
Multifunctional iron–cobalt heterostructure (FeCoHS) electrocatalysts: accelerating sustainable hydrogen generation through efficient water electrolysis and urea oxidation†
Arunagiri
Gayathri
 ,
Venkatachalam
Ashok
,
Jayaraman
Jayabharathi
,
Dhanasingh
Thiruvengadam
and
Venugopal
Thanikachalam
*
,
Venkatachalam
Ashok
,
Jayaraman
Jayabharathi
,
Dhanasingh
Thiruvengadam
and
Venugopal
Thanikachalam
*
Department of Chemistry, Material Science Lab, Annamalai University, Annamalai Nagar, Tamil Nadu 608002, India. E-mail: vtchalam2005@yahoo.com
First published on 23rd December 2024
Abstract
The urgent need to address escalating environmental pollution and energy management challenges has underscored the importance of developing efficient, cost-effective, and multifunctional electrocatalysts. To address these issues, we developed an eco-friendly, cost-effective, and multifunctional electrocatalyst via a solvothermal synthesis approach. Due to the merits of the ideal synthesis procedure, the FeCoHS@NF electrocatalyst exhibited multifunctional activities, like OER, HER, OWS, UOR, OUS, and overall alkaline seawater splitting, with required potentials of 1.48, 0.130, 1.59, 1.23, 1.40, and 1.54 V @ 10 mA cm−2, respectively. Moreover, electrolysers required only 1.40 V at 10 mA cm−2 for energy-saving urea-assisted hydrogen production, which was 190 mV lower than that of the alkaline water electrolyser. The alkaline sewage and seawater purification setup combined with the FeCoHS@NF electrolyzer led to a novel approach of producing pure green hydrogen and water. The ultrastability of the FeCoHS@NF electrocatalyst for industrial applications was confirmed using chronopotentiometry at 10 and 100 mA cm−2 over 110 h for OER, HER, UOR, and overall water splitting. The production of hydrogen using the FeCoHS@NF electrocatalyst in alkaline sewage water and seawater offers multiple benefits, including generation of renewable hydrogen energy, purification of wastewater, reduction of environmental pollutants, and low cost and low electricity consumption of the electrolyser system.
1. Introduction
The increased burning of fossil fuels has significantly elevated carbon dioxide emissions, leading to critical environmental challenges.1 As a clean energy source that only produces water vapour as a byproduct, hydrogen gas is a good choice for combating climate change, drinking water scarcity, and cutting carbon emissions. Hydrogen gas is a feasible alternative as a fuel in vehicles, and it also has a high energy density.2–5 Electrocatalytic water splitting (EWS) is an excellent route for hydrogen production, and it has zero carbon emission properties. Oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) are two half-reactions of EWS; both are tedious, although they use efficient benchmark catalysts of Pt/C (HER) and IrO2/RuO2 (OER). However, the high cost, scarcity, and low durability restrict their worldwide scalability for sustainable energy. Thus, effective, sustainable, plentiful and low-cost electrocatalysts with multifunctional activities are required for electrochemical water splitting.6,7 Nevertheless, limited clean water, kinetically slow OER, and expensive supplemental purification processes impede the growth of alkaline water splitting.Urea and sodium chloride, which are contaminants that are prevalent in water from domestic sewage, industrial processes, and agricultural operations, can readily produce N2 and CO2. Unlike freshwater, saltwater is abundant, as the oceans comprise approximately 96.5% of the Earth's water. However, direct human consumption without desalination is not practical. To solve this difficulty and clean tainted water, urea-assisted electrolysis on the anode side can be used to outperform OER. The theoretical potential of the urea oxidation reaction (UOR) is 0.37 V, which is significantly lower than that of the OER (1.23 V), resulting in a 70% power savings.8–10 For instance, combining the UOR with HER is projected to accomplish high-performance H2 generation owing to the simultaneous benefits it offers, including absence of H2/O2 explosion hazard, absence of reactive oxygen species, low electricity consumption, value-added products and effective pollutant degradation.11,12 In this sense, it is essential to investigate overall urea splitting, seawater splitting and urea-assisted seawater electrolysis.13,14
Metal glycerates have attracted the attention of researchers due to their MOF structure, LDH-like morphology with variable shapes and compositions, and high durability for electrocatalysis.15,16 Wang et al. solvothermally prepared FeNiGly microspheres as high-performance electrocatalysts for the OER, which requires 320 mV to attain 10 mA cm−2.17 Septiani et al. used the self-assembly method to synthesise NiCoTEP, which only requires 310 mV for the OER at 10 mA cm−2.18 Guo et al. prepared CoP nanospheres using a solvothermal method for the HER, which only needs 121 mV at 10 mA cm−2 in an acidic medium.19 Li et al. reported MoOx-FeCoCu as a bifunctional catalyst that needed 1.69 V to attain 10 mA cm−2.20 Previous studies on the metal glycerate system have shown that it is either only capable of a single functional activity or has a lower efficiency for bifunctional activities. Several approaches such as interface engineering and crystalline-amorphous interface are developed to enhance the inherent activity of electrocatalysts for bifunctional applications.3,11,21,22 All these techniques require high energy and more time, and are expensive. Therefore, a proper selection strategy is still required to combine the benefits of all techniques.
Hence, we prepared an iron–cobalt heterostructured multifunctional electrocatalyst by an energy-efficient solvothermal method. The benefits derived from synthetic strategies such as binder-free nature, crystalline-amorphous interface, heterostructure, synergism of bimetal and the combination of higher surface area cobalt with the fast kinetic iron in FeCoHS@NF rendered extraordinary performance for OERs, UORs, HERs and seawater splitting compared to the individual FeHS and CoHS. As a result, FeCoHS@NF has a low Tafel value, higher TOF, high surface area (152.6 cm2), low resistance (4.5 Ω) and high intrinsic activity. ECSA-normalised LSV curve indicated the outstanding intrinsic activity of FeCoHS@NF, and it only required 270 mV to attain 0.1 mA cm−2. The FeCoHS@NF demonstrated exceptional catalytic efficiency for HERs (130 mV), OERs (250 mV) and overall water splitting (1.59 V) at 10 mA cm−2 in 1 M KOH. The formation of active sites during the electrocatalytic performance was verified using post-analysis such as XRD, FT-IR spectroscopy, FESEM, and XPS. The FeCoHS@NF required only 1.23, and 0.130 V to attain 10 mA cm−2 for the UOR and HER, respectively. Furthermore, for urea-assisted hydrogen generation, the electrolyzer only needed 1.40 V at 10 mA cm−2, which was 190 mV superior to the alkaline water electrolyser. Solar-driven water electrolysis and environmental analysis provided more evidence of FeCoHS@NF effectiveness in producing sustainable hydrogen. Moreover, the selection strategy and synergistic effect of iron with cobalt may be used for constructing more affordable, multifunctional electrocatalysts for numerous electrochemical applications.
2. Results and discussion
Developing efficient and eco-friendly heterogeneous electrocatalysts for green hydrogen production is an urgent need to meet global energy demands and alleviate environmental concerns. It is very important to choose the appropriate starting materials and synthetic strategy, and the material must be non-toxic, readily available and highly efficient for atom utilization.23 Glycerol is a low-cost, renewable, non-toxic source and also affords wettability to the catalyst.15 Cobalt and iron nitrate are highly efficient, easily available, and do not affect the environment. Fig. 1 depicts the schematic representation of the FeCoHS@NF synthesis.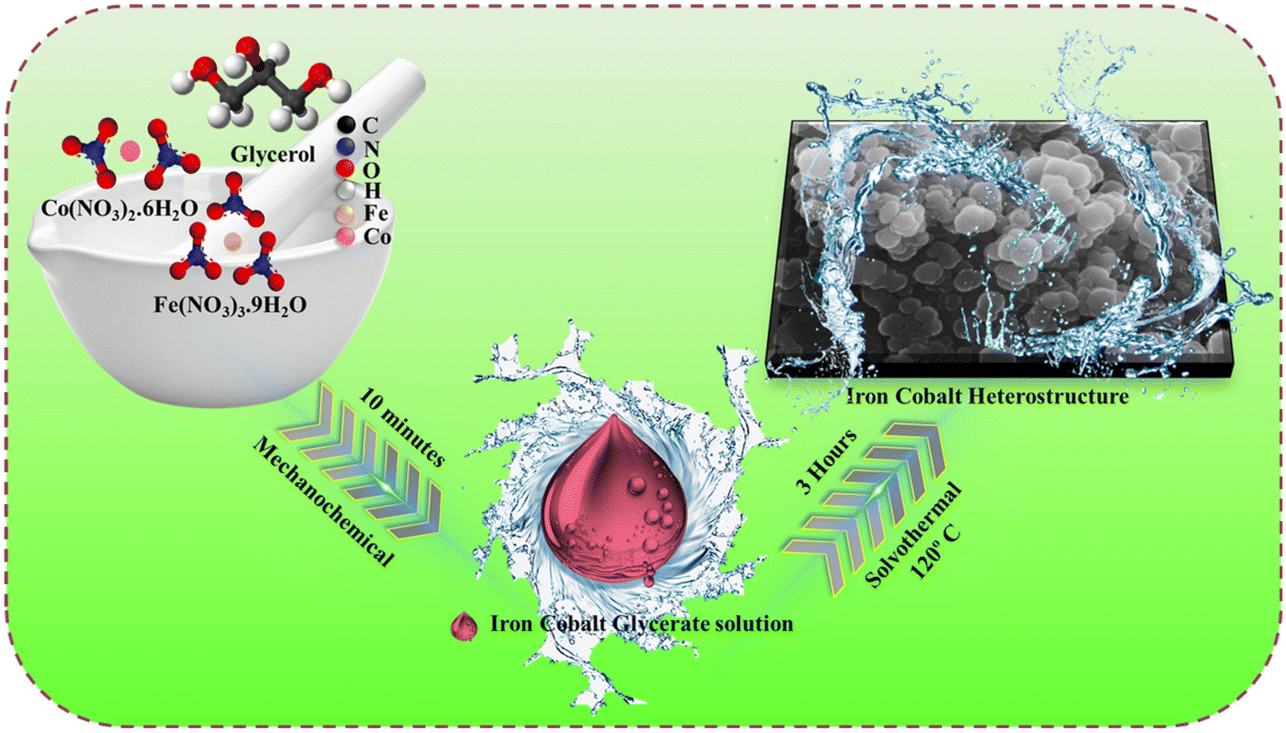 | ||
| Fig. 1 Synthetic route of FeCoHS@NF. | ||
Comprehensive synthetic procedure is provided in the ESI.† During the mechanochemical reaction, the hydroxide groups of glycerol react with the metal ions due to strong coordination affinity, forming an iron–cobalt glycerate solution. Mechanochemical reaction lowers the activation energy; hence, the reaction proceeds quickly at a lower temperature, consumes less energy and gives good yields. The OH− ions released from water at elevated temperatures react with iron–cobalt glycerate. At 80 °C, rapid hydrolysis occurs, results in the formation of Fe(OH)2 and Co(OH)2 and also provides metal glycerates.16,24–27 Compared to the conventional synthetic method, the ideal energy-efficient mechanochemical combined with a solvothermal method has several benefits. It has the advantages of reduced byproduct formation, higher starting material utilization, high selectivity, abundant active site, heterostructure, amorphous–crystalline interface, spherical nanoparticles with high surface area, low electricity utilisation, low cost and less time-consuming.
2.1. Physical characterization
XPS, XRD, FT-IR spectroscopy, FESEM and HRTEM were used to analyse the formation of targeted catalysts. XPS was employed to identify the oxidation state, constituents, and bonding nature of FeCoHS@NF, which are key parameters influencing catalytic performance (Fig. 2). The presence of C, O, Co, and Fe elements in FeCoHS@NF was established by the survey spectrum (Fig. S1a†). The spectrum of C 1s shows peaks at 284.7, 286.2 and 288.3 eV ascribed to C–C, C–O and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O fragments, respectively (Fig. 2a). The O 1s spectrum revealed signals at 529.8, 531.4, 532.2 and 533.3 eV attributed to M–O, M–OH, M–O–C (M = Co, Fe), and adsorbed water, respectively (Fig. 2b). The high-resolution Fe 2p spectra showed peaks at 711.2 eV (Fe 2p3/2) and 725.0 eV (Fe 2p1/2), and the presence of Fe3+ and Fe2+ is proven by the binding energy difference of 13.8 eV among the two peaks. It may also be fitted into shakeup satellites and spin–orbit doublets via the Gaussian fitting strategy. The peaks at 710.8 and 724.2 eV corresponded to Fe2+ and those at 713.3 and 727.2 eV were assigned to Fe3+ with satellite peaks at 717.8 and 732.4 eV, respectively (Fig. 2c). The Co 2p spectrum has peaks at 781.9, 783.8 and 785.9 eV attributed to Co3+, Co2+ and satellite peaks of Co 2p3/2 signals, respectively and peaks at 798.0, 798.9 and 803.0 eV assigned to Co3+, Co2+ and satellite peaks of Co 2p1/2 signals, respectively (Fig. 2d). High-resolution Ni 2p spectra have peaks at a low intensity, which shows the contribution of nickel from NF (Fig. S1b†). Thus, the XPS report also affirmed the construction of FeCoHS@NF.16,19,26,28–30 Fig. S2† presents the XPS spectra of FeHS and CoHS, along with a comparative analysis of FeCoHS. The incorporation of Fe induces a significant shift in the binding energy of the Co 2p XPS peak in FeCoHS compared to CoHS. This shift underscores strong electronic coupling between Co and Fe, thereby confirming the synergistic interaction and construction of FeCoHS.31
C–O fragments, respectively (Fig. 2a). The O 1s spectrum revealed signals at 529.8, 531.4, 532.2 and 533.3 eV attributed to M–O, M–OH, M–O–C (M = Co, Fe), and adsorbed water, respectively (Fig. 2b). The high-resolution Fe 2p spectra showed peaks at 711.2 eV (Fe 2p3/2) and 725.0 eV (Fe 2p1/2), and the presence of Fe3+ and Fe2+ is proven by the binding energy difference of 13.8 eV among the two peaks. It may also be fitted into shakeup satellites and spin–orbit doublets via the Gaussian fitting strategy. The peaks at 710.8 and 724.2 eV corresponded to Fe2+ and those at 713.3 and 727.2 eV were assigned to Fe3+ with satellite peaks at 717.8 and 732.4 eV, respectively (Fig. 2c). The Co 2p spectrum has peaks at 781.9, 783.8 and 785.9 eV attributed to Co3+, Co2+ and satellite peaks of Co 2p3/2 signals, respectively and peaks at 798.0, 798.9 and 803.0 eV assigned to Co3+, Co2+ and satellite peaks of Co 2p1/2 signals, respectively (Fig. 2d). High-resolution Ni 2p spectra have peaks at a low intensity, which shows the contribution of nickel from NF (Fig. S1b†). Thus, the XPS report also affirmed the construction of FeCoHS@NF.16,19,26,28–30 Fig. S2† presents the XPS spectra of FeHS and CoHS, along with a comparative analysis of FeCoHS. The incorporation of Fe induces a significant shift in the binding energy of the Co 2p XPS peak in FeCoHS compared to CoHS. This shift underscores strong electronic coupling between Co and Fe, thereby confirming the synergistic interaction and construction of FeCoHS.31
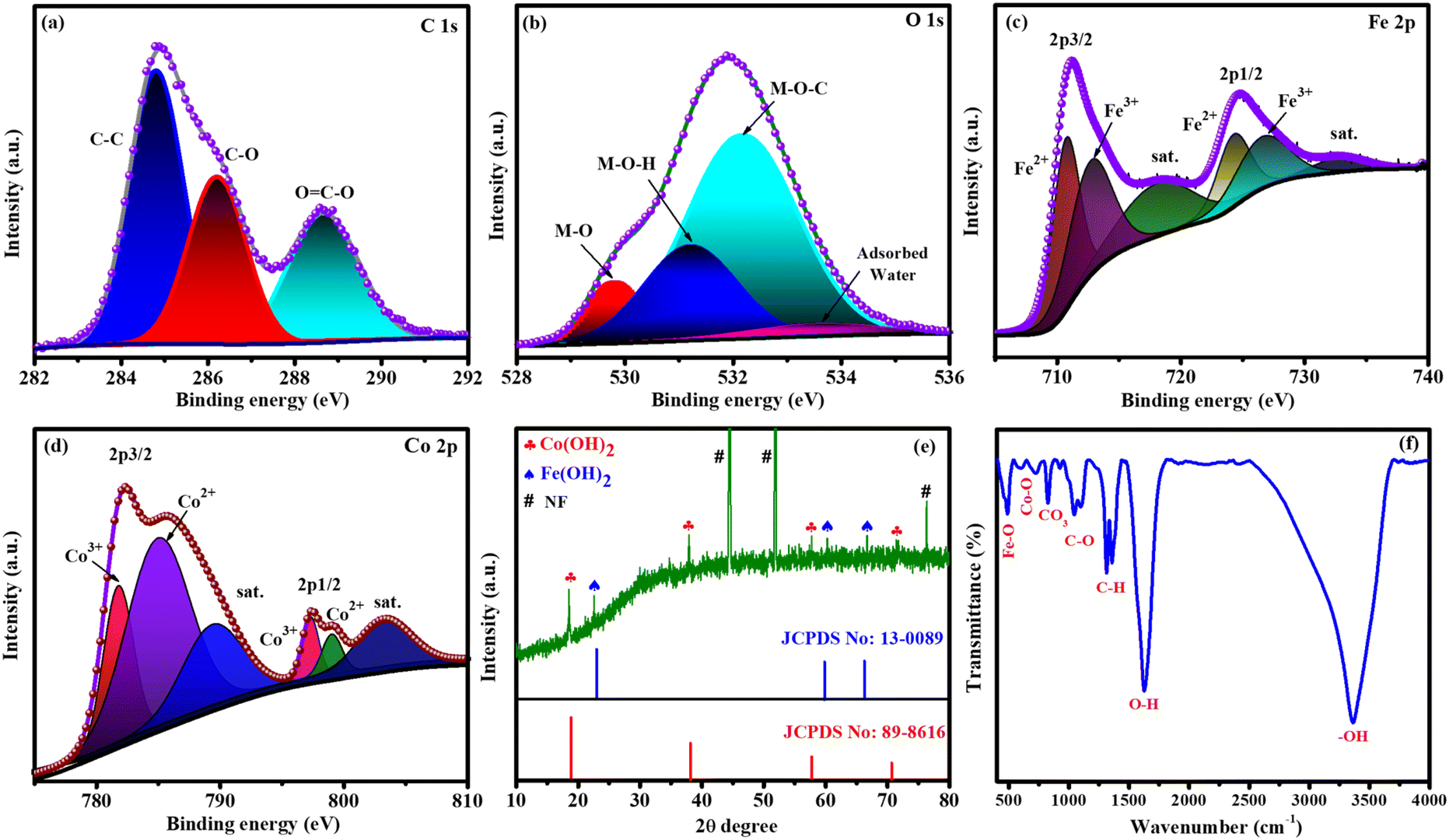 | ||
| Fig. 2 XPS spectra of FeCoHS@NF: C 1s (a), O 1s (b), Fe 2p (c) and Co 2p (d). XRD pattern (e) and FT-IR spectrum of FeCoHS@NF (f). | ||
Fig. 2e shows the XRD pattern of the FeCoHS@NF electrocatalyst, where no discernible peaks were observed, which may be due to the ferromagnetic properties of cobalt and iron. However, corresponding iron and cobalt hydroxides have been clearly identified with HR-TEM fringes and SAED patterns. The peaks are positioned at 19.0° (001), 37.8° (011), 57.8° (110), and 71.7° (112) for Co(OH)2, according to JCPDS No. 89-8616. Additional peaks at 22.4° (001), 60.6° (102) and 66.6° (110) correspond to Fe(OH)2, regarding JCPDS No. 13-0089. Notably, cobalt hydroxide exhibits higher crystallinity compared to iron hydroxide, which may be attributed to variations in the synthetic procedure. The XRD pattern also displays several unidentifiable reduced peaks, probably due to the dominant amorphous nature resulting from our synthetic method and the potential formation of metal-alkoxides, such as Co and Fe glycerates. The absence of identifiable XRD peaks for metal alkoxides suggests their amorphous state. This amorphous character of metal alkoxides has been reported in previous studies,15–20 and it aligns with our XRD pattern, which will be further confirmed using HR-TEM. Additionally, the presence of metal glycerates can be identified through FT-IR and XPS results. The presence of Fe(OH)2, Co(OH)2 crystalline phases and amorphous metal glycerates indicates the formation of a crystalline-amorphous interface and the FeCoHS@NF heterostructure nature of the electrocatalyst.15,16,32
Fourier transform infrared (FT-IR) spectroscopy was performed to analyse the functional groups of the prepared electrocatalyst. Fig. 2f shows the FT-IR spectrum of FeCoHS@NF. The peaks at ∼460, ∼576–663, ∼831, ∼850–1000, ∼1000–1200, and ∼1300–1400 cm−1 belong to the Fe–O bending, Co–O stretching, CO32− stretching, C–H bending, C–O stretching, and C–C stretching vibrations, respectively. The peaks at 1628 and 3334 cm−1 were attributed to the bending and stretching vibration of H2O and –OH groups in LDH, respectively. The presence of C–H bending and metal peaks shows the presence of metal glycerate molecules in FeCoHS@NF. Except for the splitting broad nature of C–O and CO32− stretching vibrations, CoHS has CO32− stretching, C–O stretching, C–C stretching and C–H bending vibrations similar to FeCoHS@NF (Fig. S3a†). FeHS also has all the stretching and bending vibrations similar to FeCoHS@NF due to more oxidation behaviour of iron nitrate than cobalt nitrate (Fig. S3b†). The calcination of FeCoHS@NF leads to the decomposition of metal glycerates and hydroxides and the removal of interfacial water molecules from FeCoHS@NF. Hence, the calcined FeCoHS@NF has no stretching and bending peaks (Fig. S3c†). The changes in spectral patterns clearly show the formation of catalysts with different structural properties.16,33–37
FESEM images examined the morphology of FeCoHS@NF at different resolutions. Fig. 3a–c display the development of spherical nanoparticles on a nickel foam. This nanosphere offers ample space for catalytic sites, which is beneficial for the quick diffusion of ions, electrons and O2 and H2 bubbles during the OER, UOR and HER, respectively. The existence of the component elements and their uniform distribution were verified by EDX elemental mapping (Fig. 3d–g). The lack of additional elements demonstrates the purity of FeCoHS@NF, with atomic proportions of 38.78% (C), 42.3% (O), 12.53% (Fe) and 6.39% (Co) (Fig. 3h). The FESEM images and EDX elemental mapping of calcined FeCoHS@NF agglomerated morphology with irregular particle size and non-uniform elemental distribution are shown in Fig. S4.† It may be due to the decomposition of metal glycerates, hydroxides and oxyhydroxides in FeCoHS@NF, which is consistent with the FT-IR results. The decomposition of glycerate leads to the agglomeration of particles, which results in shape change with uneven metal distribution. There are several drawbacks to nanoparticle aggregation including higher resistance, low surface area, and restricted electrolyte interaction. It shows the importance of glycerol; the formation of metal glycerates gives an even distribution of metals with unique morphologies.7,26,27 They underwent fast surface reconstruction under OER/HER conditions, resulting in rapid active site oxyhydroxide/hydroxide formation. Furthermore, because of the layered and open structure, the reactants may be integrated into the interlayer gap of metal glycerate, resulting in numerous accessible catalytic sites and quick transportation. It perfectly illustrates the significance of the synthetic strategy for heterointerface formation.15,16
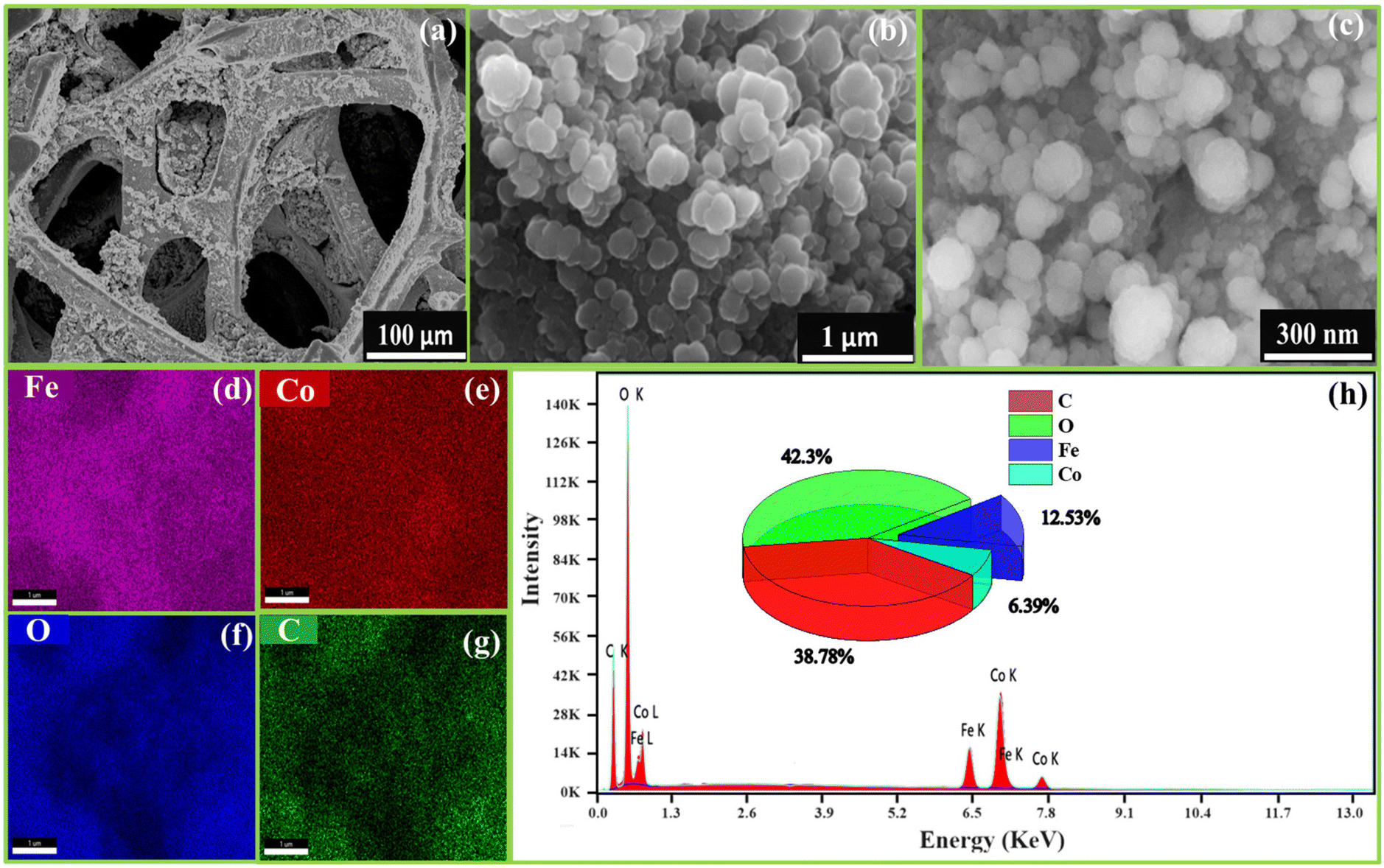 | ||
| Fig. 3 FE-SEM images of FeCoHS@NF at different magnifications (a–c), elemental mapping (d–g) and corresponding EDX spectra (h). | ||
The HR-TEM images are acquired using the electrocatalyst scraped off from the NF. The HR-TEM image was used to further confirm the nanosphere morphology, composition, and crystalline-amorphous interface of FeCoHS@NF. Fig. 4a shows the lower magnification HR-TEM image of FeCoHS@NF. Fig. 4b and c show that FeCoHS@NF has nanospherical morphology with a crystalline-amorphous interface, and the white marking in the image represents the formation of crystalline-amorphous interface, which is in line with the XRD. The yellow marking represents the presence of amorphous carbon. Fig. 4c exhibits the lattice fringes with d-space values of 4.6530, 2.3732 and 1.7820 Å, which was ascribed to the (001) and (011) planes of Co(OH)2 and the (102) planes of Fe(OH)2. It verified the heterostructure construction, consistent with the XRD findings. Fig. 4d shows the SAED pattern of FeCoHS@NF with diffused rings consisting of the (001), (011), and (102) planes which are in line with XRD results, which also confirm the crystalline-amorphous interface nature. The EDX spectrum exhibits the presence of respective (Fe, Co, C and O) elements and the lack of additional components demonstrates the pure state of the catalyst (Fig. 4e). The atomic proportions of FeCoHS@NF were determined to be 5.66% (Fe), 3.96% (Co), 49.40% (C), and 40.98% (O). Fig. 4b–d demonstrate the successful formation of heterostructures, that enhanced electron transport and provided additional catalytic sites at the heterointerfaces for overall water splitting, HERs, UORs and OERs.3,11 All the characterization confirmed the formation of an electrocatalyst with expected benefits.
 | ||
| Fig. 4 HR-TEM images of FeCoHS@NF (a and b), lattice fringes (c), SAED pattern (d) and EDX spectrum (e). | ||
2.2. Electrochemical analysis
After the successful confirmation of the formation of the desired electrocatalyst, we investigated the electrocatalytic characteristics of the catalyst. The working electrode was an electrocatalyst coated onto a nickel foam, while the reference and counter electrodes were a graphite rod and a saturated Hg/HgO electrode, respectively.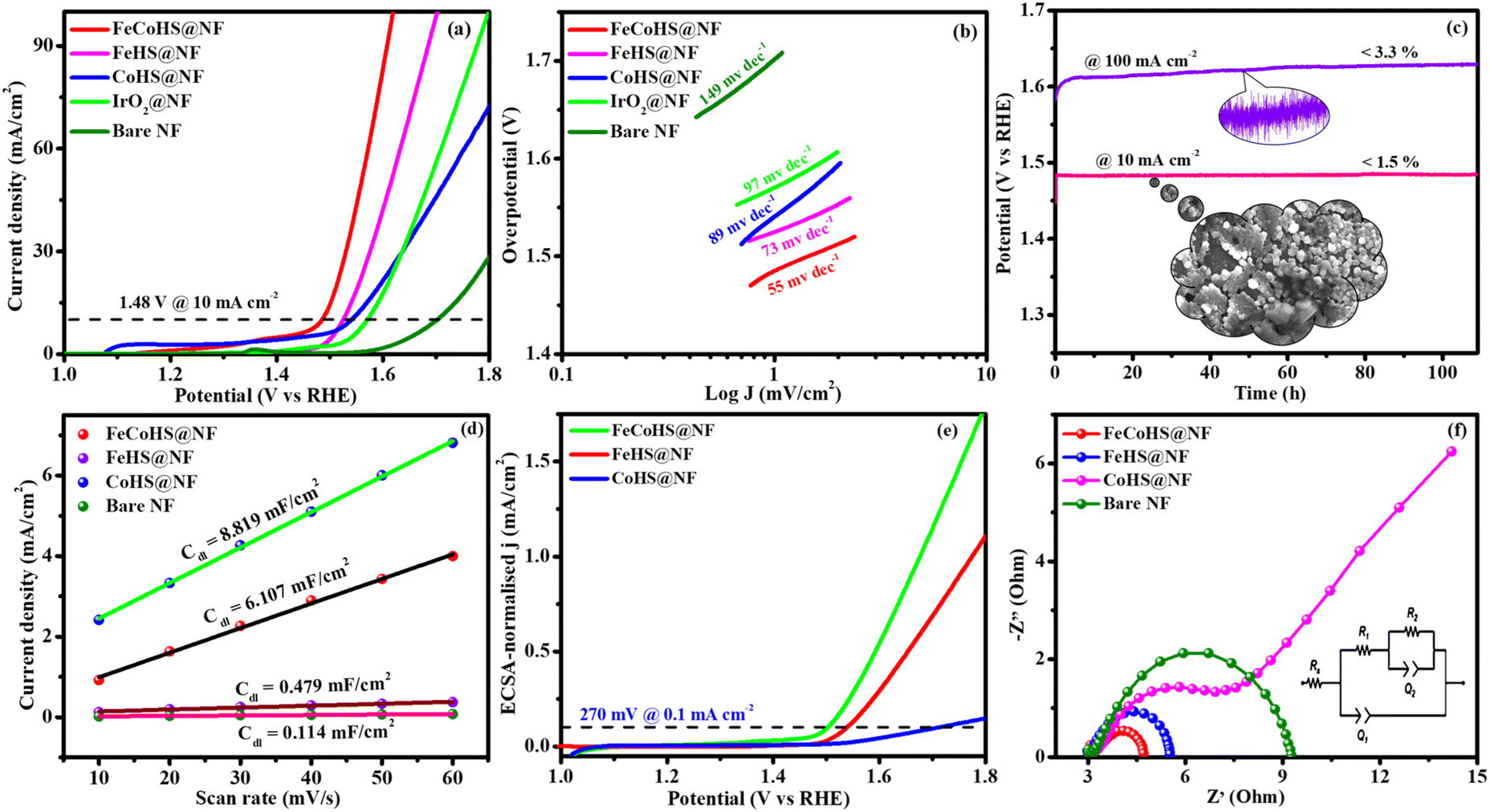 | ||
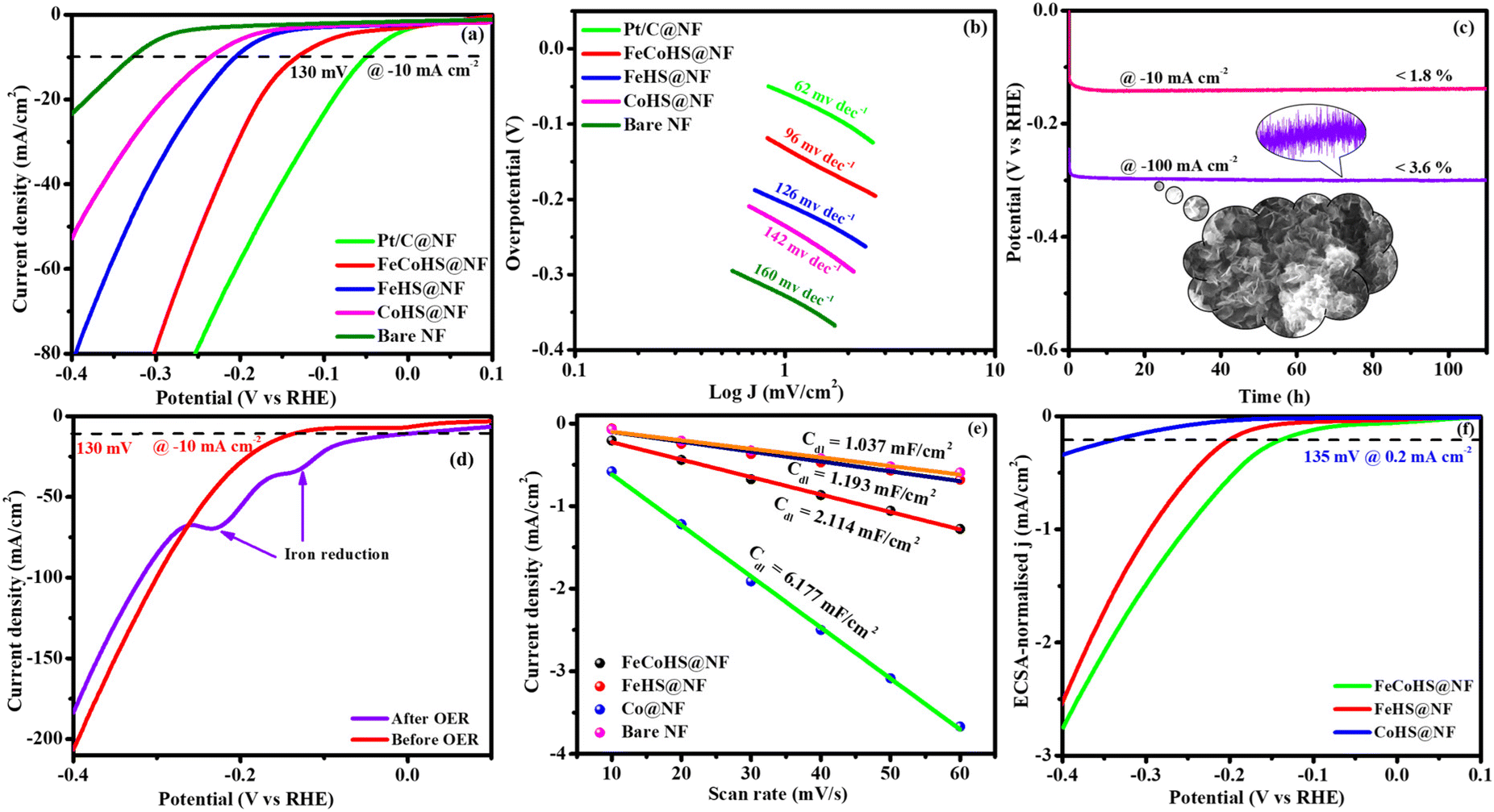
| Fig. 5 OER polarization curves of electrocatalysts: (a) LSV (without iR-correction); (b) Tafel plots; (c) chronopotentiometry durability test (inset: post OER image); (d) double-layer capacitance; (e) ECSA-normalized LSV curve and (f) Nyquist plot. | ||
The FeCoHS@NF electrocatalyst followed the common OER mechanism: M* + OH− → M–OH* + e−: M–OH* + OH− → M–O* + H2O + e−: M–O* + OH− → M–OOH* + e−: M–OOH* + OH− → O2 + H2O + e−. The presence of Fe ions as Fe2+/Fe3+ (Fe(OH)2/FeOOH) and cobalt as Co2+/Co3+ (Co(OH)2/CoOOH) oxidation states during the OER process was supported by post-analysis SEM, XPS, XRD and IR.38,39 Fig. S5a† shows the cyclic voltammetry of the as-prepared electrocatalyst in the range of 1.0–1.8 V. Fig. S5b† shows the redox behaviour of all electrocatalysts up to 10 mA cm−2, all having different behaviours. CoHS@NF has Co2+/Co3+ strong oxidation behaviour at 1.1 V, but FeCoHS@NF does not have the redox peak. This may be due to the influence of iron over cobalt. However, FeHS@NF iron does not exhibit redox behaviour due to its fast kinetics. Fe addition plays a crucial role in the OER process in cobalt electrocatalysts. At the same time, the actual function of iron and the properties of the catalytic sites in iron catalysts remain controversial. Fe3+ facilitates the development of LDH in cobalt hydroxide, providing additional accessible catalytic sites than bulk pure hydroxides; additionally, Fe3+ has much faster OER kinetics than the Co electrocatalyst because it has the ideal binding energies for the intermediate's adsorption during the OER. Fe3+ having strong Lewis acidity alters the electrical characteristics of its cobalt catalyst, lowering the activation barrier and improving the OER activity by oxidising hydroxyl protons coupled with the cobalt.7,16
Furthermore, to shed light on the role of the structure–activity relationship and electrochemical surface area (ECSA), we measured the double-layer capacitance for each electrocatalyst by running a cyclic voltammogram (CV).40 CV curves are harvested at different scan rates of 10–60 mV s−1 within a range of 1.15–1.25 V vs. RHE, excluding non-faradaic phenomena (Fig. S6†). The calculated double layer capacitance values of FeCoHS@NF, FeHS@NF, CoHS@NF and bare NF are 6.107 mF cm−2, 0.479 mF cm−2, 8.819 mF cm−2 and 0.114 mF cm−2, respectively. FeCoHS@NF has a greater Cdl of 6.107 mF cm−2 compared to FeHS@NF (0.479 mF cm−2) and NF (0.114 mF cm−2), as shown in Fig. 5d, when plotting current density change (Δj) at 1.20 V versus RHE against scan rate. The calculated ECSA and RF values of FeCoHS@NF, FeHS@NF, CoHS@NF and bare NF are 152.6/610.4, 11.9/47.9, 220.4/881.6 and 2.85/11.4, respectively. To confirm the intrinsic activity of the FeCoHS@NF catalyst, the ECSA-normalized LSV curve was analysed (Fig. 5e). The overpotential required for a current density of 0.1 mA cm−2 ECSA was 270, 310 and 470 mV for FeCoHS@NF, FeHS@NF, and CoHS@NF, respectively. The excellent OER performance of FeCoHS@NF is owing to the greater ECSA, but also because of the increased intrinsic performance of the catalyst due to the improved electronic interaction.7
Fig. 5f shows the results of the electrochemical impedance analysis during the OER together with a circuit model fitting assessment (inset: Randles equivalent circuit diagram, where Rs is the electrolyte resistance and R1 and R2 are charge transfer resistance; Q1 and Q2 are double-layer capacitance).41 The lower Rct value of FeCoHS@NF(4.5 Ω) than FeHS@NF (5.6 Ω), CoHS@NF (25.6 Ω), and bare NF (10.3 Ω) may be due to the synergistic interaction of iron and cobalt in FeCoHS@NF, which provides an extremely active structure that facilitates fast response kinetics by transferring electrons across the electrode.
CoHS@NF has a higher Cdl (8.819 mF cm−2) value, which suggests that it contains more catalytic sites that are suitable for electrochemical processes. However, because of its significantly larger Rct (25.6 Ω), it has a lower catalytic activity. FeHS@NF exhibited better catalytic activity than CoHS@NF due to its much lower Rct value (5.6 Ω), although having a lower Cdl (0.479 mF cm−2) value. CoHS@NF has a higher double-layer capacitance value and FeHS@NF has a higher intrinsic activity than each other. All these results confirmed that the as-prepared FeCoHS@NF using the ideal synthetic strategy has lower charge transfer resistance, higher double-layer capacitance and excellent intrinsic activity.42 Collectively, the outcomes demonstrated that FeCoHS@NF had the benefits of elevated catalytic sites and improved electron transfer due to the modulated electron transfer at the interface, resulting in enhanced reaction kinetics and drastically lower overpotentials when compared to the individual FeHS@NF and CoHS@NF.11,21,43
The exact half-cell ability to Pt/C, FeCoHS@NF, FeHS, CoHS, and bare NF for HER performance was investigated using LSV in a three-electrode setup (without iR correction). The LSV was detected in 1 M KOH at a potential of −0.4 to 0.1 V. FeCoHS@NF had a lower overpotential (130 mV) and a Tafel value (96 mV dec−1) than those of FeHS@NF (204 mV; 126 mV dec−1), CoHS@NF (233 mV; 142 mV dec−1), and bare NF (328 mV; 160 mV dec−1), according to the LSV and Tafel slope. Associated with the newly reported low-cost catalysts, FeCoHS@NF exhibits exceptional activity @ −10 mA cm−2, despite Pt/C having a lower Tafel slope (62 mV dec−1) and overpotential (50 mV) (Table S1†). The inferior Tafel and overpotential values exhibit excellent performance and rapid kinetics of FeCoHS@NF (Fig. 6a and b). The computed TOF at −0.25 V of 0.1371/s (FeCoHS@NF), 0.0532/s (FeHS), 0.0322/s (CoHS), 0.2036/s (Pt/C) and 0.0081/s (NF) displays the outstanding HER performance of FeCoHS@NF in a given period. The FeCoHS@NF displays stable performance and extraordinary durability over 110 h @ 10/100 mA cm−2 with 1.8% and 3.6% activity degradation (Fig. 6c). The synergistic interaction of crystalline-amorphous interface and Fe(OH)2/Co(OH)2/NF heterostructure significantly improves the HER activity by increasing the charge transfer rates, potential catalytic sites, and charge dispersion from the heterointerface.22 The HER comparative LSV before and after the OER is displayed in Fig. 6d. Following the OER, the two iron reduction peaks (Fe3+/2+ & Fe2+/1+) in the HER LSV support the formation of different active sites during the reactions.42 The common HER mechanism followed by FeCoHS@NF in alkaline electrolytes may be Volmer–Heyrovsky: [Mcat + e− + H2O ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) OH− + McatHchem (Volmer); McatHchem + e− + H2O H2↑ + OH− + Mcat (Heyrovsky)].39 The post-analysis confirmed that the formation of the surface CoFe(OH)2 layer on FeCoHS@NF may aid as the active centre for HERs (Fig. S10–S18†). The evaluation of HER/OER performance of FeCoHS@NF, FeHS and CoHS exhibits that the outstanding performance of FeCoHS@NF may be attributed to the synergistic influence of the fast kinetics of iron to cobalt.
OH− + McatHchem (Volmer); McatHchem + e− + H2O H2↑ + OH− + Mcat (Heyrovsky)].39 The post-analysis confirmed that the formation of the surface CoFe(OH)2 layer on FeCoHS@NF may aid as the active centre for HERs (Fig. S10–S18†). The evaluation of HER/OER performance of FeCoHS@NF, FeHS and CoHS exhibits that the outstanding performance of FeCoHS@NF may be attributed to the synergistic influence of the fast kinetics of iron to cobalt.
 | ||
| Fig. 6 HER polarization curves of electrocatalysts: (a) LSV curve; (b) Tafel slopes; (c) chronopotentiometry durability test (inset: post HER image); (d) HER-LSV before and after OER; (e) double-layer capacitance and (f) ECSA-normalized LSV curve. | ||
The CV curves at 10–60 mV s−1 scan rates within 0.20–0.00 V vs. RHE (excluding non-faradaic contributions) were analysed (Fig. S7†). The calculated Cdl values for FeCoHS@NF, FeHS@NF, CoHS@NF, and bare NF are 2.114, 1.193, 6.177, and 1.037 mF cm−2, respectively (Fig. 6e). The corresponding ECSA and RF values are 52.8/211.2, 29.8/119.2, 154.4/617.6, and 25.9/103.6, respectively. The intrinsic activity of the FeCoHS@NF catalyst was confirmed by ECSA-normalized LSV analysis (Fig. 6f). The overpotential for achieving 0.2 mA cm−2 ECSA was 135, 206, and 335 mV for FeCoHS@NF, FeHS@NF, and CoHS@NF, respectively. The superior HER performance of FeCoHS@NF is attributed to its larger ECSA and enhanced intrinsic activity due to improved electronic interactions.
Electrolysis was also performed using GC to determine the true catalytic activity of the electrocatalysts and understand the advantages of nickel foam (NF) over glassy carbon (GC). The electrochemical properties of the as-prepared catalysts towards OERs and HERs were evaluated in GC, and they are depicted in Fig. 7a and b, where the founded activity phenomena reflect the actual OER/HER performance. The overpotentials of FeCoHS@GC, FeHS@GC, CoHS@GC and IrO2@GC for OERs at 10 mA cm−2 are 290, 440, 310 and 390 mV, respectively (Fig. 7a). The overpotentials of FeCoHS@GC, FeHS@GC, CoHS@GC and Pt/C@GC for HER at −10 mA cm−2 are 255, 385, 347 and 107 mV, respectively (Fig. 7b). The OER activity of CoHS was unchanged in NF and GC, while the activity of FeCoHS and FeHS were lowered in GC 40 and 150 mV respectively. Glassy carbon (GC) is an inert substrate that can explore the intrinsic activity of electrocatalysts and only a lower amount of catalyst can be loaded onto GC. Consequently, electrocatalysts supported on GC often exhibit reduced catalytic performance. In Fig. 7a, the inset image shows resistance created by the O2 gas formation on the GC surface, whereas NF has good conductivity and 3D architecture beneficial for the fast release of generated gas.16 Hence, the activity of FeCoHS@NF and FeHS is a combination of iron with the nickel foam substrate, possibly due to the synergistic interaction of iron with the nickel foam substrate.44–46 We included this different substrate concept to reveal the actual catalytic activity; however, the manuscript primarily focused on large-scale hydrogen production, hence we referenced catalytic activities reported with the NF substrate.
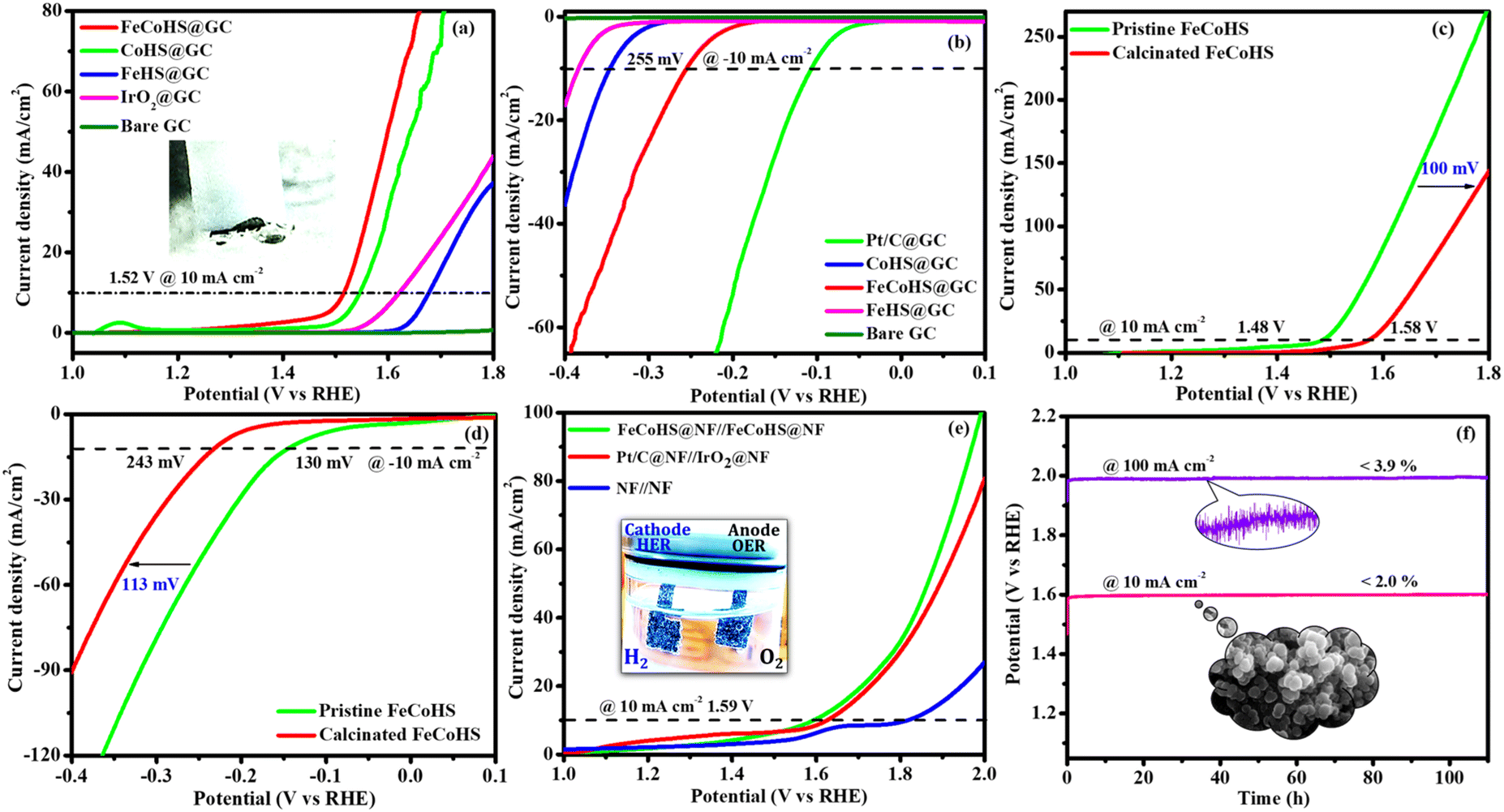 | ||
| Fig. 7 OER-HER activity comparison: LSV of different electrocatalyst in GC (a and b), OER-HER activity comparison LSV of pristine and calcinated FeCoHS@NF (c and d), overall water splitting LSV and Stability in KOH (e and f). | ||
To understand the importance of iron–cobalt heterostructure (oxide, hydroxide, and oxyhydroxide), we compared the activities of as-prepared FeCoHS@NF with calcined FeCoHS@NF. At 350 °C, the decomposition of metal glycerates, hydroxides, and oxyhydroxides and the removal of interfacial water molecules only occurred, but carbonization did not occur at <400 °C. Hence, the low activity may be due to the decomposition of metal glycerates, hydroxides, and oxyhydroxides and the removal of interfacial water molecules from FeCoHS@NF, which play a more optimistic role on the improvement of catalytic activity toward the OER process, under this condition. The strong coupling of crystalline-amorphous interface and heterostructure plays a remarkable role in optimising OER kinetics.16,18 For the OER, calcined FeCoHS@NF (350 mV) has a lower catalytic activity of 100 mV than that of as-prepared FeCoHS@NF (250 mV) @ 10 mA cm−2, whereas for the HER, calcined FeCoHS@NF (243 mV) has 113 mV lower catalytic activity than as-prepared FeCoHS@NF (130 mV) @ −10 mA cm−2, which confirms the importance of heterostructure for the excellent electrocatalytic performance (Fig. 7c and d).
Inspired by the FeCoHS@NF electrode's higher double-layer capacitance, lower resistance, excellent intrinsic activity, outstanding performance, and stability against HERs and OERs, we investigated the feasibility of using it as a cathode and anode (FeCoHS@NF||FeCoHS@NF) in a full-cell system. The polarization curve of the FeCoHS@NF||FeCoHS@NF electrolyser in 1.0 M KOH is displayed in Fig. 7e. To achieve 10 mA cm−2, only 1.59 V of cell voltage is required for the FeCoHS@NF||FeCoHS@NF electrolyser, which is less than the Pt/C||IrO2 benchmark electrolysers and bare NF||bare NF, which are 1.62 V and 1.81 V, respectively. In 1 M KOH, the FeCoHS@NF electrocatalyst demonstrated superior bifunctional activity to previously published non-precious catalysts (Table S1†). Chronopotentiometry stability was used for 110 hours at 10 and 100 mA cm−2 to confirm the stability of the electrocatalyst for industrial applications. The results demonstrated ultra-low potential loss and outstanding stability of the electrocatalyst (Fig. 7f). FeCoHS@NF produced 1.499 μmol min−1 of O2 and 3.06 μmol min−1 of H2, indicating a faradaic efficiency of about 96.5% (Fig. S8†).47
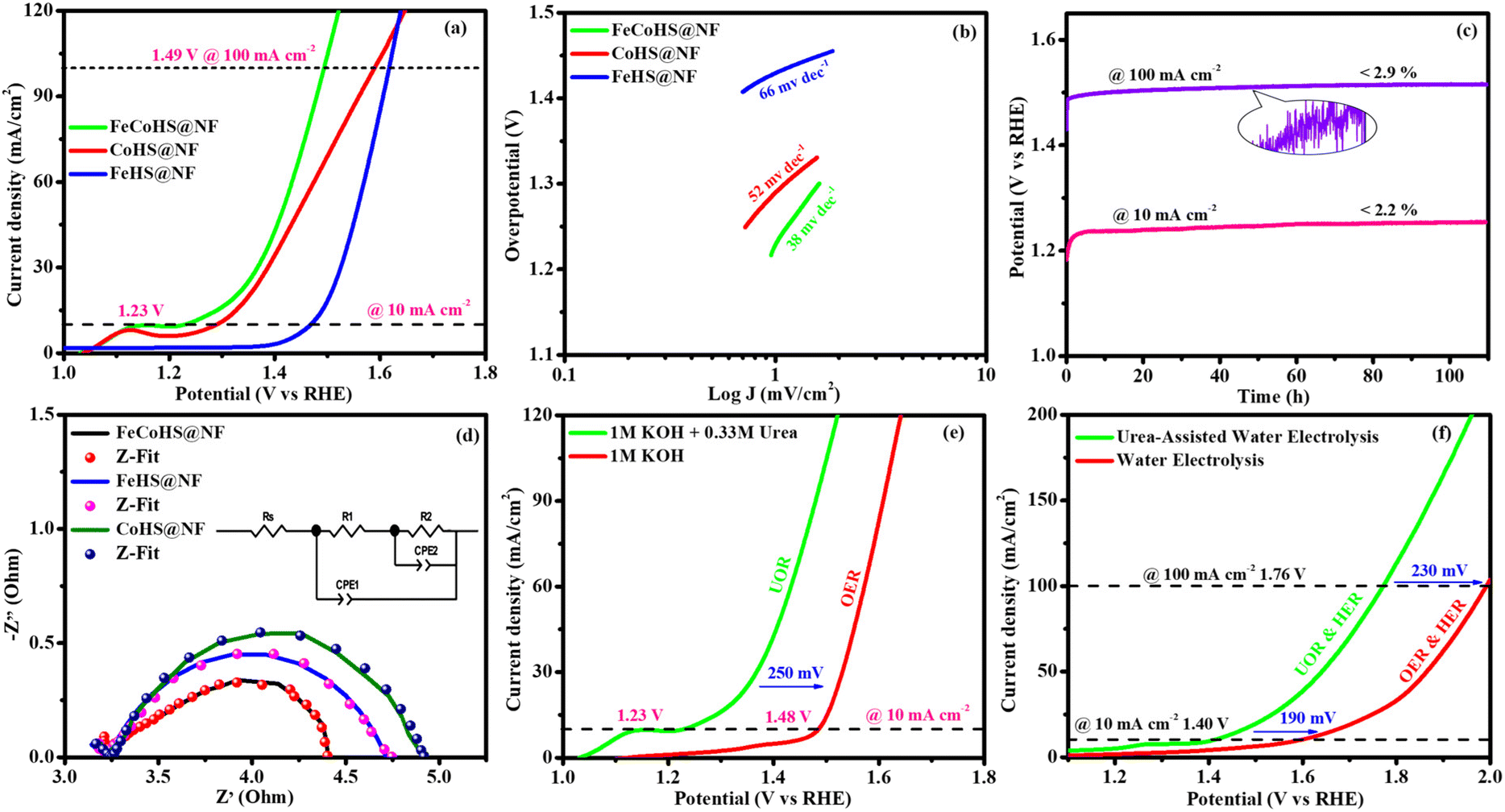 | ||
| Fig. 8 UOR polarization curves (without iR correction) of electrocatalysts: (a) LSV curve; (b) Tafel slopes; (c) chronopotentiometry durability test (inset: post HER image); (d) Nyquist plots (inset: Randles equivalent circuit); and (e and f) comparison of the LSV curve for the OER and electrolyzer with KOH. | ||
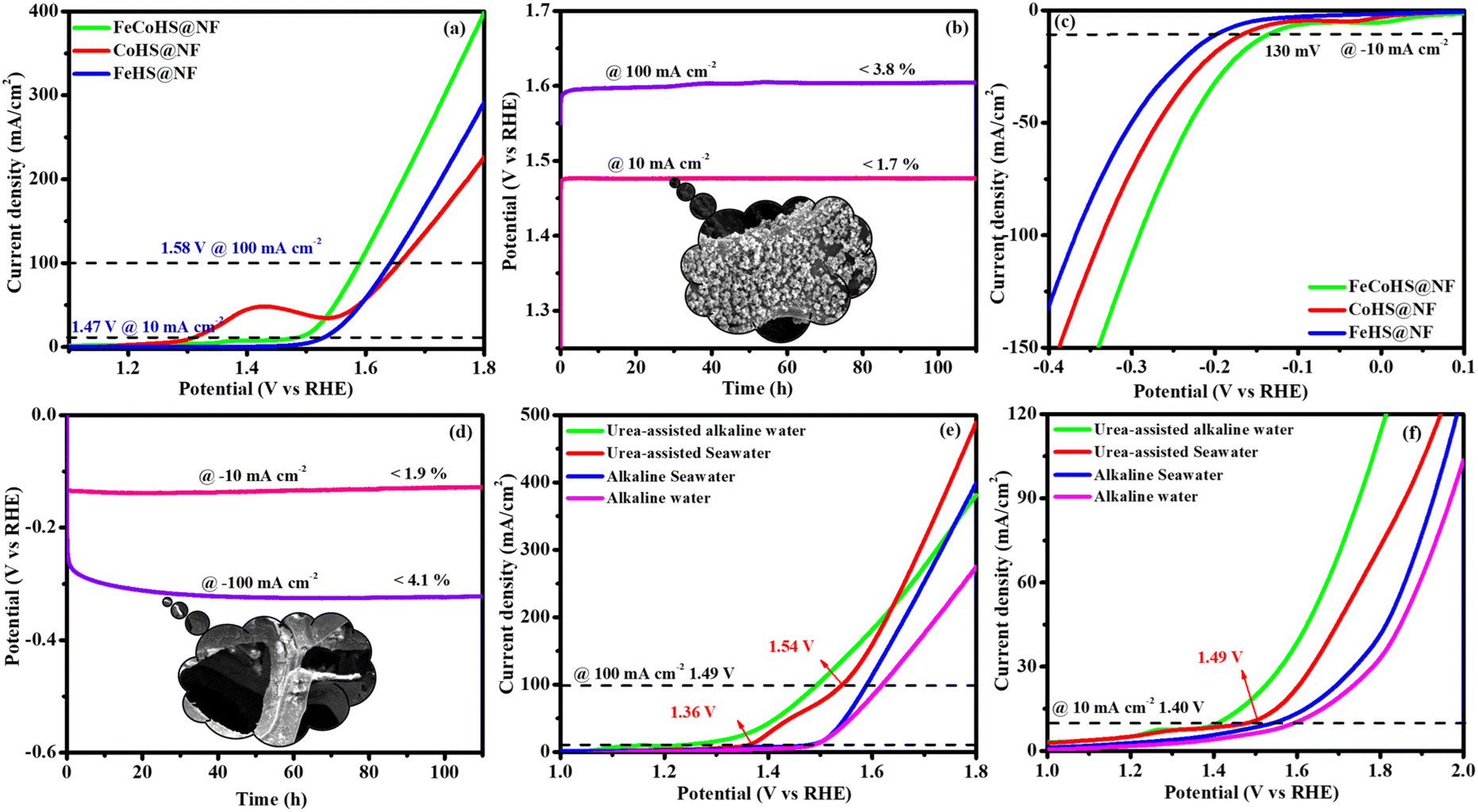 | ||
| Fig. 9 OER and HER in alkaline seawater without iR correction: OER-LSV and stability (inset: post OER image) (a and b); HER-LSV and stability (inset: post HER image) (c and d); and comparison of FeCoHS@NF activity in different electrolytes for half-cell and full-cell (e and f). | ||
2.3. Post analysis
To acquire a better understanding of the reaction process, the FeCoHS@NF electrode following HERs and OERs was characterised by XPS, FT-IR spectroscopy, XRD and FESEM. The post-FESEM images of FeCoHS@NF display that the nanospherical morphology was changed after electrocatalytic performance, and it demonstrates the generation of metal oxyhydroxide/hydroxide during the OER/HER. Fig. S10a and d† demonstrate the excellent adhesion of the electrocatalyst after 110 hours of intense H2/O2 gas development, demonstrating the ultra-durability of FeCoHS@NF. Fig. S10b and c† clearly show the heteromorphology of the catalyst with separated hexagonal cobalt oxyhydroxide nanosheets (red circle) and metal oxide nanospheres (blue circle) and the spherical nanoparticles are covered by hexagonal cobalt oxyhydroxide nanosheets (yellow circle) in the post-OER FESEM.16,44 The Post-HER FESEM images showed the interconnected nanosheet morphology attributed to the development of the metal hydroxide on the surface phase (Fig. S10e and f†).52EDX elemental mapping validated the uniform dispersion of component elements as well as their existence after 110 h of stability (Fig. S11 and S12†). The lack of additional components demonstrates the purity of FeCoHS@NF, and it comprises the atomic proportions of 6.36% (Fe), 3.73% (Co), 10.49% (Ni), 50.59% (O) and 28.83% (C) after the OER (Fig. S11g†). The increment of oxygen in EDX and smooth surface confirms the development of oxyhydroxide during the OER. After the HER, FeCoHS@NF has atomic percentages of 9.77 (Fe) 5.53 (Co), 1.84 (Ni), 54.80 (O) and 28.06 (C) (Fig. S12g†). During the HER, FeCoHS@NF only endured surface reconstruction, resulting in the development of Co(OH)2 on the FeCoHS@NF surface. During the OER, the FeCoHS@NF phase was entirely transformed into amorphous CoOOH and spherical nanoparticles, indicating significant reconstruction, where the metal glycerate was aided as a sacrificial agent for the fast conversion of metal sites to highly active site oxyhydroxides and hydroxides, thereby boosting the OER and HER activities.7,53,54 Fig. S13† shows the FESEM images of post-UOR, and it shows the excellent stability of the catalyst after 110 h of urea oxidation and the nanocluster morphology. Fig. S14† shows the FESEM images of post-OER and HER in alkaline seawater. It confirmed the excellent chlorine corrosion tolerance and good adhesion of the catalyst after seawater electrolysis.
Fig. S15† shows the post-XRD patterns of FeCoHS@NF, and the crystalline-amorphous nature of the electrocatalyst changed due to the development of amorphous metal hydroxide/oxyhydroxide surface layer. The sharp crystalline peaks become broader and fade after electrolysis due to the formation of an amorphous layer and “#” represents the NF substrate.8 The XRD patterns shown in Fig. S15a† (post-OER) and Fig. S15d† (post-UOR) reveal diffuse peaks in the range of 20° to 60°, which may indicate the formation of MOOH (M = Fe3+ and Co3+).16 The post-HER XRD pattern preserved some new small Fe(OH)2/Co(OH)2 peaks, indicating that metal hydroxides are prominent catalytic sites for the HER (Fig. S15b†). Post-analysis shows that FeCoHS@NF is elevated with the surface of hydroxides/oxyhydroxides during the OER and hydroxides during the HER. Fig. S15c† shows that the nickel foam substrate forms strong peaks at 44.3°, 51.6°, and 76.5° in the bare NF XRD (JCPDS No. 87-0712).7
The post-FTIR spectra of FeCoHS@NF were changed and showed new peaks at ∼1000–1200, ∼1360, ∼1647 and ∼3400 cm−1 attributed to the C–O stretching vibration, interlayer symmetric stretching vibration of CO32−, bending vibration of H2O molecules in the interlayers, and stretching vibration of OH− groups present in the layers of FeCo-LDH, respectively (Fig. S16†).35,36 The C–H bending vibration disappeared after the electrocatalysis. The disappearance of the C–H group mainly contributes to the formation of electrochemically active sites. Fig. S16a† shows the post-OER FTIR spectra of FeCoHS@NF, with new bands in the 428–784 and 847 cm−1 region attributed to the stretching vibrations of M–O and O–M–O (M = Fe and Co) and bending modes of OH– of FeOOH.55,56 Fig. S16b† displays the post-HER FTIR spectra of FeCoHS@NF, and the new bands in the range of 525–640, 614 and 520–810 cm−1 can be attributed to the vibrations of M–O, M–OH and O–Fe–O (M = Fe and Co) respectively.26,36,54
The post-XPS analysis confirmed the oxidation state and composition changes of FeCoHS@NF after 110 h of prolonged HER/OER catalytic activity. The survey spectrum revealed the existence of components in the electrocatalyst, including C, O, Co, and Fe after stability (Fig. S17a†). High-resolution Fe 2p spectra shows peaks at the same position similar to the pristine electrocatalyst even after 110 h of OER/HER catalytic performance. It shows the highest stability of iron in iron cobalt oxide under alkaline conditions for the OER/HER (Fig. S17b†).57 The spectrum of C 1s slightly sharpened and the intensity lowered, but their peak positions are unchanged (Fig. S17c†). The post-XPS of oxygen was completely changed after 110 h of OER/HER catalytic performance. The peak at ∼533.3 eV for adsorbed water disappeared and the peak intensity of M–O–C (∼532.2 eV) was lowered, exhibiting that most of the metal glycerate was converted into metal oxides/hydroxides/oxyhydroxides (Fig. S18a and c†). Post-OER oxygen XPS shows a peak at ∼529.8, ∼530.7, ∼531.4 and ∼532.2 eV attributed to M–O, M–OOH, M–OH and M–O–C (M = Co and Fe), respectively (Fig. S18a†).7,26,29,30 The post-OER XPS Co 2p spectrum has peaks at 780.6, 782.3 and 786.1 eV attributed to Co3+ and Co2+ and satellite peaks of Co 2p3/2 signals, respectively and peaks at 796.4, 798.2 and 802.6 eV assigned to Co3+, Co2+ and satellite peaks of Co 2p1/2 signals, respectively (Fig. S18b†).33
The formation of Co3+ and M–OOH new peaks after the OER confirmed the development of metal oxyhydroxides during the OER performance. The post-HER O 1s exhibits that the peak intensity of M–O (∼529.8 eV) and M–OH (∼531.4 eV) was increased, showing that both metal oxides/hydroxides equally contribute to the HER process (Fig. S18c†). The Co 2p spectrum has peaks at 781.0 and 785.7 eV attributed to Co2+ and satellite peaks of Co 2p3/2 signals, respectively and peaks at 796.7 and 802.8 eV assigned to Co2+ and satellite peaks of Co 2p1/2 signals, respectively (Fig. S18d†).44 Here cobalt is in a Co2+ oxidation state as cobalt hydroxide. Thus, the XPS inference further explored the development of Co(Fe)(OH)2 and Co(Fe)OOH, which has been observed as the electrocatalytically active phase during the HER/OER performance. All these findings conclude that Co(Fe)OOH and Co(Fe)(OH)2 formation may be responsible for the best catalytic activity of FeCoHS@NF during the OER/HER performance. The presence of metal glycerate leads to the dissolution of free metal ions during the HER/OER process, which facilitates the easy conversion of Co(Fe)(OH)2 and Co(Fe)OOH.7,21,22,37,54
2.4. Highlights of multifunctional FeCoHS@NF
The FeCoHS@NF electrode demonstrates very effective bifunctional catalytic activity for overall water splitting. The enhanced catalytic activity of FeCoHS@NF may be related to the following properties: (1) FESEM and HR-TEM show the nanosphere morphology of FeCoHS@NF; it offers ample space for catalytic sites, which is beneficial for the quick diffusion of ions, electrons, and O2/H2 bubbles during the OER, UOR, and HER.37 (2) The HR-TEM and XRD confirmed the strong coupling crystalline-amorphous interface of FeCoHS@NF, which significantly improves the OER, UOR, and HER activity through quicker charge transfer rates, more possible active sites, and enhanced conductivity.22 (3) ECSA and EIS explore how a combination of the high surface area of cobalt and fast kinetics of iron synergistically improved the performance of FeCoHS@NF for HERs, OERs, UORs, and overall water splitting.42 (4) The primary problem with chlorine oxidation in seawater splitting is that it causes electrode corrosion. This problem is mitigated by the presence of a newly formed FeOOH layer on the electrocatalysts during the OER,46 as evidenced by FESEM and FT-IR spectroscopy. (5) The electrochemical studies demonstrated that the incorporation of cobalt provides abundant active sites for the UOR, while iron offers excellent tolerance towards chlorine-induced electrode corrosion. These findings highlight the effectiveness of the strategic selection of FeCoHS@NF, showcasing its multifunctional activity and stability. (6) The heterostructure exhibited the lowest charge transfer resistance, attributed to the modulated electronic transition at the interface. This was evidenced by the reduced resistance, lower Tafel slope and decreased overpotential. (7) The proposed ideal synthetic strategy offers numerous benefits, including reduced waste formation, high efficiency, enhanced selectivity, the formation of an amorphous-crystalline interface, unique morphology, low electricity consumption, cost-effectiveness, and a rapid implementation timeframe. (8) The demonstrated synthetic technique is highly reproducible for catalytic performance and durability for the UOR, OUS, OER, HER, OWS, and seawater splitting.3,11,22,25 (9) Furthermore, the electrochemical performance of FeCoHS@NF using urea-contaminated water and seawater is more beneficial for energy-saving hydrogen production and wastewater purification.3. Application
3.1. Solar-driven water splitting
Solar-driven water electrolysis has emerged as a cost-effective and environmentally friendly technique for producing hydrogen. Fig. 10 depicts the graphical representation of solar-driven water electrolysis of the FeCoHS@NF electrolyzer. The sunlight was directed into the solar panel (5.63 V), which was linked with the FeCoHS@NF electrolyzer in 1 M KOH using crocodile clips to produce electricity.58 The potential of the electrolyzer was monitored and controlled using a multimeter. The solar-driven water electrolyzer only needs 1.59 V for constant O2 and H2 gas formation at the anode and cathode, respectively, demonstrating its efficiency in producing hydrogen (Fig. S19: Movie S1†). Accordingly, the solar-driven water electrolyzer proposed in this study is highly advised for cost-effective, large-scale hydrogen generation. | ||
| Fig. 10 Schematic of solar-assisted water splitting. | ||
3.2. Purification of water
4. Environmental impact assessment
Following the electrochemical technical evidence, environmental impact assessment provides the economic and environmental factors for large-scale hydrogen production. Using widely accepted scientific advancements centred on mass-relevant socioeconomic and sustainability components, the level of sustainability of the synthetic strategy of FeCoHS@NF was assessed qualitatively and quantitatively. The environmental effect was compared to previous efforts (Table S3†). The related measurements were carried out using the formulas included in SI-S6.4.1. Raw materials and their toxicity potential
FeCoHS@NF was prepared using the least amount of Co(NO3)2·6H2O, Fe(NO3)2·6H2O, and glycerol. The minimum amount of water used for washing resulted in a water intensity of 38.7. With a reaction mass efficiency (RME) of 77.6%, the mass intensity (excluding water) was calculated to be 1.29 kg kg−1. NFPA 704: standard system for the identification of hazardous materials for emergency response guidelines states that iron nitrates and glycerol are non-toxic, whereas cobalt nitrate is less hazardous to people and aquatic species. Glycerol is widely available in the market as a food additive (E 422) approved by the European Food Safety Authority (EFSA). The laboratory and industrialised processes must be carried out following simple precautionary measures.13,26,59–614.2. Energy consumption
Standard filtration and solar energy were used for filtering and drying, but a minimum amount of electricity was used for heating and stirring, even though it was very low compared to the previous report. It does not require any advanced laboratory setup for industrial synthesis. In laboratory-scale synthesis, the energy intensity was found to be 2.5 kW h per kilogramme of FeCoHS@NF. An increase in manufacturing levels can lead to a significant reduction in energy intensity.4.3. Resulting emission and disposal of waste
The FeCoHS@NF characterisation does not reveal any harmful byproducts. The calculated environmental factor (0.3) shows that nearly no emissions were created. A small amount of H2O was used for washing and the inactive salts that were recovered after many items of washing may be reused. A trustworthy garbage removal company eventually removed the spent nickel foam. The spent catalyst-coated nickel foam is mixed with a flammable solvent and incinerated in a chemical incinerator equipped with an afterburner and a scrubber.4.4. Affordability and social acceptability
The demonstrated ideal synthetic approach for preparing FeCoHS@NF provides clean hydrogen gas with significant positive social impacts and is inexpensive, user-friendly, and feasible as a consumer product. For both construction and service, the suggested method does not require highly qualified workers, making it more appropriate for industrial hydrogen production. Here, we used an efficient solvothermal method to synthesise an iron–cobalt heterostructure (FeCoHS@NF) with a crystalline amorphous interface. Based on solar-driven water electrolysis, urea oxidation, seawater splitting and eco-friendly green synthesis of FeCoHS@NF electrocatalysts, this research offers a cost-effective and efficient way to accelerate hydrogen generation and pollutant degradation in the water splitting/treatment industry.5. Conclusion
To overcome the challenges in synthesizing low-cost catalysts with excellent stability and multifunctional activity, a novel eco-friendly ideal strategy was used to synthesize FeCoHS@NF with a crystalline-amorphous interface. FeCoHS@NF has higher catalytic sites, fast reaction kinetics, variable oxidation state, optimized electron redistribution, and fast electron transfer, which leads to greater electrical conductivity, higher turnover frequency, lower Tafel and overpotential value. The FeCoHS@NF electrode delivers a prominent performance that requires a potential of 1.48, 0.130, 1.23, 1.59 and 1.54 V @ 10 mA cm−2 for the OER, HER, UOR, OWS and overall alkaline seawater splitting, respectively. FeCoHS@NF exhibits excellent UOR/OUS activity, requiring only a potential of 1.23/1.40 V to attain 10 mA cm−2, which is 250/190 mV lower than that of OER/OWS. FeCoHS@NF also demonstrated excellent activity (OWS: 1.54 V, OER: 1.47 V, and HER: 130 mV) and stability over 110 hours in alkaline seawater, highlighting its potential for direct seawater splitting application. The solar-driven FeCoHS@NF electrolyser needs only 1.59 V for water splitting. The environmental impact studies and solar-driven water electrolysis proved that FeCoHS@NF has excellent greenness and efficiency, respectively. Hence, FeCoHS@NF is an outstanding electrode for large-scale H2 generation. Moreover, the selection strategy and synergistic effect of iron with cobalt can also be utilised to create alternative multifunctional, cost-effective electrocatalysts for various applications.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The author has no conflicts of interest to declare.Acknowledgements
The author A. Gayathri acknowledges DST-INSPIRE, Govt. of India for the award of Junior Research Fellowship (DST INSPIRE-JRF IF210159/DST/INSPIRE Fellowship/2021).References
- G. An, K. Wang, Z. Wang, M. Zhang, H. Guo and L. Wang, ACS Appl. Mater. Interfaces, 2024, 16, 29060–29068 CrossRef CAS PubMed.
- M. S. S. Danish, RSC Sustainability, 2023, 1, 2180–2196 RSC.
- Z. Chen, W. Wei, H. K. Shon and B. J. Ni, Green Chem., 2024, 26, 631–654 RSC.
- S. Aslam, S. Rani, K. Lal, M. Fatima, T. Hardwick, B. Shirinfar and N. Ahmed, Green Chem., 2023, 25(23), 9543–9573 RSC.
- D. Guan, B. Wang, J. Zhang, R. Shi, K. Jiao, L. Li, Y. Wang, B. Xie, Q. Zhang and J. Yu, Energy Environ. Sci., 2023, 16, 4926–4943 RSC.
- L. Huang, R. Yao, X. Wang, S. Sun, X. Zhu, X. Liu, M. G. Kim, J. Lian, F. Liu and Y. Li, Energy Environ. Sci., 2022, 15, 2425–2434 RSC.
- A. Gayathri, V. Ashok, M. Sangamithirai, J. Jayabharathi and V. Thanikachalam, Green Chem., 2024, 26, 5326–5338 RSC.
- T. Wei, G. Meng, Y. Zhou, Z. Wang, Q. Liu, J. Luo and X. Liu, Chem. Commun., 2023, 59, 9992–9995 RSC.
- Z. Xiao, Y. Qian, T. Tan, H. Lu, C. Liu, B. Wang, Q. Zhang, M. T. Sarwar, R. Gao and A. Tang, J. Mater. Chem. A, 2023, 11, 259–267 RSC.
- Y. Wang, L. Chen, H. Zhang, M. Humayun, J. Duan, X. Xu, Y. Fu, M. Bououdina and C. Wang, Green Chem., 2023, 25(20), 8181–8195 RSC.
- J. T. Ren, L. Chen, H.-Y. Wang, W.-W. Tian and Z.-Y. Yuan, Energy Environ. Sci., 2024, 17, 49–113 RSC.
- X. Gao, S. Zhang, P. Wang, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Chem. Soc. Rev., 2024, 53, 1552–1591 RSC.
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- J. Jack, W. Zhu, J. L. Avalos, J. Gong and Z. J. Ren, Green Chem., 2021, 23(20), 7917–7936 RSC.
- J. M. Gonçalves, A. L. Hennemann, J. G. Ruiz-Montoya, P. R. Martins, K. Araki, L. Angnes and R. Shahbazian-Yassar, Coord. Chem. Rev., 2023, 477, 214954 CrossRef.
- M. Moradi, F. Hasanvandian, M. G. Afshar, A. Larimi, F. Khorasheh, E. Niknam and S. R. Setayesh, Mater. Today Chem., 2021, 22, 100586 CrossRef CAS.
- M. Wang, J. Jiang and L. Ai, ACS Sustainable Chem. Eng., 2018, 6, 6117–6125 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, K. Kani, A. E. Allah, B. Yuliarto, Nugraha, H. K. Dipojono, Z. A. Alothman, D. Golberg and Y. Yamauchi, Chem. Mater., 2020, 32, 7005–7018 CrossRef CAS.
- X. Guo, J. Liang, L. Wang, Z. Feng, T. Yu, Z. Zhang, Y. Shao, C. Hao and G. Li, Int. J. Hydrogen Energy, 2018, 43, 2034–2042 CrossRef CAS.
- J. Li, X. Gu, J. Chang, D. Wu, F. Xu, K. Jiang and Z. Gao, J. Colloid Interface Sci., 2022, 606, 1662–1672 CrossRef CAS PubMed.
- Y. Du, B. Li, G. Xu and L. Wang, InfoMat, 2023, 5, e12377 CrossRef CAS.
- J. Hou, Y. Sheng, D. Bi, N. Chen, Q. Lai and Y. Liang, J. Alloys Compd., 2024, 977, 173447 CrossRef CAS.
- R. Christoph, B. Schmidt, U. Steinberner, W. Dilla and R. Karinen, Ullmann's Encycl. Ind. Chem., 2006, 17, 67–82 Search PubMed.
- A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- C. Jiménez-González, D. J. Constable and C. S. Ponder, Chem. Soc. Rev., 2012, 41, 1485–1498 RSC.
- M. Zhu, Q. Luo, Q. Chen, W. Wei, Q. Zhang and S. Li, Mater. Chem. Front., 2021, 5, 2758–2770 RSC.
- M. J. Da Silva, N. A. Liberto, L. C. D. A. Leles and U. A. Pereira, J. Mol. Catal. A:Chem., 2016, 422, 69–83 CrossRef.
- S. Anantharaj, P. N. Reddy and S. Kundu, Inorg. Chem., 2017, 56, 1742–1756 CrossRef CAS PubMed.
- V. K. Singh, B. Malik, R. Konar, E. S. Avraham and G. D. Nessim, Electrochem, 2024, 5, 70–83 CrossRef CAS.
- T. Yang, L. Meng, S. Han, J. Hou, S. Wang and X. Wang, RSC Adv., 2017, 7, 34687–34693 RSC.
- J. Liu, Y. Li, L. Zheng, R. Tang, W. Zhou, J. Gao and H. Wu, J. Solid State Electrochem., 2021, 25, 1623–1631 CrossRef CAS.
- T. X. Nguyen, Y. Su, C. Lin, J. Ruan and J. Ting, Adv. Sci., 2021, 8, 2002446 CrossRef CAS PubMed.
- S. S. S. Afghahi, R. Peymanfar, S. Javanshir, Y. Atassi and M. Jafarian, J. Magn. Magn. Mater., 2017, 423, 152–157 CrossRef.
- H. Wang, E. Feng, Y. Liu and C. Zhang, J. Mater. Chem. A, 2019, 7, 7777–7783 RSC.
- F. Zhang, C. Yuan, X. Lu, L. Zhang, Q. Che and X. Zhang, J. Power Sources, 2012, 203, 250–256 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, Y. Guo, B. Yuliarto, X. Jiang, Y. Ide, N. Nugraha, H. K. Dipojono, A. Yu, Y. Sugahara, D. Golberg and Y. Yamauchi, ChemSusChem, 2020, 13, 1645–1655 CrossRef CAS PubMed.
- Z. Dong, W. Zhang, Y. Xiao, Y. Wang, C. Luan, C. Qin, Y. Dong, M. Li, X. Dai and X. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 5464–5477 CrossRef CAS.
- A. Gayathri, S. Mathi, M. Vijayarangan, J. Jayabharathi and V. Thanikachalam, ChemistrySelect, 2022, 7, e202203616 CrossRef CAS.
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- B. H. Suryanto, Y. Wang, R. K. Hocking, W. Adamson and C. Zhao, Nat. Commun., 2019, 10, 5599 CrossRef CAS PubMed.
- A. Gayathri, M. Vijayarangan, M. Sangamithirai, V. Ashok, J. Jayabharathi and V. Thanikachalam, Energy Fuels, 2023, 37, 19812–19821 CrossRef CAS.
- Z. Lu, J. Wang, P. Zhang, W. Guo, Y. Shen, P. Liu, J. Ji, H. Du, M. Zhao and H. Liang, Appl. Catal. B: Environ. Energy, 2024, 353, 124073 CrossRef CAS.
- M. Zhong, M. Xu, S. Ren, W. Li, C. Wang, M. Gao and X. Lu, Energy Environ. Sci., 2024, 17, 1984–1996 RSC.
- P. Wang, R. Wang, Q. Xu, Z. Xu, L. Wan, Y. Lin, P.-F. Liu and B. Wang, ACS Appl. Energy Mater., 2021, 4, 9487–9497 CrossRef CAS.
- Y. Wei, C.-H. Shin, E. B. Tetteh, B.-J. Lee and J.-S. Yu, ACS Appl. Energy Mater., 2020, 3, 822–830 CrossRef CAS.
- J. Liu, Z. Yu, J. Huang, S. Yao, R. Jiang, Y. Hou, W. Tang, P. Sun, H. Huang and M. Wang, J. Colloid Interface Sci., 2023, 650, 1182–1192 CrossRef CAS PubMed.
- V. Ashok, A. Gayathri, M. Vijayarangan and J. Jayabharathi, Sustainable Energy Fuels, 2024, 8(15), 3452–3464 RSC.
- C. Alex, G. Shukla and N. S. John, Electrochim. Acta, 2021, 385, 138425 CrossRef CAS.
- W. Liu, J. Zhao, L. Dai, Y. Qi, K. Liang, J. Bao and Y. Ren, Inorg. Chem., 2024, 63(13), 6016–6025 CrossRef CAS PubMed.
- K. Jiang, W. Liu, W. Lai, M. Wang, Q. Li, Z. Wang, J. Yuan, Y. Deng, J. Bao and H. Ji, Inorg. Chem., 2021, 60, 17371–17378 CrossRef CAS PubMed.
- S. Pal, K. Shimizu, S. Khatun, S. Singha, S. Watanabe and P. Roy, J. Mater. Chem. A, 2023, 11(23), 12151–12163 RSC.
- V. Ashok, M. Sangamithirai, M. Vijayarangan, A. Gayathri and J. Jayabharathi, ChemNanoMat, 2023, 9, e202300338 CrossRef CAS.
- J. N. Hausmann and P. W. Menezes, Angew. Chem., Int. Ed., 2022, 61, e202207279 CrossRef CAS PubMed.
- B. Vishnu, S. Sriram and J. Jayabharathi, Sustainable Energy Fuels, 2023, 7, 4638–4653 RSC.
- J. S. Kirar, S. Khare and N. Tiwari, React. Kinet., Mech. Catal., 2021, 132, 1025–1046 CrossRef CAS.
- M. Amini, Y. Mousazade, Z. Zand, M. Bagherzadeh and M. M. Najafpour, Sci. Rep., 2021, 11, 6642 CrossRef CAS PubMed.
- B. Wang, X. Chen, Y. He, Q. Liu, X. Zhang, Z. Luo, J. V. Kennedy, J. Li, D. Qian and J. Liu, Appl. Catal. B: Environ. Energy, 2024, 346, 123741 CrossRef CAS.
- V. Ashok, S. Mathi, M. Sangamithirai and J. Jayabharathi, Energy Fuels, 2022, 36, 14349–14360 CrossRef CAS.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković and Q. Tu, Green Chem., 2018, 20, 1929–1961 RSC.
- R. W. Bukowski, R. W. Bukowski, W. W. Jones and C. L. Forney, Technical references guide for the HAZARD I fire hazard assessment method, US Department of Commerce, National Institute of Standards and Technology, 1989 Search PubMed.
- N. R. Council, Fires in Mass Transit Vehicles: Guidelines for the Evaluation of Toxic Hazards: Report, National Academies Press, 1991, vol. 462 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, details of the instruments used for the investigation of FeCoHS@NF (FT-IR spectroscopy, FESEM, XRD, ICP-OES, XPS and HRTEM), electrode preparation, and electrochemical characterization of HER/OER performance; the equations used for the calculation of environmental impact assessment, electricity consumption for H2 production, faradaic efficiency, overpotential, Tafel plot, RF, ECSA, ECSA-normalized LSV, O2/H2 generation and TOF; the images of colour comparison of various electrocatalysts, FT-IR spectroscopy, XPS survey spectrum, FESEM, OER CV comparison, ECSA, faradaic efficiency, and post-analysis of XPS, FT-IR spectroscopy, XRD and FESEM; solar-powered water-splitting system setup image; comparison table for the OER, HER, OWS, OUS, energy consumed and electrolytes used for various electrolysis systems and environmental assessments. Additionally, a solar-powered water-splitting system (MP4) demonstrated the development of O2 (anode) and H2 (cathode) gas. See DOI: https://doi.org/10.1039/d4nr04382k |
| This journal is © The Royal Society of Chemistry 2025 |