
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rapid synthesis of magnetic-core mesoporous silica-shell nanostructures – as potential theranostic agents – by means of microwave irradiation and the atrane method†
M. Dolores
Garrido
 *ab,
Bejan
Hamawandi
b,
José Francisco
Serrano-Claumarchirant
b,
Giovanni Marco
Saladino
b,
Adem B.
Ergül
b,
M. Dolores
Marcos
c,
José Vicente
Ros-Lis
d,
Pedro
Amorós
a and
Muhammet S.
Toprak
*b
*ab,
Bejan
Hamawandi
b,
José Francisco
Serrano-Claumarchirant
b,
Giovanni Marco
Saladino
b,
Adem B.
Ergül
b,
M. Dolores
Marcos
c,
José Vicente
Ros-Lis
d,
Pedro
Amorós
a and
Muhammet S.
Toprak
*b
aInstitut de Ciència dels Materials (ICMUV), Universitat de València, Catedrático José Beltrán 2, 46980 Paterna, Valencia, Spain. E-mail: maria.d.garrido@uv.es
bKTH Royal Institute of Technology, Department of Applied Physics, SE106 91 Stockholm, Sweden. E-mail: toprak@kth.se
cInstituto Interuniversitario de Investigación de Reconocimiento Molecular y Desarrollo Tecnológico (IDM), Universitat Politècnica de València, Valencia, Spain
dInstituto Interuniversitario de Investigación de Reconocimiento Molecular y Desarrollo Tecnológico (IDM), Universitat de València, C/Dr. Moliner 50, 46100 Burjassot, Valencia, Spain
First published on 5th February 2025
Abstract
Nowadays, the interest in the design of particles that combine therapy and diagnosis simultaneously to obtain a theranostic material has increased. One of the most used materials for MRI diagnosis is iron oxide, where clusters of superparamagnetic iron oxide (SPIONs) are noteworthy candidates. These particles are of high interest due to their broad range of applications, such as contrast agents, use in magnetic separation processes, and in hyperthermia therapy, among others. One of the major problems with their use is maintaining superparamagnetism while having the highest magnetization-to-particle ratio. In this work, microwave-assisted synthesis of clusters formed by SPIONs has been investigated. This synthesis strategy allows for significant reduction in the time and energy required to obtain SPION clusters. Also, the magnetization-to-particle ratio has been increased in comparison with single SPIONs. Subsequently, the clusters are coated with amorphous silica using the Stöber method, followed by mesoporous (MS) silica using the atrane method, which offers high and conformal coating homogeneity over the clusters. Surfactant extraction was done using a simple mixture of water, ethanol, and sodium chloride – avoiding the use of other organic solvents. Finally, as a proof of concept, the loading and release of a model molecule were studied to confirm that the SPION-NCs@MS presented in this work have great potential as theranostic agents.
Introduction
In recent years, there has been an increasing interest in theranostic materials based on the simultaneous combination of therapy and diagnosis. This interest stems from developing advanced methods for more effective and selective detection and treatment of malignant tumor agents.1 Additionally, diagnostic techniques can be highly varied, with imaging techniques such as photoacoustic imaging (PAI), fluorescence imaging (FLI), magnetic resonance imaging (MRI), and computed tomography imaging (CT) being available.2 In the case of contrast agents for MRI, the most commonly used materials are gadolinium-based compounds (as T1 contrast agents)3 and iron-based compounds (as T2 contrast agents).4 Among the iron-based compounds, superparamagnetic iron oxide nanoparticles (SPIONs) are noteworthy candidates for use in medicine.5 These nanoparticles (NPs) are widely valued for their broad applications, including use as contrast agents, in magnetic separation, and for drug delivery, cancer detection, hyperthermia, and biosensing.6–9 The main characteristics that make these NPs particularly attractive are their biocompatibility, low toxicity, chemical stability, low cost of synthetic precursors, and superparamagnetic character.10,11 This last property is related to the size of the NP, and is only exhibited in the presence of a magnetic field.12Significant efforts have recently been made to control NP size, morphology, surface functionality, and dispersibility. However, the primary limitation of superparamagnetic NPs is their low magnetic response in biomedical applications or magnetic separation due to a low magnetization-to-particle ratio.10,13 Therefore, although the synthesis of monodisperse isolated NPs with sizes between 5 and 25 nm is well documented, it is difficult to effectively separate them from the reaction solution. In some cases, relatively large magnetite particles in the 40–200 nm size range have been stabilized.14 However, these larger particles are mainly formed by the aggregation of some smaller NPs (of around 20 nm). One way to address this issue is by aggregating a large number of small NPs into larger structures. Clustering SPIONs into secondary structures can increase the magnetization-to-particle ratio and, consequently, the magnetic response.10,11,13 Moreover, the formed cluster is able to maintain its superparamagnetic character regardless of size, as this property depends on the size of the primary NPs constituting the cluster.15,16
Different strategies exist for obtaining clusters, which are mainly divided into two principal groups: two-step and one-pot methods.10 In the two-step method, NPs are first synthesized and subsequently assembled.17 In contrast, in the one-pot method, the formation and aggregation of NPs occur simultaneously, often via solvothermal synthesis, where the solvent acts as a reducing agent. Additionally, the growth and aggregation of SPIONs can be controlled by adding a chelating agent. Finally, the primary particles aggregate into secondary structures to reduce surface energy.11,18–20
Further key parameters in developing SPION clusters are their stability and functionality. To this end, strategies have been developed to functionalize their surface with polymers, organic molecules, surfactants, biomolecules, silica, carbon, or other metal oxides.9 Among these, silica is an ideal candidate due to its biocompatibility and ease of post-functionalization.21,22 Furthermore, using templating agents to obtain mesoporous silica (MS) increases its applicability due to its large surface area and ordered porous structure. This porous structure provides cavities that can be filled with molecules to be released later.23,24 Different strategies have been established to cover both isolated magnetite NPs and larger SPION nanoclusters (SPION-NCs) with amorphous and/or porous silica.25–27 In all cases, core–shell-type organizations are generated with a magnetic core and a silica shell. A highly effective strategy for coating nanomaterials with MS is the “atrane method”.28,29 This method uses complexes derived from triethanolamine (atrane complexes) as silica precursors, and hydrolysis and condensation reactions occur in a homogeneous medium, resulting in a more homogeneous MS coating.30,31
In this work, microwave (MW)-assisted synthesis of a cluster formed from SPIONs has been developed. This synthesis strategy has allowed for significant reductions in energy and time required to obtain the desired material, while also using ethylene glycol (EG) as a biorenewable solvent (as an abundant renewable biomass resource). Additionally, various synthetic parameters, such as the reaction temperature and reaction time, ramping time, and citrate concentration have been studied to observe their effects on the final material. Subsequently, the clusters were coated with MS using the atrane method, which offers high coating homogeneity over the clusters. Surfactant extraction was done using chemical extraction with a mixture of water, ethanol, and sodium chloride. This extraction method is relatively unexplored but is of great interest due to its simplicity, speed, and the eco-friendliness of the reagents used. Finally, as a proof of concept, the loading and release of a model molecule were studied to confirm that the SPION-NCs@MS presented in this work have great potential as theranostic agents.
Experimental
Reagents and materials
All the synthesis reagents were analytically pure, and were used as received from Aldrich: iron(III) chloride hexahydrate [FeCl3·6H2O], sodium citrate tribasic [C6H9Na3O9], ethylene glycol [EG, C2H6O2], sodium acetate anhydrous [NaOAc, C2H3NaO2], ethanolamine [EA, C2H7NO], toluene, tetraethyl orthosilicate [TEOS, C8H20O4Si], 2,2′,2′′-nitrilotriethanol or triethanolamine [TEAH3, N(CH2–CH2–OH)3], cetyltrimethyl ammonium bromide [CTAB, C19H42NBr], sodium chloride [NaCl], methylene blue [MB, C16H18ClN3S], (3-mercaptopropyl)trimethoxysilane [MPTMS, C6H16O3SSi], phosphate buffered saline [PBS], and ethanol.Synthesis of SPION clusters
The SPION clusters were synthesized using a polyol solvothermal method previously described32 with some modifications in order to optimize the final material (Fig. 1). Key parameters such as temperature, citrate concentration, and synthesis time (ramp and aging) have been explored (Table 1). In a typical synthesis, a solution containing 30 mL of EG, 2.5 g of FeCl3, and 0.454 g of sodium citrate was prepared by magnetic stirring until complete dissolution. At the same time, a solution containing 2.7 g of sodium acetate and 15 mL of EG was prepared and added to the previous solution dropwise. The mixture was then transferred to a Teflon microwave (MW) vessel being filled to 1/3rd its volume; the synthesis was performed by heating the reaction mixture to various temperatures (180 °C, 200 °C, 220 °C, and 250 °C) for various aging times (10, 20 and 30 min), using MW-assisted heating, with varying ramp times (1 min, 10 min, 20 min).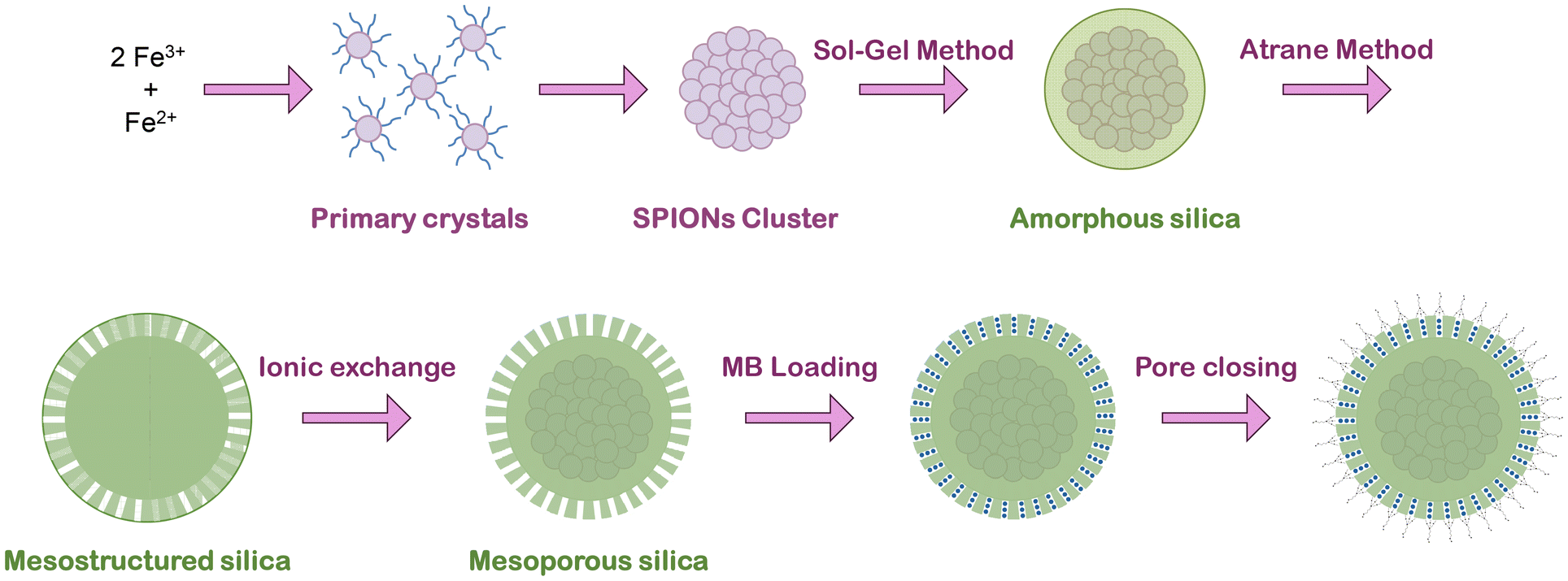 | ||
| Fig. 1 Scheme of the general preparative protocol of the SPION-NCs@MS particles, their loading with MB and finally pore closure. | ||
| Sample | Mass, m (citrate) (g) | Ramping time (min) | Aging time (min) | Temperature (°C) |
|---|---|---|---|---|
| A | 0.454 | 1 | 10 | 180 |
| B | 0.454 | 1 | 10 | 200 |
| C | 0.454 | 1 | 10 | 220 |
| D | 0.454 | 1 | 20 | 220 |
| E | 0.454 | 1 | 30 | 220 |
| F | 0.454 | 10 | 10 | 180 |
| G | 0.454 | 10 | 10 | 200 |
| H | 0.114 | 10 | 20 | 220 |
| I | 0.227 | 10 | 20 | 220 |
| J | 0.454 | 10 | 20 | 220 |
| K | 0.908 | 10 | 20 | 220 |
| L | 0.454 | 20 | 20 | 180 |
| M | 0.454 | 20 | 20 | 200 |
| N | 0.454 | 20 | 10 | 220 |
| O | 0.454 | 20 | 20 | 220 |
| P | 0.454 | 20 | 30 | 220 |
| Q | 0.454 | 20 | 20 | 250 |
| R | 0.908 | 20 | 20 | 220 |
Mesoporous silica coating
Before the MS coating, the SPION clusters were coated with an amorphous silica layer (Fig. 1) to protect the core.32 The SPION clusters were dispersed in 38 mL of a mixture of EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (3.75:1 in volume), with a final iron concentration of 250 μg mL−1, in an ultrasonic bath. Then, 50 μL of TEOS and 200 μL of EA were added. The mixture was shaken for 2 h, and then the product was centrifuged and washed with EtOH and water. Finally, the particles were dispersed in water by vortexing.
:water (3.75:1 in volume), with a final iron concentration of 250 μg mL−1, in an ultrasonic bath. Then, 50 μL of TEOS and 200 μL of EA were added. The mixture was shaken for 2 h, and then the product was centrifuged and washed with EtOH and water. Finally, the particles were dispersed in water by vortexing.
The MS coating (Fig. 1) was performed through the “atrane method”28,29 combined with modifying a previously reported method for obtaining mesoporous siliceous spheres.31 The silatrane complex was first prepared as follows: 11 mL of TEOS and 23 mL of TEAH3 were typically mixed and heated until 140 °C for 5 min, followed by cooling down to room temperature. The SPION clusters were dispersed in 40.9 mL of water by sonication (2 min) and mixed with 22.7 mL of a CTAB solution (0.414 g, EtOH), leading to the final iron concentration of 1.5 mg mL−1. Then, 1.56 mL of atrane was added and mixed for 24 hours at room temperature. The resulting product was centrifuged and washed with EtOH and water and re-dispersed in water.
For CTAB extraction (Fig. 1), we used a very simple, fast, and scalable method: the sample was suspended in 40 mL of an EtOH:water (3:1 in volume) mixture containing 0.05 g of NaCl. The suspension was heated under reflux at 80 °C for 20 min, finally collected by centrifugation, washed with ethanol and water, and dried under vacuum at 40 °C.
Methylene blue loading and release
The methylene blue (MB) was introduced inside the pores by impregnation (Fig. 1). For that purpose, 0.015 g of the sample was impregnated with 30 μL of a solution of MB (20 mg mL−1, water) and dried (45 °C in a vacuum oven overnight) to remove the water. Afterwards, it was suspended in 1.5 mL of toluene, and 33.3 μL of MPTMS was added. It was then kept under constant stirring for 24 h, finally collected by magnetic separation, washed with ethanol, and dried in an oven. For the MB release, the sample was suspended in a solution of PBS with a concentration of 1 mg mL−1. Aliquots were collected at different times and the particles were collected by magnetic separation, and the supernatant was then examined by fluorescence spectroscopy using an excitation wavelength of 600 nm to reveal the amount of MB released.Characterization techniques
The morphology of the samples was analyzed using scanning electron microscopy (SEM, FEI Nova 200, 10 kV) and transmission electron microscopy (TEM and STEM images were acquired on a JEOL-2100F microscope). The dry size of the SPION clusters was estimated by analyzing SEM images with different fields of view using ImageJ 1.52q software.33 For TEM analysis, the samples were dispersed in ethanol, and a 100 μL aliquot was placed onto a carbon-coated copper microgrid and left to dry before observation. TEM images were acquired at 200 kV. Nitrogen adsorption–desorption isotherms were recorded in an automated Micromeritics ASAP2020 instrument. Before the adsorption measurements, the samples were outgassed in situ in a vacuum (10−6 Torr) at 120 °C for 15 hours to remove adsorbed gases. X-ray photoelectron spectroscopy (XPS) measurements were collected with a SPECS spectrometer equipped with a Phoibos 150 MCD 9 analyzer and a non-monochromatic Al Kα (1486.6 eV) X-ray source. Spectra were recorded at 25 °C using an analyzer pass energy of 30 eV, an X-ray power of 50 W, and under an operating pressure of 10–9 mbar. Thermogravimetric analysis (TGA) was carried out with a TGA550 instrument working under nitrogen at a heating rate of 10 °C min−1. The presence of the capping agent on the SPION-NC surface, and the elimination of CTAB, were confirmed by Fourier transform infrared spectroscopy (FTIR, Thermo Fisher Scientific) in transmission mode using the KBr pellet method, prepared by blending 2 mg of sample with 100 mg of KBr. The iron content in the samples was determined by ICP measurements by using an ICP-MS instrument equipped with an Agilent 7900 mass detector. The hydrodynamic sizes were measured using dynamic light scattering (DLS) in water using the Zetasizer Nano ZS90 system (Malvern, UK). Fluorescence spectroscopy (FP-8300, Jasco) was employed to follow the MB release using an excitation wavelength of 600 nm. Magnetization measurements were performed at room temperature using a vibrating sample magnetometer (VSM) (PPMS, DynaCool, Quantum Design). A capillary tube was filled with the sample, the weight of the sample was recorded, followed by the insertion of the capillary directly into the VSM. To obtain the magnetization curves, the magnetic field was varied up to ±5 kOe. X-ray powder diffraction (XRD) was carried out using a Bruker D8 Advance diffractometer equipped with a monochromatic Cu Kα source at 40 kV and 40 mA. Patterns were collected in steps of 0.02° (2θ) over the angular range 20–60° (2θ), with an acquisition time of 3 s per step.Results and discussion
Monodisperse SPION nanoclusters (SPION-NCs) were prepared by an MW-assisted polyol method to quickly obtain an eco-friendlier product while retaining its promising properties. The Fe3+ was partially reduced in the presence of EG, which acted as solvent and reductant simultaneously, and sodium acetate as an alkali source.11,20 For the stabilization of the primary particles, sodium citrate was chosen because its three carboxylate groups have strong affinity for the Fe3+ ions, which favors the binding of the citrate to the surface of the iron oxide nanocrystals and prevents them from growing into larger particles.20 Also, the citrate molecules provide an electrostatic repulsive force between the NPs, but they aggregate into larger secondary structures (nanoclusters) to minimize their surface energy.10,11 Among the different options described in the literature for the use of capping ligands in conventional synthesis of SPION clusters, we have chosen to use citrate in our MW-assisted procedure for two main reasons: the high water dispersibility of the clusters formed (due to the presence of citrate groups on the surface)34 and the biocompatible character conferred by the citrate groups, which makes them particularly interesting for biomedical applications.20Microwave-assisted synthesis
The effect of various reaction parameters on the formation of SPION-NCs was studied, including reaction temperature, sodium citrate concentration, MW ramping time, and aging time. The conditions used for various samples are presented in Table 1.Previously, we reported on SPION-NCs synthesized via conventional and MW-assisted heating methods using biomolecules as capping agents, along with their thorough characterization.32 In this work, experiments were conducted to identify the conditions for achieving the highest population of SPION-NCs near 200 nm via MW-assisted heating, thoroughly examining factors including reaction temperature, citrate concentration, ramping time and aging time. The SPION-NCs displayed a Gaussian size distribution, in contrast to the log–normal trend that is commonly observed for nanocrystals synthesized via colloidal chemistry.35 A series of SEM micrographs, DLS plots and size distribution histograms obtained from SEM micrographs are presented in Fig. S1 to S5.†Table 1 summarizes the experimental conditions, with the results detailed in Table S1,† where NC sizes determined from DLS and Gaussian-multimodal fittings are presented. NCs undergo dynamic agglomeration and separation during DLS, resulting in larger cluster sizes than those observed in SEM micrographs. Therefore, we have based our discussion mainly on the size determined from the SEM micrographs through Gaussian fits.
To study reaction temperature, experiments at 180, 200, 220, and 250 °C were conducted with fixed citrate content, and varying ramping and aging times (Exp. A, B, F, G, L, M). No clusters formed below 220 °C. At 220 °C, large clusters reached a high population (70%), but these decreased in size and population (36%) at 250 °C. It is likely that the carbonaceous component surrounding the primary particles is influenced by the elevated temperatures, leading to its partial elimination (Fig. S1†). For the effect of citrate content, four experiments were conducted at 220 °C, with a 10 min ramping time, and 20 min aging time (Exp. H, I, J, K). As the citrate content increased from 0.114 g to 0.454 g, large clusters formed with increasing proportions from 50% to 66% (Fig. S2†). A further increase in citrate reduced the cluster size and increased the proportion of smaller NCs significantly (70%). Another control experiment was performed for the synthesis performed at 20 min ramping and 20 min aging times, with the citrate content doubled (Exp, O, R). As citrate increased from 0.454 g to 0.908 g, the size and proportion of large clusters formed decreased from 70% (213 nm) to 37% (140 nm) (Table S1†). Citrate plays a role in holding the NPs together, and beyond a certain amount, it increases the repulsion between the NPs, adversely influencing the size of NCs. This observed trend follows that of limited ligand protection (LLP),36 where the surface of the NPs is not completely covered with the capping agent, making them unstable, and the formation of clusters is more favorable. However, if there is too much capping agent, the NPs are stabilized, and their aggregation is avoided due to the sufficient ligand protection (SLP) process.36
The reaction time (ramping and aging) influences the shape and size dispersion of the SPION-NCs. To evaluate the effect of aging time on nanocluster size we performed two sets of experiments, where we selected a citrate content of 0.454 g, a reaction temperature of 220 °C, and a ramping time of (i) 1 minute (Exp. C, D, E) and (ii) 20 min (Exp. N, O, P). First, the results of 1 min aging will be presented, followed by 20 min aging. At 10 minutes, large clusters dominated (51%). Extending aging to 20 minutes increased the cluster size and their proportion (66%), while cluster size increased with a further increase to 30 minutes, whereas the proportion of smaller NCs increased (79%) (see Fig. S3†). Samples aged for 20 min showed the same trend. At 10 min, large clusters dominated (55%). Extending aging to 20 min increased the cluster size and their proportion (70%), while cluster size and the proportion of large NCs decreased (50%) after 30 min (see Fig. S4†). Synthesis of the material with a longer aging time resulted in an increase in augmented surface roughness. The observations align with previous findings regarding temperature optimization, indicating that prolonged aging periods and/or elevated temperatures may lead to the partial elimination of the carbonaceous component. Examining ramping time (1, 10, and 20 min) at 220 °C with a fixed citrate content and 20-minute aging (Exp. D, J, O), the cluster size steadily increased, with an increased proportion of large clusters (70%) at 20 min ramping (Fig. S5†). The progress of the nucleation process to obtain the primary particles is fast, but aggregating these particles to produce clusters is slower. Increasing the ramping time gives enough time for the particles to aggregate to obtain a higher population of larger clusters. Size evaluation results based on the Gaussian-multimodal fits are graphically summarized in Fig. 2, and the DLS results are presented in Fig. S6.† The largest NC size in high proportion is obtained under the conditions of 220 °C, 0.454 g citrate, and 20-minute ramping and aging times (Fig. 2c, Exp. O), which was then selected for scale-up and further studies.
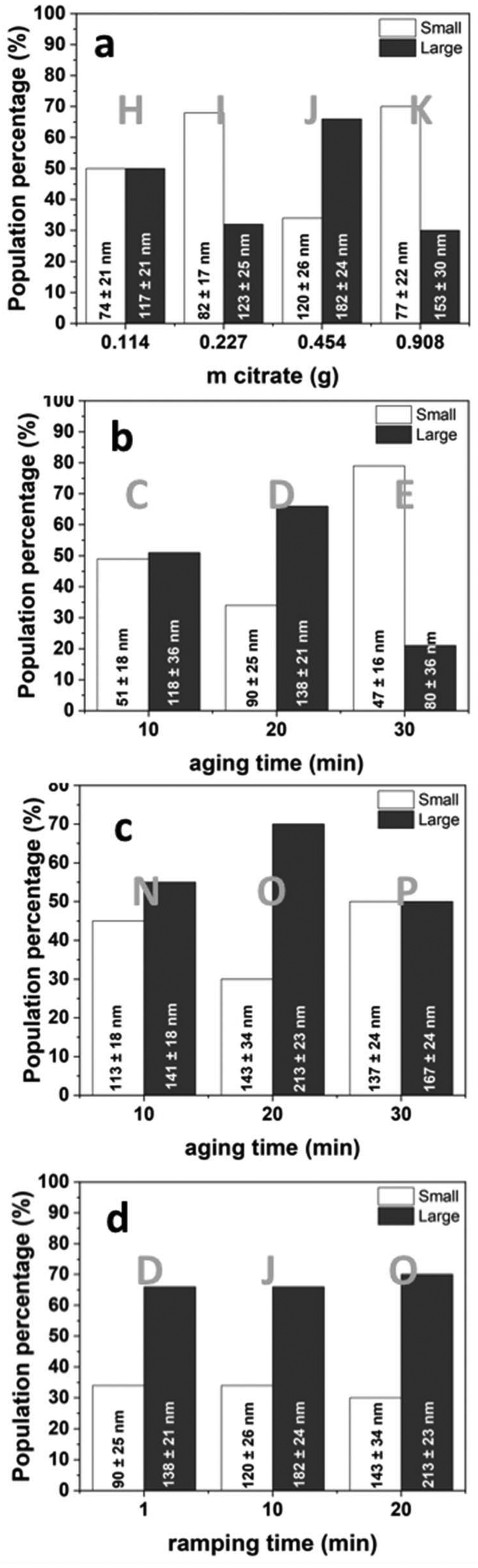 | ||
| Fig. 2 Size evolution of SPION-NCs, obtained from Gaussian fits, for the MW-assisted syntheses performed at 220 °C, with different reaction parameters – changing one parameter at a time: (a) amount of citrate, (b) aging time for 1 min and (c) 20 min ramping time, and (d) ramping time. Experimental details are summarized in Table 1. | ||
The SPION-NCs displayed a bimodal distribution. The causes of a bimodal particle size distribution are likely multifactorial and could involve factors like differences in nucleation and growth kinetics. Nucleation of SPIONs and nanocluster formation are two distinct stages in the formation of SPION-NCs, where citrate ions play a significant role in the latter process. The exact balance between stabilizing and aggregating effects of citrate ions could give rise to two different cluster populations: smaller clusters formed through limited aggregation and larger clusters formed through extensive aggregation. Alternatively, during the thermolysis process, the system may undergo kinetic trapping, where the small clusters form first and become trapped in a metastable state before fully aggregating into larger clusters. If the particles undergo partial agglomeration (forming smaller clusters) before fully aggregating (forming larger clusters), the system can end up with two distinct populations of clusters. This process might be enhanced by slow diffusion due to the high viscosity of ethylene glycol, leading to clusters that do not fully reach equilibrium in a single process.
The FT-IR spectra of the SPION-NCs reveal absorption bands at 1633 and 1380 cm−1 (Fig. 3) associated with carboxylate, indicating the presence of carboxyl groups. Thermogravimetric analysis (Fig. S7†) showed a weight loss of about 15.3% in the 100–850 °C range; the sample presents high organic matter content. XPS spectra obtained for C 1s (Fig. S8a†) was in accordance with the presence of a non-negligible amount of organic matter. Then, deconvolution of the carbon spectrum showed three contributions that can be assigned to C–C aliphatic carbon (285.2 eV), C–O (286.5 eV), and carboxylate groups (288.7 eV).34,37,38 The partial thermal degradation of the organic components was not complete, and the carbonaceous component that maintains the link between primary NPs retains an important proportion of carboxylic groups from the citrate. The total carbon content in the cluster was 56% aliphatic carbons generated during the synthesis by partial degradation of the reagents. It was estimated that approximately 21% and 23% of the citrate and EG groups, respectively, have been preserved. The XPS spectrum corresponding to the Fe 2p region was subjected to deconvolution, resulting in the identification of six distinct signals, as illustrated in Fig. S8b.† The bands observed at 710.6 and 724.1 eV can be attributed to the 2p3/2 and 2p1/2 states of Fe2+ species, respectively. The signals observed at binding energies of 711.9 and 726.1 eV can be attributed to the 2p3/2 and 2p1/2 states of the Fe3+ species, respectively. The remaining two peaks of lower intensity, at 718.6 eV and 732.6 eV, were attributed to satellite peaks. The determined molar proportion of Fe3+/Fe2+ was 2.1, a value that is very close to the stoichiometric value of magnetite.34,38,39 The XPS spectrum signals for Fe confirmed the success in obtaining the magnetite phase with high purity. Finally, the XPS O 1s spectrum (Fig. S8c†) was consistent with spectra of Fe and C. Deconvolution of the spectrum reveals three signals at 530.3, 531.6, and 532.5 eV, which can be attributed to O2− anions (present in magnetite), OH groups and carboxylate group oxygens, respectively.20 The XRD pattern also corroborates the formation of magnetite (Fig. S9†). Although typical peaks are clearly discernible, their relatively low intensity and broad width suggest the presence of nanocrystals. The HRTEM images (Fig. 4 and Fig. S10†) also reveal that the NC is constituted by primary particles of around 8–9 nm. This value range is very similar to that determined from the XRD data through the application of the modified Scherrer equation (10.3 ± 0.5 nm), which is indicative of the nearly single-crystalline character of the primary magnetite particles.6
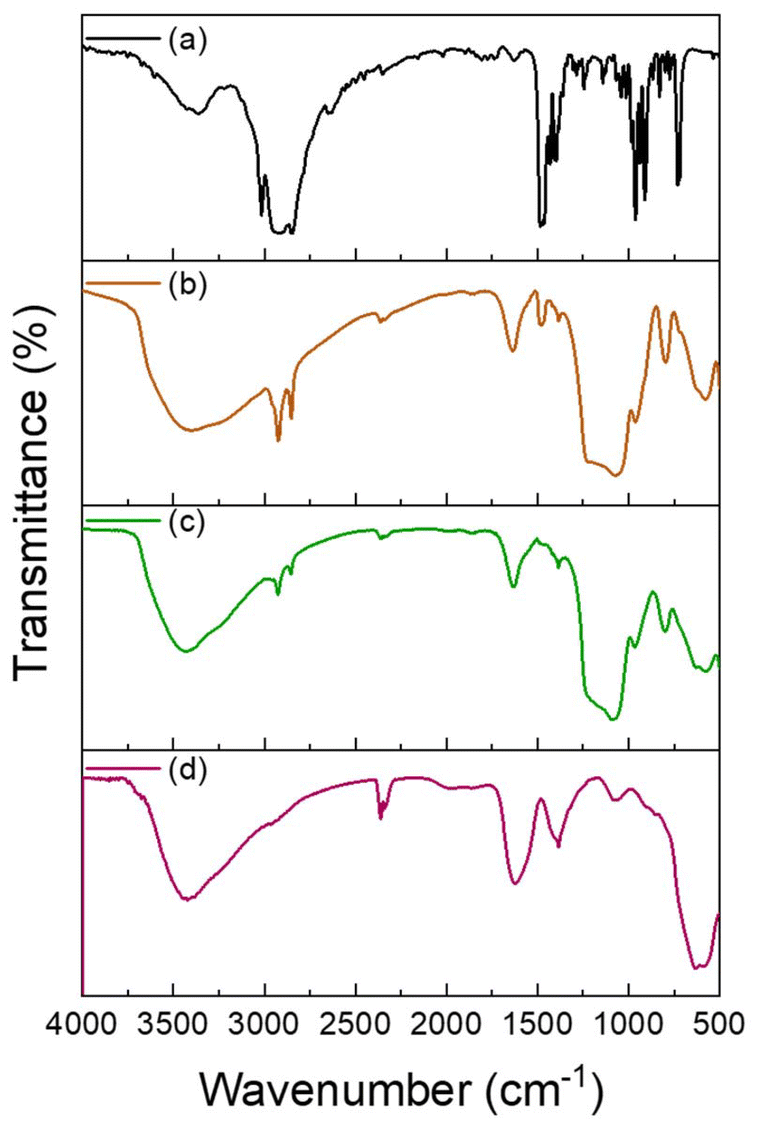 | ||
| Fig. 3 FTIR spectra of (a) CTAB, and SPION-NCs@MS (b) before and (c) after the extraction of CTAB, and (d) SPION-NCs. | ||
 | ||
| Fig. 4 HRTEM of (a) SPION-NCs and (b) SPION-NCs coated with amorphous silica. (c) SPION-NCs@MS, and (d) STEM mapping of SPION-NCs@MS particles. | ||
Mesoporous silica coating
The SPION-NC synthesized according to Exp O (see Table 1) was used for the experiments from hereon. The NCs were covered with amorphous silica to prevent the SPION-NC core from oxidation in the subsequent steps. There are several works reporting on the use of silica coating on various types of NPs, placing silica coating as a cornerstone in a wide array of applications.40 For example, iron oxide NPs coated with a thin silica shell (Fe3O4/SiO2 NPs) showed lower dissolution in cell cultures as compared to bare Fe3O4 NPs.41 A modified Stöber method using EA as the base was utilized for this purpose.32 In our case, we reduced the amount of TEOS and EA in order to prepare NCs with a thinner siliceous layer. Then, this layer was covered with an MS layer using the “atrane method”,28,29 which combines the atrane complexes as inorganic hydrolytic precursors and the CTAB as a supramolecular template. Both layers were obtained in a mixture of water and ethanol, which provides the conditions for obtaining a layer with minimal surface roughness and a conformal coating.30 There are differences in the water/ethanol ratio used for each layer synthesis. In the case of the first layer, silica formed due to the condensation of hydrolyzed TEOS on the surface of SPION-NCs. Water is used as a reagent, so its volume is smaller than that of ethanol. In the other case, the MS layer grows thanks to the interaction between the anionic silica oligomers and cationic surfactants. In this case, the water is used as a reagent and a solvent; for that reason, its volume is higher than that of ethanol. In the atrane method, TEAH3 was used as a chelating agent, to obtain the silatrane complex, and as a basic catalyst for silica hydrolysis and condensation. The atrane method allows all the reactions involved in the process (hydrolysis, condensation, nucleation, and growth) to occur in a homogeneous medium. This ensures parameters like agitation, reactor size, etc., have less influence in the process, so the reproducibility of the synthesis is increased.29The onion-like morphology of the final nanostructure (SPION-NCs@MS) is shown in Fig. 4, from the cluster (Fig. 4a) to the amorphous silica layer (Fig. 4b) and, finally, the MS layer (Fig. 4c). All the clusters presented a well-defined spherical morphology with uniform silica layers with thickness of 12 ± 2 nm for the amorphous silica and 27 ± 2 nm for the MS. The ordered nature of the primary magnetite NPs can be seen in Fig. S10.† The radial growth of the mesopores in the outermost layer can be seen in Fig. 4c. The radial organization of mesopores had also been observed when other rigid templating agents were coated with MS using the atrane method and with similar concentrations of reagents. The onion-type modular organization was also evident in the STEM and mapping images (Fig. 4d). The core was observed to present a high content of iron, and a siliceous cover was also present. Furthermore, oxygen was present throughout the volume of the particles, as expected. The clusters were characterized in terms of magnetization as their core property (Fig. 5). The uncoated SPION-NCs showcased a saturation magnetization of 65.5 emu g−1 magnetite when considering only the weight of inorganic content (Fig. 5a), corresponding to about 71% of the bulk magnetization of magnetite. The clusters exhibited limited coercivity (Hc = 30.04 Oe), indicating slightly ferrimagnetic character, possibly due to interactions between the NPs. The magnetic diameter was estimated as 8.3 nm by fitting the experimental curve with the Langevin function.42 The estimated magnetic domain size is slightly smaller than the crystallite size from XRD data, which is ascribed to the presence of a magnetically dead layer on the nanocrystals. The onion-like coated SPION-NCs also showed the same trend in magnetic behavior, and the same coercivity as the pristine SPION-NCs, indicating weakly ferrimagnetic character (Fig. 5b), with an expected decrease in saturation magnetization, owing to the added magnetically inert layers (i.e. amorphous and mesoporous silica).32 Normalized to the overall weight, the magnetization gradually decreases from 54.0 emu g−1 (clusters with citrate capping), to 42.5 emu g−1 (amorphous silica-coated clusters), and finally to 28.1 emu g−1 after mesporous silica coating (SPION-NCs@MS). The elimination of CTAB from the sample was investigated by FTIR (Fig. 2). The CTAB presents strong bands at 3012, 2947, 1483, 1431, and 960 cm−1 (CH3–N+), 2913, and 2832 cm−1 (C–H stretching), and 1393 cm−1 (–CH3 bending).43 The same signals were present in the sample before the extraction, which also displayed the signals corresponding to the silica (1069 cm−1)44 and the iron oxide (577 cm−1).45 After the extraction of CTAB, the signal corresponding to the ammonium group, which was revealed in the sample before the extraction with the band at 1484 cm−1,43 were absent. Removal of CTAB via a simple ion exchange process has also been confirmed by the FTIR analysis of a calcined mesoporous silica sample alone. The data presented in Fig. S11† indicate that there are no residues of CTAB (typical signal at 1483 cm−1 due to (CH3–N+) groups) in the FTIR spectrum for the ion-exchanged sample. The removal of the cationic surfactant inevitably entails the introduction of positive charges derived from the Na+ cations, as illustrated in Fig. 4d. These findings substantiate that the template elimination process is a simple ion exchange. To facilitate the exchange of CTA+ for Na+, a water–alcohol mixture (hydroalcoholic medium) comprising a high ethanol concentration has been used to promote the dissolution of NaCl and the stabilization of the surfactant CTACl. It is well known that the critical micelle concentration value of an alkylammonium halide-type surfactant exhibits a notable increase in the presence of ethanol,46,47 which serves to enhance the extraction of CTA+ and, consequently, the emptying of the mesopores. As far as we know, it is surprising that such a simple and intuitive method has not been described in the literature.48 We can thus conclude that the extraction was satisfactorily executed. The signals corresponding to the carbonaceous structure are present, but their intensity is reduced. As the SPION-NCs were synthesized using citrate as the capping agent, its corresponding bands (3420 cm−1 (–OH), 2963 cm−1 (C–H stretching), 1617 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and 1387 cm−1 (–CH3 bending)) were preserved on the obtained clusters. In short, STEM and spectroscopic studies allow us to determine from a chemical point of view the composition of the different layers of the onion-like SPION-NCs@MS.
O), and 1387 cm−1 (–CH3 bending)) were preserved on the obtained clusters. In short, STEM and spectroscopic studies allow us to determine from a chemical point of view the composition of the different layers of the onion-like SPION-NCs@MS.
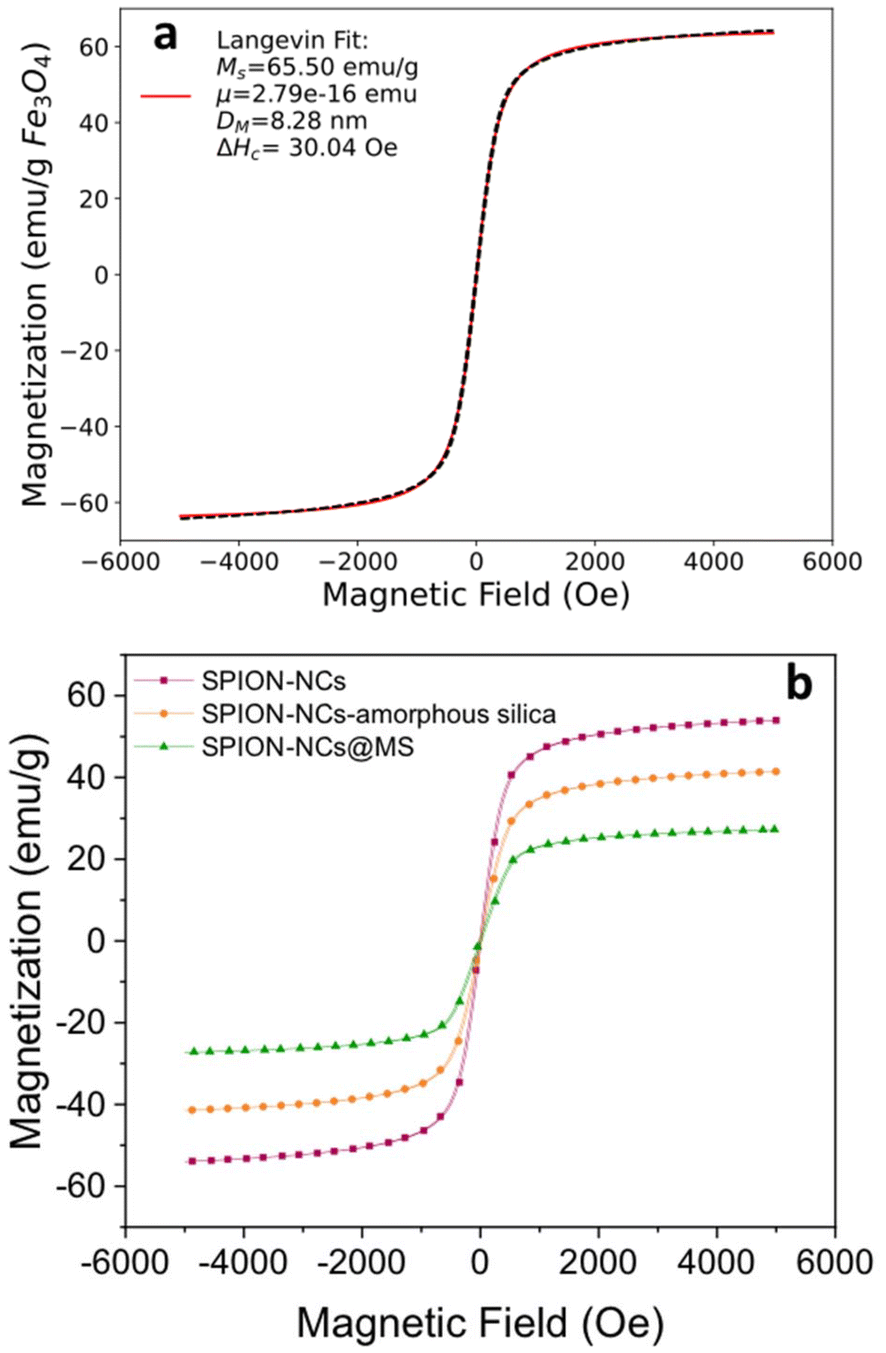 | ||
| Fig. 5 Magnetization curves of (a) the SPION-NCs normalized to the weight of Fe3O4 and (b) the SPION-NCs alone and after coating with amorphous silica, followed by mesoporous silica (MS) normalized to overall sample weight. | ||
The nitrogen adsorption–desorption isotherms and the pore size distribution (Fig. 6) evidenced the porous nature of the sample. The isotherm (Fig. 6a) displayed one abrupt adsorption step at an intermediate relative pressure (0.3 < P/P0 < 0.7), characteristic of type IV isotherms and can be related to the capillary condensation of the N2 inside the pores generated by the micelle. Also, a second adsorption step was observed at a high relative pressure value (P/P0 > 0.9), which was attributed to the capillary filling of the voids formed when the spherical SPION-NCs@MS particles are stacked. It is important to note that the external MS layer is only 27 nm thick and that the core contains heavier elements, such as iron. Given these factors, the BET area and pore volume values obtained (223.6 m2 g−1 and 0.22 cm3 g−1, respectively) are significant. However, it is evident that these values are lower than those of mesoporous siliceous Stöber particles. The pore size distribution (Fig. 6b) shows a maximum centered at 3.13 nm and a significant shoulder expanded to 6 nm. The value of the maximum was observed to correlate well with the size of the surfactant micelles. The existence of a wide distribution in pore size indicates the generation of larger mesopores during the ion exchange stage. The thickness of the MS layer amplified the surface inhomogeneities formed during the ion exchange, which may not be evident in fully mesoporous particles. This increase in mesopore size can also be explained by the partial degradation that occurs in MS when treated in saline solutions. Therefore, PBS or another medium has been used to increase the mesopore size and associated pore volume.49,50 Thus, the NaCl treatment allows us to simultaneously extract the surfactant and generate mesopore expansion using a simple protocol (without using complex surfactants or toxic reagents, avoiding multi-step processes).49 The final result was a composite with a magnetic core and an outer layer containing expanded mesopores, which is attractive for the uptake/delivery of biomolecules.
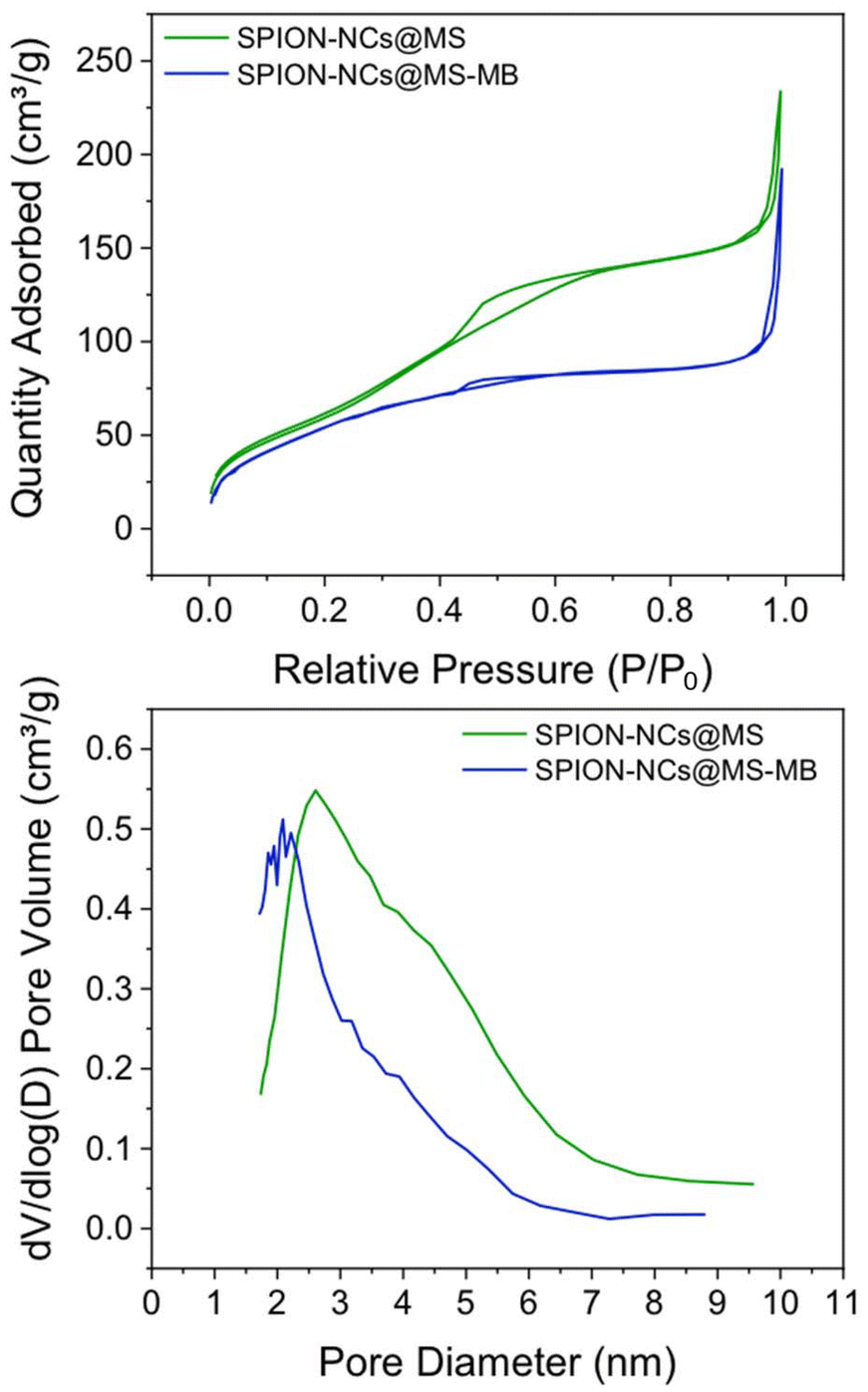 | ||
| Fig. 6 (a) Nitrogen adsorption–desorption isotherms and (b) pore size/diameter distribution of SPION-NCs@MS and SPION-NCs@MS-MB particles. | ||
There is a clear interest in these types of core–shell materials for biomedical applications.25 The possibility of modulating the properties (nature of the magnetic core, thickness of the mesoporous layer to modify its loading capacity, possibility of incorporating molecular gates to control drug release, etc.) makes these NPs excellent platforms for the design of potential theranostic materials. In any case, and although silica and magnetite are non-toxic compounds, it must be recognized that there is a long way to go between the preparation of these particles and their clinical use. However, from a chemical point of view, a first test is to check that the subsequent modifications to be carried out (loading of drugs or functionalization of the external surface) do not alter the nature of the material. The stability or possible degradation of the material may be due not only to its composition but also to the preparation method used. For example, magnetite cores or porous silica shells may show marked differences in stability depending on the specific preparation method used.
Methylene blue loading
In order to achieve this objective, a preliminary investigation has been conducted into the loading of a model molecule and its subsequent functionalization, as well as the release of the resulting product. In this case, the particles were loaded with methylene blue (MB) as a drug mimic molecule, and the surface was functionalized with MPTMS. The MB was chosen for two principal reasons: the first is its fluorescent character, making it easy to follow its release by fluorescence spectroscopy; second, it is a hydrophilic molecule making it easier to load into the hydrophilic porous silica. Once MB has been trapped inside the pores, its release under conditions that mimic biological environments can be studied using a PBS buffer. The quantity of MB that is encapsulated in the pores per gram of sample is estimated as 6.87 μg MB per mg. A typical UV-absorption spectrum of MB and a detailed procedure for its calibration curve are presented in the ESI, in Fig. S12† and the preceding discussion.The MB-loaded particles (SPION-NCs@MS-MB) retain their spherical morphology. A TEM micrograph showing a large number of SPION-NCs@MS-MB particles is presented in Fig. S13.† It is important to note that the hydrodynamic size of the MB-impregnated particles (SPION-NCs@MS-MB) is not significantly different from that for the mesoporous silica-coated (SPION-NCs@MS) particles, as shown in Fig. S14.† The TEM micrograph (Fig. S15a†) demonstrates that the external mesoporous layer, its thickness, and the radial arrangement of the mesopore system remain unaltered. The images obtained from the STEM and mapping techniques are indistinguishable from those observed for the initial material (Fig. S15b†). The sole distinction is the appearance of a small quantity of homogeneously distributed S, directly correlated with the uptake of MB and the pores closing following treatment with MPTMS. Also, there was a smaller quantity of homogeneously distributed N, which was directly correlated with the uptake of MB. On the other hand, the XRD pattern shows typical signals of magnetite (Fig. S16†), indicating that the magnetic core is not altered. It can, therefore, be concluded that the dye charge and functionalization have not affected the core particles, yielding similar crystallite sizes to those in the original NCs. Furthermore, magnetic characteristics of the samples were studied at various stages. VSM analysis of the SPION-NCs@MS, SPION-NCs@MS-MB samples, along with the MB loaded sample 24 h after incubation in PBS, is presented in Fig. S17.† It is observed that the saturation magnetization normalized to the overall sample weight decreases upon MB loading. This is due to the increasing weight provided by the MB and pore closure process, adding more non-magnetic components. The magnetization of the SPION-NCs@MS-MB sample after 24h incubation in PBS showed the same magnetic behavior, with a further decrease in the saturation magnetization, which is attributed to the partial dissolution and/or oxidation of the magnetic core, leading to the reduction in the amount of magnetic constituents. The isotherm (Fig. 6a) shows one adsorption step at an intermediate relative pressure value (0.3 < P/P0 < 0.7), which is less abrupt than that in the sample analyzed before the MB loading. This is an appropriate test for the incorporation of MB and the blocking of mesopores with MPTMS. As previously observed, a second adsorption step occurs at a high relative pressure value (P/P0 > 0.9), resulting from the formation of gaps between particles during the packing process. Given that the microparticle size remains consistent before and after the incorporation of MB, this second adsorption step in the isotherm is not affected compared to the that for the initial material. The most notable difference is in the pore size distribution (Fig. 6b), which has shifted to a smaller diameter (less than 5 nm) and a reduced pore volume (0.15 cm3 g−1). These findings align with the introduction of the dye and partial pore closure with the MPTMS. The SPION-NCs@MS particles synthesized using the atrane method for the formation of the external porous layer have demonstrated resilience to typical charge and functionalization treatments, maintaining their structural integrity after stirring periods of up to 24 h. MB release over time is shown in Fig. 7. Rapid release, corresponding to about <6% of loaded MB, is observed within the first 5 hours, probably due to weakly retained MB molecules on the external surface, followed by a slower, sustained release over time.
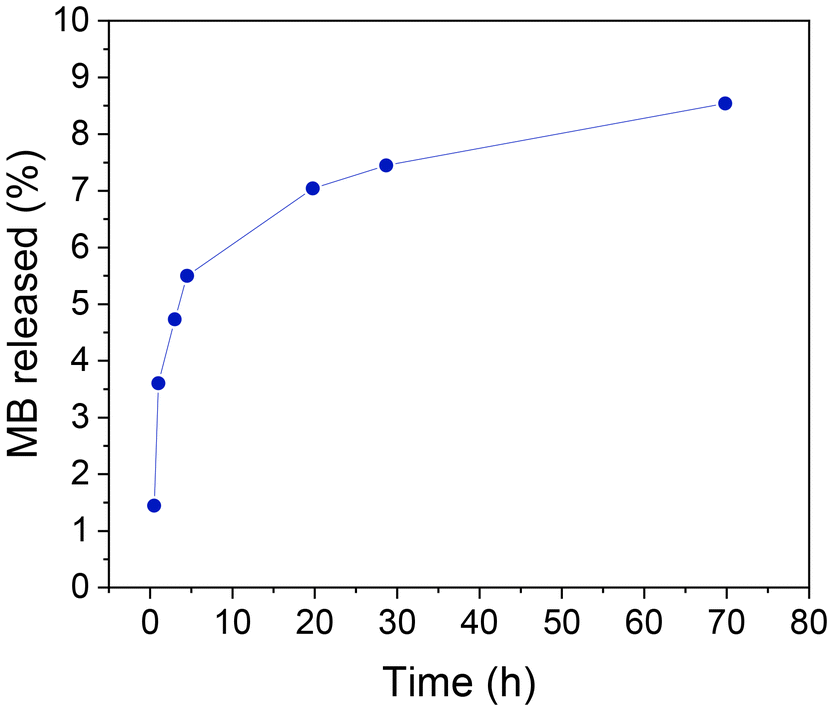 | ||
| Fig. 7 Methylene blue (MB) release from SPION-NCs@MS-MB particles as a function of time as determined by measuring the fluorescence intensity of the medium after magnetic decantation. | ||
Conclusions
In this work, we presented the design, synthesis, and characterization of onion-like particles with a magnetic core and a capped mesoporous silica shell, which can be filled with bioactive molecules. The SPION nanoclusters, with their high magnetization-per-particle, were obtained by an MW-assisted polyol process, by studying the effect of various reaction parameters, including reaction temperature, citrate content, ramping time and aging time. The synthesis time was drastically reduced to <40 min compared to that of previous works, while keeping the superparamagnetic properties and near-spherical morphology of the particles. It has been demonstrated that the organic content of the SPION-NCs, which originates from the citrate capping, has a strong influence on cluster formation. The amorphous silica layer was made using a modified sol–gel method in order to isolate the particles from the next synthetic steps. Afterwards, this layer was covered with mesoporous silica by the atrane method, whereupon a highly homogeneous conformal shell was obtained. The CTAB was then removed by a simple chemical extraction, and the pore size was slightly increased by the degradation of the silica by Na+. This expanded pore is attractive for the loading and release of molecules. Finally, as a proof of concept, the particles were loaded with methylene blue, and its release was studied in PBS. The particles presented in this work possess high potential for theranostics applications.Author contributions
M. Dolores Garrido: conceptualization, investigation, formal analysis, methodology, writing – original draft and writing – review & editing. Bejan Hamawandi, J.F. Serrano-Claumarchirant, Adem B. Ergül: investigation, formal analysis, and methodology. Giovanni Marco Saladino: conceptualization, writing – review & editing. M. Dolores Marcos: investigation. José Vicente Ros-Lis: conceptualization, project administration, supervision, validation, funding acquisition, writing – review & editing. Pedro Amorós: conceptualization, project administration, supervision, validation, funding acquisition, writing – review & editing. Muhammet S. Toprak: conceptualization, project administration, supervision, validation, funding acquisition, writing – original draft and writing – review & editing.Data availability
The data supporting this article have been included as part of the manuscript and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Knut and Alice Wallenberg Foundation (KAW 2016.0057) and the project PID2021-126304OB-C43 funded by MCIN/AEI/10.13039/50110001103 3/ and by the European Regional Development Fund - A way of doing Europe. We also thank the AGROALNEXT program supported by MCIN with funding from the European Union NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana grant number EUAGROALNEXT/2022/065. M. Dolores Garrido thanks the University of Valencia for a pre-doctoral fellowship from the program “Atracció de Talent” (UV-INV_PREDOC21-1914738) and the economic support received for research stays abroad associated with it.References
- P. Zhang, Y. Li, W. Tang, J. Zhao, L. Jing and K. J. McHugh, Nano Today, 2022, 42, 101335 CrossRef CAS.
- Y. Cai, X. Chen, J. Si, X. Mou and X. Dong, Small, 2021, 17, 2103072 Search PubMed.
- H.-K. Kim, G. H. Lee and Y. Chang, Future Med. Chem., 2018, 10, 639–661 CrossRef CAS PubMed.
- P. Mathieu, Y. Coppel, M. Respaud, Q. T. Nguyen, S. Boutry, S. Laurent, D. Stanicki, C. Henoumont, F. Novio, J. Lorenzo, D. Montpeyó and C. Amiens, Molecules, 2019, 24, 4629 CrossRef CAS PubMed.
- S. M. Dadfar, D. Camozzi, M. Darguzyte, K. Roemhild, P. Varvarà, J. Metselaar, S. Banala, M. Straub, N. Güvener, U. Engelmann, I. Slabu, M. Buhl, J. van Leusen, P. Kögerler, B. Hermanns-Sachweh, V. Schulz, F. Kiessling and T. Lammers, J. Nanobiotechnol., 2020, 18, 22 CrossRef CAS PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- A. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS.
- S. Liu, B. Yu, S. Wang, Y. Shen and H. Cong, Adv. Colloid Interface Sci., 2020, 281, 102165 CrossRef CAS PubMed.
- M. Nedylakova, J. Medinger, G. Mirabello and M. Lattuada, Adv. Colloid Interface Sci., 2024, 323, 103056 CrossRef CAS PubMed.
- W. Wang, B. Tang, B. Ju and S. Zhang, RSC Adv., 2015, 5, 75292–75299 RSC.
- A. Demortière, P. Panissod, B. P. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. Bégin-Colin, Nanoscale, 2011, 3, 225–232 RSC.
- Z. Xiao, L. Zhang, V. L. Colvin, Q. Zhang and G. Bao, Ind. Eng. Chem. Res., 2022, 61, 7613–7625 CrossRef CAS.
- A. Gandon, C. C. Nguyen, S. Kaliaguine and T. O. Do, Can. J. Chem. Eng., 2021, 99, 479–488 CrossRef CAS.
- G. Xi, C. Wang and X. Wang, Eur. J. Inorg. Chem., 2008, 2008, 425–431 CrossRef.
- J. Ge, Y. Hu, M. Biasini, W. P. Beyermann and Y. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342–4345 CrossRef CAS PubMed.
- J. K. Stolarczyk, S. Ghosh and D. F. Brougham, Angew. Chem., Int. Ed., 2009, 48, 175–178 CrossRef CAS.
- F. Dong, W. Guo, J. Bae, S. Kim and C. Ha, Chem. – Eur. J., 2011, 17, 12802–12808 CrossRef CAS.
- J. Gao, X. Ran, C. Shi, H. Cheng, T. Cheng and Y. Su, Nanoscale, 2013, 5, 7026 RSC.
- J. Liu, Z. Sun, Y. Deng, Y. Zou, C. Li, X. Guo, L. Xiong, Y. Gao, F. Li and D. Zhao, Angew. Chem., Int. Ed., 2009, 48, 5875–5879 CrossRef CAS PubMed.
- Y. Huang, P. Li, R. Zhao, L. Zhao, J. Liu, S. Peng, X. Fu, X. Wang, R. Luo, R. Wang and Z. Zhang, Biomed. Pharmacother., 2022, 151, 113053 CrossRef CAS.
- T. Asefa and Z. Tao, Chem. Res. Toxicol., 2012, 25, 2265–2284 Search PubMed.
- M. Manzano and M. Vallet-Regí, Adv. Funct. Mater., 2020, 30, 1902634 Search PubMed.
- M. Vallet-Regí, M. Colilla, I. Izquierdo-Barba and M. Manzano, Molecules, 2017, 23, 47 CrossRef PubMed.
- A. Adam and D. Mertz, Nanomaterials, 2023, 13, 1342 CrossRef CAS PubMed.
- Q. Yue, J. Sun, Y. Kang and Y. Deng, Angew. Chem., Int. Ed., 2020, 59, 15804–15817 Search PubMed.
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Chem. Mater., 2012, 24, 4572–4580 CrossRef CAS.
- S. Cabrera, J. El Haskouri, C. Guillem, J. Latorre, A. Beltrán-Porter, D. Beltrán-Porter, M. D. Marcos and P. Amorós, Solid State Sci., 2000, 2, 405–420 CrossRef CAS.
- M. D. Garrido, M. Benítez, J. V. Ros-Lis and P. Amorós, Nano Select, 2024, 1–26 Search PubMed.
- M. D. Garrido, D. Vie, J. F. Serrano-Claumarchirant, J. El Haskouri, J. V. Ros-Lis and P. Amorós, Microporous Mesoporous Mater., 2024, 378, 113244 Search PubMed.
- M. D. Garrido, J. El Haskouri, D. Vie, A. Beltrán, J. V. Ros-Lis, M. D. Marcos, N. Moliner and P. Amorós, Microporous Mesoporous Mater., 2022, 337, 111942 CrossRef CAS.
- G. M. Saladino, R. Kakadiya, S. R. Ansari, A. Teleki and M. S. Toprak, Nanoscale Adv., 2023, 5, 1323–1330 RSC.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- A. Atrei, B. Lesiak-Orlowska and J. Tóth, Appl. Surf. Sci., 2022, 602, 154366 CrossRef CAS.
- C. G. Granqvist and R. A. Buhrman, Solid State Commun., 1976, 18, 123–126 CrossRef CAS.
- A. Narayanaswamy, H. Xu, N. Pradhan and X. Peng, Angew. Chem., Int. Ed., 2006, 45, 5361–5364 CrossRef CAS PubMed.
- T. Radu, A. Petran, D. Olteanu, I. Baldea, M. Potara and R. Turcu, Appl. Surf. Sci., 2020, 501, 144267 CrossRef CAS.
- A. Bunge, A. S. Porav, G. Borodi, T. Radu, A. Pîrnău, C. Berghian-Grosan and R. Turcu, J. Mater. Sci., 2019, 54, 2853–2875 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- C. I. C. Crucho, Appl. Mater. Today, 2024, 38, 102179 CrossRef.
- M. A. Malvindi, V. De Matteis, A. Galeone, V. Brunetti, G. C. Anyfantis, A. Athanassiou, R. Cingolani and P. P. Pompa, PLoS One, 2014, 9, e85835 CrossRef PubMed.
- G. M. Saladino, B. Hamawandi, C. Vogt, G. K. Rajarao and M. S. Toprak, Appl. Nanosci., 2020, 10, 1861–1869 CrossRef CAS.
- T. Bezrodna, G. Puchkovska, V. Styopkin and J. Baran, Thin Solid Films, 2009, 517, 1759–1764 CrossRef CAS.
- R. Ellerbrock, M. Stein and J. Schaller, Sci. Rep., 2022, 12, 11708 CrossRef CAS PubMed.
- L. Yu. Novoselova, Appl. Surf. Sci., 2021, 539, 148275 CrossRef CAS.
- W. Li, Y. C. Han, J. L. Zhang, L. X. Wang and J. Song, Colloid J., 2006, 68, 304–310 CrossRef CAS.
- W. Li, Y.-C. Han, J.-L. Zhang and B.-G. Wang, Colloid J., 2005, 67, 159–163 CrossRef CAS.
- H. Ghaedi and M. Zhao, Energy Fuels, 2022, 36, 2424–2446 CrossRef CAS.
- Y. Zhang, Q. Yue, M. M. Zagho, J. Zhang, A. A. Elzatahry, Y. Jiang and Y. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 10356–10363 CrossRef CAS PubMed.
- K. Braun, A. Pochert, M. Beck, R. Fiedler, J. Gruber and M. Lindén, J. Sol-Gel Sci. Technol., 2016, 79, 319–327 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04572f |
| This journal is © The Royal Society of Chemistry 2025 |