
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanosized core–shell bio-hybrid microgels and their internal structure†
Pia
Lenßen
 a,
Rebecca
Hengsbach
b,
Anne
Frommelius
b,
Samira
Cammeraat
a,
Koen
Linssen
a,
Ulrich
Simon
b and
Dominik
Wöll
*a
a,
Rebecca
Hengsbach
b,
Anne
Frommelius
b,
Samira
Cammeraat
a,
Koen
Linssen
a,
Ulrich
Simon
b and
Dominik
Wöll
*a
aInstitute of Physical Chemistry, RWTH Aachen University, Landoltweg 2, 52074 Aachen, Germany. E-mail: woell@pc.rwth-aachen.de; Tel: +49 241 80 98624
bInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1a, 52074 Aachen, Germany
First published on 30th December 2024
Abstract
Microgels are versatile materials with applications across biomedicine, materials science, and beyond. Their controllable size and composition enables tailoring specific properties, yet characterizing their internal structures on the nanoscale remains challenging. Super-resolution fluorescence microscopy (SRFM) effectively analyzes sub-μm structures, including microgels, offering a tool for investigating more complex systems such as core–shell microgels. Understanding their internal structure, in particular interpenetration at the soft–soft interface between core and shell and accessibility for guest molecules, is vital for rationally designing predictable functionalities. This study examines the core–shell morphology and the accessibility for guest molecules of bio-hybrid DNA-poly(N-isopropylmethacrylamide) microgels at three stages of shell polymerization using SRFM. Covalent fluorescence labeling probes the core polymer, co-polymerized with N,N′-bis(acryloyl)cystamine, which provides visual insight into core and shell compartmentalization. The results demonstrate core polymer interpenetration into the shell without compromising its original structure, and additionally allow us to determine the size- and hydrophobicity dependent accessibility of the microgel core. This, offering new perspectives on the internal architecture of core–shell microgels, contributes to the in-depth understanding of their complex behavior, potentially guiding the rational design of new microgel drug delivery systems, taking into account the complex interplay of polarity, size and charge of guest molecules.
1. Introduction
Microgels have gained interest in a variety of research fields, as they have versatile structure, tunable properties and can be responsive to external stimuli.1,2 The polymer networks cover a size range from 100 nm up to 100 μm, and can be varied in their monomer choices, shape and structural architecture.1–4 Additional functionalization, assemblies and supporting materials extend the available possibilities for microgels to be adjusted to the respective applications. The plentitude of variations in structures and properties elevate them to important materials in the research for biotechnological and medical applications (e.g. in drug delivery),5–8 catalysis,9–13 and even cleansing and separating applications.14–16 A great interest lies in the uptake and release of small to medium-sized molecules and complexes.5,7,17–19 The introduction of oligonucleotides, as an example, enables temperature sensitive uptake and release regardless of the volume phase transition (VPT) of the microgels.17,18 In drug delivery research, core–shell microgel systems are often employed due to their ability to protect and stabilize loaded drugs,6,20 reduce cytotoxicity,21,22 and enhance uptake and controlled release.23–27 These systems offer flexibility, with the shell providing additional functions such as encapsulation, surface modification or alteration of the VPT.For precise applications, the detailed characterization of microgels is essential. Classical scattering techniques such as dynamic light scattering (DLS) and small-angle neutron scattering (SANS)28–30 have been extended a few years ago by real-space 3D imaging with super-resolution fluorescence microscopy (SRFM) to probe sub-micrometer microgel structures.31 Characterizing complex systems like core–shell microgels is particularly challenging. Core–shell structures often consist of two distinct polymer domains. When their physical properties converge, distinguishing between core and shell can become challenging. This typically is the case if the shell is synthesized onto a core that is a microgel itself.32–34 Detailed structural analysis of the local environment, like the positioning of functional groups, incorporated (bio-)macromolecules, nanoparticles, etc. and their availability is thus often neglected.
One possibility to study the morphology of core–shell microgels is offered by super-resolution fluorescence microscopy (SRFM). It requires the polymer network to be labeled with fluorescent dyes, either covalently through copolymerization or functionalization,35–38 or non-covalently via ionic or van der Waals interactions.39–41 Covalent labeling specifically targets parts of the polymer network that contain functional groups, which are either inherent to the monomer or introduced through copolymerization.35,42–44 Thus, one polymer network can be visualized individually from the other. Since precipitation-polymerized microgels are typically sub-micrometer in size, SRFM is necessary for resolving their features beyond the diffraction limit.31,45 Localization-based SRFM techniques allow for investigating the polymer structure in detail, if desired, in three spatial dimensions.31 Various techniques are available, for example direct stochastic optical reconstruction microscopy (dSTORM),46 point accumulation in nanoscale topography (PAINT),47 and PAINT via deoxyribonucleic acid (DNA), so-called DNA PAINT.48 The key difference between these techniques is their labeling approach: dSTORM applies covalent fluorescence labeling of the polymer network, while DNA PAINT involves labeling microgels with single stranded and non-fluorescent oligonucleotides (oligo-ssDNA), so-called anchor strands. Fluorescence signals are obtained via fluorescence labeled complementary oligo-ssDNA (imager strands) that diffuse into the microgels and temporarily bind to the anchor strands via hybridization.48 That makes DNA PAINT sensitive to only the DNA functionalized parts of the microgels, while diffusion still plays a major role. It is often also useful to probe the complete polymer network with SRFM. The widely used Nile Red PAINT47 approach utilizes the solvatochromic dye Nile Red which is non-fluorescent in water and becomes fluorescent in the less polar polymer network.
Here, we synthesized and investigated novel bio-hybrid core–shell microgels with DNA single strands covalently attached to the core. The microgel architecture allowed us to address fundamental questions regarding the design of effective drug delivery systems for a variety of molecules that can be loaded into the microgels. We explored the influence of the polymer shell on molecular transport of a small hydrophilic molecule, a hydrophobic dye and a DNA single strand. As shown in Fig. 1, both parts of our microgels were polymerized using N-isopropylmethacrylamide (NIPMAM), with the core incorporating N,N′-bis(acryloyl)cystamine (BAC) for covalent labeling. The position of the thiol groups yielded after reduction of the dithiol can be visualized using super-resolution fluorescence microscopy, thus enabling us to show how much the core polymer expands into the shell for different shell thicknesses. The comparison of the two super-resolution techniques dSTORM and DNA PAINT allowed us to address the size-dependent accessibility of various microgels regions, which is highly relevant for applications such as drug-delivery. In addition, Nile Red PAINT exhibited that rather hydrophobic molecules cannot readily access the core of the microgels.
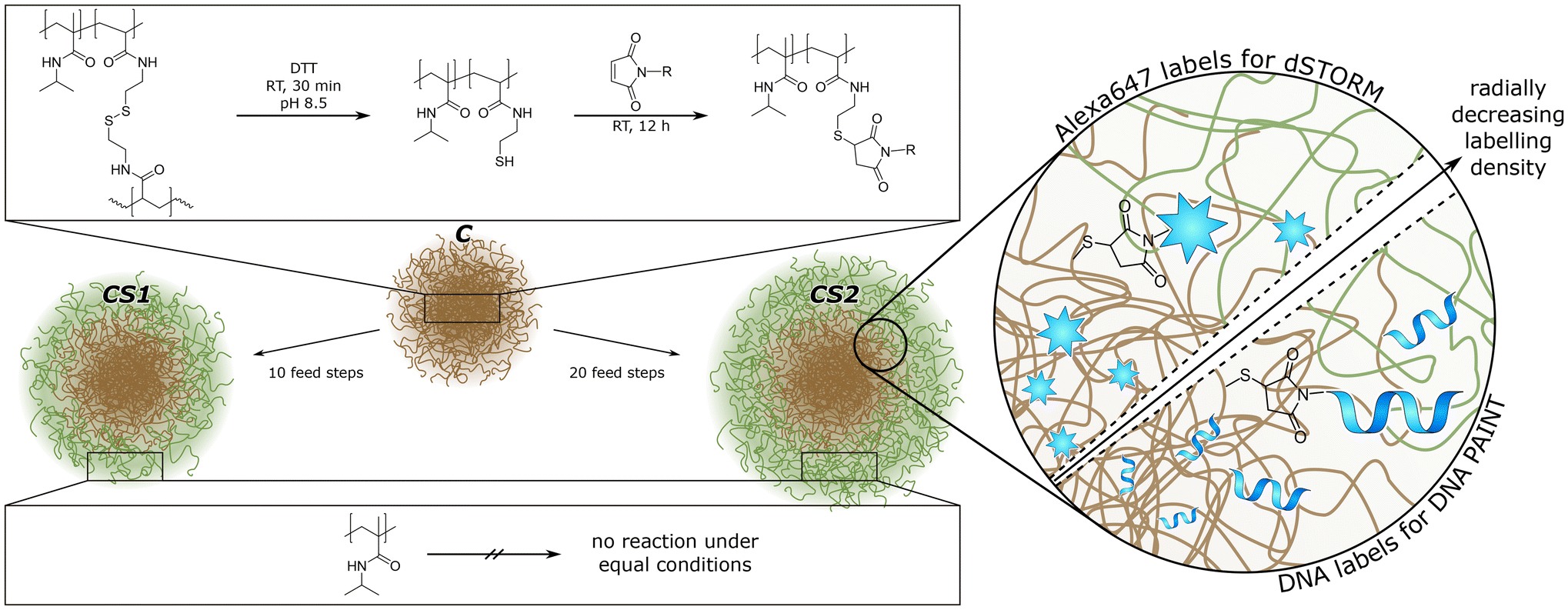 | ||
| Fig. 1 Schematic illustration of our microgel design. The microgels contain all the same base-structure, the “core” C, which can be functionalized via reduction and click reaction. The shell of core–shell microgels (CS1 and CS2) is not available for functionalization. | ||
2. Experimental
In all cases ultrapure water was used, either from the Elga Purelab System Plus (0.057–0.059 μS cm−1, pH 4.5) or the MembraPure Astacus2 RE BI (0.055–0.060 μS cm−1, pH 5.2).2.1. Microgel synthesis
Poly(N-isopropylmethacrylamide)-co-N,N′-bis(acryloyl)-cystamine (pNIPMAM-co-BAC) microgels were synthesized via precipitation polymerization. The monomer (NIPMAM, 5.1150 g, 40.2 mmol, Sigma-Aldrich, 99%), the comonomer (BAC, 0.165 g, 0.63 mmol, Alfa Aesar, 98%), the crosslinker N,N′-methylenebis(acrylamide) (BIS, 0.195 g, 1.26 mmol, Sigma Aldrich, 99%) and the surfactant cetrimonium bromide (CTAB, 0.0135 mg, 0.04 mmol, Fluka, 96%) were dissolved in H2O (250 mL) and degassed for 30 min with argon. After heating to 70 °C, the initiator V50 (0.18 g, 0.66 mmol in 50 mL H2O, degassed, Sigma-Aldrich, 99%) was added to the mixture. After 3.5 h, the mixture was cooled down to room temperature and continued stirring overnight. The microgels were purified by filtration through glass wool and centrifugation with H2O (3 × 20.6 krcf, 20 min, at 36 °C).49For core–shell microgels, the pNIPMAM-co-BAC microgels were used as core. For shell polymerization, three solutions were prepared: (A) core microgel (0.15 g) and surfactant (CTAB, 0.0015 g, 0.004 mmol) in H2O (10 mL) (B) shell-monomer (NIPMAM, 0.327 g, 2.57 mmol) and crosslinker (BIS, 0.024 g, 1.56 mmol) in H2O (17 mL) (C) initiator V50 (0.109 g, 0.8 mmol) in 1 mL H2O. Solutions B and C were degassed for 30 min with argon and heated to 70 °C. To solution A, solution B (1.7 mL) and C (0.1 mL) were added every 5 minutes, 10 times for a thinner shell and 20 times for a thicker shell (the total amount of B and C is then doubled). The mixture was stirred for 3.5 h at 70 °C and overnight at room temperature. The purification steps are equal to the initial core synthesis.49 The three samples were referred to as C (core-only), CS1 (thin shell), and CS2 (thick shell).
2.2. Microgel functionalization
To functionalize the core, the disulfide bonds of BAC were reduced. The microgel dispersion (0.4 mg, in 0.2 mL H2O, 2 mg mL−1, pH 8–9, nBAC ∼ 0.05 μmol, 1 eq.) was centrifuged (9.5 krcf, 10 min) and the supernatant was replaced by a solution of dithiothreitol (DTT, 7–8 mg, ∼0.05 mmol, Sigma Aldrich, ≥99%, in 0.5 mL H2O, pH 8–9, ∼1 × 103 eq.). After 30 min, the reduced microgels were purified by centrifugation (3 × 9.5 krcf, 10 min), and washed with H2O. After the last centrifugation, the microgels were redispersed in H2O containing the labeling agent (mixture of AlexaFluor™647 maleimide (5%) and N-ethylmaleimide (95%) (AxMal + NEM, 40 μL, 0.1 μmol, 2.5 mM, 2 eq.)) or DNA with maleimide (DNA-Mal, 40 μL, 0.1 μmol, 2.5 mM, 2 eq.) and left to shake overnight. The labeled microgels were purified by centrifugation (5 × 9.5 krcf, 10 min), exchanging the solvent twice by NaCl (0.1 M, in H2O) and then by H2O.Characterization of the non-functionalized microgels was performed via dynamic light scattering (DLS) (Zetasizer ZS, Malvern), scanning transmission electron microscopy (STEM), and atomic force microscopy (AFM) (NanoScope IIIa-Atomic Force Microscope by Digital Instruments), details are provided in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 49 Characterization of the functionalized microgels was performed via angle dependent DLS (ALV/LSE-5003 with PerkinElmer™ Photon counting module, Soliton He–Ne-Laser (λ = 633 nm), and ALV correlator software). At 22.5 °C, the hydrodynamic radius of the Alexa647 functionalized microgels is rh (C) = 293 ± 4 nm, rh (CS1) = 379 ± 7 nm, and rh (CS2) = 434 ± 7 nm. Also at 22.5 °C, the hydrodynamic radius of the DNA functionalized microgels is rh (C) = 322 ± 8 nm, rh (CS1) = 382 ± 6 nm, and rh (CS2) = 455 ± 11 nm.
49 Characterization of the functionalized microgels was performed via angle dependent DLS (ALV/LSE-5003 with PerkinElmer™ Photon counting module, Soliton He–Ne-Laser (λ = 633 nm), and ALV correlator software). At 22.5 °C, the hydrodynamic radius of the Alexa647 functionalized microgels is rh (C) = 293 ± 4 nm, rh (CS1) = 379 ± 7 nm, and rh (CS2) = 434 ± 7 nm. Also at 22.5 °C, the hydrodynamic radius of the DNA functionalized microgels is rh (C) = 322 ± 8 nm, rh (CS1) = 382 ± 6 nm, and rh (CS2) = 455 ± 11 nm.
2.3. Super-resolution fluorescence microscopy
Three SRFM methods are used to investigate the microgels: dSTORM, PAINT with Nile red, and DNA PAINT. dSTORM was performed on the Alexa647 functionalized microgels, PAINT and DNA PAINT on the DNA functionalized microgels. The measurements were conducted on the Inverted Research Microscope Nikon Eclipse Ti with a Nikon CFI HP Plan Apo VC 100× oil objective with numerical aperture of 1.4 and Leica Immersol™ 518 F immersion oil. Detection was achieved with the Photometrics Prime 95B® Scientific CMOS camera, and the whole system was operated with VisiView® by Visitron. A 640 nm Toptica iBeam smart 640CD laser was used for dSTORM and DNA PAINT and a 561 nm Cobolt Jive TEM 00 Mode laser was used for PAINT measurements, each applied in highly inclined and laminated optical shield (HILO) illumination. Blinking signals were obtained using cysteamine (50 mM, in H2O) for dSTORM, a literature-reported buffer system (5 mM Tris, 10 mM MgCl2, 1 mM EDTA, 0.05 vol% Tween20)48 containing the oligo-ssDNA imager strand ((4–7) × 10−10 M) for DNA PAINT, or a Nile red solution (10−10 M, in H2O with 1% MeOH) for PAINT. Phase modification for 3D reconstruction was performed with the DoubleHelix phasemask DH-580-1949 for NR PAINT and DH-670-1949 for dSTORM and DNA PAINT, in combination with the DoubleHelix Spindle®. Video analysis was performed using the 3DTRAX® plug-in for Fiji.50 Further data analysis was conducted using the thunderSTORM51 plug-in for Fiji50 and custom Matlab scripts. Via Matlab scripts, the individual microgel point clouds were separated from each other, the radial localization density was calculated for each point cloud, the mean radial localization density was calculated for each sample set, and the point clouds were visualized as concatenations of many. The exact number of concatenated point clouds depends on the number of microgels in the field of view of the microscopy measurements. Besides, only non-aggregated microgels yield point clouds that can be extracted for individual analysis.3. Results and discussion
We synthesized three different pNIPMAM microgels: one “core-only” microgel (C) and two different core–shell microgels with different shell thickness, where C serves as core. As illustrated in Fig. 1, both, core and shell, consist of NIPMAM as main monomer. The core contains additional N,N′-bis(acryloyl)-cystamine (BAC) co-monomer to enable covalent labeling after reduction of the disulfide bridge to thiols. The shells were synthesized by feed precipitation polymerization with 10 feed steps to obtain a thin shell (CS1) and 20 feed steps to obtain a thick shell (CS2), respectively. Other shell thicknesses are easily accessible via an adaptable number of feed steps. The thiol groups introduced during the core synthesis can be functionalized with oligo-ssDNA or covalently labeled with functional dyes, respectively (see Fig. 1).Characterization of the microgels was performed by DLS, atomic force microscopy (AFM) and scanning transmission electron microscopy (STEM) (see the ESI†). DLS measurements show that in the swollen state at 20–25 °C the shell synthesis increases the overall size of 350 nm for C microgels to 485 nm for CS1 and 595 nm for CS2, respectively, while the corresponding polydispersity index remains small with 0.11 for C and CS1, and 0.20 for CS2. The volume increase between C and CS1 is very similar to the one between CS1 and CS2, i.e. each shell synthesis step seems to increase the size by a constant volume. The presence of a core–shell structure could be verified by means of AFM and STEM on the dry sample adsorbed to an interface. The height of the dry microgels is determined by AFM measurements, while STEM yields information about the polymer density (see Fig. 2). For each method, we calculated the radial profiles and fitted them to a customized logistic curve function (see the ESI†). The inflection points of these profiles were taken to evaluate the core–shell morphology (see Tables S2 and S3 in the ESI†). Both methods exhibit only minor changes in the core size after adding the shell. A significant size difference is only observed for the core-only microgels, which appear larger with a smoother peripheral transition in AFM compared to the sharper transition seen in STEM. This is most likely caused either by the different surfaces used for the measurements, which in turn altered the spreading behavior of the microgels, or by the difference between high vacuum conditions in STEM versus measurements under air (with some remaining moisture52) in AFM.
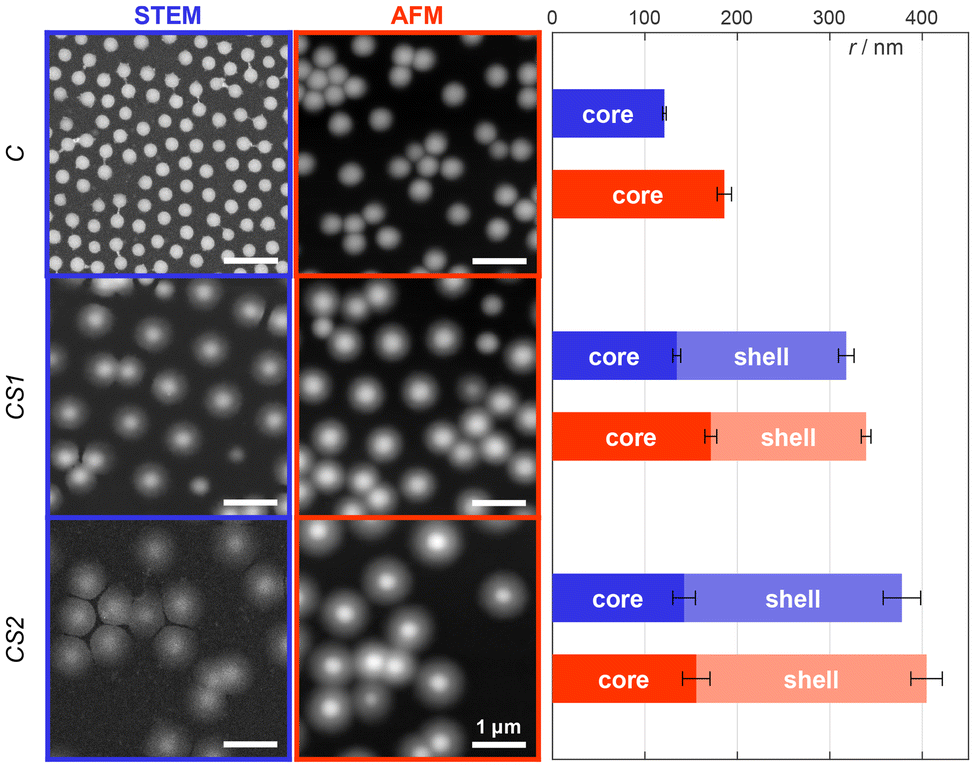 | ||
| Fig. 2 STEM and AFM images for the different microgel types C, CS1, and CS2 in their dry collapsed state. The core and shell sizes were determined by analysis of the radial intensity/height profiles using a customized logistic curve function as outlined in the ESI.† The inclination points of these logistic functions were defined as the respective radii. All scale bars are equal to 1 μm. | ||
The thiol groups can be used for the functionalization with oligo-ssDNA to form biohybrid microgels. The distribution of ssDNA can be investigated with DNA PAINT using complementary ATTO647N-labeled oligonucleotides. Furthermore, a direct labeling of the thiol groups with Alexa647 maleimide is possible for dSTORM imaging. The comparison between both super-resolution methods allows for a qualitative evaluation of size-dependent accessibility of microgels regions, which is highly relevant for applications such as drug-delivery. Relatively large dye-labeled DNA oligomers have to reach the corresponding DNA strands in the case of DNA PAINT, whereas solely the small molecule cysteamine is required to induce blinking in dSTORM.
For dSTORM measurements, we found that suitable blinking behavior, and thus super-resolution imaging, is achieved when approx. 5% of the thiol groups are labeled with Alexa647. This was achieved by mixing 5% of Alexa647 maleimide with 95% of N-ethylmaleimide as non-fluorescent capping agent. The dSTORM measurements for C, CS1, and CS2 (see Fig. 3a–c) reveal rather similar radial localization density distributions. The majority of labeling can be found in the core, even for the microgels with thin and thick shell. Yet, also the shells exhibit a significant number of localizations, which by far exceeds the number of possible mislocalizations. This points to a certain amount of interpenetration of the core polymer network into the shell, which is important information considering recent literature. Comparability of our microgels to literature is limited by the incorporation of charged molecules, though major effects are unlikely due to charge screening by counterions.18,53 The findings of Brändel et al.54 suggest that pNIPMAM is not prone to forming highly interpenetrating networks. Also, neither Berndt et al.29 nor Maccarrone et al.30 could observe significant amounts of core polymer in the shell in core–shell microgels by means of SANS measurements. Yet, their results do not contradict our findings, since small amounts of core polymer are likely obscured by their employed methods. SRFM might resolve structural details that are currently inaccessible through other techniques, providing a complementary perspective.
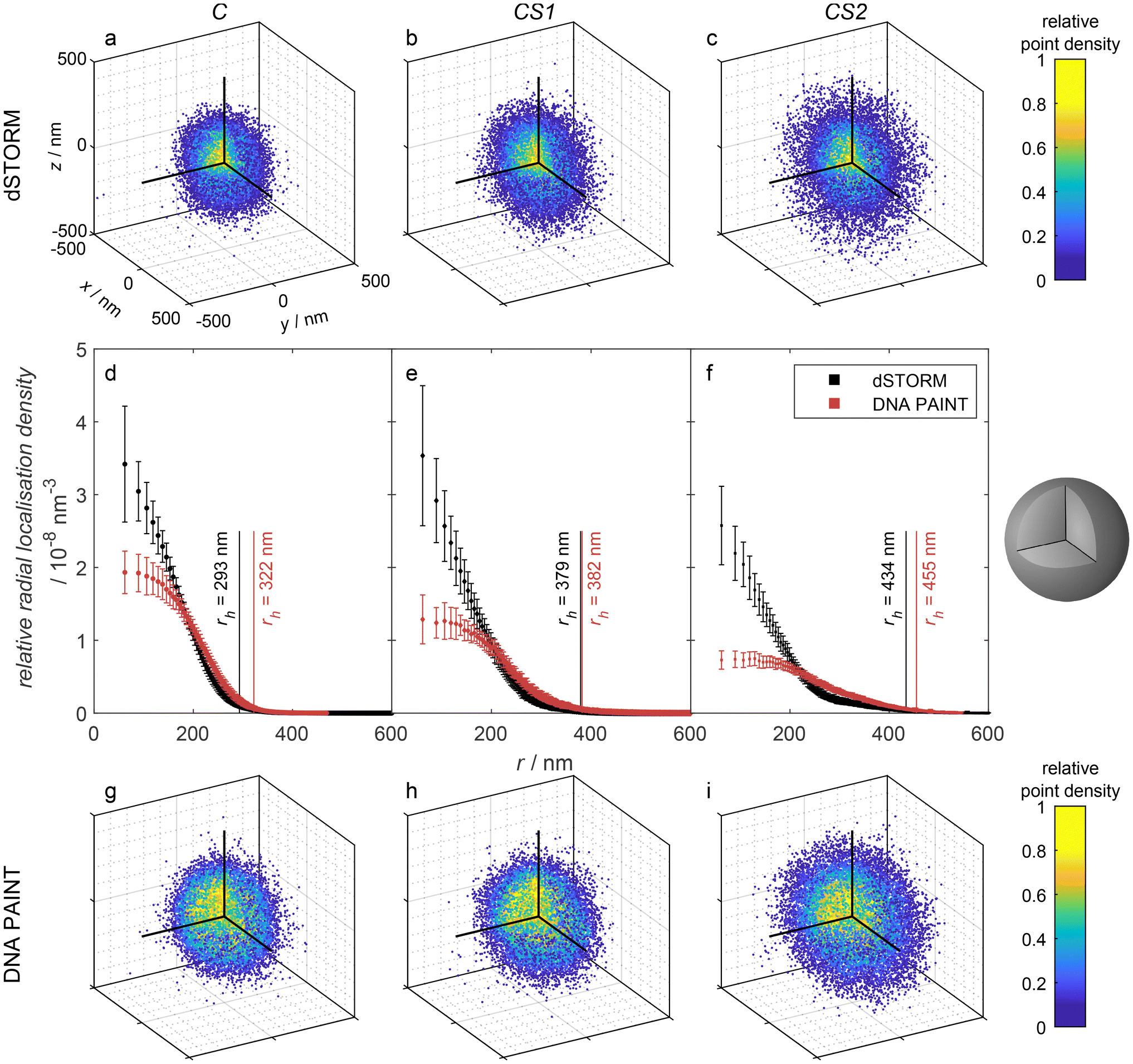 | ||
| Fig. 3 Point clouds obtained by (a–c) 3D dSTORM and (g–i) DNA PAINT measurements of the microgel samples C, CS1, and CS2, respectively. The depicted point clouds are overlays of a: 107, b: 84, c: 48, g: 103, h: 18, and i: 20 point clouds of individual microgels, respectively, shifted by their median center in each dimension. The upper left octant is cut as depicted in the sketch on the right to visualize the internal point density. For better visibility, the number of shown points was randomly reduced to 2 × 104. (d–f) Relative radial localization density of the samples, averaged over multiple measurements. Contributing number of measurements and included microgel point clouds: C – DNA PAINT: 3 measurements, 168 microgels, dSTORM: 3 measurements, 576 microgels; CS1 – DNA PAINT: 3 measurements, 61 microgels, dSTORM: 3 measurements, 389 microgels; CS2 – DNA PAINT: 5 measurements, 116 microgels, dSTORM: 3 measurements, 194 microgels. The binning was chosen so that each spherical shell presented by the bins contains the same volume. The number of localizations in each bin is divided by that bin volume to obtain the absolute localization density. Since the localizations per microgel vary statistically, the absolute density was divided by the total number of localizations, yielding the relative radial localization density. Additionally, the hydrodynamic radii rh for the three microgel types with Alexa647 functionalization (black) and oligo-ssDNA (red) functionalization, respectively, as measured by DLS are indicated. | ||
For DNA PAINT measurements, the thiol groups were stoichiometrically functionalized with oligo-ssDNA. The resulting 3D point clouds are shown in Fig. 3g–i and the corresponding 3D radial localization density distributions as a function of the distance from the microgel centers in Fig. 3d–f. Also here, a significant amount of localizations can be found in the shell, which correspond to binding events of complementary DNA strands. The relative number of localization events in the shell versus the core is, however, significantly higher than in the dSTORM measurements.
In both methods, dSTORM and DNA PAINT, the fluorescence localizations appear at positions which were originally thiol groups. The difference in the radial localization distribution between the methods can have different reasons. First, it has to be considered, that the localization distribution indicates a different distribution of the dye Alexa647 and the oligo-ssDNA docking strands, respectively. However, we assume that the reaction of thiols with maleimides does not depend on the position within the microgels. Furthermore, due to the sufficiently long reaction time of several hours of the maleimides with the thiol groups, we can assume that all positions are generally reached by the dye Alexa647 and by the DNA docking strands. Thus, a different distribution of those functionalities within the microgel cannot be the reason for the variation in the distribution of localizations. As a consequence, we attribute the higher relative amount of localizations in the shell found in DNA PAINT to a restricted diffusion of DNA imager strands within the microgel. In other words, several imager DNA strands bind to the few DNA docking strands in the shell and bleach before they can reach the core. The localization events in the core still dominate the distribution, but less pronounced than in dSTORM, as seen in the relative radial localization density. DNA PAINT requires the diffusion of oligonucleotides through the meshes of the microgel network. In contrast, the blinking events in dSTORM are induced by cysteamine, which is a small molecule that presumably can diffuse rather unhindered through the polymer and which also does not photobleach or react/disappear irreversibly. This hypothesis relates to findings from Gelissen et al.,55 who demonstrated the uptake and release of polyelectrolytes to polyampholyte microgels in dependence on the polyelectrolyte's chain length. Short chains exhibited higher penetration depth as long chains. Due to the differences to our system in microgel size, morphology and binding strategy,55,56 the results are not directly comparable, but indicate the same phenomenon, overall.
In addition to the dSTORM and DNA PAINT measurements, we conducted Nile Red PAINT measurements to visualize the overall microgel structure, which was equivalent to the approach we had previously employed for several other microgel systems.57–59 Exemplarily, we have chosen the microgels C, CS1, and CS2 functionalized with DNA since the emission of the Alexa647-functionalized microgels would partially overlap with the Nile Red emission. The solvatochromic dye Nile Red is non-fluorescent in water and only starts to fluoresce in less polar environments. Thus, emission appears when Nile Red binds to the polymer of the microgels. Typically, for simple microgels, we find Nile Red distributions which resemble the local polymer densities within the microgels, as the one shown for C in Fig. 4. Surprisingly, as obvious from Fig. 4, for the case of the core–shell microgels CS1 and CS2 we found radial distributions with a maximum at a distance which corresponds to the radius of the core-only microgel C and significantly fewer localizations in the microgel center. This points to a significant hindrance of the diffusion of the rather hydrophobic dye Nile Red through the shell or through the core–shell interface of the microgels. For a careful evaluation, it has to be taken into account that Nile Red only emits in a microgel environment when a lot of water around it is replaced by polymer. In this emitting state, it also photobleaches rather rapidly and will not be detected afterward. Ultimately, the balance between diffusion within the polymer network and photobleaching determines the radial distribution of localizations. The comparison between the three different radial localization densities in Fig. 4 points to the conclusion that the diffusion of the small rather hydrophobic dye Nile Red is significantly hindered in the shell and the transition region of the core–shell microgels CS1 and CS2 whereas it can more readily reach the microgels center in the core-only microgel C. The reduced diffusion in the shell points to a certain interpenetration of the core into the shell. A similar behavior was found by Otto et al.39 for microgels with a N-n-propylacrylamide shell synthesized onto the collapsed core of pNIPMAM. In their case, even though significantly different from our system, the dye rhodamine 6G could not reach the microgel core.
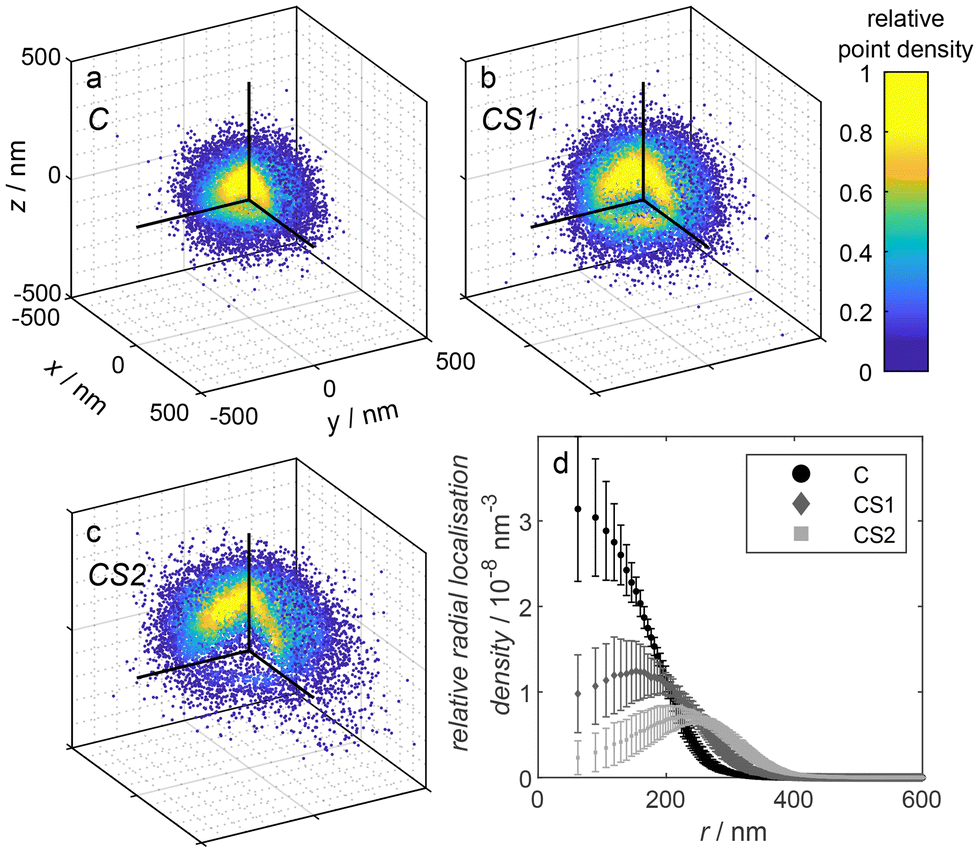 | ||
| Fig. 4 (a–c) Point clouds obtained by Nile Red PAINT measurements of the microgel samples C, CS1, and CS2, respectively. The depicted point clouds are overlays of a: 62, b: 31, c: 17 point clouds of individual microgels, respectively, shifted by their geometrical center obtained by a circle fit in each dimension. The upper left octant is cut as depicted in the sketch in Fig. 3 to visualize the internal point density. For better visibility, the number of shown points was randomly reduced to 2 × 104. (d) Relative radial localization density of the three different DNA-functionalized microgels C, CS1, and CS2, respectively, as determined via Nile Red PAINT averaged over multiple measurements. Contributing number of measurements and included microgel point clouds: C – 2 measurements, 67 microgels, CS1 – 2 measurements, 45 microgels; CS2 – 2 measurements, 27 microgels. | ||
4. Conclusions
With the study presented here, we analyzed the internal structure of bio-hybrid DNA-pNIPMAM core–shell microgels with different shell-thicknesses. Super-resolution fluorescence microscopy was established to characterize microgels with core–shell architecture and compared with other characterization methods. With dSTORM, the distribution of functional groups from the core polymer was determined. It was found that the core polymer partially extends to the shell, i.e. the shell polymer interpenetrates the core to some extend. The DNA PAINT measurements on DNA functionalized microgels resulted in a different distribution of localizations with a higher relative localization density in the shell. After careful consideration of different possibility causing this observation, we conclude that the diffusion of DNA through the polymer network of the shell or shell–core transition region is limited. This limitation in accessibility of (bio-)macromolecules is an important factor to be considered when working on e.g. uptake and release experiments. Moreover, our findings indicate that the apolar dye Nile Red is capable of labeling core-only microgels in accordance with their polymer density. However, it is unable to readily reach the centre of core–shell microgels, since most of it is instead absorbed in the shell region.With respect to the application of such complex structures, e.g. for drug-encapsulation and release, our findings provide new insights into the complex interplay of polarity, size and charge of guest molecules. This will be relevant for the future design of new drug delivery systems, that can be adjusted to specific drug compositions, ranging from hydrophobic drug candidates, to structurally more complex charged molecules, such as RNA. The new insights into the polarity features, in particular at the soft–soft interface region between core and shell and its permeability will be important for the future development of microgels with engineered encapsulation and release features.
Author contributions
P. L. – data curation, formal analysis, investigation, software, visualization, validation, writing; R. H. – conceptualization, data curation, formal analysis, investigation, validation, writing (review); A. F. – investigation, writing; S. C. – investigation, validation, writing (review); K. L. – investigation, validation, writing (review); U. S. – conceptualization, funding acquisition, project administration, supervision, writing; D. W. – conceptualization, funding acquisition, project administration, software, supervision, writing.Data availability
Data are stored according to the guidelines of the DFG. Additional data is available upon request.The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the German Research Foundation DFG for financial support within the Collaborative Research Center SFB 985 (Project A6). Furthermore, the authors thank Ali Cetin for synthesizing the microgels used in this publication.References
- F. A. Plamper and W. Richtering, Acc. Chem. Res., 2017, 50, 131–140 CrossRef CAS.
- F. Scheffold, Nat. Commun., 2020, 11, 4315 CrossRef CAS.
- M. Karg, A. Pich, T. Hellweg, T. Hoare, L. A. Lyon, J. J. Crassous, D. Suzuki, R. A. Gumerov, S. Schneider, I. I. Potemkin and W. Richtering, Langmuir, 2019, 35, 6231–6255 CrossRef CAS PubMed.
- ‘microgel’, The IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry, 2006 Online version 3.0.1, 2019.
- A. Pepe, P. Podesva and G. Simone, Sci. Rep., 2017, 7, 6014 CrossRef CAS.
- Y. Pan, Y. Qi, X. Li, S. Luan and Y. Huang, Adv. Funct. Mater., 2021, 31, 2105742 CrossRef CAS.
- M. Dirksen, T. A. Kinder, T. Brändel and T. Hellweg, Molecules, 2021, 26, 3181 CrossRef CAS PubMed.
- B. Li, H. Li, H. Chen, Y. Liu, J. Chen, Q. Feng, X. Cao and H. Dong, Adv. Funct. Mater., 2023, 33, 2302793 CrossRef CAS.
- V. Khoo, S.-F. Ng, C.-Y. Haw and W.-J. Ong, Small, 2024, 20(31), 2401278 CrossRef CAS.
- V. Sabadasch, M. Dirksen, P. Fandrich, J. Cremer, N. Biere, D. Anselmetti and T. Hellweg, ACS Appl. Mater. Interfaces, 2022, 14, 49181–49188 CrossRef CAS PubMed.
- V. Sabadasch, M. Dirksen, P. Fandrich and T. Hellweg, Front. Chem., 2022, 10, 889521 CrossRef CAS.
- T. Brändel, V. Sabadasch, Y. Hannappel and T. Hellweg, ACS Omega, 2019, 4, 4636–4649 CrossRef PubMed.
- T. Chen, M. Qiu, Y. Peng, C. Yi and Z. Xu, Small, 2023, 19, 2303282 CrossRef CAS PubMed.
- M. Dirksen, T. Brändel, S. Großkopf, S. Knust, J. Bookhold, D. Anselmetti and T. Hellweg, RSC Adv., 2021, 11, 22014–22024 RSC.
- M. Dirksen, P. Fandrich, L. Goett-Zink, J. Cremer, D. Anselmetti and T. Hellweg, Langmuir, 2022, 38, 638–651 CrossRef CAS.
- C. C. Cutright, J. L. Harris, S. Ramesh, S. A. Khan, J. Genzer and S. Menegatti, Adv. Funct. Mater., 2021, 31(47), 2104164 CrossRef CAS.
- S. Eisold, L. Hoppe Alvarez, K. Ran, R. Hengsbach, G. Fink, S. Centeno Benigno, J. Mayer, D. Wöll and U. Simon, Nanoscale, 2021, 13, 2875–2882 RSC.
- R. Hengsbach, G. Fink and U. Simon, Soft Matter, 2024, 20, 330–337 RSC.
- M. Deloney, K. Smart, B. A. Christiansen and A. Panitch, J. Controlled Release, 2020, 323, 47–58 CrossRef CAS.
- B. Lei, M. Chen, Y. Wang, J. Zhang, S. Xu and H. Liu, Colloids Surf., B, 2020, 193, 111022 CrossRef CAS.
- C. Li, W. Huang, L. Zhou, P. Huang, Y. Pang, X. Zhu and D. Yan, Polym. Chem., 2015, 6, 6498–6508 RSC.
- R. Jenjob, T. Phakkeeree and D. Crespy, Biomater. Sci., 2020, 8, 2756–2770 RSC.
- T. Jing, T. Li, Z. Ruan and L. Yan, J. Mater. Sci., 2018, 53, 14933–14943 CrossRef CAS.
- R. Raju, S. Bandyopadhyay, A. Sharma, S. V. Gonzalez, P. H. Carlsen, O. R. Gautun and W. R. Glomm, Polymers, 2018, 10, 309 CrossRef PubMed.
- W. Wu, J. Liu, P. Gong, Z. Li, C. Ke, Y. Qian, H. Luo, L. Xiao, F. Zhou and W. Liu, Small, 2022, 18, e2202510 CrossRef PubMed.
- I. K. Sommerfeld, H. Malyaran, S. Neuss, D. E. Demco and A. Pich, Biomacromolecules, 2024, 25, 903–923 CrossRef CAS.
- A. Ghosh Majumdar, B. Pany, S. S. Parua, D. Mukherjee, A. Panda, M. Mohanty, B. Das, S. Si and P. S. Mohanty, BioNanoScience, 2024, 3496–3521 CrossRef.
- C. D. Jones and L. A. Lyon, Macromolecules, 2000, 33, 8301–8306 CrossRef CAS.
- I. Berndt, J. S. Pedersen and W. Richtering, J. Am. Chem. Soc., 2005, 127, 9372–9373 CrossRef CAS.
- S. Maccarrone, A. Ghavami, O. Holderer, C. Scherzinger, P. Lindner, W. Richtering, D. Richter and R. G. Winkler, Macromolecules, 2016, 49, 3608–3618 CrossRef CAS.
- O. Nevskyi and D. Wöll, Annu. Rev. Phys. Chem., 2023, 74, 391–414 CrossRef CAS.
- J. Oberdisse and T. Hellweg, Colloid Polym. Sci., 2020, 298, 921–935 CrossRef CAS.
- M. Hildebrandt, S. Lazarev, J. Pérez, I. A. Vartanyants, J.-M. Meijer and M. Karg, Macromolecules, 2022, 55, 2959–2969 CrossRef CAS.
- A. Scotti, Soft Matter, 2021, 17, 5548–5559 RSC.
- E. Siemes, O. Nevskyi, D. Sysoiev, S. K. Turnhoff, A. Oppermann, T. Huhn, W. Richtering and D. Wöll, Angew. Chem., Int. Ed., 2018, 57, 12280–12284 CrossRef CAS PubMed.
- G. M. Conley, P. Aebischer, S. Nöjd, P. Schurtenberger and F. Scheffold, Sci. Adv., 2017, 3, e1700969 CrossRef PubMed.
- L. Hoppe Alvarez, A. A. Rudov, R. A. Gumerov, P. Lenssen, U. Simon, I. I. Potemkin and D. Wöll, Phys. Chem. Chem. Phys., 2021, 23, 4927–4934 RSC.
- E. Gau, D. M. Mate, Z. Zou, A. Oppermann, A. Töpel, F. Jakob, D. Wöll, U. Schwaneberg and A. Pich, Biomacromolecules, 2017, 18, 2789–2798 CrossRef CAS.
- P. Otto, S. Bergmann, A. Sandmeyer, M. Dirksen, O. Wrede, T. Hellweg and T. Huser, Nanoscale Adv., 2020, 2, 323–331 RSC.
- O. Wrede, S. Bergmann, Y. Hannappel, T. Hellweg and T. Huser, Soft Matter, 2020, 16, 8078–8084 RSC.
- T. Schmidt, M. Hohenschutz, M. Becker, F. Grabowski, A. Jupke, A. Pich and D. Wöll, Colloid Polym. Sci., 2023, 345–354 Search PubMed.
- A. P. H. Gelissen, A. Oppermann, T. Caumanns, P. Hebbeker, S. K. Turnhoff, R. Tiwari, S. Eisold, U. Simon, Y. Lu, J. Mayer, W. Richtering, A. Walther and D. Wöll, Nano Lett., 2016, 16, 7295–7301 CrossRef CAS PubMed.
- G. M. Conley, S. Nöjd, M. Braibanti, P. Schurtenberger and F. Scheffold, Colloids Surf., A, 2016, 499, 18–23 CrossRef CAS.
- L. Hoppe Alvarez, S. Eisold, R. A. Gumerov, M. Strauch, A. A. Rudov, P. Lenssen, D. Merhof, I. I. Potemkin, U. Simon and D. Wöll, Nano Lett., 2019, 19, 8862–8867 CrossRef CAS PubMed.
- A. Pich and W. Richtering, Chemical design of responsive microgels, Springer, Berlin, 2010, vol. 234, pp. 1–37 Search PubMed.
- M. Sauer, Far-Field Optical Nanoscopy, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, vol. 14, pp. 65–84 Search PubMed.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS PubMed.
- J. Schnitzbauer, M. T. Strauss, T. Schlichthaerle, F. Schueder and R. Jungmann, Nat. Protoc., 2017, 12, 1198–1228 CrossRef CAS PubMed.
- R. Hengsbach, PhD thesis, RWTH Aachen University, 2023.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- M. Ovesný, P. Křížek, J. Borkovec, Z. Svindrych and G. M. Hagen, Bioinformatics, 2014, 30, 2389–2390 CrossRef PubMed.
- S. Jana and D. Wöll, Phys. Chem. Chem. Phys., 2024, 26, 23250–23255 RSC.
- H. Kobayashi and R. G. Winkler, Sci. Rep., 2016, 6, 19836 CrossRef CAS PubMed.
- T. Brändel, M. Dirksen and T. Hellweg, Polymers, 2019, 11, 1269 CrossRef.
- A. P. H. Gelissen, A. Scotti, S. K. Turnhoff, C. Janssen, A. Radulescu, A. Pich, A. A. Rudov, I. I. Potemkin and W. Richtering, Soft Matter, 2018, 14, 4287–4299 RSC.
- A. A. Rudov, A. P. H. Gelissen, G. Lotze, A. Schmid, T. Eckert, A. Pich, W. Richtering and I. I. Potemkin, Macromolecules, 2017, 50, 4435–4445 CrossRef CAS.
- A. Purohit, S. P. Centeno, S. K. Wypysek, W. Richtering and D. Wöll, Chem. Sci., 2019, 10, 10336–10342 RSC.
- R. Azad, P. Lenßen, Y. Jia, M. Strauch, B. A. Bener, D. Merhof and D. Wöll, Nano Lett., 2024, 24, 4447–4453 CrossRef CAS.
- L. Schubert, C. Nenninger, M. Nöth, T. Belthle, R. D. de Lange, A. Pich, U. Schwaneberg and D. Wöll, Nanoscale, 2024, 16, 20194–20201 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04677c |
| This journal is © The Royal Society of Chemistry 2025 |