
Dynamic regulation of ion transport through a bis(1,3-propanediol)-based channel via allosteric azobenzene photoswitching†
Manzoor
Ahmad‡
 a,
Susmita
Sarkar
b,
Ravindra
Bhogade
a,
Abhishek
Mondal
ac,
Debashis
Mondal§
a,
Jagannath
Mondal
b and
Pinaki
Talukdar
*a
a,
Susmita
Sarkar
b,
Ravindra
Bhogade
a,
Abhishek
Mondal
ac,
Debashis
Mondal§
a,
Jagannath
Mondal
b and
Pinaki
Talukdar
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Pune, Dr Homi Bhabha Road, Pashan, Pune 411008, Maharashtra, India. E-mail: ptalukdar@iiserpune.ac.in
bCenter for Interdisciplinary Sciences, Tata Institute of Fundamental Research, Hyderabad 500046, Telangana, India
cLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands
First published on 21st November 2024
Abstract
The transportation of ions across cell membranes is vital in biological functions and is frequently controlled by external triggers like light, ligands, and voltage. Synthetic ion transport systems, particularly those featuring gating mechanisms, have attracted considerable interest. In this research, we engineered self-assembled barrel rosette ion channels using a photoresponsive azobenzene integrated at an allosteric site. Morphological studies verified more effective self-assembly of the trans form in contrast to the cis form. The restricted self-assembly of the cis form can be ascribed to the nonplanar structure of cis azobenzene moieties, which inhibits favorable π–π stacking interactions. The ion transport studies demonstrated the formation of ion channels by the trans form with anion antiport as the primary transport mechanism. In contrast, the cis form exhibited lower efficiency. Based on these observations, dynamically gated ion transport was achieved by employing two sets of electromagnetic radiation at 365 nm and 450 nm, respectively. Molecular dynamics simulation studies demonstrated that the channel formed by assembling trans monomers exhibited greater stability when compared to the channel formed by cis monomers. Additionally, the trans channel was found to recognize and transport chloride effectively.
Introduction
Artificially designed synthetic ion transport systems have emerged as an important class of compounds that have shown potential applications as therapeutics for cancer treatment and diseases that occur due to the malfunctioning of naturally occurring ion channels like cystic fibrosis.1–4 Although a wide variety of synthetic mimics in the form of ion channels5–7 and ion carriers8,9 have been developed, gated systems that promise better applications are very rare. Stimuli like light,10 enzymes,11 pH,12 voltage,13 ligands,14etc. are utilized to generate artificially gated ion transport systems. Light, because of its spatiotemporal control, remote addressability, tunable intensity, and less toxicity, is considered to be more effective compared to other stimuli.15 Different kinds of photosystems that have been employed include the use of photoswitches (e.g., azobenzene, acylhydrazone, phenylhydrazone, stilbene, spiropyrans, etc.)16–22 or photocleavable groups (e.g., o-nitro aromatics, bodipy, etc.).23,24 Azobenzene is one of the versatile photoswitches that is mostly used because of its spatial and temporal control, significant change in length and dipole moment upon cis–trans photoisomerization, and very quick response time of action.25 Most of these systems developed are photoresponsive ion carriers due to the lack of sophisticated designs for the construction of ion channels. Self-assembly is one of the ways to construct artificial ion channels by employing different non-covalent interactions like hydrogen bonding, π–π interactions, halogen bonding interactions, etc.26 Precisely, the incorporation of a photoswitch like azobenzene,27 acylhydrazone,28etc. into these motifs can provide an opportunity to externally control the self-assembly pattern and thereby control the ion transport activity across the lipid bilayer membrane. In this context, Linyong Zhu and colleagues employed an azobenzene subunit linked to crown ether groups to create cation channels arranged in parallel stacks within the lipid bilayer membrane, demonstrating photoresponsive characteristics.29 However, the construction of self-assembled photoresponsive anion channels poses a challenge due to the necessity for intricate design strategies and the absence of sophisticated functional units capable of forming anion channel structures within the lipid bilayer membrane.Our laboratory has recently revealed self-assembled ion channels originating from bis(1,3-diol) connected to a 1,3-diethynylbenzene core.30 These channels demonstrate noteworthy anion transport activity across the lipid bilayer membrane. The system employed intra- and inter-molecular hydrogen bonding interactions of 1,3-dihydroxyl moieties and π–π stacking interactions of 1,3-diethynylbenzene in stabilizing the ion channel. We predicted that customizing the core to include a photoswitch, specifically in the form of azobenzene, would offer light-responsive control over ion transport activity. Furthermore, introducing the azobenzene moiety at an allosteric site rather than within the channel cavity would not impede the ion transport process. The planarity of the trans form of 1,3-diethynylazobenzene was anticipated to enhance self-assembly, forming a stable ion channel through strong π–π stacking interactions among the azobenzene units. The assembly of such a channel could be disrupted upon exposure to 365 nm photoirradiation, primarily due to the photoisomerization of the planar trans-azobenzene to the nonplanar cis-azobenzene form (Fig. 1). The active form would further be reverted back upon photoirradiation at a different wavelength of light (450 nm). The incorporation of aliphatic chains into the azobenzene subunit was expected to provide lipophilic control and further strengthen the self-assembly behavior because of hydrophobic interactions.
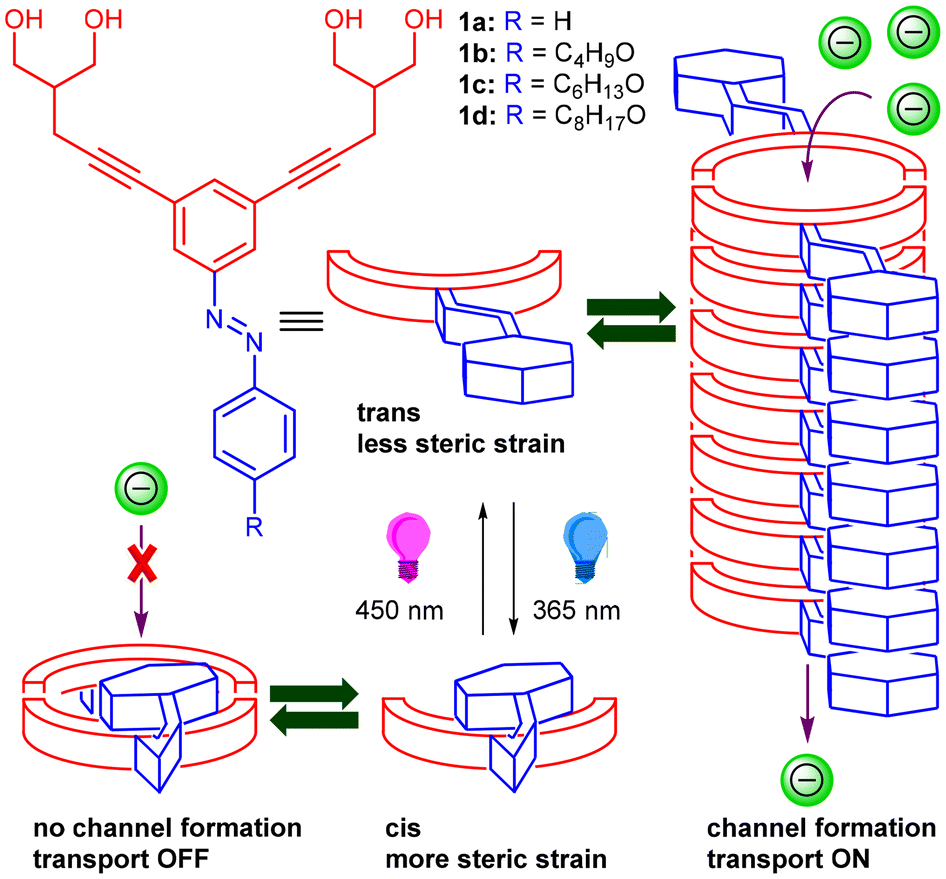 | ||
| Fig. 1 Schematic representation of the structures of the designed molecules and photoresponsive ion transport through azobenzene-based ion channel structures. | ||
Results and discussion
Synthesis
In order to synthesize the desired compounds 1a–1d, initially, 3,5-diiodoaniline 2 was coupled with nitrosobenzene 3 in the presence of glacial acetic acid to form diiodo azobenzene derivative 4 (Scheme 1). On the other hand, the diazotization reaction of 2 with phenol furnishes hydroxyl azobenzene 5 that, upon further reaction with different alkyl bromide compounds, furnishes azobenzene derivatives 6a–6c. Finally, diiodo compounds 4 and 6a–6c are coupled with 2-(prop-2-yn-1-yl) propane-1,3-diol 7 through the Sonagashira coupling reaction in the presence of Pd(PPh3)4 to furnish the desired final compounds 1a–1d in 68–80% yields. All the compounds were fully characterized by 1H NMR, 13C NMR, IR, and HRMS techniques (Fig. S25–S46†).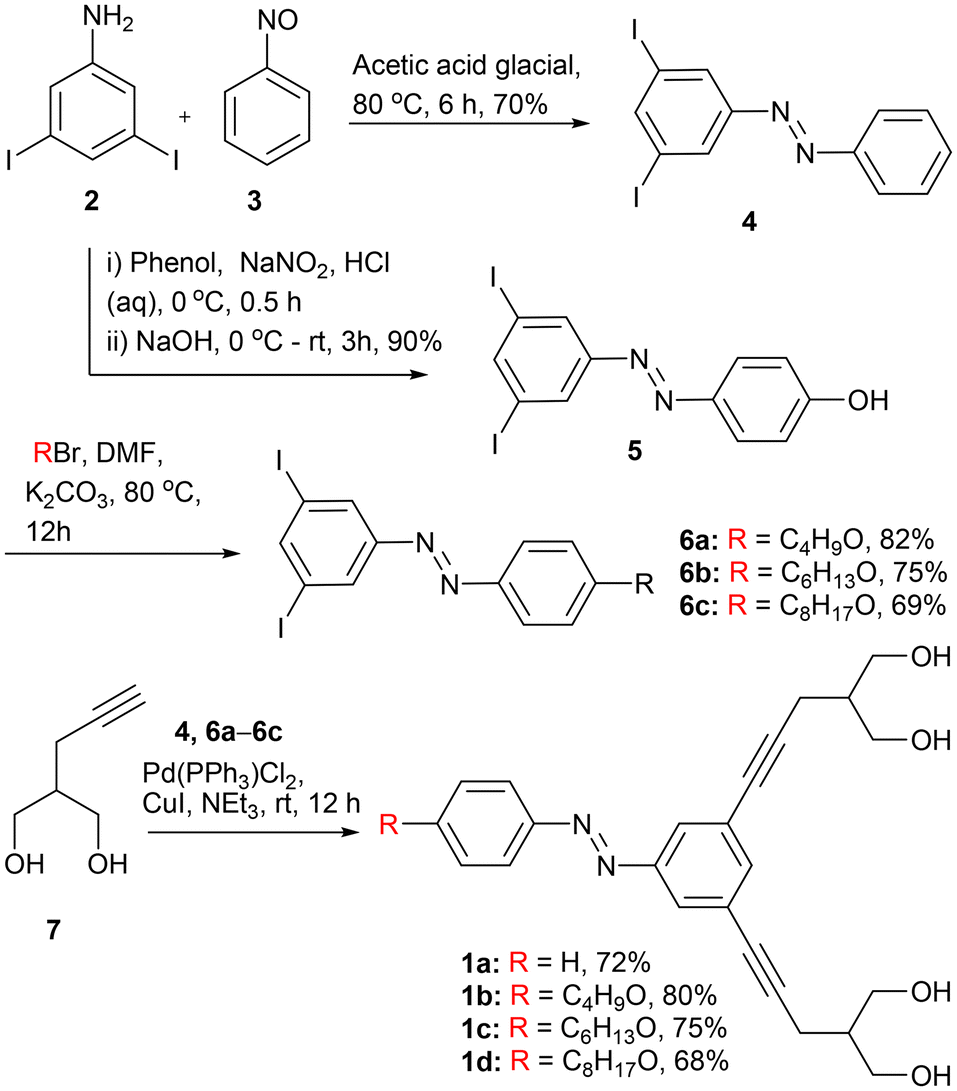 | ||
| Scheme 1 Chemical synthesis of channel forming molecules 1a–1d. | ||
Ion transport studies
After the synthesis of the desired compounds, the ion transport activity was evaluated. Initially, large unilamellar vesicles (LUVs) from egg-yolk phosphatidylcholine (EYPC) lipid entrapping 8-hydroxypyrene-1,3,6-trisulfonic acid trisodium salt (HPTS), 1 mM HPTS dye, and 100 mM NaCl were prepared and buffered at pH 7.0 using 10 mM HEPES buffer.31 The ion transport activity was monitored by recording the change in the fluorescence intensity of the HPTS dye λem = 510 nm (λex = 450 nm) occurring due to the disruption of the pH gradient created by adding an aqueous solution of sodium hydroxide in the external buffer. The ion transport activity sequence recorded at 10 μM concentration was found to be 1d > 1c > 1b > 1a (Fig. 2A). The above sequence suggests that long chain alkyl groups because of their higher lipophilicity would most likely result in better lipid permeation which eventually results in better ion transport activity. Dose-dependent studies furnished EC50 values of 1.74 μM and 2.39 μM for compounds 1d and 1c, respectively. The Hill coefficient was found to be n ≈ 1, indicating the formation of the stable ion channel structures. The EC50 values of compounds 1a and 1b could not be evaluated because of the precipitation issues at higher concentrations. | ||
| Fig. 2 Activity comparison of 1a–1d across EYPC-LUVs⊃HPTS (10.0 μM) (A). Transport activity of 1d (5.0 μM) by varying external anions across EYPC-LUVs⊃HPTS; each bar graph represents mean ion transport activity, calculated from three independent experiments (B). Transport activity of 1d (0.3 μM) measured by varying external cations (M+ = Li+, Na+, K+, Rb+, Cs+) across EYPC-LUVs⊃HPTS (C). Concentration-dependent activity of compound 1d across EYPC-LUVs⊃Lucigenin (D). Schematic representation of lucigenin-encapsulated antiport assay (E). Efflux of the Cl− ion by 1d (10.0 μM) in the presence of extravesicular SO42− or extravesicular NO3− ions (F). Normalized Cl− influx by 1d in the presence and absence of valinomycin (G). | ||
Mechanistically, the ion transport in the above HPTS-based studies could occur through (a) H+/X− symport, (b) OH−/X− antiport, (c) H+/M+ antiport, or (d) OH−/M+ symport modes. On varying the cations in the external buffer using different MCl salt solutions (M+ = Li+, Na+, K+, Rb+, and Cs+), no change in the ion transport activity was observed, indicating no role of cations in the transport process (Fig. 2C). However, upon varying anions in the external buffer, by using different NaX salt solutions (X− = Cl−, Br−, ClO4−, CN−, NO3− and I−), a significant change in the ion transport activity was observed with the activity sequence of Cl− > NO3− > Br− > I− > CN− > ClO4− (Fig. 2B and S5B†) indicating the active role of anions in the transport process. These studies ruled out H+/M+ antiport or OH−/M+ symport ion transport modes and indicate that the ion transport could potentially occur in either H+/X− symport or OH−/X− antiport mode.
Subsequently, chloride transport for the most active compound 1d was monitored across EYPC-LUVs⊃Lucigenin. Vesicles entrapping a lucigenin dye were prepared, and a Cl−/NO3− gradient was created across the lipid membrane.32 The transport of chloride was monitored by recording the time-dependent fluorescence quenching of the lucigenin dye at λem = 535 nm (λex = 455 nm). Significant fluorescence quenching was observed upon the addition of 1d (Fig. 2D). Hill analysis could not be performed because of the precipitation issues at higher concentrations. Mechanistically, the ion transport in the lucigenin assay could occur through (a) H+/Cl− symport or (b) Cl−/NO3− antiport mode, as the other modes involving the role of cations have been ruled out in the HPTS studies. A modified lucigenin assay was performed to obtain further insights into the mechanism of ion transport. The chloride influx across the lipid vesicles containing intravesicular NaNO3 (200 mM) and extravesicular KCl (200 mM) was monitored in the presence and absence of valinomycin (a highly selective K+ transporter).33 A manifold enhancement in the ion transport activity suggests the cooperative effect of valinomycin and 1d and thus indicates the Cl−/NO3− antiport mode as the primary mode of ion transport (Fig. 2G). No such cooperativity would have been possible in the case of H+/Cl− symport mode. The antiport mechanism was further validated by performing the modified lucigenin assay. A lucigenin dye was encapsulated within the liposomes containing NaCl (200 mM) buffered at a pH of 7.0. The effect of ion transport activity was monitored by the addition of either NaNO3 (200 mM) or Na2SO4 (200 mM) in the external buffer. Significant transport activity was observed in the presence of NaNO3, and no significant activity was observed in the presence of Na2SO4 (Fig. 2F). The above results suggest Cl−/NO3− antiport mode of ion transport rather than H+/Cl− symport because, in the case of H+/Cl− symport, no change should have occurred by changing the external buffer to either NaNO3 or Na2SO4. In the case of the antiport mechanism, SO42− being very hydrophilic and highly hydrated is not transported easily.
Planar bilayer conductance studies
In order to confirm the formation of an active ion channel across the lipid bilayer, electrical conductance was measured for the most active compound 1d.34,35 For that, a lipid composed of 1,2-diphytanoyl-sn-glycerol-3-phosphocholine (DPhPC) was used, which separates the 1.0 M KCl solution-filled cis and trans chambers. The treatment of 1d (2.0 μM) in the trans chamber, followed by the application of various holding potentials between the two compartments, produced significantly long single channel opening and closing events, demonstrating the development of stable ion channels by 1d at the planar bilayer (Fig. 3A, B and S21†). The single-channel conductance value, calculated using Hille's equation including the Sansom correction factor 5.61,36 was found to be 17.5 ± 0.8 pS, and the diameter of the channel was calculated to be 4.09 ± 0.19 Å. In the presence of the KCl symmetric solution (1.0 M each), current–voltage (I–V) graphs showed linear trends, indicating an ohmic characteristic for A (Fig. 3C). The non-dipolar structure of the channels accounts for this linearity. On the other hand, asymmetric KCl solutions were used to assess the ion selectivity of 1d (0.5 M in the trans chamber and 1.0 M KCl in the cis chamber). The calculation of the permeability ratio revealed PCl−/PK+ = 3.24, confirming the anion-selective nature of 1d. | ||
| Fig. 3 Current traces recorded with 1d (c = 2.0 μM) at −200 mV (A) and +200 mV (B) in a symmetrical KCl solution. Current–voltage relationship of 1d in both symmetrical and asymmetrical KCl concentrations (C). | ||
Photoisomerization studies
The photoisomerization studies of compounds 1a–1d were carried out through 1H NMR spectroscopy and UV-Vis absorption studies. Initially, 1H NMR spectra of the trans samples of 1a–1d were recorded in DMSO-d6. After that, each sample was photoirradiated at 365 nm for 20 min and NMR spectra were recorded again. The photoirradiation led to significant changes in the proton signals, indicating a trans-to-cis photoisomerization process with a trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis ratio of 08:92, 10:90, 12:88, and 11:89 for compounds 1a, 1b, 1c, and 1d, respectively at a photo-stationary state (PSS) (Fig. S14–S17†). UV-Vis absorption studies showed a distinct band at 355 nm corresponding to π–π* transition, which substantially decreased upon photoirradiation at 365 nm with the appearance of a new band at 450 nm corresponding to n–π* transition (Fig. S10A, S11A, S12A, and S13A†). These changes were consistent with the trans-to-cis photoisomerization process of the azobenzene subunit. The reverse cis-to-trans process was achieved by photoirradiating the cis compound at 450 nm of electromagnetic radiation (Fig. S10B, S11B, S12B, and S13B†). This reversible photoisomerization process was also achieved over three repeated cycles without any loss of efficiency (Fig. 4C, S10D, S11D, S12D and S13D†). Moreover, the cis form of 1d showed excellent thermal stability while keeping in the dark for 9 h. The thermal relaxation from cis-to-trans follows a first-order rate law with a half-life period of 113 ± 5 h (Fig. 4D and S18†).
:cis ratio of 08:92, 10:90, 12:88, and 11:89 for compounds 1a, 1b, 1c, and 1d, respectively at a photo-stationary state (PSS) (Fig. S14–S17†). UV-Vis absorption studies showed a distinct band at 355 nm corresponding to π–π* transition, which substantially decreased upon photoirradiation at 365 nm with the appearance of a new band at 450 nm corresponding to n–π* transition (Fig. S10A, S11A, S12A, and S13A†). These changes were consistent with the trans-to-cis photoisomerization process of the azobenzene subunit. The reverse cis-to-trans process was achieved by photoirradiating the cis compound at 450 nm of electromagnetic radiation (Fig. S10B, S11B, S12B, and S13B†). This reversible photoisomerization process was also achieved over three repeated cycles without any loss of efficiency (Fig. 4C, S10D, S11D, S12D and S13D†). Moreover, the cis form of 1d showed excellent thermal stability while keeping in the dark for 9 h. The thermal relaxation from cis-to-trans follows a first-order rate law with a half-life period of 113 ± 5 h (Fig. 4D and S18†).
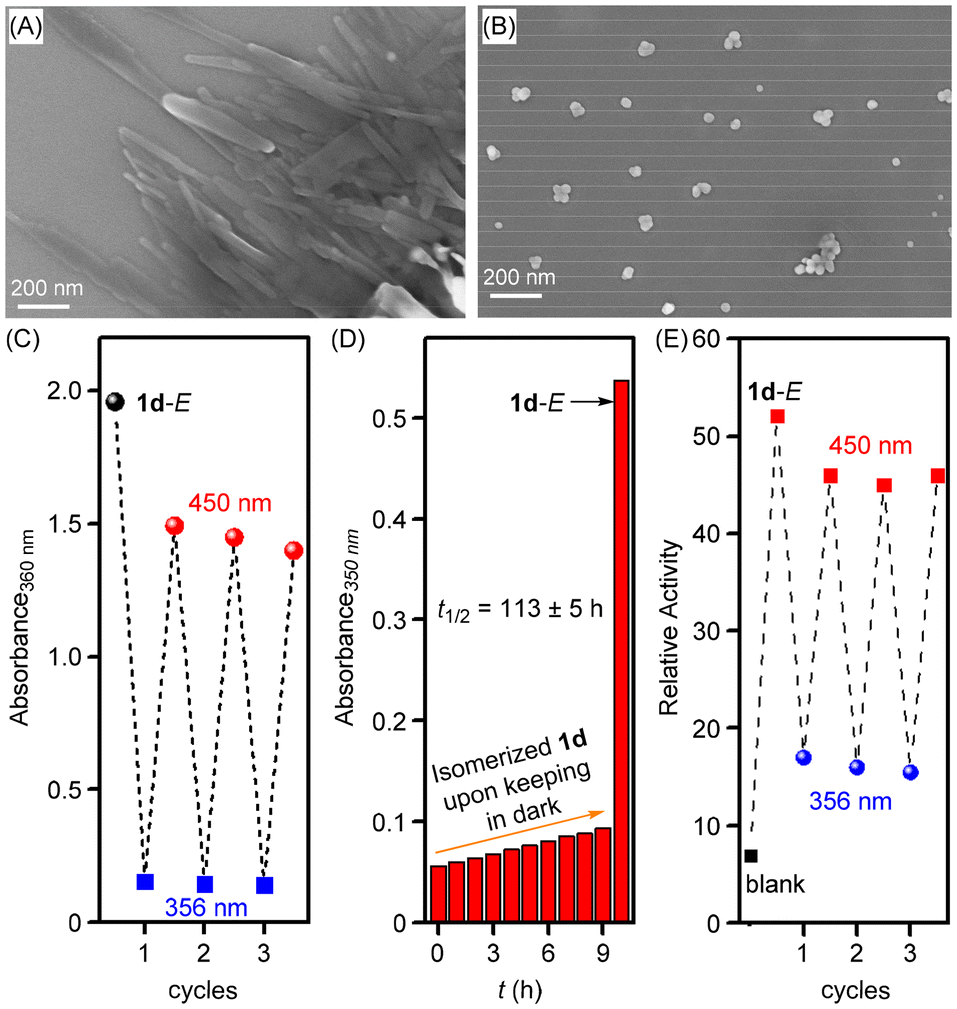 | ||
| Fig. 4 FESEM images of 1d in trans (A) and cis (B) forms showing distinct aggregation patterns recorded in methanol. Switching cycles for 1d (20 μM) under alternating irradiation with 365 nm (10 s) and 450 nm (20 s) light, respectively (C). Change in absorption at 350 nm of 1d (20 μM) upon keeping in the dark for 9 h (D). Photoregulated ion transport activity of 1d (5.0 μM) taken at t = 280 s under alternating photo-irradiation at two different wavelengths of 365 nm (15 s) and 450 nm (20 s) across EYPC-LUVs⊃HPTS (E). | ||
Furthermore, the solid-state morphological studies were performed using the field emission scanning electron microscopy (FESEM) technique in order to see the self-assembly behavior in the trans form and photoisomerized cis form of the channel-forming molecule 1d. Initially, the sample of 1d in methanol (100 μM) was prepared, dropcast on a silicon wafer, dried, and then used for morphology studies. The FESEM analysis shows rod-shaped structures in the trans form of 1d in the solid state (Fig. 4A and S20A†). These rod-shaped structures are most likely the outcome of strong self-assembly assisted with strong π–π interactions of the azobenzene subunits and inter-molecular hydrogen bonding interactions of the bis(1,3-diol) moiety. However, when the trans sample of 1d in methanol was initially photoirradiated at 365 nm of light for 5 min and then used for the FESEM analysis, vesicular shaped structures were obtained (Fig. 4B and S20B†). This clearly indicates that the long-range self-assembled structures are getting disrupted in the cis form upon photoirradiation likely because of the formation of non-planar cis azobenzene with poor self-assembly as compared to the trans form.
After photoisomerization studies, photoresponsive ion transport studies were performed using HPTS-based vesicles. Compound 1d showed efficient ion transport activity in the trans form at 0.25 μM concentration (used from 2 mM stock solution of 1d in DMSO). However, upon photoirradiation at 365 nm using LEDs (3 × 1/3 watt) for 15 s, a significant decrease in the ion transport activity of 1d was observed, likely due to the trans to cis photoisomerization process that disrupts the self-assembly and hence stops the ion channel formation (Fig. S19A†). On the other hand, when the photoisomerized solution (initially photoirradiated at 365 nm for 15 s) was photoirradiated at 450 nm of light using LEDs (3 × 1/3 watt) for 20 s, the ion transport activity was regained due to the cis to trans photoisomerization process (Fig. S19B†). This photoresponsive ion transport activity, regulated reversibly through alternating photoirradiation at 365 nm and 450 nm, was sustained over multiple cycles without any loss of efficiency (Fig. 4E).
Molecular modeling of the ion channel
To explore the underlying mechanism of ion passage through the channel and to assess the stability of the self-assembled channel on or before the ion transport, atomistic computer simulations were performed. Initially, we estimated the stability of each of the channel structures (trans and cis) and compared the time profile of root mean square deviation (RMSD) during equilibrium simulation for both channels. It was observed that the RMSD of the channel constituted with the cis monomers is higher compared to that of the trans channel and suggests that the trans channel structure remains relatively more stable compared to that of the cis structure (Fig. 5C). The snapshots in Fig. 5A and B also refer to the relatively more stable channel made up of the trans monomers than that of the cis form. We further proceeded to compute the free energy of the anion transport through the stable trans structure. From the free energy profile (Fig. 5E), it is evident that the free energy becomes maximum when the chloride ion remains in the middle of the channel, while the free energy decreases gradually when the anion moves towards either end of the channel structure, which is reflected by the fairly symmetric nature of the free energy profile on either side. The progress of the anion slows down in the center of the channel. Conversely, as the ion approaches the interface with bulk water, the free energy barrier considerably decreases due to its favorable interaction with an ample number of water molecules, which decreases toward the center (Fig. S22†). The presence of a sufficient amount of water within the channel core assists the ion in getting transported through the channel by maintaining a favorable solvent-rich environment, thereby not letting the free energy barrier increase to an unaffordable high limit. Furthermore, to gain mechanistic insights into chloride ion transport and to understand how the transport process is sensitive to the chemical nature of the channel, the interactions that are key for the ion movement through the channel structure were characterized. The contact of the chloride ion successively increases as the ion progresses through the channel, and it was observed that the favorable interactions with the channel monomers with the anion make the transport process easier (Fig. S23, S24† and Fig. 5F). | ||
| Fig. 5 Representative snapshots of self-assembled 1d, i.e., trans (A) and the corresponding cis (B) channel structures embedded within the lipid membrane during equilibrium simulation. Time profile of root mean square deviation (RMSD) of both the channels (cis and trans) during equilibrium simulation (C). Snapshot of trans channel displaying the collective variable, d, used in umbrella simulation (D) and free energy profile (calculated employing umbrella sampling simulation) of the Cl− transport through the more stable trans channel represented corresponding to cv and d (E). The possible interactions between the Cl− ion and the channel residues within the trans channel during ion progression (F). | ||
Conclusion
In conclusion, we developed azobenzene-fused bis(1,3-diol) molecules 1a–1d for photo-responsive self-assembled ion channel formation utilizing intra- and inter-molecular hydrogen bonding interactions and π–π stacking interactions. Compound 1d, with a long alkyl chain connected to the azobenzene subunit, furnished the highest ion transport activity with an EC50 value of 1.74 μM. The Hill coefficient value of ∼1 indicates the formation of a stable ion channel structure inside the lipid bilayer membrane. The planar bilayer conductance measurement studies also supported the formation of stable ion channels. Detailed mechanistic studies confirmed Cl−/NO3− antiport mode as the primary mode of ion transport. The dynamically gated ion transport process was achieved using two different sets of electromagnetic radiation at 365 nm and 450 nm, respectively, across HPTS-based lipid vesicles. Theoretical studies provided further insights into the self-assembly behaviour of channel forming molecules inside the lipid bilayer membrane and chloride transport through the channel lumen.Author contributions
P. T. conceived the project and directed the experimental studies. M. A. performed the ion transport studies and photoirradiation studies. S. S. and J. M. performed the theoretical studies. R. B. performed the synthesis and characterization of the compounds. A. M. performed the ion channel conductance studies. D. M. performed the morphological studies.Data availability
Data for this paper, including synthesis, compound characterization, experimental procedures, and theoretical calculations, are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. T. acknowledges the financial support from the Science and Engineering Research Board, Government of India (Grant No. CRG/2022/001640) and IISER Pune. J. M. acknowledges the support of the Department of Atomic Energy, Government of India, under Project Identification No. RTI 4007. M. A. and D. M. thank UGC, Government of India for research fellowships. A. M. thanks the Prime Minister's Research Fellowship.References
- H. Li, H. Valkenier, L. W. Judd, P. R. Brotherhood, S. Hussain, J. A. Cooper, O. Jurček, H. A. Sparkes, D. N. Sheppard and A. P. Davis, Nat. Chem., 2016, 8, 24–32 CrossRef CAS PubMed.
- K. A. Muraglia, R. S. Chorghade, B. R. Kim, X. X. Tang, V. S. Shah, A. S. Grillo, P. N. Daniels, A. G. Cioffi, P. H. Karp, L. Zhu, M. J. Welsh and M. D. Burke, Nature, 2019, 567, 405–408 CrossRef CAS.
- S.-K. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. Van Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885 CrossRef CAS PubMed.
- T. Saha, M. S. Hossain, D. Saha, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 7558–7567 CrossRef CAS.
- T. Saha, A. Gautam, A. Mukherjee, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 16443–16451 CrossRef CAS PubMed.
- S.-P. Zheng, L.-B. Huang, Z. Sun and M. Barboiu, Angew. Chem., Int. Ed., 2021, 60, 566–597 CrossRef CAS.
- N. Sakai, K. C. Brennan, L. A. Weiss and S. Matile, J. Am. Chem. Soc., 1997, 119, 8726–8727 CrossRef CAS.
- S. J. Moore, C. J. E. Haynes, J. González, J. L. Sutton, S. J. Brooks, M. E. Light, J. Herniman, G. J. Langley, V. Soto-Cerrato, R. Pérez-Tomás, I. Marques, P. J. Costa, V. Félix and P. A. Gale, Chem. Sci., 2013, 4, 103–117 RSC.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed.
- L. Lien, D. C. J. Jaikaran, Z. Zhang and G. A. Woolley, J. Am. Chem. Soc., 1996, 118, 12222–12223 CrossRef CAS.
- Y. R. Choi, B. Lee, J. Park, W. Namkung and K.-S. Jeong, J. Am. Chem. Soc., 2016, 138, 15319–15322 CrossRef CAS.
- E. N. W. Howe, N. Busschaert, X. Wu, S. N. Berry, J. Ho, M. E. Light, D. D. Czech, H. A. Klein, J. A. Kitchen and P. A. Gale, J. Am. Chem. Soc., 2016, 138, 8301–8308 CrossRef CAS.
- Y. Kobuke, K. Ueda and M. Sokabe, Chem. Lett., 2006, 24, 435–436 CrossRef.
- M. M. Tedesco, B. Ghebremariam, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 1999, 38, 540–543 CrossRef CAS PubMed.
- M. R. Banghart, M. Volgraf and D. Trauner, Biochemistry, 2006, 45, 15129–15141 CrossRef CAS.
- M. Ahmad, S. Metya, A. Das and P. Talukdar, Chem. – Eur. J., 2020, 26, 8703–8708 CrossRef CAS.
- M. Ahmad, D. Mondal, N. J. Roy, T. Vijayakanth and P. Talukdar, ChemPhotoChem, 2022, e202200002 CrossRef CAS.
- M. Ahmad, S. Chattopadhayay, D. Mondal, T. Vijayakanth and P. Talukdar, Org. Lett., 2021, 23, 7319–7324 CrossRef CAS PubMed.
- A. Kerckhoffs and M. J. Langton, Chem. Sci., 2020, 11, 6325–6331 RSC.
- T. G. Johnson, A. Sadeghi-Kelishadi and M. J. Langton, J. Am. Chem. Soc., 2022, 144, 10455–10461 CrossRef CAS PubMed.
- Y. R. Choi, G. C. Kim, H.-G. Jeon, J. Park, W. Namkung and K.-S. Jeong, Chem. Commun., 2014, 50, 15305–15308 RSC.
- W.-Z. Wang, L.-B. Huang, S.-P. Zheng, E. Moulin, O. Gavat, M. Barboiu and N. Giuseppone, J. Am. Chem. Soc., 2021, 143, 15653–15660 CrossRef CAS PubMed.
- C. Bao, M. Ma, F. Meng, Q. Lin and L. Zhu, New J. Chem., 2015, 39, 6297–6302 RSC.
- M. Ahmad, N. J. Roy, A. Singh, D. Mondal, A. Mondal, T. Vijayakanth, M. Lahiri and P. Talukdar, Chem. Sci., 2023, 14, 8897–8904 RSC.
- H. Fliegl, A. Köhn, C. Hättig and R. Ahlrichs, J. Am. Chem. Soc., 2003, 125, 9821–9827 CrossRef CAS PubMed.
- J. A. Malla, M. Ahmad and P. Talukdar, Chem. Rec., 2022, 22, e202100225 CrossRef CAS.
- R.-Y. Yang, C. Bao, Q.-N. Lin and L.-Y. Zhu, Chin. Chem. Lett., 2015, 26, 851–856 CrossRef CAS.
- Y. Zhou, Y. Chen, P.-P. Zhu, W. Si, J.-L. Hou and Y. Liu, Chem. Commun., 2017, 53, 3681–3684 RSC.
- T. Liu, C. Bao, H. Wang, Y. Lin, H. Jia and L. Zhu, Chem. Commun., 2013, 49, 10311–10313 RSC.
- R. Sharma, A. Vijay, S. Chattopadhayay, A. Mukherjee and P. Talukdar, Chem. Commun., 2023, 59, 3602–3605 RSC.
- A. Roy, T. Saha, M. L. Gening, D. V. Titov, A. G. Gerbst, Y. E. Tsvetkov, N. E. Nifantiev and P. Talukdar, Chem. – Eur. J., 2015, 21, 17445–17452 CrossRef CAS.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2002, 37, 68–83 RSC.
- D. Mondal, A. Sathyan, S. V. Shinde, K. K. Mishra and P. Talukdar, Org. Biomol. Chem., 2018, 16, 8690–8694 RSC.
- T. M Fyles, R Knoy, K Müllen and M Sieffert, Langmuir, 2001, 17, 6669–6674 CrossRef CAS.
- T. Saha, A. Gautam, A. Mukherjee, M. Lahiri and P. Talukdar, J. Am. Chem. Soc., 2016, 138, 16443–16451 CrossRef CAS.
- O. S. Smart, J. Breed, G. R. Smith and M. S. Sansom, Biophys. J., 1997, 72, 1109–1126 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01711k |
| ‡ Present Address: Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK |
| § Present Address: Faculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Zwirkii Wigury 101, Warsaw 02-089, Poland |
| This journal is © The Royal Society of Chemistry 2025 |