
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum single atoms on titania aid dye photodegradation whereas platinum nanoparticles do not†
Claudio Maria
Pecoraro‡
ab,
Hanna
Sopha‡
 cd,
Siming
Wu‡
a,
Hyesung
Kim
a,
Yue
Wang
a,
Jan
Macak
cd,
Monica
Santamaria
b and
Patrik
Schmuki
*ae
cd,
Siming
Wu‡
a,
Hyesung
Kim
a,
Yue
Wang
a,
Jan
Macak
cd,
Monica
Santamaria
b and
Patrik
Schmuki
*ae
aDepartment of Materials Science and Engineering, Chair for Surface Science and Corrosion (WW4-LKO), Friedrich-Alexander-Universität Erlangen-Nürnberg, Martensstraße 7, 91058 Erlangen, Germany. E-mail: schmuki@ww.uni-erlangen.de
bDipartimento di Ingegneria, Università degli Studi di Palermo, Viale delle Scienze Edificio 6, 90128 Palermo, Italy
cCenter of Materials and Nanotechnologies, Faculty of Chemical Technology, University of Pardubice, Nam. Cs. Legii 565, 53002 Pardubice, Czech Republic
dCentral European Institute of Technology, Brno University of Technology, Purkyňova 123, 61200 Brno, Czech Republic
eRegional Centre of Advanced Technologies and Materials, Šlechtitelů 27, 78371 Olomouc, Czech Republic
First published on 3rd January 2025
Abstract
The photocatalytic degradation of unwanted organic species has been investigated for decades using modified and non-modified titania nanostructures. In the present study, we investigate the co-catalytic effect of single atoms (SAs) of Pt and Pt nanoparticles on titania substrates on the degradation of the two typical photodegradation model pollutants: Acid Orange 7 (AO7) and Rhodamine B (RhB). For this, we use highly defined sputter deposited anatase layers and load them with Pt SAs at different loading densities or alternatively with Pt nanoparticles. We find that the Pt SAs have strong accelerating effects (already for a low loading density of ∼105 SAs μm−2) on the photodegradation of AO7, whereas Pt nanoparticles do hardly have an effect on the decay kinetics. The main beneficial effect of SA Pt is facilitated superoxide formation, which for SAs is significantly enhanced. Overall, the work demonstrates that Pt SA co-catalysts can have a beneficial effect not only for the well-studied use of H2 generation, but also in the photocatalytic degradation of pollutants—this is particularly the case if the degradation is dominated by a conduction band electron transfer to dissolved O2 in the solution.
1. Introduction
Since the pioneering work of Fujishima and Honda on TiO2 photoelectrodes in 1972,1 semiconductor photocatalysis has been used for a wide range of applications, including H2 generation,2–5 degradation of pollutants,6–9 and more complex reactions such as selective organic synthesis10–14 or CO2 reduction.15–18 Over many decades, pollution degradation involved various classes of pollutants, e.g., polychlorinated biphenyls or halogenated hydrocarbons,19–21 where hydrocarbons ultimately were reduced to CO2 and water. In this field, TiO2 represents the benchmark semiconductor material, due to its high photocatalytic activity, excellent stability, low cost, and non-toxic nature.22–25In the photocatalytic reaction, photoinduced electrons (e−) and holes (h+) are generated and migrate to the surface of the photocatalyst and are transferred to the environment, leading to reduction and oxidation reactions, respectively. Pollutant degradation processes can either take place in a direct reaction of the excited charge carriers (e− and h+) with the target pollutant or by the formation of various reactive oxygen species (ROS), such as hydroxyl radicals (˙OH) and superoxide anion radicals (˙O2−), through reactions with H2O or O2. Such reactive species can be formed either by the reductive reaction of O2 (if present) with electrons from the conduction band or by an oxidation reaction of valence band holes with water. The formed ROS then often play a crucial role in the transformation of organic pollutants into less harmful products.
The evaluation of photocatalysts’ photodegradation performance has been a subject of extensive investigation, often utilizing dyes such as Acid Orange 7 (AO7) and Rhodamine B (RhB) as model pollutants. These dyes offer the advantage of being readily detectable even at low concentrations, and their intricate chemical composition, often characterized by conjugated aromatic compounds, presents a challenging scenario for their conversion into less environmentally hazardous compounds.26 Due to their wide use as model pollutants, the decay cascade of AO7 and RhB on TiO2 is relatively well known.27–29 A main difference between the degradation sequences of Acid Orange 7 (AO7) and Rhodamine B (RhB) on TiO230–33 is that for AO7 the decay is reported to follow an electron-dominated reaction pathway, triggered by ˙O2− that is formed on TiO2 by the reaction of O2 with conduction band electrons, whereas for RhB usually it is assumed that the formation of ˙OH radicals by a valence band hole-dominated reaction is the key decay pathway.34,35 The behavior of these different dyes, along with other findings, was used to gain insight into the catalysts’ ability to generate these ROS. In TiO2 photocatalysis, often the application of co-catalysts is investigated. In particular, for the reaction of conduction band electrons with H+ ions in the solution – in photocatalytic H2 generation – the use of noble metal catalysts and their highly beneficial effect on the reaction have been extensively studied,36–38 interestingly their use was rarely explored for photodegradation processes. One may speculate that most common co-catalysts (e.g., Pt, Pd, etc.) have been explored, but were found to be ineffective. Nevertheless, noble metal co-catalysts are typically employed in the form of nanoparticles on TiO2 surfaces, but in recent years, the use of single atoms (SAs) of Pt as co-catalysts for H2 generation has attracted wide interest,39–42 not only because they represent minimum material usage with a maximum gain situation, but also because unusual reaction sequences become possible43–49 and exhibit an exceptional ability to enhance ROS production.50–53 Noble metal SA co-catalysts in photodegradation are limited to a few reports involving noble metal SAs on C3N4,54–56 and investigations on TiO2-based SA photocatalysts for pollution degradation are notably lacking.
We employed a Pt SA decorated sputtered TiO2 layer as a model catalyst for pollutant degradation studies. This system was synthesized using a reaction deposition method previously reported41,57–59 and has been thoroughly characterized in previous works using techniques such as X-ray Absorption Near Edge Structure (XANES), Extended X-ray Absorption Fine Structure (EXAFS), and Fourier Transform Infrared Spectroscopy (CO-FTIR), as well as investigations on the loading and distribution of Pt SA, etc.60–63 That is, the preparation procedure is well characterized and loading and dispersion control has been established.40,58,61,64,65 The findings indicate that Pt SAs can significantly accelerate the degradation of AO7, whereas they have virtually no effect on the decomposition of RhB. We attribute this to the fact that the decay of AO7 is primarily driven by a conduction band electron transfer process (specifically the formation of ˙O2−). While SA Pt has a substantial impact, the acceleration effect is only minor for Pt nanoparticles. The NBT assay confirmed this behavior, as Pt SA was significantly more effective in generating ˙O2− compared to Pt NPs and bare TiO2.
2. Experimental section
2.1. Materials
H2PtCl6·6H2O (Metakem), Rhodamine B (Sigma-Aldrich), Acid Orange 7 (Sigma-Aldrich), and nitrotetrazolium blue chloride (NBT, Sigma-Aldrich) were used as received without additional purification.2.2. Synthesis of Pt TiO2 samples
TiO2 supports were synthesized through direct current sputtering onto fluorine-doped tin oxide (FTO, 7 W sq.−1, Pilkington), and the sputtering time was 4 hours. A detailed description of the procedure can be found elsewhere.57 TiO2 supports were annealed in air at a temperature of 450 °C for 1 hour.For Transmission Electron Microscopy (TEM) investigations, supports consisting of an 8 nm SiO2 membrane with a Si3N4 mesh on a 200 μm thick silicon substrate, procured from Plano GmbH, were employed. 7 nm thin TiO2 layers were deposited onto them using DC magnetron sputtering for a duration of 12 minutes, and subsequently, they were annealed in ambient air at 450 °C for 1 hour.66
The surface of TiO2 was decorated with Pt SAs using a simple reactive deposition method.41,58 The TiO2 support was immersed in a 10 ml solution containing H2PtCl6·6H2O, with concentrations ranging from 0.005 to 5 mM, shortly after a 15 minute purging with argon. The sample was then subjected to a reactive deposition process for 1 hour. Afterward, the sample was immersed in water twice for 15 minutes and dried using a flow of N2. These samples were named “X mM Pt SA–TiO2”, where X represents the concentration expressed in mM of H2PtCl6·6H2O. For example, in 1 mM Pt SA–TiO2, a 1 mM precursor solution was used to decorate TiO2 through the reactive deposition method.
The photo-deposition method was used to load Pt nanoparticles onto TiO2 supports. The TiO2 support was placed in a 10 ml solution containing H2PtCl6·6H2O. Subsequently, the samples underwent a photo-deposition process when exposed to an LED light source (λmax = 365 nm, Imax = 600 mW cm−2) for a duration of 1 hour. Following this, the sample was immersed in water twice for 15 minutes each time and then dried using a stream of N2. Considering the same approach described above, these samples were identified as “X mM Pt NP–TiO2,” with X representing the concentration of H2PtCl6·6H2O expressed in mM.
2.3. Characterization
Field-emission Scanning Electron Microscopy (SEM, Hitachi S-4800) and Scanning Transmission Electron Microscopy (STEM, Thermo Fisher Scientific Spectra 200C-FEG) were employed to analyze the surface morphology of Pt-loaded TiO2. The surface chemical composition was also examined using X-ray Photoelectron Spectroscopy (XPS, PHI 5600).The absorbance spectra of the dye-containing solutions in the wavelength range of 200–800 nm were recorded using a UV/VIS/NIR spectrometer (Lambda XLS+, PerkinElmer). The spectrometer was equipped with a tungsten–halogen lamp for UV light. The solutions were placed in transparent quartz cuvettes (Hellma Analytics), and the relative color intensity was correlated with the dye's maximum absorbance peak. The maximum absorbance peak was observed at 554 nm for RhB, 485 nm for AO7, and 260 nm for NBT.
NBT assays were carried out to assess the ˙O2− production in bare TiO2 and TiO2 decorated with Pt as SA or NP. The concentration of the Pt precursor was 0.005 mM in both cases. The NBT maximum absorption peak is situated far from the wavelength of the monochromatic 365 nm LED light source, resulting in negligible absorption at this wavelength. Hence, employing NBT for superoxide detection does not conflict with light absorption issues. Notably, the reaction between NBT and ˙O2− resulted in the formation of a dark precipitate insoluble in aqueous solutions.67 The comparison of the ˙O2− production of various samples was executed by measuring the decreased intensity of the absorbance of NBT in the solution. Directly after the NBT assay, the dark precipitate on the TiO2 samples was analyzed through diffuse reflectance spectral analysis. This analysis was conducted using a fiber-based UV-VIS-IR spectrophotometer (Avantes, ULS2048) equipped with an integrating sphere (AvaSphere-30) using an AvaLight-DH-S-BAL balanced power light source. A BaSO4 standard whiteboard was employed as a reference material. Additionally, bright-field optical microscopy (Nikon, Japan) was used to capture images of the samples.
Linear sweep voltammetry (LSV) curves were obtained in 0.1 M Na2SO4, using a conventional three-electrode setup, with Pt foil serving as the counter electrode and a saturated Ag/AgCl electrode as the reference. The LSV measurements were carried out at a scan rate of 5 mV s−1.
2.4. Photodegradation tests
The photodegradation of dyes, 10 μM RhB or 25 μM AO7 as the starting concentration, was carried out in a quartz tube using TiO2 samples without Pt or loaded with Pt by employing various concentrations of H2PtCl6·6H2O. A schematic illustration of the setup for the photodegradation test is shown in Fig. S1.† The reaction volume was 10 ml. The experiments were conducted either in an air saturated system or under an argon atmosphere. To ensure the establishment of an adsorption/desorption equilibrium between the sample and the dye, the sample was immersed in the dye solutions for 1 hour before irradiation. During the photodegradation test, 2.5 ml of the dye solution was taken every 20 minutes of irradiation under vigorous stirring and analyzed using a UV/VIS/NIR spectrometer. After analysis, the extracted portion was returned to maintain the original reaction volume, and the total irradiation time was 120 minutes.In the case of photodegradation conducted under an argon atmosphere, the solution was purged for 15 minutes, and the cell was sealed, soon after immersing the sample. Following the aliquot analysis, the cell was purged with argon for a duration of 5 minutes before being sealed again.
The NBT assay, with 10 μM as the starting concentration, was performed in an air saturated system. The experimental setup and procedure followed were identical to those used for the photodegradation of dyes.
An LED light source (λmax = 365 nm, Imax = 65 mW cm−2) was used for illumination.
Notably, in all photodegradation experiments, a TiO2 layer was consistently used as the catalyst, prepared by sputtering to ensure uniform thickness across all samples. This ensured that the substrates were identical, with the only difference being the Pt loading. The dosage and other experimental conditions were carefully controlled and kept consistent throughout the study.
3. Results and discussion
For our investigations, we used DC magnetron deposited anatase layers on FTO substrates or alternatively on TEM grids that were then decorated with Pt SA as described in previous work.57,58,61To illustrate the process, an SEM image of a cross-section of such a layer is shown in Fig. 1a, confirming the presence of a flat layer with an approximate thickness of 200 nm. Then these surfaces were loaded either with Pt nanoparticles (Fig. 1b) using classic photodeposition as reported in the Experimental section or with Pt SAs (Fig. 1c and d) using the reactive SA deposition method described in the Experimental section and in ref. 41 and 58.
 | ||
| Fig. 1 SEM images of (a) cross-section of a 200 nm sputtered TiO2 layer on FTO; the top view is shown as the inset. (b) 0.005 mM Pt NP–TiO2; examples of NP are circled in blue, (c) 0.005 mM Pt SA–TiO2, and (d) 2 mM Pt SA–TiO2. TEM images of 0.005 mM Pt SA–TiO2 and 2 mM Pt SA–TiO2 are shown in (e) and (f), respectively, and examples of Pt as a SA and cluster are circled in red and yellow. (g) Statistics of size distribution percentage of Pt SA at 0.005 am 2 mM loading. (h) XPS spectra in the Pt 4f region. | ||
Fig. 1c and d show the SEM images of TiO2 loaded with SA Pt from a 0.005 mM Pt solution or a 2 mM Pt solution, respectively. In these high-resolution SEM images, there is no noticeable change in the TiO2 surface morphology after Pt decoration (see also Fig. S2†), and, as expected, no signs of Pt nanoclusters or nanoparticles are observed on TiO2. The XRD patterns of TiO2, TiO2 loaded with Pt SAs, and TiO2 loaded with Pt NPs show no obvious difference, i.e., only the diffraction patterns of anatase TiO2 and FTO, and no reflections associated with metallic Pt (Fig. S3†). However, the presence of Pt as SAs on the TiO2 surface can be clearly revealed through High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy (HAADF-STEM) images. Fig.†1e and f show the anatase films on the TEM stage from two precursor concentrations (0.005 mM and 2 mM Pt). For the loading from 0.005 mM precursor solution, HAADF-STEM shows exclusively well dispersed Pt atoms (examples are circled in red), whereas for the 2 mM Pt SA–TiO2 sample, also some 2D rafts are present, as indicated by yellow circles in Fig. 1f. The Pt single atom density was obtained by counting individual Pt atoms and clusters within a known area in a series of HAADF-STEM images, where both individual Pt atoms and clusters were considered as individual active sites. This resulted in SA densities of 3.3 × 105 μm−2 for the 0.005 mM Pt SA and 1.4 × 106 μm−2 for the 2 mM Pt SA loaded TiO2 sample, well in line with previous studies.61Fig. 1g presents a statistical analysis of the size distribution of Pt SAs after treatment in 0.005 mM and 2 mM Pt solutions. The analysis reveals that in the first case, a substantial portion of Pt, specifically 89%, is present on the TiO2 surface as individual atoms, and a minor amount as 2D assemblies in the range of 0.3 nm. In contrast, for the 2 mM Pt SAs sample, only 62% of Pt is present as individual atoms, while 2D rafts up to 2 nm in size are observable.
The quantitative total amount of Pt loading and the chemical state of the deposited Pt were then investigated by XPS. Fig. 1h shows the Pt 4f XPS spectra of 0.005 mM Pt SAs and 2 mM Pt SAs on the substrate, as well as for the NP samples. In the case of Pt SAs, the Pt 4f spectra exhibit a doublet positioned at ∼72.4 eV and 75.6 eV. This observation is consistent with SA Pt coordination with oxygen atoms of the TiO2 surface, where the peak position corresponds to a formal charge (δ+) on Ptδ+ with a value of δ ≈ 2.68 Meanwhile, more pronounced peaks for the 2 mM Pt SA–TiO2 sample compared to those of 0.005 mM Pt SA–TiO2 can be observed due to a higher Pt loading. Quantitative evaluation yields a Pt loading amount of 0.49 ± 0.05 at% and 0.24 ± 0.05 at% for 2 mM SAs and 0.005 mM SAs, respectively (Table S1†). In contrast, the Pt NP sample shows a doublet located at ∼70.5 eV and 74.1 eV, which is typical of Pt0 in a metallic nanoparticle configuration.62 For the NP sample, the total loading amount is 0.6 ± 0.05 at% as determined from XPS (Table S1†).
We then used the different Pt loaded samples (as well as non-loaded samples) for the photodegradation of AO7 and RhB (Fig. 2a and b). In these experiments, we used SA deposition from a range of Pt precursor concentrations (ranging from 0.005 to 5 mM). The resulting degradation of the dyes over time is shown in a ln(C/C0) plot (assuming a first-order decay mechanism) – the original UV-vis absorption spectra are reported in Fig. S4 and S5.† From a linear fit of the data, the corresponding rate constants can be extracted (in Fig. 2c, the error bars represent the average deviation between experimental data and the fitting). Besides, the UV-vis spectral data before the photodegradation experiments (Fig. S4–S6†) indicate minimal dye adsorption on all samples.
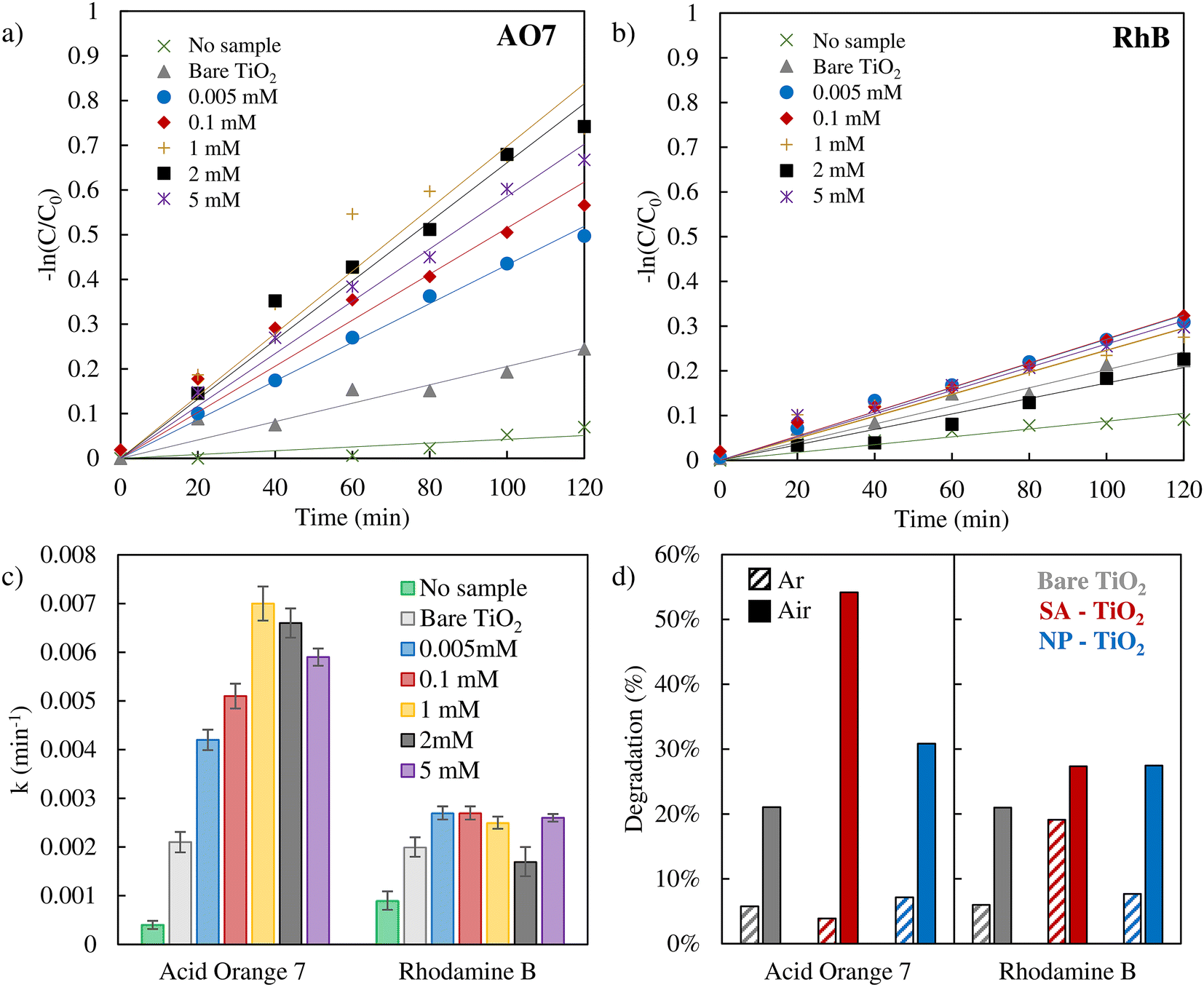 | ||
| Fig. 2 Photodegradation tests carried out in an air saturated system of AO7 (a) and RhB (b) with various Pt SA samples. The degradation rate constants are shown in (c). (d) Photodegradation percentages of AO7 and RhB after 2 hours of irradiation in an air saturated system (full bars) or under an argon atmosphere (dashed bars). 2 mM Pt SA–TiO2 was employed for AO7 degradation, and 0.005 mM Pt SA–TiO2 for RhB degradation. For both dyes, bare TiO2 and 0.005 mM Pt NP–TiO2 were used. | ||
It should be noted that in the literature apart from a true photocatalytic degradation, also self-sensitization of RhB and AO7 has been investigated as a possible degradation mechanism in dye degradation experiments.69,70 However, for our experimental configuration (flat TiO2 layer), we show that photosensitization has no measurable effect on the degradation of AO7 and RhB, as evidenced by the experimental data in Fig. S7.† In both cases, the inclusion of the TiO2 catalyst results in almost no change in degradation percentage compared to the control experiment where no catalyst was added (Fig. S7†).
Remarkably for AO7, the presence of the Pt SA co-catalyst can strongly enhance the degradation rate – the efficiency enhancement increases significantly at a low concentration of 0.005 mM with a loading of 0.24 at% and then saturates at 1–2 mM (∼0.45 ± 0.05 at%, Table S1†). Even more remarkable is the finding that the presence of nanoparticles of Pt has only a minor impact on the degradation rate of AO7 (Fig. 2d, blue bars). Furthermore, for RhB degradation, the presence or absence of Pt SAs has hardly any effect on the photodegradation rate.
The results thus show that Pt SAs can enhance the AO7 photodegradation efficiency strongly and evidently the influence of Pt SAs is very different for the two dyes – this may reflect a different critical step in their respective decay mechanisms. As mentioned before, for AO7 and RhB, the literature reports on differences in the decay initiating species that is formed either by a valence or a conduction band mechanism.30–33 In the conduction band mechanism, the key reaction is the sequence of electron transfer to O2 present in the environment (solution) and the formation of ˙O2−,71 while in the valence band mechanism, a hole is transferred to water forming ˙OH.72 In order to probe the need for O2 in the reaction sequence, we used argon purging to remove O2 from the solution.73 The results of AO7 and RhB dyes’ photodegradation after Ar purging are presented in Fig. 2d, with dashed bars (absorption spectra are reported in Fig. S6†). Clearly, the photodegradation of AO7 is found to be strongly reduced in the absence of O2. This finding shows that the presence of O2 is critical for the degradation of AO7 and thus the formation of ˙O2− may be the key. This is in stark contrast to RhB where for the SA decorated case the presence or absence of O2 only minorly affects the degradation rate.
Notably, under an air atmosphere, the presence of O2 may induce trapped electrons to undergo a reaction resulting in the generation of ˙O2−. However, while ˙O2− serves as the principal reactive species responsible for AO7 degradation, in the RhB case, although it is highly reactive and unstable, it does not react significantly with RhB.
This is in line with reports that ascribe valence band hole generated ˙OH a major role in the photodegradation of RhB.32 The AO7 results suggest that the oxygen reaction to form superoxide (˙O2−) is strongly enhanced in the presence of Pt SAs.
In order to verify the role of ˙O2− formation, we performed the NBT assay which is a comparably straightforward color indicator reaction for the presence of ˙O2− species, for experiments with plain TiO2, TiO2 decorated with Pt SAs, and TiO2 decorated with Pt nanoparticles under an air-saturated atmosphere and Ar atmosphere (Fig. 3a and S8, 9†). A reduction in degradation under an Ar atmosphere is observed in all samples compared to an air-saturated system, suggesting that a significant portion of the ˙O2− is generated through the direct reduction of O2. Evidently, NBT degrades under irradiation in all samples. But clearly, the activity for the generation of ˙O2− follows the order of SA > NP > bare TiO2. This trend can be seen in both the UV-VIS spectrophotometer data (Fig. 3b) and optical microscopy images (Fig. 3c–e). The dark precipitate that forms at the center of the irradiated region of the samples is the NBT degradation product. As depicted in the surface reflectance data in Fig. 3b, Pt SA–TiO2 samples provide clearly the highest absorption intensity for the formation of the NBT reaction product, whereas Pt NP–TiO2 shows a significantly lower activity, and bare TiO2 exhibits the lowest. Furthermore, the optical microscopy images in Fig. 3c–e also illustrate that the Pt SA sample exhibits the most pronounced formation of the dark reaction product. These observations indicate that Pt SAs have a strongly promoting effect on the generation of ˙O2− radicals, whereas Pt nanoparticles have only a minor effect.
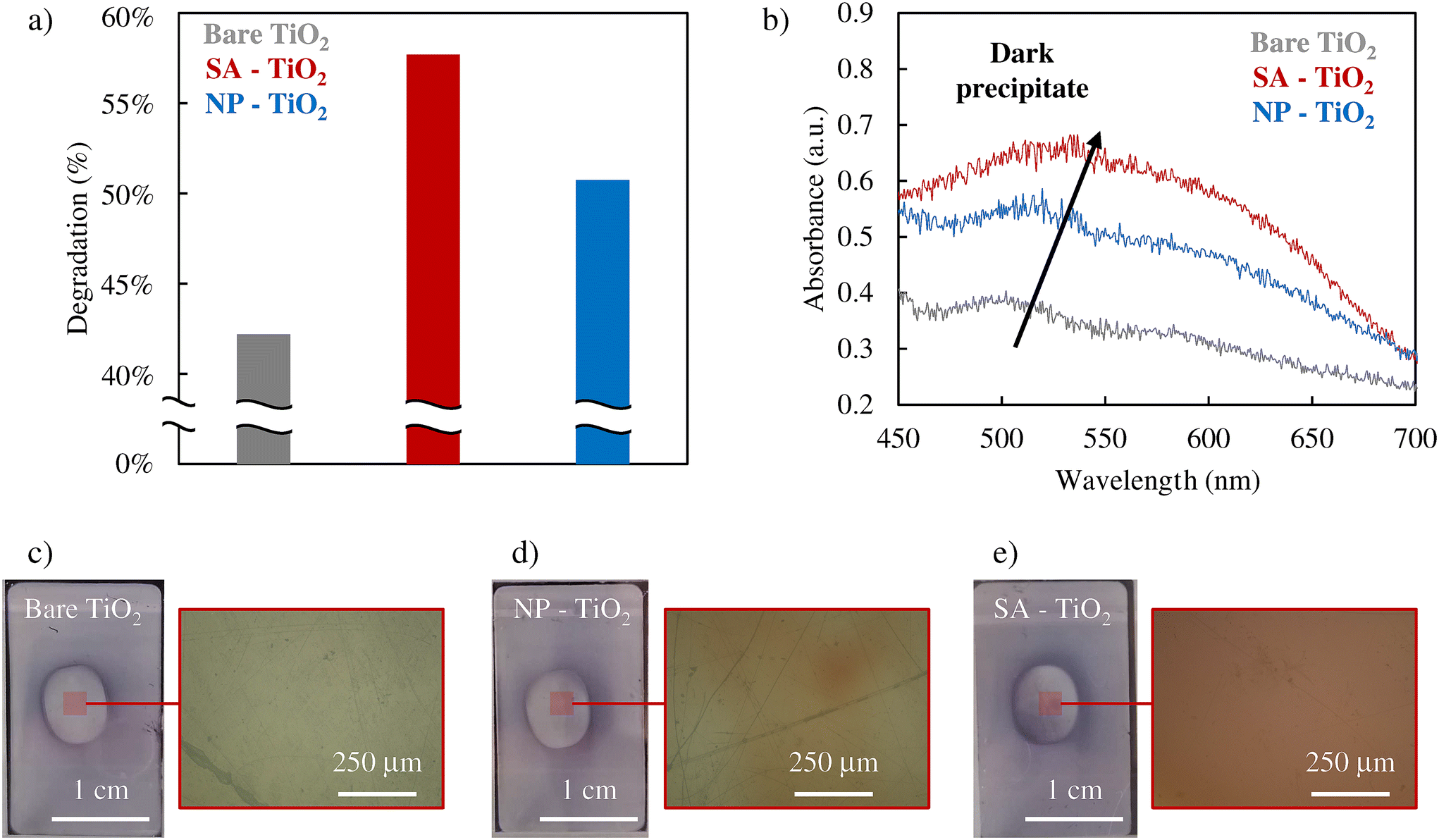 | ||
| Fig. 3 (a) NBT assay results, (b) UV-Vis spectra, and (c)–(e) pictures of the samples and the respective magnifications obtained through an optical microscope soon after the NBT assay of 0.005 mM Pt SAs–TiO2, 0.005 mM Pt NPs–TiO2 and bare TiO2. | ||
To further support these findings, we obtained linear sweep voltammetry curves comparing bare TiO2 with TiO2 decorated with Pt SAs, as shown in Fig. S10.† The LSV curves reveal that TiO2 with Pt SAs exhibits a significantly higher cathodic current density compared to bare TiO2. This indicates that the addition of Pt SAs enhances electron transfer, likely contributing to the more efficient reduction of O2 to ˙O2−. Notably, according to the existing literature, Pt SAs are recognized for their ability to act as electron-capturing centers, facilitating the conversion of O2 into ˙O2− radicals.74 Additionally, Pt SAs may improve the adsorption and activation of O2, further enhancing ˙O2− formation.75 This capacity to generate reactive oxygen species likely explains the superior performance of Pt SAs in the ˙O2− mechanism compared to Pt NPs. These findings are in line with the literature—DFT calculations confirm that single-atom catalysts are more effective than nanoparticles in generating radicals during photocatalytic oxidation and reduction reactions.52,53,75,76
Based on these findings, one can postulate the photodegradation mechanisms relevant to AO7 and RhB and the beneficial effect of Pt SAs in the case of AO7 as schematically illustrated in Fig. 4a and b. In line with the literature, ˙O2− radicals through a conduction band process play a crucial role in the photodegradation of AO7, whereas, in contrast, for RhB photodegradation this process matters only in a minor way.
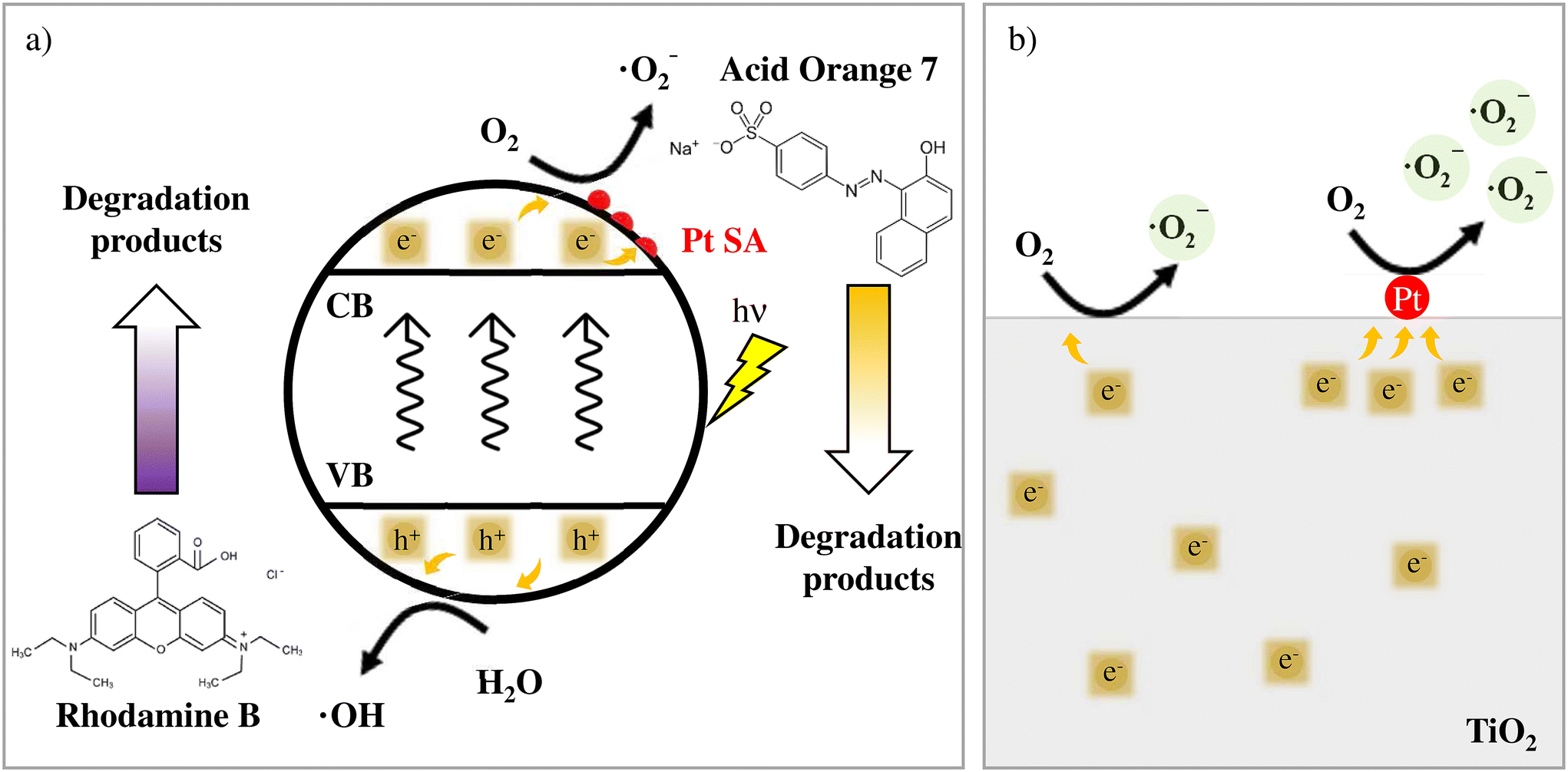 | ||
| Fig. 4 (a) Reaction mechanism proposed for Acid Orange 7 and Rhodamine B. The details of the O2 reduction to generate ˙O2− on the Pt SA surface are shown in (b). | ||
For RhB, the degradation is controlled by the oxidation process via the valence band process that is either direct or proceeds via ROS produced through the reaction of h+ with water (Fig. 4a, bottom).32 Consequently, conduction band processes and the promoting effect of Pt SAs on the electron transfer have a minor impact on the degradation of RhB. Conversely, for AO7 photodegradation the main role is the formation of superoxide ˙O2− from water dissolved oxygen (Fig. 4a. up) via conduction band electrons.
Overall the data clearly show that Pt in the form of SAs (but much less as NPs) is a highly active co-catalyst for the photocatalytic degradation of unwanted species if the reduction of solution dissolved O2 is key to the decay mechanism. Notably, additional cycling tests for AO7 degradation using 2 mM Pt SA TiO2 demonstrate that the degradation percentage remains similar at around 30% over the three cycles (Fig S11†), confirming good long-term stability.
4. Conclusions
In conclusion, this study investigates the efficacy of Pt SAs as co-catalysts on TiO2 in the photocatalytic degradation of the two most classic model pollutants (AO7 and RhB dyes). We find that Pt SAs cause a strongly enhanced degradation of AO7 – this is ascribed to the SA Pt promoted formation of ˙O2−. NBT assays show that Pt SAs are significantly more effective in generating ˙O2− compared with Pt NPs. For RhB this enhancement effect is not effective as the degradation process is conduction band controlled and seems typically not to follow a ˙O2− formation mechanism.In summary, this study describes the promising potential of Pt SA–TiO2 photocatalysts in the photodegradation of pollutants and thereby opens up new strategies for environmental remediation and sustainable technologies.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the DFG and the Operational research program, Development and Education (European Regional Development Fund, Project No. CZ.02.1.01/0.0/0.0/15_003/0000416 of the Ministry of Education, Youth and Sports of the Czech Republic), and the Czech Science Foundation for project 23-08019X for financial support. H. S. thanks the COST action (CA20126, “NETPORE”) for the financial support through the Short-Term Scientific Mission Grant. S. W. acknowledges the Emerging Talents Initiatives (ETI) project for financial support. C. M. P. acknowledges support from the University of Palermo and Friedrich-Alexander-Universität Erlangen-Nürnberg.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- N. Liu, S. Mohajernia, N. T. Nguyen, S. Hejazi, F. Plass, A. Kahnt, T. Yokosawa, A. Osvet, E. Spiecker, D. M. Guldi and P. Schmuki, ChemSusChem, 2020, 13, 4937–4944 CrossRef CAS.
- N. Kunthakudee, T. Puangpetch, P. Ramakul, K. Serivalsatit and M. Hunsom, Int. J. Hydrogen Energy, 2022, 47, 23570–23582 CrossRef CAS.
- S. Nishioka, F. E. Osterloh, X. Wang, T. E. Mallouk and K. Maeda, Nat. Rev. Methods Primers, 2023, 3, 1–15 CrossRef.
- V. M. Daskalaki and D. I. Kondarides, Catal. Today, 2009, 144, 75–80 CrossRef CAS.
- S. Ruan, W. Huang, M. Zhao, H. Song and Z. Gao, Mater. Sci. Semicond. Process., 2020, 107, 104835 CrossRef CAS.
- K. Vinodgopal, D. E. Wynkoop and P. V. Kamat, Environ. Sci. Technol., 1996, 30, 1660–1666 CrossRef CAS.
- Y. Shangguan, Y. Zhou, R. Zheng, X. Feng, Q. Ge, R. Wang, D. Yang, W. Wei, X. Wu, J. Lin and H. Chen, Chin. Chem. Lett., 2021, 32, 3450–3456 CrossRef CAS.
- M. A. I. Molla, I. Tateishi, M. Furukawa, H. Katsumata, T. Suzuki and S. Kaneco, Open J. Inorg. Non-Met. Mater., 2017, 07, 1–7 CAS.
- M. R. Karimi Estahbanati, M. Feilizadeh, F. Attar and M. C. Iliuta, React. Chem. Eng., 2021, 6, 197–219 RSC.
- M. Bellardita, D. Virtù, F. Di Franco, V. Loddo, L. Palmisano and M. Santamaria, Chem. Eng. J., 2022, 431, 134131 CrossRef CAS.
- C.-L. Tan, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2021, 298, 120541 CrossRef CAS.
- C. M. Pecoraro, M. Bellardita, V. Loddo, D. Virtù, F. Di Franco and M. Santamaria, Appl. Catal., A, 2023, 650, 118987 CrossRef CAS.
- C. M. Pecoraro, M. Bellardita, V. Loddo, F. Di Franco, L. Palmisano and M. Santamaria, J. Ind. Eng. Chem., 2023, 118, 247–258 CrossRef CAS.
- M. Bellardita, A. Di Paola, E. García-López, V. Loddo, G. Marcì and L. Palmisano, Curr. Org. Chem., 2013, 17, 2440–2448 CrossRef CAS.
- D. Li, M. Kassymova, X. Cai, S. Q. Zang and H. L. Jiang, Coord. Chem. Rev., 2020, 412, 213262 CrossRef CAS.
- G. Wang, Z. Chen, T. Wang, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2022, 61, e202210789 CrossRef CAS PubMed.
- H. Huang, R. Shi, Z. Li, J. Zhao, C. Su and T. Zhang, Angew. Chem., 2022, 134, e202200802 CrossRef.
- H. J. M. Verhaar, C. J. van Leeuwen and J. L. M. Hermens, Chemosphere, 1992, 25, 471–491 CrossRef CAS.
- S. Manzetti, E. R. Van Der Spoel and D. Van Der Spoel, Chem. Res. Toxicol., 2014, 27, 713–737 Search PubMed.
- E. Brillas and S. Garcia-Segura, J. Environ. Chem. Eng., 2023, 11, 109635 Search PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- G. Ognibene, D. A. Cristaldi, R. Fiorenza, I. Blanco, G. Cicala, S. Scirè and M. E. Fragalà, RSC Adv., 2016, 6, 42778–42785 RSC.
- H. Lachheb, E. Puzenat, A. Houas, M. Ksibi, E. Elaloui, C. Guillard and J. M. Herrmann, Appl. Catal., B, 2002, 39, 75–90 CrossRef CAS.
- A. M. Djaballah, M. Bellardita, L. Palmisano, V. Loddo, M. Umair, C. M. Pecoraro, R. Bagtache and M. Trari, Mol. Catal., 2023, 546, 113251 CrossRef CAS.
- A. A. Yaqoob, N. H. B. M. Noor, A. Serrà and M. N. M. Ibrahim, Nanomaterials, 2020, 10, 932 CrossRef CAS PubMed.
- R. Fiorenza, M. Bellardita, T. Barakat, S. Scirè and L. Palmisano, J. Photochem. Photobiol., A, 2018, 352, 25–34 CrossRef CAS.
- M. H. Abdel-Khalek, M. A. Ahmed, M. F. Abdel-Messih and F. El-Shahat, Mater. Sci. Energy Technol., 2022, 5, 334–343 CAS.
- Y. Chen, S. Yang, K. Wang and L. Lou, J. Photochem. Photobiol., A, 2005, 172, 47–54 CrossRef CAS.
- M. Stylidi, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2004, 47, 189–201 CrossRef CAS.
- J.-H. Shen, H.-Y. Chuang, Z.-W. Jiang, X.-Z. Liu and J.-J. Horng, Chemosphere, 2020, 251, 126380 CrossRef CAS PubMed.
- J. Zhuang, W. Dai, Q. Tian, Z. Li, L. Xie, J. Wang, P. Liu, X. Shi and D. Wang, Langmuir, 2010, 26, 9686–9694 CrossRef CAS PubMed.
- S. Fang, K. Lv, Q. Li, H. Ye, D. Du and M. Li, Appl. Surf. Sci., 2015, 358, 336–342 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- P. Fernández-Castro, M. Vallejo, M. F. San Román and I. Ortiz, J. Chem. Technol. Biotechnol., 2015, 90, 796–820 CrossRef.
- M. Zhang, C. Lai, B. Li, F. Xu, D. Huang, S. Liu, L. Qin, Y. Fu, X. Liu, H. Yi, Y. Zhang, J. He and L. Chen, Chem. Eng. J., 2020, 396, 125343 CrossRef CAS.
- G. Zhao and X. Xu, Nanoscale, 2021, 13, 10649–10667 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- U. Kerketta, A. B. Tesler and P. Schmuki, Catalysts, 2022, 12, 1223 CrossRef CAS.
- Y. Wang, I. Hwang, Z. Wu and P. Schmuki, Electrochem. Commun., 2021, 133, 107166 CrossRef CAS.
- S. M. Wu, I. Hwang, B. Osuagwu, J. Will, Z. Wu, B. B. Sarma, F. F. Pu, L. Y. Wang, Z. Badura, G. Zoppellaro, E. Spiecker and P. Schmuki, ACS Catal., 2023, 13, 33–41 CrossRef CAS.
- B. Zhang, T. Fan, N. Xie, G. Nie, H. Zhang, B. Zhang, T. Fan, H. Zhang, N. Xie and G. Nie, Adv. Sci., 2019, 6, 1901787 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- X. Li, X. Yang, Y. Huang, T. Zhang and B. Liu, Adv. Mater., 2019, 31, 1902031 CrossRef CAS PubMed.
- Y. Wang, G. Zhuang, J. Zhang, F. Luo, X. Cheng, F. Sun, S. Fu, T. Lu and Z. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202216592 CrossRef CAS PubMed.
- J. Huang, W. Shi, S. Xu, H. Luo, J. Zhang, T. Lu and Z. Zhang, Adv. Mater., 2024, 36, 2306906 CrossRef CAS PubMed.
- S. Xu, W. Shi, J. Huang, S. Yao, C. Wang, T. Lu and Z. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202406223 CrossRef CAS PubMed.
- Z. Xue, M. Yan, Y. Zhang, J. Xu, X. Gao and Y. Wu, Appl. Catal., B, 2023, 325, 122303 CrossRef CAS.
- S. Zhao, Y. Wen, X. Liu, X. Pen, F. Lü, F. Gao, X. Xie, C. Du, H. Yi, D. Kang and X. Tang, Nano Res., 2020, 13, 1544–1551 CrossRef CAS.
- X. Wei, S. Song, W. Cai, Y. Kang, Q. Fang, L. Ling, Y. Zhao, Z. Wu, X. Song, X. Xu, S. M. Osman, W. Song, T. Asahi, Y. Yamauchi and C. Zhu, ACS Nano, 2024, 18, 4308–4319 CrossRef CAS PubMed.
- W. Yan, J. Sun, T. Hu, S. Tian, J. Feng and Y. Xiong, Appl. Catal., B, 2023, 323, 122143 CrossRef CAS.
- J. Wang, Y. Xie, G. Yu, L. Yin, J. Xiao, Y. Wang, W. Lv, Z. Sun, J. H. Kim and H. Cao, Environ. Sci. Technol., 2022, 56, 17753–17762 CrossRef CAS PubMed.
- L. Sun, Y. Feng, K. Ma, X. Jiang, Z. Gao, J. Wang, N. Jiang and X. Liu, Appl. Catal., B, 2022, 306, 121106 CrossRef CAS.
- X. Liu, S. Wang, W. Yu, J. Zhang, S. Fang, J. Zhang, J. Qiu, F. Kong and X. Duan, Chem. Eng. J., 2022, 446, 137426 CrossRef CAS.
- G. Vilé, P. Sharma, M. Nachtegaal, F. Tollini, D. Moscatelli, A. Sroka-Bartnicka, O. Tomanec, M. Petr, J. Filip, I. S. Pieta, R. Zbořil and M. B. Gawande, Sol. RRL, 2021, 5, 2100176 CrossRef.
- H. Kim, Y. Wang, N. Denisov, Z. Wu, Š. Kment and P. Schmuki, J. Mater. Sci., 2022, 57, 12960–12970 CrossRef CAS.
- S. Qin, N. Denisov, J. Will, J. Kolařík, E. Spiecker and P. Schmuki, Sol. RRL, 2022, 6, 2101026 CrossRef CAS.
- S. Qin, N. Denisov, B. B. Sarma, I. Hwang, D. E. Doronkin, O. Tomanec, S. Kment and P. Schmuki, Adv. Mater. Interfaces, 2022, 9, 2200808 CrossRef CAS.
- N. Denisov, S. Qin, J. Will, B. N. Vasiljevic, N. V. Skorodumova, I. A. Pašti, B. B. Sarma, B. Osuagwu, T. Yokosawa, J. Voss, J. Wirth, E. Spiecker and P. Schmuki, Adv. Mater., 2023, 35, 2206569 CrossRef CAS PubMed.
- S. Qin, J. Will, H. Kim, N. Denisov, S. Carl, E. Spiecker and P. Schmuki, ACS Energy Lett., 2023, 8, 1209–1214 CrossRef CAS.
- S. Hejazi, S. Mohajernia, B. Osuagwu, G. Zoppellaro, P. Andryskova, O. Tomanec, S. Kment, R. Zbořil and P. Schmuki, Adv. Mater., 2020, 32, 1908505 CrossRef CAS PubMed.
- Y. Wang, S. Qin, N. Denisov, H. Kim, Z. Bad'ura, B. B. Sarma and P. Schmuki, Adv. Mater., 2023, 35, 2211814 CrossRef CAS PubMed.
- Z. Wu, I. Hwang, G. Cha, S. Qin, O. Tomanec, Z. Badura, S. Kment, R. Zboril, P. Schmuki, Z. Wu, I. Hwang, G. Cha, S. Qin, P. Schmuki, O. Tomanec, Z. Badura, S. Kment and R. Zboril, Small, 2022, 18, 2104892 CrossRef CAS PubMed.
- G. Cha, A. Mazare, I. Hwang, N. Denisov, J. Will, T. Yokosawa, Z. Badura, G. Zoppellaro, A. B. Tesler, E. Spiecker and P. Schmuki, Electrochim. Acta, 2022, 412, 140129 CrossRef CAS.
- Y. Wang, S. Qin, N. Denisov, H. Kim, Z. Bad'ura, B. Sarma, P. Schmuki, Y. Wang, S. Qin, N. Denisov, H. Kim, P. Schmuki, Z. Bad'ura and B. B. Sarma, Adv. Mater., 2023, 35, 2211814 CrossRef CAS PubMed.
- H. Goto, Y. Hanada, T. Ohno and M. Matsumura, J. Catal., 2004, 225, 223–229 CrossRef CAS.
- S. M. Wu, L. Wu, N. Denisov, Z. Badura, G. Zoppellaro, X. Y. Yang and P. Schmuki, J. Am. Chem. Soc., 2024, 146, 16363–16368 CrossRef CAS PubMed.
- Y. Wu, J. Zhang, L. Xiao and F. Chen, Appl. Surf. Sci., 2010, 256, 4260–4268 CrossRef CAS.
- L. Pan, J. J. Zou, S. Wang, Z. F. Huang, X. Zhang and L. Wang, Appl. Surf. Sci., 2013, 268, 252–258 CrossRef CAS.
- J. M. Burns, W. J. Cooper, J. L. Ferry, D. W. King, B. P. DiMento, K. McNeill, C. J. Miller, W. L. Miller, B. M. Peake, S. A. Rusak, A. L. Rose and T. D. Waite, Aquat. Sci., 2012, 74, 683–734 CrossRef CAS.
- M. Saeed, M. Muneer, A. ul Haq and N. Akram, Environ. Sci. Pollut. Res., 2021, 29, 293–311 CrossRef PubMed.
- D. I. Petkowicz, S. B. C. Pergher, C. D. S. da Silva, Z. N. da Rocha and J. H. Z. dos Santos, Chem. Eng. J., 2010, 158, 505–512 CrossRef CAS.
- Y. Wang, J. Dai, M. Wang, F. Qi, X. Jin and L. Zhang, J. Colloid Interface Sci., 2023, 636, 577–587 CrossRef CAS PubMed.
- S. Zhao, Y. Wen, X. Liu, X. Pen, F. Lü, F. Gao, X. Xie, C. Du, H. Yi, D. Kang and X. Tang, Nano Res., 2020, 13, 1544–1551 CrossRef CAS.
- M. J. Eslamibidgoli and M. H. Eikerling, ACS Catal., 2015, 5, 6090–6098 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02450h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |