
Nickel single atoms anchored on a bipyridine-based covalent organic framework: boosting active sites for photodegradation of acetaminophen†
Guijiao
Wen
a,
Qianqian
Peng
a,
Chen
Yuan
 a,
Juan
He
*a and
Xiandeng
Hou
*ab
a,
Juan
He
*a and
Xiandeng
Hou
*ab
aAnalytical & Testing Centre, Sichuan University, Chengdu, Sichuan 610064, China. E-mail: hejuan1117@scu.edu.cn
bCollege of Chemistry, Sichuan University, Chengdu, Sichuan 610064, China. E-mail: houxd@scu.edu.cn
First published on 4th November 2024
Abstract
It remains crucial but challenging to construct single-atom photocatalysts based on covalent organic framework (COF) materials in a simple, fast, and controllable manner and to clarify their structure–efficacy relationship. Here, a single-atom photocatalyst (Ni-TpBpy) featuring atomically dispersed Ni sites with a high loading content and a specific tetra-coordinated N2–Ni–Cl2 environment in a bipyridine-based COF was for the first time rapidly synthesized using dielectric barrier discharge (DBD) plasma and a wet chemical method. Visible light-driven Ni-TpBpy can achieve 97.8% photodegradation efficiency of acetaminophen at 0.177 min−1 in 30 min, outperforming other advanced photocatalysts. Experimental studies and density-functional theory (DFT) calculation clarified the role of well-dispersed Ni active sites in enhanced photodegradation, which not only narrowed the bandgap, facilitating carrier separation and migration, but also promoted the generation of reactive superoxide radicals. This study represents the first use of single-atom Ni-TpBpy in the efficient photocatalytic degradation of emerging pollutants with remarkable stability and universality, bringing new insights into the application of COF-based single-atom materials in environmental remediation.
1 Introduction
In recent decades, the extensive use of pharmaceuticals and personal care products (PPCPs) has resulted in their pervasive dissemination within the global water environment, presenting a substantial concern for water safety on a worldwide scale.1,2 In particular, during the COVID-19 epidemic, there was a global surge in self-medication practices, leading to an overaccumulation of acetaminophen (ACT) in aquatic systems worldwide.3 The excessive use and discharge of ACT have engendered profound ecotoxicity and environmental persistence, leading to intractable environmental challenges.4 Conventional approaches for processing pollutant-containing wastewater, such as adsorption, electrocatalysis, and bio-catalysis, have been hindered in practical applications due to the limitations of high cost, low reproducibility, weak degradation efficacy, and low mineralization.5–9 Nowadays, photocatalytic technology offers a promising green method for the degradation of organic environmental pollutants by making full use of renewable clean energy sources such as solar energy or molecular oxygen.10 As the foundation and core of photocatalytic reactions, photocatalysts play an important role in the reaction process. However, existing photocatalysts still have some drawbacks, including low photogenerated carrier yield, short lifetimes, high carrier recombination rates, etc., which greatly hinder the widespread application of photocatalysts. Therefore, the rational design of efficient photocatalysts with enhanced light trapping capability, an adjusted band gap structure,11 creation of efficient active sites, promotion of mass transfer and improved photocatalyst durability is crucial for the practical application of photocatalysts.12–15Approaches aimed at enhancing photocatalytic efficacy, such as introducing heterojunctions, incorporating co-catalysts,16 and anchoring single atoms,17 have proven effective in regulating the energy band structure, facilitating charge transfer, and augmenting photocatalytic activity. Currently, there is burgeoning interest in single-atom catalysts (SACs), as one of the most vigorously explored fields.18 The special geometric and electronic properties they possess greatly boost their atomic utilization efficiency, active site density, and thus their excellent photocatalytic performance.19 Particularly for the photocatalytic degradation of pollutants in the environmental clean-up zones, suitable single-atom catalysts can adsorb reactants more effectively than clusters, promote the electron exchange between the catalysts and reactants, and activate the precursors (e.g., molecular oxygen) to produce active radicals, thus greatly improving the photodegradation performance.20 Although studies on SACs have progressed rapidly, there are still issues related to the stability and aggregation of SACs, low loading content, and high fabrication costs.21–23 Therefore, it is crucial to construct SACs with isolated and uniform metal atoms, high stability, high metal loading, and light-harvesting capability.
COFs are crystalline organic materials with long-range ordered structures, consisting of low-density elements (e.g., C, N, and O) connected by covalent bonds.24 They possess distinct advantages, including abundant active sites, a precisely tunable porous structure, and high stability,25 which fulfill the prerequisites for stabilizing single atoms on their surfaces. However, certain COFs lack post-metallation sites, resulting in the deactivation of single atoms by aggregating into clusters or even nanoparticles during synthesis. Fortunately, bipyridine-containing structures are preferred substrates because their abundant N,N-chelation sites can provide the basis for the uniform and stable incorporation of single atoms. Most importantly, anchoring transition-metal single atoms into COFs is considered efficient for narrowing the energy band, accelerating the transport and separation of photogenerated carriers, and enhancing the strong synergism of metal-COF carriers.26,27 For example, Mo single atoms stabilized by bipyridine-based COFs and Au single atoms anchored on isostructural porphyrin-rich COFs have exhibited excellent activity for CO2 conversion28 and photocatalytic N2 fixation.29 Therefore, the construction of COF-based SACs presents a significant opportunity for improving the photocatalytic efficiency of COFs in wastewater treatment, particularly for the degradation of PPCPs. However, as far as we know, the implantation of atomically dispersed Ni atoms into COF materials to enhance the photodegradation performance and the mechanism of isolated nickel active sites in improving the photocatalytic performance still wait to be explored. Moreover, most COF materials used to anchor single atoms are synthesized using solvothermal synthesis, making the preparation of single-atom catalysts labor-intensive and time-consuming.
Taking the above considerations, 2,2′-bipyridine-linked TpBpy was easily synthesized by using the dielectric barrier discharge (DBD) plasma method, and atomically dispersed single Ni sites were successfully anchored on TpBpy (denoted as Ni-TpBpy) using a wet chemistry method. The structure–efficacy relationship was fully investigated via a combined experimental and DFT calculation approach, and the constructed Ni-TpBpy bearing single Ni sites could greatly improve the separation and migration efficiency of photogenerated carriers and promote the adsorption and activation of oxygen to produce reactive oxygen species (ROS). Consequently, Ni-TpBpy exhibits enhanced photocatalytic performance for the photodegradation of emerging pollutants. This study highlights the first use of single-atom Ni-TpBpy in the photocatalytic degradation of ACT, bringing new insights into the application of COF-based SACs in environmental remediation.
2 Experimental
Note that the materials, characterization and tests, intermediate measurements, and detailed steps of the theoretical calculation are given in the ESI.†2.1 Synthesis of TpBpy
TpBpy was prepared via a novel DBD plasma strategy. In brief, precursors containing 42 mg triformylphloroglucinol (Tp) and 55.8 mg 2,2′-bipyridine-5,5′-diamine (Bpy) were ultrasonically dispersed in 8 mL of a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) solution mixture of N,N-dimethylacetamide and o-dichlorobenzene, with the addition of 0.4 mL of 6 mol L−1 acetic acid aqueous solution (AcOH). This mixture was then introduced into a DBD reactor under nitrogen protection,30 followed by reaction at a specified voltage (32 V) and current (1.2 A) for 40 min. The resultant red TpBpy material was subsequently filtered, washed with tetrahydrofuran, and finally dried at 60 °C overnight in a vacuum oven.
:1 (v/v) solution mixture of N,N-dimethylacetamide and o-dichlorobenzene, with the addition of 0.4 mL of 6 mol L−1 acetic acid aqueous solution (AcOH). This mixture was then introduced into a DBD reactor under nitrogen protection,30 followed by reaction at a specified voltage (32 V) and current (1.2 A) for 40 min. The resultant red TpBpy material was subsequently filtered, washed with tetrahydrofuran, and finally dried at 60 °C overnight in a vacuum oven.
2.2 Synthesis of Ni-TpBpy
Ni-TpBpy was synthesized using a modified wet chemistry method.31 Specifically, 100 mg of TpBpy and 60 mg of nickel(II) perchlorate hexahydrate were dispersed in acetonitrile (40 mL) and shaken for 8 h. Ni-TpBpy was then obtained through filtration separation, acetonitrile washing, and vacuum dried at 60 °C for 8 h.2.3 Photocatalytic experiments
The photocatalytic activity of Ni-TpBpy was evaluated by the degradation of the model pollutant ACT. Typically, a xenon arc lamp with a UV cutoff filter (λ ≥ 400 nm) was used as the visible light source, and a double-walled beaker jacketed with cooling water was applied as a photoreactor. Ni-TpBpy (10 mg) was dispersed in ACT aqueous solution (30 mL, 20 mg L−1). The mixture was first kept under dark conditions for 30 min to reach adsorption equilibrium. The photodegradation test began by switching on the xenon arc lamp. Then, at every 5 min, 1 mL of the suspension was taken and filtered through a microporous filter membrane (0.22 μm) and measured using a UV-vis spectrophotometer at 244 nm. The ACT degradation efficiency can be obtained using the following equation: Degradation efficiency (%) = (C0 − Ct)/C0 × 100%, where C0 and Ct were the concentrations of ACT at 0 and t min, respectively.3 Results and discussion
3.1 Synthesis and characterization
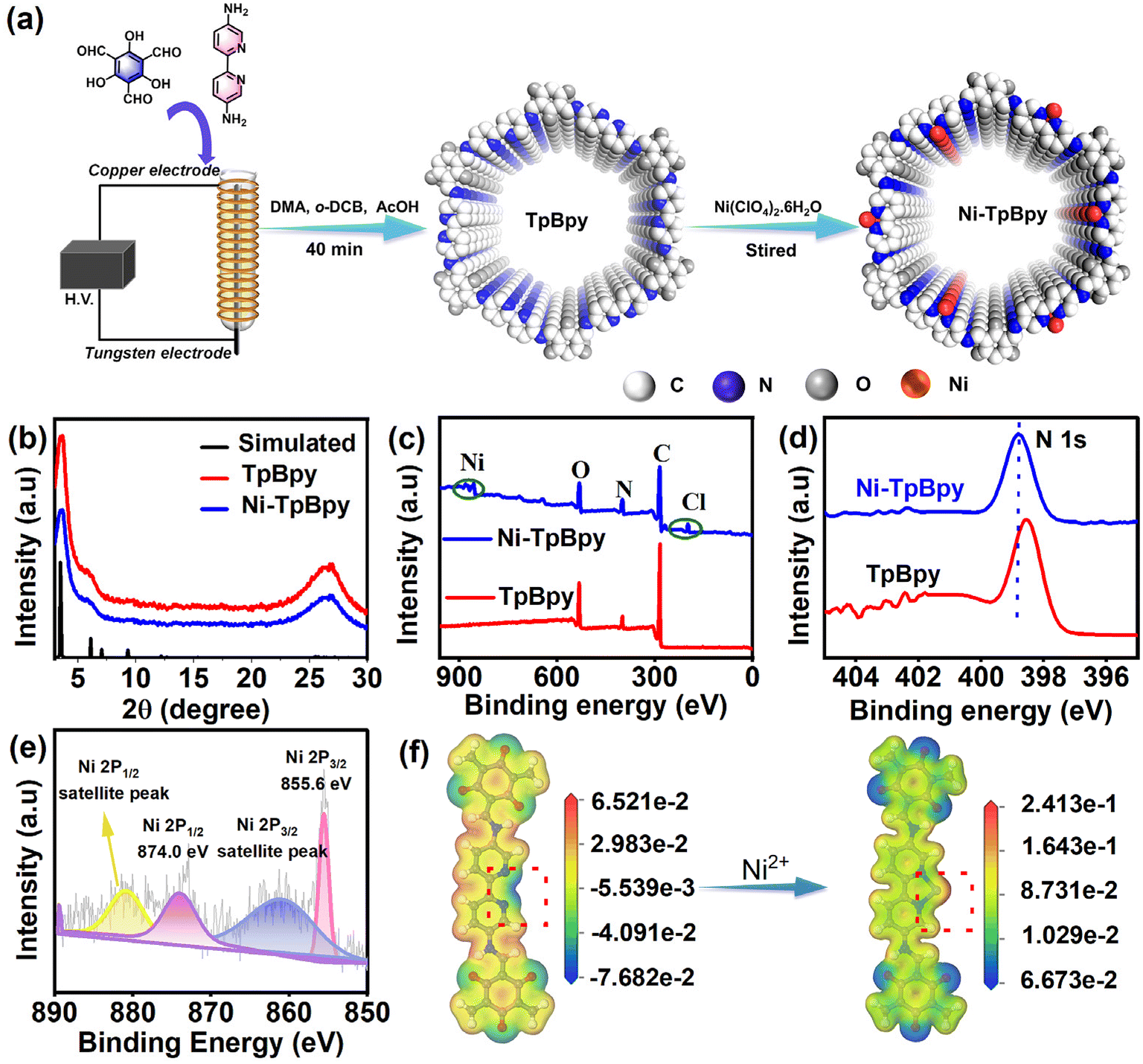
Dielectric barrier discharge plasma strategy as a rapid, facile, and energy-saving method32 was used to synthesize Ni-TpBpy for the first time. TpBpy was yielded in 40 min via the DBD plasma treatment under mild conditions, significantly improving reaction efficiency compared to conventional methods that require 3 days.33 The well-defined bipyridine construction unit with a special duple coordinative bonding microenvironment enabled the accommodation and atomic anchoring of single metal active sites in COFs in a precise manner. As shown in Fig. 1a, the single dispersed Ni atoms were effectively implanted on the pre-designed bipyridine structure by a simple chemical impregnation method.31 The weight percentage of Ni in TpBpy was measured and found to be 4.6 wt% using an inductively coupled plasma optical emission spectrometer (ICP-OES), revealing the outstanding coordination capability of TpBpy to Ni. The powder X-ray diffraction (PXRD) patterns of Ni-TpBpy (Fig. 1b) at 3.6° and 26.9° correspond to the (hkl) values of (100) and (001)34 respectively, similar to the diffraction peaks of pristine TpBpy. Although the crystallinity of Ni-TpBpy decreased slightly (from 66% to 62%), the overall order of the COF backbone is not affected during the anchoring of the nickel atoms. Noticeably, TpBpy was formed by layer-by-layer stacking through π–π interactions, resulting in a broad diffraction peak at 26.9°.35 This periodic π-conjugated channel is conducive to the separation and migration of photogenerated electrons and holes, thus contributing to the improvement of photocatalytic performance.36 Moreover, no PXRD peaks of crystalline Ni species were observed in Ni-TpBpy, which can be attributed to the absence of crystalline nickel oxides.37 In the Fourier transform infrared (FT-IR) spectra (Fig. S1†), the characteristic absorption bands for the amino group of Bpy (3200–3419 cm−1) and the aldehyde group of Tp (1640 cm−1) were absent in TpBpy, verifying the completion of the condensation reaction. In particular, the emerging peaks at 1580 cm−1 and 1610 cm−1 are derived from the stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO groups, whereas the peaks near 1261 cm−1 belong to the C–N bond, indicating the occurrence of keto–enol tautomerism in the TpBpy structure.38,39 After the implantation of Ni2+, the corresponding FT-IR spectra showed negligible changes, except for a slight redshift and broadening of the νC–N peaks, and this suggests that the Ni ions should be coordinated to the bipyridine N atoms in the TpBpy framework.40
C and CO groups, whereas the peaks near 1261 cm−1 belong to the C–N bond, indicating the occurrence of keto–enol tautomerism in the TpBpy structure.38,39 After the implantation of Ni2+, the corresponding FT-IR spectra showed negligible changes, except for a slight redshift and broadening of the νC–N peaks, and this suggests that the Ni ions should be coordinated to the bipyridine N atoms in the TpBpy framework.40
 | ||
| Fig. 1 Schematic illustration of the fabrication of Ni-TpBpy (a), PXRD patterns (b), XPS spectra (c), high-resolution N 1s XPS spectrum (d), Ni 2p XPS spectra of Ni-TpBpy (e), and the electrostatic potential of TpBpy and Ni-TpBpy calculated by DFT (f). | ||
X-ray photoelectron spectroscopy (XPS) measurements were applied to further reveal the structural interaction between Ni ions and the COF frameworks. The full XPS analysis confirmed the elemental composition of Ni-TpBpy, mainly including C, N, O, Cl, and Ni elements (Fig. 1c); and consistent with the PXRD and FT-IR findings, no impurity peaks could be assigned to other nickel species (e.g. nickel oxide and nickel metal). A subtle upward shift of the N 1s peaks in Ni-TpBpy compared to that of TpBpy (Fig. 1d) further validated the existence of a metal coordination–chelation interaction between the bipyridine N units and Ni2+.31 The high-resolution Ni 2p spectra of Ni-TpBpy showed that the binding energies located at 855.6 eV and 874.0 eV (Fig. 1e) were assigned to Ni 2p3/2 and Ni 2p1/2 states,41 respectively, suggesting that isolated Ni atoms have been effectively anchored to TpBpy. Moreover, DFT simulations revealed that before Ni impregnation, the two N atom sites displayed a more negative electrostatic potential (Fig. 1f), suggesting that they should have a high capacity for electron-donation and coordination with metal ions/metal. However, the electrostatic potential became more positive after Ni anchoring, also demonstrating the successful anchoring of single Ni atoms through the metal coordination–chelation interaction.
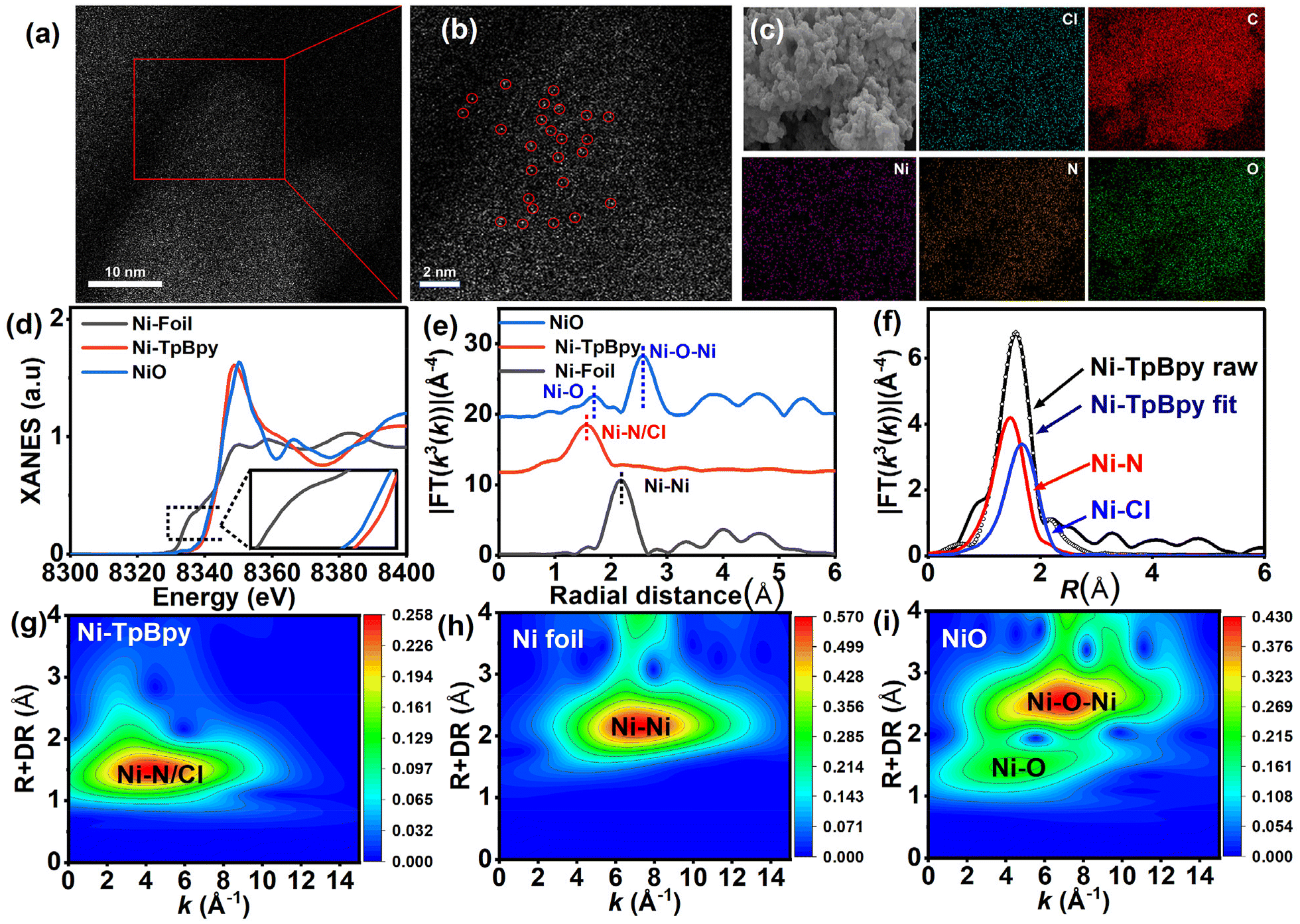
N2 (77 K) adsorption–desorption experiments were conducted to investigate the porosity of Ni-TpBpy. As depicted in Fig. S2,† both TpBpy and Ni-TpBpy exhibited typical reversible type-I isotherms, implying their microporous nature. Following Ni impregnation, the surface area based on the Brunner–Emmett–Teller model decreased from 857 m2 g−1 to 720 m2 g−1, accompanied by a reduction in pore size from 1.26 nm to 1.18 nm (Fig. S3†). This decrease is due to the anchoring of Ni atoms to the pore walls of TpBpy, occupying part of the interior cavities. Scanning electron microscopy (SEM) images showed no significant alteration in the porous morphology of Ni-TpBpy compared to the original structure (Fig. S4†). HRTEM was applied to further demonstrate the absence of any metal nanoparticles or metal oxide particles in Ni-TpBpy (Fig. S5†). More authoritative evidence comes from the aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) images, where the bright spots in Ni-TpBpy correspond to single nickel sites (Fig. 2a and b), visually demonstrating the successful formation of atomically dispersed Ni atoms in Ni-TpBpy. The absence of nickel nanoparticles observed in the AC HAADF-STEM images may be attributed to the inhibition of cluster and metal oxide generation by the coordination of nickel with well-defined bipyridine structural units. EDS mapping intuitively revealed the homogeneous distribution of C (red), N (orange), O (green), Cl (blue), and Ni (purple) in Ni-TpBpy (Fig. 2c), further verifying the presence of Ni element in the Ni-TpBpy matrix, which was consistent with the XPS results. The confinement of single atom Ni sites in the TpBpy substrate provides an ideal platform for facilitating the separation of e− and h+, thus significantly enhancing the mobility of photogenerated carriers.42 X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were further characterized to identify the local coordination environment of atomic Ni sites. Comparing the Ni K-edge XANES spectra of Ni-TpBpy to NiO and Ni foil (Fig. 2d), the absorption edge of Ni-TpBpy was near NiO, suggesting that Ni was in an ortho-divalent state, which was consistent with the XPS characterization. In the Fourier transform k3-weighted EXAFS plot of Ni-TpBpy (Fig. 2e), the main peak aligns with the first coordination layer of Ni-N/Cl at 1.57 Å. The absence of the Ni–Ni peak at 2.18 Å suggested the isolation state of the Ni site in Ni-TpBpy rather than metallo–Ni species, which was also supported by HAADF-STEM characterization. For the analysis of the quantitative compositional parameters of the single Ni sites, EXAFS curve fitting (Fig. 2f) was performed, with the first-shell peaks positioned at 1.47 and 1.67 Å, attributable to the Ni–N and Ni–Cl bonds, respectively. The detailed structural EXAFS data fitting results are shown in Fig. S6 and Table S1,† suggesting that the single-atom Ni was anchored to the double nitrogen structure of bipyridine, which is in agreement with the result of electrostatic potential calculations. Furthermore, with sufficiently high resolution, WT-EXAFS can provide both radial distance resolution (R-space) and k-space resolution. Compared to Ni foil (Fig. 2h) and NiO (Fig. 2i), the maximum intensity at 4.0 Å−1 (k-space) was fully resolved at 1.57 Å (R-space) for the wavelet transform (WT) of Ni-TpBpy (Fig. 2g), which was attributed to the N2–Ni–Cl2 coordination structure in line with the above observations. In addition, the maximum intensities at 7.3 and 6.9 Å−1 (k-space) were not recorded in Ni-TpBpy, which corresponded to the Ni–Ni and Ni–O–Ni coordination of the Ni foil and NiO reference samples, respectively. The results provided further evidence that Ni in Ni-TpBpy was an isolated state rather than a metal monomer or metal oxide, which reinforced the attribution of the main peak at 1.57 Å to the N2–Ni–Cl2 coordination structure. Therefore, the above analysis confirmed the existence of metallic single atoms of nickel in Ni-TpBpy.
 | ||
| Fig. 2 AC HAADF-STEM images (a and b) of Ni-TpBpy, EDS mapping of Ni-TpBpy (c), normalized XANES (d) and Fourier transform (FT) EXAFS spectra (e) at the Ni K-edge, the fit in R-space at the Ni K-edge of Ni-TpBpy (f), and the WT-EXAFS of Ni-TpBpy (g), Ni foil (h) and NiO (i). | ||
3.2 Photoelectrochemical properties
To understand the effect of Ni loading on the photocatalytic activity of TpBpy, the photochemical properties of Ni-TpBpy and TpBpy, including charge transfer, current density, and photocurrent response, were systematically examined. Compared with TpBpy, Ni-TpBpy exhibited higher current density (Fig. S7†), stronger photocurrent response strength (Fig. 3a), and smaller Nyquist radius (Fig. 3b), which indicated that the Ni loading reduced the resistance value of TpBpy, and improved the transport and separation efficiency of carriers.43,44 Furthermore, the photoluminescence spectrum of Ni-TpBpy was significantly attenuated compared with that of TpBpy (Fig. 3c), which revealed that Ni-TpBpy effectively suppressed the photogenerated electrons–holes recombination rate,45 and thus greatly favored the photocatalytic activity. In addition, the time-resolved PL decay spectra (Fig. 3d) showed that the average fluorescence lifetime (τave = Σαiτi, where τ is the fluorescence lifetime and α the pre-exponential factor, Table S2†) of Ni-TpBpy (0.46 ns) was longer than that of TpBpy (0.32 ns), indicating that the nickel loading in Ni-TpBpy can effectively boost the carrier separation efficiency and reduce the e−–h+ recombination rate.46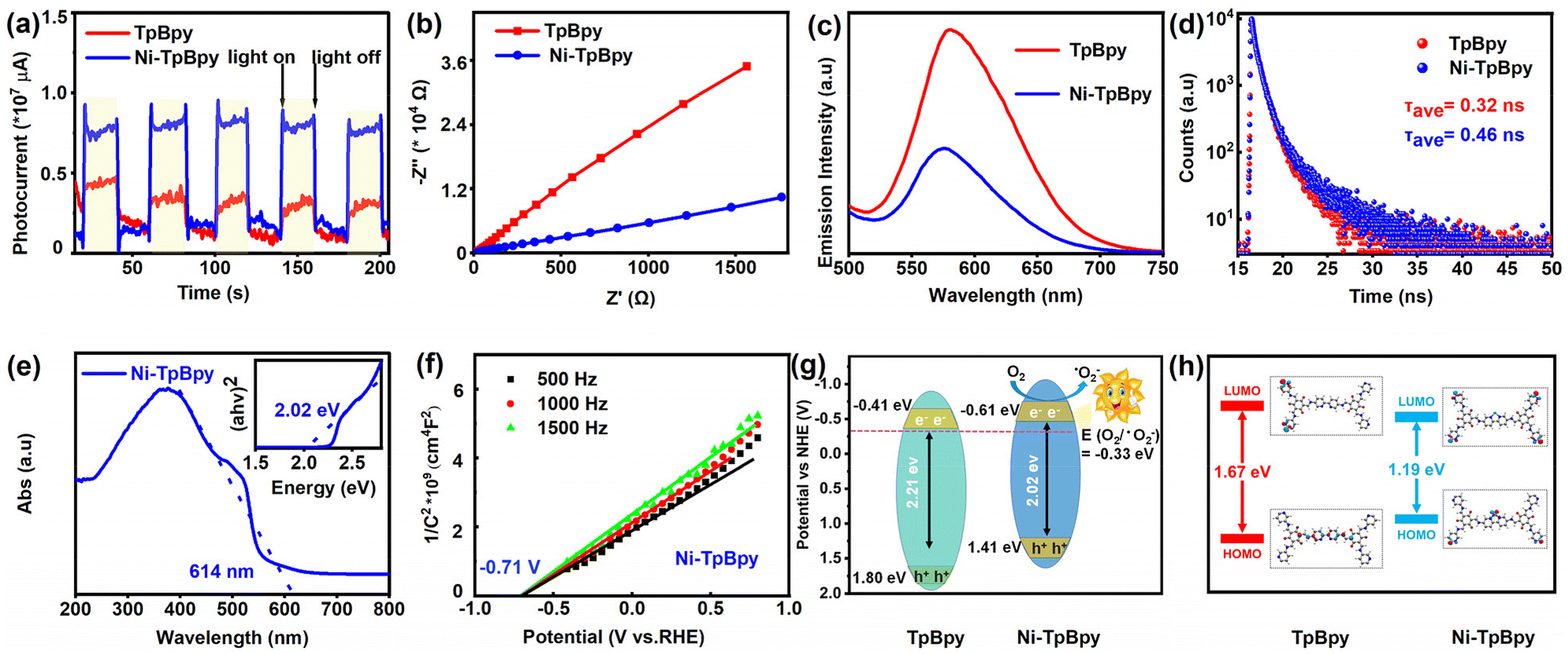 | ||
| Fig. 3 Transient photocurrent (a), EIS Nyquist plots (b), photoluminescence spectra (c), time-resolved PL decay spectra (d), UV-vis DRS spectra and Tauc plot diagrams (e), Mott–Schottky plot (f), energy band structures (g), and LUMO and HOMO energy sketch maps of TpBpy and Ni-TpBpy (h). | ||
Meanwhile, the absorption capability of the catalysts for visible light is a crucial factor in the photodegradation process. UV-vis DRS analysis revealed that Ni-TpBpy displayed an excellent visible light response, with a broadened absorption edge at 614 nm compared to TpBpy (Fig. 3e and S8†), and the corresponding optical bandgap energy based on the Tauc plots (inset of Fig. 3e and Fig. S9†) was narrowed from 2.21 to 2.02 eV due to the increased delocalization.47 Subsequently, the Mott–Schottky plots (Fig. 3f and Fig. S10†) demonstrated that TpBpy and Ni-TpBpy were both identified as n-type semiconductors judging from their positive slopes, and the flat-band potential (Efb) is positively shifted by 0.1 eV compared to the conduction band (ECB).48 The Efb value was determined by extrapolating the linear region of the M–S plot to the x-axis (potential). Thus, the ECB value of TpBpy and Ni-TpBpy conversions to standard hydrogen electrodes is −0.41 and −0.61 eV (vs. NHE) respectively. Furthermore, the valence band potentials (EVB) of TpBpy and Ni-TpBpy were evaluated according to EVB = ECB + Eg,49 resulting in their EVB values of 1.80 and 1.41 eV (vs. SHE), respectively. Consequently, the energy levels of the photocatalysts are illustrated in Fig. 3g. The introduction of the single Ni atom resulted in a more negative ECB level and a narrower band gap compared to TpBpy, implying that Ni-TpBpy exhibits a stronger reducing capability and better electron–hole separation, leading to superior photocatalytic performance.50 In addition, photocatalytic performance was described with electronic stability, determined from HOMO and LUMO orbital gaps,51 and the HOMO–LUMO gaps of TpBpy and Ni-TpBpy were calculated using the PBE method to be 1.67 eV and 1.19 eV, respectively (Fig. 3h). The theoretical HOMO–LUMO gaps also reinforced the experimental bandgap trends of TpBpy and Ni-TpBpy, with the narrower frontier orbital gaps favoring the excitation of Ni-TpBpy under visible-light irradiation. These results indicate that Ni-TpBpy has superior photochemical properties, which is predictive of its efficient photocatalytic performance.
3.3. Evaluation of the photocatalyst performance
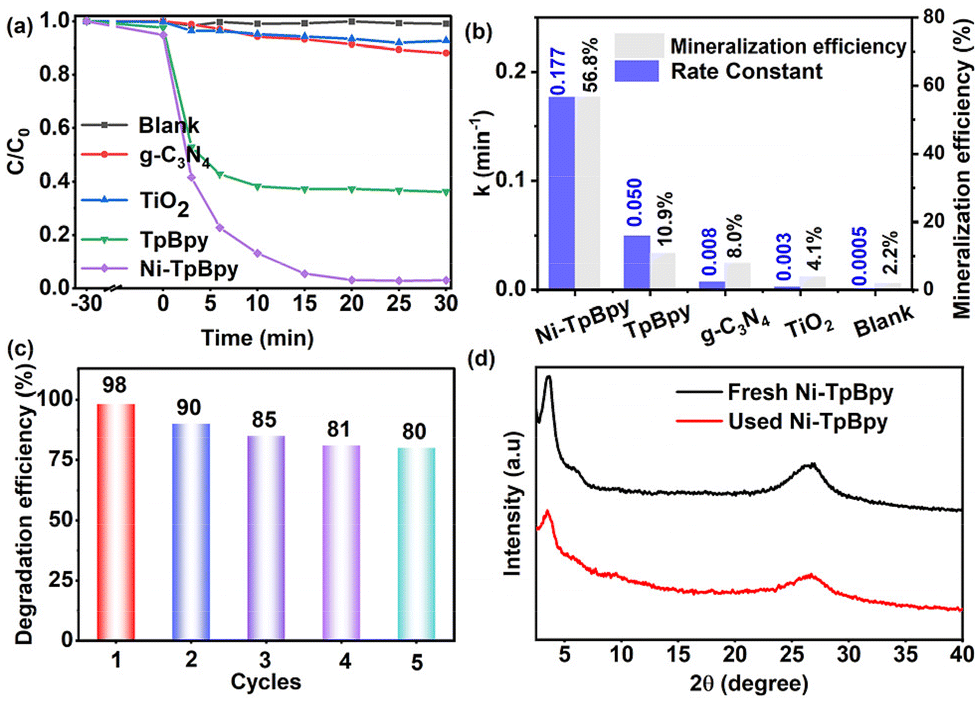
To assess the photodegradation performance of Ni-TpBpy, ACT, a common emerging organic contaminant, was chosen as the model. As demonstrated in Fig. 4a, ACT remained stable and was barely degradable in the absence of a photocatalyst. In comparison, Ni-TpBpy significantly increased the degradation efficiency of ACT to 97.8% after 30 min, which was significantly higher than that of TpBpy (64.0%), and other typical commercial photocatalysts such as TiO2 (7.4%) and g-C3N4 (12.0%). The photodegradation reaction of ACT follows pseudo-first-order kinetics (eqn (1)):| ln(C0/Ct) = k·t | (1) |
 | ||
| Fig. 4 ACT degradation efficiency (a) and corresponding pseudo-first-order kinetic constants and mineralization efficiency of Ni-TpBpy, TpBpy, g-C3N4 and TiO2 for the degradation of ACT (b). Recyclability of Ni-TpBpy for the photocatalytic degradation of ACT (c). XRD pattern of Ni-TpBpy before and after 5 cycling experiments (d). | ||
The ACT degradation efficiency of Ni-TpBpy was found to be closely related to the reaction parameters. The optimal catalyst concentration and Ni loading for the degradation of ACT by Ni-TpBpy were determined to be 300 mg L−1 and 4.6 wt%, respectively (Fig. S12, S13 and Table S4†). A noticeable enhancement in the degradation efficiency was observed as the amount of Ni-TpBpy increased from 100 mg L−1 to 300 mg L−1. This increase was attributed to a higher probability of the photocatalyst absorbing photons, thus generating more ROS to promote the surface reaction.53 As the loading of single-atom Ni increased from 4.6 wt% to 5.6 wt%, the degradation efficiency remained constant. However, the k-value decreased (Fig. S14†), possibly due to the partial agglomeration of the single atoms, occupying the catalytically active site. When the pH ranged from 9 to 12, the removal rate of ACT decreased due to the enhanced repulsion between Ni-TpBpy and ACT (Fig. S15†). However, the degradation efficiency remained consistently above 90% with minimal fluctuations across a pH range of 3 to 9, implying the remarkable adaptability of Ni-TpBpy to the variation of pH.54 As depicted in Fig. S16,† the optimal ACT concentration was 20 mg L−1. However, as the ACT concentration went up to 30 mg L−1, the photodegradation rate diminished attributed to the increased number of ACT molecules and their intermediates absorbing a large amount of light, thus reducing the overall degradation rate.55 Moreover, the impact of co-existing anions (e.g., Cl−, NO3−, SO42−, and CO32−) and varied concentrations of humic acid (HA) on the ACT photodegradation was also investigated (Fig. S17 and S18†). The results indicated that Ni-TpBpy possesses the advantage and potential for efficient degradation of ACT in complex matrices.
3.4 Universality on photocatalytic degradation
Sulfonamide antibiotics, including sulfamethoxazole (SMX), sulfisoxazole (SIZ), and sulfathiazole (STZ), are an emerging class of environmental pollutants. With the widespread use of antibiotics, the degradation and removal of these sulfonamide pollutants are crucial for the protection of aquatic organisms and human health. Therefore, these three substances were further used as models to assess the universality of Ni-TpBpy for visible photocatalytic degradation of organic pollutants. Significantly, Ni-TpBpy exhibited excellent adsorption and visible-light-driven photocatalytic degradation performance for SMX, STZ and SIZ, achieving a removal efficiency of 92%, 92% and 90% within 30 min, respectively (Fig. S19†). Apparently, the results demonstrate the potential of Ni-TpBpy as an effective solution for the treatment of organic contaminants in water, indicating the high versatility of Ni-TpBpy for the photocatalytic degradation of organic pollutants.3.5. Photocatalytic degradation pathway
It is widely acknowledged that photo-induced reactive species, such as trapped h+, ˙OH, and ˙O2−, play a pivotal role in the photocatalytic process.56 To investigate the predominant active species generated in the reaction system, scavengers, including 1,4-benzoquinone (BQ), ethylenediaminetetraacetate (EDTA-2Na), AgNO3, and isopropanol (IPA) were used to quench ˙O2−, h+, e−, and ˙OH, respectively.57 As shown in Fig. 5a and b, the introduction of EDTA-2Na and BQ can both remarkably inhibit the degradation efficiency of ACT from 97.8% to 59.9% and 50.9%, with the corresponding degradation rate decreasing from 0.17 min−1 to 0.04 min−1 and 0.02 min−1, respectively, verifying the dominant roles of h+ and ˙O2− in the ACT degradation process. In contrast, IPA and AgNO3 showed negligible inhibition, indicating that ˙OH and e− had marginal effects on the degradation of ACT. Additionally, the EPR results showed the characteristic signal corresponding to the DMPO-˙O2− adduct (AN = 13.0 G, AH = 10.2 G, AH = 1.5 G, Fig. 5c) and TEMPO-h+ (Fig. S20†), further confirming the participation of ˙O2− and h+ in the photodegradation process.58 A faint ˙O2− signal was produced on Ni-TpBpy under dark conditions, whereas the capability of Ni-TpBpy to produce ˙O2− radicals was significantly enhanced under visible light irradiation for 1 min. TpBpy showed a similar pattern, but its ˙O2− production capacity was still much lower than that of Ni-TpBpy, consistent with its photodegradation ACT performance. The characteristic peaks of DMPO-˙OH and TEMP-1O2 were not detected under identical conditions (Fig. S20†). The results revealed that the degradation process was mainly driven by ˙O2− and h+, while ˙OH and 1O2 had a negligible effect.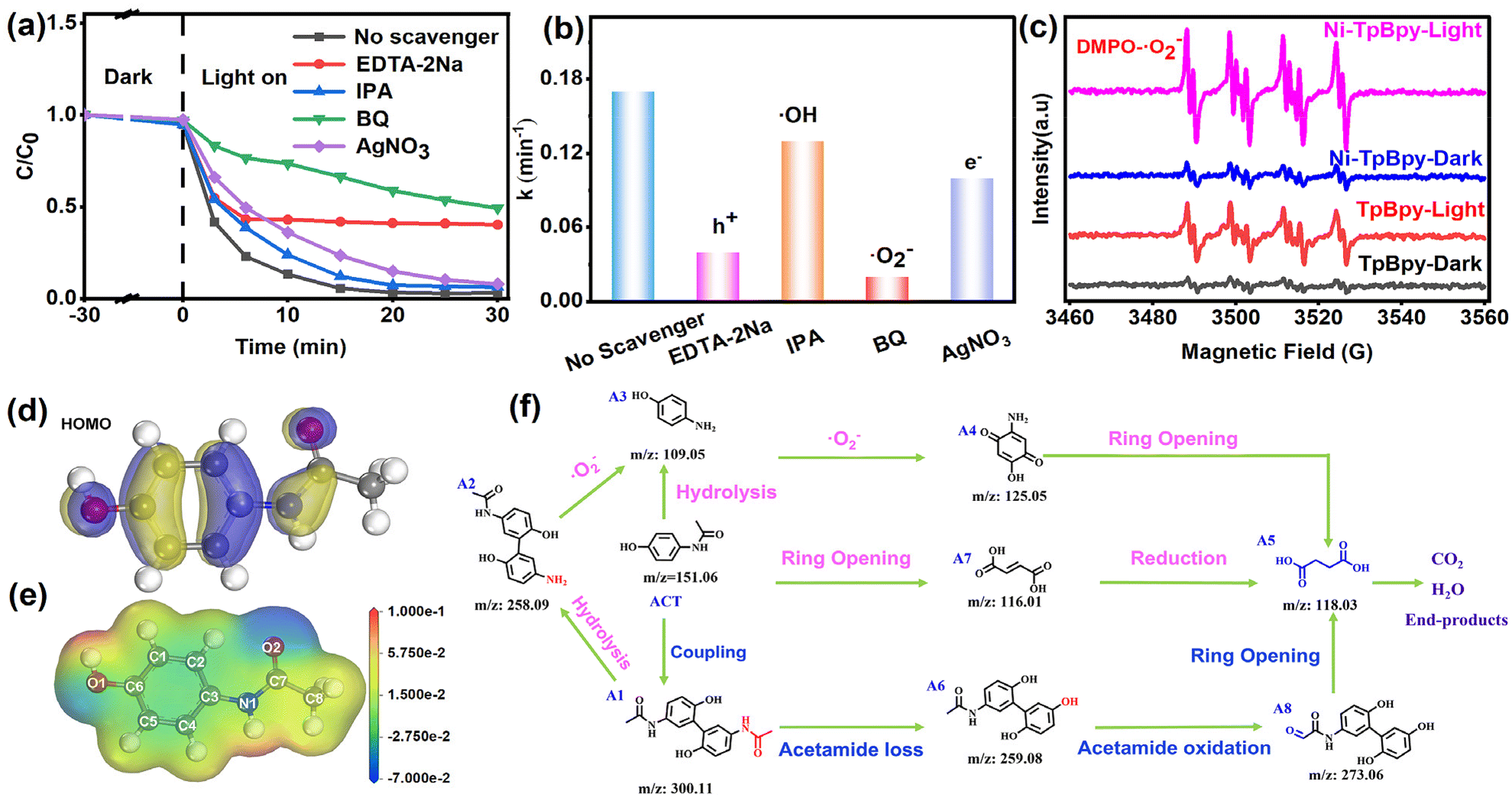 | ||
| Fig. 5 Effect of various free radical scavengers on the photocatalytic degradation of ACT (a). The k value of the ACT degradation systems with different trapping reagents (b). EPR spectra of DMPO-˙O2-generated by TpBpy and Ni-TpBpy in the dark and under visible-light irradiation (c). Chemical structure and surface electrostatic potential (d) and HOMO (e) of ACT. Photocatalytic degradation pathways of ACT (f). | ||
In addition, the Fukui index (Fig. S21†) (f0), electrostatic potentials (Fig. 5d), the HOMO (Fig. 5e), and LUMO (Fig. S22†) of ACT were calculated to identify the reactive sites in the ACT molecule that were susceptible to free radical attack.59 The calculation results showed that the HOMO of ACT was mainly centered on the benzene ring and the C–O, N–C, and C–O bonds. These regions are vulnerable to ROS attack due to easy electron escape from HOMO. Prior investigations have indicated that ROS preferentially targets sites with high f0 values. Notably, during ACT degradation, the sites exhibiting augmented f0 values and the most positive electrostatic potentials were situated in proximity to the C5 and N1 atoms, thus rendering them vulnerable to further attack by negatively charged ˙O2−.
To facilitate the understanding of the degradation pathway, eight crucial intermediates in ACT degradation were identified using liquid chromatography-mass spectrometry (LC-MS), as presented in Table S5.† Therefore, the potential degradation pathway of Ni-TpBpy to ACT was elucidated based on the above experimental and DFT calculations, as depicted in Fig. 5f. Firstly, the ˙O2− radicals would attack the C5 atom (C5, f0 = 0.07) of ACT to give the dimer isomer A1 (m/z: 300.11). Alternatively, the C−N (N1, f0 = 0.043) bond in ACT could also be directly hydrolyzed to yield product A3 (m/z: 109.05). Additionally, ring-opening within ACT might produce byproduct A7 (m/z: 116.01). Subsequently, the hydrolysis of the C–N bond in byproduct A1 could lead to the formation of byproduct A2 (m/z: 258.09), which might subsequently transform into byproduct A3 (m/z: 109.05). Simultaneously, the loss of an acetamide group from A1 resulted in the formation of A6 (m/z: 259.08). A6 was subsequently oxidized, catalyzing the conversion into the corresponding A8 (m/z: 273.06). Upon prolonged exposure to ˙O2− radicals, intermediate A3 could convert into a quinone-like structure, denoted as intermediate A4 (m/z: 125.05). Both intermediate A4 and A8 can undergo further transformation into byproduct A5 (m/z: 118.08) through a ring-opening process. Ultimately, product A5 could undergo further conversion into carbon dioxide and water. Consequently, Ni-TpBpy can efficiently photodegrade the emerging pollutant ACT into small molecules or achieve complete mineralization. The degradation of these intermediates was monitored through total organic carbon (TOC) analysis. After 30 min of reaction, the evolution of TOC demonstrated 56.8% mineralization, indicating the successful degradation of the transformation products of ACT.
3.6. Theoretical insights into the photodegradation mechanism
DFT calculation is a valid method to evaluate the capability of a catalyst to adsorb O2 and produce ˙O2−.60–62 According to the calculation, the adsorption energy of O2 on the bipyridine double N-site of TpBpy was −1.30 eV, which decreased to −3.04 eV after the single Ni atom anchoring (Fig. 6a), indicating a higher adsorption affinity of Ni-TpBpy for O2. In addition, the energy barriers for the adsorption of O2 to generate ˙O2− radicals were 0.89 eV and 0.36 eV for TpBpy and Ni-TpBpy, respectively (Fig. 6b), and this substantial decrease in energy barriers suggests that introducing Ni single atoms into TpBpy facilitates the generation of ˙O2− radicals. The production of the reactive species ˙O2− was further elucidated using the first-principles approach,63 where the Ni-TpBpy surface was considered to have a series of inserted electrons with 0, 1, 2, and 3 e−. In the absence of electron introduction, the O2 molecule is physically tilted to the Ni-TpBpy surface with a free-state configuration. Upon the introduction of the electron from 0 to 3 e−, the O–O bond length progressively elongated from 1.28881 to 1.30617 Å (Fig. 6c). This elongation suggests a gradual activation of the O2 molecule, ultimately leading to the formation of ˙O2−. Furthermore, the charge density difference and Bader charge analysis also showed that the charge of the adsorbed O2 increased from −0.7773 e− to −0.8980 e− with the insertion of electrons (Fig. 6d), where the O2 molecules gained additional e− from the active Ni atomic sites, thereby favoring the ˙O2− generation. In conclusion, both experimental and theoretical calculations demonstrated that anchoring single-atom Ni in TpBpy was advantageous for enhancing the generation of ˙O2− radicals, which significantly improves the visible light degradation performance of ACT.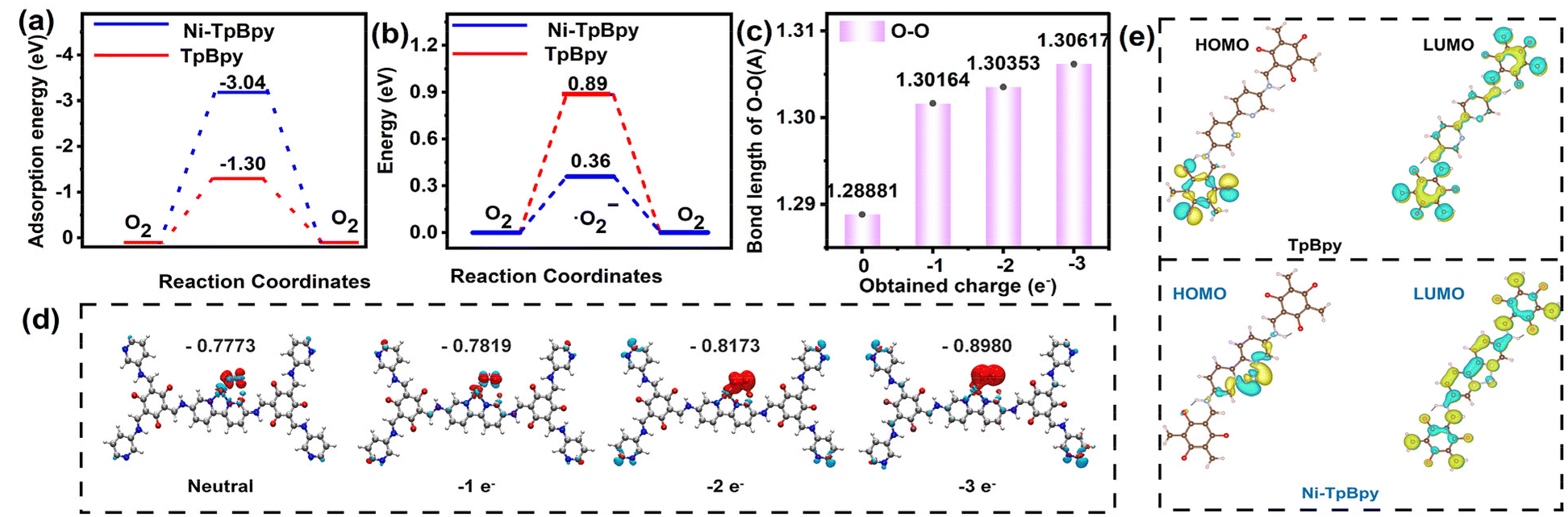 | ||
| Fig. 6 Adsorption energy of O2 on TpBpy and Ni-TpBpy (a). Comparison of energy barriers between TpBpy and Ni-TpBpy in absorbing O2 to produce ˙O2− (b). The O–O bond length (c) and differential charge density contour (d) of adsorbed O2 after the insertion of electrons. Note: the red and cyan contours represent increased and decreased charge density, respectively (d). Calculated ground state HOMO and LUMO of TpBpy and Ni-TpBpy (e). | ||
Furthermore, to gain insight into whether nickel single atoms can efficiently regulate the charge distribution, DFT simulations were performed to investigate the electron and hole distributions in the first excited states of TpBpy and Ni-TpBpy. As shown in Fig. S23,† the electrons (green regions) and holes (brown regions) in TpBpy were uniformly distributed, while the electrons in Ni-TpBpy were mainly concentrated on the Ni atoms, which suggests that the anchoring of individual nickel atoms should accelerate the massive separation of electron–hole pairs. Moreover, from the two-dimensional electrostatic potential map (Fig. S24†), the blue region around the two N atom positions in TpBpy displays a negative electrostatic potential, signifying that this area should be enriched with electrons. When Ni was anchored in TpBpy, the yellow-green region around Ni implied a more positive electrostatic potential, indicating that the electrons in Ni-TpBpy were mainly concentrated on the Ni atoms, which can easily act as electron acceptors. The LUMO of TpBpy showed no significant changes before and after anchoring the single atom Ni. However, a comparison of the HOMO revealed that the electrons in the HOMO of TpBpy were distributed at the edge positions, while the electrons in the HOMO of Ni-TpBpy were concentrated in the single atom of Ni (Fig. 6e). Therefore, the single-atom Ni acted as an electron acceptor, facilitating the rapid separation and transfer of electron holes. These calculation results indicated that the implantation of single-atom Ni not only significantly inhibits the recombination of photo-induced charge carriers but also serves as an electron collector, promoting the migration of e− from TpBpy to single-atom Ni.64 Consequently, Ni-TpBpy exhibited excellent intra-molecular charge transfer properties during the photocatalytic process, thereby demonstrating superior photodegradation performance for emerging pollutants such as ACT.
Based on the above findings, an elucidation of the photodegradation mechanism of ACT on Ni-TpBpy is illustrated in Fig. 7. Initially, Ni-TpBpy produced electron-hole pairs under visible light irradiation, the electrons moved from HOMO to LUMO and promptly transferred to Ni2+, whereas the holes remained on HOMO, leading to efficient separation of electron-hole pairs. Subsequently, Ni acting as a catalytically active site reacted with adsorbed O2 and H2O to form abundant active ROS for the photocatalytic degradation of ACT. Meanwhile, the h+ after acquiring e− can also degrade ACT directly. The reaction steps are shown in the following equations:
| Ni-TpBpy + hν → e−(LUMO) + h+(HOMO) | (2) |
| O2 + e− → ˙O2− | (3) |
| ˙O2−/h+ + ACT → CO2 + H2O | (4) |
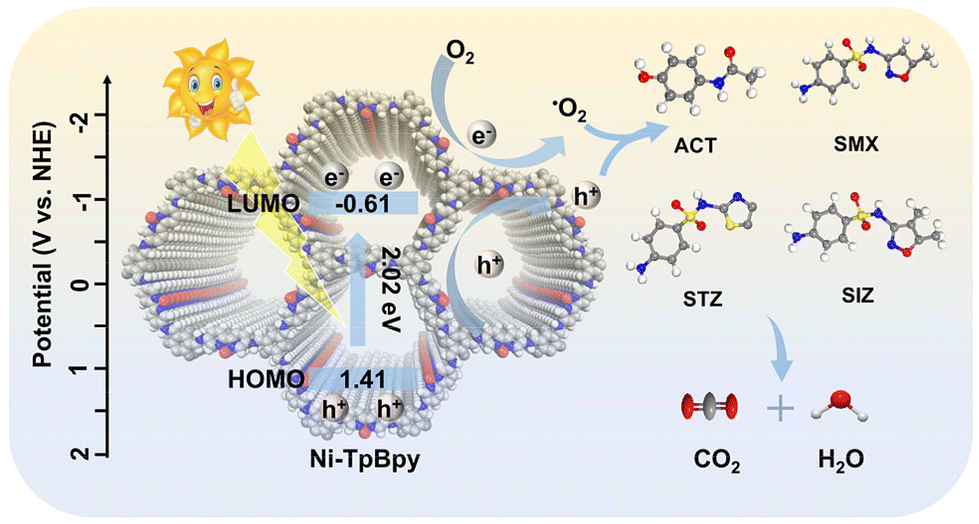 | ||
| Fig. 7 Illustration of PPCP photodegradation mechanism using Ni-TpBpy. Note: C, N, O and Ni are shown as light grey, blue, dark grey and red spheres, respectively; H atoms have been omitted for clarity. | ||
Therefore, Ni-TpBpy represents a unique prototype, which simultaneously meets the various demands of high-performance photocatalysis through the powerful metal-carrier synergistic interactions between the single transition metal atoms and COFs.
4 Conclusion
In this work, interconnected pyridine-based COF materials were synthesized in an innovative manner using DBD-based green technology under a nitrogen atmosphere, which greatly reduced the synthesis time. Meanwhile, atomically dispersed Ni atoms with high loading content were successfully anchored to bipyridine TpBpy via a specific tetra-coordinated N2–Ni–Cl2 structure between nickel and unsaturated coordinating bis-nitrogen atoms on pyridine. Experimental results and DFT theoretical calculations demonstrated a strong synergistic effect between the nickel catalytic sites and carriers, endowing Ni-TpBpy with stronger visible light absorption, more efficient photogenerated carrier separation and migration, a narrower bandgap, and easier generation of reactive superoxide radicals, thus possessing the capability to rapidly degrade and strongly mineralize emerging pollutants. The optimized Ni-TpBpy can achieve photodegradation of 97.8% ACT with 56.8% mineralization under 30 min of visible light irradiation, outperforming most advanced photocatalysts. In addition, EPR measurements and free radical scavenging experiments verified the pivotal role of h+ and ˙O2− as the primary reactive oxygen species in ACT degradation. Combined with LC-MS, a putative photodegradation pathway for ACT was proposed. Notably, the exceptional stability and versatility of Ni-TpBpy make it a promising candidate for practical applications. Crucially, this investigation marks the first application of single-atom Ni-TpBpy in photocatalytic degradation of emerging pollutants, highlighting the immense potential of single-atom photocatalysts constructed from COF materials in the realms of water treatment and environmental remediation.Author contributions
G. J. Wen: methodology, formal analysis, investigation, and writing – original draft. Q. Q. Peng: validation. C. Yuan: software, data curation, and validation. J. He: conceptualization, writing – review & editing, and funding acquisition. X. D. Hou: conceptualization, funding acquisition, writing – review & editing, and supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (No. 22204110), the 111 Center (B17030) and the Fundamental Research Funds for the Central Universities, China. We also thank Dr Aiqun Gu, Hanjiao Chen, Yunfei Tian, Shanling Wang and Xi Wu from the Analytical & Testing Centre of Sichuan University for their technical support in XRD, EPR, XPS, AC HAADF-STEM and ICP-OES, respectively.References
- M. Sun, H.-H. Liu, Y. Zhang and L.-F. Zhai, Chem. Eng. J., 2020, 387, 124124–124130 CrossRef CAS
.
- Y. Chen, W. Lu, H. Shen, Y. Gu, T. Xu, Z. Zhu, G. Wang and W. Chen, Appl. Catal., B, 2019, 258, 117960–117971 CAS
- V. K. Parida, D. Sikarwar, A. Majumder and A. K. Gupta, J. Environ. Manage., 2022, 308, 114609–114633 CrossRef CAS PubMed
- G. Fan, X. Zheng, J. Luo, H. Peng, H. Lin, M. Bao, L. Hong and J. Zhou, Chem. Eng. J., 2018, 351, 782–790 CrossRef CAS
- D. R. Baker and B. Kasprzyk-Hordern, Sci. Total Environ., 2013, 454–455, 442–456 CrossRef CAS PubMed
- Z. Wang, Z. Xu and X. Li, J. Hazard. Mater., 2018, 359, 356–364 CrossRef CAS PubMed
- A. Y.-C. Lin, X.-H. Wang and C.-F. Lin, Chemosphere, 2010, 81, 562–570 CrossRef CAS
- X. Zhang, P. Sun, K. Wei, X. Huang and X. Zhang, Chem. Eng. J., 2021, 412, 128525–128526 CrossRef CAS
- N. Goyal, P. Gao, Z. Wang, S. Cheng, Y. S. Ok, G. Li and L. Liu, J. Hazard. Mater., 2020, 392, 122494–122509 CrossRef CAS PubMed
- M. Chen, C. Guo, S. Hou, J. Lv, Y. Zhang, H. Zhang and J. Xu, Appl. Catal., B, 2020, 266, 118614–118626 CrossRef CAS
- S. R. Mishra, V. Gadore, K. R. B. Singh, S. S. Pandey and M. Ahmaruzzaman, Environ. Res., 2024, 259, 119435 CrossRef CAS PubMed
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050–17066 CrossRef CAS
- J. Yuan, H. Li, G. Wang, C. Zhang, Y. Wang, L. Yang, M. Li and J. Lu, Appl. Catal., B, 2022, 303, 120892–120901 CrossRef CAS
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694–1601713 CrossRef
- V. Gadore, S. R. Mishra and M. Ahmaruzzaman, Sci. Rep., 2023, 13, 7708 CrossRef CAS
- X. Gao, L. An, D. Qu, W. Jiang, Y. Chai, S. Sun, X. Liu and Z. Sun, Sci. Bull., 2019, 64, 918–925 CrossRef CAS
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS
- H. Jeong, S. Shin and H. Lee, ACS Nano, 2020, 14, 14355–14374 CrossRef CAS PubMed
- C. Z. Lv, P. C. Jiao, H. Xin, L. Wu, G. F. Ouyang and X. D. Hou, Appl. Catal., B, 2024, 340, 123248–123259 CrossRef CAS
- V. Hasija, S. Patial, P. Raizada, A. A. P. Khan, A. M. Asiri, Q. V. Le, V.-H. Nguyen and P. Singh, Coord. Chem. Rev., 2022, 452, 214298–2142312 CrossRef CAS
- S. S. A. Shah, M. S. Javed, T. Najam, M. A. Nazir, A. Ur Rehman, A. Rauf, M. Sohail, F. Verpoort and S.-J. Bao, Mater. Today, 2023, 67, 229–255 CrossRef CAS
- G. Huang, Q. Niu, J. Zhang, H. Huang, Q. Chen, J. Bi and L. Wu, Chem. Eng. J., 2022, 427, 131018–131027 CrossRef CAS
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1–1 CrossRef CAS
- Y. An, S. Tan, Y. Liu, K. Zhu, L. Hu, Y. Rong and Q. An, Energy Storage Mater., 2021, 41, 354–379 CrossRef
- K. Qi, M. Chhowalla and D. Voiry, Mater. Today, 2020, 40, 173–192 CrossRef CAS
- M. Kou, W. Liu, Y. Wang, J. Huang, Y. Chen, Y. Zhou, Y. Chen, M. Ma, K. Lei, H. Xie, P. K. Wong and L. Ye, Appl. Catal., B, 2021, 291, 120146–120153 CrossRef CAS
- T. He, Z. Zhao, R. Liu, X. Liu, B. Ni, Y. Wei, Y. Wu, W. Yuan, H. Peng, Z. Jiang and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 6057–6066 CrossRef CAS PubMed
- J. He, X. Jiang, F. Xu, C. Li, Z. Long, H. Chen and X. Hou, Angew. Chem., Int. Ed., 2021, 60, 9984–9989 CrossRef CAS PubMed
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS
- X. Jiang, Z. E. Lin, X. Zeng, J. He, F. Xu, P. Deng, J. Jia, X. Jiang, X. Hou and Z. Long, Chem. Commun., 2019, 55, 12192–12195 RSC
- Y. Huang, H. Gu, H. Zhang, X. Wang, L. Gao, Y. Cui, B. Zong, H. Li and W.-L. Dai, ACS Catal., 2024, 14, 12541–12550 CrossRef CAS
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 1856–1862 CrossRef CAS PubMed
- A. Jati, K. Dey, M. Nurhuda, M. A. Addicoat, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2022, 144, 7822–7833 CrossRef CAS PubMed
- L. Zhang, Z. Liu, Q. Deng, Y. Sang, K. Dong, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2020, 60, 3469–3474 CrossRef PubMed
- J. Kou, J. Fang, J. Li, H. Zhao, M. Gao, G. Zeng, W. D. Wang, F. Zhang, J. Ma and Z. Dong, Chem. Eng. J., 2023, 463, 142255–142265 CrossRef CAS
- X. Sun, Y. Hu, Y. Fu, J. Yang, D. Song, B. Li, W. Xu and N. Wang, Small, 2023, 2305978–2305985 Search PubMed
- W.-R. Cui, C.-R. Zhang, W. Jiang, R.-P. Liang and J.-D. Qiu, ACS Appl. Nano Mater., 2019, 2, 5342–5349 CrossRef CAS
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS
- Z. Li, S. Qiu, Y. Song, S. Huang, J. Gao, L. Sun and J. Hou, Sci. Bull., 2022, 67, 1971–1981 CrossRef CAS PubMed
- L. Yang, Z. Chen, Q. Cao, H. Liao, J. Gao, L. Zhang, W. Wei, H. Li and J. Lu, Adv. Mater., 2023, 2306758–2306770 Search PubMed
- F. He, Y. Lu, G. Jiang, Y. Zhang, P. Dong, X. Liu, Y. Wang, C. Zhao, S. Wang, X. Duan, J. Zhang and S. Wang, Appl. Catal., B, 2024, 341, 123307–123319 CrossRef CAS
- S. Sun, J. Li, H. Ding, B. Zhang, H. Huang, Z. Xu, Y. Tu, D. Chen and X. Duan, Chem. Eng. J., 2023, 460, 141718–141728 CrossRef
- C.-C. Wang, X.-H. Yi and P. Wang, Appl. Catal., B, 2019, 247, 24–48 CrossRef CAS
- Z. Dong, L. Zhang, J. Gong and Q. Zhao, Chem. Eng. J., 2021, 403, 126383–126392 CrossRef CAS
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467–2476 CrossRef
- H. Sepehrmansourie, H. Alamgholiloo, N. N. Pesyan and M. A. Zolfigol, Appl. Catal., B, 2023, 321, 122082–122094 CrossRef CAS
- L. Yang, J. Yuan, G. Wang, Q. Cao, C. Zhang, M. Li, J. Shao, Y. Xu, H. Li and J. Lu, Adv. Funct. Mater., 2023, 33, 2300954–2300963 CrossRef CAS
- F. Liu, Z. Ma, Y. Deng, M. Wang, P. Zhou, W. Liu, S. Guo, M. Tong and D. Ma, Environ. Sci. Technol., 2021, 55, 5371–5381 CrossRef CAS PubMed
- Y. Hou, F. Liu, B. Zhang and M. Tong, Environ. Sci. Technol., 2022, 56, 16303–16314 CrossRef CAS
- V. K. Parida, S. K. Srivastava, S. Chowdhury and A. K. Gupta, Chem. Eng. J., 2023, 472, 144969–144989 CrossRef CAS
- Z. Chu, J. Li, H. Y. Sohn, C. Chen, X. Huang, Y. Lan, A. Murali and J. Zhang, Composites, Part B, 2023, 257, 110689–110701 CrossRef CAS
- S. Moradi, A. A. Isari, F. Hayati, R. Rezaei Kalantary and B. Kakavandi, Chem. Eng. J., 2021, 414, 128618 CrossRef CAS
- L. Jiang, X. Yuan, G. Zeng, Z. Wu, J. Liang, X. Chen, L. Leng, H. Wang and H. Wang, Appl. Catal., B, 2018, 221, 715–725 CrossRef CAS
- T. Li, C. Wang, T. Wang and L. Zhu, Appl. Catal., B, 2020, 268, 118442–118451 CrossRef CAS
- G. R. Buettner, Free Radicals Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed
- A. Mehmood, S. I. Jones, P. Tao and B. G. Janesko, J. Chem. Inf. Model., 2018, 58, 1836–1846 CrossRef CAS PubMed
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- C. Jin, D. Sun, Z. Sun, S. Rao, Z. Wu, C. Cheng, L. Liu, Q. Liu and J. Yang, Appl. Catal., B, 2023, 330, 122613–122621 CrossRef CAS
- S. Cao, H. Li, T. Tong, H. C. Chen, A. Yu, J. Yu and H. M. Chen, Adv. Funct. Mater., 2018, 28, 1802169–1802177 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02759k |
| This journal is © The Royal Society of Chemistry 2025 |