
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards 1D supramolecular chiral assemblies based on porphyrin–calixarene complexes†
Massimiliano
Gaeta
 *a,
Chiara M. A.
Gangemi
b,
Matteo
Barcellona
a,
Gabriele
Travagliante
a,
Marco
Milone
b,
Anna
Notti
*b,
Maria E.
Fragalà
a,
Ilenia
Pisagatti
b,
Melchiorre F.
Parisi
b,
Roberto
Purrello
a and
Alessandro
D'Urso
*a
*a,
Chiara M. A.
Gangemi
b,
Matteo
Barcellona
a,
Gabriele
Travagliante
a,
Marco
Milone
b,
Anna
Notti
*b,
Maria E.
Fragalà
a,
Ilenia
Pisagatti
b,
Melchiorre F.
Parisi
b,
Roberto
Purrello
a and
Alessandro
D'Urso
*a
aDipartimento di Scienze Chimiche, Università degli Studi di Catania, Viale A. Doria 6, 95125 Catania, Italy. E-mail: adurso@unict.it; gaetamassimiliano@libero.it
bDipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali, Università degli Studi di Messina, V.le F. Stagno d'Alcontres 31, 98166 Messina, Italy. E-mail: anotti@unime.it; chiaramariaantonietta.gangemi@unime.it
First published on 7th February 2025
Abstract
The design of functional chiral nanostructures in aqueous solution represents one of the most exciting challenges in supramolecular chemistry, offering potential applications in catalysis, sensing, and materials science. In this scenario, it has already been shown that the hierarchical step-by-step addition of porphyrins to calix[4]arene aqueous solutions yields porphyrin–calixarene supramolecular complexes with exact and tuneable stoichiometries and defined dimensionality. The present study reports the formation of novel 1D porphyrin–calix[4]arene assemblies, achieved through a hierarchical and stoichiometrically controlled self-assembly process in water using host–guest interactions between the anionic trisulfonated porphyrin, H2DPPS3, and the cationic bis-calix[4]arene, BC4. In addition, to obtain chiral 1D noncovalent assemblies, the copper(II) porphyrin, CuDPPS3, and the enantiomerically pure bis-calix[4]arenes, (R,R)- and (S,S)-BC4, were also used in aqueous solution. The stepwise formation of linear noncovalent and chiral assemblies, based on porphyrin–calixarene complexes, was demonstrated by a number of different techniques such as: UV-vis spectroscopy, circular dichroism (CD), resonance light scattering (RLS) and scanning electron microscopy (SEM), revealing precise stoichiometries, sequence, dimensionality and induction of chirality.
Introduction
In recent decades, the synthesis of large chiral supramolecular architectures derived from the self-assembly of simple molecular synthons has gained considerable interest owing to the increasing use of these species in various fields,1–7 ranging from light-harvesting8,9 and sensing10,11 to catalysis12,13 and photodynamic processes.14 In this context, porphyrins represent a relevant class of organic compounds exhibiting unique photophysical properties and synthetic versatility.15–19 Furthermore, under specific experimental conditions, porphyrins can self-organize into well-defined nanostructures with tuneable sizes, morphologies and properties.20–23 However, obtaining discrete porphyrin-based supramolecular complexes in aqueous medium requires careful selection of the reactants and rigorous control of the reaction conditions (e.g., the sequence of the reactants addition) to prevent undesired aggregation phenomena driven by electrostatic interactions, π–π stacking, and other weak forces.24–31 To this end, the choice of an appropriate molecular template is crucial for the successful synthesis of well-ordered self-assembled architectures.32–38In a pioneering study published in 1994,39 M. C. Drain and J.-M. Lehn introduced an innovative one-pot synthetic method that took advantage of kinetically inert metal–ligand coordination between platinum or palladium ions and the pyridyl moieties of meso-substituted phenyl–pyridyl porphyrins. This approach allowed for rapid and efficient formation of porphyrin arrays with controlled stoichiometry and geometry, achieving yields close to 100%. Their work paved the way for metal ion-mediated synthesis of porphyrin assemblies with precise structural control.
More recently, host–guest chemistry has emerged as a compelling approach to achieve precise control on the stoichiometry and sequence of these assemblies.40–46 In this context, the host molecule replaces the metal ion, contributing to the structural and electronic features of the resulting complexes. Cyclodextrins, for example, have been employed to form binary host–guest complexes with porphyrins, initially synthesised to mimic natural enzymes like myoglobin in aqueous environments.47
The ability to form stable complexes in aqueous solutions is an intrinsic advantage of host–guest chemistry, enabling the simultaneous use of water-soluble and hydrophobic molecular components. In this regard, calixarenes have emerged as promising molecules capable of templating discrete porphyrin arrays in aqueous solutions with defined stoichiometry, sequence and high stability.36,48
Studies have demonstrated that the interaction between meso-tetrakis(4-N-methylpyridyl)porphyrin and a tetracarboxylic p-sulfonate calix[4]arene results in a significant hypochromism and broadening of the Soret band, indicating strong host–guest interactions. By Job plot analyses and spectrophotometric titrations, the formation of discrete species at specific stoichiometric ratios has been revealed, highlighting the control of the assembly sequence of porphyrin units as well.43,49 The stability and kinetic inertness of these supramolecular complexes have also been confirmed by diffusion NMR and dynamic light-scattering experiments.50 In addition it has been demonstrated that by using ditopic51,52 or tritopic53 bis- or triscalix[n]arene it is possible to control the dimensionality of the assembly, hierarchically forming 2D and 3D noncovalent architectures of considerable size.
Such control over the stoichiometry, sequence and dimensionality of porphyrin assemblies is achieved by following rigid hierarchical rules and exploiting electrostatic interactions as driving force and solvophobic interactions as “non-covalent glue” to stabilize the complexes. Noteworthy, an electronic communication between the external components and the central core of the supramolecular system has been detected, opening new avenues for the development of functional materials with tailored optical and electronic properties. Previously, by exploiting the electronic communication between porphyrins, assembled via the calixarene-templated approach, we have induced chirality in the resulting porphyrin–calix[4]arene assemblies. In addition, we were also able to increase the distance between the central and peripheral porphyrin, demonstrating in such a way the presence of a long-range chirality transfer effect.54,55
A further step towards a more complete understanding of the above-mentioned self-assembly processes involves the use of new molecular building blocks.
In the present study, we demonstrate that our approach, based on the hierarchical step-by-step addition of porphyrin to a calix[4]arene solution, allows the design of new architectures with desired properties. To this end, we have selected as building blocks the ditopic bis-calix[4]arene, 1,6-bis[5,11,17,23-tetrakis(trimethylammonium)-25,26,27-tripropoxy-28-(oxy)calix[4]arene]hexane octachloride52 (BC4, Fig. 1b), and the novel anionic trisulfonated porphyrin, H2DPPS3, (Fig. 1a) to assemble 1D noncovalent structures with tuneable properties. In addition, to induce chirality onto 1D porphyrin/bis-calixarene complexes we have synthetised a new chiral bis-calix[4]arene, N,N′-bis(5,11,17,23-tetraamino-25,26,27-tripropoxy-28-[3-carbonylpropoxy]calix[4]arene)-1,2-diaminocyclohexane octahydrochloride in both enantiomeric forms ((1R,2R)- and (1S,2S)-BC4 respectively, Fig. 1c). With this study, we aim to contribute to the development of new chiral functional materials based on porphyrin systems, expanding their applicability in the field of molecular devices, catalysis, and materials science.
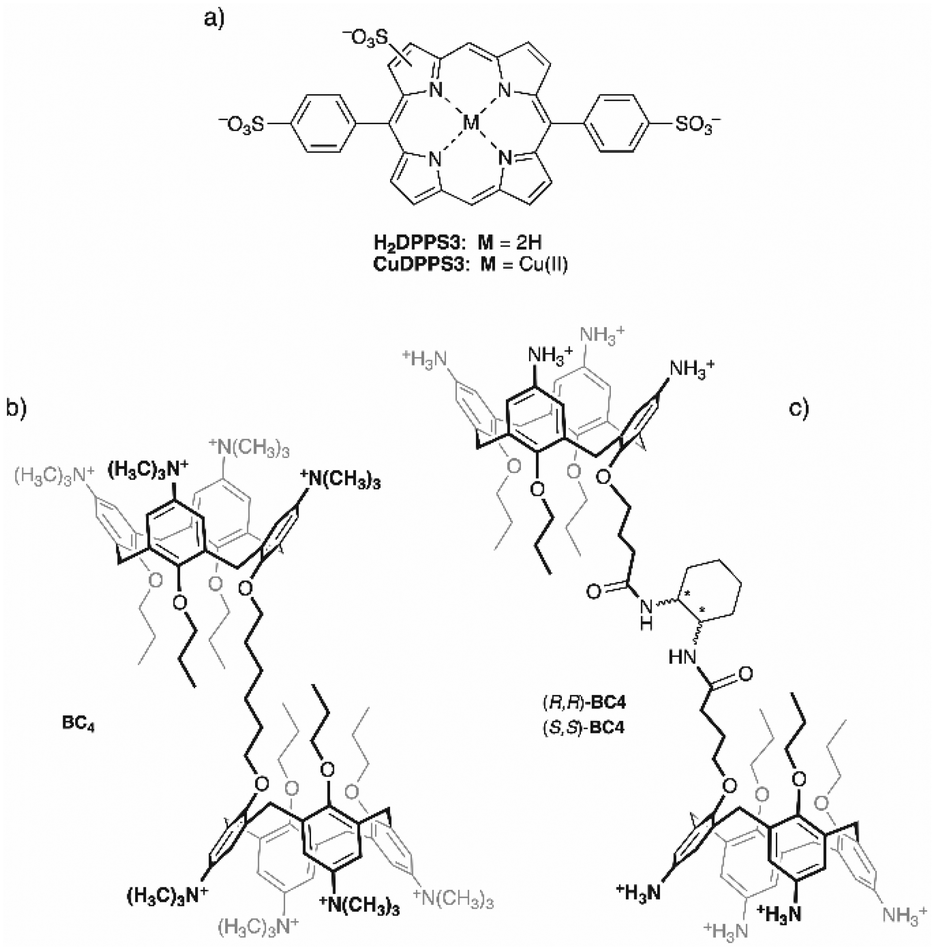 | ||
| Fig. 1 Molecular structures of (a) H2DPPS3 and its Cu(II) derivative, (b) BC4, and (c) (S,S)- or (R,R)-BC4. | ||
Experimental section
Commercial reagent grade chemicals were used as received without any further purification. Solvents were dried by standard methods.Trisulfonated porphyrin H2DPPS3 was synthesised according to a slight modification of Ribo's procedure,56 using 5,15-diphenylporphine57 as the starting material. The detailed synthetic procedure is reported in the ESI.†
The copper(II) derivative of H2DPPS3, CuDPPS3, was synthesised from the corresponding free-base by heterogeneous metal-insertion from copper(II) oxide (CuO) in water, according to a previous report.58
BC4 was available from previous studies.52
The water soluble chiral bis-calix[4]arenes (R,R)-BC4 and (S,S)-BC4 were synthetised starting from the known acid chloride 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 59 according to Scheme 1. Calix[4]arene 1 was reacted in turn with enantiomerically pure (1S,2S)-(+)- and (1R,2R)-(–)-1,2-cyclohexanediamine to produce C2-symmetric enantiomerically pure octanitro-bis-calix[4]arene diamides (R,R)-8NO2-BC4 and (S,S)-8NO2-BC4, in good yields. The reduction of the octanitro enantiomers afforded the corresponding octaamino derivatives (R,R)-8NH2-BC4 and (S,S)-8NH2-BC4, which were directly converted to the corresponding hydrochlorides (R,R)-BC4 and (S,S)-BC4 (see the ESI for details†).
59 according to Scheme 1. Calix[4]arene 1 was reacted in turn with enantiomerically pure (1S,2S)-(+)- and (1R,2R)-(–)-1,2-cyclohexanediamine to produce C2-symmetric enantiomerically pure octanitro-bis-calix[4]arene diamides (R,R)-8NO2-BC4 and (S,S)-8NO2-BC4, in good yields. The reduction of the octanitro enantiomers afforded the corresponding octaamino derivatives (R,R)-8NH2-BC4 and (S,S)-8NH2-BC4, which were directly converted to the corresponding hydrochlorides (R,R)-BC4 and (S,S)-BC4 (see the ESI for details†).
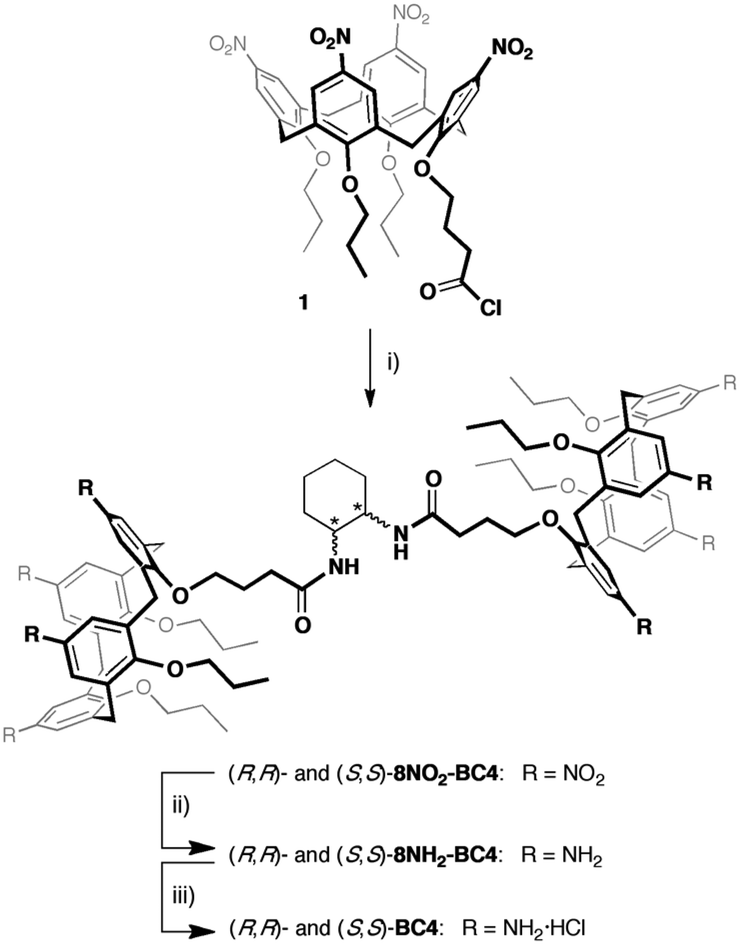 | ||
| Scheme 1 (i) (1S,2S)-(+)- or (1R,2R)-(–)-1,2-cyclohexanediamine, Et3N, CH2Cl2, 0 °C, 12 h; (ii) H2, Ni/RANEY®, THF, r.t., 18 h; (iii) HCl/dioxane. | ||
Ultrapure water at room temperature (18.2 MΩ cm, TOC 1 ppb), obtained from the PURELAB Flex 3 system (Elga Veolia company), was used to prepare all samples and stock solutions.
H2DPPS3 and CuDPPS3 stock solutions (about 4 × 10−4 M) were prepared by dissolving a given amount of the solid powder in ultrapure water. The concentrations of the porphyrin stock solutions were spectrophotometrically calculated (UV-vis in H2O) at neutral pH, by means of the corresponding molar extinction coefficients at the maximum of the Soret band: λmax(H2O)/nm 405.5 nm (ε/dm3 mol−1 cm−1 113600) for H2DPPS3, and λmax(H2O)/nm 405.5 nm (ε/dm3 mol−1 cm−1 110000) for CuDPPS3.
Sample and titration solutions were acidified or basified by HCl 6 M or NaOH 6 M, respectively.
Porphyrin/bis-calixarene complexes were obtained at room temperature by adding increasing aliquots of porphyrin (so that the concentration of the porphyrin in the titrating solution was 0.25 μM higher after each addition) to a 2.5 μM aqueous solution (at pH = 7.0 for BC4, at pH = 2.0 for (S,S)- and (R,R)-BC4) of the bis-calixarene, up to the desired molar ratio [porphyrin]/[bis-calixarene]. Each titration experiment was replicated at least three times.
UV-vis and Circular Dichroism (CD) measurements were carried out at room temperature on a JASCO V-530 spectrophotometer and a JASCO-810 spectropolarimeter, respectively. The Resonance light scattering (RLS) measurements were collected on a Jobin Yvon Horiba FL11 spectrofluorimeter. A quartz cuvette with 1 cm path-length was used in all measurements.
The values of the dissymmetry g-factor of the chiral complexes (g = Δε/ε = ΔA/A) were obtained from the corresponding CD and UV-vis data by dividing ΔA (ΔA = θ/32980, where θ is the total amplitude ellipticity in mdeg of the negative and positive component of the bisegnated band at 400 nm) by the absorbance A (in absorbance unit) of the chiral porphyrin/bis-calixarene complexes at 400 nm and a given molar ratio (2:1 and 4:1 [CuDPPS3]/[(R,R)- or (S,S)-BC4]).
The morphology of the nanosticks porphyrin/calixarene complexes was examined using Field Emission Scanning Electron Microscopy (FE-SEM, ZEISS SUPRA-55 VP). Images were captured with an electron beam energy of 15 kV. To mitigate surface charging effects, a clean silicon substrate was employed for the depositions. The films were analysed in their as-deposited state on silicon, with no additional treatments.
Results and discussion
The H2DPPS3 is a synthetic anionic porphyrin with two sulfonatophenyl substituents in trans–meso-positions and one sulfonate group directly bonded to the porphyrin ring. H2DPPS3 is highly soluble in water and its aqueous solutions shows, at basic pH (pH = 10.00), an intense Soret band at 405.5 nm and four Q-bands at 507, 543, 572 and 624 nm (Fig. 2, black traces). Nevertheless, under strong acid condition (pH = 1.50) core protonation occurs, leading to the corresponding protonated form, H4DPPS3, characterised by a red-shift of the Soret band at 420 nm along with the appearance of two Q bands at 570 nm and 614 nm (Fig. 2, red traces).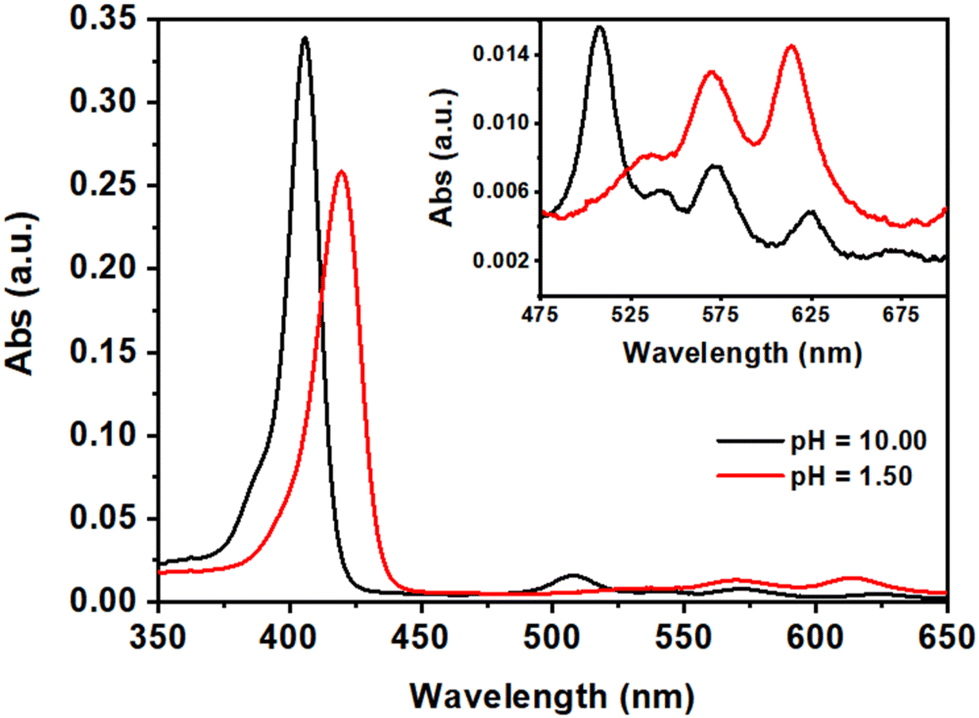 | ||
| Fig. 2 UV-vis spectra of aqueous solutions of H2DPPS3 (3 μM) at pH = 10.00 (black traces) and at pH = 1.50 (red traces). The magnification of the Q-band region is reported in the inset. | ||
Known the aggregation tendency of water-soluble porphyrins, a preliminary spectroscopic characterization of our porphyrin system was carried out to determine the optimal experimental conditions under which the system is protonated or tends to self-assemble.
To collect relevant information on the protonation equilibria and determine the pKa values of H2DPPS3, we prepared several independent aqueous solutions of H2DPPS3 (2 μM) at various pH values (by adding variable amounts of aqueous HCl or NaOH solutions) ranging from 10.00 to 0.40. This experiment, namely independent solutions experiment, allowed us to minimize the porphyrin self-aggregation effects.60
The absorption spectra of the independent H2DPPS3 solutions at different pH values (Fig. S10†) display the characteristic shape of a porphyrin core-protonation curve.60 As the pH decreases, the band at 405.5 nm, associated with the deprotonated form of H2DPPS3, undergoes a hypochromic effect, giving rise to a new band at 420 nm which is consistent with the formation of the core-protonated form of H2DPPS3 (i.e., H4DPPS3). In detail, by plotting the absorbance values at 405.5 nm and 420 nm against the pH values of each independent solution, it is possible to evaluate the pKa values of H2DPPS3 (Fig. 3). We observe only the equilibrium corresponding to the protonation step of the nitrogen core atoms of H2DPPS3, with an estimated pKa value of about 2.8. The protonation of the sulfonate groups cannot be obtained under this conditions, because it generally occurs in the presence of strong acidic solutions ([HCl] > 4 M).24,61
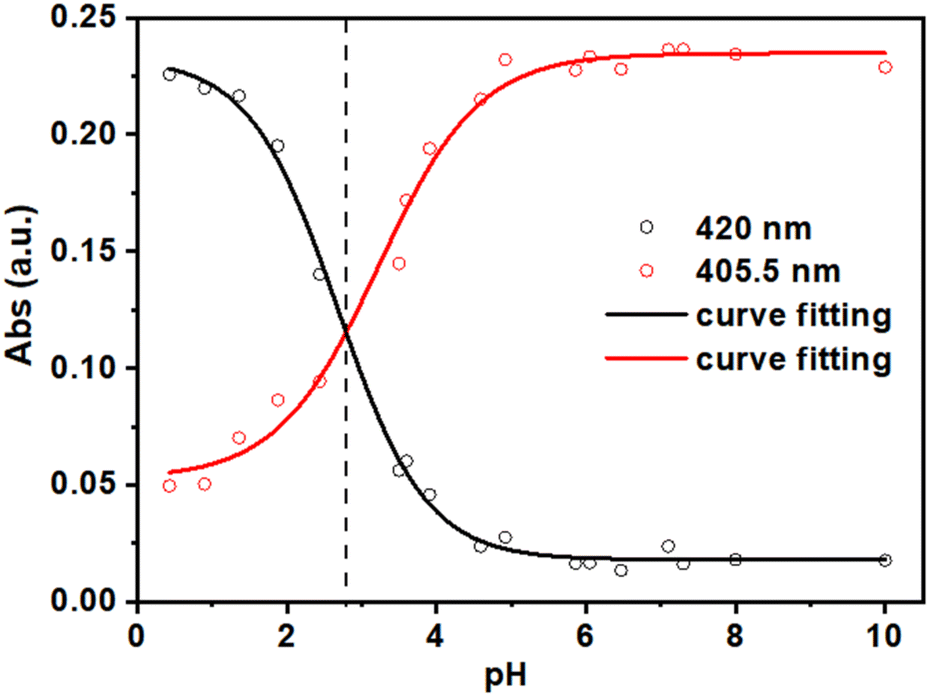 | ||
| Fig. 3 Plot and experimental fit of the absorbance values at λ = 405.5 nm (red circles) and λ = 420 nm (black circles) as function of pH values (pH = 10.00; 8.00; 7.30; 7.10; 6.47; 6.05; 5.86; 4.92; 4.59; 3.91; 3.60; 3.50; 2.44; 1.88; 1.36; 0.9) during the independent solutions experiment of H2DPPS3 (2 μM). | ||
On the contrary, by slowly lowering the pH of a H2DPPS3 solution (2 μM), in the 10.00–2.40 range, we were able to collect useful information on the porphyrin self-assembly process (Fig. S11†). During the pH titration, the absorption band of the deprotonated porphyrin (λ = 405.5 nm) decreases in intensity and it becomes gradually broader. Moreover, Rayleigh scattering effect tends to increase over the titration course. Ultimately these phenomena can reasonably be ascribed to the formation of self-aggregates during the pH titration.60
To test whether such intrinsic self-aggregation tendency of H2DPPS3 could be controlled in the presence of an appropriate templating agent, we turned our attention to BC4. Namely, a homodytopic bis-calix[4]arene (Fig. 1b) bearing eight permanent cationic trimethylammonium groups and, consequently, fully soluble in water even at neutral pH and capable, in principle, of interacting with anionic counterparts such as the sulfonated porphyrins.
To evaluate the potential of ditopic BC4 as a templating agent for the noncovalent assembly of porphyrin-based supramolecular structures, a 2.5 μM aqueous solution of BC4 at neutral pH was gradually titrated (Fig. S13†) with increasing aliquots of an aqueous solution of H2DPPS3 (0.25 μM per aliquot). The absorption data recorded over the course of the titration displays hypochromicity and a broadening of the H2DPPS3 Soret band as compared to the experiment performed in the absence of BC4 (Fig. S14†). According to our previous observations,43,49 this represents a clear-cut evidence in favour of porphyrin/bis-calixarene interactions. Moreover, our earlier reports43,49,52 revealed the importance of electrostatic and hydrophobic interactions in the formation and stabilization of porphyrin/calix[4]arene assemblies. On this ground, we can reasonably state that the formation of porphyrin–calix[4]arene host–guest complexes is primarily driven by strong electrostatic interactions between the cationic moieties of BC4 (host) and the anionic meso-phenyl sulfonate groups of H2DPPS3 (guest). The additional sulfonate group present on one of the pyrrole moiety of H2DPPS3 increases the water solubility and improves the charge balance, contributing to the formation and stabilization of such discrete complexes. Finally, the host–guest complexes are further stabilized by π–π stacking between the aromatic cores of the porphyrins and by solvophobic forces.
Several studies on other water-soluble calix[4]arenes,43,49 bis-calix[4]arenes,52,55 and tris-calix[4]arenes,53 have demonstrated that discrete calixarene/porphyrin assemblies form in a stepwise, hierarchical fashion, as evidenced by distinct spectral changes in absorption or emission. More specifically, the formation of complexes or assemblies with well-defined stoichiometry are consistently marked by specific break-points in a plot of the absorbance values of the porphyrin Soret band vs. the [porphyrin]/[calixarene] ratio. On a titration curve, these break-points are highlighted by changes in slope greater than 10%. The presence of different slopes indicates distinct assemblies in solution, each characterised by its own molar extinction coefficient. Noteworthy, the occurrence of break-points confirms that the species formed are not in equilibrium with each other; otherwise, a continuous straight line would be observed throughout the titration experiment.62
In analogy with other examples reported in the literature, the self-assembly process of H2DPPS3 and BC4 can be most effectively analysed by plotting the porphyrin absorbance values at 405.5 nm against the [H2DPPS3]/[BC4] ratio (Fig. 4).
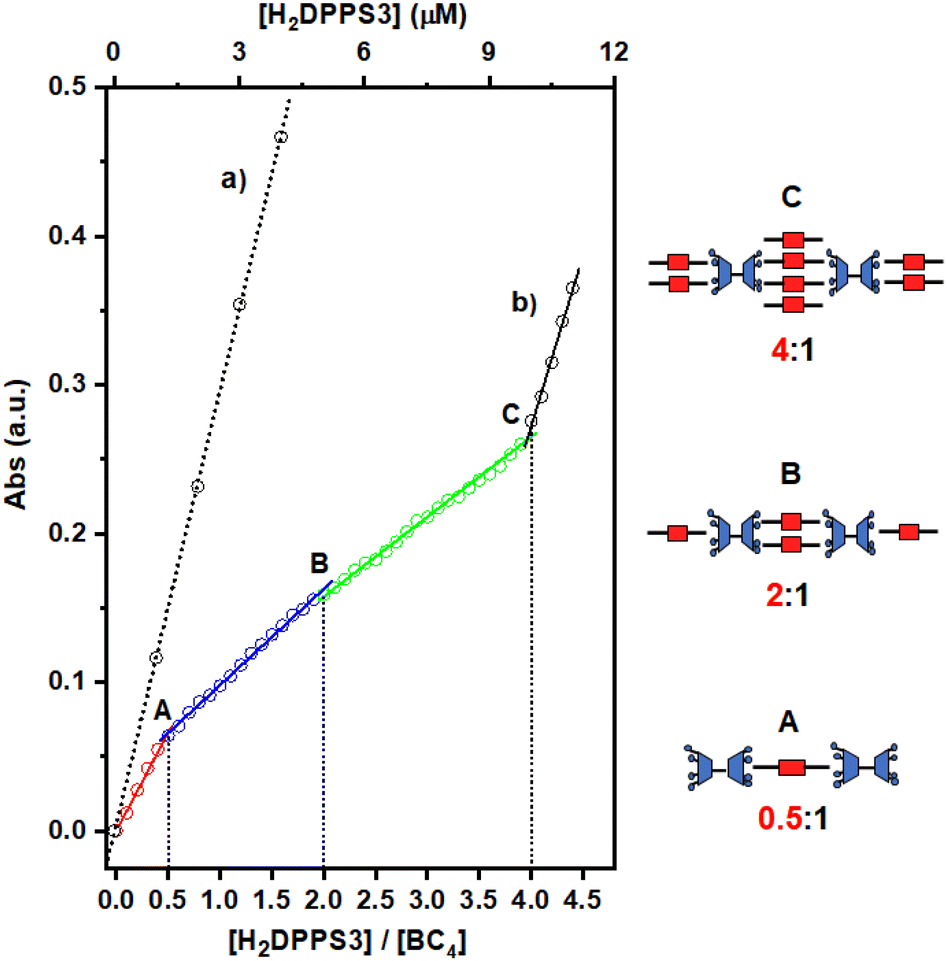 | ||
| Fig. 4 Variation in the absorbance of the H2DPPS3 Soret band (λ = 405.5 nm) observed upon: (i) increase of the porphyrin concentration in water at pH = 7.0 (dotted black trace labelled as (a)); (ii) portion-wise addition (0.25 μM) of H2DPPS3 to a 2.5 μM aqueous solution of BC4 at pH = 7.0 (multi-coloured trace labelled as (b)). Break-points are labelled as A, B, and C. The cartoons sketched on the right hand-side represent the likely structures of the complexes A, B and C, formed at the corresponding break-points. | ||
The titration plot reveals that the assembly process proceeds under stoichiometric control up to a H2DPPS3 concentration of 10 μM. This is unambiguously demonstrated by the presence of several break-points (labelled as A, B, and C in Fig. 4, trace b), which correspond to H2DPPS3/BC4 complexes with precise stoichiometries—specifically, 0.5:1, 2:1, and 4:1 (refer to Fig. 4, trace b).‡ These observations, coupled with the hypochromicity and broadening of the Soret band, indicate the presence of strong host–guest interactions.
As the titration progresses beyond the addition of the fourth equivalent of porphyrin (i.e., for [H2DPPS3] > 10 μM), the absorbance sharply increases and becomes almost identical to that observed for H2DPPS3 on its own (compare trace (a) with the last segment of trace (b) in Fig. 4). These findings indicate that the excess of porphyrin molecules now present in the solution are no longer interacting with the supramolecular complex.
Such porphyrin/bis-calixarene complexes (Fig. 4) could further self-assemble to generate discrete 1D nanostructures. To promote the linear growth of porphyrin/bis-calix[4]arene assemblies, a solution of the 2:1 H2DPPS3/BC4 complex (structure B in Fig. 4) was initially treated with BC4 (up to 5 μM) and then, after 1 hour and in a step-wise manner, with H2DPPS3 (up to 26.25 μM) to produce a 10.5:2 H2DPPS3/BC4 complex where the interacting negative and positive charges of the porphyrin and the bis-calix[4]arene are roughly balanced. Finally, approximately 200 μL of an aqueous solution containing the 10.5:2 H2DPPS3/BC4 complex were drop-cast onto a silicon substrate. After a slow evaporation in the air, the substrate surface was subjected to SEM investigation (Fig. 5a and b). A low-magnification micrograph shows the presence of nanosticks on the silicon substrate surface (Fig. 5a). The darker appearance of the nanosticks is likely due to the lower conductivity of these nanostructures compared to that of the plain silicon substrate. Poorly conductive samples accumulate charges and, as a result, the signal output is reduced. Close examination of the micrograph at high magnification indicates the presence of nanosticks of similar shape and size (Fig. 5b), with an average length and width of 3.3 ± 0.8 μm and 0.7 ± 0.2 μm, respectively. The length and width distribution of the nanosticks is shown in Fig. 5c and d, respectively.
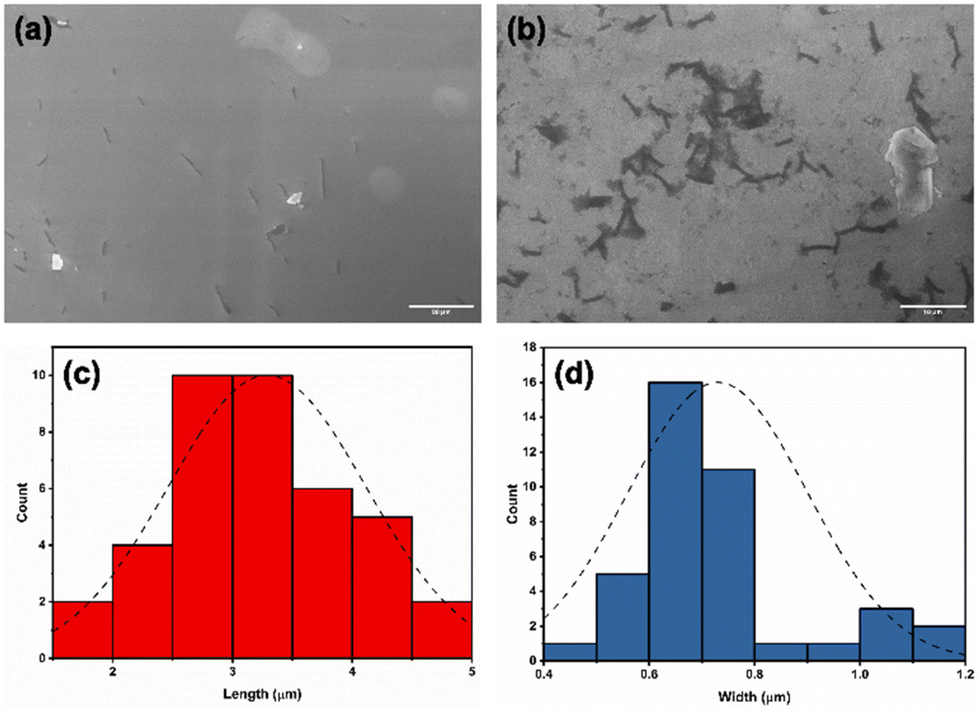 | ||
| Fig. 5 SEM micrographs, at (a) low (scale bar = 20 μm) and (b) high magnification (scale bar = 10 μm), of nanosticks formed upon deposition of H2DPPS3/BC4 complexes onto a silicon substrate surface. Nanostick length (c) and width (d) distribution bars. Distribution curves are shown as black dashed lines. | ||
Given that the sizes of H2DPPS3 and BC4 are about 0.002 μm, the length of the 2:1 complex should be around 0.01 μm, it is reasonable to assume that an average of about 300 2:1 species are assembled to form a nanostick.
The successful formation of such porphyrin/bis-calix[4]arene assemblies not only demonstrates that self-assembly is hierarchically controlled and takes place in a stepwise manner, but also provides an opportunity to transfer electronic properties to the whole complex. In this context, recent studies have shown that stereogenic agents are able to induce chirality in porphyrin–calixarene assemblies, indicating electronic communication among the porphyrins in both 2D and 3D assemblies.53–55 For this reason, the use of novel enantiomerically pure bis-calix[4]arenes, namely (R,R)-BC4 and (S,S)-BC4 (Fig. 1c), as compared to the achiral BC4 (Fig. 1b) might induce chirality to the entire supramolecular complex.
In water, bis-calix[4]arenes (R,R)- and (S,S)-BC4 are soluble only under acid conditions upon protonation of the amino groups at the wider rims. For instance, acid aqueous solutions (pH = 2.0) of these chiral bis-calix[4]arenes (50 μM) exhibit two absorption peaks at about 275 nm and 300 nm (Fig. S16†). Consequently, an optical activity appears in their absorption range (see the inset of Fig. S16†).
Moreover, the spectrophotometric pH titration in aqueous solution (in the 1.50–10.70 pH range) confirmed that, at low pH values (i.e., pH = 2.0), (R,R)- and (S,S)-8NH2-BC4, are converted to their fully protonated octaammonium forms (R,R)- and (S,S)-BC4, respectively (pKa ≈ 5, see Fig. S17†).
Thus, aqueous solutions of (R,R)- or (S,S)-BC4 are stable only under acid conditions (pH < 4) and this evidence suggests that a free-base porphyrin, such as H2DPPS3, cannot be employed for the formation of supramolecular complexes. Under acidic conditions, H2DPPS3 would firstly undergo core-protonation, which would subsequently trigger self-assembly. On the contrary, the use of a metal-derivative of H2DPPS3 would likely “protect” the porphyrin by avoiding the protonation of its core. Since demetallation of Cu(II) porphyrin derivatives hardly occurs in acid aqueous solutions, our attention was turned to CuDPPS3 (Fig. 1a), easily obtained from H2DPPS3via heterogeneous metal-insertion reaction (see the Experimental section for details). Compared to free-base H2DPPS3, CuDPPS3 displays a similar Soret band at 405.5 nm and two Q-bands at 532 nm and 570 nm (Fig. S18†). The remarkable stability of CuDPPS3 under acid conditions (pH = 2.0) is shown in Fig. S19† in which a 3 μM aqueous solution of copper(II) porphyrin was kept at pH = 2.0 for 24 hours: the CuDPPS3 absorption spectrum recorded at pH 2.0 is almost identical to that recorded at pH 10.0 (Fig. S19†).
The UV-vis spectrophotometric titration of a 2.5 μM aqueous solution (pH = 2.0) of (R,R)- or (S,S)-BC4 with increasing amounts of CuDPPS3 (0.25 μM per aliquot) causes, on the porphyrin Soret band (λ = 405.5 nm), a progressive hypochromic effect modulated by the relative stoichiometry of the two complementary components present in solution (Fig. S20†), thus revealing host–guest interactions similar to those observed for the H2DPPS3/BC4 complexes.
Moreover, a close inspection of the titration spectra obtained by plotting the CuDPPS3 absorbance vs. the [CuDPPS3]/[(R,R)- or (S,S)-BC4] ratio (Fig. 6) reveals the formation of complexes of discrete stoichiometry. The straight black dotted line (Fig. 6, trace a) accounts for the absorbance (at λ = 405.5 nm) of CuDPPS3 on its own (Fig. S21†) at increasing concentrations, whereas, the multi-coloured line (Fig. 6, trace b) refers to the absorbance measured, at 405.5 nm, upon titration of a 2.5 μM (R,R)- or (S,S)-BC4 solution with increasing amounts of CuDPPS3 (0.25 μM) at pH = 2.0.
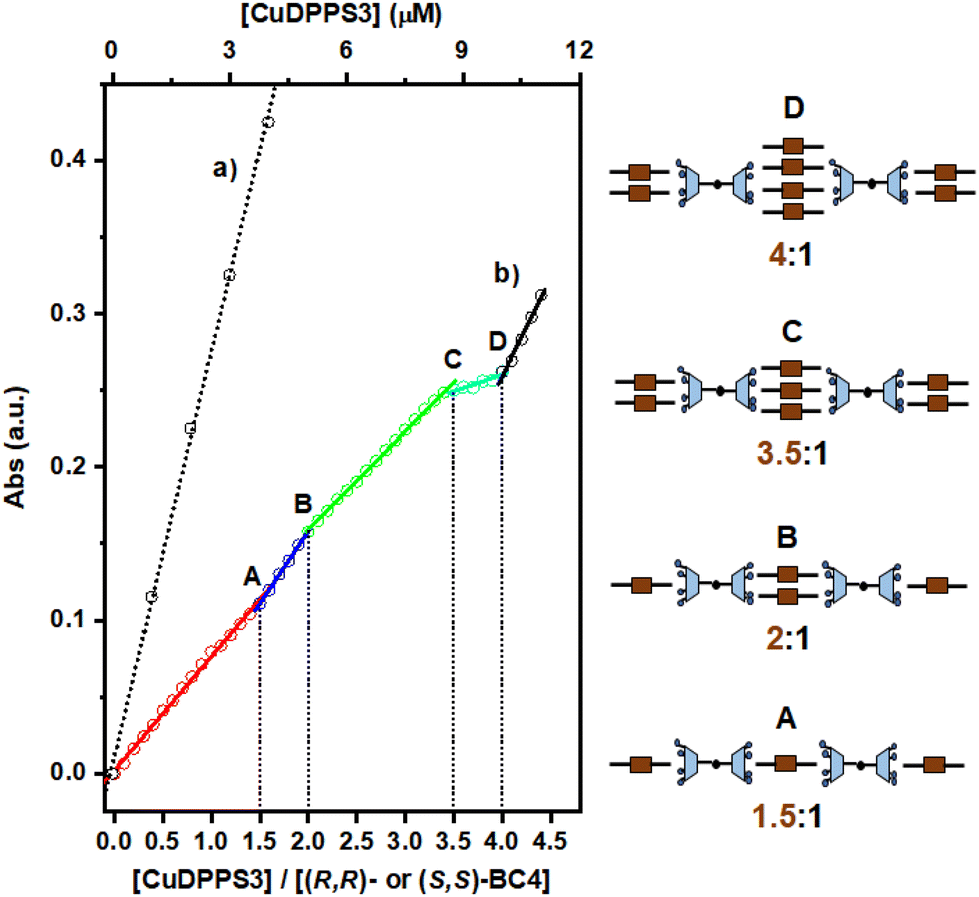 | ||
| Fig. 6 Variation in the absorbance of the CuDPPS3 Soret band (λ = 405.5 nm) observed upon: (i) increase of the porphyrin concentration in water at pH = 2.0 (dotted black trace labelled as (a)); (ii) portion-wise addition (0.25 μM) of CuDPPS3 to a 2.5 μM aqueous solution of (R,R)- or (S,S)-BC4 at pH = 2.0 (multi-coloured trace labelled as (b)). The break-points are labelled as A, B, C, and D. The cartoons sketched on the right hand-side represent the likely structures of the complexes A, B and C, formed at the corresponding break-points. | ||
Stepwise formation of four discrete species – namely a 1.5:1, 2:1, 3.5:1 and 4:1 ([CuDPPS3]/[(R,R)- or (S,S)-BC4]) complexes is indicated by the presence of four distinct break-points (labelled in Fig. 6 as A, B, C, D, respectively) characterised by a change in the titration-curve slope (at least 10%). Again, after addition of the fourth equivalent of porphyrin to (R,R)- or (S,S)-BC4, the slope of the titration curve becomes almost identical to that detected in the case of CuDPPS3 on its own (compare trace (a) with the black segment of trace (b)), indicating that once the 4:1 (CuDPPS3/(R,R)- or (S,S)-BC4) complex has quantitatively formed, the porphyrin molecules added in excess remain unbound in solution.
In analogy with the H2DPPS3/BC4 system (Fig. 4), in the presence of (R,R)- or (S,S)-BC4 and CuDPPS3 discrete complexes are also formed with similar structure (Fig. 6).§
In addition, the presence of an equilibrium similar to the one depicted in Fig. S15,† may also affect the stoichiometries of the assemblies formed.
In agreement with the UV-vis titration studies, the formation of discrete porphyrin/bis-calix[4]arene complexes is also confirmed by the enhancement of the resonance light scattering (RLS) response observed in the case of 1.5:1, 2:1, 3.5:1 and 4:1 [CuDPPS3]/[(R,R)- or (S,S)-BC4] solutions (Fig. S22†). The chiral nature of (R,R)- and (S,S)-BC4 offers a straightforward method to transfer chirality to the complexes by exploiting chiral induction processes. The step-wise formation of specular CuDPPS3/(R,R)-BC4 and CuDPPS3/(S,S)-BC4 complexes was followed by monitoring the induced circular dichroism (ICD) signals (Fig. 7).
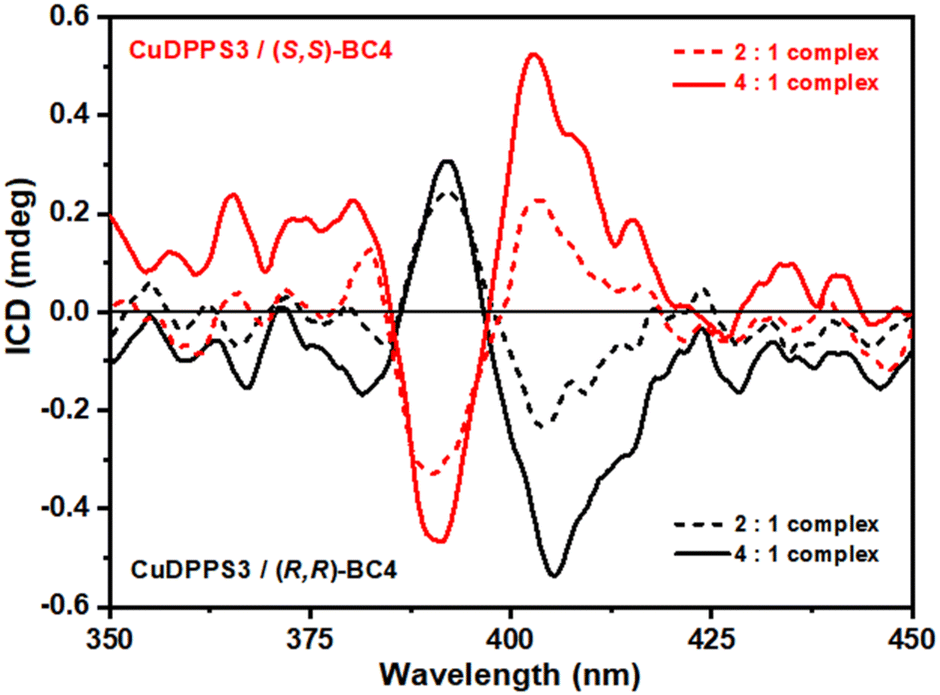 | ||
| Fig. 7 ICD spectra of aqueous solutions at pH 2.0 of (i) CuDPPS3 (5 μM dashed red trace, 10 μM solid red trace) in the presence of (S,S)-BC4 (2.5 μM), and (ii) CuDPPS3 (5 μM dashed black trace, 10 μM solid black trace) in the presence of (R,R)-BC4 (2.5 μM). | ||
No significant ICD signals were detected below 2:1 porphyrin/bis-calixarene molar ratio, likely due to a very low absorbance value. However, ICD signals appear at the 2:1 ratio, becoming even more evident at the 4:1 molar ratio (Fig. 7), highlighting the presence of an exciton coupling as well. These data are consistent with our proposed structures (see Fig. 6) where the porphyrin-stacking leads to a strong electric dipole–dipole coupling, which is also responsible for the CD couplet in the porphyrin absorbance region.64 Finally, the efficiency of chirality induction was further quantified by calculating the dissymmetry g-factor which is related to the quality of the CD rotational oscillator and uncoupled with respect to the molar concentration.65 The calculated g-factor is comparable for both enantiomeric complexes and it increases from ≈7.9 × 10−5 to ≈9.2 × 10−5 at a porphyrin/bis-calix[4]arene molar ratio of 2:1 and 4:1, respectively. This increase further confirms the efficient transfer of chirality and supports the structural model proposed in Fig. 6. At higher porphyrin/bis-calix[4]arene molar ratios, the structural arrangement of the porphyrins promotes, via π–π stacking, a stronger exciton coupling amplifying the asymmetry of the supramolecular complexes and thus their g-factor.
Conclusions
By exploiting the versatility of porphyrin–calixarene complexes we have formed chiral linear noncovalent polymer in aqueous solution. Indeed, by selecting the appropriate building blocks with suitable functionalities, it is possible to drive the self-assembly towards the formation of a given desired structure with specific characteristics. In this work we employed a tri-anionic porphyrin and two cationic bis-calix[4]arenes to take advantage of the electrostatic forces to firstly induce the self-assembly; then, the hydrophobic interactions contribute to yield kinetically inert porphyrin/bis-calixarene assemblies. Moreover, the rigid hierarchical rules apply, so as only a step-wise addition of small aliquots of porphyrin to the solution of chiral bis-calix[4]arene provides complexes with precise stoichiometry, sequence, dimensionality and chirality.Our approach shed further light on how to exploit noncovalent interactions in driving the self-assembly process and how to induce chirality through the electronic communication between external components and the central core of the supramolecular system. These data confirm the high potentiality of supramolecular chemistry and how hierarchical self-assembly allow to design functional supramolecular chiral material for several applications.
Author contributions
Conceptualization: A. D., M. G., R. P.; funding acquisition: A. D. A. N.; investigation: M. G., M. B., C. M. A. G., M. M., I. P., M. F. P., A. N., G. T.; methodology: A. D., R. P., M. E. F., M. F. P.; project administration: A. D., M. G.; resources: M. P., A. N., C. M. A. G.; supervision: A. D., M. G.; validation: M. B., M. G.; visualization; A. D., M. G.; writing – original draft: A. D., M. G.; writing – review & editing: M. F. P., A. N., R. P., M. E. F., C. M. A. G.Data availability
Data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Unione Europea – Next Generation EU, Missione 4 Componente 1 CUP E53D23010030006 PRIN 2022 2022R9WCZS and CUP E53D23020520001 PRIN 2022 PNRR P2022; UniCT for programma ricerca di ateneo UNICT 2020-22 linea 2 and Finanziamento Attività di Base della Ricerca di Ateneo (FFABR 2022) UniME.References
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- A. S. Ovsyannikov, M. N. Lang, S. Ferlay, S. E. Solovieva, I. S. Antipin, A. I. Konovalov, N. Kyritsakas and M. W. Hosseini, CrystEngComm, 2016, 18, 8622–8630 RSC.
- J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, J. Mater. Chem., 2003, 13, 2661–2670 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- D. B. Amabilino, Chem. Soc. Rev., 2009, 38, 669–670 RSC.
- M. K. Panda, K. Ladomenou and A. G. Coutsolelos, Coord. Chem. Rev., 2012, 256, 2601–2627 CrossRef CAS.
- T. S. Balaban, H. Tamiaki and A. R. Holzwarth, Top. Curr. Chem., 2005, 258, 1–38 CrossRef CAS.
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Chem. Rev., 2017, 117, 2517–2583 CrossRef CAS PubMed.
- G. Travagliante, M. Gaeta, R. Purrello and A. D'Urso, Chemosensors, 2021, 9, 204 CrossRef CAS.
- C. Deraedt and D. Astruc, Coord. Chem. Rev., 2016, 324, 106–122 CrossRef CAS.
- Z. Wang, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 15954–15955 CrossRef CAS PubMed.
- J. Tian, B. Huang, M. H. Nawaz and W. Zhang, Coord. Chem. Rev., 2020, 420, 213410 CrossRef CAS.
- H. N. Fonda, J. V. Gilbert, R. A. Cormier, J. R. Sprague, K. Kamioka and J. S. Connolly, J. Phys. Chem., 1993, 97, 7024–7033 CrossRef CAS.
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2017, 117, 2910–3043 CrossRef CAS PubMed.
- K. Ladomenou, M. Natali, E. Iengo, G. Charalampidis, F. Scandola and A. G. Coutsolelos, Coord. Chem. Rev., 2015, 304–305, 38–54 CrossRef CAS.
- U. Siggel, U. Bindig, C. Endisch, T. Komatsu, E. Tsuchida, J. Voigt and J.-H. Fuhrhop, Ber. Bunsenges. Phys. Chem., 1996, 100, 2070–2075 CrossRef CAS.
- M. Jurow, A. E. Schuckman, J. D. Batteas and C. M. Drain, Coord. Chem. Rev., 2010, 254, 2297–2310 CrossRef CAS PubMed.
- Z. El-Hachemi, J. Crusats, C. Troyano and J. M. Ribó, ACS Omega, 2019, 4, 4804–4813 CrossRef CAS PubMed.
- M. De Napoli, S. Nardis, R. Paolesse, M. G. H. Vicente, R. Lauceri and R. Purrello, J. Am. Chem. Soc., 2004, 126, 5934–5935 CrossRef CAS PubMed.
- C. J. Medforth, Z. Wang, K. E. Martin, Y. Song, J. L. Jacobsen and J. A. Shelnutt, Chem. Commun., 2009, 7261–7277 RSC.
- M. A. Castriciano, A. Romeo, V. Villari, N. Micali and L. Monsù Scolaro, J. Phys. Chem. B, 2003, 107, 8765–8771 CrossRef CAS.
- R. Zagami, M. A. Castriciano, A. Romeo, M. Trapani, R. Pedicini and L. Monsù Scolaro, Dyes Pigm., 2017, 142, 255–261 CrossRef CAS.
- M. A. Castriciano, A. Romeo, R. Zagami, N. Micali and L. Monsù Scolaro, Chem. Commun., 2012, 48, 4872–4874 RSC.
- Z. El-Hachemi, C. Escudero, O. Arteaga, A. Canillas, J. Crusats, G. Mancini, R. Purrello, A. Sorrenti, A. D'Urso and J. M. Ribo, Chirality, 2009, 21, 408–412 CrossRef CAS PubMed.
- Z. El-Hachemi, C. Escudero, F. Acosta-Reyes, M. T. Casas, V. Altoe, S. Aloni, G. Oncins, A. Sorrenti, J. Crusats, J. L. Campos and J. M. Ribó, J. Mater. Chem. C, 2013, 1, 3337–3346 RSC.
- J. M. Short, J. A. Berriman, C. Kübel, Z. El-Hachemi, J. V. Naubron and T. S. Balaban, ChemPhysChem, 2013, 14, 3209–3214 CrossRef CAS PubMed.
- M. Gaeta, R. Randazzo, C. Costa, R. Purrello and A. D'Urso, Chem. – Eur. J., 2023, 29, e202202337 CrossRef CAS PubMed.
- A. Di Mauro, R. Randazzo, S. F. Spanò, G. Compagnini, M. Gaeta, L. D'Urso, R. Paolesse, G. Pomarico, C. Di Natale, V. Villari, N. Micali, M. E. Fragalà, A. D'Urso and R. Purrello, Chem. Commun., 2016, 52, 13094–13096 RSC.
- A. Sorrenti, Z. El-Hachemi, O. Arteaga, A. Canillas, J. Crusats and J. M. Ribò, Chem. – Eur. J., 2012, 18, 8820–8826 CrossRef CAS PubMed.
- M. Gaeta, R. Randazzo, D. A. Cristaldi, A. D'Urso, R. Purrello and M. E. Fragalà, J. Porphyrins Phthalocyanines, 2017, 21, 426–430 CrossRef CAS.
- M. Gaeta, D. Raciti, R. Randazzo, C. M. A. Gangemi, A. Raudino, A. D'Urso, M. E. Fragalà and R. Purrello, Angew. Chem., Int. Ed., 2018, 57, 10656–10660 CrossRef CAS PubMed.
- A. Li, L. Zhao, J. Hao, R. Ma, Y. An and L. Shi, Langmuir, 2014, 30, 4797–4805 CrossRef CAS PubMed.
- A. D'Urso, M. E. Fragalà and R. Purrello, Chem. Commun., 2012, 48, 8165–8176 RSC.
- A. S. R. Koti and N. Periasamy, Chem. Mater., 2003, 15, 369–371 CrossRef CAS.
- N. Tuccitto, G. Trusso Sfrazzetto, C. M. A. Gangemi, F. P. Ballistreri, R. M. Toscano, G. A. Tomaselli, A. Pappalardo and G. Marletta, Chem. Commun., 2016, 52, 11681–11684 RSC.
- C. M. Drain and J.-M. Lehn, J. Chem. Soc., Chem. Commun., 1994, 2313–2315 RSC.
- J. W. Steed, D. R. Turner and K. Wallace, Core Concepts in Supramolecular Chemistry and Nanochemistry: From Supramolecules to Nanotechnology, John Wiley & Sons, 2007 Search PubMed.
- P. J. Cragg, A Practical Guide to Supramolecular Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2005 Search PubMed.
- C. D. Gutsche, Calixarenes, The Royal Society of Chemistry, Cambridge, UK, 2008 Search PubMed.
- L. Di Costanzo, R. Purrello, R. Lauceri, F. G. Gulino, V. Pavone, S. Geremia, D. Sciotto and L. Randaccio, Angew. Chem., Int. Ed., 2001, 40, 4245–4247 CrossRef CAS PubMed.
- C. Bonaccorso, G. Brancatelli, G. Forte, G. Arena, S. Geremia, D. Sciotto and C. Sgarlata, RSC Adv., 2014, 4, 53575–53587 RSC.
- C. Sgarlata, G. Brancatelli, C. G. Fortuna, D. Sciotto, S. Geremia and C. Bonaccorso, ChemPlusChem, 2017, 82, 1341–1350 CrossRef CAS PubMed.
- C. Bonaccorso, R. Migliore, M. A. Volkova, G. Arena and C. Sgarlata, Thermochim. Acta, 2017, 656, 47–52 CrossRef CAS.
- K. Kano, Colloid Polym. Sci., 2008, 286, 79–84 CrossRef CAS.
- R. De Zorzi, N. Guidolin, L. Randaccio, R. Purrello and S. Geremia, J. Am. Chem. Soc., 2009, 131, 2487–2489 CrossRef CAS PubMed.
- G. Moschetto, R. Lauceri, F. G. Gulino, D. Sciotto and R. Purrello, J. Am. Chem. Soc., 2002, 124, 14536–14537 CrossRef CAS PubMed.
- F. G. Gulino, R. Lauceri, L. Frish, T. Evan-Salem, Y. Cohen, R. De Zorzi, S. Geremia, L. Di Costanzo, L. Randaccio, D. Sciotto and R. Purrello, Chem. – Eur. J., 2006, 12, 2722–2729 CrossRef CAS PubMed.
- D. S. Guo, K. Chen, H. Q. Zhang and Y. Liu, Chem. – Asian J., 2009, 4, 436–445 CrossRef CAS PubMed.
- A. D'Urso, D. A. Cristaldi, M. E. Fragalà, G. Gattuso, A. Pappalardo, V. Villari, N. Micali, S. Pappalardo, M. F. Parisi and R. Purrello, Chem. – Eur. J., 2010, 16, 10439–10446 CrossRef PubMed.
- A. D'Urso, N. Marino, M. Gaeta, M. S. Rizzo, D. A. Cristaldi, M. E. Fragalà, S. Pappalardo, G. Gattuso, A. Notti, M. F. Parisi, I. Pisagatti and R. Purrello, New J. Chem., 2017, 41, 8078–8083 RSC.
- A. D'Urso, P. F. Nicotra, G. Centonze, M. E. Fragalà, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi and R. Purrello, Chem. Commun., 2012, 48, 4046–4048 RSC.
- M. Gaeta, G. Sortino, R. Randazzo, I. Pisagatti, A. Notti, M. E. Fragalà, M. F. Parisi, A. D'Urso and R. Purrello, Chem. – Eur. J., 2020, 26, 3515–3518 CrossRef CAS PubMed.
- R. Rubires, J. Crusats, Z. El-Hachemi, T. Jaramillo, M. López, E. Valls, J. A. Farrera and J. M. Ribó, New J. Chem., 1999, 23, 189–198 RSC.
- F. Gou, X. Jiang, R. Fang, H. Jing and Z. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 6697–6703 CrossRef CAS PubMed.
- O. Herrmann, S. H. Mehdi and A. Corsini, Can. J. Chem., 1978, 56, 1084–1087 CrossRef CAS.
- G. Gattuso, G. Grasso, N. Marino, A. Notti, A. Pappalardo, S. Pappalardo and M. F. Parisi, Eur. J. Org. Chem., 2011, 5696–5703 CrossRef CAS.
- R. Randazzo, A. Savoldelli, D. A. Cristaldi, A. Cunsolo, M. Gaeta, M. E. Fragalà, S. Nardis, A. D'Urso, R. Paolesse and R. Purrello, J. Porphyrins Phthalocyanines, 2016, 20, 1272–1276 CrossRef CAS.
- R. Zagami, A. Romeo, M. A. Castriciano and L. M. Scolaro, J. Mol. Liq., 2021, 332, 115801 CrossRef CAS.
- M. Gaeta, E. Rodolico, M. E. Fragalà, A. Pappalardo, I. Pisagatti, G. Gattuso, A. Notti, M. F. Parisi, R. Purrello and A. D'Urso, Molecules, 2021, 26, 704 CrossRef CAS PubMed.
- C. J. Kingsbury and M. O. Senge, Coord. Chem. Rev., 2021, 431, 213760 CrossRef CAS.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- N. Berova, K. Nakanishi and R. W. Woody, Circular Dichroism: Principles and Applications, Wiley-VCH, New York, NY, USA, 2nd edn, 2000 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04288c |
| ‡ The absence of break-points at the 1:1- and 3:1-(H2DPPS3/BC4) ratios could probably be explained with the presence of a simultaneous equilibrium between two species with the same stoichiometry but different arrangement. For instance, in the 1:1-(H2DPPS3/BC4) complex two porphyrin could occupy the central core or, alternately, one porphyrin could be located between two bis-calixarene molecules while the second is bound to one of the external cavity of a bis-calixarene (Fig. S15†). |
| § Notably, in the latter case the titration curve does not show break-points at 0.5, 1.0 and 3.0 CuDPPS3/(R,R)- or (S,S)-BC4 ratio. Since copper(II) porphyrin derivatives tend to preserve a square planar arrangement of the core,63 this behaviour may be reasonably due to substantial differences between the two bis-calixarenes used (Fig. 1). |
| This journal is © The Royal Society of Chemistry 2025 |