
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein–surface interactions in nano-scale biosensors for IL-6 detection using functional monolayers†
Serena
Giberti
a,
Sutapa
Dutta
a,
Stefano
Corni
b,
Marco
Frasconi
 *b and
Giorgia
Brancolini
*a
*b and
Giorgia
Brancolini
*a
aInstitute Nanoscience – CNR-NANO, Center S3, via G. Campi 213/A, 41125, Modena, Italy. E-mail: giorgia.brancolini@nano.cnr.it
bDepartment of Chemistry, University of Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: marco.frasconi@unipd.it
First published on 16th January 2025
Abstract
A multiscale approach is employed to investigate the interaction dynamics between interleukin-6, a key cancer biomarker, and alkyl-functionalized surfaces, with the ultimate goal of guiding biosensor design. The study integrates classical molecular dynamics, Brownian dynamics simulations, and binding experiments to explore the adsorption dynamics and energetics of IL-6 on surfaces modified with self-assembled monolayers (SAMs). The comparative analysis reveals a dramatic effect on the interaction strength of IL-6 with a SAMs comprising a mix of charged and hydrophobic ligands. Solvent accessible surface area analysis shows enhanced exposure of charged terminal groups on the mixed SAM surface. Experimental investigations using surface plasmon resonance reveal that IL-6 interactions enhance with increased charged ligand content in mixed SAMs, retaining high binding affinity even under high ionic strength conditions. Computational studies further highlight hydrophobic and electrostatic interactions as key factors driving the high affinity of IL-6 on the mixed SAMs surface. This research offers insights into optimizing surfaces for enhanced IL-6 recognition, which can be extended to other protein biomarkers, by combining experimental and computational approaches to improve biosensing performance.
1. Introduction
Nanoscale biosensors have attracted considerable interest in the field of medical diagnostic analysis due to their sensitive, label-free detection capabilities for biomolecular recognition. These biosensors, which can be finely tuned for various applications, are often constructed by functionalizing noble metal surfaces with appropriate bio-receptors that can interact with target analytes in solution.1 Among the various methods available, self-assembled monolayers (SAMs) on gold surfaces have emerged as a pivotal technology in the development of biosensors due to their ability to create well-defined and stable interfaces for the immobilization of biomolecules.2,3 SAMs are typically formed by the spontaneous adsorption of thiol-containing molecules onto gold surfaces, resulting in a densely packed and highly ordered monolayer.4 This method offers several advantages, including ease of preparation, versatility in chemical modification, and robustness under various experimental conditions.5–7 The use of mixed SAMs, incorporating different functional groups, further allows for the modulation of surface properties to optimize the interaction with biological targets.8–10The interaction between proteins and SAMs plays a fundamental role in optimizing the surface properties necessary for biosensor applications. The use of mixed SAMs, which incorporate different functional groups, allows for fine-tuning the surface characteristics to enhance interactions with biological targets.11 In particular, the ability to control the density, orientation, and binding affinity of biomolecules, such as proteins, is critical for improving the performance of SAM-based systems. Given these properties, SAMs with mixed functional groups provide an excellent platform for studying the behavior of proteins at the molecular level. For example, the presence of amine head groups on a mixed SAMs with alkyl-terminated alkanethiols significantly enhanced the strength of hydrophobic interactions with a helical β-peptide, compared to a SAM composed solely of the alkyl component.12 Furthermore, the subnanoscale distance between the ionic head groups and the hydrophobic surface on a mixed SAM comprising ionic functional groups and alkyl-terminated alkanethiols has been shown to significantly influence interactions with biomolecules in solution.13 Although substantial progress over the past decade has advanced our understanding of hydrophobic interactions in simple model systems,14–16 including SAM-modified surfaces, the role of nanometer-scale chemical heterogeneity, such as structures containing non-polar domains in close proximity to polar and charged functional groups, remains not fully understood, particularly regarding their interactions with peptides and proteins.
In this study, we exploit a combination of computational modelling and experimental investigations to understand the interactions between interleukin-6 (IL-6) protein and gold surfaces functionalized with SAMs. IL-6, a critical biomarker associated with immune responses and various pathological conditions, including cancers and inflammatory diseases,17 is a protein of clinical interest. Detecting IL-6 at low concentrations in biological fluids requires surfaces that promote specific and stable interactions with the target protein.18,19 The surface chemistry of SAMs, especially in terms of how they influence protein adsorption, is crucial in enabling these interactions.20
The focus is on understanding how mixed SAMs, composed of alternating alkyl thiols and ammonium alkyl thiols, influence protein binding and stability with respect to single-component SAM. Detailed molecular simulations reveal information of protein–surface interactions that experiments alone cannot access. Simulations provide information about the dynamics, energetics and stability of such interactions, allowing to probe the molecular mechanisms underlying protein adsorption, orientation and binding on functionalized surfaces. Atomistics simulations are directly compared with protein-binding experiments obtained by surface plasmon resonance (SPR), providing valuable insights into the diffusion dynamics, binding energetics, and stability of IL-6 on SAM-functionalized surfaces. These surface-binding analyses are essential for validating key aspects of the simulation results, strengthening confidence in the theoretical models and advancing our understating of these complex systems. By integrating experimental data with computational modeling, the design of nanoscale biosensors can be optimized more effectively, providing also insights across different scales.21 This approach provides reliable tools for creating new technologies based on rational design of nano–bio interactions.22,23
2. Results and discussion
2.1 Simulations results
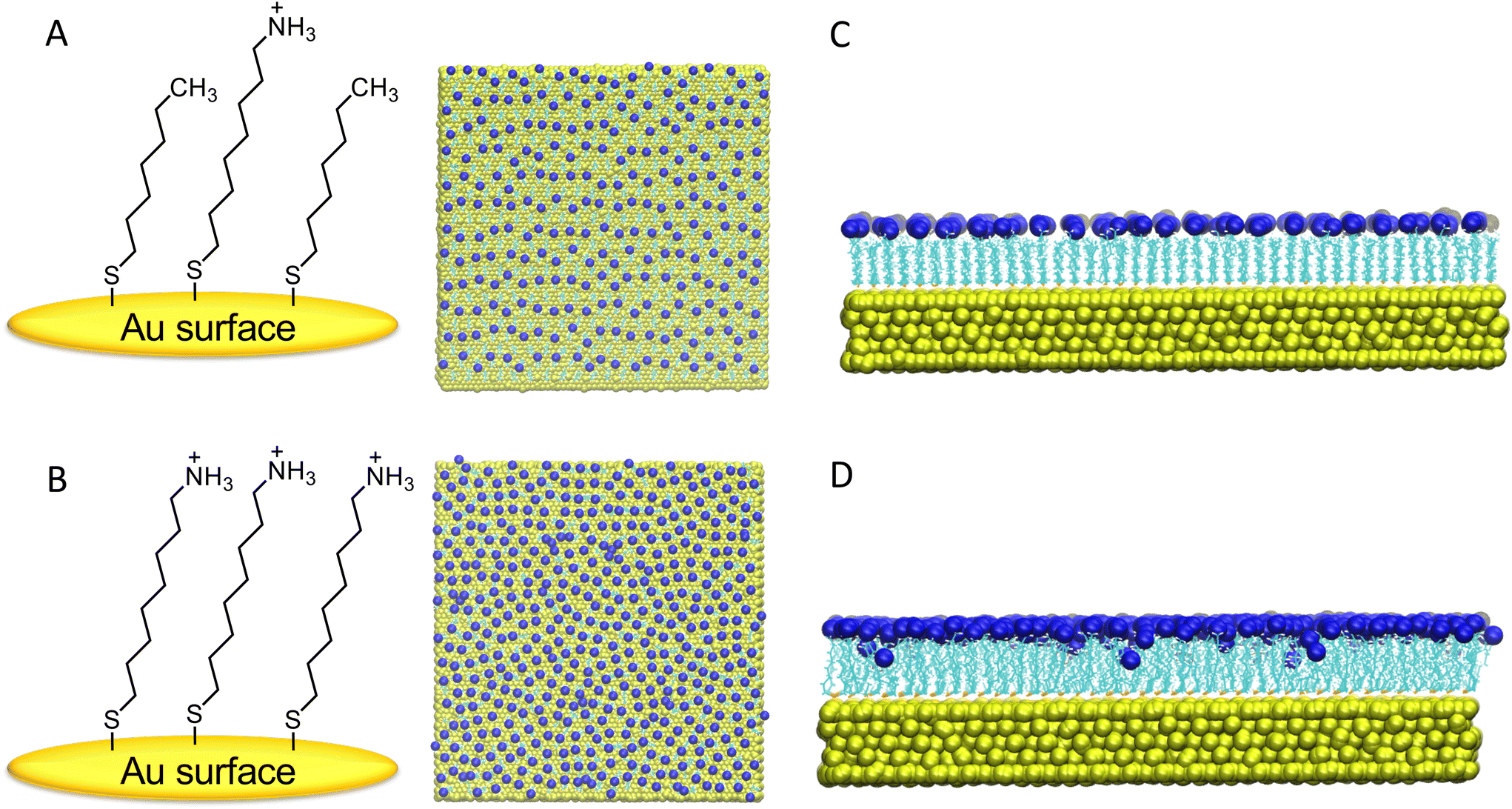 | ||
| Fig. 1 (A) Chemical sketch of mixed-component self-assembly monolayer (M-SAM) and (B) single-component self-assembly monolayer (S-SAM). (C) shows the structure of the M-SAM surface after 300 ns of relaxation, and (D) shows the structure of the S-SAM. Each surface structure is depicted from both the top and side views, with Au atoms in yellow, alkyl chains in cyan, and terminal NH3+ charged groups in blue. | ||
After initial relaxation with 300 ns of fully atomistic MD, on the individual surfaces in the presence of explicit solvent and counterions, a notable distinction was observed in the behavior of the M-SAM (shown in panel C of Fig. 1) compared to the S-SAM (shown in panel D of Fig. 1).
In the S-SAM, some ligands bent inward toward the gold surface, primarily due to strong electrostatic repulsion between neighboring positively charged NH3+ terminal groups. In contrast, the M-SAM did not exhibit this behavior because of the alternation of charged ligands with neutral ligands, namely terminated with hydrophobic CH3 groups. The latter thus resulting in elongated ligands, with a “brush-like” distribution.
To quantify the stability of the ligands, RMSD values were calculated. The RMSD analysis of the 300 ns trajectories is shown below, with the reference structure being the first frame is reported in ESI, Fig. S1† panel A. The plots indicate that the SAM-based surfaces achieved stability throughout the simulation, with RMSD remaining constant around 0.125 nm after 5 ns and 0.145 nm after 150 ns for M-SAM and S-SAM, respectively. The counterions located near the surface became trapped between ligands of varying heights, resulting in tighter packing and reduced ligand mobility in the M-SAM. In contrast, the S-SAM exhibited the highest RMSD due to increased chain mobility.
The GROMACS tool gmx sasa was employed to calculate the solvent accessible surface area (SASA). This is crucial for understanding molecular interactions in a solvent environment, particularly for protein–surface interactions. The average SASA values were found to be 316.660 nm2 for M-SAM and 276.673 nm2 for S-SAM, indicating a 14.52% difference. This difference is due to the presence of two types of ligands with varying heights in M-SAM, one of which is shorter, allowing more accessible area for the solvent (see Fig. S1† panel B).
Furthermore, the SASA per ligand value was calculated for the NH3+ terminal groups in both systems. As shown in panel C of Fig. S1,† the SASA per ligand for the M-SAM system is notably higher than for the S-SAM system. This suggests that the NH3+ groups in M-SAM are more exposed to the solvent compared to those in S-SAM. The greater exposure of these terminal groups implies that the M-SAM modified surface is more prone to interact with the target protein. Thus, the SASA of NH3+ spacers is increased in the mixed surface compared to the non-mixed one. Since the overall SASA of amino groups in the second and third groups is larger for the mixed-SAM compared to the S-SAM, fully functionalized high-density surfaces are expected to reach saturation earlier than their half-functionalized counterparts.
The analysis also determined the number density of Cl− counterions along the z-axis using the GROMACS tool gmx density. This involved computing the number density of particles by dividing the box into slices and calculating the temporal average over time. A layer of 217 Cl− ions formed near the ligands of the M-SAM, and 432 Cl− ions formed near the S-SAM. The peak of the S-SAM appears slightly right-shifted compared to the M-SAM peak, as the S-SAM comprises a single type of ligand, leading to a consistent ion monolayer formation, see Fig. S2.†
Additionally, ions were observed within 2.3 nm of the surface due to ligand repulsion, causing the ligands to bend inwards and allowing Cl− ions to approach the gold surface closely in the S-SAM. In contrast, the M-SAM structure had ligands at different heights, resulting in ions near the hydrophobic (CH3) ligands being relatively closer to the gold surface, though not as significantly as in the S-SAM due to the absence of bent ligands. This distinction is evident as the M-SAM exhibits two peaks at different z-coordinates.
The stability of the proteins in each system was assessed using the root mean square deviation (RMSD) calculated for their structures over time, relative to the initial structure at the start of the production simulation. RMSD calculations were performed for the backbone atoms of each structure. Panel A, Fig. S3† shows the RMSD of the two protein structures over a 500 ns MD simulation. For IL-6-noTER, the average RMSD value was 0.304 ± 0.013 nm, while for IL-6, it was 0.290 ± 0.010 nm. IL-6-noTER reached a plateau faster than IL-6, indicating that IL-6 stabilized after 400 ns.
The total radius of gyration (Rg) was calculated for both protein structures to assess their compactness. As shown in panel B of Fig. S3,† IL-6-noTER (red graph) was slightly less compact compared to IL-6 (violet line). The average Rg for IL-6-noTER was 1.681 ± 0.007 nm, and for IL-6, it was 1.662 ± 0.006 nm. The smaller Rg for IL-6 suggests that the protein structure is more compact in the presence of the N-TER tail, which could imply some stabilization of the protein.
The root mean square fluctuation (RMSF) of the protein backbones was analyzed to identify which residues deviated most from the starting structure and which maintained rigidity during the dynamics. The RMSF analysis, shown in panel C of Fig. S3,† compared the two protein structures, IL-6-noTER and IL-6. The red line represents IL-6-noTER, and the violet line represents IL-6. Three regions were highlighted: the first region (residues ALA1 to PRO19) showed fluctuations of the flexible tail of the IL-6 structure, ranging from 0.13 nm to 1.22 nm. The AB-loop (residues SER48 to ASN80) and the CD-loop (residues LYS132 to GLN155) showed slightly higher fluctuations, with IL-6 exhibiting larger fluctuations in the AB-loop and IL-6 in the CD-loop. The E α-helix (within the CD-loop) did not show substantial differences in fluctuations between the two structures.
The GROMOS clustering algorithm, based on an RMSD cutoff, was used to determine similar structures sampled during the MD simulation. Clustering analysis with different cutoffs ranging from 0.08 nm to 0.2 nm was performed, and the corresponding number of clusters is shown in Fig. S4.† A cutoff of 0.13 nm for IL-6-noTER and 0.18 nm for IL-6 was chosen as the best cutoff, providing a reasonable number of clusters (less than 40), with 90% of the trajectory contained in a few clusters, and avoiding many single-member clusters.
Overall, these analyses provided comprehensive insights into the stability, flexibility and compactness of the protein structure upon inclusion of its N-terminal showing that the addition of the N-TER tail has a minor influence on the overall structure of the protein. Since the tail is making the structure slightly more compact and stable, the complete structure of IL-6, including a N-terminal tail, is used to perform all the following simulations.
At low ionic strength (10 mM), two clusters were identified as the final docking arrangement for M-SAM (Fig. 2, panel A), sharing similar contacting residues at the nano-bio-interface (distances <3.5 Å). Conversely, only one cluster was identified for the S-SAM. Surprisingly, the M-SAM exhibited a more negative electrostatic interaction energy, approximately −1.12 × 103 kJ mol−1, compared to −9.40 × 102 kJ mol−1 for the S-SAM. This difference is primarily due to the difficulty of accommodating the protein in different orientations on S-SAM, due to the rigidity of the fully charged residues resulting in slightly larger protein–surface center-to-center distances compared to M-SAM. Additionally, M-SAM exhibited a larger non-polar hydrophobic contribution to the binding energy due to a stronger attraction to hydrophobic regions, characterized by terminal CH3 groups present in the mixed surface but absent in the S-SAM.
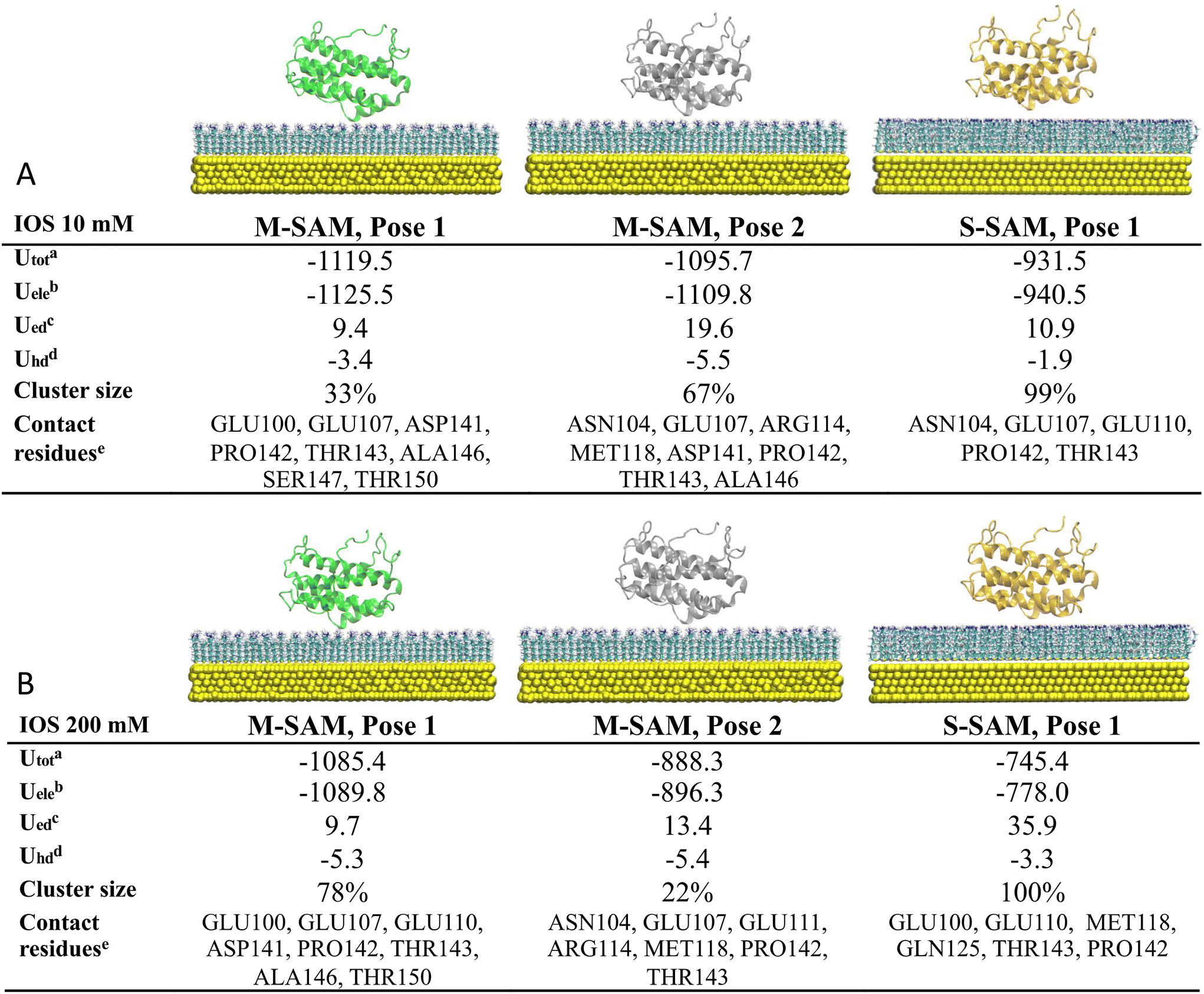 | ||
Fig. 2 Most populated encounter complexes of IL-6 on M-SAM and S-SAM obtained by BD simulation are reported. In panel A results from protein–surface docking at low ionic strength (10 mM) and in panel B at high ionic strength (200 mM). The structures of most representative complexes for are shown, ordered by decreasing energy. The tables in panel A and panel B show: a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) total cluster interaction energy in kT with T = 300 K; btotal electrostatic energy, in kT; csurface desolvation energy, in kT; dnonpolar (hydrophobic) desolvation energy, in kT; eresidues with atoms in contact with surfaces at distances <3.5 Å. total cluster interaction energy in kT with T = 300 K; btotal electrostatic energy, in kT; csurface desolvation energy, in kT; dnonpolar (hydrophobic) desolvation energy, in kT; eresidues with atoms in contact with surfaces at distances <3.5 Å. | ||
The results indicate that the interaction between the protein and both surfaces is predominantly electrostatic. However, the M-SAM binds with both negatively and positively charged residues of the protein, whereas the S-SAM primarily binds with negatively charged residues. Hydrophobic interactions are important in shaping the overall binding affinity (see Fig. 2 for more details).
At high ionic strength (200 mM), two clusters were identified for the M-SAM with similar binding poses, while only one cluster was identified for the S-SAM (Fig. 2 panel B). The screening effect of high ionic strength decreased the electrostatic contribution to the binding energy, especially for the highly charged S-SAM surface. Specifically, the electrostatic energy decreased from the order of 103 to 102 kJ mol−1 when comparing the low to high ionic strength simulations of IL-6 with S-SAM.
Consistent with previous results, the M-SAM surface demonstrates greater binding robustness compared to S-SAM. The S-SAM exhibited slightly stronger electrostatic desolvation energy, indicating that an amount of energy is required to remove solvent molecules due to the strong stabilization by electrostatic interactions in the solvation shell. Notably, the binding between IL-6 and M-SAM involves both hydrophilic and hydrophobic components, with stability preserved at high ionic strength due to compensatory hydrophobic interactions when electrostatic interactions are weakened by ion screening.
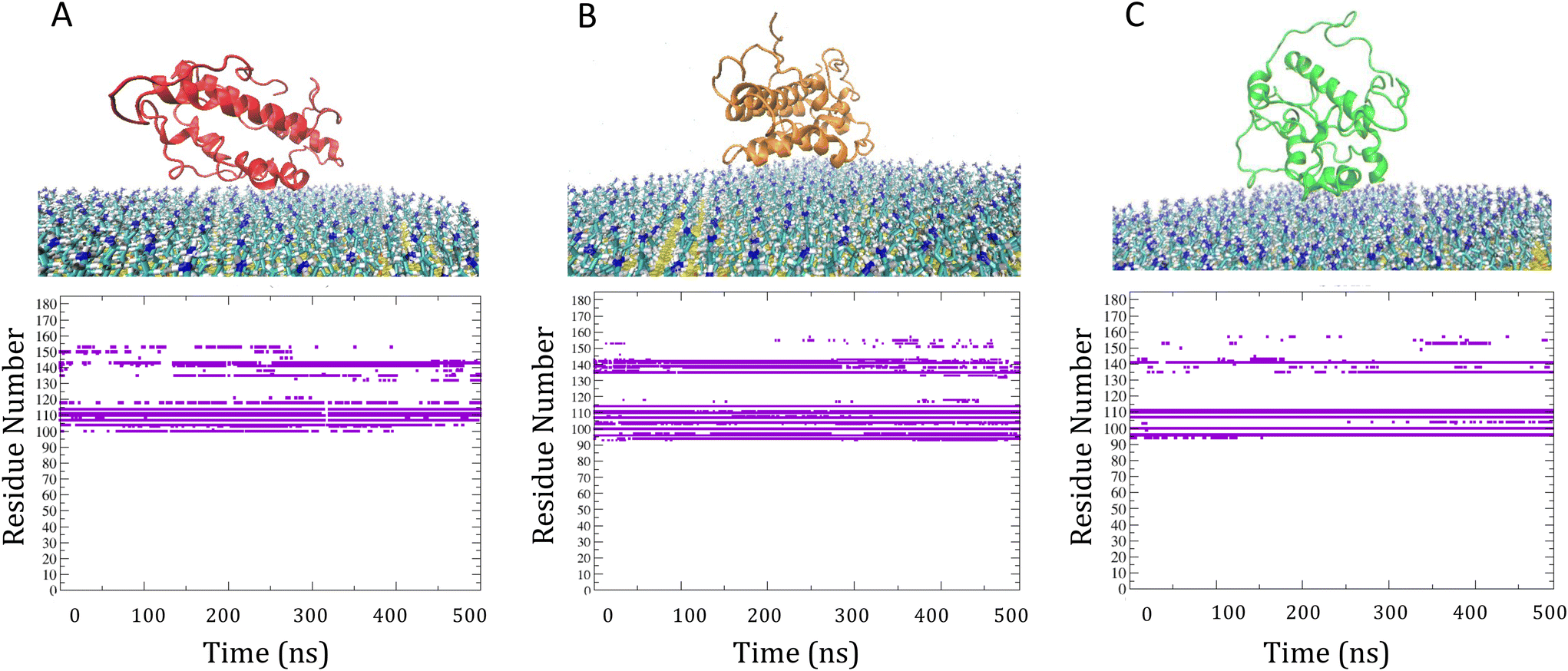 | ||
| Fig. 3 Panel A and B: final most representative association complexes of IL-6 on M-SAM and time evolution of contacting residues (i.e. protein residues within 3 A from the surface), extracted from the total 500 ns MD. Binding to the M-SAM is conserved during the entire length of MD since the protein remains anchored through the following key residues, GLU94, GLU96 (B α-helix), GLU100, ASN104, GLU107, GLU110, GLU111, ARG114 (C α-helix), ASP135, ASP141 (CD loop). Panel C: final most representative association complexes of IL-6 on S-SAM and time evolution of contacting residues. For S-SAM, key residues are all negatively charged indicating a dominant electrostatic interaction with the surface and the protein remains anchored through the B-α helix residues (GLU94, GLU96) and C-α helix residues (GLU100, GLU110, GLU111) and partially with CD-loop (ASP135, ASP141). | ||
RMSF analysis showed how binding to the SAM surfaces affected protein flexibility with respect to solvent (Fig. 4). For M-SAM, rigidity increased in specific regions, notably from SER22 to LEU40, THR90 to THR120, and GLN160 to ARG180, with fluctuations below 0.20 nm. The second cluster of M-SAM displayed additional rigidity from LYS130 to PRO140. In contrast, on S-SAM the protein exhibited higher fluctuations, up to 0.7 nm, indicating possible unfolding and therefore a tendency of irreversible binding to the surface. In order to deepen our understanding of how surface charge density influences protein dynamics and binding reversibility, in Fig. S5† we have compared the adsorption energy terms extracted from 500 ns molecular dynamics (MD) simulations of IL-6 protein interactions with S-SAM and M-SAM surfaces. Adsorption energy profiles derived from simulations confirm that Coulomb interactions dominate protein binding. For M-SAMs, the simulations (cluster 1 and cluster 2) showed stable yet reversible adsorption energies, aligning with koff values obtained from SPR measurements. This agreement highlights how a balanced surface charge and hydrophobicity in M-SAMs moderate protein binding, allowing stable yet reversible adsorption (Fig. S5†).
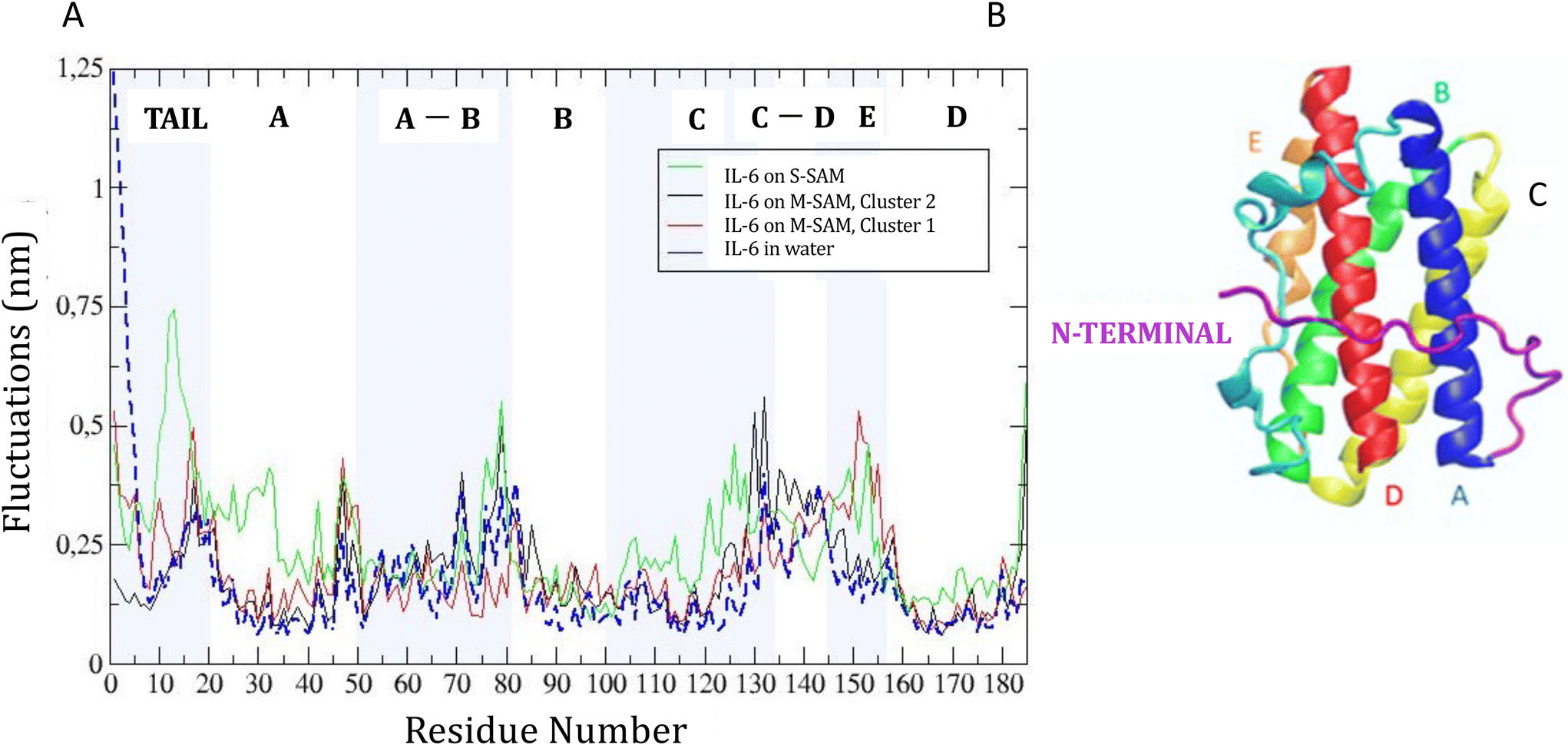 | ||
| Fig. 4 Root mean square fluctuations (RMSF) of the IL-6 protein on M-SAM cluster 1, cluster 2, on S-SAM and in water. | ||
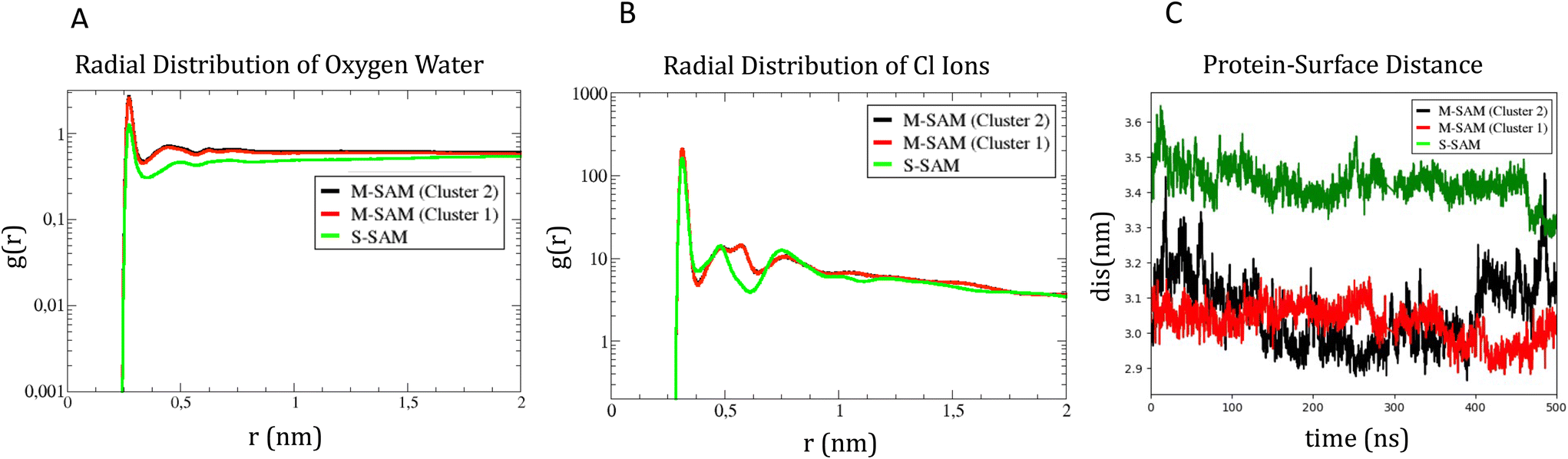 | ||
| Fig. 5 Panel A, radial distribution function of water oxygen atoms around the NH3+ terminal groups and panel B, radial distribution function for Cl− ions (in logarithmic scale). Panel C, protein center of mass distance to the M-SAM and S-SAM surfaces along the z axis. | ||
To identify differences in the adsorption of the protein on the different SAM, the distance fluctuation over time between the z coordinate of the center of mass of the protein to the M-SAM and S-SAM surfaces were computed in the three cases, as reported in panel C of Fig. 5. The distance between the center of mass (COM) of the protein and the M-SAM is shorter compared to the S-SAM, as observed over time. This is expected due to the different length and chemistry composition of the two investigated surfaces.
To summarize, the behavior of the target protein greatly influences the detection limit of the biosensor. The results demonstrate that the adsorption stability of IL-6 on the M-SAM and S-SAM persists over extended timescales, indicating a robust interaction. MD refinement simulations confirm that once the protein is adsorbed, it forms stable interactions with both SAMs, with minimal changes in orientation.
However, in the case of S-SAM, the interaction leads to a partial unfolding heralding irreversible binding, which makes this surface less suitable for biosensor applications since irreversible binding can result in faster saturation of the biosensor surface. On the contrary, promising adsorption stability on M-SAM highlights the potential for practical applications in biosensors for capturing IL-6 at the surface-liquid interface.
2.2 Experimental investigation of the interaction of IL-6 on SAM
The interaction between IL-6 and the SAMs of alkanethiolates on gold was investigated using SPR spectroscopy. SPR is particularly well-suited for these studies as it enables label-free detection of binding events in real time.25 We prepared SAMs with different ratios of alkanthiols with terminal ammonium group (S(CH2)8NH3+) and alkylthiols with terminal methyl group (S(CH2)7CH3). Fig. 6A shows the sensorgrams obtained when solutions of IL-6 were passed over mixed SAMs with different molar fractions of S(CH2)8NH3+. The amount of protein adsorbed onto the mixed SAMs increased as the mole fraction of alkanethiols with terminal ammonium groups (S(CH2)8NH3+) increased, whereas SAMs composed solely of alkylthiols with terminal methyl groups (S(CH2)7CH3) showed only minimal adsorption. Upon removing the protein from the buffer flowing over the surface, the signal largely recovered (>85%) for SAMs with molar fractions of S(CH2)8NH3+ at 0.2 and 0.5, indicating that protein binding to these mixed SAMs was largely reversible. In contrast, adsorption of IL-6 on SAMs with higher molar fractions of S(CH2)8NH3+ was mostly irreversible, as confirmed by the low signal recovery after flushing the surface with PBS. The dependence of IL-6 adsorption on S-SAM with ammonium groups aligns with MD simulations, which demonstrate partial protein unfolding over a highly charged SAM surface likely leading to the irreversibility of binding.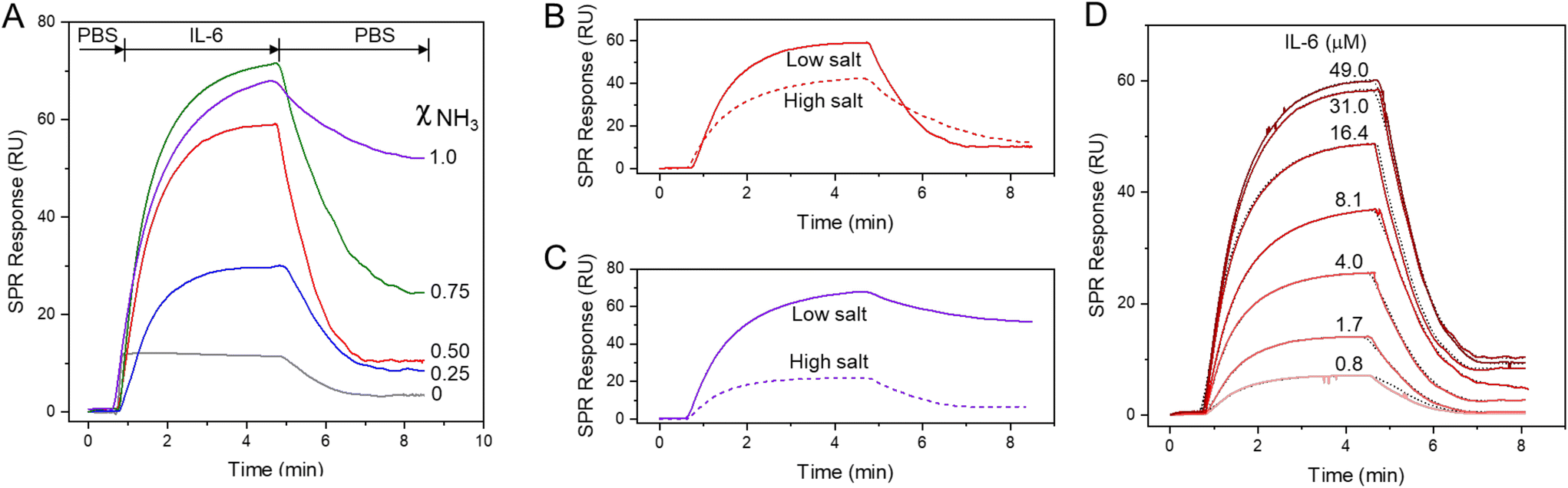 | ||
| Fig. 6 Panel A. Sensorgrams obtained by passing IL-6 (31 μM) over mixed SAMs with different molar fractions of S(CH2)8NH3+ (χNH3) in PBS (pH 7.2, 10 mM). Panels B and C. Effect of the ionic strength (low salt: 10 mM PBS; high salt: 10 mM PBS with 200 mM NaCl) on the binding of IL-6 (31 μM) over SAMs with χNH3 0.50 (B) and 1.0 (C). Panel D. Sensorgrams obtained by passing IL-6 at different concentrations over a mixed SAM (χNH3 = 0.50). Best fits (dotted black lines) were performed by using nonlinear regression analysis to a reversible bimolecular interaction model. | ||
Given that the charges of the SAM strongly influences IL-6 binding, as demonstrated by our model, we investigated the effect of ionic strength of the solution on IL-6 adsorption to SAMs with terminal NH3+ groups and mixed SAMs, namely M-SAM with a 0.5 molar fraction of S(CH2)8NH3+ alkanethiol. The adsorption of the protein on the S-SAM decreased significantly with increasing ionic strength of the buffer, accompanied by improved binding reversibility, as evidenced by the higher signal recovery after washing with the buffer. IL-6 adsorption on the M-SAM was only slightly affected by the increased ionic strength, with signal recovery comparable to that observed at lower ionic strength.
By following the time varying SPR angle shift due to binding molecular processes occurring at the sensor surface in real time, it is possible to evaluate the association and dissociation kinetics of the interaction. The SPR sensorgrams, shown in Fig. 6D, have been obtained by injecting IL-6 at different concentrations, ranging from 0.8 to 49 mM, onto the SAM with a 0.5 molar fraction of S(CH2)8NH3+ alkanethiol. The SPR response increased with increasing IL-6 concentration up to 31 mM, and the binding observed in all cases was largely reversible. The rate for association (kon) and dissociation (koff) of the protein on the SAM surface was evaluated according to a predefined bimolecular interaction model,26 obtained from the kinetic analysis of the SPR sensorgram. A koff of 0.082 s−1 and a kon of 1920 M s−1 is determined from the fitting, yielding a dissociation constant (Kd) of 0.42 mM. For comparison, the adsorption rate of cytochrome c was measured using quartz crystal microbalance with dissipation (QCM-D) on SAMs with terminal arginine and guanidine groups. The guanidine-terminated SAM showed a four-fold increase in adsorption rate compared to the hydrophobic arginine-terminated SAM, which had an adsorption rate of 1 × 104 M−1 s−1. In contrast, the adsorption rate of α-chymotrypsin showed only a minor difference between the two SAMs.27 The adsorption of bovine carbonic anhydrase II on a mixed SAM with a benzenesulfonamide terminal group yielded a Kd of 0.68 mM.28 The binding of IL-6 to nucleic acid ligands, in particular aptamers, has been extensively studied to elucidate how these ligands interact with the protein surface and to develop new therapeutic agents.29 These studies revealed that aptamers with substantial hydrophobic groups overlapping the binding surfaces of IL-6 enhance binding affinity. Therefore, alternating charged and hydrophobic domains on the ligand surface can provide a means to fine-tune the ligand binding surface and enhance specificity.
3. Conclusions
The comparison between multiscale simulations and SPR experiments for the association between IL-6, a key biomarker in various cancers, and two types of SAM surfaces demonstrates a strong agreement, confirming that mixed M-SAMs outperform single-component S-SAMs in terms of stability and suitability for potential biosensing applications. M-SAMs offer a more favorable surface for protein binding, characterized by stable and reversible interactions, which is essential for the repeated use of biosensors. In contrast, while S-SAMs also show stable binding, the interaction tends to be irreversible, as evidenced both by MD simulations and SPR experiments, potentially leading to earlier saturation of the sensor surface and consequently limiting its performance.The MD simulations reveal that the M-SAM surface, due to its combination of hydrophobic and charged groups, allows for a greater surface-accessible area, promoting more effective protein docking and better exposure of binding sites. This is supported by SASA analysis and root mean square deviation (RMSD) calculations, which indicate faster stabilization of the M-SAM system compared to S-SAMs.
Furthermore, BD simulations demonstrate that IL-6 interacts with both types of surfaces primarily through electrostatic interactions. However, only the M-SAM maintains stable binding even at high ionic strength, where electrostatic forces weaken but are compensated by hydrophobic interactions and desolvation effects. These factors contribute to the superior performance of M-SAM in maintaining stable protein adsorption over time, with minimal alteration in protein conformation.
In summary, the combined experimental and computational data show that M-SAM surfaces, with their enhanced stability and reversible binding, are preferable over S-SAMs for biosensor applications. The stability and adaptability of the M-SAM surface in maintaining robust protein interactions at the surface-liquid interface highlight its promise for practical applications in detecting IL-6 and similar biomarkers. For example, M-SAMs could enhance electrochemical and optical biosensors, such as those using surface plasmon resonance or quartz crystal microbalance, by improving sensitivity and reproducibility. These surfaces could also be integrated into microfluidic systems for point-of-care diagnostics and wearable biosensors for continuous cytokine monitoring in dynamic environments. Additionally, M-SAMs hold potential for applications in environmental monitoring and food safety, where reliable protein binding and release are crucial for accurate detection.
4. Methods
4.1 Construction of the Au(111)-SAM surface model
Experimental studies have shown that the adsorption of n-alkane thiols onto gold substrates, with the general structure HS(CH2)nX, results in the formation of highly dense, oriented, and ordered films that can be indexed as (n√3 × √3) R × 30°, where n varies from 1 to 6. This is attributed to the strong affinity of the Au–S bond.4The choice of the Au(111) surface for this study is motivated by several key factors.30 This surface has been extensively studied in experimental settings, particularly for small molecule adsorption under controlled conditions. It exhibits robust stability in the presence of air and water, even if susceptible to contamination by hydrophobic molecules. The typical 22√3 reconstruction of Au(111) is well-documented in both air and aqueous environments, and this reconstruction minimally affects the surface reactivity of Au(111).31
In nanobioelectronics, Au(111) serves as a crucial substrate, commonly employed to establish contacts between nano-objects and the macroscopic world. Additionally, it plays a pivotal role in optical detection systems, notably in techniques like surface plasmon resonance (SPR) for studying biomolecular interactions.32
The Au(111) surfaces used in the model consist of four atomic layers. The force field parameters for the gold atoms were defined using the GolP classical force field,33 recognized for its suitability in modeling biomolecule-surface interactions. The ligands used in the SAM model were modeled with parameters derived from the OPLS/AA (all-atom) force field. Bonded and non-bonded parameters were adopted from previous studies, ensuring consistency and reliability in the model construction. Specifically, parameters for the covalent bond between the gold surface atoms, the sulfur atoms, and the first two carbon atoms of the alkyl chain.34,35
To assemble the self-assembled monolayer (SAM) on the Au(111) surface, a gold unit cell containing a covalently bound ligand was replicated using the GROMACS tool genconf to create the SAM's final configuration. Each ligand occupies an area of 21.6 Å2 on average. The SAM was structured on a gold substrate with dimensions of 10 × 10 nm2, forming a hexagonal √3 × √3 × R × 30° overlayer, mirroring experimental observations of thiol adsorption on Au(111).4
The interaction between the ligands and the Au(111) surface primarily occurs through a metal–sulfur interface. This interface involves covalent bonds between the gold surface atoms (AU) and the sulfur atoms (S) within the ligands. These bonds are crucial for stabilizing the surface atoms and modulating their electronic properties.1
4.2 Interleukin-6 protein model
To perform the simulations, the IL-6 structure determined by NMR (nuclear magnetic resonance) was initially downloaded (PDB code: 1IL636). This structure included 166 amino acids, numbered 20 to 185, omitting the first 19 residues of the complete 185 amino acid sequence of IL-6, named IL-6-noTER. To address the lack of the N-TER tail, a homology model was created using the I-TASSER web server.37 I-TASSER predicts the 3D structure of proteins by threading amino acid sequences onto known templates, followed by replica exchange Monte Carlo simulations. The quality of the models is estimated using a confidence score (C-score), ranging from −5 to 2, with higher scores indicating greater confidence. The model with the highest C-score for IL-6's complete sequence was chosen and named IL-6. IL-6 consists of 185 residues, numbered from 1 to 185, with the first 19 residues forming a tail at the N-terminus.
To determine the best-predicted protein IL-6 model, several criteria were used. First, five models of the IL-6-noTER protein were generated using AI-based methods, including AlphaFold 238 and Robetta39 with the RoseTTAFold method.40 These AI-predicted models (Fig. S6†) were then compared with structures obtained through traditional non-AI methods.
The first model from AlphaFold 2 was excluded from consideration because its straight tail conformation did not match the expected protein structure and because it was inconsistent with the low disorder predicted for IL-6 by IUPRED3.41 To evaluate the remaining models, the RMSD was calculated using the Gromacs tool, specifically the gmx rms function, which performs least-squares fitting of one structure onto another to assess structural similarity, results are reported in Fig. S6.†
Model 3 emerged as the best choice due to its lowest RMSD value with respect to its initial reference structure, compared to the non-AI predicted structures. Consequently, model 3 was selected for further studies based on its superior agreement with the validated protein structure and its effective representation of the IL-6 protein. This model, shown in Fig. S6,† was used as the starting structure for subsequent simulations.
4.3 Relaxation by molecular dynamics (MD)
The first step involved (i) energy minimization of the system, relaxing the structures through a protocol ensuring appropriate geometry. The steepest descent minimization algorithm was used, and the Particle-Mesh Ewald (PME) method treated long-range electrostatic interactions; (ii) subsequently, an NVT equilibration step was performed. Initially, a 1 ns equilibration involved only the solvent molecules, with both the gold surfaces and the ligands kept fixed. The solvent and the functionalized surfaces were separately weakly coupled to an external temperature bath set to 300 K. Pressure coupling was not performed, as NPT is not suitable for rigid surfaces. (iii) After completing the two equilibration phases, the systems were well-equilibrated, allowing the position restraints to be released and the production MD to commence for data collection. The simulations were run for 300 ns, with coordinates, velocities, and energies saved every 10 ps.
For the MD, the equations of motion were integrated using the Leap-Frog algorithm with a time step of 0.001 ps. The LINCS algorithm was used to constrain all bonds containing hydrogen atoms, and the neighbor list (non-bonded pair) was updated every 10 steps. The PME switch method was used to handle long-range electrostatic interactions with a Verlet cutoff scheme, setting both the short-range electrostatic and van der Waals cutoffs to 1.2 nm.
An initial NVT equilibration was performed, keeping all systems fixed except for water molecules and ions. The second equilibration was then performed for 1 ns, keeping only the gold surfaces fixed while allowing ligands, solvent, and ions to move.
After 300 ns of MD without restraints, the systems maintained consistent values for temperature, potential, and other properties compared to their initial configuration.
The gold surfaces coated with SAM were subjected to clustering analysis using the GROMOS clustering algorithm in GROMACS. For this analysis, cutoffs of 0.11 nm for the M-SAM and 0.10 nm for the S-SAM were selected to yield an appropriate number of clusters. The cluster structures were used as starting points for the docking simulations.
First, energy minimization was performed, followed by NVT and NPT equilibration for 100 ps each. During the equilibration runs, a velocity rescale thermostat maintained the temperature at 300 K, and a Parinello-Rahman barostat maintained the pressure close to 1 bar. The production simulations ran using NPT conditions with the Parinello-Rahman barostat applied every 2 ps, and each system was simulated for 500 ns. The equation of motion was integrated using the Leap-Frog algorithm with a time step of 2 fs. All hydrogen bond lengths were constrained with the LINCS algorithm. Electrostatic interactions were treated using the PME scheme. Short-range electrostatic and van der Waals interactions were computed up to 1.0 nm and treated with a Verlet-buffer, with the list updated every 10 time steps. Standard structural analysis on the MD trajectories as RMSD and RMSF was performed using GROMACS. Trajectories and structures were visualized using VMD44 and PyMOL.
4.4 Brownian dynamics of IL-6 onto SAM-surfaces
BD simulations were used to create encounter complexes between IL-6 and SAM-based gold surfaces, treating both as rigid bodies. Electrostatic calculations were performed with APBS (Adaptive Poisson–Boltzmann Solver) and the ProMetCS protein-metal force field. During BD simulations, only electrostatic interactions and desolvation were considered. For each system, 5000 docked binding poses were collected, encounter complexes were clustered based on their size and energy, leading to the identification of several potential binding poses.4.5 MD refinement of the docked complexes
The final, most stable protein–surface complexes obtained from protein–surface docking, were used as initial structures for MD refinements. For each gold SAM surface, MD refinements were conducted, each lasting 500 ns. The MD refinement process employed the same protocol, algorithm, and parameters as those used for MD simulations involving the individual SAM surfaces and protein, as described in paragraph 2.3. The analysis included the evaluation of the protein–surface complexes after refinements, analysis of the contacting residues at the short distances (distances <3.5 Å) to explore the stability of the contacts during the entire length of the dynamics, root mean square fluctuation (RMSF) to compare changes in the most flexible regions of the protein associated to the binding with the SAM surfaces, the SASA of M-SAM and S-SAM during the binding with the target protein and the number of solvated Cl− ions condensed to M-SAM compared to the S-SAM.4.6 Surface plasmon resonance experiments
SPR experiments were performed with Biacore 1000 instrument (GE Healthcare, Sweden). Substrates for SPR measurements were prepared by electron beam evaporation of Ti and then Au (thickness 50 nm) onto glass cover slips. Self-assembled monolayers (SAMs) were prepared by immersing the substrates into ethanolic solutions containing mixtures of the thiols (2 mM total concentration) for 18 h. Upon removal from the solution, substrates were rinsed thoroughly with ethanol and water, and then dried with nitrogen. Analysis of the SPR sensorgrams was performed by using nonlinear regression fit procedure derived from a reversible bimolecular interaction model:45where A is the IL-6 protein in solution and B is the SAM-modified surface. kon (M s−1) and koff (s−1) are the association and dissociation rate constants for protein–SAM interaction. The quality of the fits was judged by the residuals and the normalized chi square of the fitting model. The equilibrium dissociation constant (Kd, M) was determined from the ratio of the measured dissociation and association rate constants.
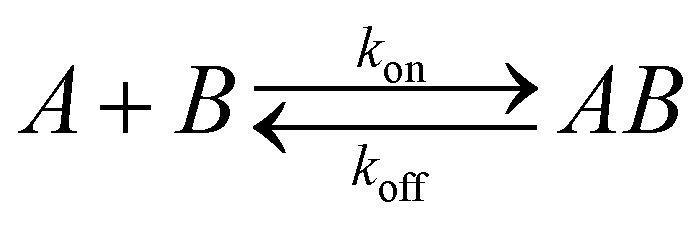
Author contributions
G. B., S. C. and M. F. designed the project. S. G. performed all multi-scale simulations and analysed the results. S. D. performed additional analysis.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. B. and S. C. acknowledge Giovanni Parolin for his assistance in setting up the force field parameters for the SAM surfaces. G. B. acknowledges the CINECA award under the ISCRA initiative, for the availability of high-performance computing resources on Marconi100 and support. G. B. acknowledges Oak Ridge National Laboratory by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy is acknowledged for the supercomputing project CNMS2020-B-00433. G. B. acknowledges Facilities of the National Energy Research Scientific Computing Center (NERSC), which is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231, are also acknowledged. M. F. acknowledges the support of Next Generation EU PRIN 2022 (M4.C2.1.1) under the Prot. 2022WN89PC. GB acknowledges the funding by MUR, PRIN2020 Project (2020LW7XWH).References
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed.
- M. Singh, N. Kaur and E. Comini, J. Mater. Chem. C, 2020, 8, 3938–3955 RSC.
- B. Wang, U. Akiba and J. I. Anzai, Molecules, 2017, 22(7), 1048–1068 CrossRef PubMed.
- L. H. Dubois, B. R. Zegarski and R. G. Nuzzo, J. Chem. Phys., 1993, 98, 678–688 CrossRef CAS.
- S. Ferretti, S. Paynter, D. A. Russell and K. E. Sapsford, TrAC, Trends Anal. Chem., 2000, 19, 530–540 CrossRef CAS.
- N. K. Chaki and K. Vijayamohanan, Biosens. Bioelectron., 2002, 17(1–2), 1–13 CrossRef CAS.
- S. Kumar and P. Huo, J. Mater. Chem. C, 2017, 5, 3879–3892 Search PubMed.
- T. Wink, S. J. van Zuilen, A. Bult and W. P. van Bennekom, Analyst, 1997, 122, 43R–50R RSC.
- F. Frederix, K. Bonroy, W. Laureyn, G. Reekmans, A. Campitelli, W. Dehaen and G. Maes, Langmuir, 2003, 19, 4351–4357 CrossRef CAS.
- N. Afara, S. Omanovic and M. Asghari-Khiavi, Thin Solid Films, 2012, 522, 381–389 CrossRef CAS.
- S. Y. Yeung, S. Yee, A. Mucha, R. Deshmukh, M. Boutrus, T. Arnebrant and B. Sellergren, ACS Cent. Sci., 2017, 3, 1198–1207 CrossRef CAS PubMed.
- C. Ma, C. Wang and C. Acevedo-Vélez, et al. , Nature, 2015, 517, 347–350 CrossRef CAS.
- S. Chen, J. Zhang and Y. Wang, et al. , Science, 2015, 348, 555–559 CrossRef CAS PubMed.
- B. C. Dallin, A. S. Kelkar and R. C. Van Lehn, Chem. Sci., 2023, 14, 1308–1319 RSC.
- N. B. Rego, A. L. Ferguson and A. J. Patel, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2200018119 CrossRef CAS.
- N. Giovambattista, P. G. Debenedetti and P. J. Rossky, J. Phys. Chem. C, 2007, 111, 1323–1332 CrossRef CAS.
- J. Scheller and S. Rose-John, Med. Microbiol. Immunol., 2006, 195, 173–183 CrossRef CAS PubMed.
- M. Majdinasab, M. Lamy de la Chapelle and J. L. Marty, Biosensors, 2023, 13, 898 CrossRef CAS PubMed.
- L. E. McCrae, W.-T. Ting and M. M. R. Howlader, Biosens. Bioelectron.: X, 2023, 13, 100288 CAS.
- M. A. Khan and M. Mujahid, Sensors, 2020, 20, 646 CrossRef.
- S. Dutta, S. Corni and G. Brancolini, Int. J. Mol. Sci., 2022, 23, 1484 CrossRef CAS PubMed.
- S. Dutta, S. Corni and G. Brancolini, Nanotechnology, 2021, 32, 095702 CrossRef CAS.
- S. Dutta, M. Gagliardi, L. Bellucci, M. Agostini, S. Corni, M. Cecchini and G. Brancolini, Front. Mol. Biosci., 2022, 9, 1000330 Search PubMed.
- J. Romanowska, D. B. Kokh, M. Ozboyaci, X. Yu, M. A. Öztürk, S. Richter, R. C. Wade, M. Martinez and N. J. Bruce, J. Comput. Chem., 2015, 36, 1669–1680 Search PubMed.
- M. A. Cooper, Nat. Rev. Drug Discovery, 2002, 1, 515–528 CrossRef CAS PubMed.
- T. A. Morton and D. G. Myszka, Methods Enzymol., 1998, 295, 268–294 CAS.
- L. Cai, T. Xiang, R. Guo, X. Hu, Y. Xie, Y. Jiang and J. Wang, Anal. Chem., 2018, 90, 2609–2617 CrossRef.
- J. Lahiri, L. Isaacs, J. Tien and G. M. Whitesides, Anal. Chem., 1999, 71, 777–790 CrossRef CAS.
- A. D. Gelinas, D. R. Davies, T. E. Edwards, J. C. Rohloff, J. D. Carter, C. Zhang, S. Gupta, Y. Ishikawa, M. Hirota, Y. Nakaishi, T. C. Jarvis and N. Janjic, J. Biol. Chem., 2014, 289, 8720–8734 CrossRef CAS.
- M. Frasconi, F. Mazzei and T. Ferri, Anal. Bioanal. Chem., 2010, 398, 1545–1564 CrossRef CAS PubMed.
- M. García-Algar, A. Costa-García and M. T. Fernández-Abedul, Electroanalysis, 2017, 29, 1131–1146 CrossRef.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS.
- F. Iori, R. Di Felice, E. Molinari and S. Corni, J. Comput. Chem., 2009, 30, 1465–1476 CrossRef CAS PubMed.
- S. Dutta, L. Bellucci, M. Agostini, M. Gagliardi, S. Corni, M. Cecchini and G. Brancolini, Nanotechnology, 2021, 32, 095702 CrossRef CAS.
- S. Dutta, L. Bellucci, M. Agostini, M. Gagliardi, S. Corni, M. Cecchini and G. Brancolini, Front. Mol. Biosci., 2022, 9, 915392 Search PubMed.
- Protein Data Bank, PDB, 1998, DOI: 10.2210/pdb1IL6/pdb.
- Y. Zhang and J. Yang, Curr. Protoc. Bioinf., 2015, 52, 5.8.1–5.8.15 Search PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS.
- D. E. Kim, D. Chivian and D. Baker, Nucleic Acids Res., 2004, 32, W526–W531 CrossRef CAS PubMed.
- N. Hiranuma, H. Park, M. Baek, I. Anishchenko, J. Dauparas and D. Baker, Nat. Commun., 2021, 12, 1340 CrossRef CAS PubMed.
- E. G. Erdős, M. Pajkos and Z. Dosztányi, Nucleic Acids Res., 2021, 49, W297–W303 CrossRef PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- R. Zangi, ACS Omega, 2018, 3, 18089–18099 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. A. Morton and D. G. Myszka, Methods Enzymol., 1998, 295, 268–294 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04199b |
| This journal is © The Royal Society of Chemistry 2025 |