermediate 191 was prepared in a sequence of steps that differ from the traditional method involving acylation of phenylacetic acids with phenols.156
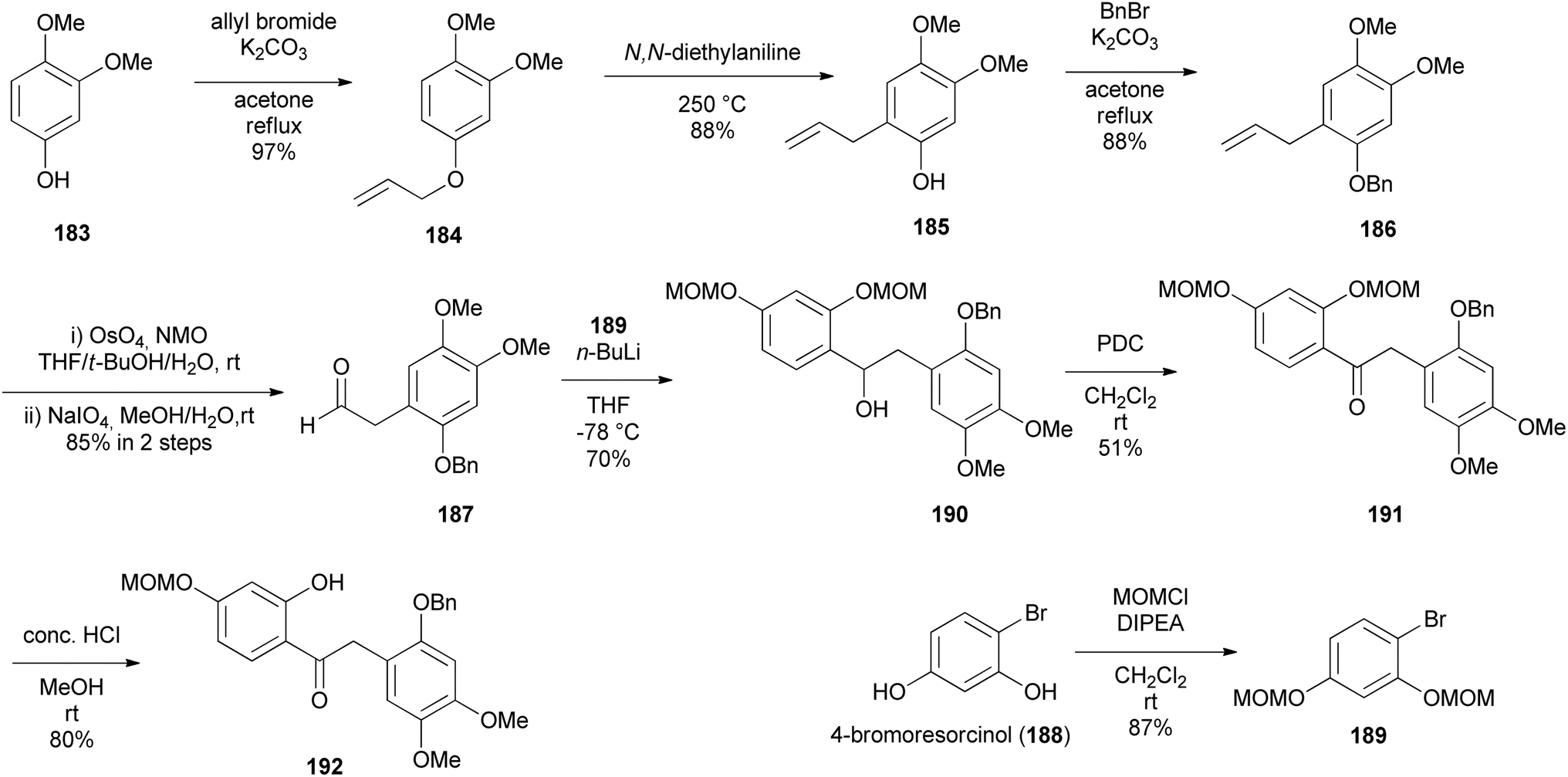
The preparation of key intermediate deoxybenzoin 192 was paramount to accessing the isoflavone scaffold 193 (Scheme 27). 1-Allyl-2-(benzyloxy)-4,5-dimethoxybenzene (186) was synthesised by allylation of commercially available 3,4-dimethoxyphenol (183) to afford O-allyl-substituted phenol 184 in 97% yield, followed by Claisen rearrangement in N,N-diethylaniline exclusively fashioning C-allylated phenol 185 in 88% yield, which was then protected to give benzyl ether 186 in 88% yield. Dihydroxylation followed by oxidative cleavage of the olefinic function of benzyl ether 186 gave phenylacetaldehyde 187 in 85% yield. Aryl bromide 189 was lithiated followed by a spontaneous nucleophilic addition to the acetaldehyde 187 giving benzyl alcohol 190 in 70% yield, which was oxidised by pyridinium dichromate (PDC) affording deoxybenzoin 191 in 51% yield, subsequently followed by selective MOM deprotection in deoxybenzoin 191 finally affording benzylketone 192 in 80% yield. To construct the isoflavanone nucleus, the efficient and scalable approach developed by Gouda et al. was used.155 It is important to note that aryl bromide 189 was obtained in good yields (87%) by protecting 4-bromoresorcinol (188) with the MOM group. Isoflavanone 193 was synthesised in 88% yield on a multi-gram scale by refluxing 2-hydroxyketone 192 in paraformaldehyde and Et2NH in MeOH (Scheme 26). Finally, successive deprotection of the MOM and Bn protecting groups in 193 gave isoflavanone 196 in 82% yield over two-steps. Reversing the sequence of deprotection failed to give the targeted compound 196 (Scheme 27).
Scheme 26 Preparation of key intermediate deoxybenzoin 192.
Scheme 27 Completion of 7,2′-dihydroxy-4′,5′-dimethoxyisoflavanone (196) synthesis.
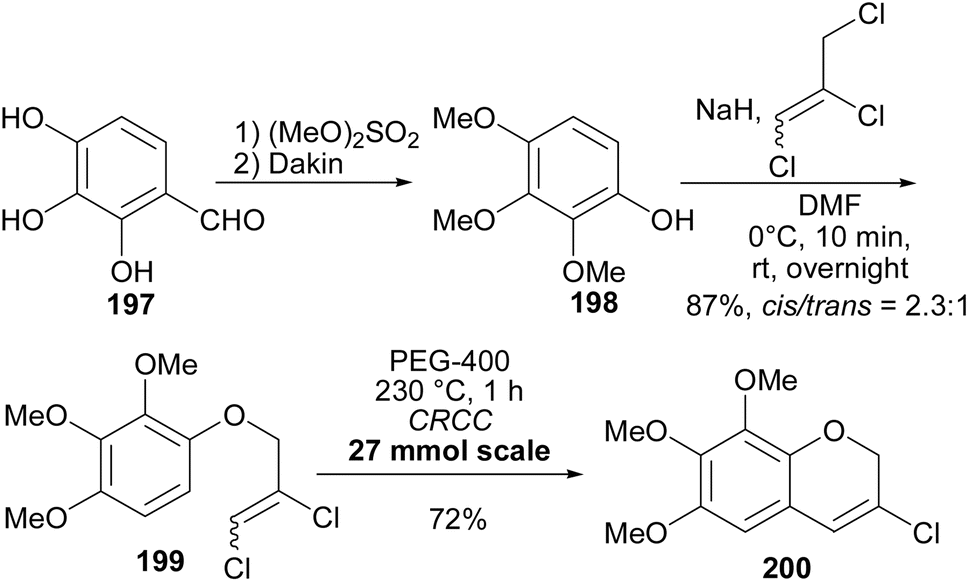
3.1.2 Isoflavans. Kang and colleagues utilised 3-chlorochromenes153,154,161 as intermediates for the divergent synthesis of several natural isoflavans153 and isoflavanquinones, abruquinones B, E, and P (217, 218, and 219).154 The synthesis of the 3-chloro-6,7,8-trimethoxy-2H-chromene 200 and aryl boronic acids needed in divergent Suzuki–Miyaura coupling reactions to form isoflavene derivatives is shown in Schemes 28 and 29. They began by synthesising 2,3,4-trimethoxyphenol 198 starting with commercially available 2,3,4-trihydroxyybenzaldehyde (197).162,163 This was followed by alkylation of phenol 198 with 1,2,3-trichloropropene (1:0.8 mixtures of cis- and trans-isomers) to give aryl 2,3-dichloroallyl ether 199 in 87% yield as a 2.3:1 mixture of cis- and trans-isomers. Lastly, compound 200 was synthesised in 72% yield in a Claisen-rearrangement–cyclisation cascade (CRCC) by heating 199 in polyethylene glycol 400 (PEG-400) at 230 °C for 1 h (Scheme 28),154 which was an improvement compared to the yields obtained in their previous study.153
Scheme 28 Synthesis of 3-chloro-2H-chromene 200.
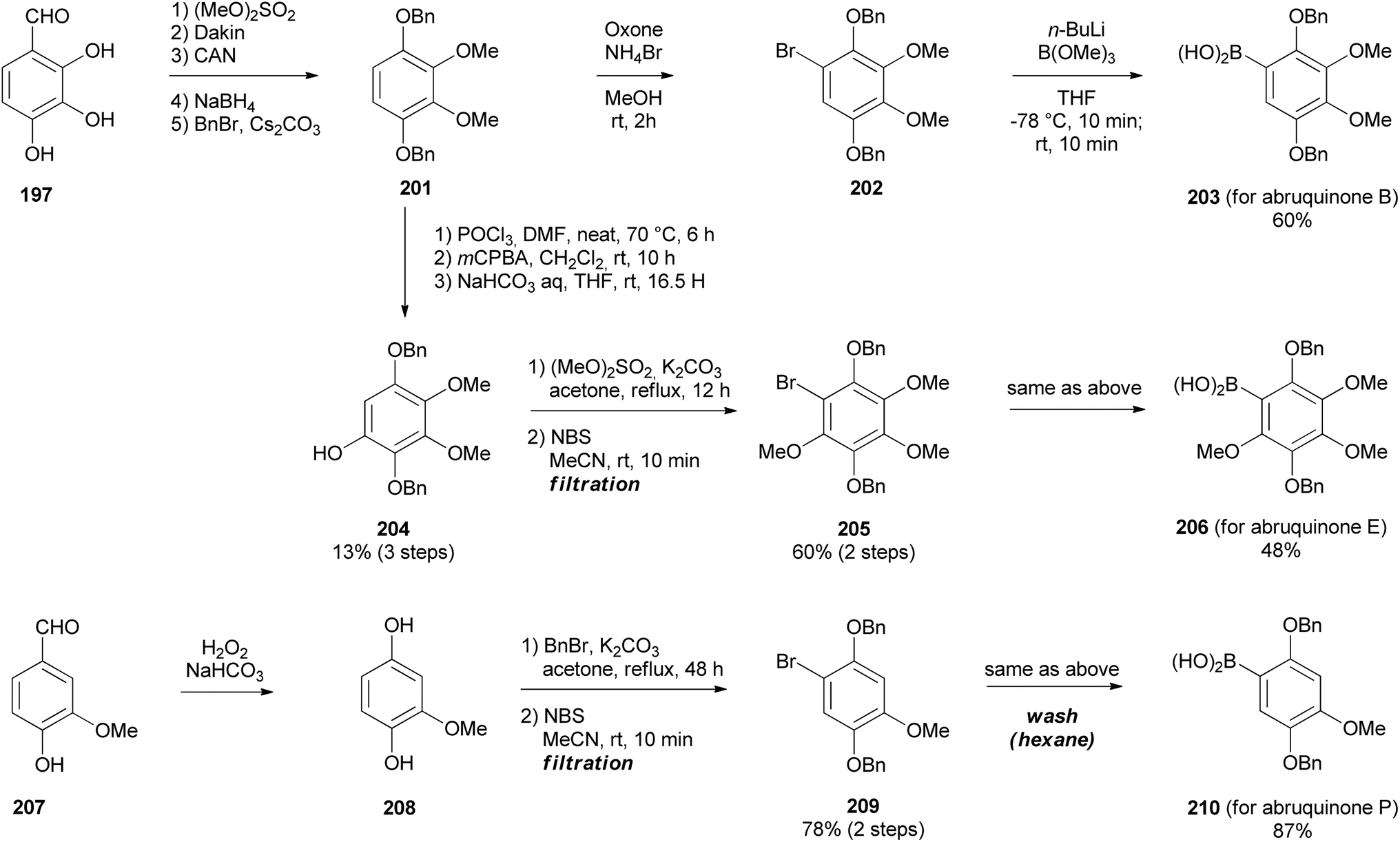
Scheme 29 Synthesis of arylboronic acids 203, 206, and 210.
Having synthesised the 3-chlorochromene 199 precursor, the next step was to prepare arylboronic acids 203, 206, and 210 (Scheme 29). The arylboronic acid 203 was synthesised by converting 2,3,4-trihydroxybenzaldehyde (197) to compound 202 over six steps.164,165 Subsequently, compound 202 was reacted with B(OMe)3 and n-BuLi to yield arylboronic acid 203 in 60% yield. The phenylboronic acid 206 was synthesised from 201 by Vilsmeier–Haack formylation followed by Dakin oxidation to give phenol 204, in low yields (13%). Methylation, bromination (68% in two steps), and borylation of phenol 204 afforded arylboronic acid 206 (48%). Borylation of 202 and 205 succeeded by pre-addition of B(OMe)3 before n-BuLi. The phenylboronic acid 210 was obtained by Dakin oxidation of vanillin (207) to give compound 208,164 which was benzylated and brominated to give highly crystalline bromobenzene 209 in 60% yield over two steps and finally, borylation of 209 gave arylboronic acid 210 in 87% yield, Scheme 29.154
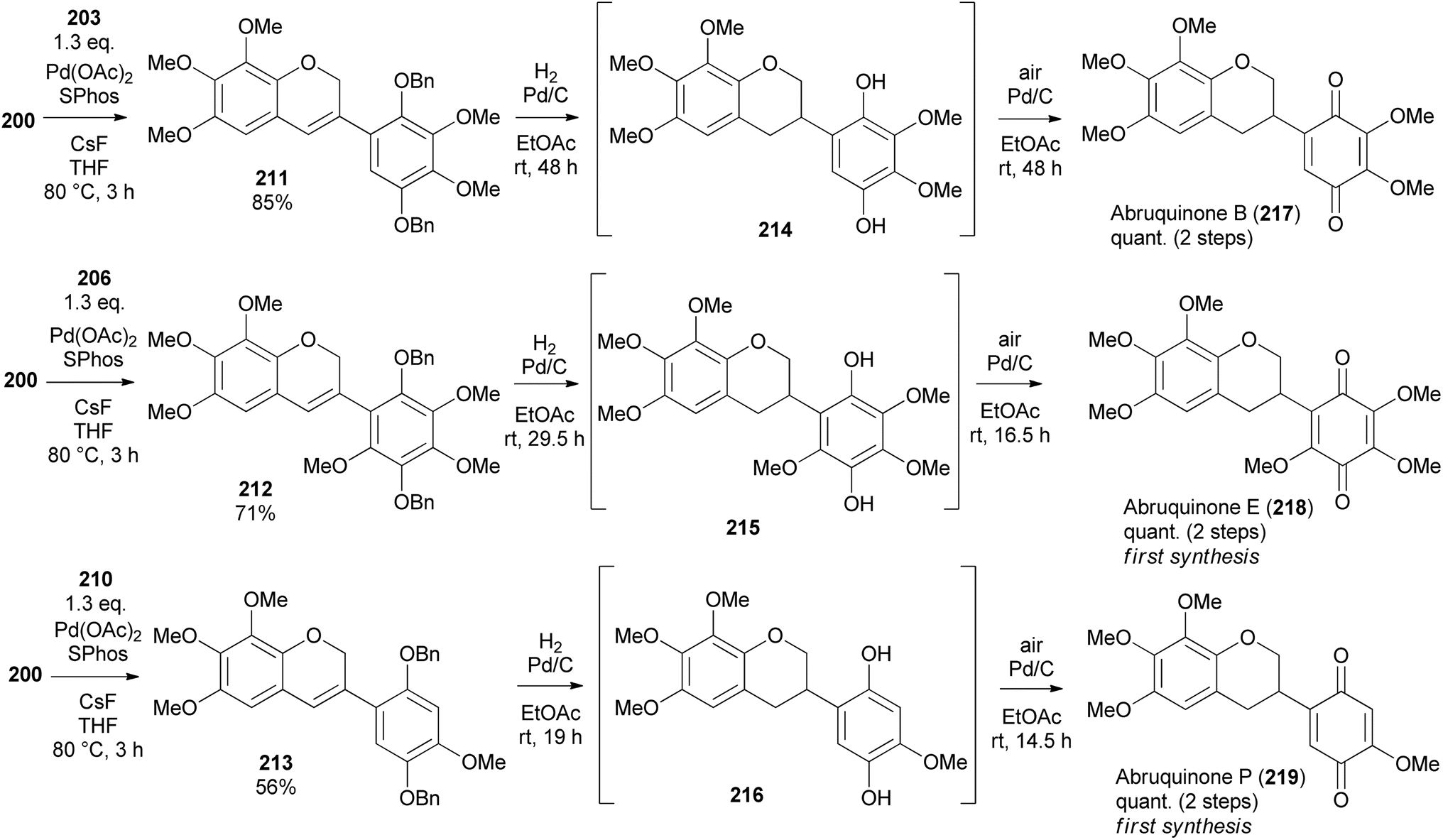
The Suzuki–Miyaura coupling of 3-chloro-2H-chromene 200 and arylboronic acids 203, 206, and 210 using SPhos ligand and Pd(OAc)2 proceeded successfully giving isoflavene derivatives 211, 212, and 213 in 85, 71, and 56% yield, respectively (Scheme 30).153 Reduction and debenzylation of 211, 212, and 213 were carried out under H2 with Pd/C catalyst affording hydroquinone intermediates 214, 215 and 216, which were subsequently exposed to Pd/C in the presence of air to give isoflavanquinone racemates, abruquinones B, E, and P (217, 218, and 219) quantitatively.
Scheme 30 Total synthesis of abruquinones B, E, and P.
3.2. Stereoselective synthesis
Several strategies have been employed for stereoselective syntheses of isoflavanones and isoflavans. These include stereoselective hydrogenation and stereoselective transfer hydrogenation promoted by palladium, ruthenium, and iridium catalysts.76–78 Pd-catalysed decarboxylative stereoselective protonation68 and organo-catalysed stereoselective syntheses of isoflavanones and isoflavans have also been reported.70,74,166 Earlier conventional studies utilised chiral auxiliaries and reagents.167–170
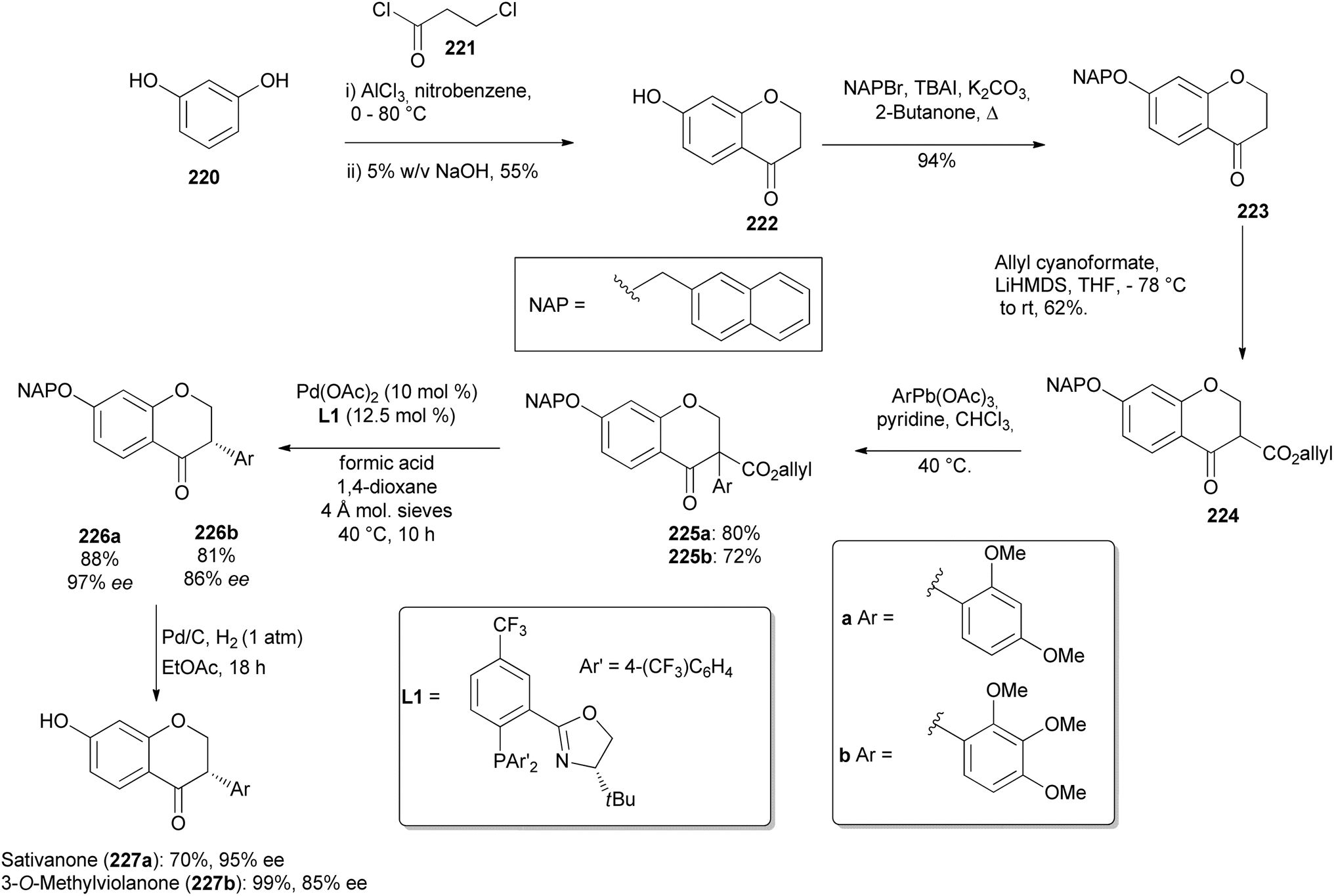
3.2.1 Isoflavanones. Doran et al. synthesised the 7-hydroxyisoflavanones including sativanone (227a) and 3-O-methylviolanone (227b) by Pd-catalysed decarboxylative stereoselective protonation68 starting from chromanone carboxylate.68,171 They synthesised isoflavanone precursors 225a and 225b by lead-mediated arylations of allyl-β-keto ester 224 in yields of 80 and 72%, respectively. To begin, a Friedel–Crafts acylation and cyclisation of resorcinol 220 and 3-chloropropionyl chloride (221) gave 7-hydroxychroman-4-one 222 in 55% yield, followed by 2-naphthylmethyl (NAP) protection to afford 223 in 94% yield and subsequent acylation with allyl cyanoformate to give 224 in 94% yield (Scheme 31). The isoflavanone precursors 225a and 225b were reacted in the decarboxylative stereoselective protonation reaction with the (S)-(CF3)3-t-Bu-PHOX ligand to give compounds 226a and 226b in 88 to 81% isolated yields and 97 to 81% ee (Scheme 31). The NAP group was removed to yield the natural compounds sativanone (227a) and 3-O-methylviolanone (227b) without erosion of enantiomeric excess by stirring the protected isoflavanones 226a and 227b overnight with Pd/C (10%) in ethyl acetate under hydrogen (1 atm), Scheme 31.68
Scheme 31 First stereoselective synthesis of sativanone (227a) and the first synthesis of 3-O-methylviolanone (227b).
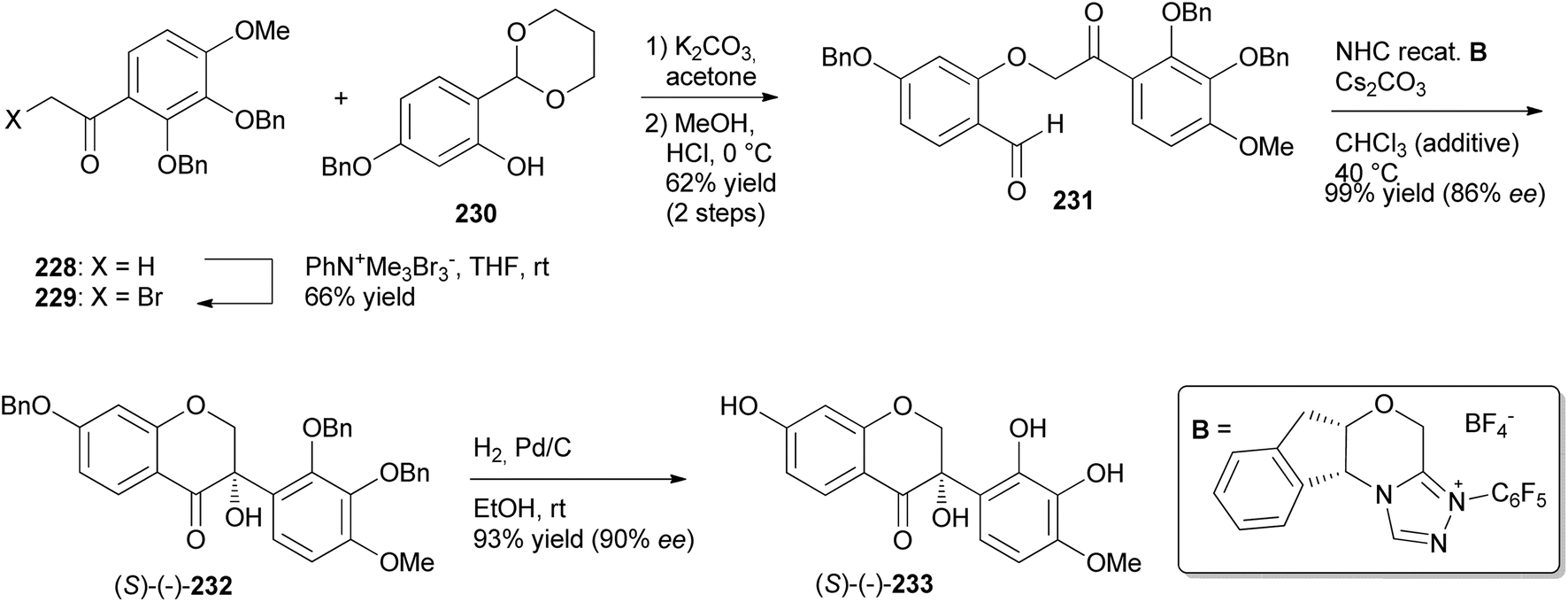
Iwai and colleagues were the first to synthesise natural product isodarparvinol B (233) using an intramolecular benzoin reaction as a key step in the presence of N-heterocyclic carbene (NHC) catalyst.70 This took advantage of C–C bond formation via polarity inversion of the formyl group into acetyl anion equivalent and high enantioselectivity of NHCs.73 Ketone 228 was α-brominated with phenyltrimethylammonium tribromide to give α-bromoketone 229 in 66% yield. Compound 230 was O-alkylated with α-bromoketone 229 in a Williamson ether synthesis reaction, followed by deprotection of the acetal group, to afford aldehyde 231 in 62% yield. Aldehyde 231 was exposed to the NHC-catalysed intramolecular benzoin reaction to furnish optically active 4-chromanone 232 in 99% yield with 86% ee. Finally, hydrogenation of 232 gave (−)-233 in 93% yield with 90% ee (38% total yield from starting material) (Scheme 32).70 When the NHC-catalysed intramolecular benzoin reaction was conducted in THF in the previous study, a racemic mixture was obtained.166
Scheme 32 Total synthesis of (−)-isodarparvinol B [(−)-233].
3.2.2 Isoflavans. One of the well-studied isoflavan compounds is equol, a metabolite of the soy isoflavone, daidzein (94). Equol was first identified from the horse urine.172 It has been reported to exhibit many biological activities, particularly oestrogenic activity.173,174 Therefore, its synthesis has been of interest to researchers. Several studies report the stereoselective synthesis of the natural enantiomer (S)-equol (237)74,77–79,169,170,175–178 and there are limited studies that include the synthesis of the non-natural enantiomer (R)-equol.75,179,180
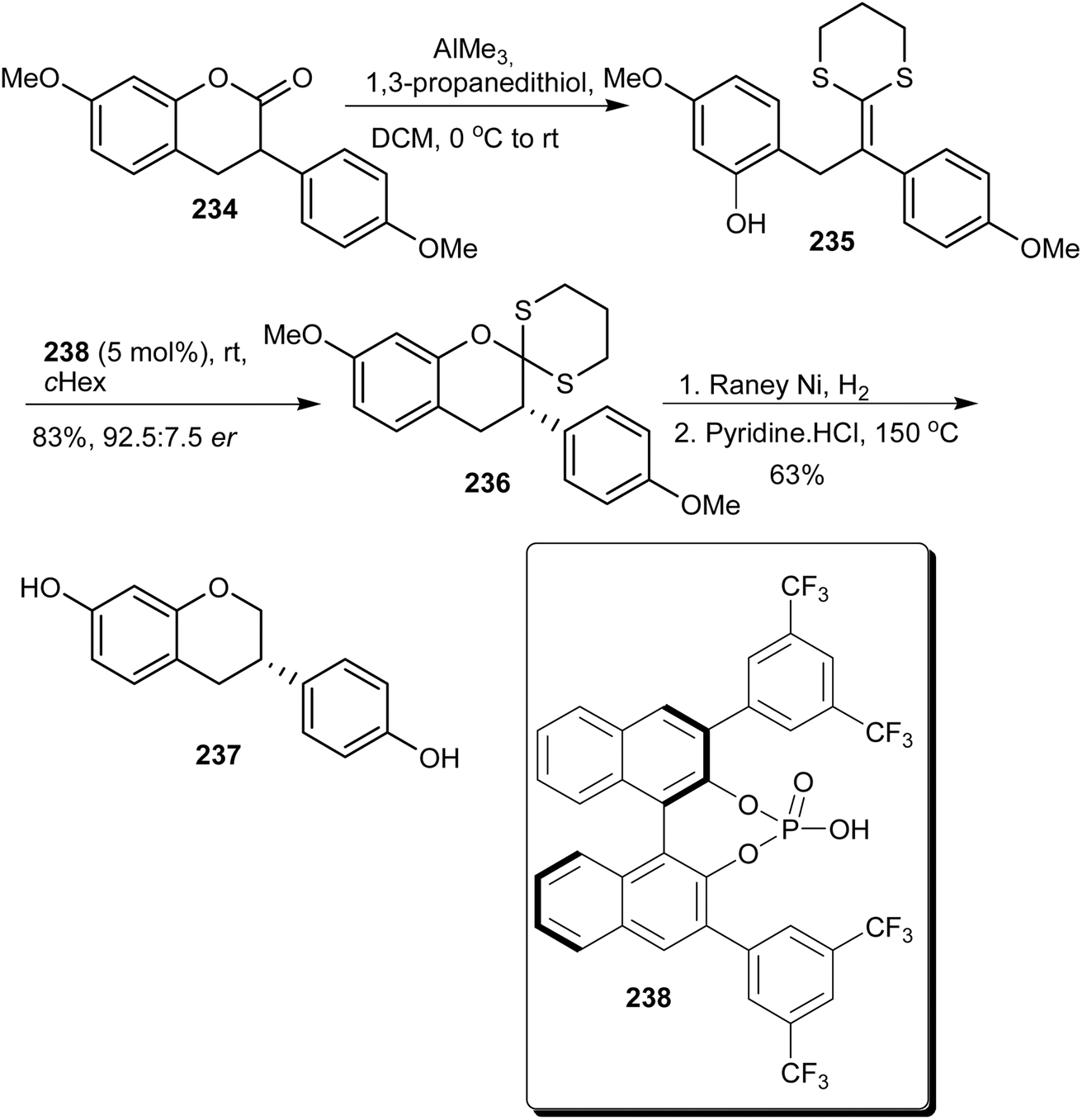
Lee and List developed a method for deracemisation of 3-arylcoumarins using chiral phosphoric acids.74 The method could be applied to afford enantioenriched 3-arylcoumarins and 3-arylchromans. Following this, (S)-equol (237) was synthesised by conversion of racemic 3-arylcoumarin 234 into ketene dithioacetal 235, which upon protonation in the presence of chiral phosphoric acid 238 afforded enantioenriched coumarin dithioacetal 236 in 83% yield and 92.5 er. Reduction of 236 and demethylation afforded (S)-equol (237), Scheme 33.74
Scheme 33 Synthesis of (S)-equol (237) using chiral phosphoric acid.
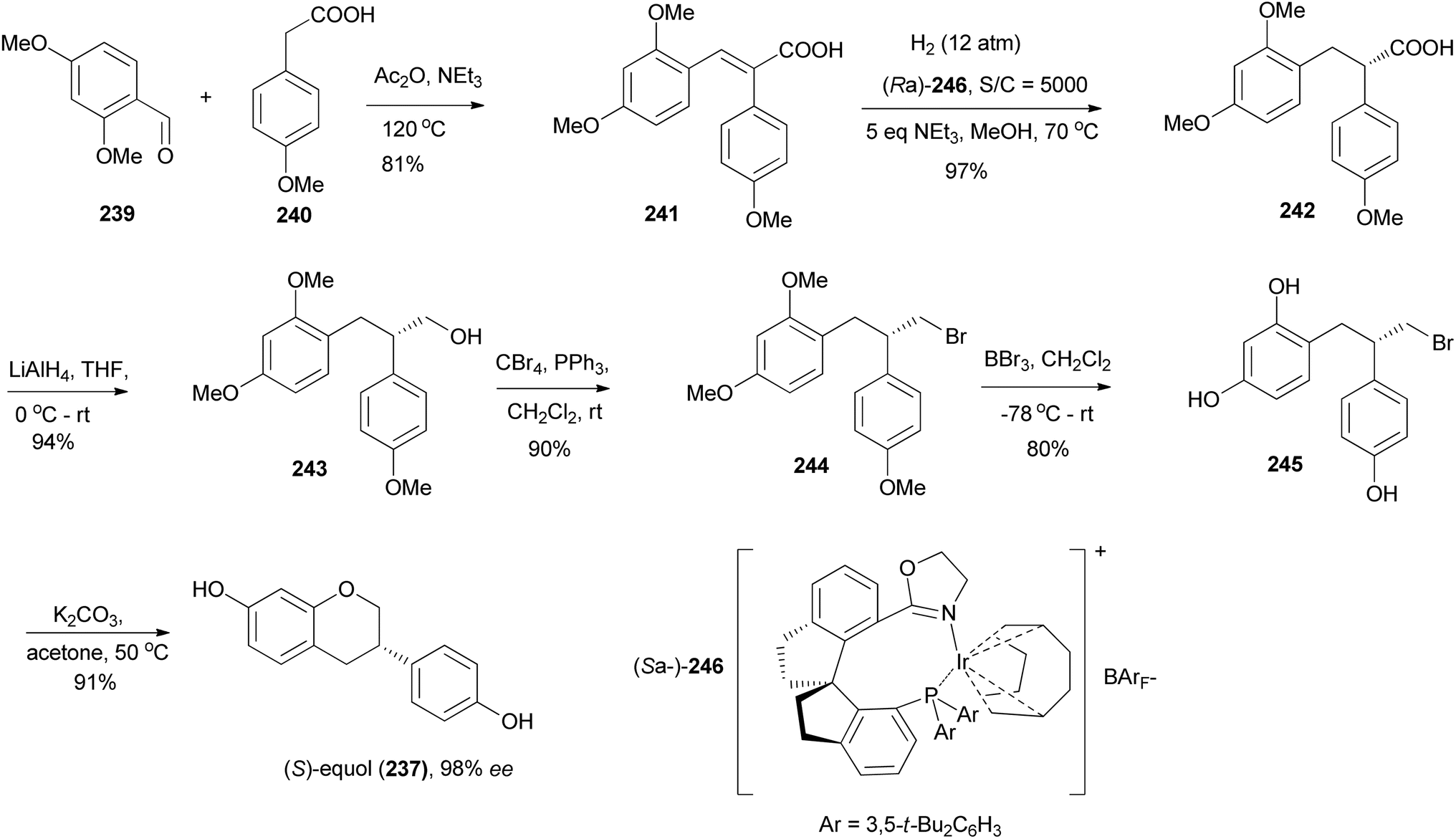
Yang and colleagues synthesised (S)-equol (237) from a chiral 2,3-diarylpropionic acid 242, obtained by stereoselective hydrogenation of cinnamic acid 241 in the presence of a chiral iridium catalyst (Ra)-246. Reduction of the carboxylic acid group of 242 gave an alcohol 243, which was converted into alkylbromide 244. Demethylation of 244 and subsequent O-cyclisation of 245 gave (S)-equol (237) in 91% yield and 98% ee, Scheme 34.79
Scheme 34 Synthesis of (S)-equol (237) by stereoselective hydrogenation using iridium catalyst.
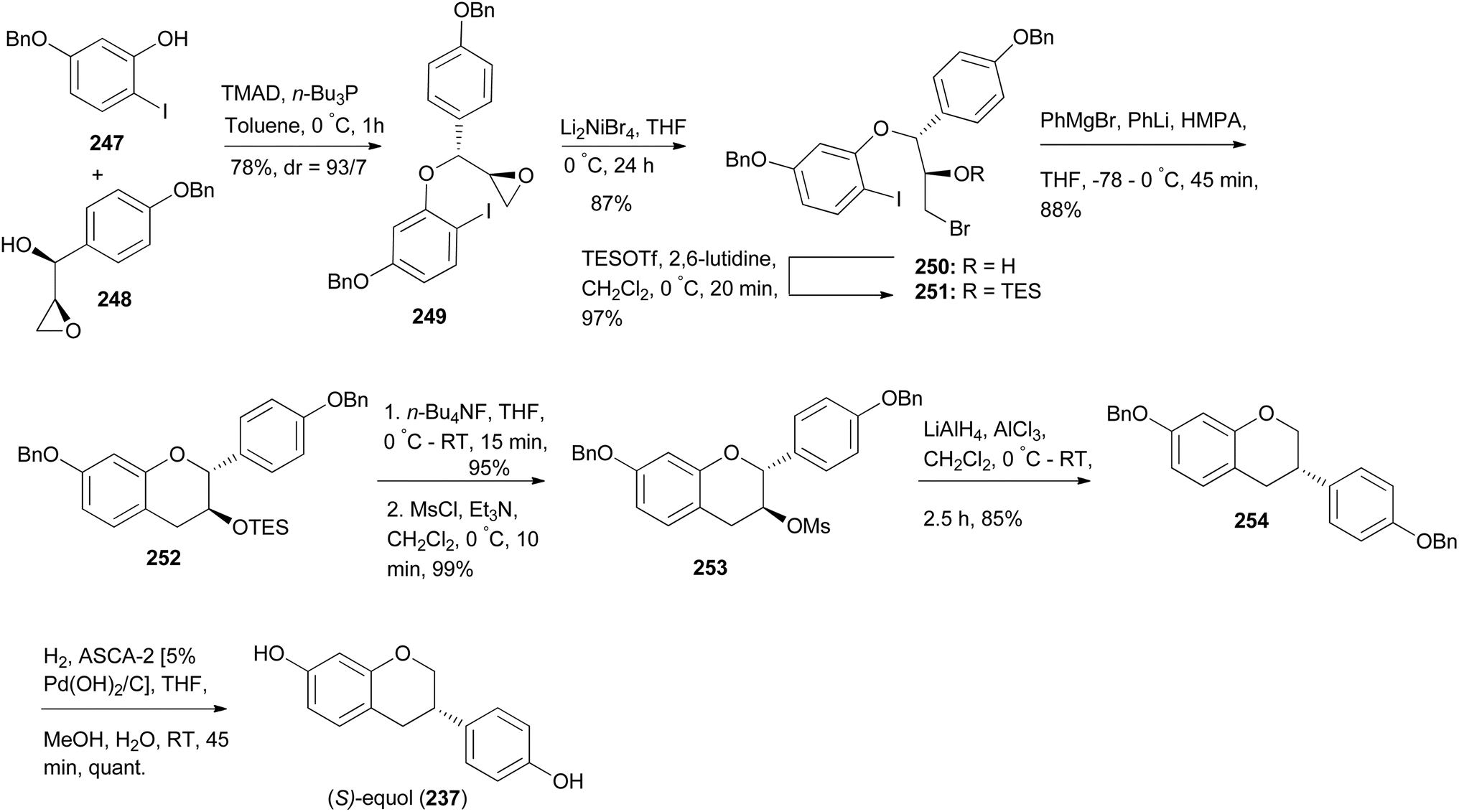
Nakamura and colleagues synthesised (S)-equol (237) by a strategy that involved 1,2-aryl migration of 3-methanesulfonylflavans as the key step.175 The synthesis was initiated by Mitsunobu reaction of iodophenol 247 with chiral epoxyalcohol 248 to afford a diaryl ether 249. Bromination of oxirane 249 gave an alkylbromide 250 in 87% yield and its epimer. Bromohydrin 250 was converted into a TES ether 251, which was assembled into an isoflavan 252 upon treatment with Ph3MgLi. Conversion of 252 into mesylate 253 followed by 1,2-aryl shift facilitated by AlH3 afforded isoflavan 254, which upon debenzylation gave (S)-equol (237), Scheme 35.175
Scheme 35 Synthesis of (S)-equol 237 by 1,2-aryl migration of a 3-methanesulfonylflavan 253.
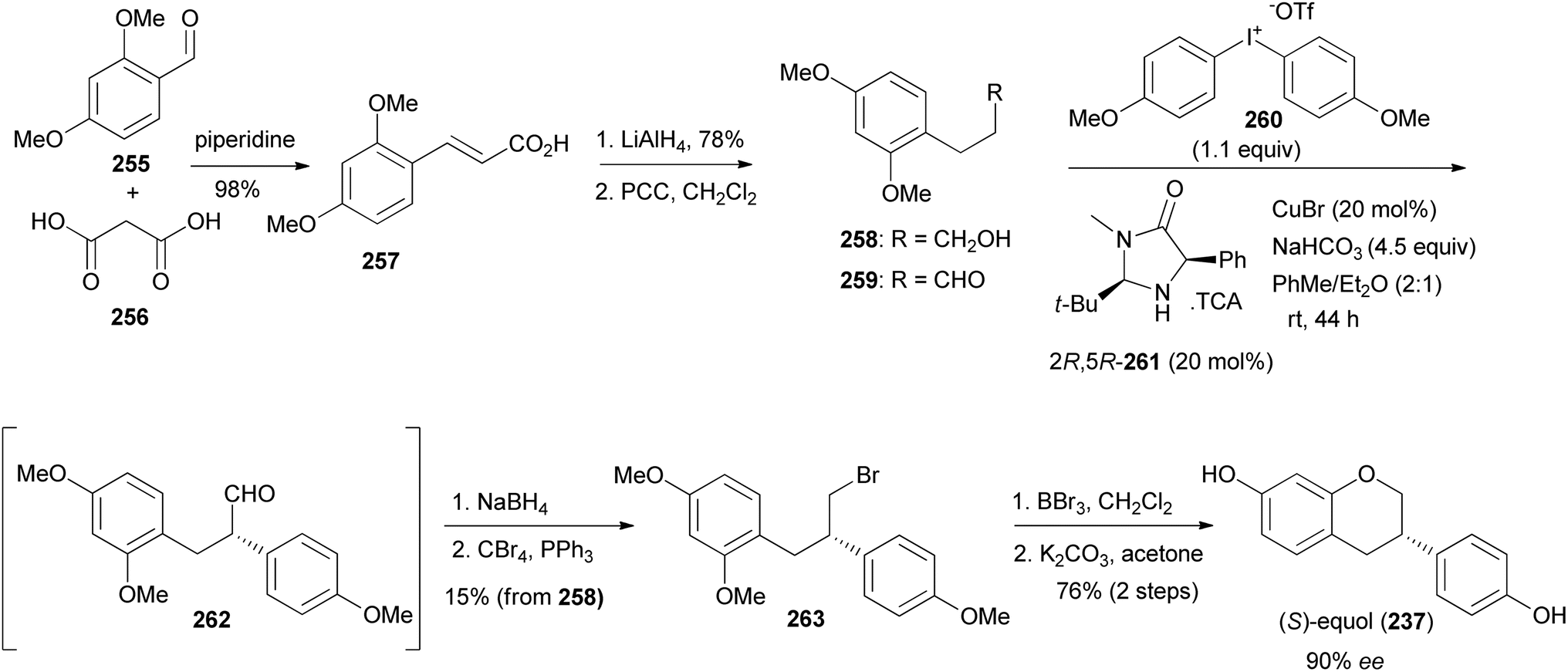
Uemura and colleagues synthesised (S)-equol (237) from enantioenriched diphenylpropanal 262.75 The diphenylpropanal (S)-262 was prepared by stereoselective α-arylation of phenylpropanal 259 with diaryliodotriflate 260 in the presence of a chiral phenylimidazolidin-4-one catalyst (2R,5R) 261 and CuBr.181,182 Compound 259 was in turn synthesised by oxidation of the alcohol 258, obtained from cinnamic acid 257. Reduction of the aldehyde 262 and bromination of the resulting alcohol gave bromodiphenylpropane 263. Demethylation and oxycyclisation rendered (S)-equol (237) in 76% yield over two steps and 90% ee (Scheme 36). The same protocol was followed for the synthesis of (R)-equol from 259, using phenylimidazolidin-4-one catalyst (2S,5S) 261.75
Scheme 36 Synthesis of (S)-equol (237) from enantioenriched diarylpropanal.
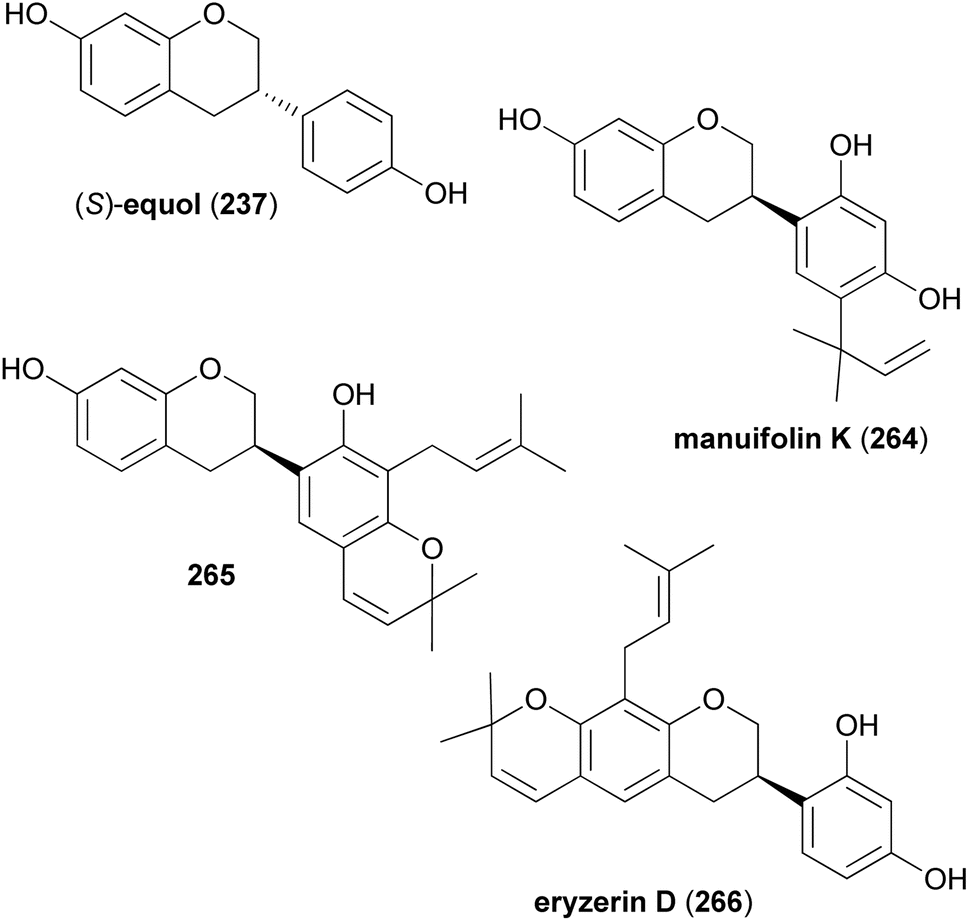
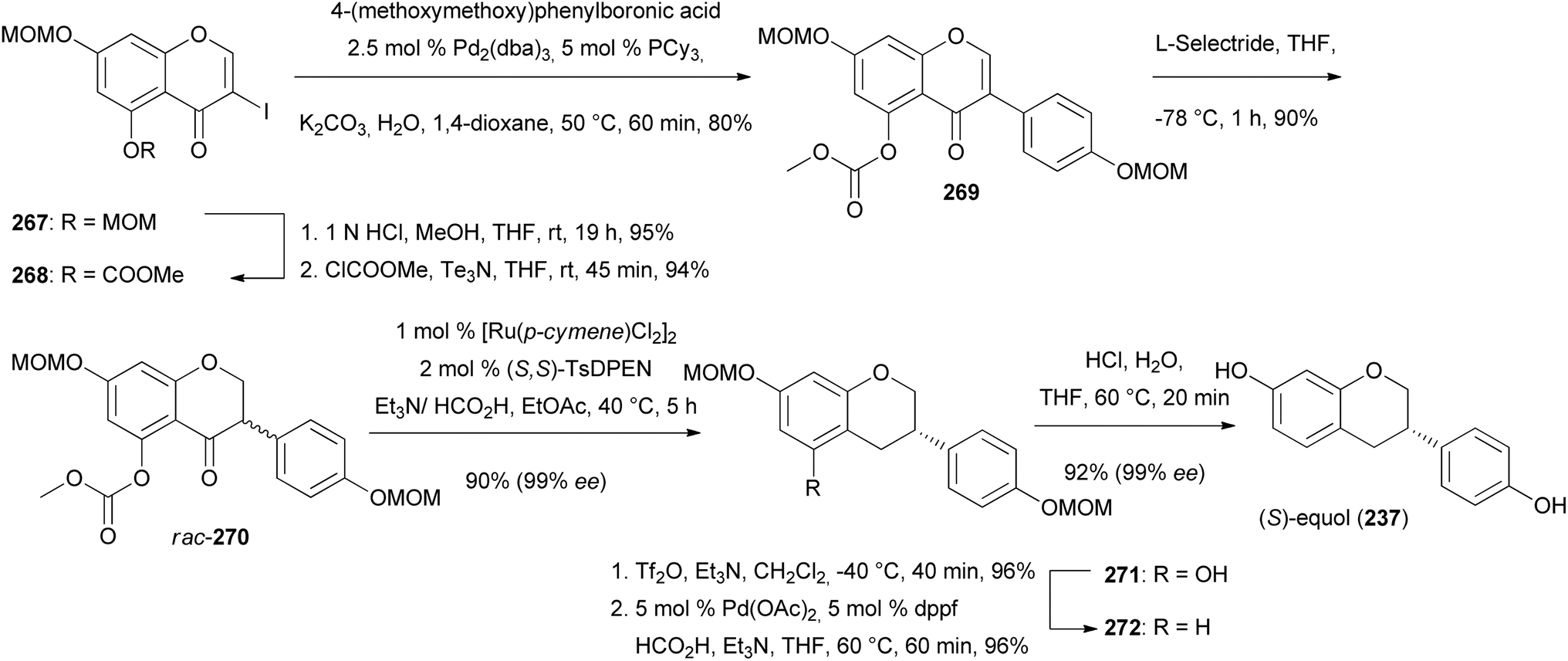
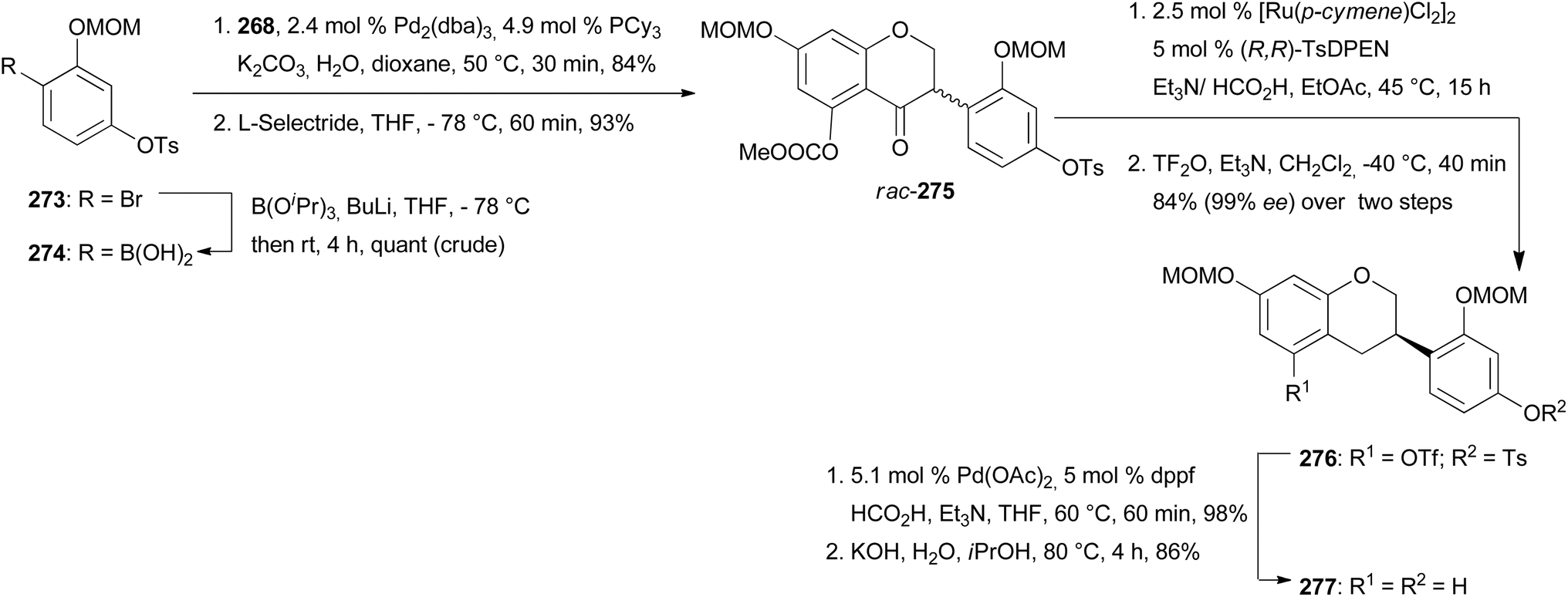
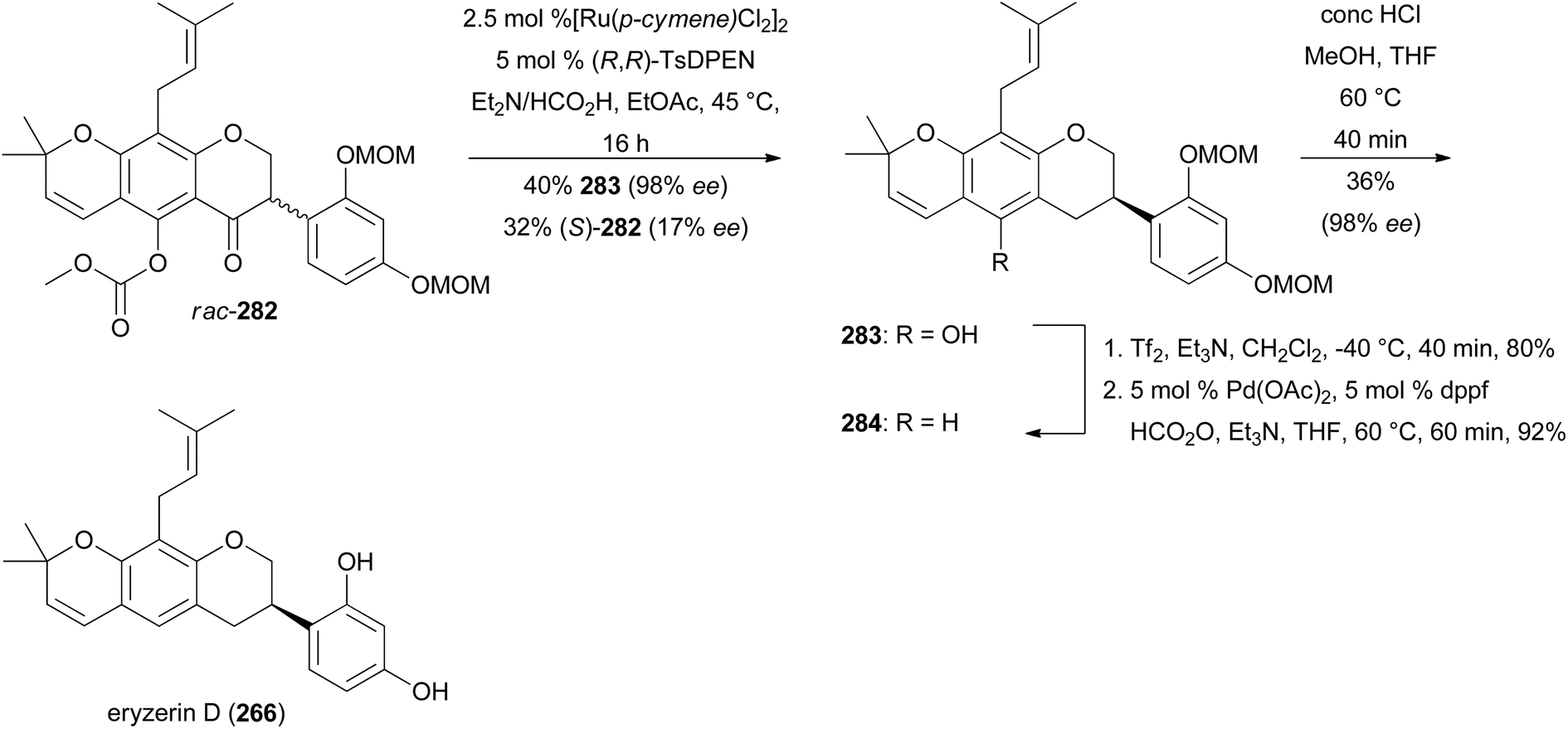
Keßberg et al. stereoselectively synthesised isoflavans (S)-equol (237), manuifolin K (264), 265 and eryzerin D (266) by key steps involving ruthenium-catalysed stereoselective transfer hydrogenation of racemic isoflavanone precursors and Pd-catalysed deoxygenation (Fig. 3).77 The synthesis of (S)-equol (237) started with chemoselective deprotection of the 5-O-MOM moiety of chromone 267, followed by acylation of the phenol with methyl chloroformate to give vinyl iodide 268 in 94% yield (Scheme 37). The Suzuki coupling of 268 with 4-(methoxymethoxy)phenylboronic acid followed by conjugate reduction of 269 resulted in isoflavanone rac-270. The stereoselective transfer hydrogenation/deoxygenation cascade using ruthenium catalyst at low catalyst loading first gave (S)-271 in 90% yield with 99% ee. The free hydroxy group was removed reductively by treatment of compound 271 with triflic anhydride followed by palladium-catalysed deoxygenation to afford product 272 in 96% yield over two steps. Finally, hydrolysis of the acetal moieties of 272 gave (S)-equol (237) in 92% yield with 99% ee.
Fig. 3 Target isoflavonoids.
Scheme 37 Synthesis of (S)-equol (237).
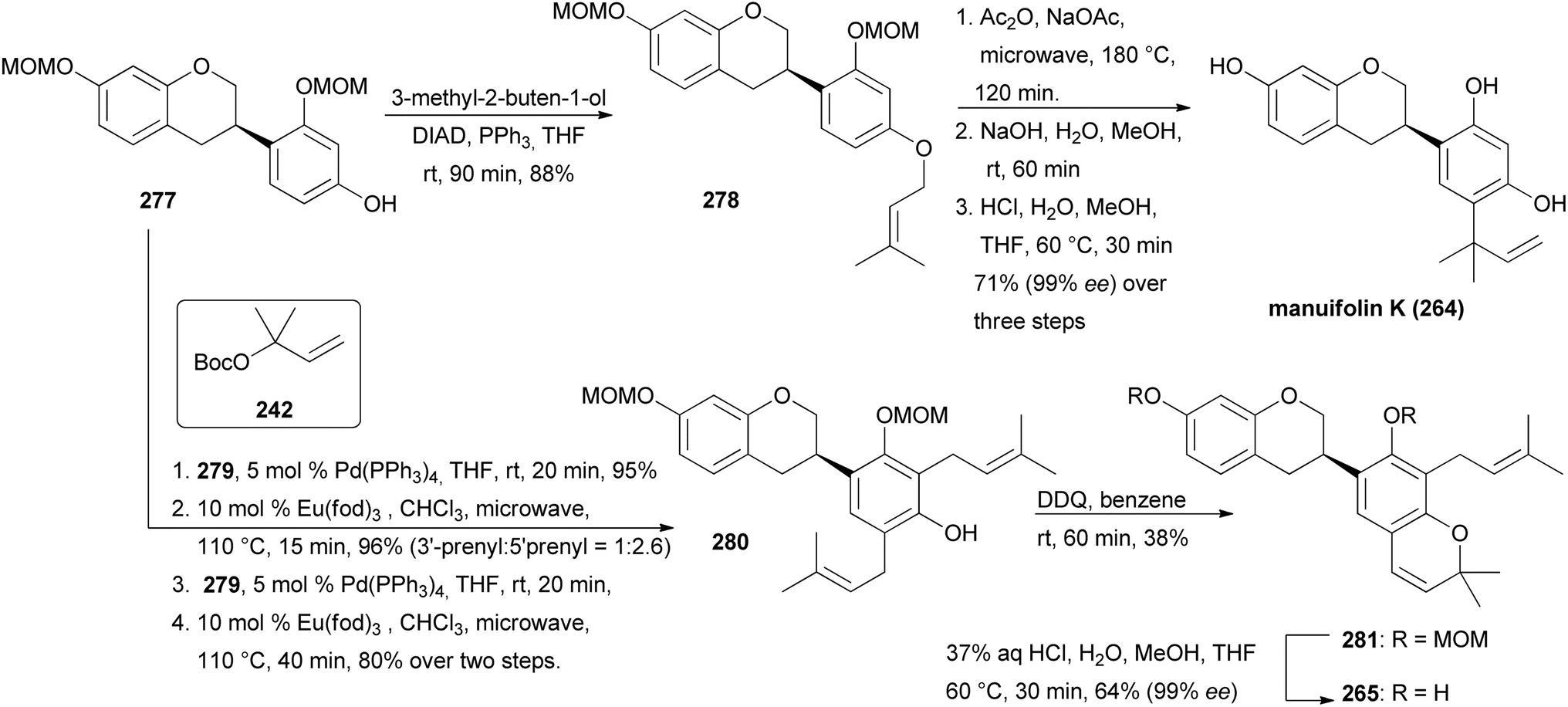
The syntheses of manuifolin K (264) and isoflavan 265 commenced by coupling of chromone 268 with boronic acid 274 and subsequent treatment of the intermediate with L-selectride to give ATH substrate rac-275 in 93% yield (Scheme 38). The domino ATH reaction of rac-275 was achieved at a slightly higher catalyst loading followed by treatment with triflic anhydride giving enantiopure isoflavan 276 in 84% yield with 99% ee over two steps. Lastly, deoxygenation at C-5 and removal of the tosylate group furnished product 277. O-Prenylation of isoflavan 277 under Mitsunobu conditions gave prenyl ether 278 in 88% yield. Lastly, Claisen rearrangement in acetic anhydride and cleavage of the acetyl and MOM groups gave manuifolin K (264) in 71% with 99% ee (Scheme 39). The isoflavan 265 was synthesised from compound 277 in six steps. The prenyl groups were installed by a Tsuji–Trost allylation with allylic carbonate in 95% yield and subsequent europium-catalysed rearrangement, favouring the 5′-position over the 3′-position due to steric constraints. Both regioisomers were transformed to 280 in 80% yield using the same two-step protocol. Benzopyran 281 was synthesised in 38% yield through oxidative cyclisation of 280 using DDQ. Finally, acid-promoted deprotection furnished the target compound 265 in 68% yield with 99% ee.77 Comparison of the analytical data of the synthesised isoflavan 265 with those of the isolated compound revealed a mismatch between the data sets,183 which revealed that Jang and colleagues had isolated eryzerin D (266) instead.77,183
Scheme 38 Synthesis of isoflavan 277.
Scheme 39 Synthesis of isoflavans 264 and 265.
To complete the synthesis of eryzerin D (266) an enantioselective reduction of the highly functionalised isoflavanone rac-282 was conducted, which yielded 40% of the unstable compound (R)-283 with 98% ee. Compound (R)-283 was immediately treated with triflic anhydride and subsequently deoxygenated to give isoflavan 284 in 86% yield over two steps. The removal of MOM groups gave eryzerin D (266) in 38% yield with 98% ee (Scheme 40).77 The NMR and MS data of the synthesised eryzerin D (266) matched those reported for the isolated compound,38,183 and its configuration was determined to be R based on ECD data.77 It is noteworthy that rac-282 was synthesised in several steps from chromone 267.184
Scheme 40 Synthesis of eryzerin D (266).
4. Synthesis of pterocarpans
A detailed review on pterocarpans was published in 2013 (ref. 185) and other recent reviews included sections on the synthesis of pterocarpans.76,186 Therefore, the syntheses of pterocarpans not included in these reviews will be discussed. They include: the synthesis of 2,3,9-trimethoxypterocarpan by Mizoroki–Heck oxyarylation reaction,41 total synthesis of neorautenol and shinpterocarpin,187 and the stereoselective total synthesis of stachyodin A.67
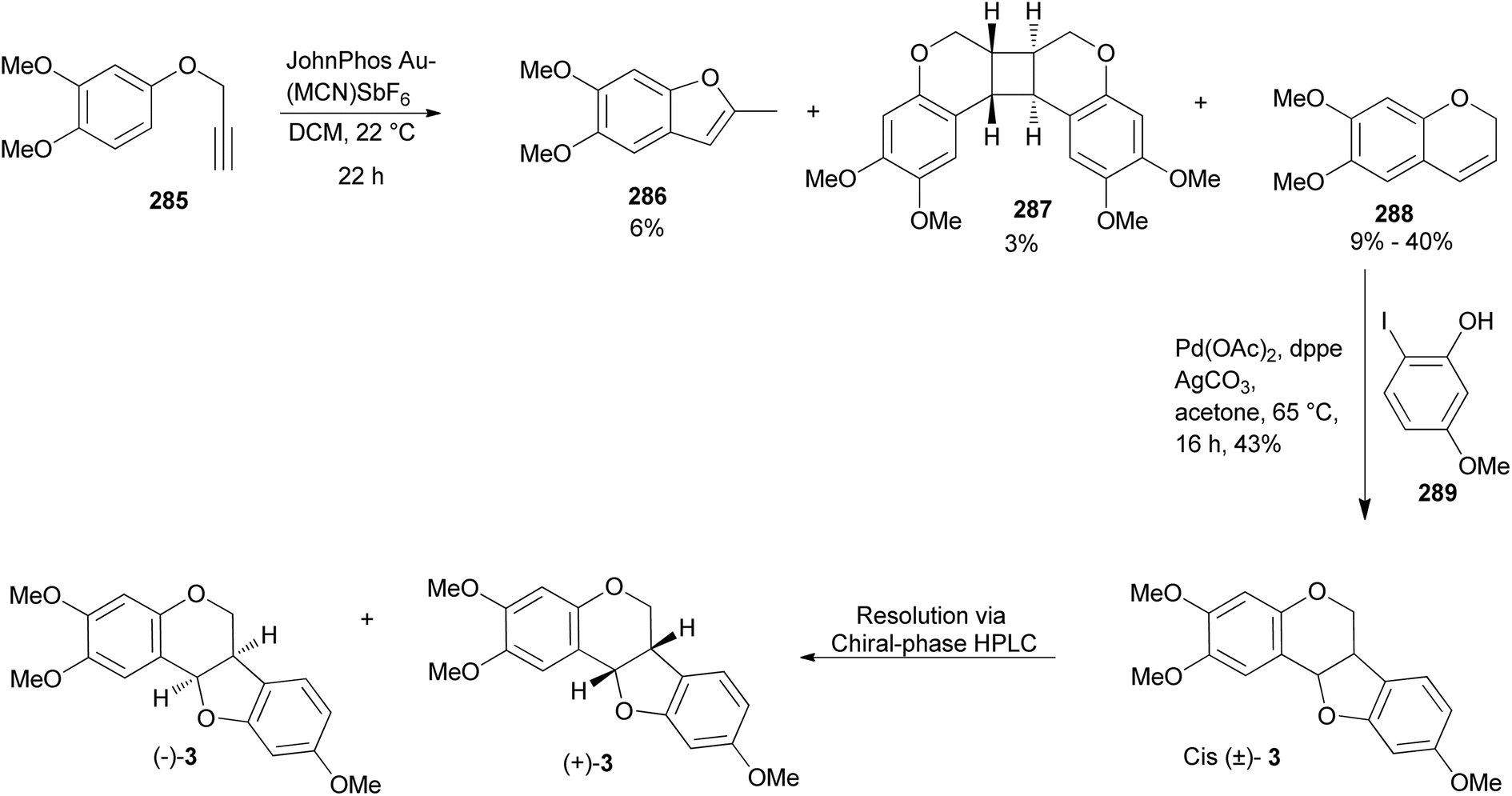
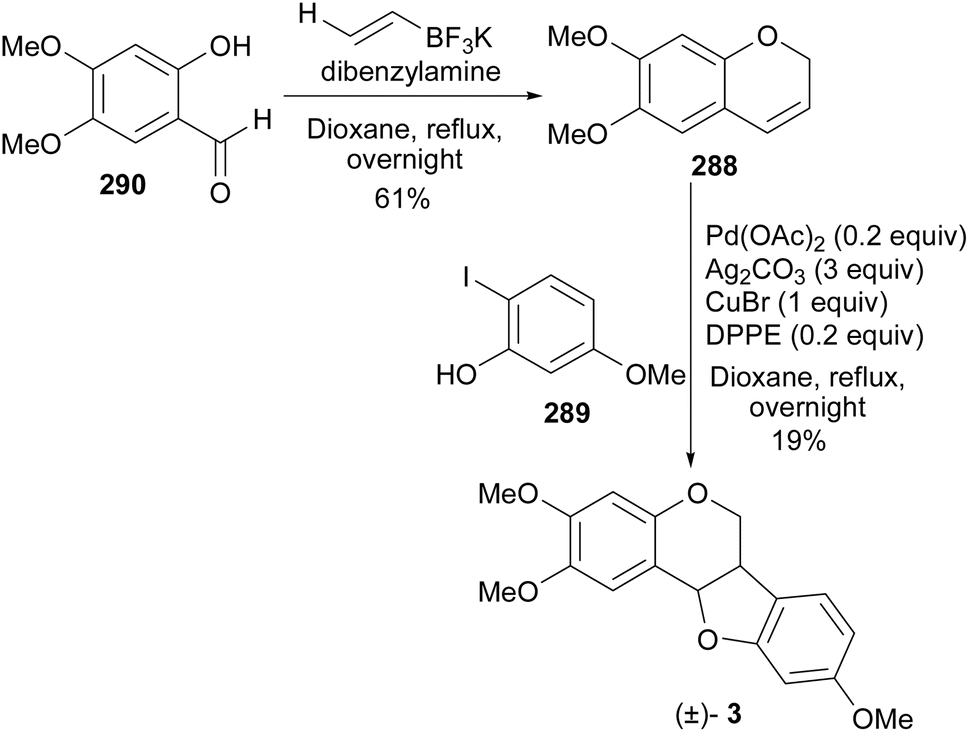
A pterocarpan, (+)-(6aS,11aS)-2,3,9-trimethoxypterocarpan [(+)-3] was isolated from the indigenous Brazilian tree Platymiscium floribundum Vogel. (Fabaceae) and the compound showed high cytotoxic activity against leukemia (HL-60 and CEM), breast (MCF-7), colon (HCT-8), and skin (B16) cancer cell-lines with IC50 values ranging from 0.1 to 2.9 μg mL−1.40 To provide additional material for further studies, Farias et al. completed the total synthesis of the racemic compound (±)-3 in 2020 and resolved it into its enantiomers, (+)- and (−)-2,3,9-trimethoxypterocarpans [(+)-3 and (−)-3].41 (−)-(6aR,11aR)-2,3,9-Trimethoxypterocarpan [(−)-3] is also a natural compound that was isolated from Fusarium solani infected Pisum sativum Linn. (Fabaceae).188 They started their synthesis by alkylation of the 3,4-dimethoxyphenol with propargyl bromide to obtain the aryl propargyl ether 285 in a 90% yield. Initially, the gold(I)-catalysed intramolecular cyclisation reaction of the aryl propargyl ether 285 afforded a mixture of benzofuran 286 (6%), chromene 288 (9%), and a [2 + 2] dimer 287 (3%), Scheme 41. The chromene 288 could be synthesised in moderate yields (40%) by changing the reaction conditions.86 Finally, the (±)-2,3,9-trimethoxypterocarpan [(±)-3] was synthesized in a 43% yield by Mizoroki–Heck oxyarylation of chromene 288 and 2-iodophenol 289. The racemic mixture was resolved into separate enantiomers using semi-preparative HPLC that was equipped with a chiral-phase column. The synthesised (±)-2,3,9-trimethoxypterocarpan [(±)-3] and enantiomers (+)-3 and (−)-3 were evaluated for antiproliferative effects against HL-60, HCT-116, OVCAR-8, and SF-295 tumor cell lines. The enantiomer (+)-3 was determined to be the most active, particularly against OVCAR-8, while the racemic mixture (±)-3 and the levorotatory enantiomer (−)-3 were less active.41
Scheme 41 Synthesis of (±)-2,3,9-trimethoxypterocarpan [(±)-3] and its resolution into enantiomers.
It is noteworthy that Kakuda et al. reported the synthesis of chromene 288 in a 61% yield from the salicylaldehyde 290 while utilising the Petasis boronic acid-Mannich reaction.189 However, by subjecting the chromene 288 to the Mizoroki–Heck oxyarylation conditions [Pd(OAc)2 (0.2 equiv.), Ag2CO3 (3.0 equiv.), CuBr (1.0 equiv.), and DPPE (0.2 equiv.)] the racemic 2,3,9-trimethoxypterocarpan [(±)-3] was obtained in 19% yield (Scheme 42).189
Scheme 42 Synthesis of the racemic 2,3,9-trimethoxypterocarpan [(±)-3].
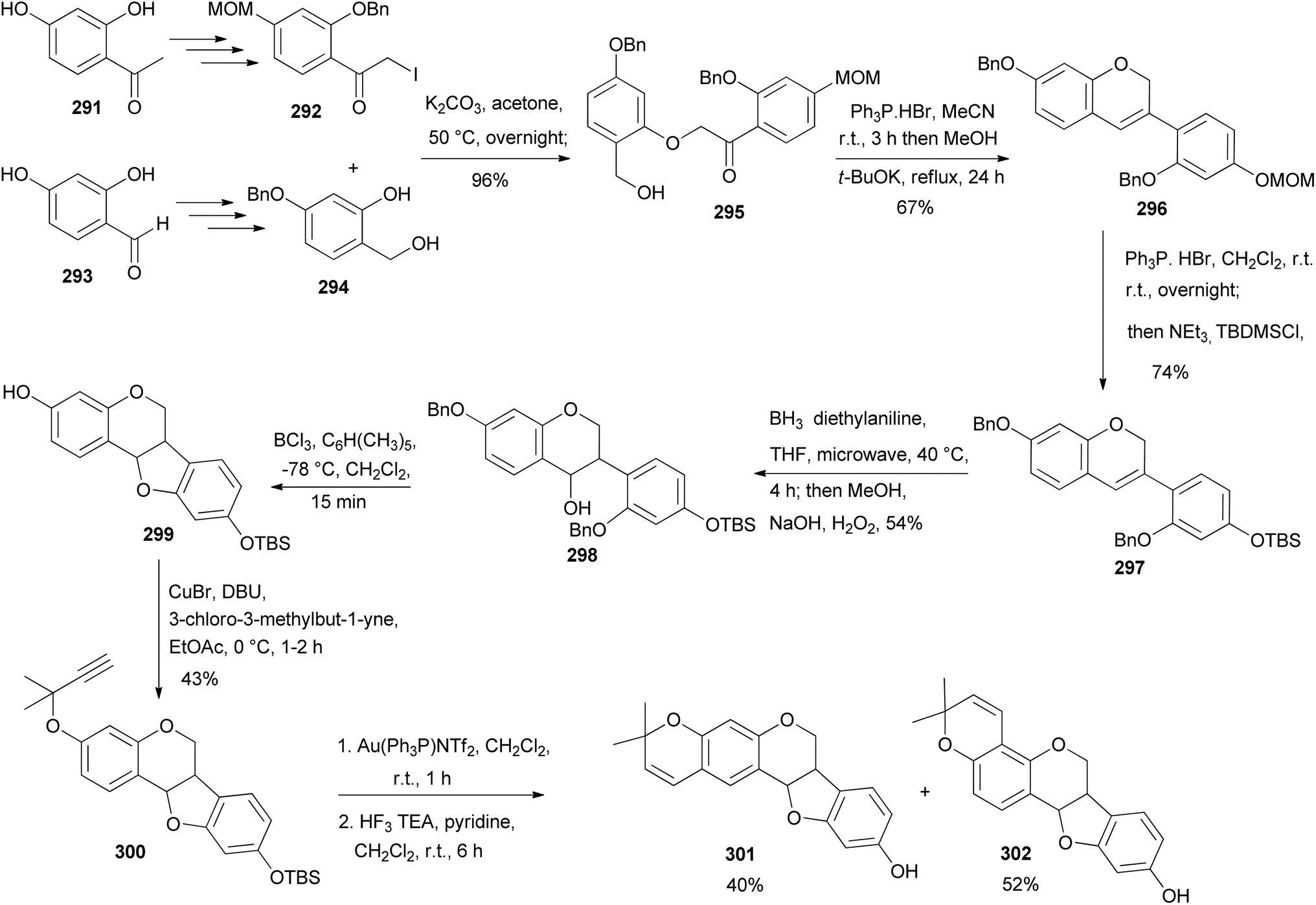
Neorautenol (301) and shinpterocarpin (302) are pyranopterocarpans which were first isolated from Neorautanenia edulis and Glycyrrhiza glabra L., respectively.190,191 The first synthesis of racemic pterocarpans neorautenol (301) and shinpterocarpin (302) together with their derivatives was conducted by Huang et al.187 This was followed by evaluation of their antitumour properties towards a panel of cancer cells. They were synthesised by a modified version of the method described by Erhardt's group.192,193 The synthesis of 301 and 302 proceeded via an intramolecular Wittig reaction of 295 that gave the highly sought-after isoflavene intermediate 296, Scheme 43. The precursor 295 was synthesised starting from 2,4-dihydroxyacetophenone (291) that was chemo-selectively protected with MOM and benzyl groups, followed by iodination using Selectfluor and molecular iodine to afford the α-iodoketone 292. In a parallel synthesis to the α-iodoketone 292, 2,4-dihydroxybenzaldehyde (293) was chemoselectively protected and reduced with NaBH4 to afford benzyl alcohol 294 that served as the coupling partner to α-iodoketone 292. The union of 292 and 294 under basic conditions gave the α-phenoxyketone 295 in excellent yields. The cyclisation of 295 afforded isoflavene 296 which was subsequently silyl protected in situ via MOM deprotection with mild acid to produce 297. Compound 297 was subjected to the hydroboration–oxidation of the chromene double bond, which favoured the anti-Markovnikov product 298 due to the steric presence of the phenyl ring at C-3 of isoflavene 297. Treatment of 298 with boron trichloride furnished pterocarpan 299 that was alkylated with 3-chloro-3-methylbut-1-yne to produce 300. The cyclisation of 300 under gold(I) catalysis gave two 2,2-dimethyl-2H-chromene regioisomers in a ratio of 4:5, which upon desilyllation afforded racemic neorautenol (301) and shinpterocarpin (302), Scheme 43.187
Scheme 43 Total syntheses of neorautenol (301) and shinpterocarpin (302).
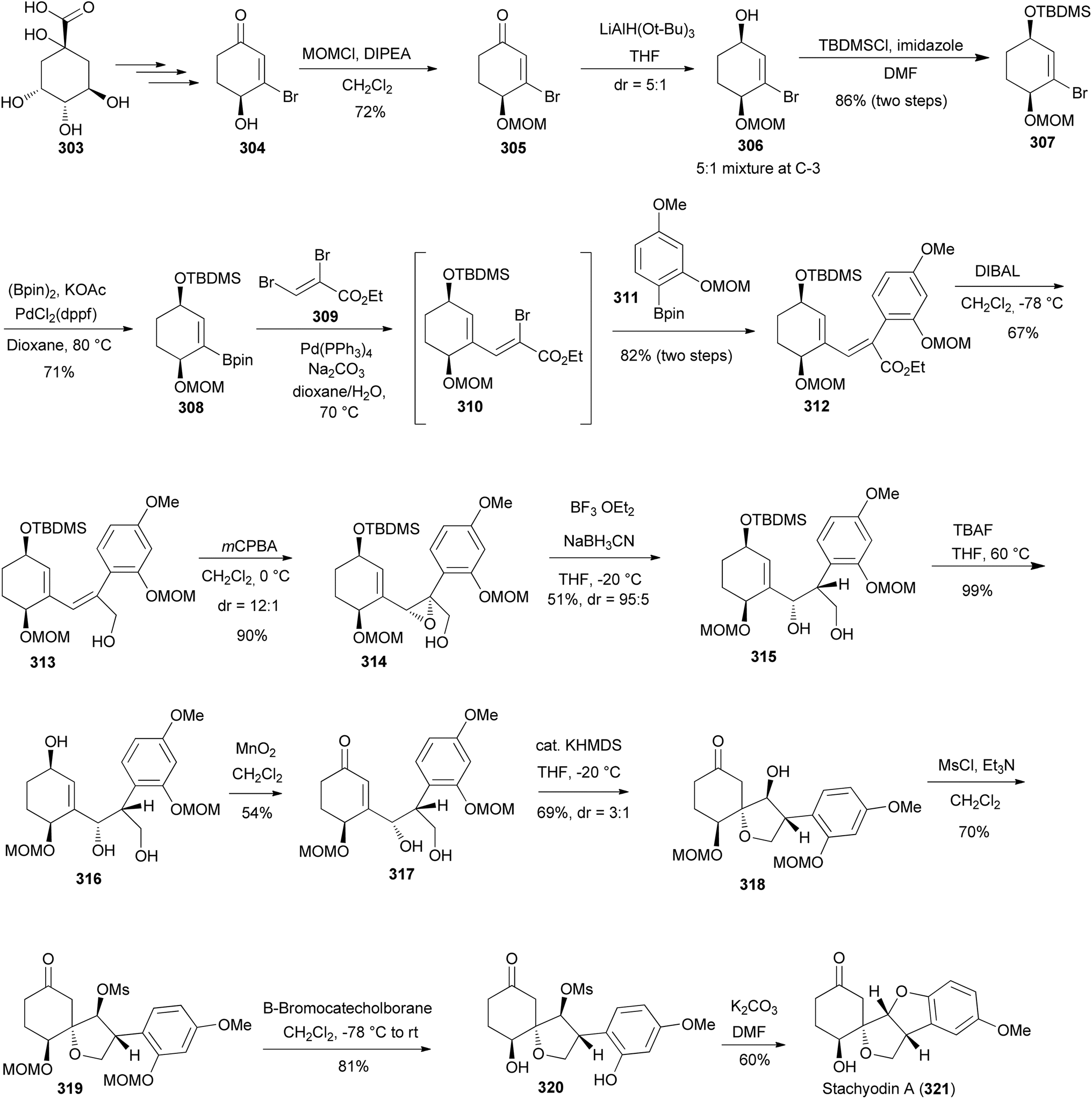
Stachyodin A (321), a pterocarpan derivative with an unusual spirotetrahydrofuran ring was isolated from the roots of Indigofera stachyodes Lindl. (Papilionoideae).194 Kawamoto et al. reported the first total synthesis of racemic 321 in 2021 and in the subsequent year developed its stereoselective total synthesis.67,195 The stereoselective total synthesis was accomplished in 14 steps from a known precursor 304 that was readily prepared from the chiral pool building block, (−)-quinic acid (303) (Scheme 44).67 The synthesis was initiated by instituting the MOM protecting group to a carbinol group of 304 to give the MOM ether 305. The reduction of the MOM ether 305 with LiAlH(O-t-Bu)3 in THF afforded 306 as a 5:1 diastereomeric ratio at C-3. The subsequent treatment of 306 with TBDMSCl in the presence of a mild base (imidazole) also afforded 307 as a mixture of diastereoisomers with a ratio of 5:1 at C-3. Compound 307 was transformed into the pinacolato borane intermediate 308 through the Suzuki coupling reaction. It is noteworthy that 308 and its coupling partner 309 serve as crucial intermediates towards achieving the stereoselective total synthesis of stachyodin B. The aryl borane 311 was prepared in three steps starting from 1-hydroxy-3-methoxybenzene.195 Therefore, the one-pot three-component Suzuki coupling reaction of pinacolato borane 308, dibromide 309, and aryl borane 311 was undertaken to render 312 through the intermediate 310. The reaction was conducted at elevated temperatures in the presence of Na2CO3 while utilising Pd(PPh3)4 as a catalyst to obtain 312 in 82% yield. Upon successfully achieving the synthesis of 312 the focus shifted to the synthesis of an optically active enone 317. The treatment of 312 with DIBAL gave allyl alcohol 313, which upon stereoselective epoxidation with mCPBA afforded 314. The subsequent reductive opening at the benzylic position afforded the diol 315 in 51% yields. Desilylation of 315 with TBAF gave the secondary alcohol 316, which was selectively oxidized with MnO2 to furnish the requisite enone 317 in a 54% yield. Treatment of 317 with KHDMS afforded compound 318 through a 1,4-intramolecular dehydration reaction. Mesylation of 318 with MsCl in the presence of Et3N produced 319. MOM deprotection with catechol borane gave 320. The intramolecular cyclisation of 320 following the SN2 inversion strategy in the presence of K2CO3 afforded stachyodin A (321) in a 60% yield after purification (Scheme 39). The specific rotation of 321 matched that of the reported natural compound.67
Scheme 44 Stereoselective total synthesis of stachyodin A (321).
5. Synthesis of rotenoids
5.1. Synthesis of dehydrorotenoids
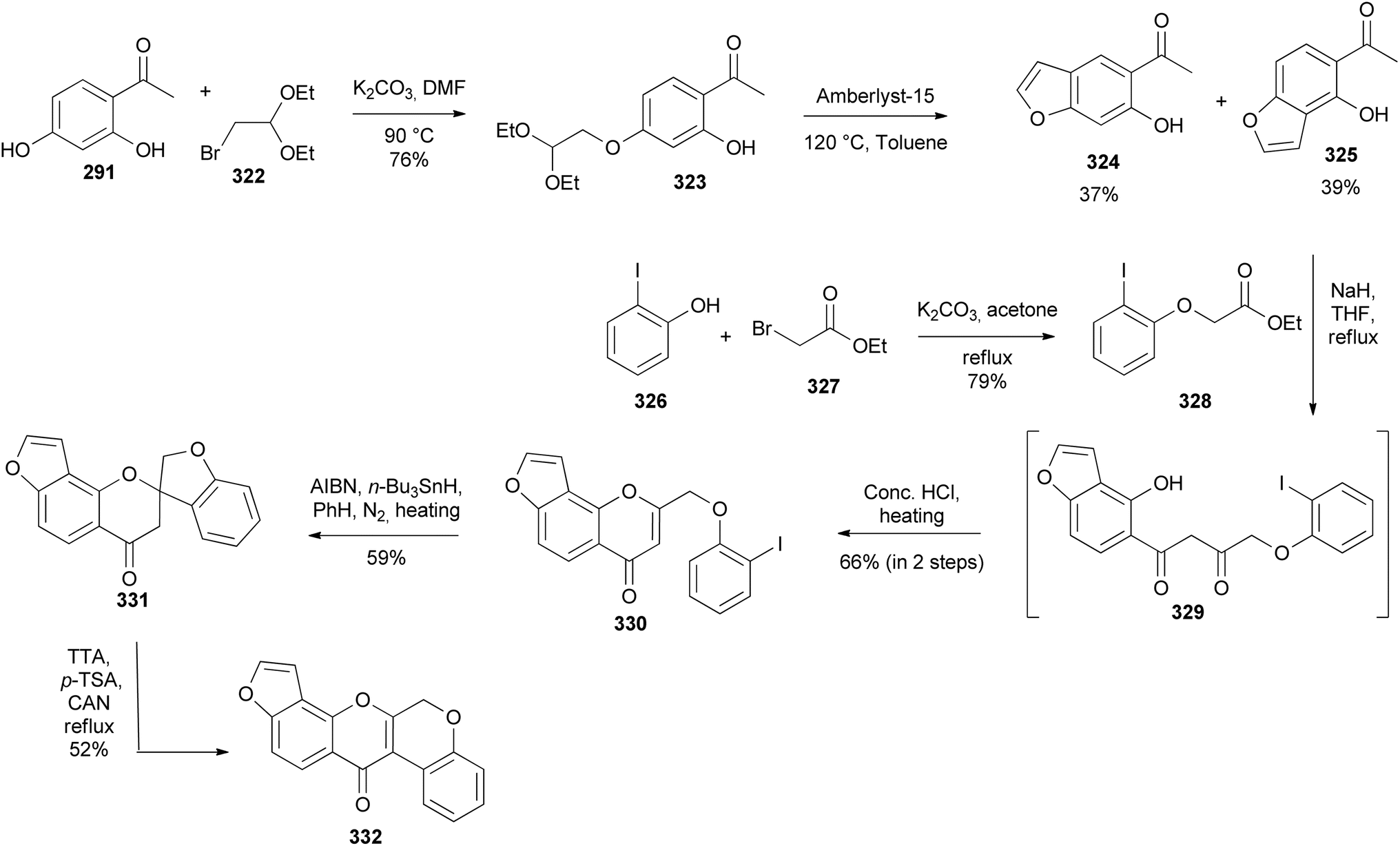
Kurapati and colleagues synthesised pongarotene (332), an antimicrobial dehydrorotenoid isolated from Pongamia pinnata (L.) Pierre.46,196 The synthesis featured oxidative aryl rearrangement of a spirocyclic flavanone. It commenced by selective alkylation of dihydroxyacetophenone 291 with bromoacetal 322 followed by cyclisation of 323 to give the targeted angular benzofuranyl ketone 325 in 39% yield together with a linear benzofuran 324 in 37% yield (Scheme 45). Condensation of acetophenone 325 with phenoxyacetate 328 obtained from the reaction of iodophenol 326 with bromoacetate 327 gave 329, which was converted into a chromone 330 by treatment with concentrated HCl. The radical-initiated cyclisation of 330 gave a spirocyclic flavanone 331, which underwent 1,2-aryl migration upon treatment with in situ-generated thallium(III) p-tosylate to give pongarotene (332) (Scheme 45).46
Scheme 45 Synthesis of pongarotene (332).
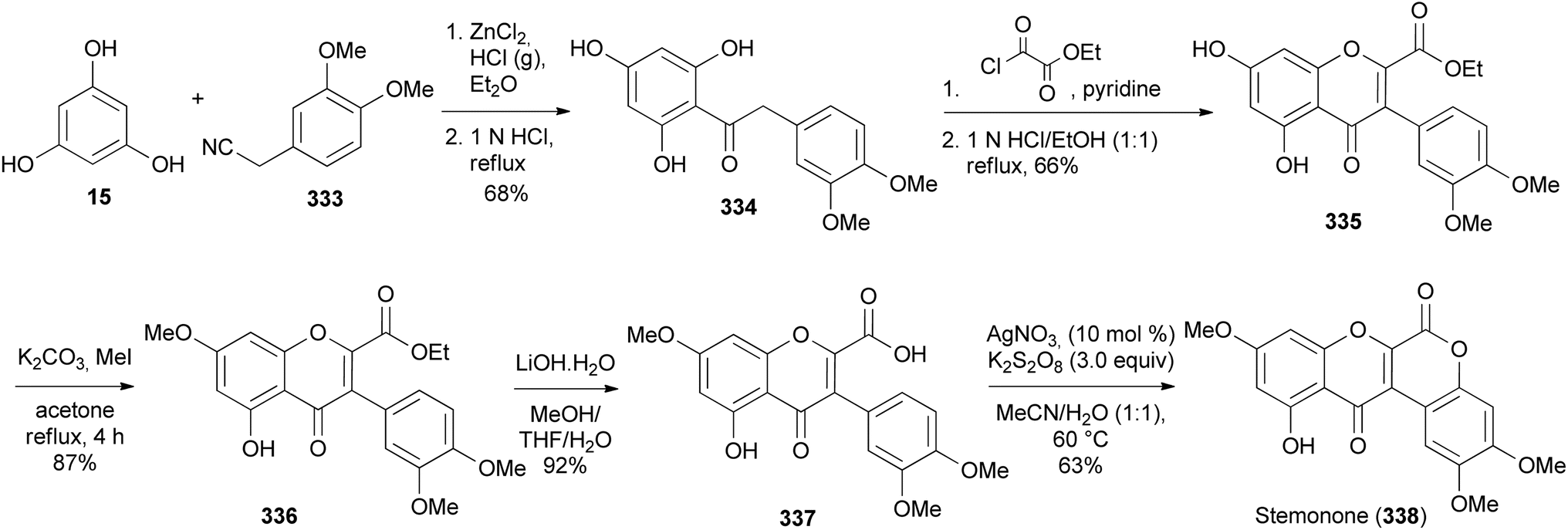
Boonsombat and colleagues developed a method for the synthesis of oxodehydrorotenoid by silver-catalysed intramolecular lactonisation of isoflavone-2-carboxylic acids.197 The method was applied to the first total syntheses of stemonone (338), 6-oxodehydroelliptone (342), 6-oxo-6a,12a-dehydrodeguelin (349), and rotenonone (346). The synthesis of stemonone (338) commenced by preparation of isoflavone-2-ethylcarboxylate 335 by Houben–Hoesch reaction of phloroglucinol (15) and phenylacetonitrile 333 to give 334 that was subsequently converted to 335 following Baker's reaction.50,198 Methylation of 335 and hydrolysis of the resulting isoflavone 336 rendered 337. Direct lactonisation of 337 using AgNO3 and K2S2O8 optimised conditions gave stemonone (338) (Scheme 46).197
Scheme 46 The total synthesis of stemonone (338).
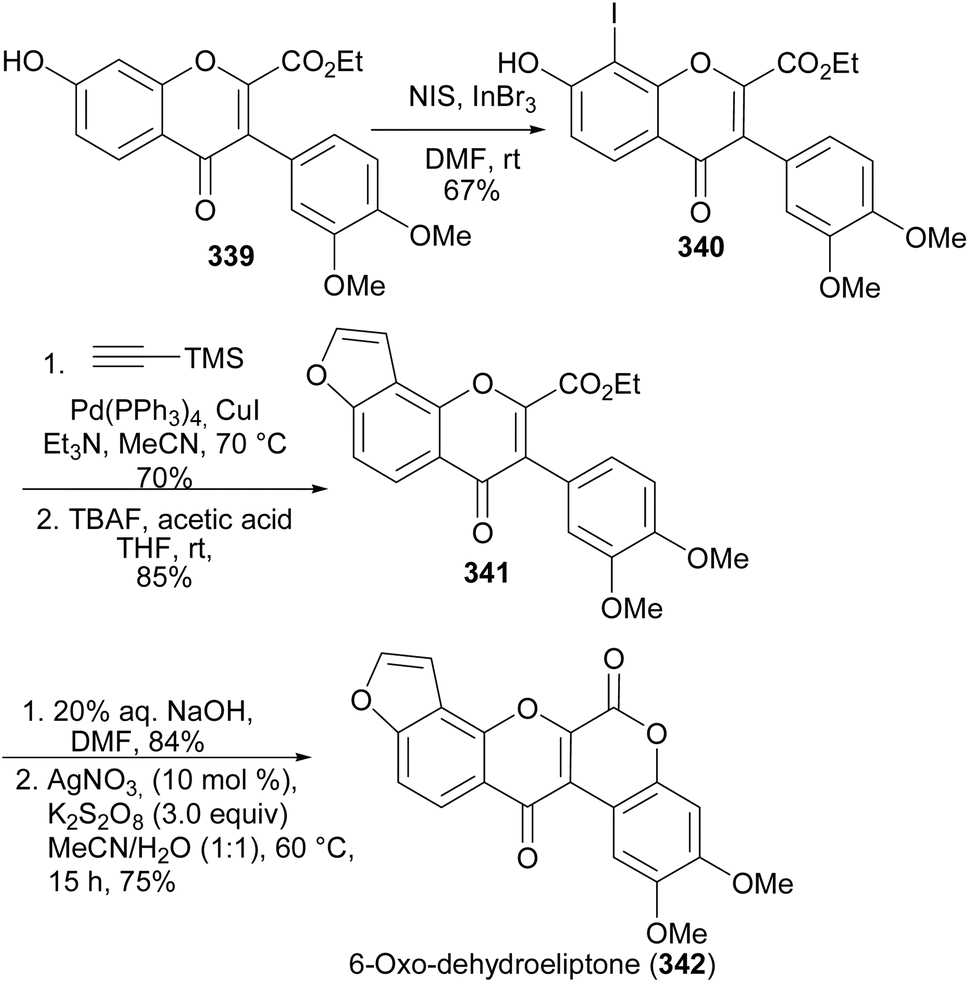
6-Oxodehydroelliptone (342) was synthesised from isoflavone-2-ethylcarboxylate 339 by a sequence of steps that involved iodination to give 8-iodoisoflavone 340, followed by Sonogashira coupling annulation with trimethylsilylacetylene and TMS deprotection to give the furanoisoflavone 341 (Scheme 47). Cleavage of the ester and silver-catalysed intramolecular esterification of the aryl Csp2–H carbon and the carboxylic group gave 6-oxodehydroelliptone (342).
Scheme 47 Total synthesis of 6-oxodehydroelliptone (342).
The two final oxodehydrorotenoids, 6-oxo-6a,12a-dehydrodeguelin (349) and rotenonone (346) were synthesised from 339 (Scheme 48) via hydrolysis of dimethylpyranoisoflavone 347 and furanoisoflavone 344 and subsequent lactonisation of 348 and 345, respectively. The requisite dimethylpyran scaffold in 347 was constructed by O-propagylation of 339 followed by microwave-assisted cyclisation at 180 °C to give 347. Alternatively, 347 could be prepared in one step by treatment of 339 with dimethylpropargyl chloride under reflux (Scheme 48). The furan ring in 344 was prepared by Heck coupling reaction of the prenylated isoflavone 343, which was in turn synthesised by O-propagylation, reduction, and Claisen rearrangement, Scheme 48.197
Scheme 48 Total syntheses of rotenonone (346) and 6-oxo-6a,12a-dehydrodeguelin (349).
5.2. Synthesis of rotenoids
Rotenoids including deguelin, tephrosin, and rotenone have attracted the interest of researchers due to their biological activities that include pesticidal, insecticidal, and anticancer activities.8,69,71 Several synthesis routes have been reported for these compounds.47,66,69,71,199–203
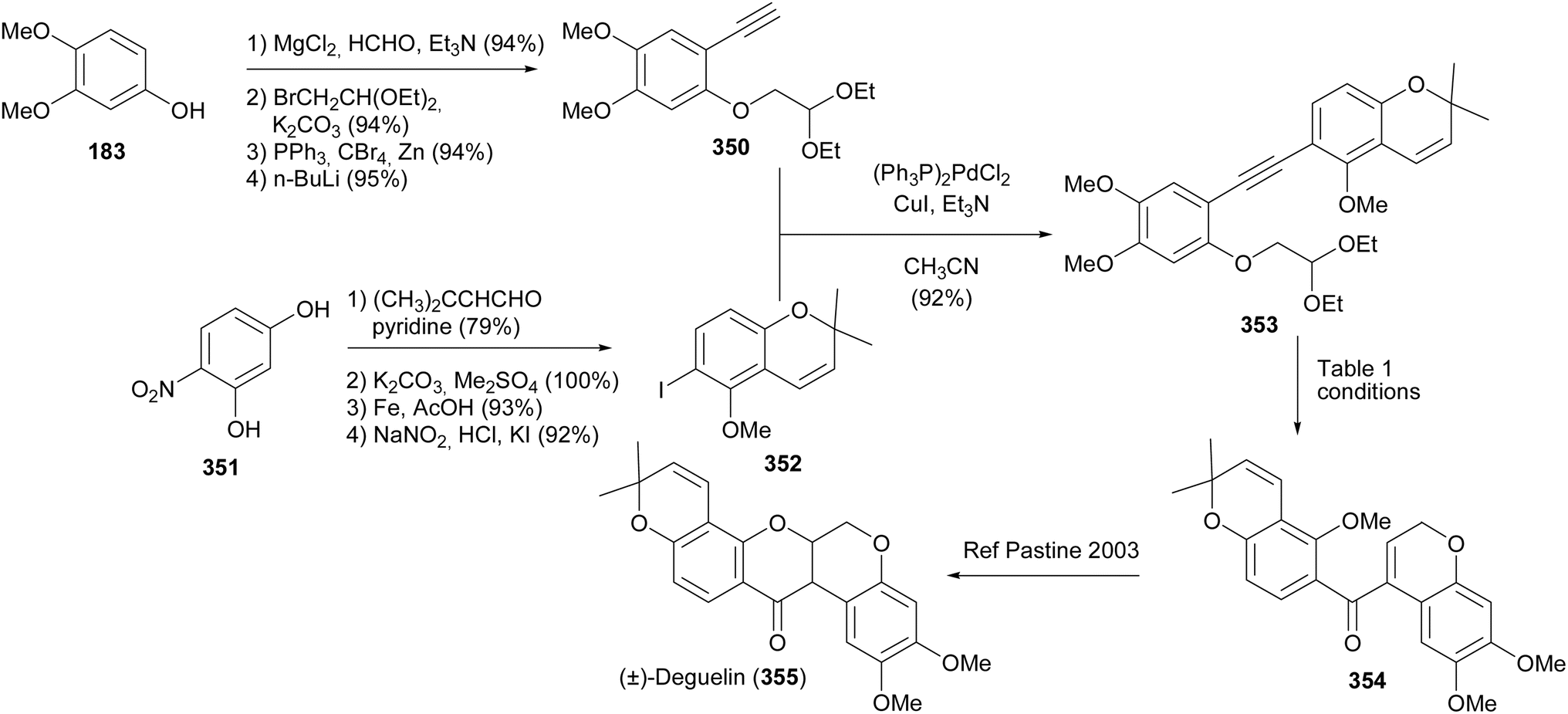
Nayak and Kim synthesised (±)-deguelin (355) and (±)-munduserone (360) by utilising benzoylchromenes as key intermediates (Schemes 49 and 50).47 The successful approach to the synthesis of (±)-deguelin (355) involved Sonogashira coupling of alkyne 350 with iodochromene 352 to give a diarylalkyne 353. Different conditions were evaluated for the intramolecular alkyne carbonyl metathesis of 353 leading to acyldichromene 354. Good yields of 354 were obtained when In(OTf)3, Sc(OTf)3, or Yb(OTf)3 were used as catalysts (Table 1). Compound 354 could be converted into (±)-deguelin (355) following the protocol of Pastine and Sames.204
aA mixture of 354 (0.1 mmol) and catalyst in solvent (1 mL) was heated at the temperature indicated above.bIsolated yield (%).cHCO2H was used as solvent.dA complex mixture.
1
HCO2H
100
12
nr
2
InCl3 (1.0)
THF/H2O (4:1)
80
12
nr
3
TFA·HCl
DCM
80
16
4
FeCl3 (1.0)
THF/H2O (4:1)
80
16
5
In(OTf)3 (1.0)
THF/H2O (4:1)
80
12
94
6
In(OTf)3 (0.4)
THF/H2O (4:1)
80
36
93.5
7
In(OTf)3 (0.2)
THF/H2O (4:1)
80
48
84.6
8
Cu(OTf)3 (0.4)
THF/H2O (4:1)
80
36
34
9
Sc(OTf)3 (0.4)
THF/H2O (4:1)
80
36
83
10
Yb(OTf)3 (0.4)
THF/H2O (4:1)
80
36
82
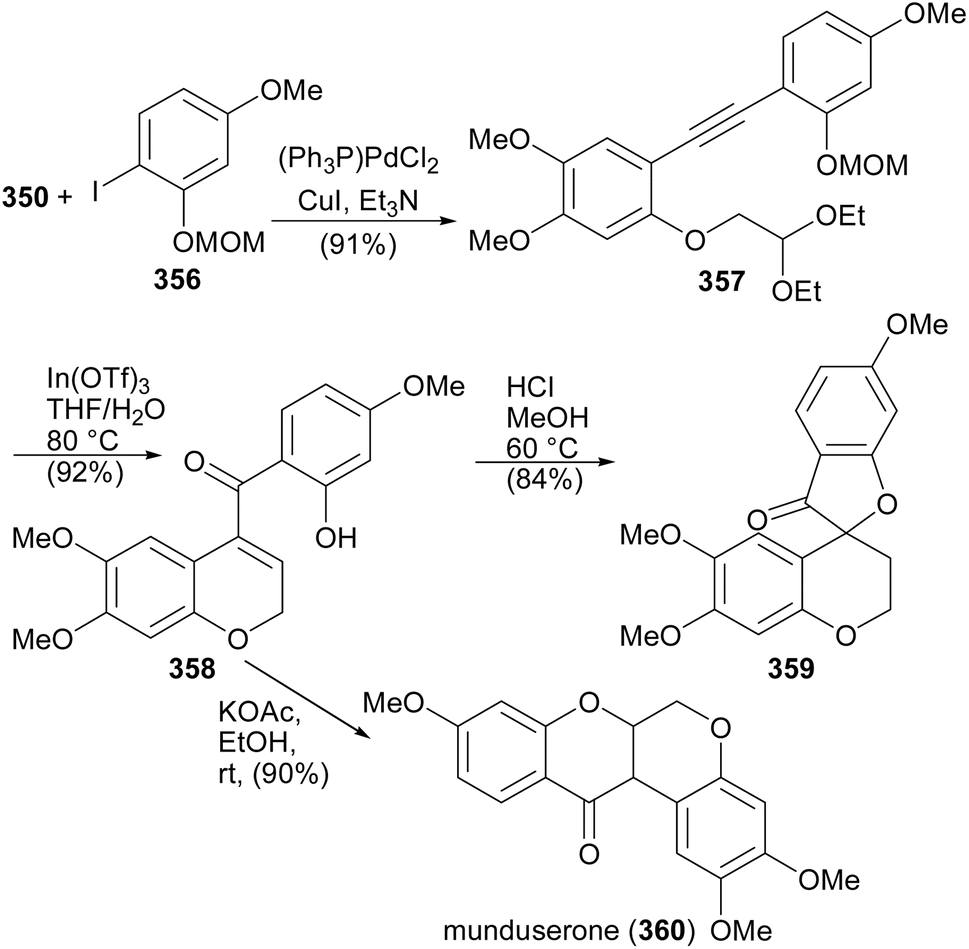
Munduserone (360) was synthesised by the Sonogashira coupling of phenylethyne 350 with iodobenzene 356 to give diphenylacetylene 357. The intramolecular alkyne carbonyl metathesis of 357 using In(OTf)3 rendered 358, which was converted into munduserone (360) by conjugate addition under basic conditions. An attempt to cyclise 358 under acidic conditions led to the formation of spirocycle 359.
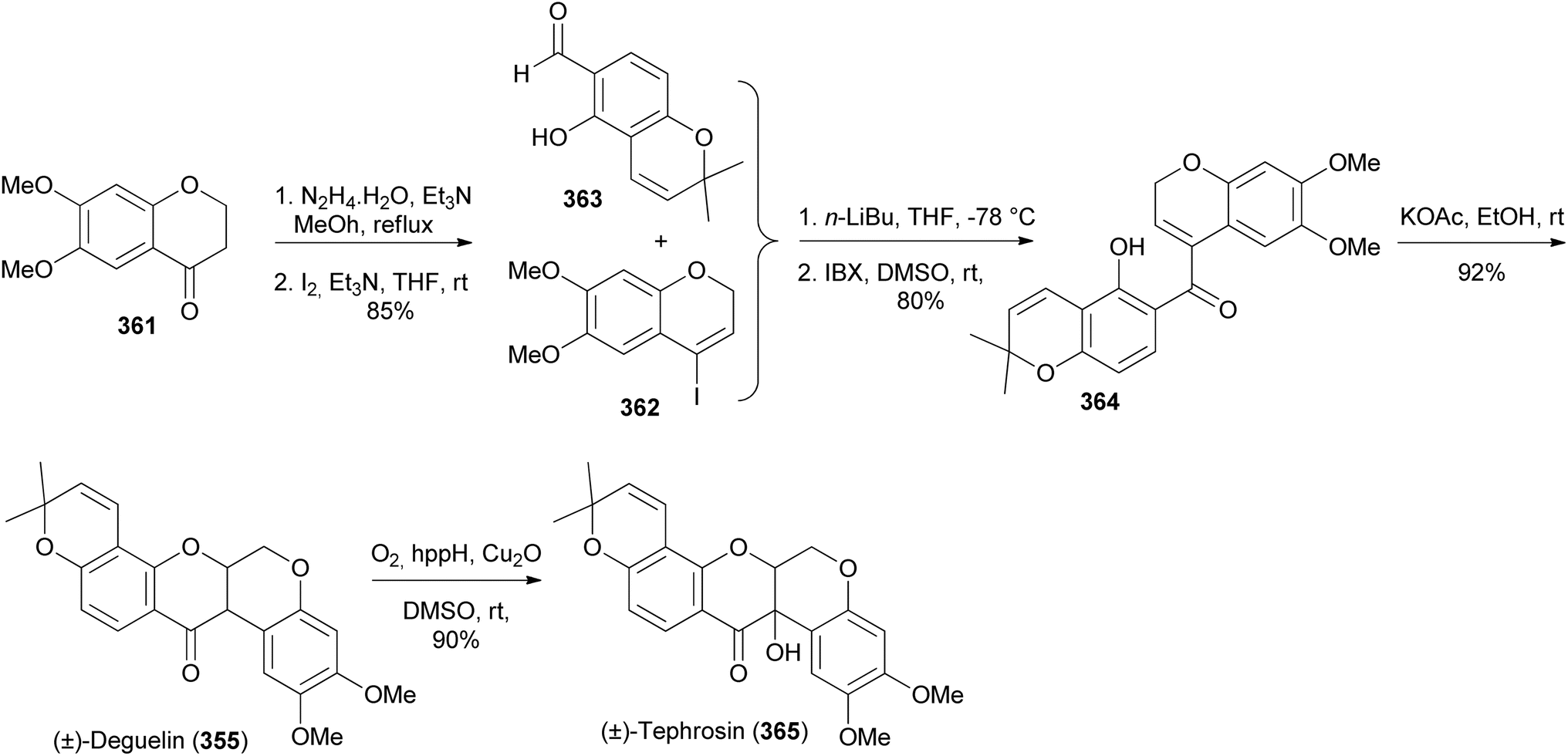
Xu and colleagues synthesised deguelin (355) and tephrosin (365) in racemic form through a protecting-group-free strategy starting from commercially available chromanone 361 and chromenecarbaldehyde 363.200 Conversion of 361 into iodochromene 362 followed by coupling of 362 with chromenecarbaldehyde 363 and oxidation gave a bischromene 364. Base-catalysed oxa-Michael addition of 364 gave deguelin (355), which was converted into tephrosin (365) by Cu-catalysed hydroxylation with molecular oxygen (Scheme 51).200,205
Scheme 51 Total syntheses of (±)-deguelin (355) and (±)-tephrosin (365).
5.3. Stereoselective synthesis
Several methods have been employed for the stereoselective synthesis of rotenoids. These include organocatalysed enantioselective synthesis,71,72 metal-catalysed reactions using chiral ligands,69 induction of configuration by Sharpless asymmetric hydroxylation,202,206 the use of chiral starting materials,65,66,203 and semisynthetic strategies starting from readily available enantiopure rotenoids.201,207
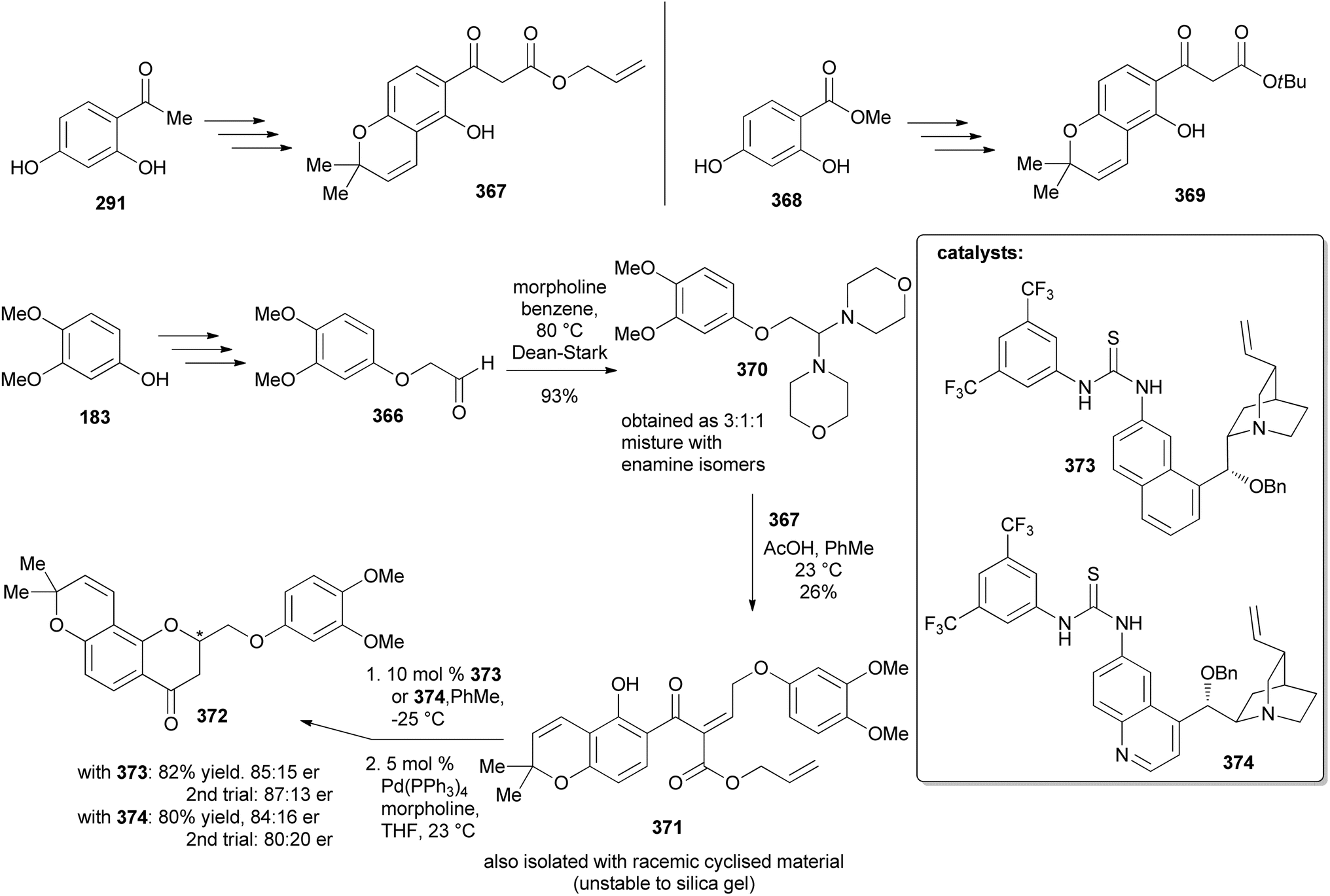
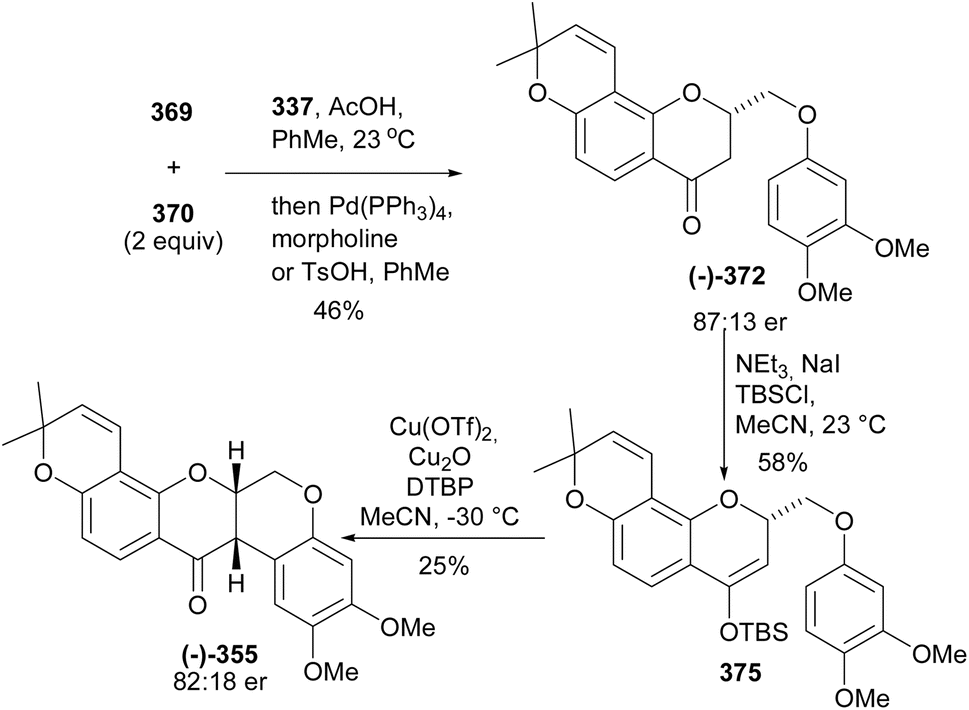
5.3.1 Organocatalysed stereoselective synthesis. Farmer and colleagues synthesised two enantiomers of deguelin by a thiourea-catalysed enantioselective cyclisation strategy.71 The synthesis was initiated by preparation of ketoesters 367 and 369 starting from 2,4-dihydroxyacetophenone (291) and 2,4-dihydroxymethyl benzoate (368), respectively (Scheme 52). The coupling partner, 2-phenoxyacetaldehyde 366 was synthesised from the reaction of 3,4-dimethoxyphenol (183) with 2-bromodimethoxyethane followed by deprotection of the resulting acetal under mild conditions using Amberlyst-15 resin. Attempts to couple the ketoester 367 with the acetaldehyde 366 led to poor yields of 371. Therefore, phenoxyethylmorpholine 370 was coupled to the oxopropanoate 367 leading to 371 in 26% yields. The low yields were attributed to cyclisation, leading to racemic chromanone. Stereoselective oxa-Michael addition facilitated by chiral thiourea catalysts 373 and 374 and subsequent decarboxylation gave the enantioenriched chromanones 372 in 85:15 and 84:16 er from 373 and 374, respectively (Scheme 52). An alternative strategy was designed for the direct conversion of 367 or 369 into 372 and better yields and enantiomeric excess were obtained from 369 after optimisation of the conditions. Therefore, the complete synthesis of the deguelin enantiomers was carried out using chromanone 372 obtained from 369 in one pot. As illustrated for the synthesis of (−)-deguelin [(−)-355] in Scheme 53, the chromanone enantiomer (−)-372 obtained using 373 as a catalyst was converted into a TBDMS-protected enol ether 375, which upon oxyarylation using Snider's procedure208 gave (−)-deguelin [(−)-355] in 25% yield (Scheme 53). The deguelin enantiomers were evaluated for antiproliferative activity against PC-3 (prostate), MCF-7 (breast), HepG2 (liver) and Jurkat (leukemia) cells. Both compounds showed potent inhibitory activities and (+)-deguelin inhibited the growth of the MCF-7 and HepG2 cell lines more effectively than the natural enantiomer (−)-deguelin [(−)-355] (IC50 = 4.64 ± 1.33 μM and 0.90 ± 0.29 μM, for (+)-deguelin; and 10.59 ± 1.38 μM and 2.95 ± 1.19 μM, for (−)-deguelin, respectively).
Scheme 52 Synthesis of enantioenriched chromanone precursors 372.
Scheme 53 Complete synthesis of (−)-deguelin [(−)-355].
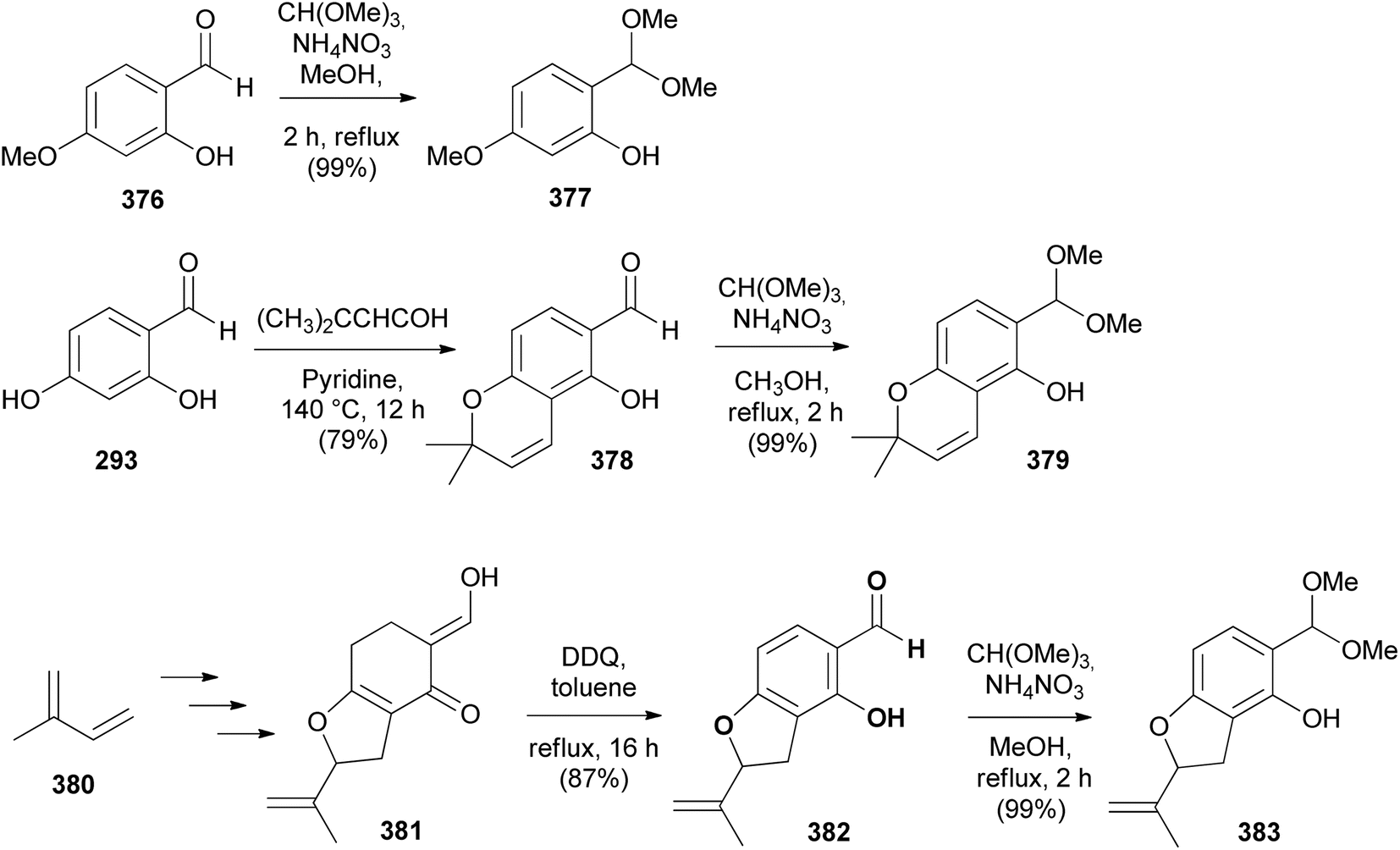
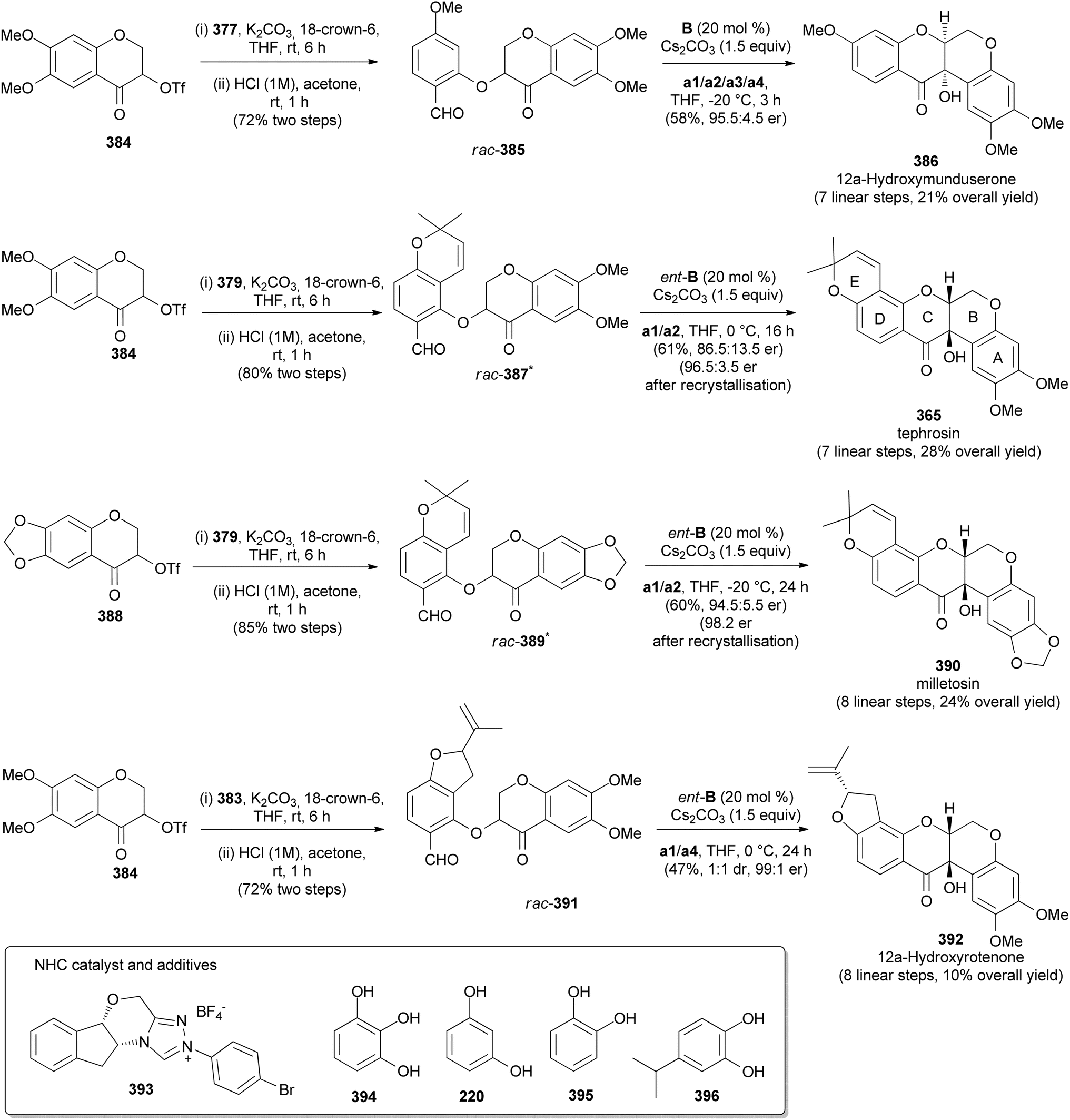
Fang's group reported the stereoselective total synthesis of several rotenoids, including 12a-hydroxymunduserone (386), tephrosin (365), milletosin (390), and 12a-hydroxyrotenone (392) by N-heterocyclic carbene (NHC) catalysis with dynamic kinetic resolution.72 Taking advantage of the similarity of the substitution patterns in the A and D rings, all the compounds were synthesised by a combination of dimethylacetals 377, 379, and 383 and chromanone-3-triflates 384 and 388 (Schemes 54 and 55). Compounds 377, 379, and 383 were prepared by acetyl protection of 376, 378 and 382, respectively. Compound 378 was in turn synthesised by condensation of 2,4-dihydroxybenzaldehyde (293) with prenal, while 382 was obtained by aromatisation of enol 381, prepared in a sequence of steps from isoprene 380 (Scheme 54). Coupling of chromanones and phenols 377 and 384; 379 and 384; 379 and 388; and 383 and 384 followed by deprotection gave racemic aryl ethers 385, 387, 389, and 391, respectively, which upon treatment with CsCO3 in the presence of chiral NHC catalyst 393 rendered 386, 365, 390, and 392, respectively with good enantiomeric excess (Scheme 55). Compound 392 was obtained together with its diastereoisomer. In addition, deguelin (355) and rotenone (406) could be synthesised from tephrosin (365) and 12a-hydroxyrotenone (392), respectively, following the established procedures.72
Scheme 54 Synthesis of acetal precursors 377, 379, and 383.
Scheme 55 NHC-catalysed stereoselective synthesis of rotenoids.
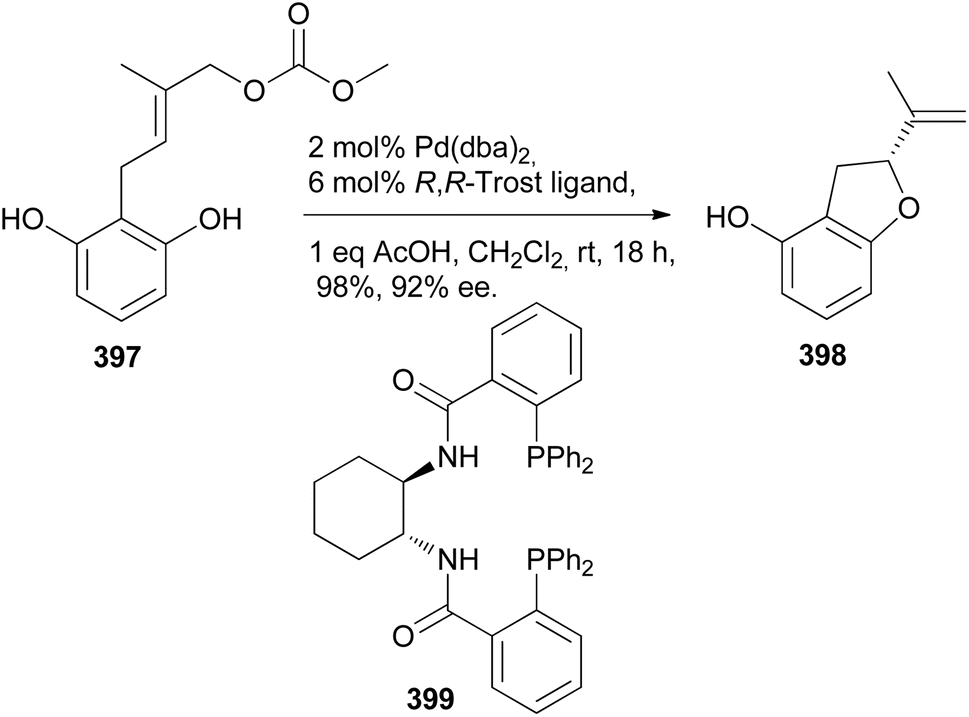
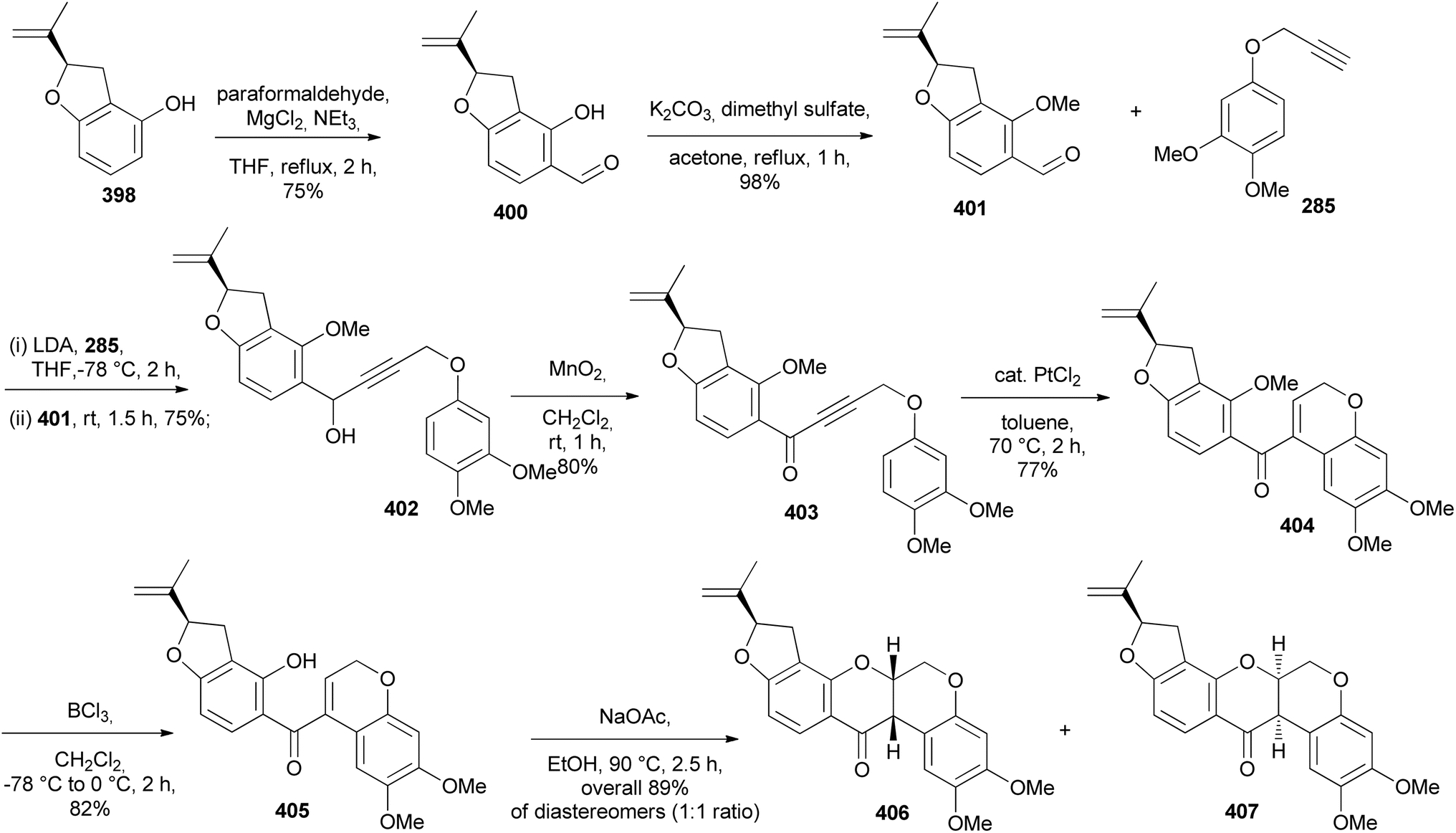
5.3.2 Chiral ligands. In 2017, De Koning's group reported the stereoselective total synthesis of rotenone (406) and the synthesis of munduserone in racemic form. The chiral dihydrobenzofuran 398, prepared by Pd-catalysed oxyarylation of 397 in the presence of the R,R′-Trost ligand 399 was used as a key building block for the stereoselective synthesis of rotenone (406), Scheme 56.69,209 The benzofuran 398 was converted into aldehyde 400 and methylated to give 401. A coupling of 401 with 285 gave alkyne 402, which was oxidised to give 403. Pt-catalysed hydroarylation and subsequent demethylation of 404 rendered 405. The final step involved intramolecular conjugate addition to give rotenone (406) and its quasi-enantiomer 407, which were separable (Scheme 57).
Scheme 56 Stereoselective synthesis of the dihydrobenzofuran 398 moiety.
Scheme 57 Synthesis of rotenone (406).
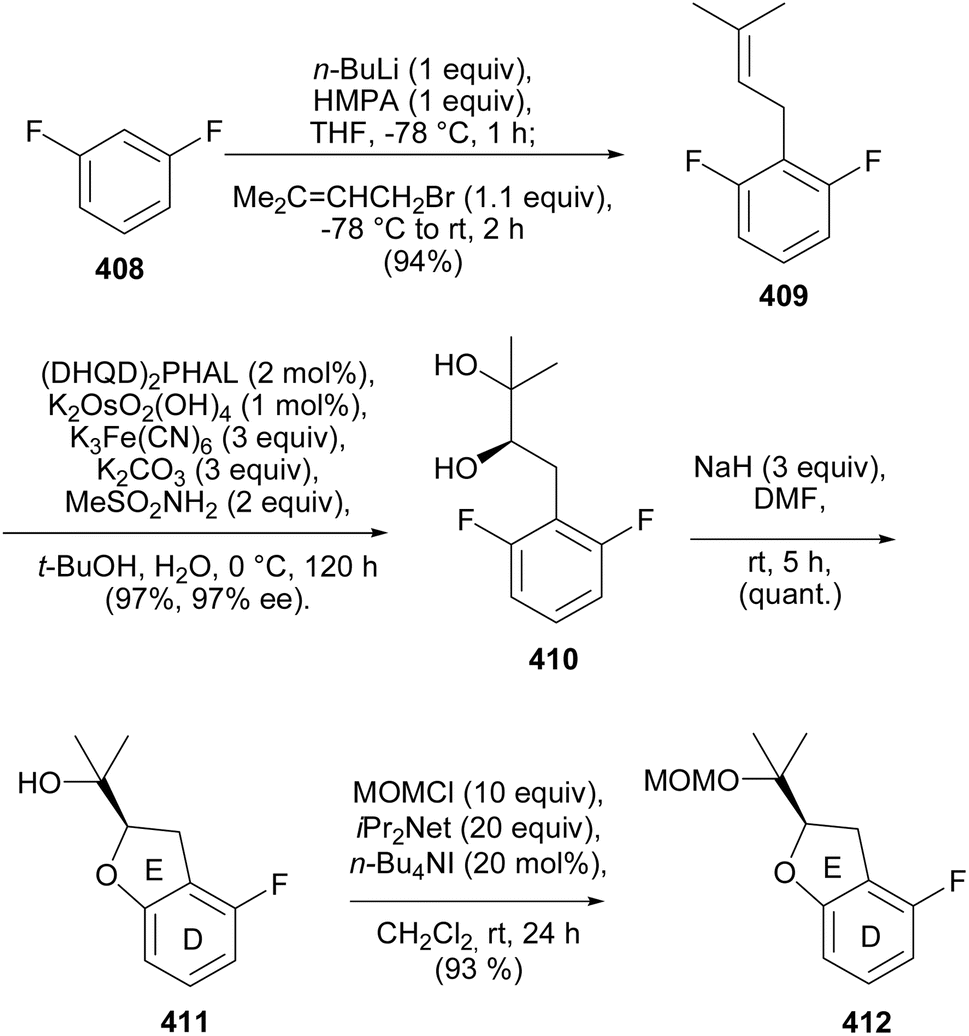
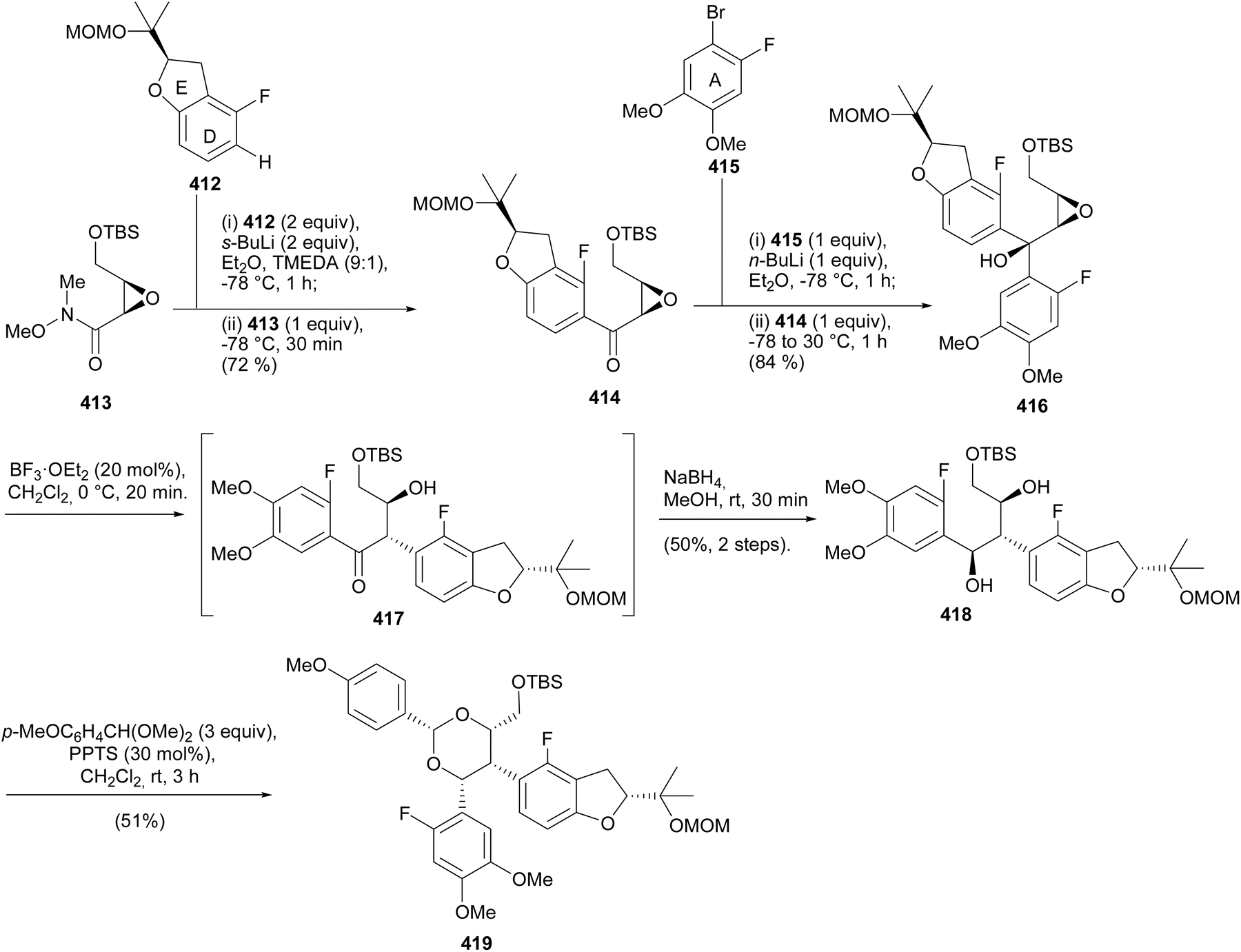
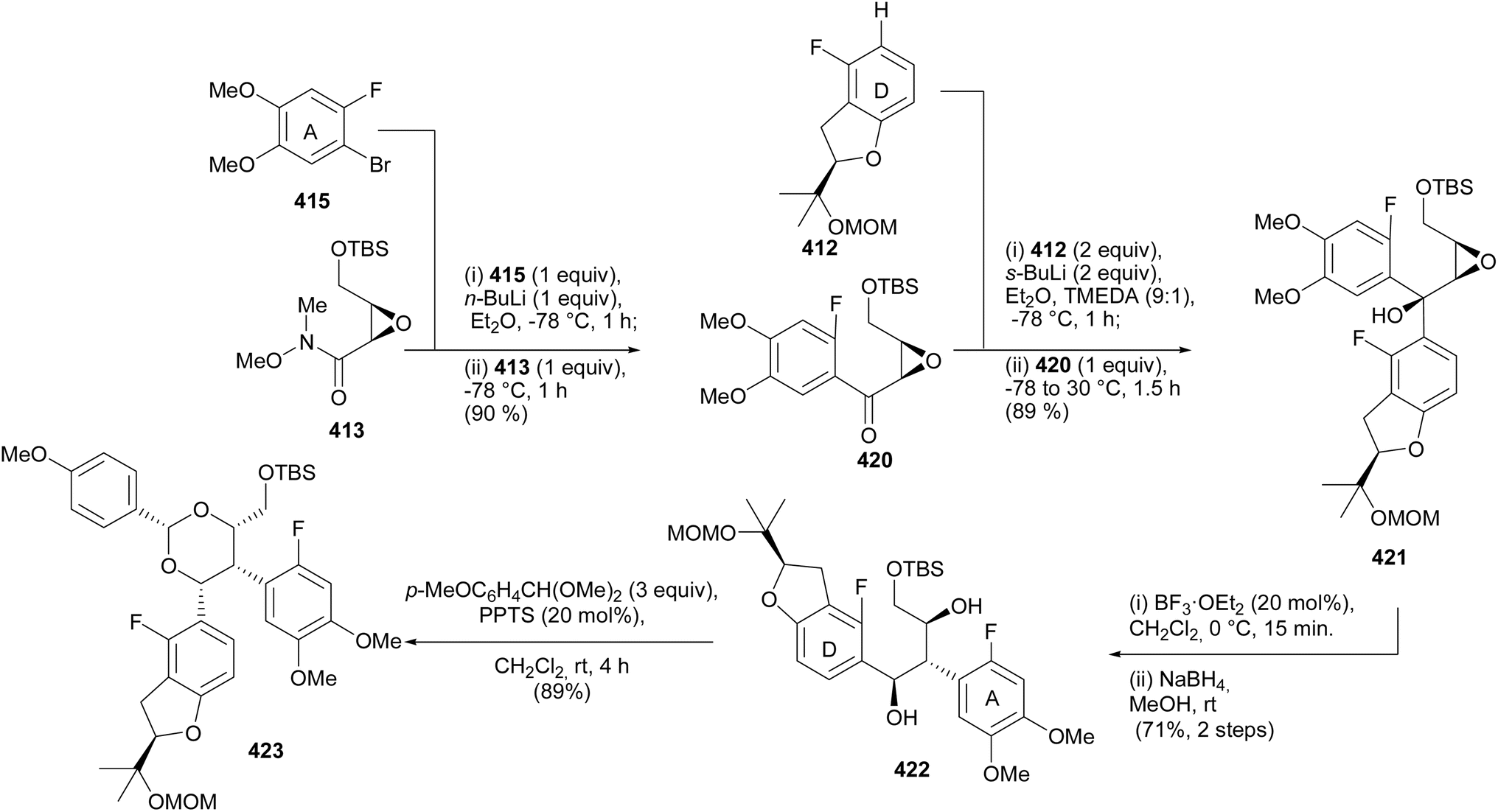
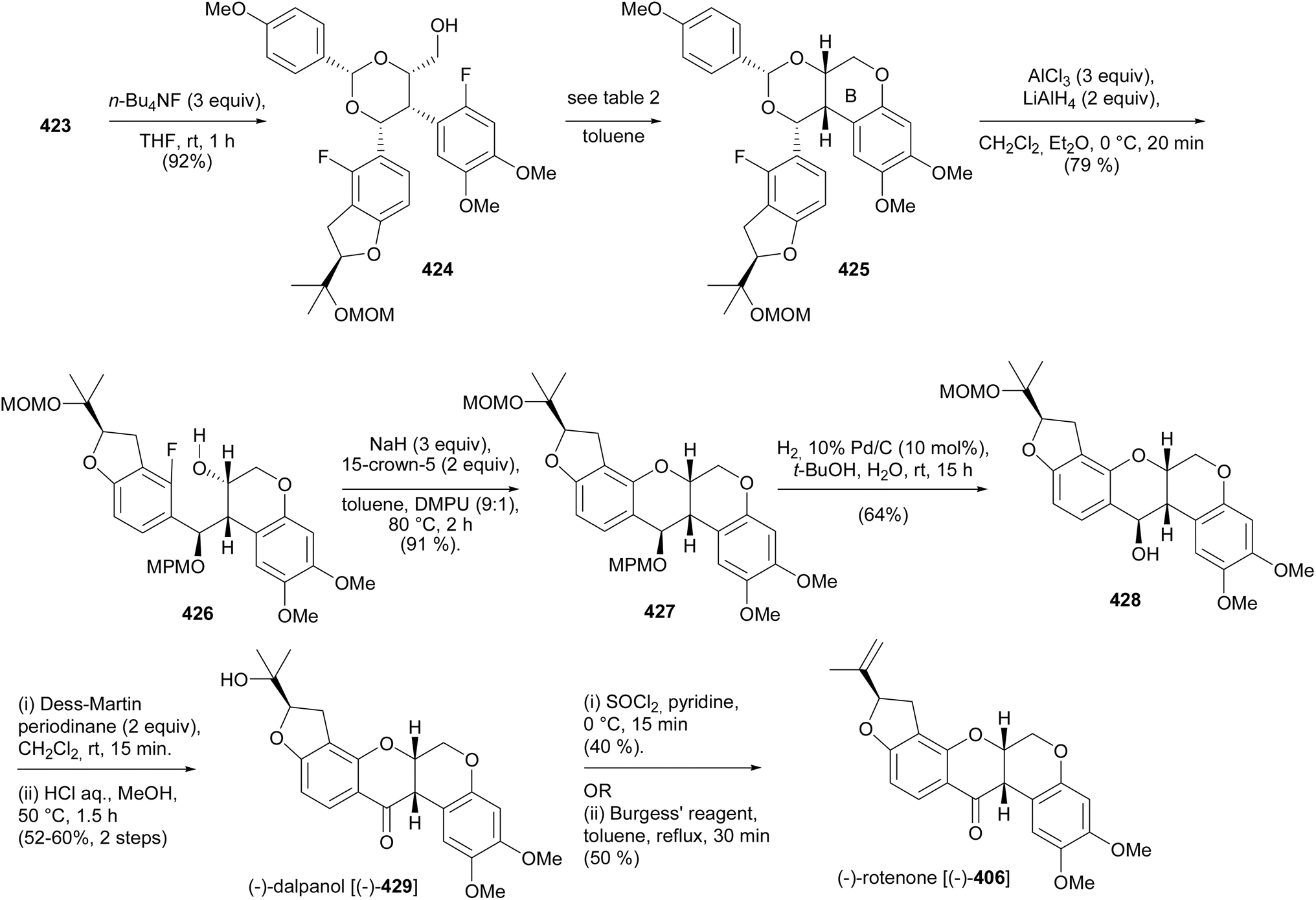
5.3.3 Chiral pool strategy. Ohmori and Suzuki's group reported the stereoselective total syntheses of (−)-rotenone [(−)-406] and (−)-dalpanol [(−)-429] that featured 1,2-aryl migration and triple SNAr O-cyclisation for the construction of the furanyl E-ring and two fused pyranyl C- and B-ring systems with the requisite stereogenicity.66 The synthesis was initiated by preparation of the chiral benzofuranyl unit 412, by prenylation of difluorobenzene 408 to afford 409, followed by asymmetric Sharpless dihydroxylation210 and nucleophilic aromatic substitution of the resulting (R)-diol 410 to give the benzofuran 411, which was protected with a MOM group (Scheme 58). The reaction of the lithiated species of 412 with chiral epoxy amide 413 gave 414. Bromine–lithium exchange of 415 and nucleophilic addition to 414 rendered a diaryl epoxy alcohol 416. A BF3·OEt2-catalysed 1,2-aryl shift followed by reduction and protection of the diol 418 gave 419. However, 418 was the untargeted intermediate that resulted from the migration of the benzofuranyl (DE) ring instead of the A-ring (Scheme 59). As a result, the sequence of assembly of substrates was altered as shown in Scheme 60. Coupling of epoxy amide 413 with the lithiated 415 gave 420, which was reacted with the lithium reagent of 412 to give epoxy alcohol 421. A 1,2-aryl shift followed by reduction gave diol 422 as a single isomer, confirmed by extensive NMR data analysis. The final steps involved diol protection, TBS deprotection, and oxycyclisation to give 423, 424, and 425, respectively. The oxycyclisation conditions are shown in Table 2. Acetal deprotection of 425 and subsequent O-cyclisation of the resulting 426 gave 427. Cleavage of the MPM group of 427 followed by oxidation and MOM deprotection gave (−)-dalpanol [(−)-429], which was dehydrated to give (−)-rotenone [(−)-406] (Scheme 61).66
Scheme 58 Synthesis of the DE-ring fragment 412.
Scheme 59 Synthesis of the epoxy alcohol 416 and the first attempted 1,2-shift-reduction sequence.
Scheme 60 Synthesis of the epoxy alcohol 421 from altered sequence of precursors and 1,2-aryl migration.
Table 2Conditions for the conversion of 424 into 425
Scheme 61 Stereoselective total synthesis of (−)-rotenone [(−)-406] and (−)-dalpanol [(−)-429].
6. Synthesis of coumaronochromones
Coumaronochromones constitute a rare subclass of isoflavonoids.3 They have been determined to exhibit biological activities that include anti-HIV,211 immunosuppressive,212 cytotoxicity against cancer cells213 and anti-neuroinflammatory.214 A few synthetic strategies have been developed for coumaronochromones62,215–218 and the most widely employed procedure involves oxidative cyclisation of 2′-hydroxyisoflavones.217–221
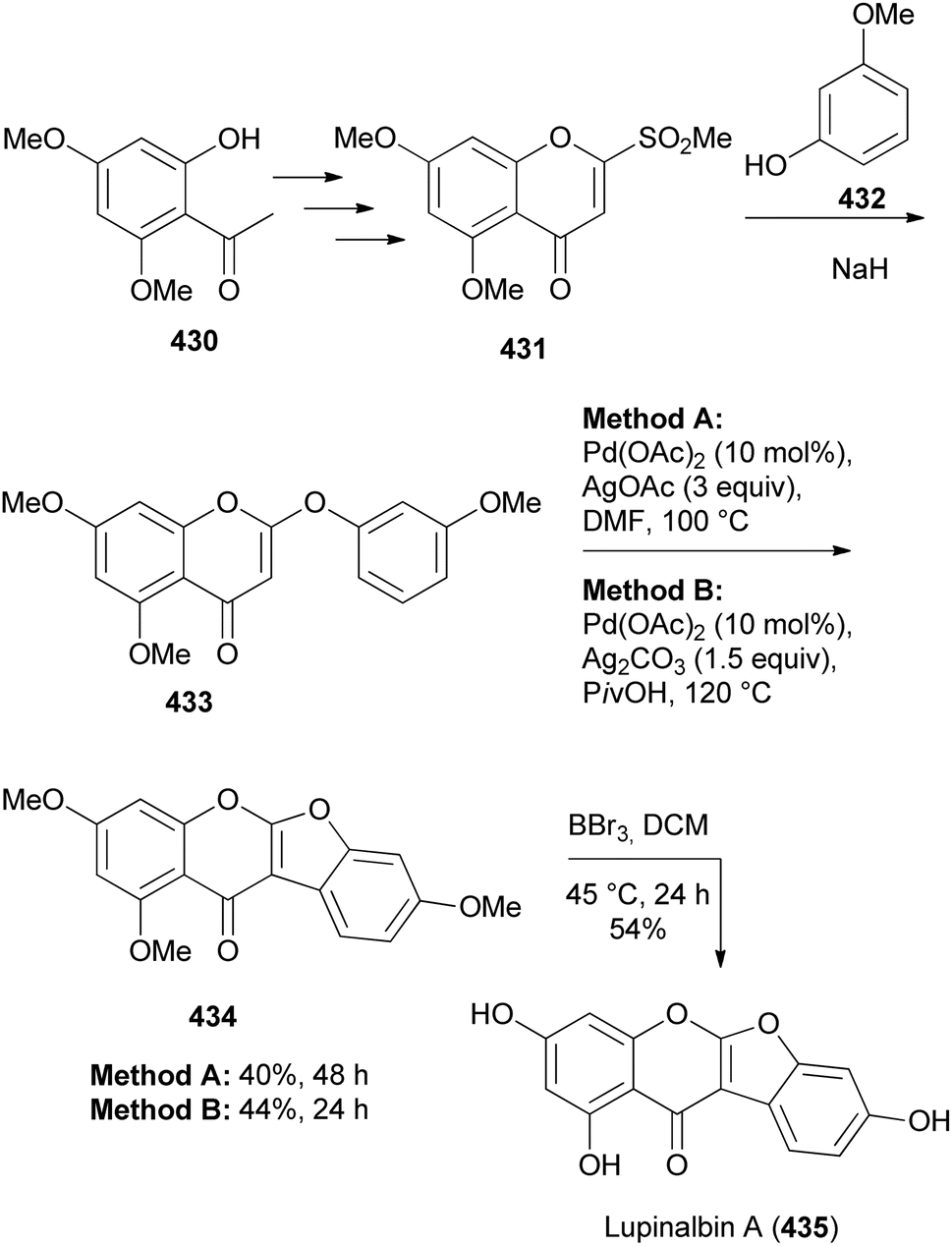
Recently, Gu and co-workers developed a method for the synthesis of coumaronochromones by direct oxidative double C–H activation of 2-phenoxychromones using palladium catalyst and silver oxidants.62 The application of the method was demonstrated in the synthesis of a natural coumaronochromone, lupinalbin A (435). Cross dehydrogenative coupling of phenoxychromone 433 using Pd(OAc)2 with AgOAc or Ag2CO3 gave trimethoxycoumaronochromone 434 in 40–44% yield, which was demethylated using BBr3 to give lupinalbin A (435) in 54% yield (Scheme 62). The phenoxychromone 433 was prepared by displacement of the sulfonyl group in 2-methylsulfonyl-4H-4-chromenone 431 by 3-methoxyphenol (432), which was in turn synthesised from 2-hydroxyacetophenone 430.62,222
Scheme 62 Synthesis of lupinalbin A (435).
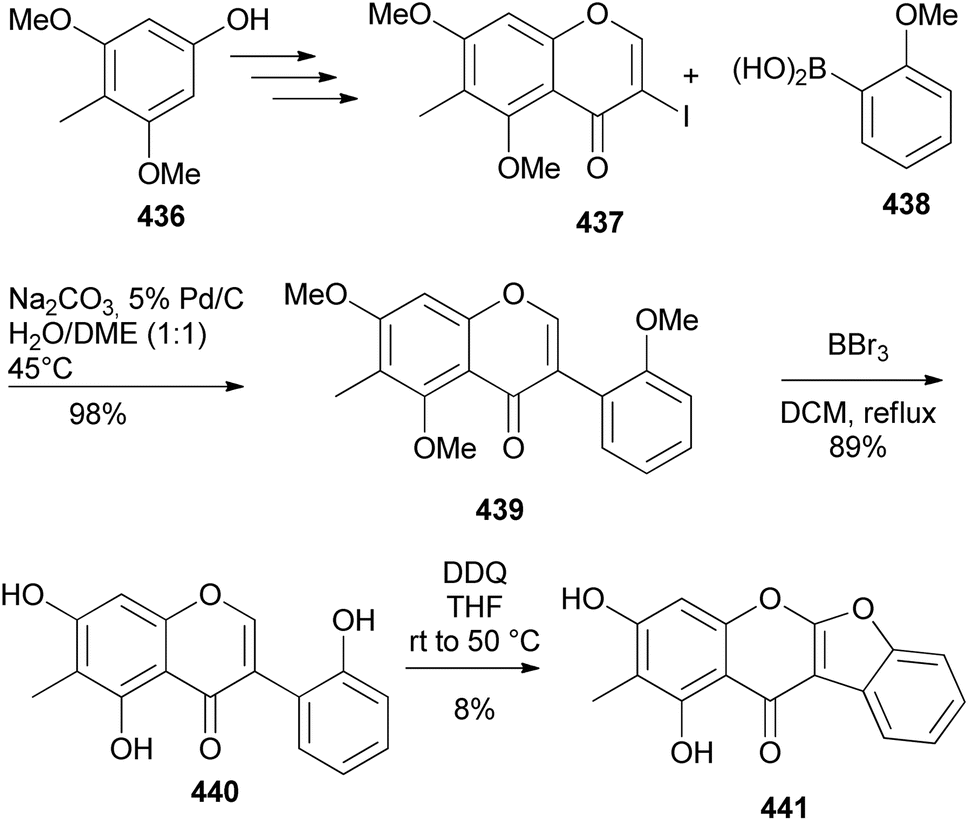
Lee and co-workers isolated a coumaronochromone, antiboeravinone Y (441) with antisepsis properties from Abronia nana suspension cultures.221 The structure of 441 was confirmed by synthesis that involved the preparation of isoflavone 439 by the Suzuki–Miyaura reaction of chromone 437 with boronic acid 438, demethylation and oxidative cyclisation of the 2′-hydroxyisoflavone 440 with DDQ (Scheme 63). The oxidative cyclisation step was however low-yielding.221
Scheme 63 Confirmation of the structure of antiboeravinone Y (441).
Hirtellanine A (5) was isolated from Campylotropis hirtella (Franch.).212 It strongly inhibited proliferation of concanavalin A-induced T-cells (IC50 = 0.92 μM) and lipopolysaccharide-induced B-cell splenocytes (IC50 = 0.06 μM), and showed low cytotoxicity on splenic lymphocytes (CC50 = 3.03 μM).212 The synthesis of hirtellanine A (5) was reported by Zheng and Shen in 2010.32 The first synthesis involved the Suzuki–Miyaura reaction for the construction of the isoflavone, followed by oxidative deprotection to give an isoflavonequinone, and acid-mediated O-cyclisation.32 Another synthesis route, based on the deoxybenzoin intermediate was reported in 2013.45 Paramount to the total synthesis, was regioselective installation of the dimethylpyran scaffold that was achieved by O-allylation of isoflavone 442 to give 443 followed by Claisen rearrangement to afford pyranoisoflavone 444. Several conditions were screened for the Claisen rearrangement, and optimum conditions that yielded the linear chromene isomer 444 in excellent yields were when xylene was used with NaH as a base at 130 °C. Methylation of the 5-hydroxy group followed by oxidative demethylation of 445 using CAN afforded an isoflavonequinone 446, which was cyclised under acidic conditions to give the coumaronochromone 447. The last steps involved the second oxidative deprotection and reduction to give the target compound, hirtellanine A (5), Scheme 64.45
Scheme 64 Total synthesis of hirtellanine A (5).
7. Synthesis of isoflavenes and 3-arylcoumarins
Isoflavenes are synthesised by strategies that include ring-closing metathesis,223,224 conjugate addition of phenyllithium to chromenesulphones followed by elimination of sulphonyl benzene,225 reduction of 2-morpholinoisoflav-3-ene precursors,226 and Suzuki–Miyaura coupling of halochromenes with boronic acids,161 they serve as synthetic precursors to other isoflavonoid compounds that include 3-arylcoumarins, isoflavans, pterocarpans, and complex isoflavonoids.153,161,224
The 3-arylcoumarin, santalin AC (453) and isoflavene-bearing condensed derivatives, santalins and santarubins were synthesised by Strych and colleagues.227,228 Santalins and santarubins are chemical entities that belong to the red sandalwood (Pterocarpus santalinus tree and related species). They were first isolated by Pelletier in the early 1800's and due to their complex structures, their structural elucidation remained an unfinished quest.227–232 In the early 1970's, studies towards the elucidation of santalins and santarubins attracted attention as the structures for santalin A (449) and B (450) were proposed. The structural elucidation of compounds 449 and 450 was later established by Arnone et al. in 1975.232 Although the structures of santalins A (449), B (450) and santarubins A (451), B (452) share a similar core (Fig. 4), they have different hydroxylation, methylation, oxidation, and substitution patterns and these features made it impossible for many years to properly elucidate the structures of these compounds. For example, the phenyl substituent at C-5 for santalin A (449) bears the resorcinol substitution pattern and the phenyl substituent at C-5 for santarubin A (451) features the catechol substitution pattern. Interestingly, a contrasting substitution pattern of the benzyl substituent is observed for both structures at C-6.233 The structural assignments of these compounds were made possible by improving the sensitivity of the NMR and X-ray crystallography machines. In 1995, Kinjo et al. isolated and elucidated the structures of a simple isoflavonoid santalin AC (453), which is a coumarin and that of a yellow pigment santalin Y (454), which is a rather complex structure from red sandalwood, Fig. 4.234
Fig. 4 Structures for santalins and santarubins.
The advent of santalin AC (453) and santalin Y (454) made it possible to propose the biosynthetic route for santalins and santarubins. The proposed biosynthetic route places the isoflavylium as the central core structure for obtaining the santalins and santarubins.227,234 Satalin Y (454) is a racemic natural product that exhibits a unique [6,6,6,5]-oxafenestrane framework that is comprised of a catechol ring as well as pyrogallol and resorcinol moieties that are partially methylated. Following the synthesis of santalins A (449), B (450), and santarubins A (451), B (452),227 Strych et al. reported a biomimetic total synthesis of santalin Y (454).228 The synthesis started by first preparing the isoflavylium 463 and anhydrobase base 464 in seven and eight linear steps from malic acid (456) and 1,2,4-triacetoxybenzene (455) (Scheme 65). Thus, the union of 455 and 456 under acid-catalysed conditions gave coumarin 457 (52%), which was subsequently subjected to TBDMS protection to afford compound 458 in 96% yield. The key step utilised to synthesise 463 and 464 was the zinc-mediated Negishi-coupling reaction developed by the group of Prof. Knochel.57,227,228,235 Therefore, the zincation of 458 delivered intermediate 459 that was subjected to Negishi-cross coupling reaction with aryl bromide 460 to afford the isoflavonoid derivative 461. The silyl deprotection of isoflavonoids 461 gave access to santalin AC (453). Reduction of 461 with DIBAL-H afforded lactol 462 and subsequent protonation and dehydration of lactol 462 using acetic acid and perchloric acid rendered isoflavylium 463 in a 76% yield. Finally, the deprotonation of 463 with non-nucleophilic base afforded anhydrobase 464.
Scheme 65 Synthesis of intermediate isoflavylium 463, anhydrobase 464, and santalin AC (453).
The sequential monoallylation of resorcinol (220), protection of the hydroxy group by the silyl group, Lewis acid-catalysed Claisen rearrangement of 465, mild methylation with MeO3BF4, and subsequent desilylation afforded the allylresorcinol 466 in a 73% overall yield from four synthesis steps. The styrene 468 which is a coupling partner of allylresorcinol 466 was synthesised in two steps from 3,4-dihydroxybenzaldehyde (467) by silyl protection of the hydroxy groups and subsequent Wittig olefination.227,228 Therefore, with the successful synthesis of allylresorcinol 466 and styrene 468, the intermediate benzylstyrene 469 was synthesised employing olefin cross metathesis (Scheme 66). Thus, the union of anhydrobase 464 with benzylstyrene 469 while utilising Et3N as a base in trifluoroethanol led to the formation of santalin Y (454) in 67% isolatable yields. This reaction proceeds through the concerted oxidopyrylium [2 + 3]-cycloaddition reaction and subsequent in situ Friedel–Crafts alkylation facilitated by the keto–enol tautomerisation and activation of the carbonyl functionality by the Brønsted base. It is important to mention that these reactions were happening asynchronously as confirmed by the computations done to model the reaction sequence. The use of bifunctional organocatalysts 470 (Takemoto catalyst) and 471 (Shreiner's catalyst) provided moderate to no formation of santalin Y (454). Interestingly, when Takemoto's catalyst 470 was used in conjunction with Et3N santalin Y(454) was obtained in 45% yield and Shreiner's catalyst did not work at all.228
Scheme 66 Biomimetic synthesis of santalin Y (454).
8. Synthesis of coumestans
Coumestans are tetracyclic isoflavonoids characterised by fused coumarin and benzofuran ring systems. They have mainly been isolated from the Leguminosae and Asteraceae families,21,236,237 and have been reported to exhibit anticancer,238,239 antiarthritic,28 immunosuppressive,240 anti-inflammatory,241 anti-snake-venom,242 antiosteoporsis,42 estrogenic,243 and other activities.21 Several synthetic strategies have been developed for the coumestan scaffold and their applications have been demonstrated on the synthesis natural coumestans that include coumestrol,236,237,244 4′-O-methylcoumestrol,237,244,245 flemichapparin C,245,246 medicagol,245 aureol,236 wedelolactone,247 plicadin,237,248 hirtellanine B54 and lespeflorin I1.249 The coumestans have mainly been constructed from 3-arylcoumarin,236,237,244,245,250 4-phenoxycoumarin,63,64 4-hydroxycoumarin54,246,251,252 and benzofuran precursors.247,248,253,254 Other strategies involved oxidative rearrangement of chalcones and intramolecular cyclisation of oxodiphenylpropanoate.43,249
8.1. Synthesis from 3-arylcoumarin precursors
Coumestans have been synthesised by oxidative coupling of 2′-hydroxy-3-arylcoumarins using reagents that include Pb(OAc)4,255 DDQ,256 PdCl2,257 I2 (ref. 258) and Cu(OAc)2.244,245 Recently, Yan and colleagues developed a method for the synthesis of coumestans using DBU as a catalyst and applied it to the synthesis of 4′-O-methylcoumestrol (478), coumestrol (479), and plicadin (482).237 The reaction is postulated to proceed via dehydrogenation and oxa-Michael addition. The syntheses of the compounds are shown in Schemes 67 and 68. The reaction of 3-bromophenol 472 with oxoacetic acid gave 4-hydroxyphenylglycolic acid 473, which was reduced and hydroxylated to give phenylacetic acids 474 and 475, respectively. Perkin condensation of 475 with 2-hydroxy-4-methoxybenzaldehyde (376), followed by deacetylation gave 3-phenylcoumarin 477. Treatment of 477 with DBU rendered 4′-O-methylcoumestrol (478), which upon demethylation afforded coumestrol (479), Scheme 67. Similarly, plicadin (482) was synthesised by BDU-catalysed cyclisation of 2′,4′-dihydroxy-3-phenylcoumarin 481, which was in turn prepared by Perkin condensation of pyranobenzaldehyde 480 with phenylacetic acid 475. Compound 480 was synthesised by condensation of benzaldehyde 293 with prenal, Scheme 68.237
Scheme 67 Synthesis of 4′-O-methylcoumestrol (478) and coumestrol (479).
Scheme 68 Synthesis of plicadin (482).
Alternative to 2′-hydroxy-3-arylcoumarin precursors, Sheng and co-workers synthesised coumestrol (479) and aureol (485) by a one-pot Cu(II)-catalysed hydroxylation and oxidative coupling of 2′-bromo-3-arylcoumarins 484a and 484b, which were obtained by Perkin condensation of benzaldehydes 293 and 483 with bromophenylacetic acid 474. Bromophenylacetic acid 474 was derived from 3-bromophenol (472), Scheme 69.236
Scheme 69 Synthesis of coumestrol (479) and aureol (485).
8.2. Synthesis from 4-hydroxy- and 4-phenoxycoumarins precursors
Examples of methods used for the synthesis of coumestans that proceed through the 4-hydroxy- and 4-phenoxycoumarin precursors include: Pd-catalysed cascade reaction of 4-hydroxycoumarins with in situ generated arynes,246 coupling of 4-hydroxycoumarins with catechols using Rh/AlPO4 nanoparticle catalyst252 or K3Fe(CN)6 (ref. 54) and Pd-catalysed double C–H activation of phenoxycoumarins.63,64
The synthesis of coumestans has also been attained from 4-phenoxycoumarin or 4-hydroxycoumarin precursors.54,63,64,246,252,259 From 4-phenoxycoumarins, the coumestans were synthesised by direct arylation involving double C–H activation using palladium catalysts and silver salts as oxidants, thereby avoiding the utilisation of pre-derivatised building blocks.63,64 The natural coumestan, flemichapparin C (489a) was synthesised by this protocol by McGlacken's group in 2016.63 As shown in Scheme 70, the synthesis commenced with the preparation of the 4-phenoxychromone 488 by bromination of 4-hydroxycoumarin 486 and coupling of 4-bromocoumarin 487 with sesamol. The intramolecular double dehydrogenative cyclisation of the phenoxycoumarin 488 using Pd(OAc)2, AgO, and NaO-t-Bu gave the targeted flemichapparin C (489a) and its isomer 489b in ratio of 84:16 and 82% yield, Scheme 70.63
Scheme 70 Synthesis of flemichapparin C (489a).
Xu's group concurrently reported a similar protocol and demonstrated its application in the synthesis of coumestrol (479) and flemichapparin C (489a). Intramolecular cross dehydrogenative coupling of phenoxycoumarins 490 and 488 using Pd(OAc)2 catalyst, AgOAc as oxidant and CsOAc as a base exclusively rendered coumestan 491 and flemichapparin C (489a), respectively (Scheme 71). The compounds were obtained in high yields and good regioselectivity. Demethylation of 491 and flemichapparin C (489a) gave coumestrol (479) and demethylflemichapparin C (492), respectively.64
Scheme 71 Synthesis of coumestrol (479) and flemichapparin C (489a).
4-Hydroxycoumarins have been successfully employed for the synthesis of coumestans.54,246,252 Neog and colleagues synthesised coumestans including flemichapparin C (489a) by palladium-catalysed coupling of 4-hydroxycoumarins with arynes generated in situ from o-trimethylsilylphenyl triflates.246,260 The reaction proceeded via tandem C–H activation followed by C–O and C–C bond formations.246 As shown in Scheme 72, the reaction of 4-hydroxycoumarin 486 with o-trimethylsilylphenyl triflate 493 under optimised conditions gave flemichapparin C (489a).
Scheme 72 Synthesis of flemichapparin C (489a).
Maeno and co-workers synthesised flemichapparin C (489a) by oxidative annulation of 4-hydroxycoumarin 486 and o-quinones generated from catechol (395) using AlPO4-supported Rh nanoparticle and O2 as oxidant, and subsequent reaction of the resulting coumestan 494 with bromochloromethane, Scheme 73.252
Scheme 73 Synthesis of flemichapparin C (489a).
Hirtellanine B (499) was synthesised by oxidative coupling of catechol with 4-hydroxypyranocoumarin 498 using K3FeCN as oxidant.54 The 4-hydroxypyranocoumarin 498 was prepared in a sequence of steps that involved the synthesis of 3-iodochromone 495 from phloroacetophenone (38)32 and the conversion of 495 into 2-imidazolylchromone 497 (Scheme 74).261,262
Scheme 74 The synthesis of hirtellanine B (499).
8.3. Synthesis from benzofuran precursors
2-Phenylbenzofurans have proven to be important building blocks for the synthesis of coumestans. Syntheses of the 2-phenylbenzofuran derivatives have been attained by methods that include palladium-catalysed carbonylative cyclisation of diaryl alkynes,263 the Suzuki–Miyaura coupling of 2-halobenzofurans with boronate esters,247 and newly developed methods that include base-catalysed cyclisation of 2-bromobenzylketones253 and indium-catalysed restructuring of phenylethynyl pyrones.254
Wedelolactone (508) is an important coumestan, which has been synthesised by several research groups.250,258,263 Recently, Gou and colleagues synthesised wedelolactone (508) by lactonisation of ethyl 2-phenylbenzofuran-3-carboxylate 507.247 The 2-phenylbenzofuran-3-carboxylate 507 was synthesised by TBS deprotection of the benzofuran 506, which was in turn assembled by the Suzuki–Miyaura coupling of two key precursors: the 2-iodobenzofuran-3-carboxylate 504 and the boronate ester 505 (Scheme 75). The boronate ester was synthesised in a sequence of steps from phloroglucinol, while the 2-iodobenzofuran-3-carboxylate 504 was prepared by formylation of trihydroxybenzene 500 to give benzaldehyde 501. The reaction of benzaldehyde 501 with ethyl diazoacetate in the presence of boron tetrafluoride/ether and subsequent treatment with sulphuric acid rendered 3-ethoxycarbonylbenzofuran 502.264 TBS protection and iodination of the benzofuran 503 gave 504 (Scheme 75).247
Scheme 75 Total synthesis of wedelolactone (508).
Zhang and co-workers developed a method for the synthesis of 2-arylbenzofurans that involved t-BuOK-catalysed cyclisation of 2-bromobenzylketones.253 The utility of the method was demonstrated in the synthesis of coumestrol (479). The synthesis proceeded via condensation of the 2-bromophenylacetate 509 with dimethoxybenzoyl chloride 510 to give 2-bromobenzylketone 511. Treatment of 511 with t-BuOK gave 2-arylbenzofuran-3-carboxylate 512. Demethylation of 512 with BBr3 followed by intramolecular esterification of the resulting intermediate 513 afforded coumestrol (479), Scheme 76.253
Scheme 76 Total synthesis of coumestrol (479).
Zhu's group developed a method that transformed kojic acid-derived alkynes into 2-phenylbenzofurans using indium triflate as a catalyst.254 The 2-phenylbenzofurans were further elaborated to afford several benzofuran-containing natural compounds, including coumestrol (479). As shown in Scheme 77, coumestrol (479) was synthesised by a sequence of steps that involved C-3 formylation of 515, followed by conversion of the formyl group into hydroxycarbonyl group by Pinnick oxidation, demethylation, and spontaneous lactonisation. The benzofuran 515 resulted from indium triflate-catalysed reassembly of phenylethynyl pyrone 514.254
Scheme 77 Synthesis of coumestrol (479).
Alternative to 2-phenylbenzofurans, Nayak and colleagues synthesised pterocarpens and coumestans from 3-phenoxymethylbenzofurans constructed by BCl3-mediated cyclisation of 1,3-diphenoxypropanones.248,265 As shown in Scheme 78, the diaryloxyacetone 519 was synthesised by consecutive O-alkylation of phenols 432 and 289 with epichlorohydrin 516 and subsequent oxidation of the resulting alcohol 518. Treatment of 519 with BCl3 gave the 3-phenoxymethylbenzofuran 520 in 65% yield.265 Intramolecular Heck coupling of 520 gave pterocarpene 521 in 86% yield, which was oxidised into coumestan 522.248 Compound 522 could be demethylated to render coumestrol (479). Other coumestans that include flemichapparin C (489a) and proposed plicadin (482) were also synthesised from appropriately substituted 1,3-diaryloxyketones following the same sequence.248
Scheme 78 Synthesis of coumestrol (479).
8.4. Other
Pahari and colleagues synthesised prenylated coumestans that include psoralidin (6), lespeflorin I1 (527) and their derivatives from oxodiphenylpropanoate precursors.43,249 As illustrated for the synthesis of lespeflorin I1 (527), LDA-mediated coupling of phenyl acetate 523 with benzoyl chloride generated from benzoic acid 524 gave oxodiphenylpropanoate 525, which upon treatment with BBr3 underwent demethylation and spontaneous intramolecular cyclisation to give the coumestan 526. Protection of the adjacent hydroxy groups with phosgene followed by metathesis protocol (catalysed by second generation Grubb's catalyst) and hydrolysis of the cyclic carbonate group gave lespeflorin I1 (527), Scheme 79.249
Scheme 79 Synthesis of lespeflorin I1 (527).
9. Conclusions
Isoflavonoids are phenolic natural compounds with diverse structural variation and important biological activities. Owing to their interesting structures and potential benefits for human health, isoflavonoids have been synthesised by several research groups. Conventional methods that include oxidative rearrangements, classical condensations (Perkin reaction, Claisen condensation, aldol condensation etc.) and sigmatropic rearrangements are still widely employed for the construction of isoflavonoid core structures and the incorporation of substituents found in natural isoflavonoids. Alternative to the classical methodologies, isoflavonoids have been conveniently accessed by transition metal-catalysed reactions of pre-functionalised building blocks that include the Suzuki–Miyaura, the Negishi, the Stille, and the Heck cross-coupling reactions, as well as cross-coupling metathesis reactions. Of these methods, the Suzuki–Miyaura reaction has been widely applied to the synthesis of different classes of isoflavonoid compounds such as isoflavones, coumarins, and isoflavenes, which also serve as precursors for other elaborate isoflavonoid compounds. Although additional steps are required for the preparation of non-commercial boronic acids/esters and their coupling partners, the Suzuki reaction continues to be the most preferred method, due to the benign conditions that enable introduction of sensitive substituents in the early and late stages of the synthesis, thereby facilitating regiocontrol. The cross-coupling metathesis using Grubs II catalyst has been employed for the synthesis of isoflavenes, and several research groups have used the reaction for the assembly of the prenyl substituents in the late stages of the synthesis. This has been particularly useful when highly acidic or oxidative conditions were employed for the synthesis of isoflavonoid scaffolds.
Advancements in the syntheses of isoflavonoids using metal catalysts have been realised through C–H activation protocols that utilise single pre-derivatised precursors. The applications of these have been demonstrated in the synthesis of isoflavones through direct arylation of 2-hydroxyenaminoketones90 and the synthesis of coumestans by tandem arylation and oxycyclisation of 4-hydroxycoumarins with arynes generated in situ from O-trimethylsilylphenyl triflates.246,260 Isoflavonoids have also been synthesised by double C–H activation involving C–C bond formation from unfunctionalised carbon atoms. Examples include the syntheses of coumaronochromones and coumestans by double dehydrogenative C–C coupling of 2-phenoxychromones and 4-phenoxycoumarins, respectively.62–64 The development of a new synthetic strategy for coumaronochromones is commendable, considering that coumaronochromones have mainly been synthesised by oxidative coupling of 2′-hydroxyisoflavones. Other methods that do not utilise pre-functionalised precursors include oxidative annulation of 4-hydroxycoumarins or chromenes with quinones to afford coumestans or pterocarpans, respectively.54,157,252
Lewis acids and superacids such as BBr3, BCl3 and In(OTf)3 have also facilitated transformations leading to the formation of different classes of isoflavonoids, particularly coumestans and isoflavones. BCl3 and BBr3 have mainly been used for tandem demethylation and intramolecular O-cyclisation in the synthesis of coumestans that include coumestrol and lespeflorin I1.248,249,253 While tandem processes involving deprotection of the methyl ether and spontaneous oxycyclisation are widely reported in the synthesis of natural compounds, a rather unexpected transformation that involved the conversion of methoxybenzoylbenzofurans into isoflavones was reported by Kunyane and colleagues.87 It proceeded through a cascade of processes that involved demethylation and intramolecular O-cyclisation, resulting in furan ring-opening and the formation of a new chromone ring of isoflavones.87 Another interesting novel method was developed by Zhu's group that transformed kojic acid-derived alkynes (with pyrone moiety) into 2-phenylbenzofurans using In(OTf)3 as a catalyst.254 The 2-phenylbenzofurans were further elaborated to afford several benzofuran-containing natural compounds, including coumestrol.254
Although several new methods have been developed for the synthesis of isoflavonoid compounds, the application of most of them has mainly been demonstrated in the synthesis of simple natural isoflavonoids. An exceptional example is the zinc-mediated Negishi coupling reaction, which facilitated the synthesis of complex isoflavene-derived santarubins and santalins.57,227,228 Conventional methods are still utilised frequently for the synthesis of isoflavonoids with intricate structures. Therefore, future studies could take advantage of the newly developed methods for the syntheses of more complex natural isoflavonoids, especially the atom-economic strategies based on C–H activation and direct annulation of non-functionalised precursors.
The syntheses of chiral isoflavonoids that include isoflavanones, isoflavans, pterocarpans, and rotenoids have been accomplished in racemic form and stereoselectively. The syntheses of racemates were accompanied by resolution using chiral-phase chromatography in several instances. The stereoselective syntheses employed chiral pool building blocks,65–67 chiral ligands,68,69 chiral organocatalysts,70–72 and chiral transition metal catalysts that facilitated stereoselective hydrogenation and hydrogen transfer.76–78 Although natural isoflavonoids could be obtained in good ee in the reported stereoselective syntheses, most of them proceeded via dynamic kinetic resolution of racemic isoflavonoids and intermediates. The exceptions were the solvent-free benzoin reaction that gave optically active 3-hydroxyisoflavanone upon intramolecular cyclisation in the presence of the chiral NHC catalyst,70,73 as well as the organo-catalysed stereoselective arylation for the synthesis of S-equol.75 The chiral pool strategy in conjunction with stereocontrolled epoxidation or Sharpless asymmetric dihydroxylation also furnished the requisite stereogenicity from the onset of the syntheses and were successfully employed in the stereoselective synthesis of complex isoflavonoids that included stachyodin A,67 a pterocarpan with a rare spirotetrahydrofuran ring and the rotenoids, (−)-rotenone and (−)-dalpanol.66
10. Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
11. Author contributions
All the authors wrote, reviewed and edited the manuscript.
12. Conflicts of interest
There are no conflicts to declare.
13. Acknowledgements
This research was supported by the National Research Foundation of South Africa through Thuthuka funding (grant numbers: 138404 & 0415594923) and the University of Pretoria. STM acknowledges the Department of Higher Education and Training (Republic of South Africa) for support through University Staff Capacity Programme (UCDP).
14. References
P. M. Dewick, in The Flavonoids Advances in Research Since 1986, ed. J. B. Harborne, Taylor & Francis, New York, 1st edn, 1994, ch. 5 Search PubMed.
N. C. Veitch, Nat. Prod. Rep., 2007, 24, 417–464 RSC.
N. C. Veitch, Nat. Prod. Rep., 2013, 30, 988–1027 RSC.
N. Al-Maharik, Nat. Prod. Rep., 2019, 36, 1156–1195 RSC.
J. Reynaud, D. Guilet, R. Terreux, M. Lussignol and N. Walchshofer, Nat. Prod. Rep., 2005, 22, 504–515 RSC.
O. Lapčík, Phytochemistry, 2007, 68, 2909–2916 CrossRefPubMed.
G. M. Boland and D. M. X. Donnelly, Nat. Prod. Rep., 1998, 15, 241–260 RSC.
K. T. Desta and A. M. Abd El-Aty, Phytochem. Rev., 2023, 22, 275–308 CrossRefCASPubMed.
S. Tahara and R. K. Ibrahim, Phytochemistry, 1995, 38, 1073–1094 CrossRefCAS.
W. G. Ma, Y. Fukushi, K. Hostettmann and S. Tahara, Phytochemistry, 1998, 49, 251–254 CrossRefCAS.
W. Szeja, G. Grynkiewicz and A. Rusin, Curr. Org. Chem., 2017, 21, 218–235 CrossRefCASPubMed.
B. C. B. Bezuidenhoudt, E. V. Brandt, J. A. Steenkamp, D. G. Roux and D. Ferreira, J. Chem. Soc., Perkin Trans., 1988, 1227–1235, 10.1039/P19880001227.
J. A. Ferreira, J. W. Nel, E. V. Brandt, B. C. B. Bezuidenhoudt and D. Ferreira, J. Chem. Soc., Perkin Trans., 1995, 1049–1056, 10.1039/P19950001049.
J. Li, L. Pan, Y. Deng, U. Muñoz-Acuña, C. Yuan, H. Lai, H. Chai, T. E. Chagwedera, N. R. Farnsworth, E. J. Carcache de Blanco, C. Li, D. D. Soejarto and A. D. Kinghorn, J. Org. Chem., 2013, 78, 10166–10177 CrossRefCASPubMed.
T. P. Banzato, J. R. Gubiani, D. I. Bernardi, C. R. Nogueira, A. F. Monteiro, F. F. Juliano, S. M. de Alencar, R. A. Pilli, C. A. d. Lima, G. B. Longato, A. G. Ferreira, M. A. Foglio, J. E. d. Carvalho, D. B. Vendramini-Costa and R. G. S. Berlinck, J. Nat. Prod., 2020, 83, 1784–1793 CrossRefCASPubMed.
R. A. Dixon and C. L. Steele, Trends Plant Sci., 1999, 4, 394–400 CrossRefPubMed.
S. Gafner, C. Bergeron, J. R. Villinski, M. Godejohann, P. Kessler, J. H. Cardellina II, D. Ferreira, K. Feghali and D. Grenier, J. Nat. Prod., 2011, 74, 2514–2519 CrossRefCASPubMed.
Y. X. Zhou, H. Zhang and C. Peng, Phytother. Res., 2014, 28, 961–975 CrossRefCASPubMed.
C. Selvam, B. C. Jordan, S. Prakash, D. Mutisya and R. Thilagavathi, Eur. J. Med. Chem., 2017, 128, 219–236 CrossRefCASPubMed.
X. Liu, R. Huang and J. Wan, Biomed. Pharmacother., 2023, 162, 114581 CrossRefCASPubMed.
Y. Tu, Y. Yang, Y. Li and C. He, Pharmacol. Res., 2021, 169, 105615 CrossRefCASPubMed.
R. Simons, H. Gruppen, T. F. H. Bovee, M. A. Verbruggen and J.-P. Vincken, Food Funct., 2012, 3, 810–827 RSC.
S.-Y. Ahn, M. S. Jo, D. Lee, S.-E. Baek, J. Baek, J. S. Yu, J. Jo, H. Yun, K. S. Kang, J.-E. Yoo and K. H. Kim, Bioorg. Chem., 2019, 83, 135–144 CrossRefCASPubMed.
B. Bueno-Silva, S. M. Alencar, H. Koo, M. Ikegaki, G. V. J. Silva, M. H. Napimoga and P. L. Rosalen, J. Agric. Food Chem., 2013, 61, 4546–4550 CrossRefCASPubMed.
S. S. Çiçek, M. Galarza Pérez, A. Wenzel-Storjohann, R. M. Bezerra, J. F. O. Segovia, U. Girreser, I. Kanzaki and D. Tasdemir, J. Nat. Prod., 2022, 85, 927–935 CrossRefPubMed.
G. Porras, F. Chassagne, J. T. Lyles, L. Marquez, M. Dettweiler, A. M. Salam, T. Samarakoon, S. Shabih, D. R. Farrokhi and C. L. Quave, Chem. Rev., 2021, 121, 3495–3560 CrossRefCASPubMed.
S. E. Drewes, M. M. Horn, O. Q. Munro, J. T. B. Dhlamini, J. J. M. Meyer and N. C. Rakuambo, Phytochemistry, 2002, 59, 739–747 CrossRefCASPubMed.
Y. Tu, K. Wang, X. Jia, L. Tan, B. Han, Q. Zhang, Y. Li and C. He, J. Agric. Food Chem., 2020, 68, 10664–10677 CrossRefCASPubMed.
J. Wu, K. Li, Y. Liu, A. Feng, C. Liu, J. Adu-Amankwaah, M. Ji, Y. Ma, Y. Hao, H. Bu and H. Sun, Food Funct., 2023, 14, 934–945 RSC.
I. Kiganda, J. Bogaerts, L. H. E. Wieske, T. Deyou, Y. Atilaw, C. Uwamariya, M. Miah, J. Said, A. Ndakala, H. M. Akala, W. Herrebout, E. Trybala, T. Bergström, A. Yenesew and M. Erdelyi, J. Nat. Prod., 2024, 87, 1003–1012 CrossRefCASPubMed.
Q. Y. Shou, Q. Tan and Z. W. Shen, Bioorg. Med. Chem. Lett., 2009, 19, 3389–3391 CrossRefCASPubMed.
Y. Zhai, Q. Wang, Y. Li, J. Cui, K. Feng, X. Kong and C. J. Xian, Biomed. Pharmacother., 2018, 102, 1015–1024 CrossRefCASPubMed.
R. Muharini, A. Díaz, W. Ebrahim, A. Mándi, T. Kurtán, N. Rehberg, R. Kalscheuer, R. Hartmann, R. S. Orfali, W. Lin, Z. Liu and P. Proksch, J. Nat. Prod., 2017, 80, 169–180 CrossRefCASPubMed.
F. A. Adem, A. T. Mbaveng, V. Kuete, M. Heydenreich, A. Ndakala, B. Irungu, A. Yenesew and T. Efferth, Phytomedicine, 2019, 58, 152853 CrossRefCASPubMed.
C. Ito, T. Matsui, K. Miyabe, C. M. Hasan, M. A. Rashid, H. Tokuda and M. Itoigawa, Phytochemistry, 2020, 175, 112376 CrossRefCASPubMed.
D. Y. W. Lee, W.-Y. Zhang and V. V. R. Karnati, Tetrahedron Lett., 2003, 44, 6857–6859 CrossRefCAS.
H. Tanaka, T. Oh-Uchi, H. Etoh, M. Sako, F. Asai, T. Fukai, M. Sato, J. Murata and Y. Tateishi, Phytochemistry, 2003, 64, 753–758 CrossRefCASPubMed.
N. J. Sadgrove, T. B. Oliveira, G. P. Khumalo, S. F. van Vuuren and B.-E. van Wyk, Antibiotics, 2020, 9, 223 CrossRefCASPubMed.
M. J. C. Falcão, Y. B. M. Pouliquem, M. A. S. Lima, N. V. Gramosa, L. V. Costa-Lotufo, G. C. G. Militão, C. Pessoa, M. Odorico de Moraes and E. R. Silveira, J. Nat. Prod., 2005, 68, 423–426 CrossRefPubMed.
K. Farias, R. F. da Costa, A. S. Meira, J. Diniz-Filho, E. M. Bezerra, V. N. Freire, P. Guest, M. Nikahd, X. Ma, M. G. Gardiner, M. G. Banwell, M. d. C. F. de Oliveira, M. O. de Moraes and C. do Ó Pessoa, ACS Med. Chem. Lett., 2020, 11, 1274–1280 CrossRefCASPubMed.
Y. Zhai, Y. Li, Y. Wang, J. Cui, K. Feng, X. Kong and L. Chen, Eur. J. Pharmacol., 2017, 801, 62–71 CrossRefCASPubMed.
P. Pahari and J. Rohr, J. Org. Chem., 2009, 74, 2750–2754 CrossRefCASPubMed.
M. A. Selepe, S. E. Drewes and F. R. van Heerden, J. Nat. Prod., 2010, 73, 1680–1685 CrossRefCASPubMed.
Q.-L. Li, Q.-L. Liu, Z.-Y. Ge and Y.-M. Zhu, Helv. Chim. Acta, 2011, 94, 1304–1309 CrossRefCAS.
V. V. Semenov, D. V. Tsyganov, M. N. Semenova, R. N. Chuprov-Netochin, M. M. Raihstat, L. D. Konyushkin, P. B. Volynchuk, E. I. Marusich, V. V. Nazarenko, S. V. Leonov and A. S. Kiselyov, J. Nat. Prod., 2016, 79, 1429–1438 CrossRefCASPubMed.
K. Mackey, L. M. Pardo, A. M. Prendergast, M.-T. Nolan, L. M. Bateman and G. P. McGlacken, Org. Lett., 2016, 18, 2540–2543 CrossRefCASPubMed.
C. Cheng, W.-W. Chen, B. Xu and M.-H. Xu, Org. Chem. Front., 2016, 3, 1111–1115 RSC.
M. S. Hofmayer, A. Sunagatullina, D. Brösamlen, P. Mauker and P. Knochel, Org. Lett., 2020, 22, 1286–1289 CrossRefCASPubMed.
K. Nakamura, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2017, 56, 182–187 CrossRefCASPubMed.
Y. Kawamoto, T. Kobayashi and H. Ito, Tetrahedron, 2022, 106–107, 132595 CrossRefCAS.
R. Doran, M. P. Carroll, R. Akula, B. F. Hogan, M. Martins, S. Fanning and P. J. Guiry, Chem.–Eur. J., 2014, 20, 15354–15359 CrossRefCASPubMed.
K. H. Georgiou, S. C. Pelly and C. B. de Koning, Tetrahedron, 2017, 73, 853–858 CrossRefCAS.
K. Iwai, M. Ono, Y. Nanjo and T. Ema, ACS Omega, 2020, 5, 10207–10216 CrossRefCASPubMed.
R. L. Farmer and K. A. Scheidt, Chem. Sci., 2013, 4, 3304–3309 RSC.
S. Perveen, S. Yang, M. Meng, W. Xu, G. Zhang and X. Fang, Commun. Chem., 2019, 2, 8 CrossRef.
T. Ema, Y. Nanjo, S. Shiratori, Y. Terao and R. Kimura, Org. Lett., 2016, 18, 5764–5767 CrossRefCASPubMed.
J.-W. Lee and B. List, J. Am. Chem. Soc., 2012, 134, 18245–18248 CrossRefCASPubMed.
T. Uemura, Y. Saito, M. Sonoda and S. Tanimori, Synlett, 2020, 32, 693–696 Search PubMed.
G. S. Caleffi, F. C. Demidoff, C. Nájera and P. R. R. Costa, Org. Chem. Front., 2022, 9, 1165–1194 RSC.
A. Keßberg, T. Lübken and P. Metz, Org. Lett., 2018, 20, 3006–3009 CrossRefPubMed.
J. Xia, Y. Nie, G. Yang, Y. Liu and W. Zhang, Org. Lett., 2017, 19, 4884–4887 CrossRefCASPubMed.
S. Yang, S.-F. Zhu, C.-M. Zhang, S. Song, Y.-B. Yu, S. Li and Q.-L. Zhou, Tetrahedron, 2012, 68, 5172–5178 CrossRefCAS.
J.-W. Jung, K. Damodar, J.-K. Kim and J.-G. Jun, Chin.
Chem. Lett., 2017, 28, 1114–1118 CrossRefCAS.
A. R. Jesus, C. Dias, A. M. Matos, R. F. M. de Almeida, A. S. Viana, F. Marcelo, R. T. Ribeiro, M. P. Macedo, C. Airoldi, F. Nicotra, A. Martins, E. J. Cabrita, J. Jiménez-Barbero and A. P. Rauter, J. Med. Chem., 2014, 57, 9463–9472 CrossRefCASPubMed.
M. M. Hossain, Y. Kawamura, K. Yamashita and M. Tsukayama, Tetrahedron, 2006, 62, 8625–8635 CrossRefCAS.
M. M. Hossain, J. Heterocycl. Chem., 2012, 49, 999–1003 CrossRefCAS.
G. Kwesiga, A. Kelling, S. Kersting, E. Sperlich, M. von Nickisch-Rosenegk and B. Schmidt, J. Nat. Prod., 2020, 83, 3445–3453 CrossRefCASPubMed.
E. H. Granados-Covarrubias and L. A. Maldonado, Tetrahedron Lett., 2009, 50, 1542–1545 CrossRefCAS.
R. S. Menon, A. D. Findlay, A. C. Bissember and M. G. Banwell, J. Org. Chem., 2009, 74, 8901–8903 CrossRefCASPubMed.
P. Kunyane, M. S. Sonopo and M. A. Selepe, J. Nat. Prod., 2019, 82, 3074–3082 CrossRefCASPubMed.
M. A. Selepe and F. R. Van Heerden, Molecules, 2013, 18, 4739–4765 CrossRefCASPubMed.
D. A. Vasselin, A. D. Westwell, C. S. Matthews, T. D. Bradshaw and M. F. G. Stevens, J. Med. Chem., 2006, 49, 3973–3981 CrossRefCASPubMed.
J.-P. Wan, Z. Tu and Y. Wang, Chem.–Eur. J., 2019, 25, 6907–6910 CrossRefCASPubMed.
S. Mkrtchyan and V. O. Iaroshenko, Chem. Commun., 2020, 56, 2606–2609 RSC.
S. Mkrtchyan and V. O. Iaroshenko, J. Org. Chem., 2021, 86, 4896–4916 CrossRefCASPubMed.
S. Mkrtchyan, M. Jakubczyk, S. Lanka, M. Yar, T. Mahmood, K. Ayub, M. Sillanpää, C. M. Thomas and V. O. Iaroshenko, Adv. Synth. Catal., 2022, 364, 3512–3521 CrossRefCAS.
J. M. Hastings, M. K. Hadden and B. S. J. Blagg, J. Org. Chem., 2008, 73, 369–373 CrossRefCASPubMed.
U. Hakala and K. Wähälä, Tetrahedron Lett., 2006, 47, 8375–8378 CrossRefCAS.
K. Wähälä and T. A. Hase, J. Chem. Soc., Perkin Trans., 1991, 3005–3008, 10.1039/P19910003005.
P. F. Schuda and W. A. Price, J. Org. Chem., 1987, 52, 1972–1979 Search PubMed.
N. Asebi and K.-i. Nihei, Tetrahedron, 2019, 75, 130589 Search PubMed.
A. Pelter and S. Foot, Synthesis, 1976, 1976, 326 CrossRef.
A. I. Calderón, C. Terreaux, K. Schenk, P. Pattison, J. E. Burdette, J. M. Pezzuto, M. P. Gupta and K. Hostettmann, J. Nat. Prod., 2002, 65, 1749–1753 CrossRefPubMed.
M. Ichige, E. Fukuda, S. Miida, J.-i. Hattan, N. Misawa, S. Saito, T. Fujimaki, M. Imoto and K. Shindo, J. Agric. Food Chem., 2013, 61, 2183–2187 CrossRefCASPubMed.
J. Chen, W. Huang, G. Lian and F. Lin, Carbohydr. Res., 2009, 344, 2245–2249 CrossRefCASPubMed.
D.-H. Kim, K.-U. Yu, E.-A. Bae and M. J. Han, Biol. Pharm. Bull., 1998, 21, 628–630 CrossRefCASPubMed.
F.-L. Hsu, I. M. Liu, D.-H. Kuo, W.-C. Chen, H.-C. Su and J.-T. Cheng, J. Nat. Prod., 2003, 66, 788–792 CrossRefCASPubMed.
L. Zhao, Y. Wang, J. Liu, K. Wang, X. Guo, B. Ji, W. Wu and F. Zhou, J. Agric. Food Chem., 2016, 64, 7291–7297 Search PubMed.
D. Wang, T. Bu, Y. Li, Y. He, F. Yang and L. Zou, Antioxidants, 2022, 11, 2121 CrossRefCASPubMed.
Y. Zou, T. Peng, G. Wang, X. Wen, S. Liu, Y. Sun, S. Zhang, Y. Gao and L. Wang, J. Carbohydr. Chem., 2018, 37, 461–470 CrossRefCAS.
S. A. Kagal, P. Madhavan Nair and K. Venkataraman, Tetrahedron Lett., 1962, 3, 593–597 CrossRef.
W. Baker and R. Robinson, J. Chem. Soc. Trans., 1925, 127, 1981–1986 RSC.
Y. Miki, R. Fujita and K.-I. Matsushita, J. Chem. Soc., Perkin Trans., 1998, 2533–2536, 10.1039/A803561J.
A. P. Rauter, A. Martins, C. Borges, J. Ferreira, J. Justino, M.-R. Bronze, A. V. Coelho, Y. H. Choi and R. Verpoorte, J. Chromatogr. A, 2005, 1089, 59–64 CrossRefCASPubMed.
G. Kwesiga, E. Sperlich and B. Schmidt, J. Org. Chem., 2021, 86, 10699–10712 CrossRefCASPubMed.
Z. Wei, Y. Yang, C. Xie, C. Li, G. Wang, L. Ma, M. Xiang, J. Sun, Y. Wei and L. Chen, Fitoterapia, 2014, 97, 172–183 CrossRefCASPubMed.
S. Tahara, J. L. Ingham, S. Nakahara, J. Mizutani and J. B. Harborne, Phytochemistry, 1984, 23, 1889–1900 CrossRefCAS.
C. Vilain, Phytochemistry, 1980, 19, 988–989 CrossRefCAS.
A. Yenesew, J. O. Midiwo and P. G. Waterman, Phytochemistry, 1998, 47, 295–300 CrossRefCAS.
E. Dagne and A. Bekele, Phytochemistry, 1990, 29, 2679–2682 CrossRefCAS.
A. Yenesew, S. Derese, J. O. Midiwo, H. A. Oketch-Rabah, J. Lisgarten, R. Palmer, M. Heydenreich, M. G. Peter, H. Akala, J. Wangui, P. Liyala and N. C. Waters, Phytochemistry, 2003, 64, 773–779 CrossRefCASPubMed.
H. Ye, A. Fu, W. Wu, Y. Li, G. Wang, M. Tang, S. Li, S. He, S. Zhong, H. Lai, J. Yang, M. Xiang, A. Peng and L. Chen, Fitoterapia, 2012, 83, 1402–1408 CrossRefCASPubMed.
X. Li, L. Wan, F. Wang, H. Pei, L. Zheng, W. Wu, H. Ye, Y. Wang and L. Chen, Phytother. Res., 2018, 32, 733–740 CrossRefCASPubMed.
Y. Youzhe, W. Chao and S. Jian, Chin. J. Org. Chem., 2013, 33, 159–163 CrossRef.
S. C. Bhrara, A. C. Jain and T. R. Seshadri, Tetrahedron, 1965, 21, 963–967 CrossRefCAS.
R. B. Gammill, Synthesis, 1979, 1979, 901–903 CrossRef.
G. Wei and B. Yu, Eur. J. Org Chem., 2008, 2008, 3156–3163 CrossRef.
A. Kumar and M. L. Rao, J. Chem. Sci., 2018, 130, 1–11 CrossRef.
N. Ngamrojanavanich, A. Loontaisong, S. Pengpreecha, W. Cherdshewasart, S. Pornpakakul, K. Pudhom, S. Roengsumran and A. Petsom, J. Ethnopharmacol., 2007, 109, 354–358 CrossRefCASPubMed.
V.-A. Nchiozem-Ngnitedem, E. Sperlich, V. Y. Matieta, J. R. Ngnouzouba Kuete, V. Kuete, E. A. Omer, T. Efferth and B. Schmidt, J. Nat. Prod., 2023, 86, 1520–1528 CrossRefCASPubMed.
G. Kwesiga, J. Greese, A. Kelling, E. Sperlich and B. Schmidt, J. Org. Chem., 2023, 88, 1649–1664 CrossRefCASPubMed.
T. Okada, N. Ngoc Thanh Luan, R. Arata, S. Awale and N. Toyooka, ChemistrySelect, 2022, 7, e202201136 CrossRefCAS.
G. Tsuji, M. Yusa, S. Masada, H. Yokoo, J. Hosoe, T. Hakamatsuka, Y. Demizu and N. Uchiyama, Chem. Pharm. Bull., 2020, 68, 797–801 CrossRefCASPubMed.
R. Wang, R. Ma, K. Feng, H. Lu, W. Zhao and H. Jin, Pharmaceuticals, 2024, 17, 86 CrossRefCASPubMed.
S. Chansakaow, T. Ishikawa, K. Sekine, M. Okada, Y. Higuchi, M. Kudo and C. Chaichantipyuth, Planta Med., 2000, 66, 572–575 CrossRefCASPubMed.
F. Ito, M. Iwasaki, T. Watanabe, T. Ishikawa and Y. Higuchi, Org. Biomol. Chem., 2005, 3, 674–681 RSC.
S. Sun, D. F. Dibwe, M. J. Kim, A. M. Omar, N. D. Phan, H. Fujino, N. Pongterdsak, K. Chaithatwatthana, A. Phrutivorapongkul and S. Awale, Bioorg. Med. Chem. Lett., 2021, 40, 127967 CrossRefCASPubMed.
Y. Fu, J. Chen, Y.-J. Li, Y.-F. Zheng and P. Li, Food Chem., 2013, 141, 1063–1071 CrossRefCASPubMed.
F. Bao, H.-Y. Bai, Z.-R. Wu and Z.-G. Yang, Nat. Prod. Res., 2021, 35, 562–569 CrossRefCASPubMed.
A. K. Singhal, R. P. Sharma, G. Thyagarajan, W. Herz and S. V. Govindan, Phytochemistry, 1980, 19, 929–934 CrossRefCAS.
N. T. Hiep, J. Kwon, D.-W. Kim, B. Y. Hwang, H.-J. Lee, W. Mar and D. Lee, Phytochemistry, 2015, 111, 141–148 CrossRefCASPubMed.
Q. Lu, D. S. Harmalkar, G. Quan, H. Kwon, J. Cho, Y. Choi, D. Lee and K. Lee, J. Nat. Prod., 2021, 84, 1359–1365 CrossRefCASPubMed.
W. Lwande, C. S. Greene and M. D. Bentley, J. Nat. Prod., 1985, 48, 1004–1005 CrossRefCAS.
A. Pelter and P. Stainton, J. Chem. Soc. C, 1966, 701–704, 10.1039/J39660000701.
S. Cheenpracha, R. Chokchaisiri, S. Laphookhieo, T. Limtharakul and C. Thepmalee, RSC Adv., 2022, 12, 30359–30364 RSC.
M. L. Wolfrom, W. D. Harris, G. F. Johnson, J. E. Mahan, S. M. Moffett and B. Wildi, J. Am. Chem. Soc., 1946, 68, 406–418 CrossRefCASPubMed.
G. D. Monache, R. Scurria, A. Vitali, B. Botta, B. Monacelli, G. Pasqua, C. Palocci and E. Cernia, Phytochemistry, 1994, 37, 893–898 CrossRef.
A. Raksat, W. Maneerat, N. Rujanapun, R. J. Andersen, S. G. Pyne and S. Laphookhieo, J. Nat. Prod., 2019, 82, 2343–2348 CrossRefCASPubMed.
M. Harfenist and E. Thom, J. Org. Chem., 1972, 37, 841–848 CrossRefCAS.
Q. Sun and G. Chou, PLoS One, 2015, 10, e0135893 CrossRefPubMed.
Q.-H. Sun, Y. Zhang and G.-X. Chou, Sci. Rep., 2017, 7, 46735 CrossRefPubMed.
H. Sun, Y. N. Lei, W. Deng, H. M. Wang, Y. O. Teng, H. Y. Zhao, Q. W. Yao and P. Yu, Chem. Nat. Compd., 2016, 52, 896–898 CrossRefCAS.
T. Miyase, M. Sano, K. Yoshino and K. Nonaka, Phytochemistry, 1999, 52, 311–319 CrossRefCASPubMed.
S. I. Khalivulla, B. A. K. Reddy, D. Gunasekar, M. M. Murthy, T. P. Rao, A. Blond and B. Bodo, Nat. Prod. Commun., 2007, 2, 1934578X0700201112 CrossRef.
K. H. Almabruk, J. H. Chang and T. Mahmud, J. Nat. Prod., 2016, 79, 2391–2396 CrossRefCASPubMed.
B. Kang, T. Oe, Y. Shimizu and H. Takikawa, Tetrahedron, 2022, 113, 132714 CrossRefCAS.
Y. Uchida, B. Kang and H. Takikawa, Tetrahedron, 2023, 136, 133362 CrossRefCAS.
P. Gouda and S. K. Grover, Indian J. Chem., Sect B, 2001, 40, 142–144 Search PubMed.
T. Kim, H. S. Kim, J. Jang, D.-J. Kim, J. Lee, D. Lee and S.-H. Kim, Molecules, 2022, 27, 6660 CrossRefCASPubMed.
D. K. Singh, J. Kim, J.-H. Sung and I. Kim, Bull. Korean Chem. Soc., 2018, 39, 239–243 CrossRefCAS.
Q. Mei, C. Wang, Z. Zhao, W. Yuan and G. Zhang, Beilstein J. Org. Chem., 2015, 11, 1220–1225 CrossRefCASPubMed.
S. Rajaonson, O. M. Vandeputte, D. Vereecke, M. Kiendrebeogo, E. Ralambofetra, C. Stévigny, P. Duez, C. Rabemanantsoa, A. Mol, B. Diallo, M. Baucher and M. El Jaziri, Environ. Microbiol., 2011, 13, 1236–1252 CrossRefCASPubMed.
T. A. Engler, K. D. Combrink and J. P. Reddy, J. Chem. Soc., Chem. Commun., 1989, 454–455, 10.1039/C39890000454.
Y. Kohno, M. Koso, M. Kuse and H. Takikawa, Tetrahedron Lett., 2014, 55, 1826–1828 CrossRefCAS.
Q. Shen, Y. Dai, G. Wang, F. Yao, Y. Duan, H. Chen, W. Zhang, X. Zhang and X. Yao, Bioorg. Med. Chem., 2014, 22, 2671–2677 CrossRefCASPubMed.
M. Luo, S. Delaplane, A. Jiang, A. Reed, Y. He, M. Fishel, R. L. Nyland II, R. F. Borch, X. Qiao, M. M. Georgiadis and M. R. Kelley, Antioxid. Redox Signaling, 2008, 10, 1853–1867 CrossRefCASPubMed.
A. Y. Dubovtsev, N. V. Shcherbakov, D. V. Dar'in and V. Y. Kukushkin, J. Org. Chem., 2020, 85, 745–757 CrossRefCASPubMed.
D. G. Twigg, L. Baldassarre, E. C. Frye, W. R. J. D. Galloway and D. R. Spring, Org. Lett., 2018, 20, 1597–1599 CrossRefCASPubMed.
N. Malik and P. Erhardt, Tetrahedron Lett., 2013, 54, 4121–4124 CrossRefCAS.
M. Versteeg, B. C. B. Bezuidenhoudt, D. Ferreira and K. J. Swart, J. Chem. Soc., Chem. Commun., 1995, 1317–1318, 10.1039/C39950001317.
J. L. Vicario, D. Badía and L. Carrillo, Tetrahedron: Asymmetry, 2003, 14, 489–495 CrossRefCAS.
J. M. Heemstra, S. A. Kerrigan, D. R. Doerge, W. G. Helferich and W. A. Boulanger, Org. Lett., 2006, 8, 5441–5443 CrossRefCASPubMed.
Y. Takashima, Y. Kaneko and Y. Kobayashi, Tetrahedron, 2010, 66, 197–207 CrossRefCAS.
M. P. Carroll, H. Müller-Bunz and P. J. Guiry, Chem. Commun., 2012, 48, 11142–11144 RSC.
G. F. Marrian and G. A. Haslewood, Biochem. J., 1932, 26, 1227–1232 CrossRefCASPubMed.
J. P. Yuan, J. H. Wang and X. Liu, Mol. Nutr. Food Res., 2007, 51, 765–781 CrossRefCASPubMed.
K. Nakamura, K. Ohmori and K. Suzuki, Chem. Commun., 2015, 51, 7012–7014 RSC.
H.-H. Zhang, J.-J. Zhao and S. Yu, J. Am. Chem. Soc., 2018, 140, 16914–16919 CrossRefCASPubMed.
J. Zhang, S. Zhang, H. Yang, D. Zhou, X. Yu, W. Wang and H. Xie, Tetrahedron Lett., 2018, 59, 2407–2411 CrossRefCAS.
M. Umeda, K. Sakamoto, T. Nagai, M. Nagamoto, Y. Ebe and T. Nishimura, Chem. Commun., 2019, 55, 11876–11879 RSC.
T. Qin and P. Metz, Org. Lett., 2017, 19, 2981–2984 CrossRefCASPubMed.
C. Yalamanchili, A. G. Chittiboyina, S. Chandra Kumar Rotte, J. A. Katzenellenbogen, W. G. Helferich and I. A. Khan, Tetrahedron, 2018, 74, 2020–2029 CrossRefCAS.
A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 4260–4263 CrossRefCASPubMed.
J. C. Conrad, J. Kong, B. N. Laforteza and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 11640–11641 CrossRefCASPubMed.
J. Jang, M. Na, P. T. Thuong, D. Njamen, J. T. Mbafor, Z. T. Fomum, E.-R. Woo and W. K. Oh, Chem. Pharm. Bull., 2008, 56, 85–88 CrossRefCASPubMed.
J. Zsindely and H. Schmid, Helv. Chim. Acta, 1968, 51, 1510–1514 CrossRefCAS.
A. Goel, A. Kumar and A. Raghuvanshi, Chem. Rev., 2013, 113, 1614–1640 CrossRefCASPubMed.
Z. Chen, M. Pitchakuntla and Y. Jia, Nat. Prod. Rep., 2019, 36, 666–690 RSC.
G. Huang, V.-H. Hoang, H.-Y. Min, H.-Y. Lee, J. Ann and J. Lee, Bioorg. Med. Chem. Lett., 2023, 91, 129353 CrossRefCASPubMed.
S. G. Pueppke and H. D. VanEtten, J. Chem. Soc., Perkin Trans., 1975, 946–948, 10.1039/P19750000946.
S. Kakuda, M. Ninomiya, K. Tanaka and M. Koketsu, ChemistrySelect, 2016, 1, 4203–4208 CrossRefCAS.
A. J. Brink, G. J. H. Rall and J. P. Engelbrecht, Phytochemistry, 1974, 13, 1581–1585 CrossRefCAS.
I. Kitagawa, W.-Z. Chen, K. Hori, E. Harada, N. Yasuda, M. Yoshikawa and J. Ren, Chem. Pharm. Bull., 1994, 42, 1056–1062 CrossRefCASPubMed.
R. S. Khupse and P. W. Erhardt, Org. Lett., 2008, 10, 5007–5010 CrossRefCASPubMed.
R. S. Khupse, J. G. Sarver, J. A. Trendel, N. R. Bearss, M. D. Reese, T. E. Wiese, S. M. Boue, M. E. Burow, T. E. Cleveland, D. Bhatnagar and P. W. Erhardt, J. Med. Chem., 2011, 54, 3506–3523 CrossRefCASPubMed.
Y.-F. Zhang, Z.-X. Zhu, H. Sun, H.-N. Yao, X.-N. Chen, L. Liu, S.-L. Zhang, Y.-F. Zhao, P.-F. Tu and J. Li, Tetrahedron Lett., 2018, 59, 4514–4516 Search PubMed.
Y. Kawamoto, T. Kobayashi and H. Ito, Org. Lett., 2021, 23, 3864–3867 Search PubMed.
K. Simin, Z. Ali, S. M. Khaliq-Uz-Zaman and V. U. Ahmad, Nat. Prod. Lett., 2002, 16, 351–357 Search PubMed.
J. Boonsombat, S. Thongnest and S. Ruchirawat, Eur. J. Med. Chem., 2019, 2971–2983 CAS.
W. Baker, J. B. Harborne and W. D. Ollis, J. Chem. Soc., 1953, 1860–1864, 10.1039/JR9530001860.
P. B. Anzeveno, J. Org. Chem., 1979, 44, 2578–2580 Search PubMed.
S. Xu, G. Wang, F. Xu, W. Li, A. Lin, H. Yao and J. Xu, J. Nat. Prod., 2018, 81, 1055–1059 Search PubMed.
D. A. Russell, J. J. Freudenreich, J. J. Ciardiello, H. F. Sore and D. R. Spring, Org. Biomol. Chem., 2017, 15, 1593–1596 RSC.
J. Garcia, S. Barluenga, K. Beebe, L. Neckers and N. Winssinger, Chem.–Eur. J., 2010, 16, 9767–9771 Search PubMed.
S. Lee, H. An, D.-J. Chang, J. Jang, K. Kim, J. Sim, J. Lee and Y.-G. Suh, Chem. Commun., 2015, 51, 9026–9029 RSC.
S. J. Pastine and D. Sames, Org. Lett., 2003, 5, 4053–4055 CrossRefCASPubMed.
A. S. K. Tsang, A. Kapat and F. Schoenebeck, J. Am. Chem. Soc., 2016, 138, 518–526 Search PubMed.
P. Khorphueng, J. Tummatorn, A. Petsom, R. J. K. Taylor and S. Roengsumran, Tetrahedron Lett., 2006, 47, 5989–5991 CrossRefCAS.
D. A. Russell, J. J. Freudenreich, H. L. Stewart, A. D. Bond, H. F. Sore and D. R. Spring, Org. Biomol. Chem., 2018, 16, 6395–6398 RSC.
B. B. Snider and T. Kwon, J. Org. Chem., 1992, 57, 2399–2410 CrossRefCAS.
S. C. Pelly, S. Govender, M. A. Fernandes, H.-G. Schmalz and C. B. de Koning, J. Org. Chem., 2007, 72, 2857–2864 Search PubMed.
H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRefCAS.
W.-L. Lo, C.-C. Wu, F.-R. Chang, W.-Y. Wang, A. T. Khalil, K.-H. Lee and Y.-C. Wu, Nat. Prod. Res., 2003, 17, 91–97 Search PubMed.
Q. Y. Shou, Q. Tan and Z. W. Shen, Bioorg. Med. Chem. Lett., 2009, 19, 3389–3391 CrossRefCASPubMed.
J. Yao, Z. Wang, R. Wang, Y. Wang, J. Xu and X. He, Bioorg. Chem., 2021, 113, 104996 CrossRefCASPubMed.
J. Yao, Q. Qin, Y. Wang, J. Zeng, J. Xu and X. He, Phytochemistry, 2022, 202, 113313 CrossRefCASPubMed.
M. Chubachi, M. Hamada and E. Kawano, Agric. Biol. Chem., 1983, 47, 619 Search PubMed.
A. B. C. Simas, L. F. O. Furtado and P. R. R. Costa, Tetrahedron Lett., 2002, 43, 6893–6895 CrossRefCAS.
A. J. Tilley, S. D. Zanatta, C. X. Qin, I.-K. Kim, Y.-M. Seok, A. Stewart, O. L. Woodman and S. J. Williams, Bioorg. Med. Chem., 2012, 20, 2353–2361 CrossRefCASPubMed.
S. Strych and D. Trauner, Angew. Chem., Int. Ed., 2013, 52, 9509–9512 CrossRefCASPubMed.
S. Strych, G. Journot, R. P. Pemberton, S. C. Wang, D. J. Tantillo and D. Trauner, Angew. Chem., Int. Ed., 2015, 54, 5079–5083 CrossRefCASPubMed.
E. S. B. Ferreira, A. N. Hulme, H. McNab and A. Quye, Chem. Soc. Rev., 2004, 33, 329–336 Search PubMed.
Y. Yazaki, Nat. Prod. Commun., 2015, 10, 505–512 Search PubMed.
A. Arnone, L. Camarda, L. Merlini, G. Nasini and D. A. H. Taylor, J. Chem. Soc., Perkin Trans., 1977, 2116–2118, 10.1039/P19770002116.
A. Arnone, L. Camarda, L. Merlini and G. Nasini, J. Chem. Soc., Perkin Trans., 1975, 186–194, 10.1039/P19750000186.
A. Robertson and W. B. Whalley, J. Chem. Soc., 1954, 2794–2801, 10.1039/JR9540002794.
J. Kinjo, H. Uemura, T. Nohara, M. Yamashita, N. Marubayashi and K. Yoshihira, Tetrahedron Lett., 1995, 36, 5599–5602 CrossRefCAS.
S. H. Wunderlich and P. Knochel, Angew. Chem., Int. Ed., 2007, 46, 7685–7688 CrossRefCASPubMed.
J. Sheng, T. Xu, E. Zhang, X. Zhang, W. Wei and Y. Zou, J. Nat. Prod., 2016, 79, 2749–2753 CrossRefCASPubMed.
Q. Yan, Y. Jiang, X. Song, G. Lu, Q. Zhang, Z. Du, A. S. C. Chan and Y. Zou, J. Org. Chem., 2022, 87, 5785–5794 CrossRefCASPubMed.
T. Nehybova, J. Smarda and P. Benes, Anti-Cancer Agents Med. Chem., 2014, 14, 1351–1362 CrossRefCASPubMed.
N. T. T. Thuy, J.-E. Lee, H. M. Yoo and N. Cho, J. Nat. Prod., 2019, 82, 3025–3032 CrossRefCASPubMed.
L. Li, X. Deng, L. Zhang, P. Shu and M. Qin, Fitoterapia, 2011, 82, 615–619 CrossRefCASPubMed.
Y. Tu, L. Tan, T. Lu, K. Wang, H. Wang, B. Han, Y. Zhao, H. Chen, Y. Li, H. Chen, M. Chen and C. He, Biochem. Pharmacol., 2022, 197, 114912 CrossRefCASPubMed.
L. C. Diogo, R. S. Fernandes, S. Marcussi, D. L. Menaldo, P. G. Roberto, P. V. F. Matrangulo, P. S. Pereira, S. C. França, S. Giuliatti, A. M. Soares and M. V. Lourenço, Basic Clin. Pharmacol. Toxicol., 2009, 104, 293–299 CrossRefCASPubMed.
X. Liu, J.-W. Nam, Y. S. Song, A. N. I. Viswanath, A. N. Pae, Y.-S. Kil, H.-D. Kim, J. H. Park, E.-K. Seo and M. Chang, Bioorg. Med. Chem. Lett., 2014, 24, 1403–1406 CrossRefCASPubMed.
M. M. Naik, V. P. Kamat and S. G. Tilve, Tetrahedron, 2017, 73, 5528–5536 CrossRefCAS.
X. Song, X. Luo, J. Sheng, J. Li, Z. Zhu, Z. Du, H. Miao, M. Yan, M. Li and Y. Zou, RSC Adv., 2019, 9, 17391–17398 RSC.
K. Neog, A. Borah and P. Gogoi, J. Org. Chem., 2016, 81, 11971–11977 CrossRefCASPubMed.
H. Gou, J. Zhang, P. Li, C. Li, H. Wang and W. Hong, Synth. Commun., 2023, 53, 1126–1133 CrossRefCAS.
M. Nayak, Y. Jung and I. Kim, Org. Biomol. Chem., 2016, 14, 8074–8087 RSC.
P. Pahari, U. P. Saikia, T. P. Das, C. Damodaran and J. Rohr, Tetrahedron, 2016, 72, 3324–3334 CrossRefCASPubMed.
H. Huang, J. Chen, J. Ren, C. Zhang and F. Ji, Molecules, 2019, 24, 4130 CrossRefCASPubMed.
L. Tang, Y. Pang, Q. Yan, L. Shi, J. Huang, Y. Du and K. Zhao, J. Org. Chem., 2011, 76, 2744–2752 CrossRefCASPubMed.
Z. Maeno, M. Yamamoto, T. Mitsudome, T. Mizugaki and K. Jitsukawa, ChemistrySelect, 2019, 4, 11394–11397 CrossRefCAS.
Q. Zhang, H. Nie, K. Zhang, H. Huang and C. Song, Tetrahedron, 2022, 116, 132815 CrossRefCAS.
L. Zhang, T. Cao, H. Jiang and S. Zhu, Angew. Chem., Int. Ed., 2020, 59, 4670–4677 CrossRefCASPubMed.
K. Kurosawa and K. Nogami, Bull. Chem. Soc. Jpn., 1976, 49, 1955–1957 CrossRefCAS.
R. Mali and S. Tilve, Synth. Commun., 1990, 20, 1781–1791 CrossRefCAS.
D. H. Gong, C. Z. Li and C. Y. Yuan, Chin. J. Chem., 2001, 19, 522–527 CrossRefCAS.
A. M. Prendergast, Z. Zhang, Z. Lin and G. P. McGlacken, Dalton Trans., 2018, 47, 6049–6053 RSC.
Y. Ueta, K. Mikami and S. Ito, Angew. Chem., Int. Ed., 2016, 55, 7525–7529 CrossRefCASPubMed.
Y. Sugita, S. Yin and I. Yokoe, Heterocycles, 2000, 53, 2191–2199 CrossRefCAS.
Y. Igarashi, H. Kumazawa, T. Ohshima, H. Satomi, S. Terabayashi, S. Takeda, M. Aburada and K.-i. Miyamoto, Chem. Pharm. Bull., 2005, 53, 1088–1091 CrossRefCASPubMed.
C. C. Li, Z. X. Xie, Y. D. Zhang, J. H. Chen, Z. Yang and J. Org, Chem, 2003, 68, 8500–8504 CAS.
M. E. Dudley, M. M. Morshed and M. M. Hossain, in Org. Synth., 2009, pp. 172–180, DOI:10.1002/0471264229.os086.18.
I. Kim, S.-H. Lee and S. Lee, Tetrahedron Lett., 2008, 49, 6579–6584 CrossRefCAS.

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Siyanda T.
Mthembu
b and
Molahlehi S.
Sonopo
c
*a,
Siyanda T.
Mthembu
b and
Molahlehi S.
Sonopo
c



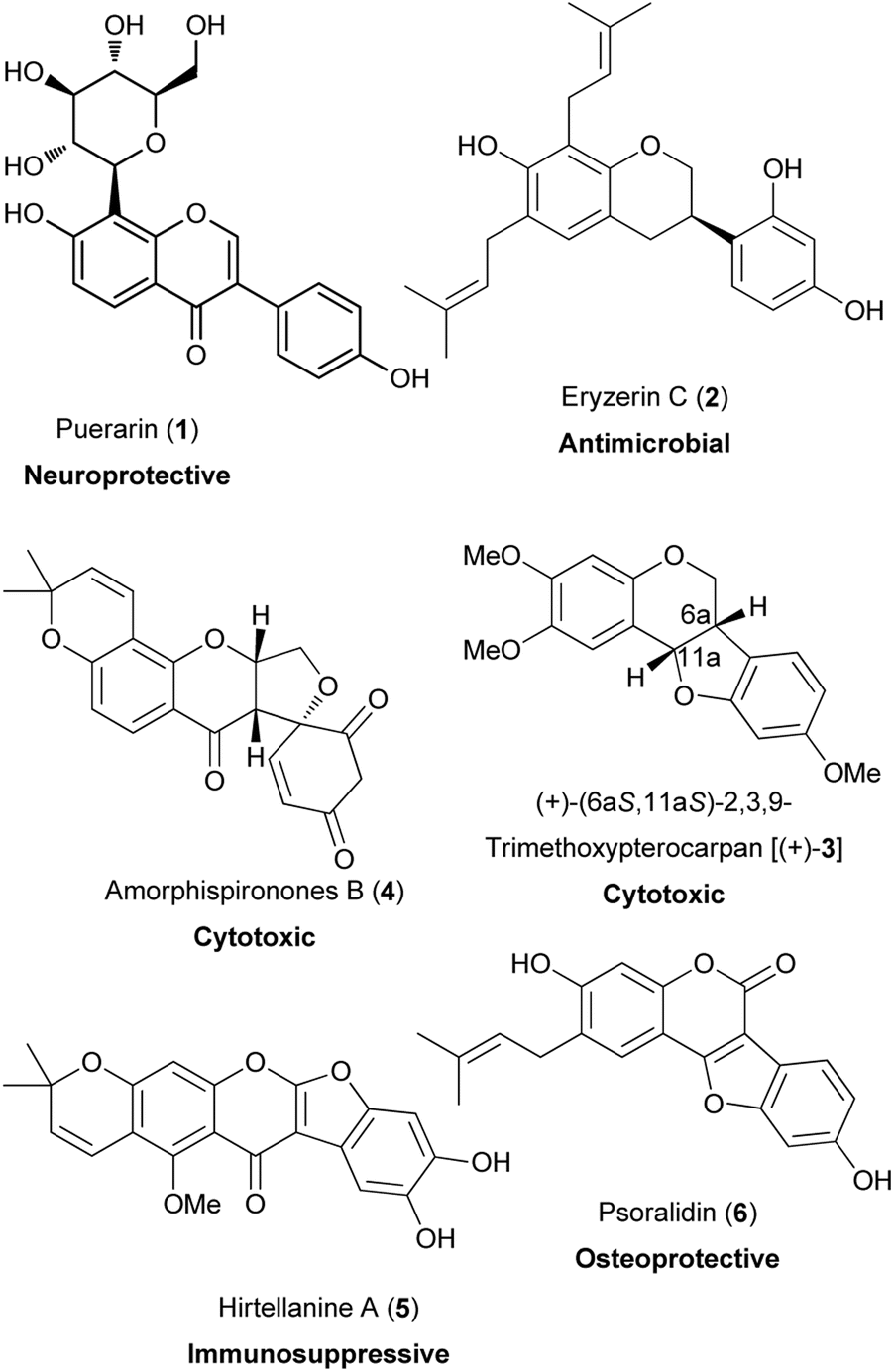
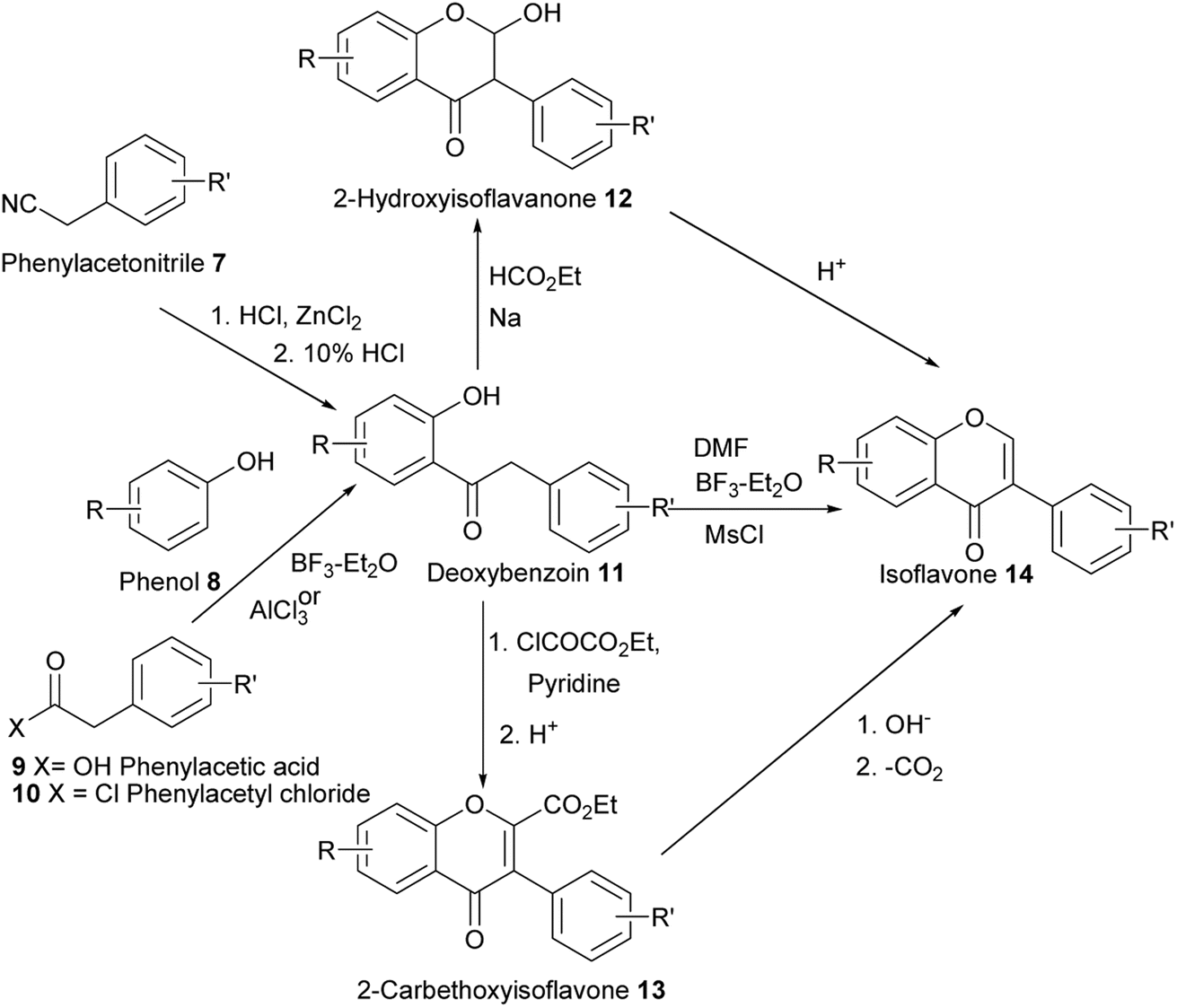
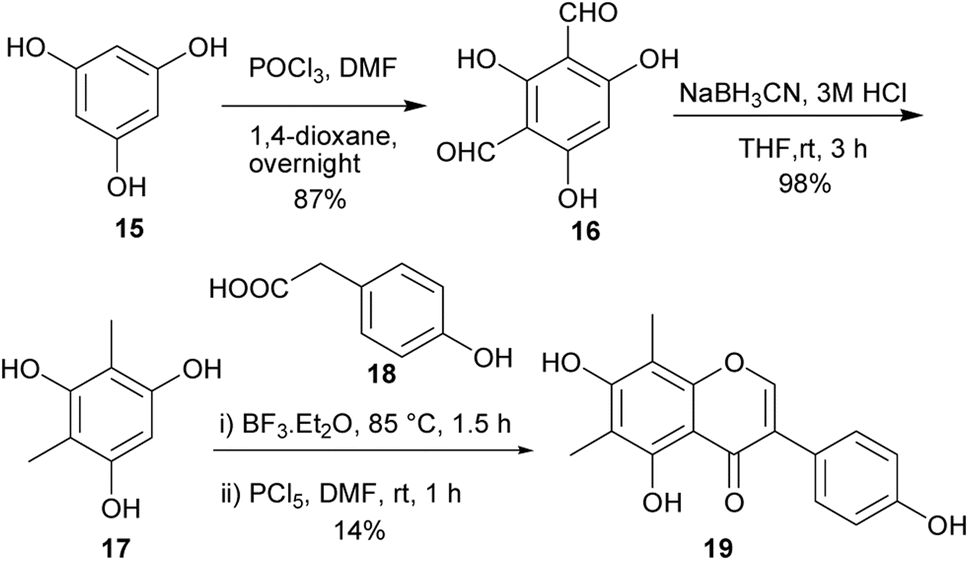
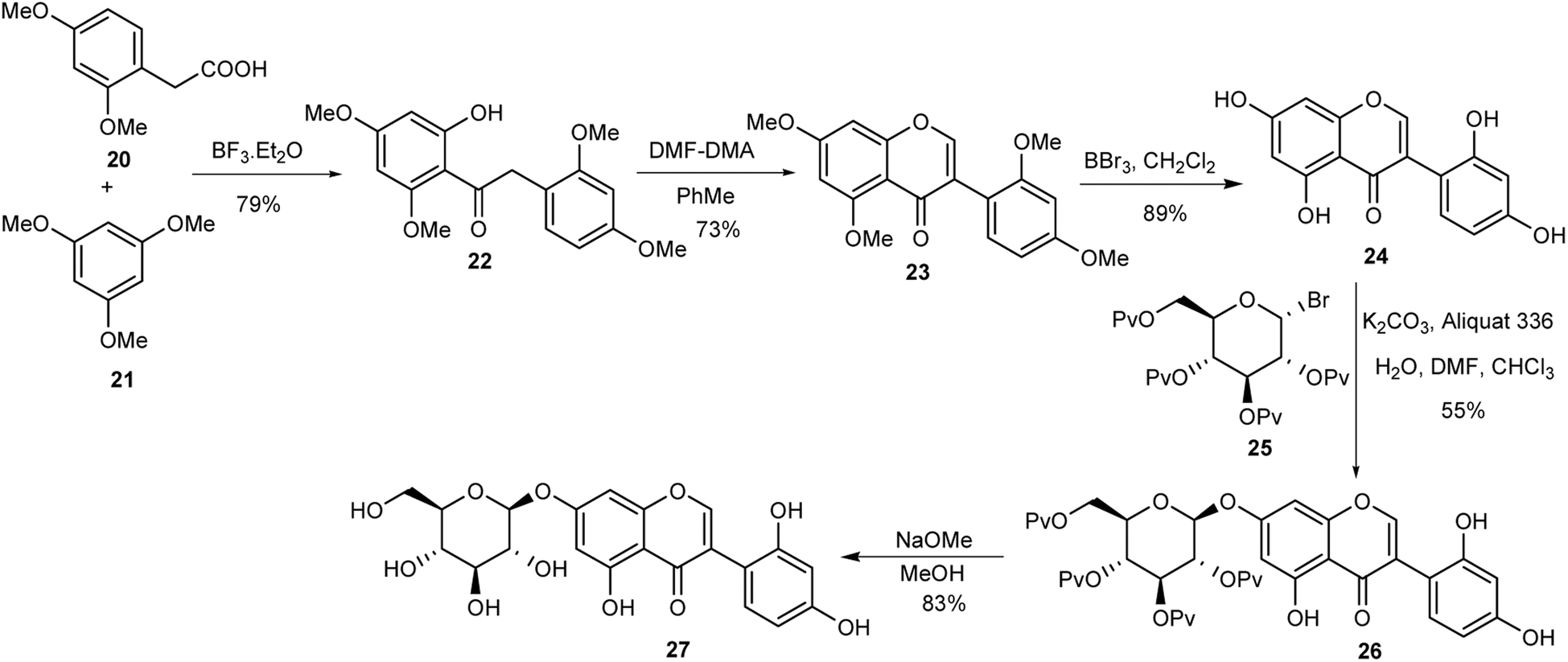
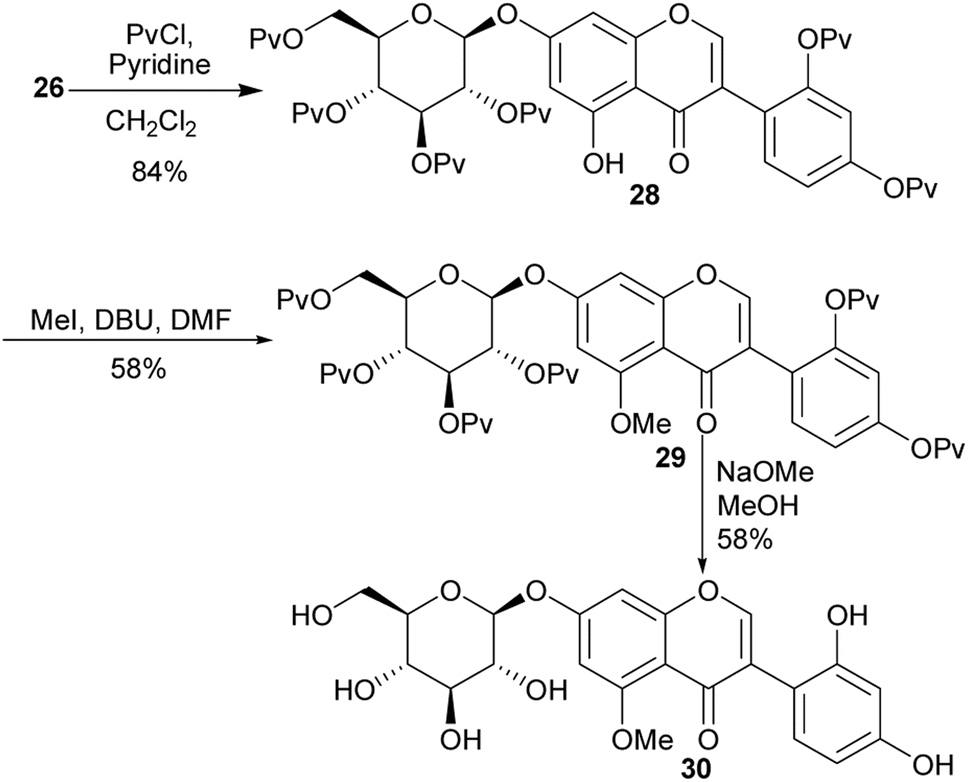
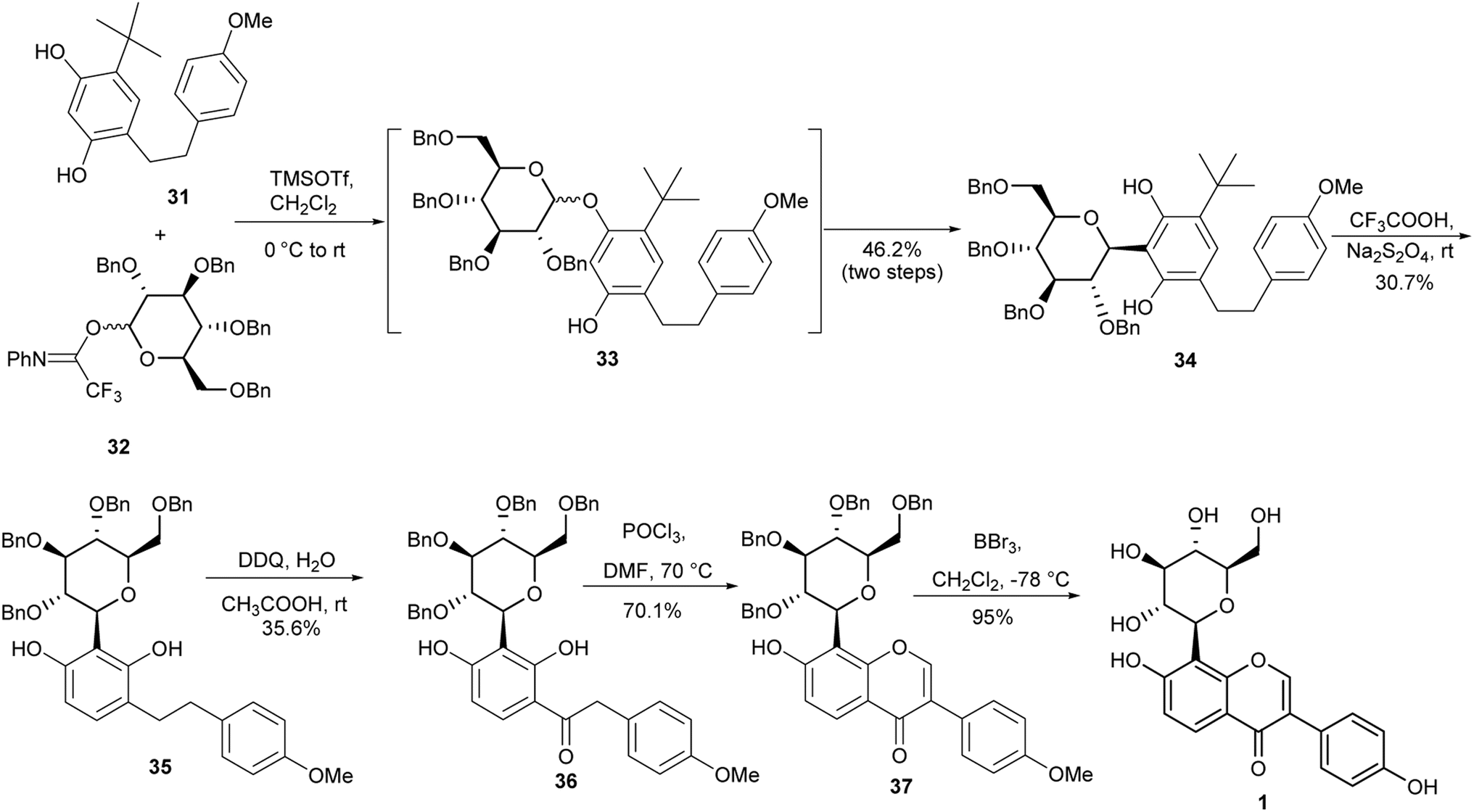
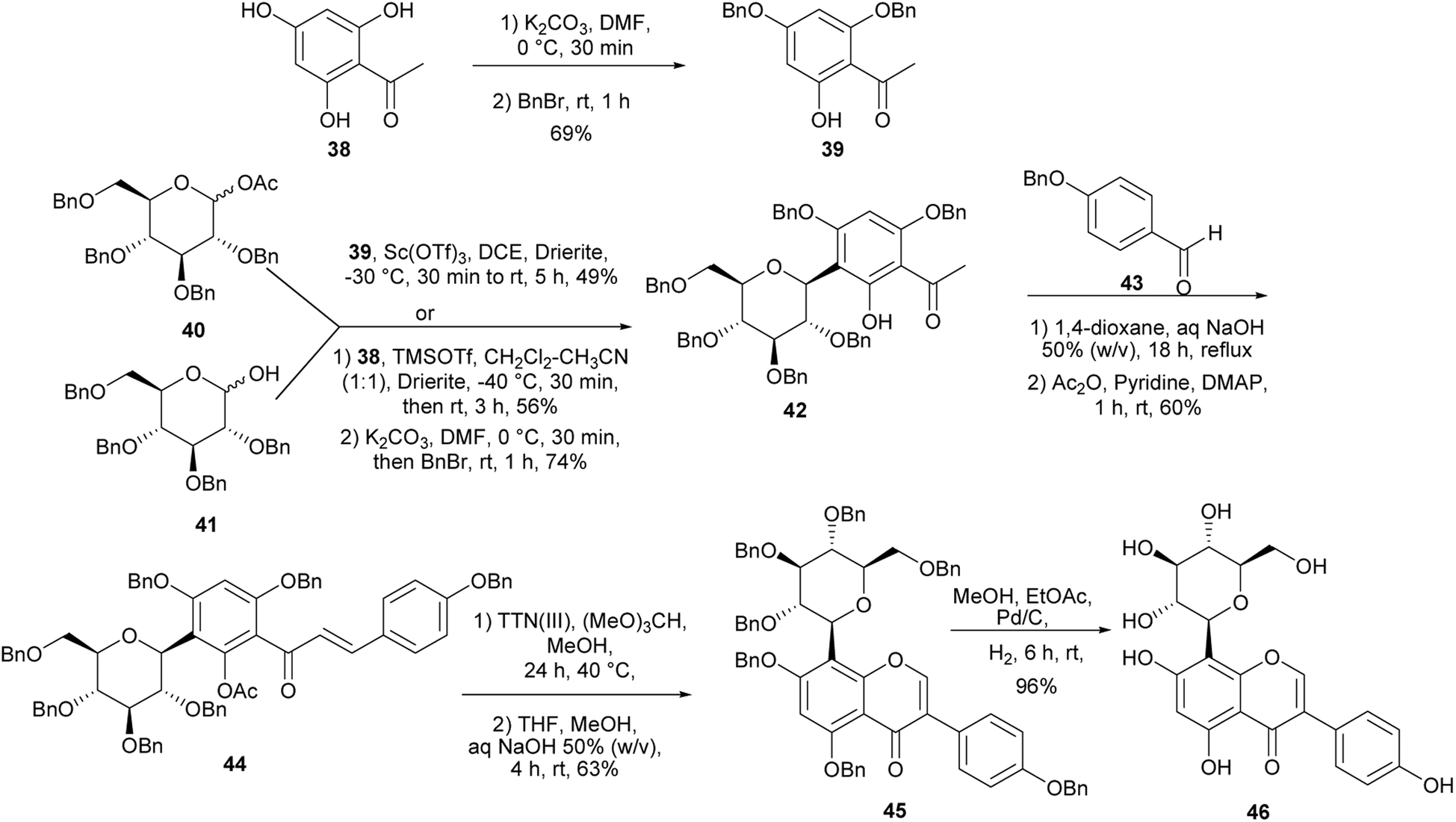
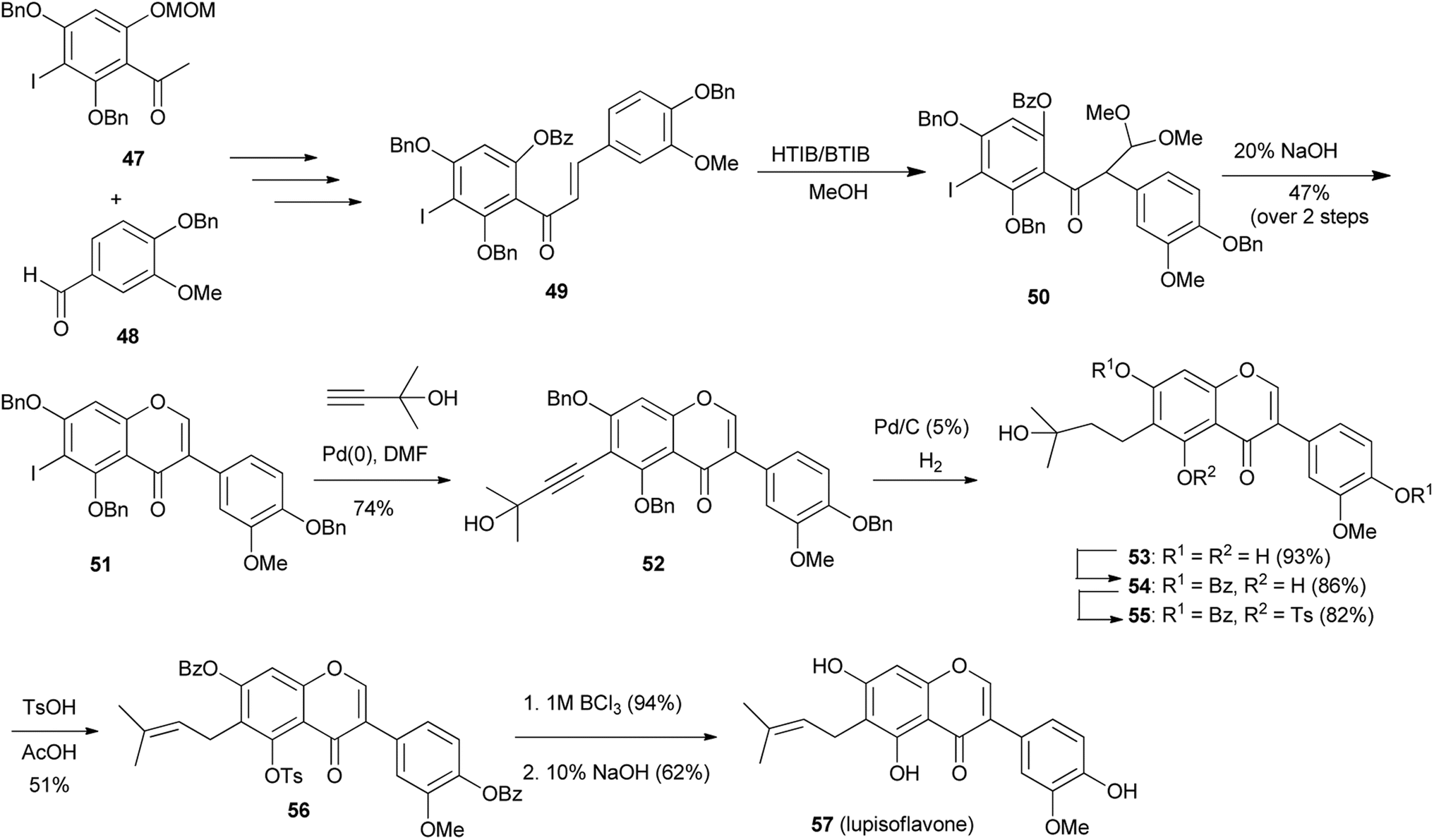
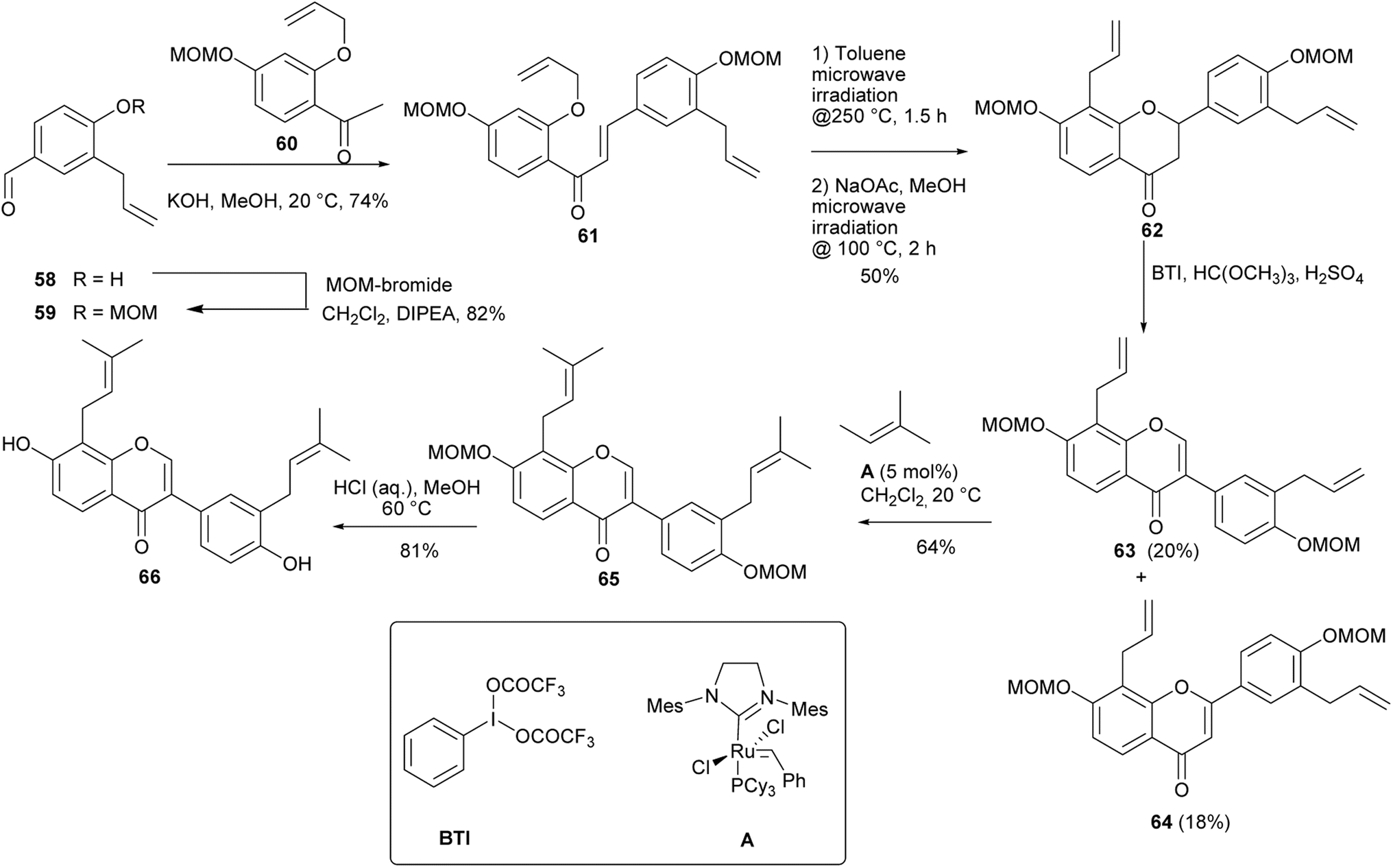
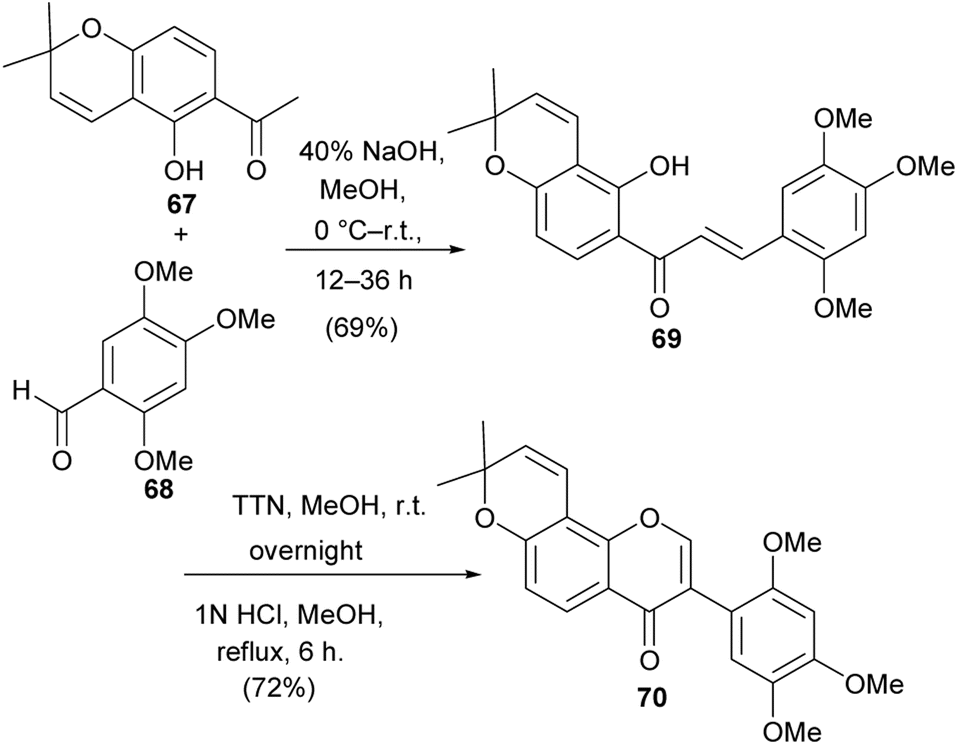
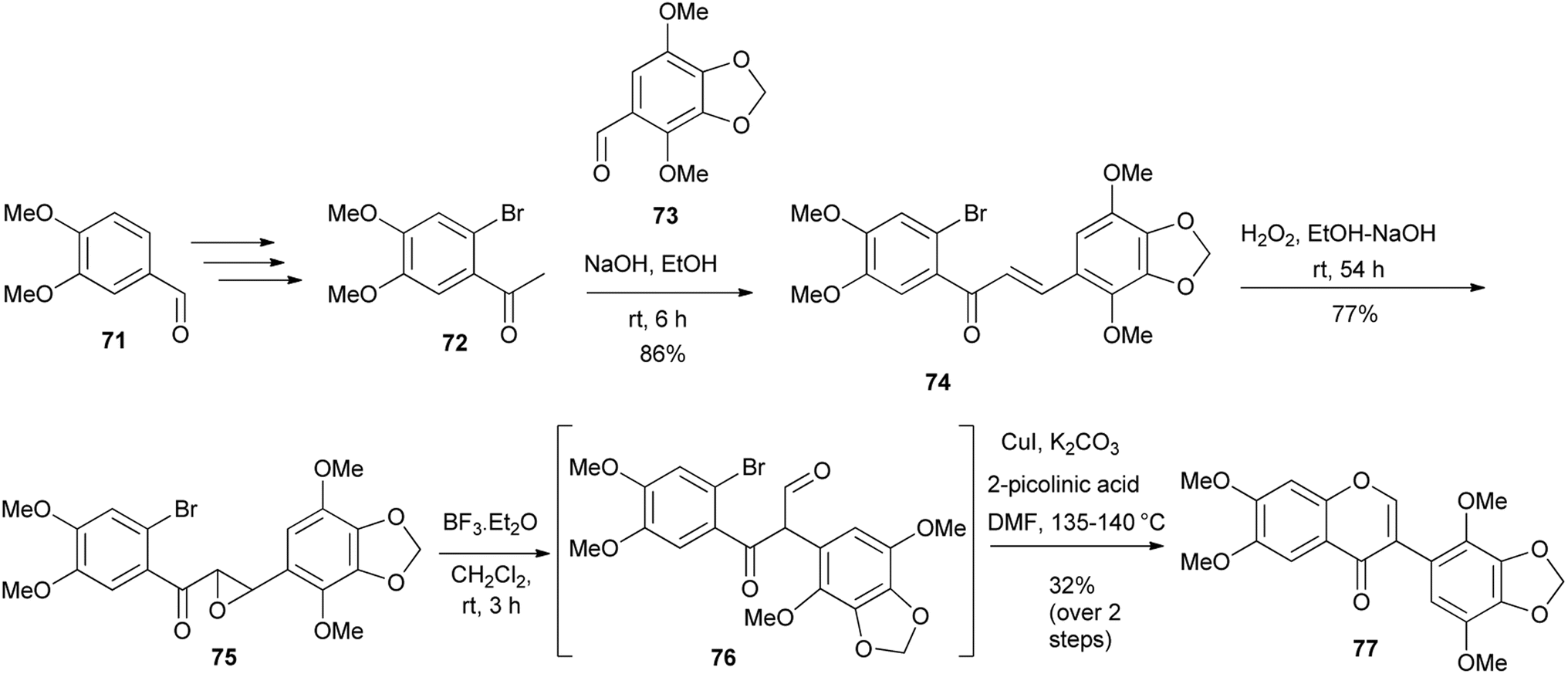
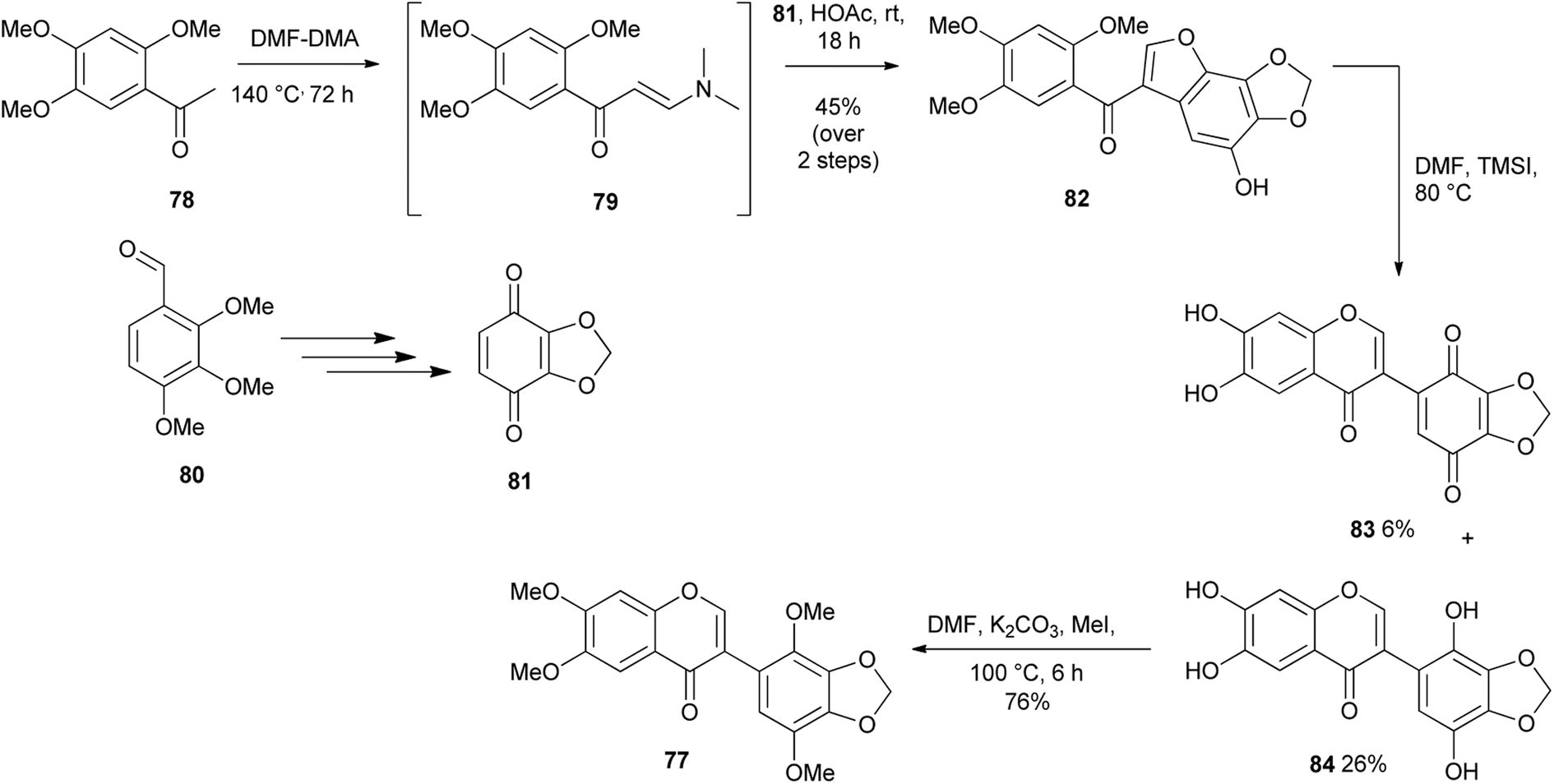
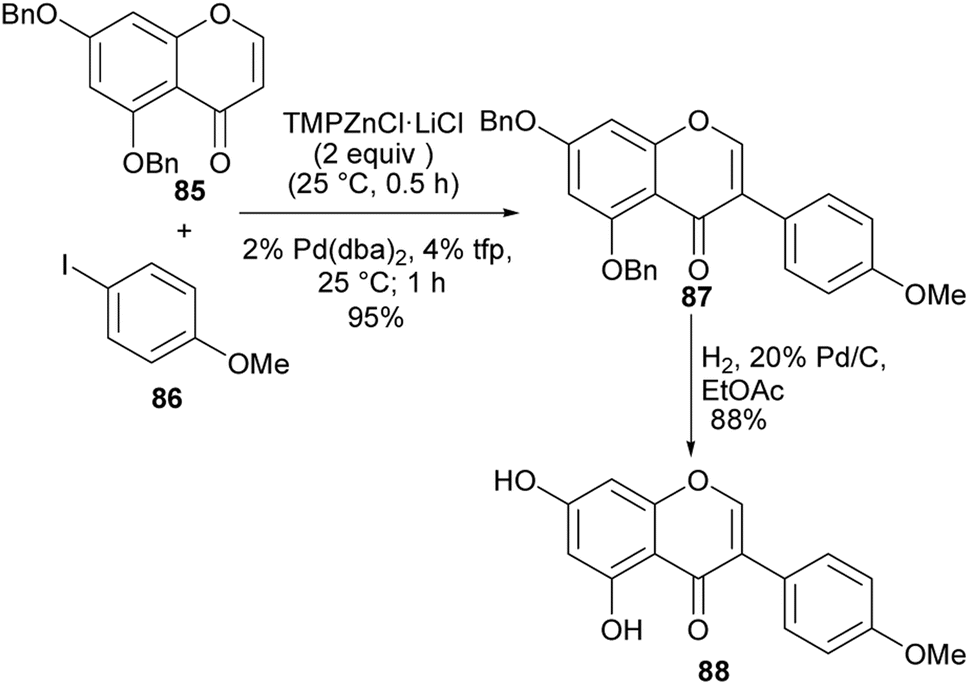
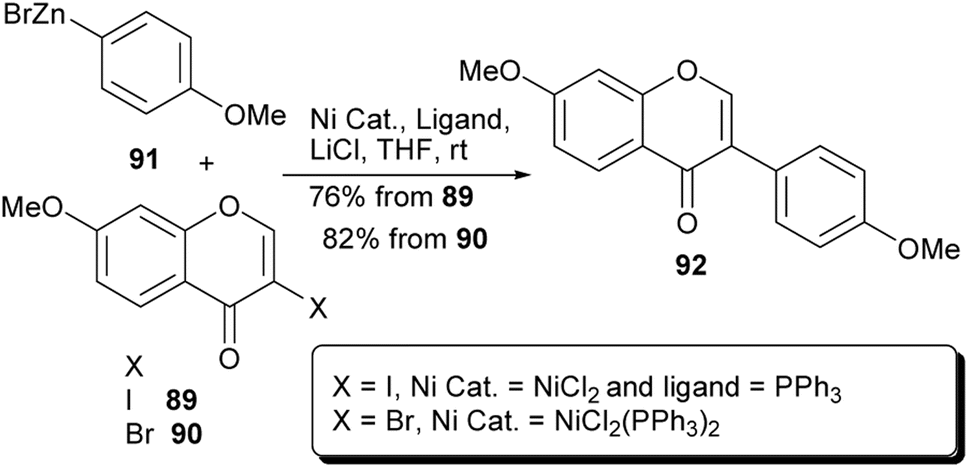
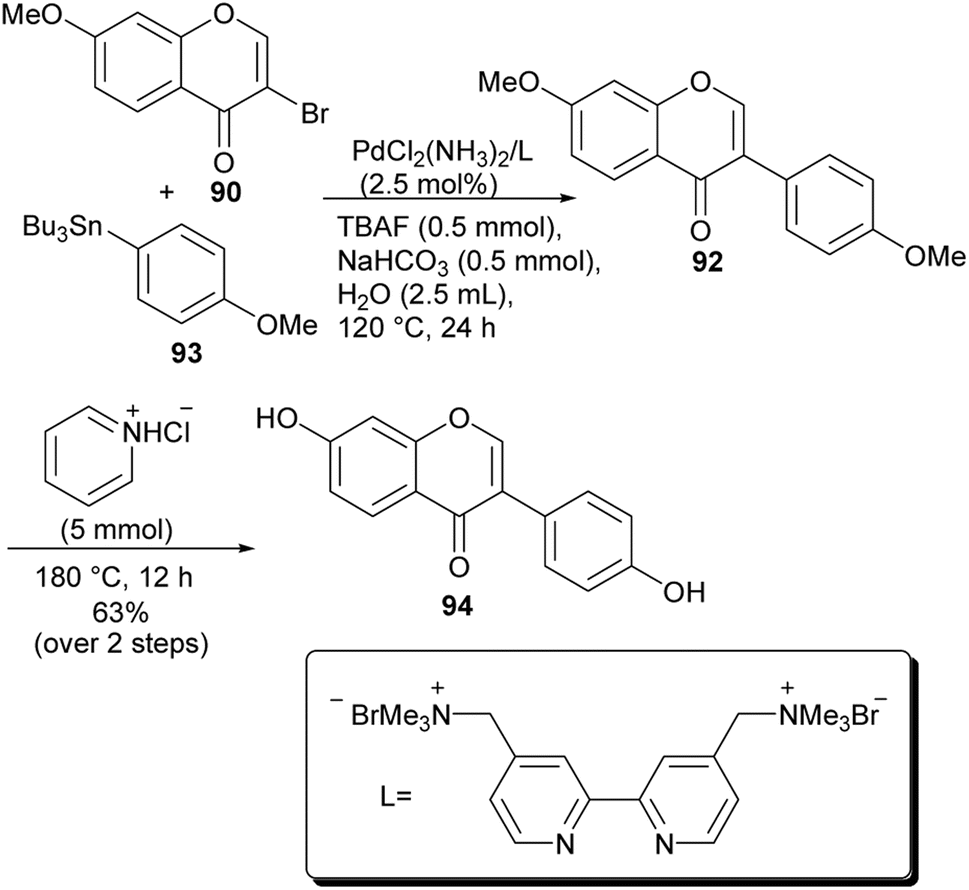
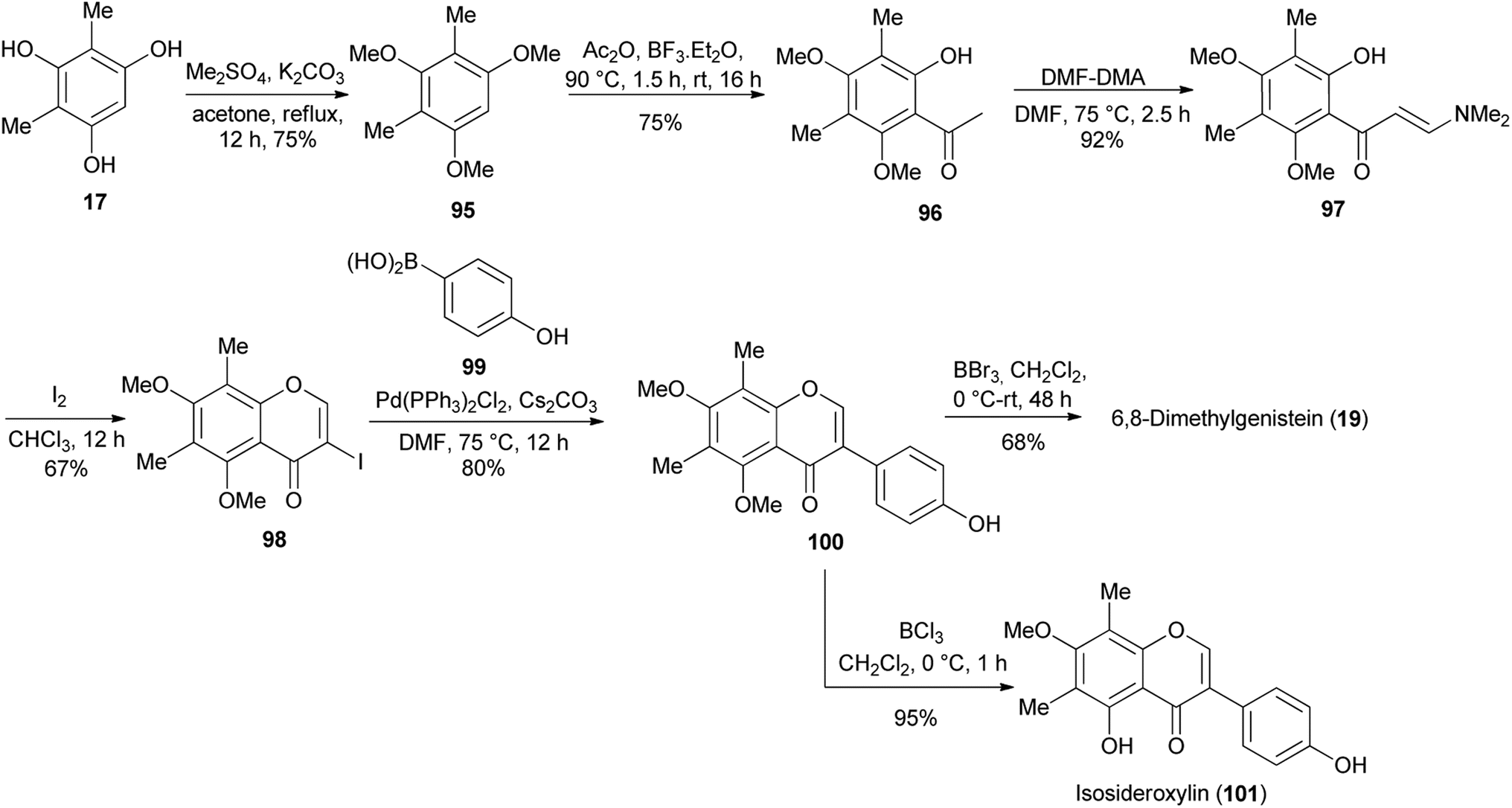
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 133 and a modified synthesis route was disclosed by Tsuji and co-workers in 2020.130 As illustrated in Scheme 16, the synthesis entailed coupling of bromophenol 105 (synthesised from aldehyde 104) with boronic acid 103 that was obtained from 3-bromochromone 102 to give isoflavone 106. Pd-catalysed O-allylation followed by Claisen rearrangement of the resulting 1,1-dimethylallyl ether 107, methylation and desilylation gave kwakhurin (108), Scheme 16.130,133
133 and a modified synthesis route was disclosed by Tsuji and co-workers in 2020.130 As illustrated in Scheme 16, the synthesis entailed coupling of bromophenol 105 (synthesised from aldehyde 104) with boronic acid 103 that was obtained from 3-bromochromone 102 to give isoflavone 106. Pd-catalysed O-allylation followed by Claisen rearrangement of the resulting 1,1-dimethylallyl ether 107, methylation and desilylation gave kwakhurin (108), Scheme 16.130,133