
Constructing protein-functionalized DNA origami nanodevices for biological applications
Chuangyuan
Zhao
ab,
Xinran
Jiang
c,
Miao
Wang
d,
Songbai
Gui
*e,
Xin
Yan
*f,
Yuanchen
Dong
 *ab and
Dongsheng
Liu
*g
*ab and
Dongsheng
Liu
*g
aCAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China. E-mail: dongyc@iccas.ac.cn
cSchool of Life Sciences Fudan University, Shanghai, 200433, China
dChemistry and chemical biology, Cornell university, 122 Baker Laboratory, Ithaca, NY 14853, USA
eDepartment of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, 100071, Beijing, China. E-mail: guisongbai@ccmu.edu.cn
fDepartment of Sports Medicine, Beijing Key Laboratory of Sports Injuries, Peking University Third Hospital, Beijing, 100191, China. E-mail: dr_yanxin@126.com
gEngineering Research Center of Advanced Rare Earth Materials, (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing, 100084, China. E-mail: liudongsheng@tsinghua.edu.cn
First published on 31st October 2024
Abstract
In living systems, proteins participate in various physiological processes and the clustering of multiple proteins is essential for efficient signaling. Therefore, understanding the effects of the number, distance and orientation of proteins is of great significance. With programmability and addressability, DNA origami technology has enabled fabrication of sophisticated nanostructures with precise arrangement and orientation control of proteins to investigate the effects of these parameters on protein-involved cellular processes. Herein, we highlight the construction and applications of protein-functionalized DNA origami nanodevices. After the introduction of the structural design principles of DNA origami and the strategies of protein–DNA conjugation, the emerging applications of protein-functionalized DNA origami nanodevices with controlled key parameters are mainly discussed, including the regulation of enzyme cascade reactions, modulation of cellular behaviours, drug delivery therapy and protein structural analysis. Finally, the current challenges and potential directions of protein-functionalized DNA origami nanodevices are also presented, advancing their applications in biomedicine, cell biology and structural biology.
 Chuangyuan Zhao | Chuangyuan Zhao is currently a Ph.D. candidate at the Institute of Chemistry, Chinese Academy of Sciences. He received his B.S. degree in Chemistry from Lanzhou University in 2021. His research focuses on the regulation of protein function based on DNA nanotechnology. |
 Songbai Gui | Songbai Gui is a professor, chief physician, doctoral supervisor, and director at the Third Ward of Neuro-Oncology at Beijing Tiantan Hospital, which is affiliated with Capital Medical University. His research focuses on the diagnosis and surgical treatment of various cranial tumors or diseases of the ventricular and cisternal systems, particularly in endoscopic minimally invasive surgery for skull base and ventricular/cisternal diseases, achieving international advanced levels. |
 Xin Yan | Xin Yan is the attending physician in the Department of Sports Medicine at Peking University Third Hospital. His research focuses on the application of DNA nanomaterials in the treatment of osteoarthritis and cartilage defects. His major representative research is the proposal of an improved traditional stem cell injection therapy for OA. |
 Yuanchen Dong | Yuanchen Dong is a professor at the Institute of Chemistry, Chinese Academy of Sciences. He received his Ph.D. degree in Polymer Chemistry and Physics from Tsinghua University in 2015. His current research interests are construction and assembly of DNA amphiphiles, structure and functions of proteins based on DNA self-assembly, construction of amphiphilic assembly with a controllable size and morphology via Frame Guided Assembly and applications of Cryo-EM in structural biology. |
 Dongsheng Liu | Dongsheng Liu is a professor from the Department of Chemistry at Tsinghua University. He received his B.S. degree in Polymer Chemistry from the University of Science and Technology of China in 1993 and Ph.D. degree from Hong Kong Polytechnic University in 2002. He joined the National Nanoscience Center in 2005 and moved to Tsinghua University in 2009. He was elected as a Fellow of the Royal Society of Chemistry in 2011. His main scientific interests are in the fields of biomolecules and their specific interaction in nanostructure fabrication and nanodevice design, including DNA nanomachines, DNA supramolecular hydrogels and Frame Guided Assembly. |
1. Introduction
Proteins are known as the fundamental material basis of life and participate in essential biological processes, such as metabolism,1,2 gene expression,3,4 and tumorigenesis.5,6 Notably, proteins often form networks or higher-order assemblies, which are defined by their numbers, spatial distances, and orientations. These structural characteristics play crucial roles in modulating cellular functions, including signaling, communication, and transportation.7–10 For example, the tumor necrosis factor (TNF) ligand and receptor superfamilies are crucial for cell proliferation, survival, and apoptosis.11 Numerous studies have indicated that the receptors, prearranged in a large hexagonal structure of antiparallel dimers, are activated upon the binding of ligands and stabilized by the crosslinking of antibodies.12,13 This clustering of proteins is essential for efficient signaling. Therefore, the structural analysis of proteins and the precise patterns of proteins to regulate cellular behaviours are of great significance in biomedicine, cell biology and structural biology. However, it is still challenged by precisely modulating molecular distributions at the nanometer scale or controlling their orientation.DNA origami, which involves the highly cooperative hybridization of a long DNA scaffold with multiple short DNA strands, has been employed to fabricate DNA origami nanodevices with complex shapes and functions.14,15 Due to the programmability, addressability, and modification capabilities of DNA origami, various biomolecules, particularly proteins, can be incorporated into the framework with controlled parameters, including copy number, distance, and orientation. This precise control enables the construction of protein-functionalized DNA origami nanodevices with a defined spatial distribution at the nanometer scale, thereby addressing limitations in the investigation and regulation of complex physiological processes.16–18
In this review, we provide a comprehensive summary of previous research studies concerning protein-functionalized DNA origami. Initially, we introduce the fundamental concepts and structural design principles of DNA origami. Following this, we outline various strategies for protein–DNA conjugation. Next, we discuss in detail the precise control of critical parameters, including the number, distance, and orientation of proteins incorporated into DNA origami, along with their successful biological applications. Finally, we highlight the existing challenges and prospective opportunities in the field of protein-functionalized DNA origami nanodevices, aiming to broaden their applications.
2. Development of DNA origami technology
Since Seeman pioneered the concept of DNA nanotechnology, various strategies for DNA nanostructure fabrication have been developed.19 Notably, DNA origami technology enables the bottom-up fabrication of well-defined nanostructures and holds significant applications across diverse research domains. In 2006, Rothemund first proposed the concept of DNA origami and employed this strategy to construct two-dimensional structures with various shapes, including squares, triangles, stars, and smiling faces (Fig. 1A and B).20 In this technology, a long single stranded DNA called scaffold (e.g., M13mp18; 7249 nucleotides) was folded with hundreds of short sequence-designed complementary DNA strands called staples. Each staple with multiple binding domains hybridized into defined regions of the scaffold, thereby bringing them into proximity and controlling their orientation through periodic crossovers. The high cooperative interactions between the scaffold and multiple staples enabled the formation of designed shapes by utilizing an excess of staples, which reduced sensitivity to the stoichiometry and purity of oligonucleotides. Consequently, the shapes of DNA origami can be programmed by manipulating the staple sequences. | ||
| Fig. 1 Schematic of the DNA origami assembly and the nanostructures. (A) Schematic of the principles of the DNA origami assembly. Reproduced with permission from ref. 20. Copyright 2006 Springer Nature. (B) Schematic and characterization of 2D DNA origami shapes. Scale bars are 100 nm unless noted otherwise. Reproduced with permission from ref. 20. Copyright 2006 Springer Nature. (C) Schematic and characterization of 3D DNA origami shapes. Scale bars are 20 nm. Reproduced with permission from ref. 21. Copyright 2009 Springer Nature. (D) Schematic and characterization of curved DNA origami with a 12-tooth gear shape. Scale bars are 20 nm. Reproduced with permission from ref. 33. Copyright 2009 The American Association for the Advancement of Science. (E) Schematic and characterization of DNA origami with a nanoflask shape. Scale bars are 75 nm. Reproduced with permission from ref. 34. Copyright 2011 The American Association for the Advancement of Science. (F) Schematic and characterization of 2D single-stranded DNA/RNA origami shapes. Scale bars are 50 nm. Reproduced with permission from ref. 36. Copyright 2017 The American Association for the Advancement of Science. | ||
In 2009, Shih and Yan expanded this approach to design dense three-dimensional (3D) nanostructures based on three fundamental building blocks: honeycomb lattices,21 square lattices,22 and hexagonal lattices.23 As a result, a series of complex structures were created, such as monoliths, square nuts and railed bridges (Fig. 1C). In addition to these densely packed nanostructures, Högberg and Yan introduced new principles for folding arbitrary polygonal digital meshes with a more open and loose conformation using the wireframe architecture, which proved to be more stable in biological environments.24,25 Furthermore, several strategies have also been developed for building large structures, primarily involving the use of longer scaffolds and base stacking or base hybridization at the ends of each designed nanostructure to assist hierarchical assembly.26–30 For instance, Rothemund and co-workers constructed hierarchical large structures by introducing diverse “bonds” at the edges of individual building blocks. The creation of these “bonds” was based on the characteristics of blunt ends (i.e., stacking polarity, symmetry, and sequence) and shape complementarity.31 In 2014, Liu and co-workers utilized two-scaffold chains to construct a designed frame with a hollow interior and localized stick chains to control the orderly assembly of four DNA tile building blocks via hybridization, facilitating the construction of large shapes.32
In addition, sophisticated structures such as twisted and curved 3D shapes could also be created with rational design of the DNA origami. In 2009, Shih and co-workers fabricated twisted 3D shapes by solely inserting or deleting base pairs per crossover chain. They also created curved 3D shapes by distributing a gradient of deletions to insertions of base pairs throughout a bundle's cross-section.33 Consequently, complex structures, such as square-toothed gears and a wireframe beach ball, were fabricated (Fig. 1D). In another work, a series of DNA nanostructures with out-of-plane curvature, including 3D spherical shells, ellipsoidal shells, and a nanoflask, were fabricated by shifting the relative positions of crossover points between adjacent double helices (Fig. 1E).34 Additionally, the complex and replicable nucleic acid-based nanostructures were also fabricated using a single DNA strand or RNA strand (Fig. 1F). In contrast to the multicomponent self-assembly strategy, this unimolecular folding strategy enabled the efficient replication of the strand and subsequent self-assembly into nanostructures both in vitro and within living cells. This advancement has significantly broadened the design space and enhanced the material scalability for bottom-up nanotechnology.35,36
In addition to the structural design, the incorporation of functional biomolecules into DNA nanostructures has broadened the range of applications in medicine, biology, and nanoscience.37–39 This integration has enabled more accurate, controllable, and quantitative outcomes in research due to programmable shape design, nanoscale addressability, and biocompatibility. In particular, protein-functionalized DNA origami nanodevices have been utilized in the spatial and temporal regulation of enzyme cascade reactions, modulation of cellular behaviours, anticancer/antibacterial treatments and construction of artificial biological macromolecules.40–42
3. Protein–DNA conjugation methods
To construct DNA origami nanostructures with precisely distributed proteins, the conjugation of DNA and proteins is critical. During the last few decades, many strategies have been developed to covalently or noncovalently conjugate proteins with DNA.43,44 The selection of an appropriate conjugation strategy depends on the size and composition of the proteins, the location of the binding or active domain of proteins, and the purification of the conjugates. Consequently, selecting an appropriate strategy for conjugating with DNA, followed by incorporation into DNA origami, can facilitate the effective manipulation of proteins across various applications.3.1. Noncovalent binding strategy
Several noncovalent strategies have been devised for the conjugation of DNA and proteins, including biotin–streptavidin, Ni-NTA–His tag, antigen–antibody, aptamer–protein, and DNA binding protein–DNA interactions. The specific interaction between streptavidin, a tetrameric protein, and biotin-conjugated DNA demonstrates high affinity and specificity (Fig. 2A).45–47 However, the large size of the tetrameric streptavidin limits its precise and dense distribution into DNA origami structures and the tetrameric configuration may result in an unpredictable stoichiometric ratio of DNA to the proteins. Another frequently employed strategy is the Ni-NTA–His tag interaction (Fig. 2B).48,49 This interaction is based on the binding of polyhistidine-tagged proteins to nitrilotriacetic acid (NTA)-labeled DNA in the presence of Ni2+ ions through chelation. The Ni2+ ions can be substituted with Co2+, Cu2+, or Zn2+,50 and this interaction is reversible when exposed to a stronger chelator like ethylenediaminetetraacetic acid (EDTA). Notably, the introduction of this small His tag, which consists of six histidine amino acids and has a molecular weight of 0.84 kDa, facilitates precise incorporation into DNA origami. | ||
| Fig. 2 Several strategies for the noncovalent protein–DNA conjugation. (A) Biotin–streptavidin interaction; (B) Ni-NTA–His tag interaction; (C) antigen–antibody interaction; (D) aptamer–protein interaction; and (E) DNA-binding protein–DNA interaction. | ||
The specific recognitions, including antigen–antibody (Fig. 2C) and aptamer–protein (Fig. 2D) interactions, have also been documented in the construction of protein-functionalized DNA origami nanodevices.51–53 Notably, aptamers, which are composed of single-stranded DNA that folds into secondary structures (i.e., stem loops, convex loops, and pockets) through base pairing, can specifically bind to various targets including small molecules, ions, and proteins. In contrast to the antigen–antibody based strategies, aptamers can directly hybridize with staple strands in DNA origami without the necessity for additional chemical modifications, thereby enhancing their applicability.
The last strategy is based on the interactions between DNA binding protein and DNA (Fig. 2E).54–56 In particular, zinc-finger proteins and other transcription factors possess specific DNA-binding domains that interact with double-stranded DNA at designated sequences. Consequently, the incorporation of specific sequences at defined positions within DNA origami can facilitate the targeted loading of proteins that are fused with DNA-binding proteins. Additionally, the orientation of these proteins can be altered by the addition or deletion of base pairs along the binding sequence.
3.2. Covalent conjugation
Although the noncovalent binding between DNA and proteins is reversible, this property hinders their interaction when protein-functionalized DNA origami nanodevices are exposed to a complex biological environment. Therefore, stable and irreversible covalent conjugation has been employed in the fabrication of protein-functionalized DNA origami nanodevices.Firstly, the well-established and typical method of covalent conjugation involves the use of heterobifunctional linkers, such as the N-succinimidyl-3-(2-pyridyldithio)-propionate (SPDP) (Fig. 3A) and the sulfo-succinimidyl-4-(N-maleimidomethyl)-cyclohexane-1-carboxylate (Sulfo-SMCC) (Fig. 3B).57,58 In most cases, these reagents react with lysine residues on the protein surface and a thiol group on DNA to produce protein–DNA conjugates. Notably, this method does not necessitate genetic manipulation of the proteins. However, the application of Sulfo-SMCC to form disulfide bonds may be compromised by bond cleavage under reductive conditions. Furthermore, these strategies exhibit a lack of regioselective conjugation due to the presence of multiple lysine residues on the proteins, resulting in a heterogeneous mixture of protein–DNA conjugates, which may inactivate proteins. This non-regioselective problem could be resolved through the application of maleimide–thiol chemistry (Fig. 3C), particularly when proteins possess a single cysteine residue either in their native state or in a genetically engineered form.59 This approach facilitates site-specific modification.
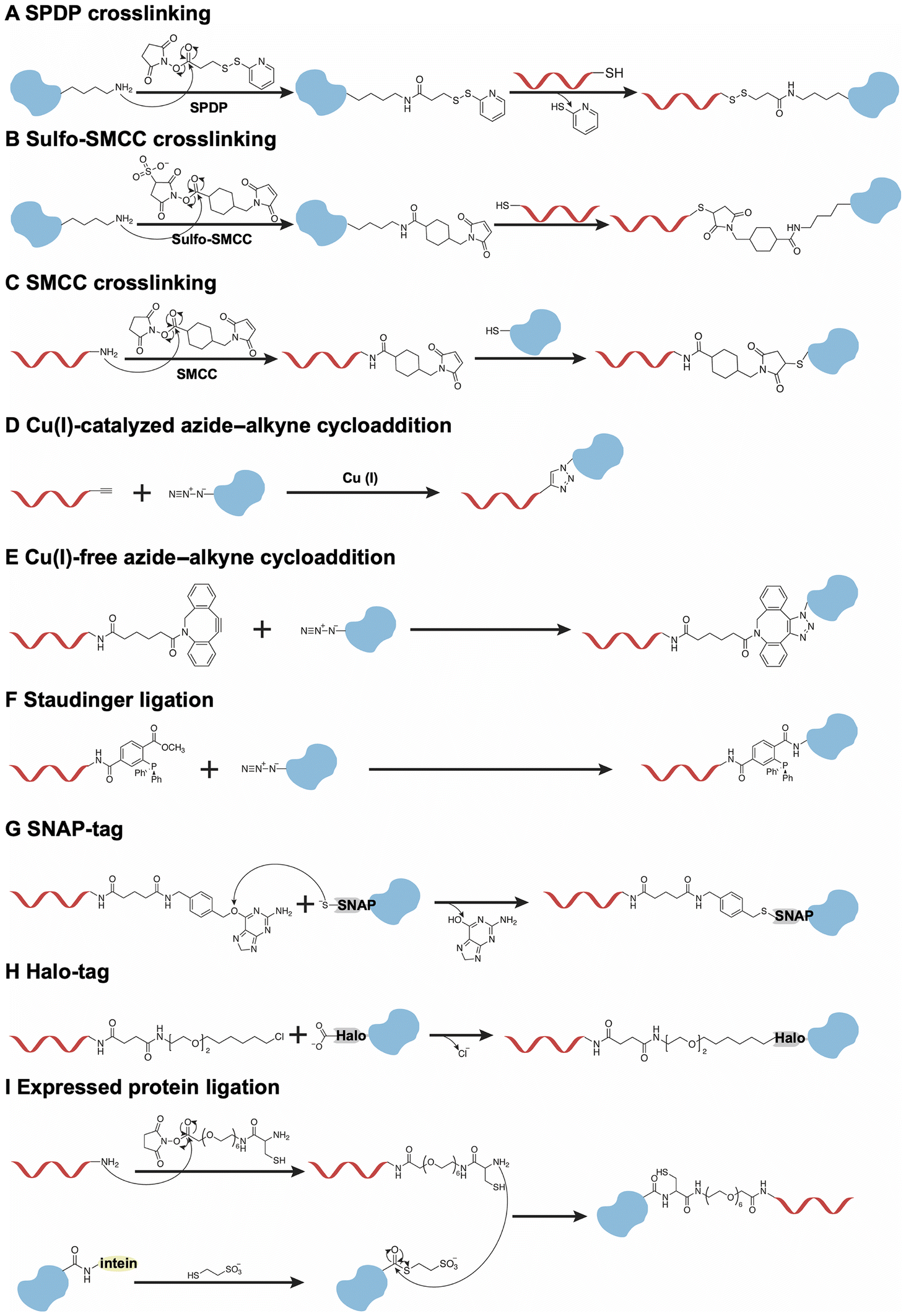 | ||
| Fig. 3 Several strategies for the covalent protein–DNA conjugation. (A) SPDP crosslinking; (B) sulfo-SMCC crosslinking; (C) SMCC crosslinking; (D) Cu(I)-catalyzed azide–alkyne cycloaddition; (E) Cu(I)-free azide–alkyne cycloaddition; (F) Staudinger ligation; (G) SNAP-tag; (H) Halo-tag; and (I) expressed protein ligation. | ||
An alternative strategy involves the expression of proteins with unique chemical handles, which facilitates site-specific modification. For example, the incorporation of unnatural amino acids at designated sites within proteins allows for subsequent orthogonal reactions with DNA, thereby enabling efficient conjugation at a single site. As illustrated in Fig. 3D and E, the proteins could be modified with an azide group at a specific site through genetic manipulation. The azide-modified proteins undergo conjugation with alkyne/DBCO-modified DNA via a Cu(I)-catalyzed or Cu-free click reaction.60–62 Additionally, Staudinger ligation could also be used to conjugate the azide-modified proteins with the phosphine-modified DNA to achieve site-specific modifications (Fig. 3F).63 While this strategy is effective for site-specific modifications due to its minimal disruption of the protein's original structure, optimizing the introduction of unnatural amino acids presents significant challenges.
Another method involves the introduction of protein tags at the C- or N-terminal of proteins, such as the human O6-alkylguanine-DNA-alkyltransferase (hAGT, referred to as SNAP-tag)64 and the haloalkane dehalogenase (referred to as Halo-tag).65 As illustrated in Fig. 3G, the formation of a thioether bond occurs between the cysteine-145 (Cys-145) residue located in the binding pocket of the SNAP-tag and the alkyl group on O6-benzylguanine-modified DNA. In addition, the Halo-tag is conjugated with chlorohexane-modified DNA through the nucleophilic displacement of the chloride group of the DNA by the aspartate-106 (Asp-106) residue on the Halo-tag (Fig. 3H). In addition to two commonly employed strategies, the expressed protein ligation approach is utilized to conjugate C-terminal or N-terminal intein-fused proteins with peptide-modified DNA (Fig. 3I).66,67 Firstly, the intein-fused protein, with an additional chitin-binding domain for affinity purification, reacts with mercaptoethanesulfonic acid to form a thioester group. Subsequently, the modified protein is ligated to the cysteine-modified DNA, resulting in a defined stoichiometric composition and site-specific linkage. However, this method encounters challenges associated with insoluble intein fusions. Generally, the orthogonal conjugations between modified DNA and the tags introduced at the terminal of proteins result in the formation of DNA–protein conjugates, thereby preventing any interference with the structure of the proteins.
4. Construction and applications of protein-functionalized DNA origami nanodevices
Following the conjugation of protein to DNA, the resulting DNA–protein conjugates hybridize with the fixed staple strands that extend from DNA origami structures. Utilizing the fundamental geometrical parameters of double stranded DNA (i.e., 2.0 nm diameter, 0.34 nm per base pair rise, and ∼34° bp−1 average twist),68 DNA origami nanostructures allow for the precise manipulation of proteins in terms of distance, number, and orientation. This capability enables the systematic study of protein structures, associations, and functions, and also holds potential applications in therapy.4.1. Spatial and temporal regulation of enzyme cascade reaction
In natural systems, metabolic pathways are typically executed efficiently and independently through synergistic cascade reactions, wherein the systematic organization of multiple enzymes is of paramount importance. Therefore, development of artificial enzyme cascades in vitro and comprehension of the synergistic interactions are critical for applications in biocatalysis and biomedicine. The precise addressability of DNA origami facilitates construction of the artificial multi-enzyme systems, thereby enabling the regulation of cascade reactions.69,70In 2012, Yan and co-workers designed a rectangular DNA origami tile with 60 nm × 80 nm.57 The glucose oxidase (GOx)/horseradish peroxidase (HRP) enzyme pairs were systematically arranged on the DNA origami with precise control over their spacing and positioning. The enzymatic activity exhibited a significant enhancement when the enzymes were positioned closely at a distance of 10 nm. However, this activity decreased markedly at a distance of 20 nm. As the distance between the enzyme pairs was further increased, the enzymatic activity showed a weak dependence on the distance. These findings indicated that the enhanced activity observed for closely spaced GOx/HRP pairs was due to the dimensionally limited diffusion of intermediates between the enzymes, whereas the altered activity for more distantly distributed GOx/HRP pairs was attributed to the Brownian diffusion of intermediates in solution. In 2016, the same research group constructed a DNA origami nanocage to confine the GOx/HRP pairs (Fig. 4A).71 The observed enhancement in catalytic activity was ascribed to the proximity of the cascade enzymes enclosed within the nanocage, as well as stabilization of their active conformations facilitated by the confined DNA nanocages. These findings were further confirmed in the research work reported by Fan and Qian.72–74 Therefore, the enzymatic cascade efficiency was dependent on the intra-enzyme distance, spatial confinement and stabilization supported by DNA origami.
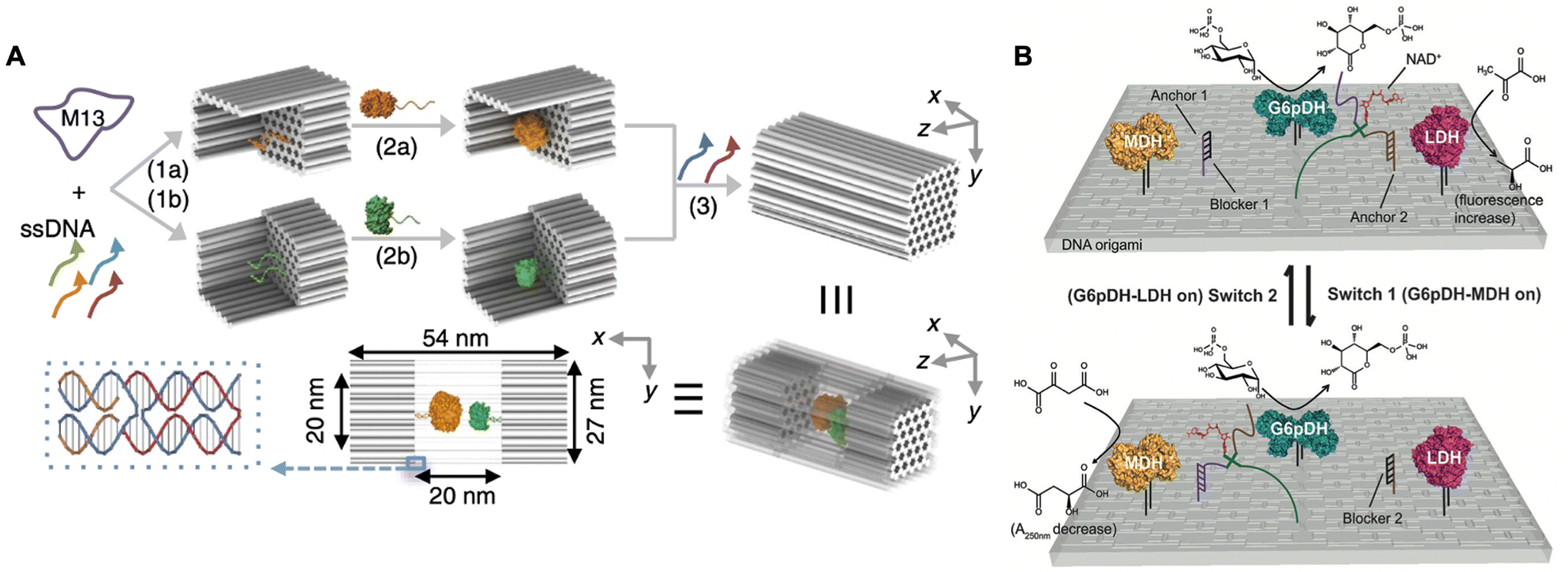 | ||
| Fig. 4 Spatial and temporal regulation of enzyme cascade reactions. (A) Schematic of the GOx/HRP enzyme cascade confined on the DNA nanocage. Reproduced with permission from ref. 71. Copyright 2016 Springer Nature. (B) Schematic of the two enzyme pathways directionally regulated by shifting NAD+ substrate channeling on rectangular DNA origami. Reproduced with permission from ref. 76. Copyright 2016 John Wiley and Sons. | ||
A comprehensive analysis of intra-enzyme distance control within cascade systems reveals that substrate channeling, which facilitates transfer of intermediates between enzymes, plays a crucial role in enhancing the efficiency of enzymatic catalysis. Consequently, several researchers have focused on modulating the activity of the enzyme cascade reactions by strategically positioning substrate channeling between pairs of enzymes.75 In 2016, Yan and co-workers designed a rectangular DNA origami platform to organize a multi-enzyme system comprising glucose-6-phosphate dehydrogenase (G6pDH), malate dehydrogenase (MDH), lactate dehydrogenase (LDH), and the cofactor nicotinamide adenine dinucleotide (NAD+). Within this artificial multi-enzyme network, the two enzymatic pathways (G6pDH–MDH and G6pDH–LDH) were directly regulated by manipulating the substrate channeling of NAD+ between the two enzyme pairs.76 As shown in Fig. 4B, the multi-enzyme network demonstrated varying activities for these two pathways in response to the introduction of different regulatory DNA single strands: switch 1 activated the G6pDH–MDH pathway, while switch 2 activated the G6pDH–LDH pathway. These findings indicated that the efficient catalytic performance of the multi-enzyme system in vivo may be facilitated by the establishment of substrate channeling from one enzyme to another, thereby mitigating the effects of Brownian diffusion and the loss of intermediates in solution.
4.2. Regulation of cellular behaviours
Some membrane proteins often exhibit a tendency to form clusters or higher-order assemblies, which play a crucial role in regulating cellular behaviours such as signaling, communication, and transportation.7–11 Therefore, the nanoscale organization of ligands to modulate the distribution of these proteins may offer an alternative approach to regulate cellular behaviours. Traditional strategies typically involve the use of magnetic bead-modified or polymer/antibody-linked ligand clusters, but the disordered arrangement of these clusters results in suboptimal efficacy in cellular regulation.77–80 In recent years, DNA origami has emerged as a technique characterized by exceptional programmability and precise addressability at the nanoscale resolution, thereby providing a robust platform for the creation of orderly patterned ligands.In 2014, Teixeira and co-workers rationally designed a hollow tube-like nanocaliper modified with Ephrin-A5 ligands, which were separated by distances of 0 nm (NC0), 42.9 nm (NC40), and 100.1 nm (NC100).81 As shown in Fig. 5A, the nanoscale spacing of Ephrin-A5 directed the activation of the EphA2 receptor in human breast cancer cells. Notably, the NC40 was more efficient in increasing the phosphorylation level and further decreasing cell invasiveness compared to NC100 in cancer cells. Furthermore, the nanocaliper containing a saturated amount of Ephrin-A5 did not yield additional increases in activation levels relative to NC40, indicating that a precisely defined distribution of ligands tailored to match the membrane protein pattern could effectively regulate cellular behaviours and contribute to cancer treatment. In addition to the nanoscale patterning of Ephrin-A5 for cancer therapy, Högberg and co-workers designed a single-layer wireframe flat sheet and a double-layer square lattice flat sheet, both of which integrated TNF-related apoptosis-inducing ligand-mimicking peptides into nanoscale patterns to induce death receptor clustering and subsequent apoptosis (Fig. 5B).82 The findings revealed that the induction of apoptosis in breast cancer cells was maximized by hexagonally patterned peptides distributed at approximately 5 nm on these two nanostructures. In another research work, the hexagonally patterned death receptor ligand FasL (CD178), with 10 nm inter-molecular spacing on a rectangular DNA origami sheet, also successfully induced cell apoptosis.83
 | ||
| Fig. 5 Nanoscale protein pattern assisted by DNA origami on regulating cellular behaviours. (A) Schematic of using the hollow tube-like nanocaliper with precisely distributed ephrin-A5 to bind the EphA2 receptor and induce the EphA2 clustering for suppressing cancer cell invasiveness. Scale bars are 10 μm. Reproduced with permission from ref. 81. Copyright 2014 Springer Nature. (B) Schematic of using the flat nanosheet with precisely distributed TNF-related apoptosis-inducing ligand-mimicking peptides to bind death receptors and induce clustering for triggering cancer cell apoptosis. Reproduced with permission from ref. 82. Copyright 2021 American Chemical Society. (C) Schematic of using icosahedral/six-helix bundle nanostructure with precisely distributed eOD-GT8 to activate B cell. Scale bars are 10 nm. Reproduced with permission from ref. 84. Copyright 2020 Springer Nature. (D) Schematic of using DNA origami flat nanosheet with precisely distributed PD-L1 to bind T cells and inhibit the T-cell signaling. Reproduced with permission from ref. 85. Copyright 2021 American Chemical Society. | ||
In the field of immunotherapy, Bathe and co-workers designed a three-dimensional (3D) icosahedral nanostructure and a 1D rigid-rod six-helix bundle, both featuring the systematically distributed immunogen eOD-GT8 (i.e., an engineered outer domain of the HIV-1 glycoprotein-120) to investigate various nanoscale distributions affecting B-cell activation (Fig. 5C).84 The results showed that B-cell signal transduction was maximized by as few as five antigens spaced up to 25–30 nm on the surface of the virus-like icosahedral nanostructure. Additionally, Teixeira and co-workers designed a DNA origami flat nanosheet modified with CD3/CD28 antibodies to stimulate T-cell activation, while another was modified with PD-L1 ligands to interact with PD-1 receptors on cell membranes (Fig. 5D).85 Notably, the two PD-L1 ligands positioned at the terminus of the nanosheet, spaced 200 nm apart, facilitated the formation of smaller PD-1 clusters and significantly reduced the IL-2 expression, thereby inhibiting T-cell signaling. Furthermore, this inhibition was enhanced by increasing the ratio of PD-L1-loaded nanosheets to CD3/CD28-loaded nanosheets. These results elucidated the roles of the spatial organization of PD-L1 ligands on PD-1 clustering and T-cell signaling, thereby promoting the development of immunomodulatory therapies.
Beyond the patterning of ligands, significant advances have also been made in programmable DNA-origami-based T-cell engagers to modulate T cell behaviours. A recent study has reported that a nano-hybrid DNA engager, which consisted of two reprogrammed domains (recognition and signaling), enabled a diverse and precise modulation of T-cell responses.86 Accordingly, Dietz and co-workers constructed a DNA origami nanocarrier featuring two modular domains, each composed of distinct antibodies designed to link target cells with T-cells.87 This innovative approach effectively directed T-cell behaviors in the context of immunotherapy.
Additionally, it has been reported that other DNA nanodevices, including pH-responsive DNA hydrogels88,89 and peptide-coated DNA helix bundles90 were also employed to regulate cellular behaviours (e.g., cell movement and cell autophagy) by modulating the lysosomal activity through mechanisms known as lysosomal interference. The regulation of cellular behaviours held the potential for cancer treatment. Given the remarkable programmability and precise addressability of DNA origami, this regulatory strategy may be effectively applied to DNA origami nanodevices, thereby augmenting their utility in the modulation of cellular behaviours.
4.3. Biomedical applications in anticancer and antibacterial therapeutics
Active proteins, such as thrombin, RNase A and lysozymes, have exhibited excellent therapeutic effects in anticancer and antibacterial treatments.91,92 However, these proteins exhibit a lack of targeted specificity when compared to antibodies, and they often lose their biological activity upon cellular delivery.93 DNA origami has also emerged as a promising candidate for the construction of intelligent nanocarriers for protein drugs, owing to its rationally designed geometries, marked biocompatibility, versatility in surface modification, and weak immune response.In 2016, Ding and co-workers constructed a stimulus-responsive DNA origami nanorobot to preserve and deliver thrombin in cancer treatment.94 As shown in Fig. 6A, a rectangular DNA nanosheet was fabricated with dimensions of 90 nm × 60 nm × 2 nm. Four thrombins were incorporated onto the inner surface of the nanosheet via DNA hybridization to protect them from circulating platelets and plasma fibrinogen. The nucleolin-targeting aptamer (i.e., AS1411), served as both a targeting domain and a molecular trigger in response to the tumor site, hybridized with its complementary strand along a defined seam, resulting in the formation of a tube-shaped DNA nanorobot with dimensions of 90 nm × 19 nm. Upon targeting nucleolin proteins present on the surface of cancer cells, the fastener strands dissociated and then the DNA nanorobot was transformed into the sheet. The thrombin was thus exposed and activated coagulation at the tumor site, which resulted in tumor necrosis and the inhibition of tumor growth. In another research work, Ding and co-workers also constructed a nanosheet loaded with cytotoxic ribonucleases (RNase A) on its inner surface and the Mucin 1 (MUC1) aptamers along its periphery.95 This nanosheet was capable of targeting MUC1 on the surface of cancer cells, facilitating effective internalization of RNase A into the cells, thereby triggering antitumor activity through the degradation of intracellular RNAs.
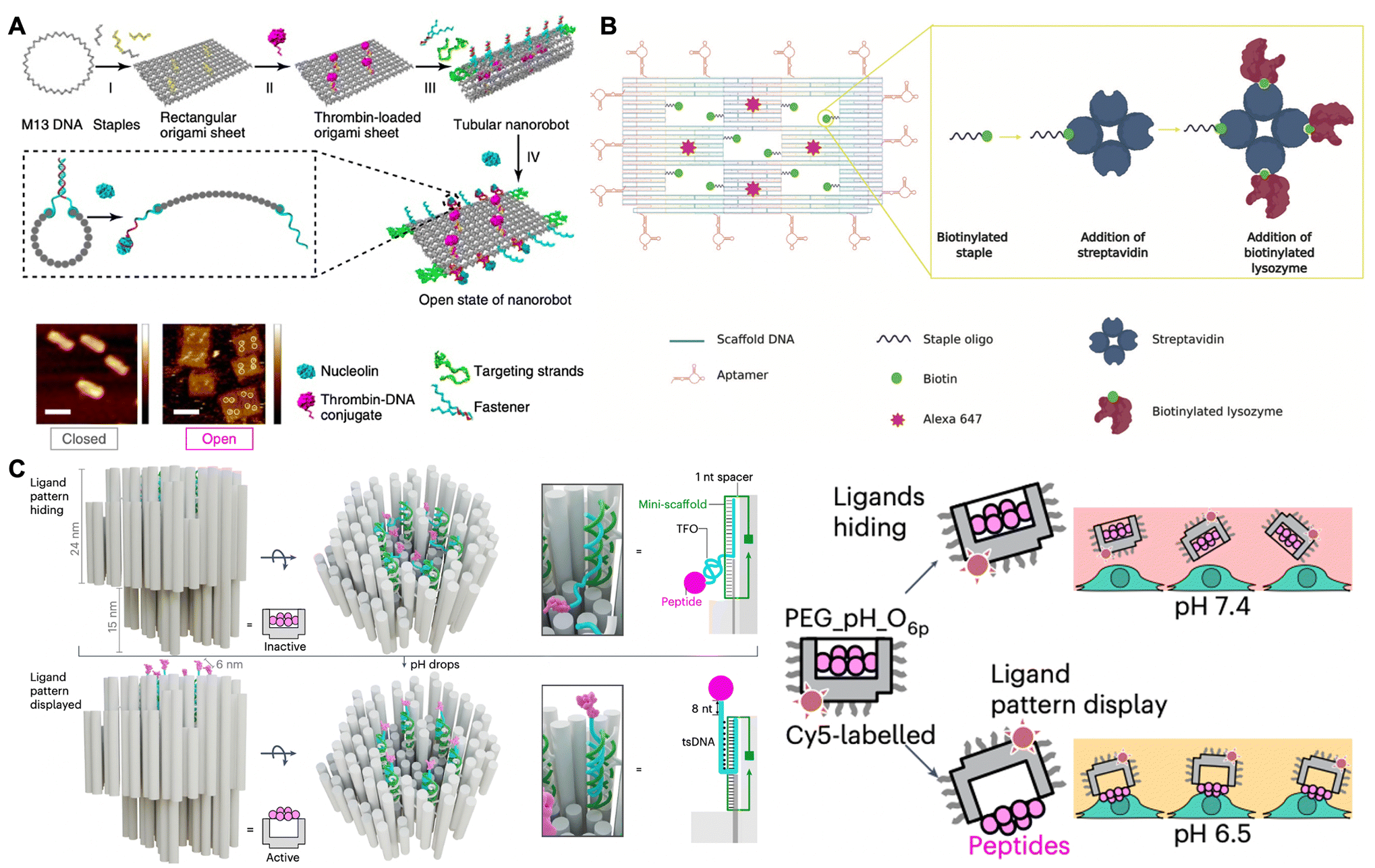 | ||
| Fig. 6 Biomedical applications in anticancer and antibacterial therapeutics. (A) Schematic of the stimulus-responsive DNA nanorobot loaded with the thrombin for targeted drug delivery in cancer treatment. Scale bars are 100 nm. Reproduced with permission from ref. 94. Copyright 2018 Springer Nature. (B) Schematic of DNA nanoframe loaded with the lysozyme for targeted drug delivery in antibacterial therapy. Reproduced with permission from ref. 96. Copyright 2020 John Wiley and Sons. (C) Schematic of the pH-responsive robotic switch nanodevices with a hexagonal pattern of six cytotoxic ligands for triggering cell apoptosis in cancer treatment. Reproduced with permission from ref. 97. Copyright 2024 Springer Nature. | ||
In addition to application in cancer treatment, Kaminski and co-workers constructed a DNA nanoframe that incorporated the antimicrobial enzyme lysozyme within five specifically designed “wells” for antibacterial therapy (Fig. 6B).96 This origami nanoframe was modified with aptamers to selectively target Gram-positive (Bacillus subtilis) and Gram-negative (Escherichia coli) bacteria. Consequently, the nanoframe could target their bacterial strains and then the exposed lysozyme came into contact with the surface of these bacteria, thereby slowing down the bacterial growth. Notably, the lysozyme-functionalized nanoframe exhibited superior therapeutic efficacy compared to free lysozyme, demonstrating the efficiency and targeted specificity of the nanoframe as an intelligent drug delivery vehicle. Therefore, the therapeutic protein-loaded DNA nanostructure may serve as a stable and effective delivery vehicle in various biomedical applications.
Recently, some researchers have focused on the development of protein-patterned DNA origami for cancer treatment. In 2024, Högberg and co-workers designed a stimulus-responsive robotic switch nanodevices featuring six cytotoxic ligands arranged in a 10 nm hexagonal pattern (Fig. 6C).97 This nanodevice was activated to display the patterned cytotoxic ligands on human breast cancer cells at a pH of 6.5, while remaining inert to healthy cells at a pH of 7.4. Consequently, it effectively clustered death receptors and triggered apoptosis in cancer cells, thereby achieving targeted cancer treatment. Additionally, Teixeira and co-workers found that the rod-like insulin-DNA nanostructures could modulate insulin receptor responses for insulin therapies.98
4.4 DNA origami nanodevice-based vaccine for cancer immunotherapy
The treatment of cancer has involved manipulation of the immune system to effectively eliminate malignant cells.99–101 Nevertheless, most anti-tumor efficacy of cancer vaccines has been relatively modest, which can be attributed in part to the insufficient transport of antigens and adjuvants to the sites where the immune response is spatially and temporally coordinated.102–104 Furthermore, achieving a controllable release of these functional components at the targeted sites of action presents a significant challenge.105–107 Consequently, the efficient delivery of vaccines, characterized by precise dosing and on-demand release, remains a formidable obstacle. Recently, development of structurally well-defined DNA origami nanodevices, which exhibit controlled size, shape, and stimulus-responsive mechanical reconfiguration, has shown considerable promise for cancer vaccination.108In 2021, Ding and co-workers designed a well-defined pH-responsive tubular DNA nanodevice that incorporated two types of molecular adjuvants and an antigen peptide within its inner cavity (Fig. 7A).109 This nanodevice was activated within the lysosomes of antigen-presenting cells through the opening of low pH-responsive DNA ‘locking strands’. As a result, the two molecular adjuvants and an antigen peptide were exposed in the subcellular environment, thereby facilitating T-cell activation and inducing cancer cytotoxicity. Consequently, the DNA nanodevice vaccine provoked a robust antigen-specific T-cell response, leading to significant tumor regression in mouse cancer models. Moreover, this vaccination strategy resulted in the generation of long-lasting T-cell responses that effectively conferred protection to the mice against tumor rechallenge. In 2024, Shih and co-workers designed a square-block DNA origami platform that utilized ovalbumin (OVA) proteins as an antigen model, in conjunction with precisely distributed CpG oligonucleotides that served as adjuvants (Fig. 7B).59 Their findings indicated that when the CpG oligonucleotides were spaced at 3.5 nm, the DC activation, antigen cross-presentation, CD8 T-cell activation, Th1-polarized CD4 activation and natural-killer-cell activation were enhanced. Synergistically treated with anti-PDL1 antibodies, this vaccine demonstrated effective cancer immunotherapy in melanoma and lymphoma models, while also inducing a long-term T-cell memory.
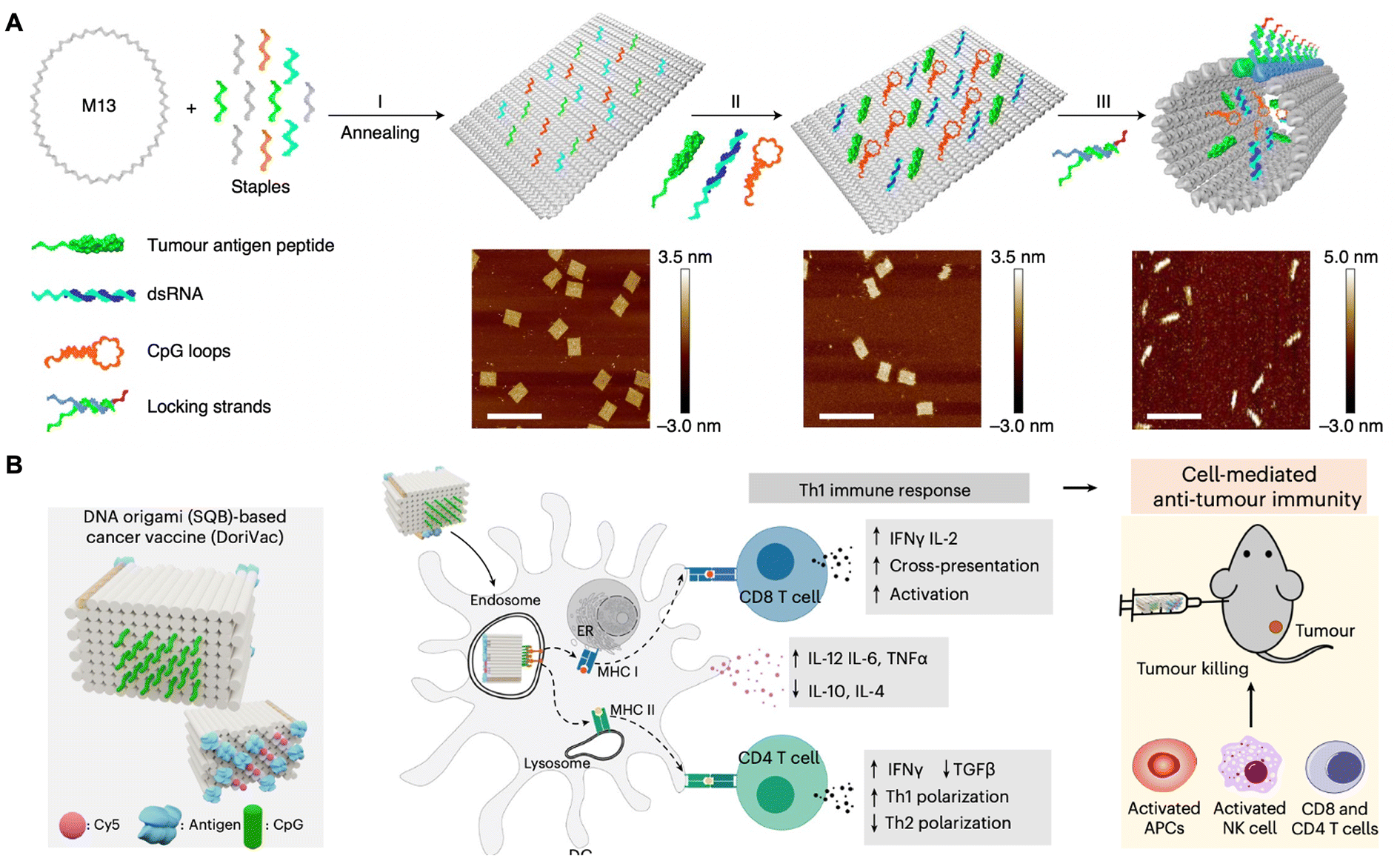 | ||
| Fig. 7 DNA origami nanodevice-based vaccine for cancer immunotherapy. (A) Schematic of tumor antigen peptide/CpG loop/dsRNA-co-loaded DNA origami nanodevice for cancer immunotherapy. Scale bars are 200 nm. Reproduced with permission from ref. 109. Copyright 2020 Springer Nature. (B) Schematic of DNA origami (SQB)-based cancer vaccine in cancer immunotherapy. Reproduced with permission from ref. 59. Copyright 2024 Springer Nature. | ||
4.5. Protein structural analysis
In recent decades, cryo-electron microscopy (cryo-EM) has emerged as a pivotal technique for structural determination of biological macromolecules with near-atomic resolution, particularly for proteins and protein complexes.110,111 Cryo-EM enables the observation of specimens in their native states at cryogenic temperatures, thereby circumventing the structural damage associated with drying during sample preparation for transmission electron microscopy and the complicated process involved in the crystallization for X-ray crystallography.112–115 However, the uncontrolled and non-random orientation of specimens in the ice layer usually results in the loss of significant structural information, which impedes 3D reconstruction.116,117 Recently, the DNA origami technique has been demonstrated to provide a powerful toolbox for manipulating proteins with tunable and controlled orientations for Cryo-EM imaging.In 2016, Dietz and co-workers designed a hollow DNA origami pillar that utilized double stranded DNA (dsDNA) to manipulate DNA-binding protein (i.e., transcription factor p53) at programmable angles.118 As shown in Fig. 8A, the dsDNA, which contained the p53 binding sequence at its center, was anchored inside the hollow pillar. The bound p53 could rotate at multiple angles along the axis of the dsDNA by adjusting the position of the binding sequence either upwards or downwards the corresponding number of base pairs. Consequently, five distinct versions of p53, covering the angles from 21.4° to 158.6°, were visualized by cryo-EM, leading to the 3D reconstruction of p53 with a resolution of 15 Å. Later on, Douglas and co-workers constructed a DNA origami goniometer with a double helix stage that precisely oriented the DNA-binding protein (i.e., BurrH) at user-defined rotation angles (Fig. 8B).119 To ensure accurate determination of the BurrH, the DNA goniometer was designed with many asymmetric features to align with the BurrH at different angles. As a result, the DNA origami goniometer could execute 14 distinct stage configurations, each characterized by unique barcode patterns, thereby enabling the classification of particles based on orientations of the BurrH. This approach successfully yielded a 6.5 Å resolution structure of BurrH. Therefore, this methodology may be extended to other DNA-binding proteins as well as proteins that are fused to DNA-binding domains.
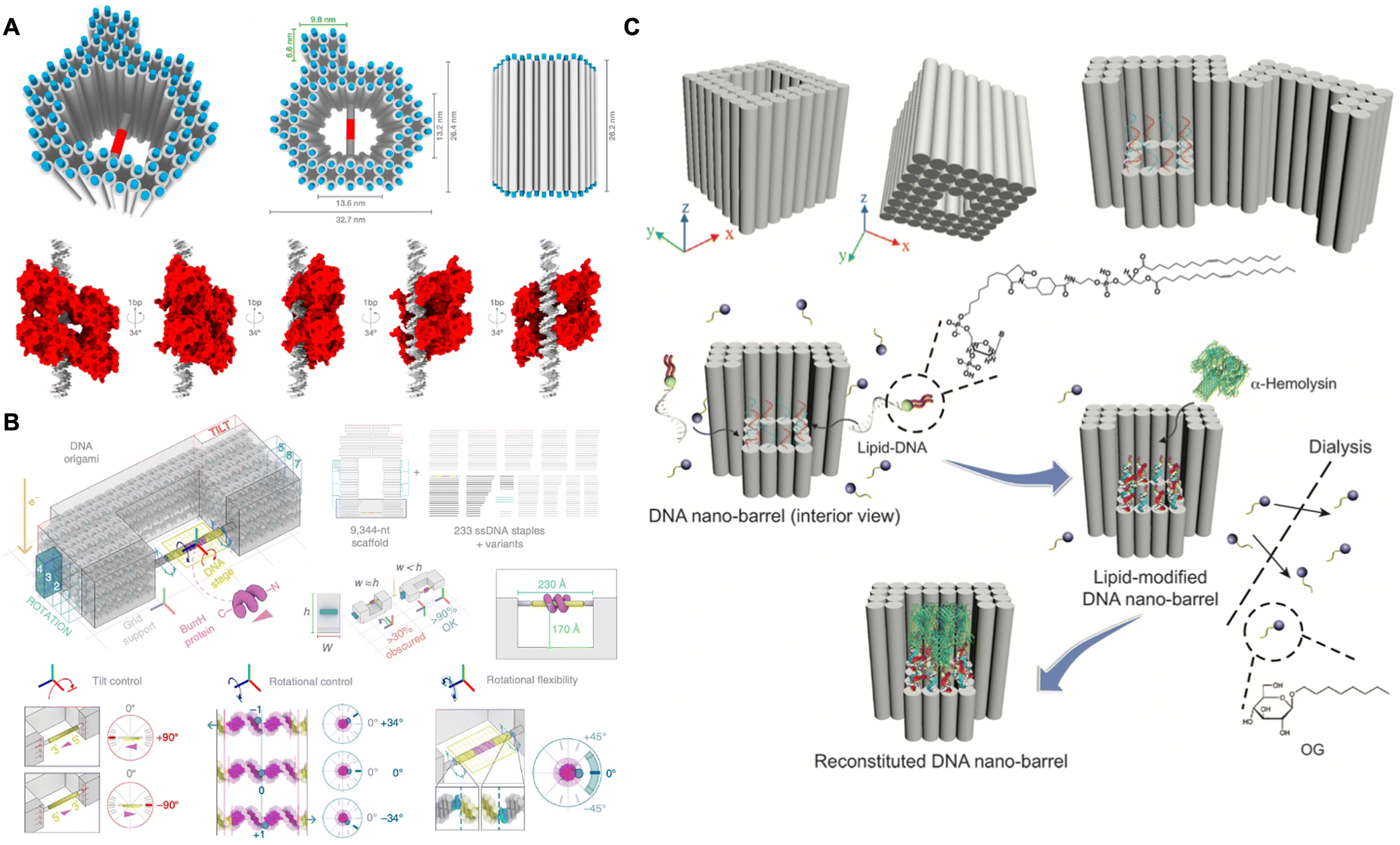 | ||
| Fig. 8 Protein structural analysis assisted by DNA origami. (A) Schematic of using the hollow DNA origami pillar to precisely orient the transcription factor p53 for structural analysis by cryo-EM. Reproduced with permission from ref. 118. Copyright 2016 National Academy of Sciences. (B) Schematic of using the DNA goniometer to precisely orient the BurrH for structural analysis by cryo-EM. Reproduced with permission from ref. 119. Copyright 2020 Springer Nature. (C) Schematic of DNA nanobarrel as a scaffold to stabilize and accommodate α-hemolysin for structural analysis by cryo-EM. Reproduced with permission from ref. 120. Copyright 2018 John Wiley and Sons. | ||
In addition to soluble proteins, the structural determination of membrane proteins can also be achieved by DNA origami. The DNA nanostructures can provide a size-controlled and stabilized lipid bilayer membrane environment to adopt and stabilize the appropriated conformation of membrane proteins, thereby achieving structural determination in their native states. In 2018, Mao and co-workers designed a nanobarrel with a supporting platform at the bottom (Fig. 8C).120 After the hybridization of lipid molecules into the platform and the constraints imposed by the peripheral DNA frame, a confined lipid bilayer was assembled. This bilayer was specifically designed to accommodate single α-hemolysin for structural determination by cryo-EM. By leveraging the defined size, enhanced stability and homogeneity of the lipid bilayer environment, the stabilized α-hemolysin was reconstructed with a resolution of 30 Å. This strategy also demonstrated a general method for structural determination of membrane proteins.
5. Conclusions
In summary, due to the programmability, precise addressability, modification, and biocompatibility, protein-functionalized DNA origami nanodevices have been highlighted as valuable tools in the systematic investigation of protein structures, interactions, functions, and therapeutic applications. However, there are still some challenges that remain to be addressed. Firstly, the activity of conjugated proteins may be reduced in comparison to their native states due to the occupation of binding sites or active sites by the non-regioselective covalent or non-covalent conjugation.70,121,122 Furthermore, the incorporation of large tags onto proteins for DNA conjugation may restrict the precise and dense arrangement of proteins within the DNA origami nanostructures. Consequently, the development of robust site-specific modifications that aim to preserve protein activity and realize precise arrangement is essential. Recent advancements, such as the incorporation of unnatural amino acids with orthogonal reactive groups at defined sites on proteins, have demonstrated the potential for efficient and specific conjugation.123,124 Additionally, the DNA-templated conjugation may serve as an alternative method for site-specific modifications.125,126 Secondly, the stability and safety of protein-functionalized DNA origami in vivo represent significant concerns. In biological environments, DNA origami is exposed to biological fluids, which can lead to denaturation induced by low physiological salt concentrations and degradation mediated by nucleases.127,128 Unenclosed proteins may also experience a loss of activity in vivo. Recent studies have shown that the introduction of oligolysine-PEG copolymers or dendritic oligonucleotides can protect DNA nanostructures and enhance pharmacokinetic bioavailability,129,130 but the more enclosed space for encapsulating proteins may disrupt their functions. Moreover, the introduction of additional DNA segments derived from DNA origami into cells may pose potential biosafety risks, such as integration into the genome and the induction of genetic mutations. Therefore, it is imperative to conduct assessments of long-term toxicity, metabolic pathways, biodistribution, and pharmacokinetics of DNA origami. Lastly, the high costs associated with DNA chemical synthesis, protein conjugation and purification, as well as the functionalization and purification of DNA origami, continue to limit their translational applications. Recent developments in bio-fabrication may help reduce these costs in constructing nanodevices.131,132 Additionally, the distinct differences between protein–DNA conjugates and DNA origami in terms of molecular weight, charge, and hydrophobicity may offer various strategies for purification, which may not necessitate prior purification of protein–DNA conjugates.The protein-functionalized DNA origami nanodevices also present numerous promising research avenues beyond challenges. Firstly, fabrication of artificial biomacromolecules in vitro allows for the investigation of their structure–function relationships.133,134 The construction of bio-macromolecules may enhance comprehensive understanding of their functions and enable various applications in sensing and quantifying biomolecules, as well as innovations in synthetic biology, including the development of artificial organelles and even artificial cells. Secondly, in contrast to protein/polymer-assisted nanodiscs, DNA scaffolds offer a promising approach for assembling lipid membranes with defined size and stability to accommodate specific membrane proteins and modulating the curvature of lipid membranes to facilitate the incorporation of curvature-dependent membrane proteins. Furthermore, DNA nanostructures can be employed to construct hierarchical assemblies that investigate protein–protein interactions within heterogeneous lipid membrane systems. Thirdly, the densely packed DNA nanostructures, which exhibit high contrast in cryo-EM imaging and are capable of manipulating proteins within confined spaces and controlling their copy numbers, allow for the enrichment of small-sized proteins using shape-defined nanostructures and follow 3D reconstruction via cryo-EM. Lastly, considering the protein–protein interactions among different cell types, the application of DNA origami to decorate functional proteins can facilitate intercellular connections and enhance communication between cells, which may be critical for immunotherapy.87
In conclusion, we anticipate that protein-functionalized DNA origami will serve as a robust tool for elucidating the molecular mechanisms underlying protein-involved cellular processes. Furthermore, it holds promise for advancing applications in structural analysis, the regulation of cellular behaviours, and the treatment of diseases.
Author contributions
All the authors contributed to writing and revision of this review.Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Basic Research Plan of China (2023YFA0915201), the Beijing Municipal Science & Technology Commission and Zhongguancun Science Park Administrative Committee (Z231100007223003), the Beijing Natural Science Foundation (JQ24007), the Natural Science Foundation of Beijing Municipality (Z180016), and the National Natural Science Foundation of China (21971248; 21821001; 8227071803). We acknowledge the Key Clinical Projects of Peking University Third Hospital No. BYSY2022065.References
- J. J. D. Ho, N. C. Balukoff, P. R. Theodoridis, M. Wang, J. R. Krieger, J. H. Schatz and S. Lee, Nat. Commun., 2020, 11, 2677 CrossRef CAS.
- K. Oliphant and E. Allen-Vercoe, Microbiome, 2019, 7, 91 CrossRef.
- T. Imoto, M. Minoshima, T. Yokoyama, B. P. Emery, S. D. Bull, H. Bito and K. Kikuchi, ACS Cent. Sci., 2020, 6, 1813–1818 CrossRef CAS PubMed.
- Y. Liu, Q. Yang and F. Zhao, Annu. Rev. Biochem., 2021, 90, 375–401 CrossRef CAS PubMed.
- W. Li, J. Hu, B. Shi, F. Palomba, M. A. Digman, E. Gratton and H. Jiang, Nat. Cell Biol., 2020, 22, 960–972 CrossRef CAS.
- N. Meng, M. Chen, D. Chen, X. H. Chen, J. Z. Wang, S. Zhu, Y. T. He, X. L. Zhang, R. X. Lu and G. R. Yan, Adv. Sci., 2020, 7, 1903233 CrossRef CAS.
- B. T. Lobingier, R. Huttenhain, K. Eichel, K. B. Miller, A. Y. Ting, M. von Zastrow and N. J. Krogan, Cell, 2017, 169, 350–360 Search PubMed.
- Q. Cong, I. Anishchenko, S. Ovchinnikov and D. Baker, Science, 2019, 365, 185–189 CrossRef CAS PubMed.
- M. C. Good, J. G. Zalatan and W. A. Lim, Science, 2011, 332, 680–686 CrossRef CAS.
- R. E. Powers, S. Wang, T. Y. Liu and T. A. Rapoport, Nature, 2017, 543, 257–260 CrossRef CAS PubMed.
- B. B. Aggarwal, Nat. Rev. Immunol., 2003, 3, 745–756 CrossRef CAS PubMed.
- É. S. Vanamee and D. L. Faustman, Sci. Signaling, 2018, 11, eaao4910 CrossRef PubMed.
- J. D. Graves, J. J. Kordich, T. H. Huang, J. Piasecki, T. L. Bush, T. Sullivan, I. N. Foltz, W. Chang, H. Douangpanya, T. Dang, J. W. O'Neill, R. Mallari, X. Zhao, D. G. Branstetter, J. M. Rossi, A. M. Long, X. Huang and P. M. Holland, Cancer Cell, 2014, 26, 177–189 CrossRef CAS.
- S. Dey, C. Fan, K. V. Gothelf, J. Li, C. Lin, L. Liu, N. Liu, M. A. D. Nijenhuis, B. Saccà, F. C. Simmel, H. Yan and P. Zhan, Nat. Rev. Methods Primers, 2021, 1, 13 CrossRef CAS.
- P. Zhan, A. Peil, Q. Jiang, D. Wang, S. Mousavi, Q. Xiong, Q. Shen, Y. Shang, B. Ding, C. Lin, Y. Ke and N. Liu, Chem. Rev., 2023, 123, 3976–4050 CrossRef CAS.
- R. F. Hariadi, R. F. Sommese, A. S. Adhikari, R. E. Taylor, S. Sutton, J. A. Spudich and S. Sivaramakrishnan, Nat. Nanotechnol., 2015, 10, 696–700 CrossRef CAS.
- A. Shaw, I. T. Hoffecker, I. Smyrlaki, J. Rosa, A. Grevys, D. Bratlie, I. Sandlie, T. E. Michaelsen, J. T. Andersen and B. Högberg, Nat. Nanotechnol., 2019, 14, 184–190 CrossRef CAS PubMed.
- B. J. H. M. Rosier, A. J. Markvoort, B. G. Audenis, J. A. L. Roodhuizen, A. den Hamer, L. Brunsveld and T. F. A. de Greef, Nat. Catal., 2020, 3, 295–306 CrossRef CAS PubMed.
- M. E. A. Churchill, T. D. Tullius, N. R. Kallenbach and N. C. Seeman, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 4653–4656 CrossRef CAS.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- Y. Ke, S. M. Douglas, M. Liu, J. Sharma, A. Cheng, A. Leung, Y. Liu, W. M. Shih and H. Yan, J. Am. Chem. Soc., 2009, 131, 15903–15908 CrossRef CAS PubMed.
- Y. Ke, N. V. Voigt, K. V. Gothelf and W. M. Shih, J. Am. Chem. Soc., 2012, 134, 1770–1774 CrossRef CAS.
- F. Zhang, S. Jiang, S. Wu, Y. Li, C. Mao, Y. Liu and H. Yan, Nat. Nanotechnol., 2015, 10, 779–784 CrossRef CAS PubMed.
- E. Benson, A. Mohammed, J. Gardell, S. Masich, E. Czeizler, P. Orponen and B. Högberg, Nature, 2015, 523, 441–444 CrossRef CAS.
- Z. Zhao, Y. Liu and H. Yan, Nano Lett., 2011, 11, 2997–3002 CrossRef CAS.
- Z. Zhao, H. Yan and Y. Liu, Angew. Chem., Int. Ed., 2010, 49, 1414–1417 CrossRef CAS PubMed.
- A. N. Marchi, I. Saaem, B. N. Vogen, S. Brown and T. H. LaBean, Nano Lett., 2014, 14, 5740–5747 CrossRef CAS PubMed.
- R. Linuma, Y. Ke, R. Jungmann, T. Schlichthaerle, J. B. Woehrstein and P. Yin, Science, 2014, 344, 65–69 CrossRef PubMed.
- K. F. Wagenbauer, C. Sigl and H. Dietz, Nature, 2017, 552, 78–83 CrossRef CAS.
- S. Woo and P. W. K. Rothemund, Nat. Chem., 2011, 3, 620–627 CrossRef CAS PubMed.
- W. Li, Y. Yang, S. Jiang, H. Yan and Y. Liu, J. Am. Chem. Soc., 2014, 136, 3724–3727 CrossRef CAS.
- H. Dietz, S. M. Douglas and W. M. Shih, Science, 2009, 325, 725–730 CrossRef CAS PubMed.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- C. Geary, P. W. K. Rothemund and E. S. Andersen, Science, 2014, 345, 799–804 CrossRef CAS.
- D. Han, X. Qi, C. Myhrvold, B. Wang, M. Dai, S. Jiang, M. Bates, Y. Liu, B. An, F. Zhang, H. Yan and P. Yin, Science, 2017, 358, eaao2648 CrossRef.
- Q. Jiang, S. Liu, J. Liu, Z. G. Wang and B. Ding, Adv. Mater., 2019, 31, 1804785 CrossRef CAS.
- M. Madsen and K. V. Gothelf, Chem. Rev., 2019, 119, 6384–6458 CrossRef CAS.
- Y. Hu and C. M. Niemeyer, Adv. Mater., 2019, 31, 1806294 CrossRef.
- A. Keller and V. Linko, Angew. Chem., Int. Ed., 2020, 59, 15818–15833 CrossRef CAS PubMed.
- J. Huang, A. Jaekel, J. van den Boom, D. Podlesainski, M. Elnaggar, A. Heuer-Jungemann, M. Kaiser, H. Meyer and B. Saccà, Nat. Nanotechnol., 2024, 19, 1521–1531 CrossRef CAS PubMed.
- I. Smyrlaki, F. Fördős, I. Rocamonde-Lago, Y. Wang, B. Shen, A. Lentini, V. C. Luca, B. Reinius, A. I. Teixeira and B. Högberg, Nat. Commun., 2024, 15, 465 CrossRef CAS.
- Y. R. Yang, Y. Liu and H. Yan, Bioconjugate Chem., 2015, 26, 1381–1395 CrossRef CAS PubMed.
- T. A. Ngo, H. Dinh, T. M. Nguyen, F. F. Liew, E. Nakata and T. Morii, Chem. Commun., 2019, 55, 12428–12446 RSC.
- L. Mallik, S. Dhakal, J. Nichols, J. Mahoney, A. M. Dosey, S. Jiang, R. K. Sunahara, G. Skiniotis and N. G. Walter, ACS Nano, 2015, 9, 7133–7141 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang, C. Fan, C. Li, Y. Li, L. Qian, Y. Fu, Y. Shi, J. Hu and L. He, Adv. Mater., 2010, 22, 2672–2675 CrossRef CAS PubMed.
- H. Li, S. H. Park, J. H. Reif, T. H. LaBean and H. Yan, J. Am. Chem. Soc., 2004, 126, 418–419 CrossRef CAS.
- W. Shen, H. Zhong, D. Neff and M. L. Norton, J. Am. Chem. Soc., 2009, 131, 6660–6661 CrossRef CAS.
- X. Ouyang, M. De Stefano, A. Krissanaprasit, A. L. B. Kodal, C. B. Rosen, T. Liu, S. Helmig, C. Fan and K. V. Gothelf, Angew. Chem., Int. Ed., 2017, 56, 14423–14427 CrossRef CAS.
- S. V. Wegner and J. P. Spatz, Angew. Chem., Int. Ed., 2013, 52, 7593–7596 CrossRef CAS PubMed.
- T. Yamazaki, J. G. Heddle, A. Kuzuya and M. Komiyama, Nanoscale, 2014, 6, 9122–9126 RSC.
- M. Tintore, I. Gallego, B. Manning, R. Eritja and C. Fàbrega, Angew. Chem., Int. Ed., 2013, 52, 7747–7750 CrossRef CAS PubMed.
- R. Chhabra, J. Sharma, Y. Ke, Y. Liu, S. Rinker, S. Lindsay and H. Yan, J. Am. Chem. Soc., 2007, 129, 10304–10305 CrossRef CAS PubMed.
- F. Praetorius and H. Dietz, Science, 2017, 355, eaam5488 CrossRef.
- M. H. Raz, K. Hidaka, S. J. Sturla, H. Sugiyama and M. Endo, J. Am. Chem. Soc., 2016, 138, 13842–13845 CrossRef CAS PubMed.
- J. H. Laity, B. M. Lee and P. E. Wright, Curr. Opin. Struct. Biol., 2001, 11, 39–46 CrossRef CAS.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef CAS.
- P. D. E. Fisher, Q. Shen, B. Akpinar, L. K. Davis, K. K. H. Chung, D. Baddeley, A. Šaric, T. J. Melia, B. W. Hoogenboom, C. Lin and C. P. Lusk, ACS Nano, 2018, 12, 1508–1518 CrossRef CAS PubMed.
- Y. C. Zeng, O. J. Young, C. M. Wintersinger, F. M. Anastassacos, J. I. MacDonald, G. Isinelli, M. O. Dellacherie, M. Sobral, H. Bai, A. R. Graveline, A. Vernet, M. Sanchez, K. Mulligan, Y. Choi, T. C. Ferrante, D. B. Keskin, G. G. Fell, D. Neuberg, C. J. Wu, D. J. Mooney, I. C. Kwon, J. H. Ryu and W. M. Shih, Nat. Nanotechnol., 2024, 19, 1055–1065 CrossRef CAS.
- G. Marth, A. M. Hartley, S. C. Reddington, L. L. Sargisson, M. Parcollet, K. E. Dunn, D. D. Jones and E. Stulz, ACS Nano, 2017, 11, 5003–5010 CrossRef CAS PubMed.
- B. P. Duckworth, Y. Chen, J. W. Wollack, Y. Sham, J. D. Mueller, T. A. Taton and M. D. Distefano, Angew. Chem., Int. Ed., 2007, 46, 8819–8822 CrossRef CAS PubMed.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- R. A. Chandra, E. S. Douglas, R. A. Mathies, C. R. Bertozzi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 896–901 CrossRef CAS PubMed.
- E. Nakata, H. Dinh, T. A. Ngo, M. Saimura and T. Morii, Chem. Commun., 2015, 51, 1016–1019 RSC.
- K. J. Koβmann, C. Ziegler, A. Angelin, R. Meyer, M. Skoupi, K. S. Rabe and C. M. Niemeyer, ChemBioChem, 2016, 17, 1102–1106 CrossRef.
- S. Takeda, S. Tsukiji and T. Nagamune, Bioorg. Med. Chem. Lett., 2004, 14, 2407–2410 CrossRef CAS.
- S. Takeda, S. Tsukiji, H. Ueda and T. Nagamune, Org. Biomol. Chem., 2008, 6, 2187–2194 Search PubMed.
- F. Diezmann and O. Seitz, Chem. Soc. Rev., 2011, 40, 5789–5801 RSC.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed.
- O. I. Wilner, Y. Weizmann, R. Gill, O. Lioubashevski, R. Freeman and I. Willner, Nat. Nanotechnol., 2009, 4, 249–254 CAS.
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CAS.
- Z. Ge, J. Fu, M. Liu, S. Jiang, A. Andreoni, X. Zuo, Y. Liu, H. Yan and C. Fan, ACS Appl. Mater. Interfaces, 2019, 11, 13881–13887 CAS.
- L. Sun, Y. Gao, Y. Xu, J. Chao, H. Liu, L. Wang, D. Li and C. Fan, J. Am. Chem. Soc., 2017, 139, 17525–17532 CAS.
- Y. Yang, S. Zhang, S. Yao, R. Pan, K. Hidaka, T. Emura, C. Fan, H. Sugiyama, Y. Xu, M. Endo and X. Qian, Chem. – Eur. J., 2019, 25, 5158–5162 CrossRef CAS PubMed.
- Y. Chen, G. Ke, Y. Ma, Z. Zhu, M. Liu, Y. Liu, H. Yan and C. J. Yang, J. Am. Chem. Soc., 2018, 140, 8990–8996 CrossRef CAS.
- G. Ke, M. Liu, S. Jiang, X. Qi, Y. R. Yang, S. Wootten, F. Zhang, Z. Zhu, Y. Liu, C. J. Yang and H. Yan, Angew. Chem., Int. Ed., 2016, 55, 7483–7486 CrossRef CAS.
- J. S. Swers, L. Grinberg, L. Wang, H. Feng, K. Lekstrom, R. Carrasco, Z. Xiao, I. Inigo, C. C. Leow, H. Wu, D. A. Tice and M. Baca, Mol. Cancer Ther., 2013, 12, 1235–1244 CrossRef CAS.
- G. Lamanna, C. R. Smulski, N. Chekkat, K. Estieu-Gionnet, G. Guichard, S. Fournel and A. Bianco, Chem. – Eur. J., 2013, 19, 1762–1768 CrossRef CAS.
- T. Jiang, W. Sun, Q. Zhu, N. A. Burns, S. A. Khan, R. Mo and Z. Gu, Adv. Mater., 2015, 27, 1021–1028 CrossRef CAS PubMed.
- H. Schneider, D. Yanakieva, A. Macarron, L. Deweid, B. Becker, S. Englert, O. Avrutina and H. Kolmar, ChemBioChem, 2019, 20, 3006–3012 CrossRef CAS PubMed.
- A. Shaw, V. Lundin, E. Petrova, F. Fordos, E. Benson, A. Al-Amin, A. Herland, A. Blokzijl, B. Högberg and A. I. Teixeira, Nat. Methods, 2014, 11, 841–846 CrossRef CAS PubMed.
- Y. Wang, I. Baars, F. Fordos and B. Högberg, ACS Nano, 2021, 15, 9614–9626 CrossRef CAS PubMed.
- R. M. L. Berger, J. M. Weck, S. M. Kempe, O. Hill, T. Liedl, J. O. Radler, C. Monzel and A. Heuer-Jungemann, Small, 2021, 17, 2101678 CrossRef CAS.
- R. Veneziano, T. J. Moyer, M. B. Stone, E. C. Wamhoff, B. J. Read, S. Mukherjee, T. R. Shepherd, J. Das, W. R. Schief, D. J. Irvine and M. Bathe, Nat. Nanotechnol., 2020, 15, 716–723 CrossRef CAS PubMed.
- T. Fang, J. Alvelid, J. Spratt, E. Ambrosetti, I. Testa and A. I. Teixeira, ACS Nano, 2021, 15, 3441–3452 CAS.
- P. Q. Ma, T. X. Liu, H. D. Li, B. C. Yin and B. C. Ye, J. Am. Chem. Soc., 2022, 144, 22458–22469 CAS.
- K. F. Wagenbauer, N. Pham, A. Gottschlich, B. Kick, V. Kozina, C. Frank, D. Trninic, P. Stömmer, R. Grünmeier, E. Carlini, C. A. Tsiverioti, S. Kobold, J. J. Funke and H. Dietz, Nat. Nanotechnol., 2023, 18, 1319–1326 CAS.
- Y. Dong, F. Li, Z. Lv, S. Li, M. Yuan, N. Song, J. Liu and D. Yang, Angew. Chem., Int. Ed., 2022, 61, e202207770 CAS.
- S. Yang, Y. Cheng, M. Liu, J. Tang, S. Li, Y. Huang, X. Kou, C. Yao and D. Yang, Nano Today, 2024, 56, 102224 CAS.
- P. Elblova, M. Lunova, S. J. W. Henry, X. Tu, A. Cale, A. Dejneka, J. Havelkova, Y. Petrenko, M. Jirsa, N. Stephanopoulos and O. Lunov, Chem. Eng. J., 2024, 498, 155633 CrossRef CAS.
- A. Fu, R. Tang, J. Hardie, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2014, 25, 1602–1608 CrossRef CAS.
- X. Liu, F. Wu, Y. Ji and L. Yin, Bioconjugate Chem., 2019, 30, 305–324 CrossRef PubMed.
- S. Mitragotri, P. A. Burke and R. Langer, Nat. Rev. Drug Discovery, 2014, 13, 655–672 CrossRef CAS PubMed.
- S. Li, Q. Jiang, S. Liu, Y. Zhang, Y. Tian, C. Song, J. Wang, Y. Zou, G. J. Anderson, J. Y. Han, Y. Chang, Y. Liu, C. Zhang, L. Chen, G. Zhou, G. Nie, H. Yan, B. Ding and Y. Zhao, Nat. Biotechnol., 2018, 36, 258–264 CrossRef CAS.
- S. Zhao, F. Duan, S. Liu, T. Wu, Y. Shang, R. Tian, J. Liu, Z. G. Wang, Q. Jiang and B. Ding, ACS Appl. Mater. Interfaces, 2019, 11, 11112–11118 CrossRef CAS PubMed.
- I. Mela, P. P. Vallejo-Ramirez, S. Makarchuk, G. Christie, D. Bailey, R. M. Henderson, H. Sugiyama, M. Endo and C. F. Kaminski, Angew. Chem., Int. Ed., 2020, 59, 12698–12702 CrossRef CAS PubMed.
- Y. Wang, I. Baars, I. Berzina, I. Rocamonde-Lago, B. Shen, Y. Yang, M. Lolaico, J. Waldvogel, I. Smyrlaki, K. Zhu, R. A. Harris and B. Högberg, Nat. Nanotechnol., 2024, 19, 1366–1374 CrossRef CAS.
- J. Spratt, J. M. Dias, C. Kolonelou, G. Kiriako, E. Engström, E. Petrova, C. Karampelias, I. Cervenka, N. Papanicolaou, A. Lentini, B. Reinius, O. Andersson, E. Ambrosetti, J. L. Ruas and A. I. Teixeira, Nat. Nanotechnol., 2024, 19, 237–245 CrossRef CAS.
- I. Mellman, G. Coukos and G. Dranoff, Nature, 2011, 480, 480–489 CrossRef CAS PubMed.
- P. A. Ott, Z. Hu, D. B. Keskin, S. A. Shukla, J. Sun, D. J. Bozym, W. Zhang, A. Luoma, A. Giobbie-Hurder, L. Peter, C. Chen, O. Olive, T. A. Carter, S. Li, D. J. Lieb, T. Eisenhaure, E. Gjini, J. Stevens, W. J. Lane, I. Javeri, K. Nellaiappan, A. M. Salazar, H. Daley, M. Seaman, E. I. Buchbinder, C. H. Yoon, M. Harden, N. Lennon, S. Gabriel, S. J. Rodig, D. H. Barouch, J. C. Aster, G. Getz, K. Wucherpfennig, D. Neuberg, J. Ritz, E. S. Lander, E. F. Fritsch, N. Hacohen and C. J. Wu, Nature, 2017, 547, 217–221 CrossRef CAS.
- U. Sahin, E. Derhovanessian, M. Miller, B. Kloke, P. Simon, M. Löwer, V. Bukur, A. D. Tadmor, U. Luxemburger, B. Schrörs, T. Omokoko, M. Vormehr, C. Albrecht, A. Paruzynski, A. N. Kuhn, J. Buck, S. Heesch, K. H. Schreeb, F. Müller, I. Ortseifer, I. Vogler, E. Godehardt, S. Attig, R. Rae, A. Breitkreuz, C. Tolliver, M. Suchan, G. Martic, A. Hohberger, P. Sorn, J. Diekmann, J. Ciesla, O. Waksmann, A. Brück, M. Witt, M. Zillgen, A. Rothermel, B. Kasemann, D. Langer, S. Bolte, M. Diken, S. Kreiter, R. Nemecek, C. Gebhardt, S. Grabbe, C. Höller, J. Utikal, C. Huber, C. Loquai and Ö. Türeci, Nature, 2017, 547, 222–226 Search PubMed.
- J. Banchereau and A. K. Palucka, Nat. Rev. Immunol., 2005, 5, 296–306 CrossRef CAS.
- J. Hamanishi, M. Mandai, M. Iwasaki, T. Okazaki, Y. Tanaka, K. Yamaguchi, T. Higuchi, H. Yagi, K. Takakura, N. Minato, T. Honjo and S. Fujii, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3360–3365 CrossRef CAS.
- G. Zhu, F. Zhang, Q. Ni, G. Niu and X. Chen, ACS Nano, 2017, 11, 2387–2392 CrossRef CAS.
- G. Zhu, L. Mei, H. D. Vishwasrao, O. Jacobson, Z. Wang, Y. Liu, B. C. Yung, X. Fu, A. Jin, G. Niu, Q. Wang, F. Zhang, H. Shroff and X. Chen, Nat. Commun., 2017, 8, 1482 CrossRef PubMed.
- Y. Min, K. C. Roche, S. Tian, M. J. Eblan, K. P. McKinnon, J. M. Caster, S. Chai, L. E. Herring, L. Zhang, T. Zhang, J. M. DeSimone, J. E. Tepper, B. G. Vincent, J. S. Serody and A. Z. Wang, Nat. Nanotechnol., 2017, 12, 877–882 CrossRef CAS PubMed.
- M. Luo, H. Wang, Z. Wang, H. Cai, Z. Lu, Y. Li, M. Du, G. Huang, C. Wang, X. Chen, M. R. Porembka, J. Lea, A. E. Frankel, Y. X. Fu, Z. J. Chen and J. Gao, Nat. Nanotechnol., 2017, 12, 648–654 CrossRef CAS PubMed.
- R. Tian, Y. Shang, Y. Wang, Q. Jiang and B. Ding, Small Methods, 2023, 7, 2201518 CrossRef CAS PubMed.
- S. Liu, Q. Jiang, X. Zhao, R. Zhao, Y. Wang, Y. Wang, J. Liu, Y. Shang, S. Zhao, T. Wu, Y. Zhang, G. Nie and B. Ding, Nat. Mater., 2021, 20, 421–430 Search PubMed.
- Y. Dong, S. Zhang, Z. Wu, X. Li, W. L. Wang, Y. Zhu, S. Stoilova-McPhie, Y. Lu, D. Finley and Y. Mao, Nature, 2019, 565, 49–55 CAS.
- Y. Lu, J. Wu, Y. Dong, S. Chen, S. Sun, Y. B. Ma, Q. Ouyang, D. Finley, M. W. Kirschner and Y. Mao, Mol. Cell, 2017, 67, 322–333 Search PubMed.
- X. Yao, X. Fan and N. Yan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18497–18503 CAS.
- M. Beckers, D. Mann and C. Sachse, Prog. Biophys. Mol. Biol., 2021, 160, 26–36 CAS.
- A. D. Bendre, P. J. Peters and J. Kumar, J. Membr. Biol., 2021, 254, 321–341 Search PubMed.
- J. Garcia-Nafria and C. G. Tate, Biochem. Soc. Trans., 2021, 49, 2345–2355 CAS.
- K. A. Taylor and R. M. Glaeser, J. Struct. Biol., 2008, 163, 214–223 CAS.
- R. M. Glaeser and B. G. Han, Biophys. Rep., 2017, 3, 1–7 CAS.
- T. G. Martin, T. A. Bharat, A. C. Joerger, X. C. Bai, F. Praetorius, A. R. Fersht, H. Dietz and S. H. Scheres, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7456–E7463 Search PubMed.
- T. Aksel, Z. Yu, Y. Cheng and S. M. Douglas, Nat. Biotechnol., 2021, 39, 378–386 CAS.
- Y. Dong, S. Chen, S. Zhang, J. Sodroski, Z. Yang, D. Liu and Y. Mao, Angew. Chem., Int. Ed., 2018, 57, 2094–2098 CrossRef.
- Y. R. Yang, Y. Liu and H. Yan, Bioconjugate Chem., 2015, 26, 1381–1395 CrossRef CAS.
- G. Kong, M. Xiong, L. Liu, L. Hu, H.-M. Meng, G. Ke, X.-B. Zhang and W. Tan, Chem. Soc. Rev., 2021, 50, 1846–1873 RSC.
- B. Rosier, G. A. O. Cremers, W. Engelen, M. Merkx, L. Brunsveld and T. F. A. de Greef, Chem. Commun., 2017, 53, 7393–7396 RSC.
- N. Stephanopoulos, M. Liu, G. J. Tong, Z. Li, Y. Liu, H. Yan and M. B. Francis, Nano Lett., 2010, 10, 2714–2720 CrossRef CAS.
- C. B. Rosen, A. L. Kodal, J. S. Nielsen, D. H. Schaffert, C. Scavenius, A. H. Okholm, N. V. Voigt, J. J. Enghild, J. Kjems, T. Torring and K. V. Gothelf, Nat. Chem., 2014, 6, 804–809 CrossRef CAS PubMed.
- A. L. Kodal, C. B. Rosen, M. R. Mortensen, T. Torring and K. V. Gothelf, ChemBioChem, 2016, 17, 1338–1342 CrossRef CAS.
- J. Hahn, S. F. J. Wickham, W. M. Shih and S. D. Perrault, ACS Nano, 2014, 8, 8765–8775 CrossRef CAS.
- C. E. Castro, F. Kilchherr, D. N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 Search PubMed.
- N. Ponnuswamy, M. M. C. Bastings, B. Nathwani, J. H. Ryu, L. Y. T. Chou, M. Vinther, W. A. Li, F. M. Anastassacos, D. J. Mooney and W. M. Shih, Nat. Commun., 2017, 8, 15654 CrossRef CAS.
- Y. Kim and P. Yin, Angew. Chem., Int. Ed., 2020, 59, 700–703 CAS.
- D. Han, X. Qi, C. Myhrvold, B. Wang, M. Dai, S. Jiang, M. Bates, Y. Liu, B. An, F. Zhang, H. Yan and P. Yin, Science, 2017, 358, eaao2648 Search PubMed.
- F. Praetorius, B. Kick, K. L. Behler, M. N. Honemann, D. Weuster-Botz and H. Dietz, Nature, 2017, 552, 84–87 CAS.
- Q. Shen, T. Tian, Q. Xiong, P. D. E. Fisher, Y. Xiong, T. J. Melia, C. P. Lusk and C. Lin, J. Am. Chem. Soc., 2021, 143, 12294–12303 CAS.
- Q. Shen, Q. Xiong, K. Zhou, Q. Feng, L. Liu, T. Tian, C. Wu, Y. Xiong, T. J. Melia, C. P. Lusk and C. Lin, J. Am. Chem. Soc., 2023, 145, 1292–1300 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |