
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular electrostatic functionalization of 1T-MoS2 based on alkylammonium salts†
Giuseppe
Misia
 a,
Michele
Cesco
b,
Maurizio
Prato
*abc and
Alessandro
Silvestri
*d
a,
Michele
Cesco
b,
Maurizio
Prato
*abc and
Alessandro
Silvestri
*d
aDepartment of Chemical and Pharmaceutical Sciences, INSTM UdR Trieste, Università Degli Studi di Trieste Trieste, 34127, Italy
bCenter for Cooperative Research in Biomaterials (CIC BiomaGUNE) Basque Research and Technology Alliance (BRTA) Paseo de Miramon 194, 20014, Donostia-San Sebastián, Spain
cIkerbasque Basque Foundation for Science Bilbao, 48009, Spain
dDepartment of Molecular Sciences and Nanosystems Ca’ Foscari University of Venice Venezia, 30170, Italy. E-mail: alessandro.silvestri@unive.it
First published on 20th February 2025
Abstract
Chemical functionalization is key for expanding the applicability and manufacturing of 2D materials. The functionalization of exfoliated molybdenum disulfide (MoS2) can allow tuning its chemical–physical properties or introducing anchoring points for post-functionalization, creating heterostructures, hybrid or stimuli-responsive materials. When choosing a functionalization strategy, key aspects are: achieving a high functionalization yield, using sustainable solvents and reactants, and preserving the favorable material's properties. In this work, we report a supramolecular functionalization strategy for 1T-MoS2 which exploits the coulombic interactions between the negatively charged material and the positively charged alkylammonium salts. The functionalization, performed in water at room temperature, addresses the requirements of green chemistry. Furthermore, the proposed approach allows high functionalization yields (with an alkylammonium![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo ratio of 1:2.6), while preserving the crystalline phase and conductivity characteristic of 1T-MoS2. Finally, the functionalization with cholamine permits the covalent post-functionalization of the material, exploiting the reactivity of the primary amine of this alkylammonium salt.
:Mo ratio of 1:2.6), while preserving the crystalline phase and conductivity characteristic of 1T-MoS2. Finally, the functionalization with cholamine permits the covalent post-functionalization of the material, exploiting the reactivity of the primary amine of this alkylammonium salt.
 Alessandro Silvestri | Dr. Alessandro Silvestri received his Ph.D. in Chemistry at the University of Milan (Italy) in 2017. He was a postdoctoral researcher at the Max-Planck Institute of Colloids and Interfaces (Potsdam, Germany) and CIC biomaGUNE (San Sebastian, Spain). Since June 2023 he has been an assistant professor in Chemistry at Ca’ Foscari University of Venice. His research interests comprise the synthesis and chemical functionalization of nanomaterials and their application in electrochemical biosensors. |
Introduction
Among 2D materials, exfoliated MoS2 is attracting growing attention due to its low production costs, unique chemical–physical properties dependent on its crystalline phase (2H or 1T), and material thickness.1 Chemically exfoliated 1T-MoS2 presents appealing properties such as metallic conductivity,2 marked catalytic and electrocatalytic properties,3 and SERS activity.4–6The functionalization of MoS2 plays a key role in its applicability. Indeed, modulating MoS2 surface chemistry can facilitate its integration in devices and manufacturing operations, provide selectivity to external stimuli, create hybrid materials and heterostructures, tune the bandgap, or modulate other physical–chemical properties.4,7–9 Several functionalization strategies are known for 2D MoS2. One diffused strategy is to exploit the sulphur vacancies and defective sites generated during the exfoliation by healing them with organic thiols.10 The functionalization can also be achieved through radical reaction using aryl diazonium salts.4,11 Pérez et al. reported a versatile mild covalent functionalization that exploits the nucleophilic character of the S in 2H-MoS2 to bind maleimide derivatives covalently.12 Hydrophobic interactions can also be used to functionalize the material while facilitating its exfoliation.13 The reactivity of MoS2 depends strongly on the material phase: not all the reactions effective on 2H work on 1T and vice versa. 1T-MoS2 can be prepared through chemical exfoliation, intercalating metal ions (Na, K, or Li) between MoS2 layers.14–16 The harsh chemical conditions used during the exfoliation can induce the formation of more defective sites, making the 1T-MoS2 more chemically reactive than 2H-MoS2.4 For instance, the healing of sulphur vacancies is more efficient on 1T-MoS2 as more defective sites are present on the basal plane. Functionalization with aryl diazonium salts is also widely exploited for 1T-MoS2, as the single electron transfer reaction required to trigger the functionalization is favoured in this material. The different reactivity also allows the use of diverse functionalization strategies such as organohalide-based reactions, as demonstrated by Voiry and coworkers.17
A key aspect to consider when choosing the most proper functionalization strategy is that the favourable properties of the material, such as the crystalline phase and conductivity, must be preserved or improved after the functionalization. For example, the filling of sulphur vacancies heals the defects of the 1T-MoS2, reducing the number of catalytically active sites and affecting its marked catalytic properties.18 On the other hand the use of diazonium salts can introduce further defects in the crystalline structure undermining the metallic conductivity of the material. In recent years, functionalization strategies that are respectful of the physicochemical properties of MoS2 have started to emerge. For instance, Pumera and co-workers demonstrated that it is possible to preserve the conducting properties of 1T-MoS2 after covalent functionalization by using barbituric acid.19
Herein we propose a supramolecular functionalization of 1T-MoS2 which allows the preservation of the material crystalline phase and conductive properties. This non-covalent functionalization strategy exploits the coulombic interactions between the negatively charged flake and positively charged alkylammonium salts. The proposed approach answers several of the requirements of green chemistry as it uses water as a solvent, room temperature, short reaction times, and non-toxic reagents. The alkylammonium salts used in this work are bifunctional ligands bearing an amino functional group that can be exploited for the post-functionalization of the material. This is a significant advancement compared to previously proposed supramolecular strategies, which exploited flat and monovalent cationic dyes to achieve electrostatic functionalization.20
Results and discussion
The 1T-MoS2 used in this work was synthesized by chemical exfoliation using butyl lithium to engineer the material crystalline phase as previously reported in the literature.21 The obtained 2D material has been extensively characterized by TEM, AFM, Raman, UV-vis, and XPS (Fig. S1–5†). The interaction between the 1T-MoS2 and different alkyl ammonium salts, namely cholamine chloride (ChNH2), N′,N′,N′-triethylethane-1,2-diaminium salt (Et3NEtNH2), and polydiallyldimethylammonium chloride (polyDADMAC) was investigated by zeta potential measurement. The pristine 1T-MoS2 is intrinsically negatively charged, with a ζ-potential of −33 mV (Fig. S6†), which provides good colloidal stability to the material in water. Adding increasing amounts of the organic salts determines a shift of the zeta potential towards less negative values, confirming the formation of coulombic interaction between the negatively charged material and the positively charged ammonium salts (Fig. 1A–C). Adding 1 equivalent of ChNH2 and Et3NEtNH2 (with respect to the molybdenum) the functionalized MoS2 reaches ζ-potential of −21.4 and −17.1 mV respectively. In both cases, the change of the material's surface charge leads to aggregation when the alkylammonium salt exceeds 0.5 equivalents, with complete precipitation after 1 equivalent (Fig. 1D).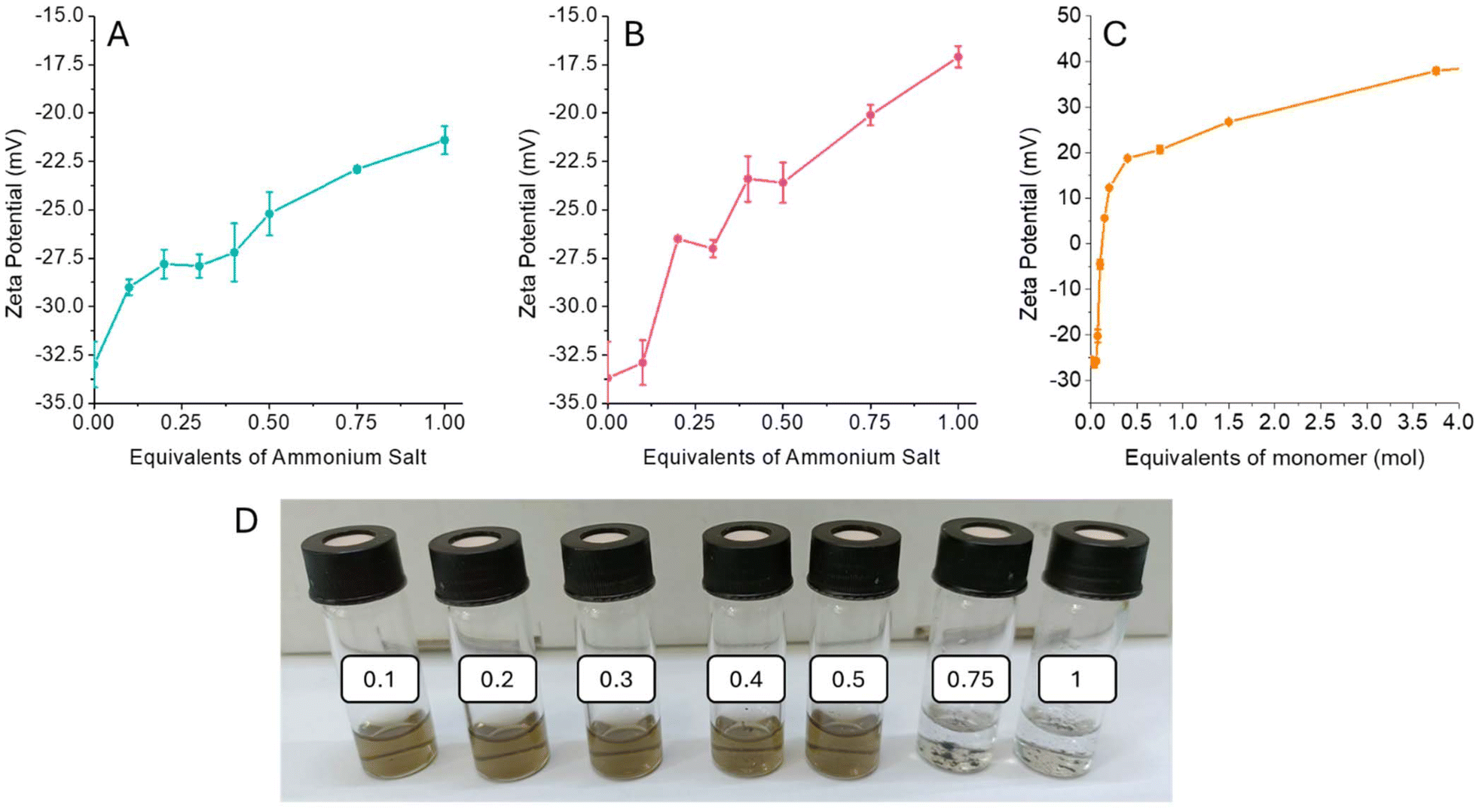 | ||
| Fig. 1 Zeta potential titration of 1T-MoS2 with ChNH2 (A), Et3NEtNH2 (B), and polyDADMAC (C). (D) Photograph of 1T-MoS2 suspensions with the addition of 0.1, 0.2, 0.3, 0.4, 0.5, 0.75 and 1 equivalent of ChNH2. | ||
A different behaviour is observed when a polyvalent alkylammonium salt, such as polyDADMAC is used (Fig. 1C). In this case, after adding 0.1 equivalents of poly-alkylammonium, the ζ-potential reaches a null value, with a complete precipitation of MoS2. If more polyDADMAC is added, ζ-potential turns positive and increases until reaching a plateau, allowing the redispersion of the material in water (Fig. S7†). In fact, for each polyDADMAC alkylammonium moiety interacting with 1T-MoS2 several others are exposed on the surface, inducing an inversion of the material ζ-potential.
After demonstrating the supramolecular interaction between 1T-MoS2 and diverse diazonium salts, attention was focused on ChNH2, which was used as a model compound to investigate the functionalization strategy further. Based on the results of the ζ-potential titration, the functionalization was carried out using 1 equivalent of organic salt to guarantee a high degree of functionalization and to facilitate purification, which was performed by precipitation and washing of the precipitate. After functionalization, the modified material was deposited on a SiO2 substrate and characterized by Raman spectroscopy. Fig. 2 compares the Raman spectra of pristine 1T-MoS2, ChNH2, and the modified material (1T-MoS2-ChNH2). In the spectra of the functionalized MoS2 the Raman modes characteristic of the 1T phase, J1 (151–154 cm−1), J2 (228 cm−1), and J3 (327–324 cm−1) E2g (380 cm−1), and A1g (405 cm−1), are preserved (Fig. 2B).4 This demonstrates the retention of the crystalline phase of the material after functionalization. Furthermore, the Raman modes of CH3 and CH2 characteristic of organic matter appear above 2800 cm−1 in the 1T-MoS2-ChNH2 spectra, proving the successful functionalization (Fig. 2A). Several of the peaks characteristic of ChNH2 appear in the 1T-MoS2-ChNH2 (Fig. 2C).22 The vibrational modes due to C–N–C stretching are visible at 1102 and 1129 cm−1. The CH3 symmetrical stretching, coupled with the CH3 deformational vibrations at 1419, 1438, and 1459 cm−1, are present at 2847 and 2881 cm−1. Finally, the antisymmetric CH3 stretching is visible at 2960 cm−1.
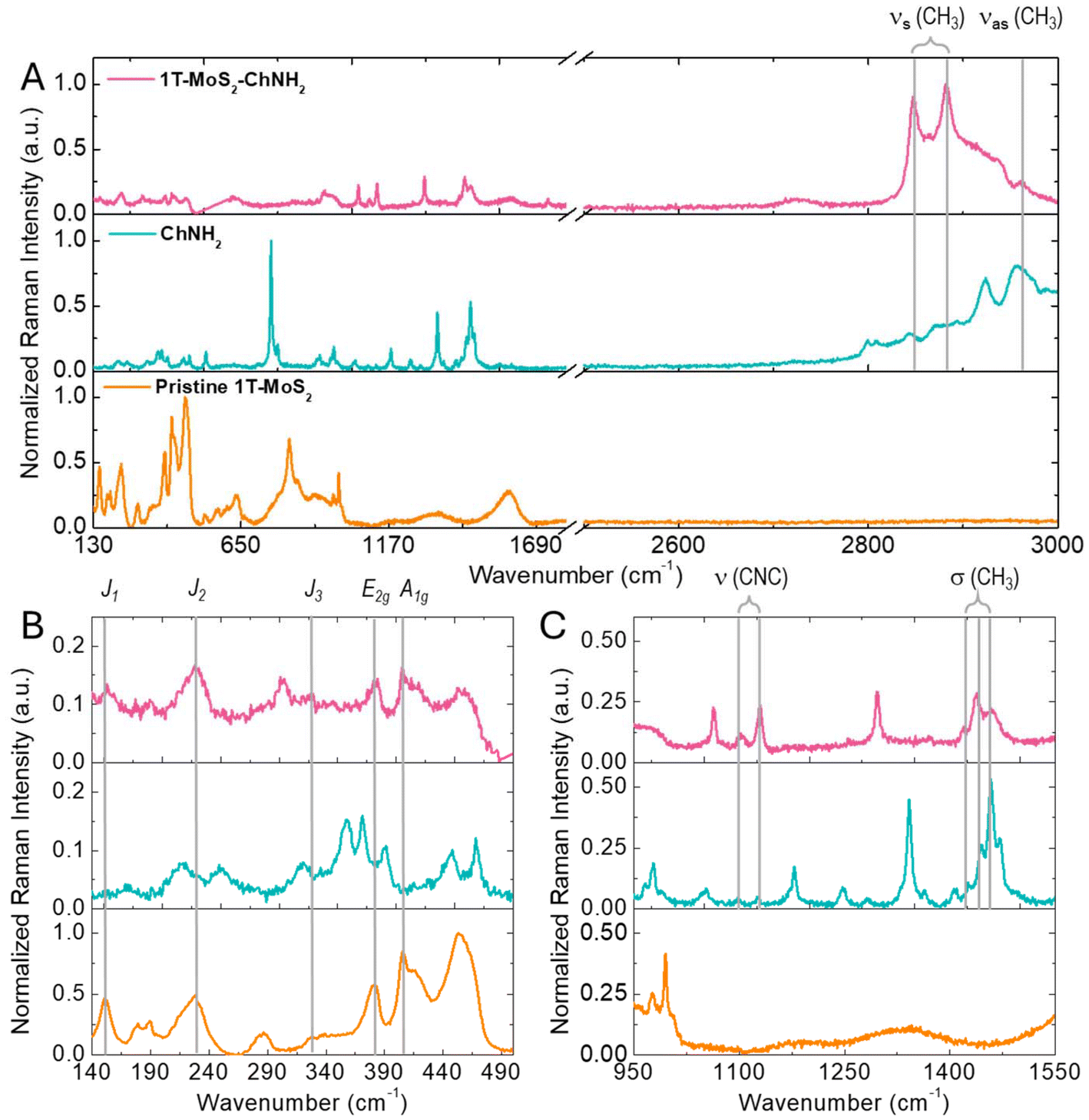 | ||
| Fig. 2 (A) Normalized Raman spectra of pristine 1T-MoS2, ChNH2, and 1T-MoS2-ChNH2; (B) magnification of the region comprised between 140 and 490 cm−1; (C) magnification of the region comprised between 950 and 1550 cm−1. All spectra were collected using a 633 nm excitation source. | ||
It is interesting to notice how some of the observed peaks experience an enhancement of their intensity in the spectrum of 1T-MoS2-ChNH2. These peaks (2847, 2881, 1102, 1129, 1419, 1438, and 1459 cm−1) are those related to the CH3 and C–N–C vibration of the methyl ammonium group. On the other hand, the vibrations associated with the in-plane (756 cm−1) and out-of-plane (1471 cm−1) bending of amino groups, pronounced in the ChNH2 spectrum, present a reduced intensity in 1T-MoS2-ChNH2. This peculiar observation can be explained by considering that MoS2 is known as SERS active material, able to enhance the intensity of the Raman modes of the functional groups closely absorbed on its surface. This evidence demonstrates that ChNH2 adopted the desired orientation on MoS2 with the methylammonium adsorbed on the surface and the amine available for further reactions.
To evaluate if the MoS2 preserved its conductive properties, we performed electrochemical impedance spectroscopy (EIS) using a glassy carbon electrode modified by drop casting the pristine material and 1T-MoS2-ChNH2 onto the carbon surface. EIS was conducted in a 5 mM solution of K3[Fe(CN6)]/K4[Fe(CN6)] using 0.1 M of PBS. Fig. 3A reports the Bode plot of pristine 1T-MoS2 and 1T-MoS2-ChNH2, showing that the impedance of the material was not affected by the functionalization in the investigated frequency range. The uncompensated resistance, corresponding to the impedance at high frequency, remained comparable: 38.89 Ω for pristine 1T-MoS2versus 40.78 Ω for 1T-MoS2-ChNH2. These data confirm that the material preserved its advantageous electrochemical and electrical properties. The Nyquist plot (Fig. 3B) of 1T-MoS2-ChNH2 shows a more marked semicircle due to the charge transfer process. This is because after the functionalization the surface charge of the 1T-MoS2 is shielded, favouring the interaction with the negatively charged [Fe(CN6)3−].
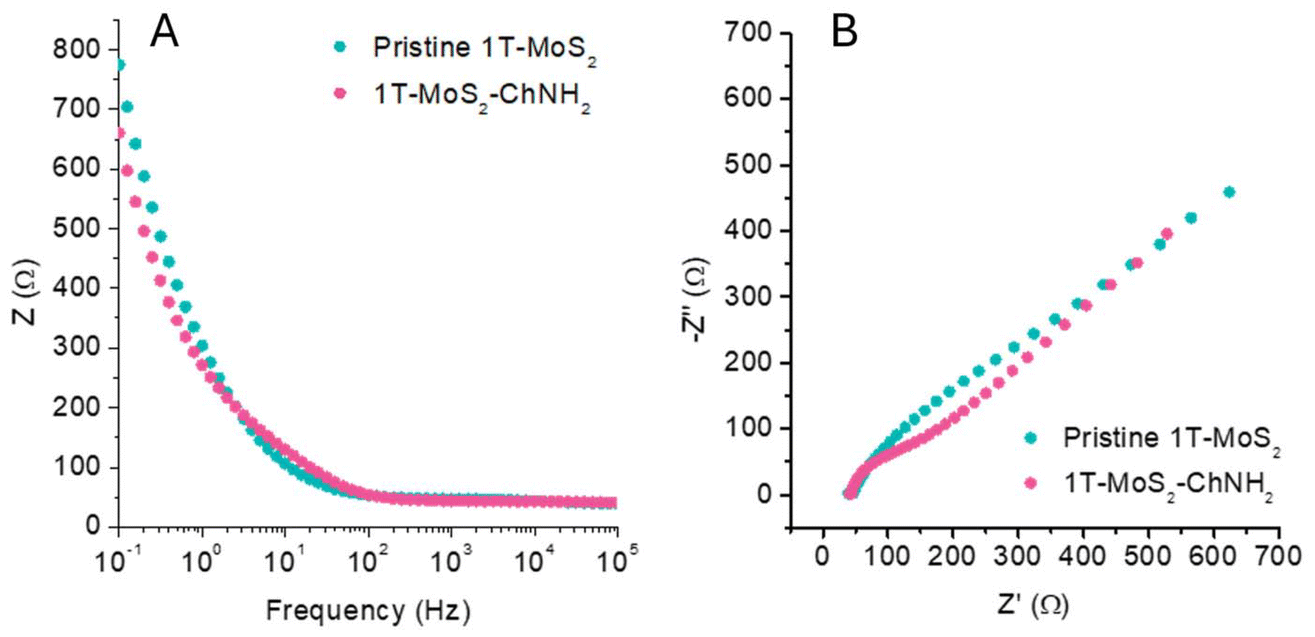 | ||
| Fig. 3 EIS measurements of a GCE electrode modified with pristine 1T-MoS2 and 1T-MoS2-ChNH2 in the presence of 5 mM K3[Fe(CN)6] in 0.1 M PBS. (A) Bode plot, (B) Nyquist plot. | ||
To demonstrate that the amino groups are exposed and reactive on the material surface we performed a post-functionalization with p-fluorobenzaldehyde (pFBA). The benzaldehyde allows the formation of an imine with the primary amino group of cholamine while, the presence of the fluorine atom facilitates the characterization of the final material, using FT-IR and STEM-EDX.
FT-IR was used to monitor the formation of the imine bond. As shown in Fig. S8† the intense peaks of MoS2 dominate the FT-IR spectra. However, after the reaction with pFBA four new peaks arise between 1200 and 1800 cm−1. Fig. 4A reports the subtraction between the 1T-MoS2-ChNH2 and 1T-MoS2-ChN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-pFB spectra, to better appreciate these peaks. The peak at 1606 cm−1 can be attributed to the CN stretching of the formed Schiff's base.23 The peaks at 1377 cm−1 (ν(C–F)), 1459 cm−1 and 1560 cm−1 (ν(CC)) further confirm the successful functionalization.24 Another evidence of the successful functionalization is the absence of CO vibrations around 1700 cm−1, which indicates that all the aldehyde reacted to form the imine and that the pFBA is not simply adsorbed on the material surface.
C-pFB spectra, to better appreciate these peaks. The peak at 1606 cm−1 can be attributed to the CN stretching of the formed Schiff's base.23 The peaks at 1377 cm−1 (ν(C–F)), 1459 cm−1 and 1560 cm−1 (ν(CC)) further confirm the successful functionalization.24 Another evidence of the successful functionalization is the absence of CO vibrations around 1700 cm−1, which indicates that all the aldehyde reacted to form the imine and that the pFBA is not simply adsorbed on the material surface.
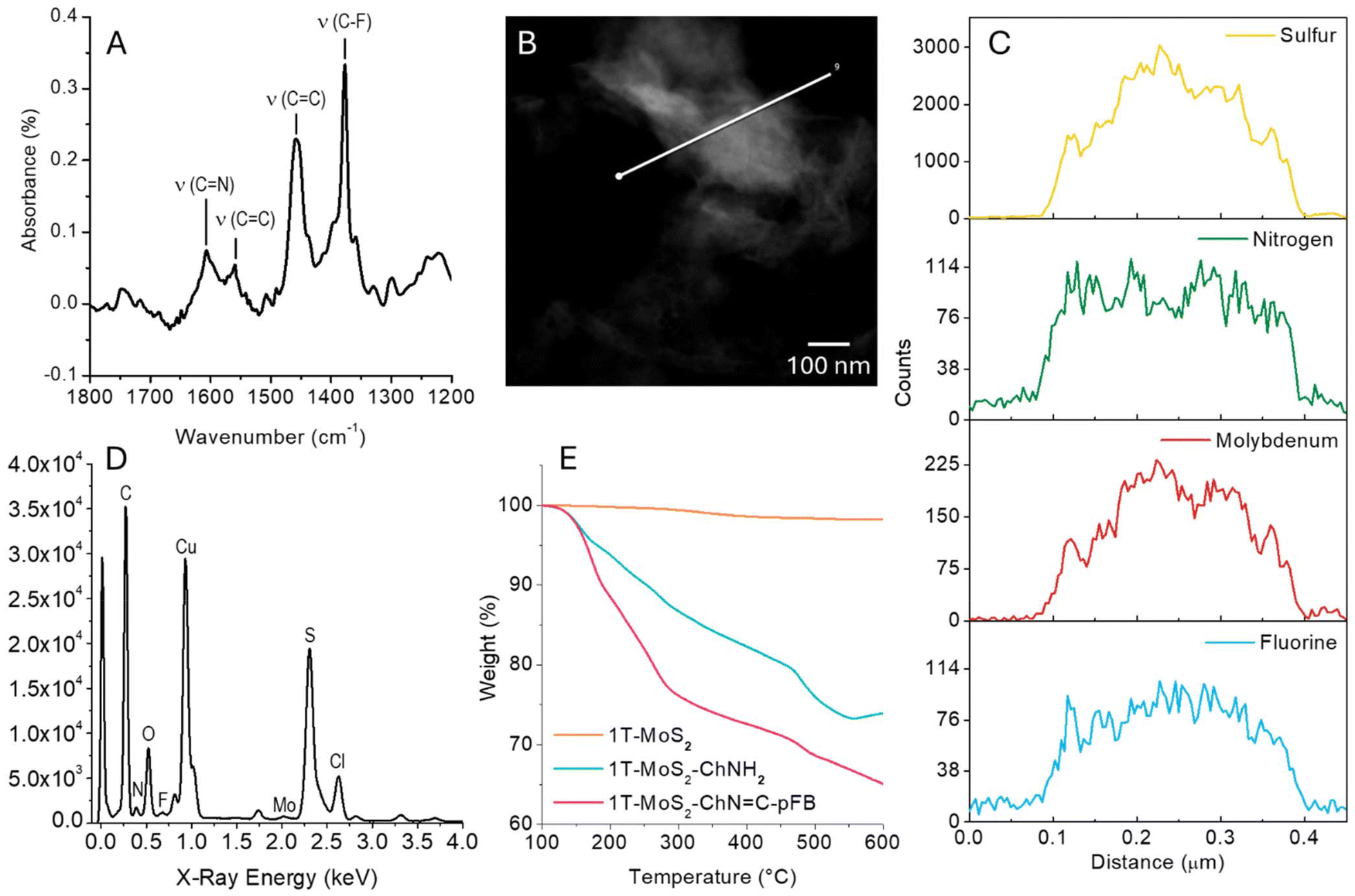 | ||
| Fig. 4 (A) Subtraction between the FT-IR spectra of 1T-MoS2-ChNH2 and 1T-MoS2-ChNC-pFB. (B) STEM micrograph of 1T-MoS2-ChNC-pFB. The white line indicates the region where the EDX line scan was performed. (C) Sulfur, nitrogen, molybdenum and fluorine profiles relative to the EDX line-scan highlighted in panel B. (D) Sum EDX spectrum of the line scan highlighted in panel B. (E) TGA of pristine 1T-MoS2, 1T-MoS2-ChNH2 and 1T-MoS2-ChNC-pFB, performed in N2 atmosphere. | ||
Once it was demonstrated that the pFBA is covalently linked to ChNH2, we used STEM-EDX mapping to observe the distribution of nitrogen and fluorine on the surface of 1T-MoS2-ChNC-pFB. Fig. 4B, C, and D show the STEM image, EDX line-scan mapping of Mo, S, N, and F and the sum spectra acquired from one 1T-MoS2-ChNH2 flake. The four elements are co-localized. N and F signals are well distributed on the flake surface, demonstrating an efficient and homogeneous material functionalization. STEM-EDX images, line scans, and spectra of the pristine MoS2 and 1T-MoS2-ChNH2 are reported in ESI (Fig. S9 and S10†).
Finally, the functionalization degree for each step was estimated using thermogravimetric analysis (TGA, Fig. 4E and Fig. S11†). The difference in weight loss between pristine 1T-MoS2, 1T-MoS2-ChNH2, and 1T-MoS2-ChNC-pFB was evaluated at 350 °C. At this temperature, under an inert atmosphere (N2), MoS2 is stable while the organic functionalization is completely degraded, without degrading the crystalline lattice.4,25,26 The functionalization with cholamine induced a variation of the weight of 17.4%, which corresponds to a functionalization degree of 1.67 μmol mg−1. This remarkable result corresponds to a functionalization yield of 26% (1 cholamine every 3.8 Mo atoms). The weight loss correlated with the pFBA functionalization equals 10.2%, corresponding to a 32.6% functionalization yield for the second step. This means 1:3.2 primary amine is available to form the imine with pFBA.
Conclusions
Herein, we present a supramolecular strategy to functionalize chemically exfoliated 1T-MoS2. This approach exploits the electrostatic interaction between the negatively charged material and positively charged alkylammonium salts. Three different ammonium salts were tested to demonstrate the method's versatility. The proposed supramolecular approach allows the achievement of a remarkable functionalization yield (with an alkylammonium:Mo ratio of 1:3.8) while preserving the material's crystalline phase and conductive properties. An additional advantage of the proposed strategy is that it can be performed in water and does not rely on organic solvents hazardous to health and the environment. Furthermore, we demonstrated that the covalent post-functionalization of the material could be achieved by exploiting the reactivity of cholamine primary amine. As a case study, we investigated the reaction with aldehydes to form a Schiff's base. The possibility of performing post-functionalization represents an important step forward compared with previously proposed approaches based on electrostatic interaction.
Experimental section
Materials and methods
All reagents were purchased from Sigma-Merck and used without further purification, with the exception of N′,N′,N′-triethylethane-1,2-diaminium salt (Et3NEtNH2), which was synthesized as reported in ESI.†DLS measurements were performed on a Zetasizer Nano (Malvern Panalytical Ltd) using a ζ-potential cuvette (Malvern Panalytical Ltd, model DTS1070).
UV-Vis spectra were collected with an Agilent Cary 5000 UV-Vis spectrophotometer at room temperature with quartz cuvettes in a wavelength range from 200 nm to 800 nm.
Raman spectra were recorded using a Renishaw inVia Raman microscope equipped with a of 633 nm laser, 1800 gr per mm gratings, Peltier-cooled front-illuminated CCD camera (1024 × 532 px), and a 100× objective. Samples were prepared by drop-casting MoS2 solution on a silicon oxide slide. Each spectrum is derived from the average of at least 100 spectra recorded in different spots of the sample for 5 s with a laser power of 1.29 mW. Data were processed using Renishaw WiRE 4 software.
AFM images were obtained with a Nanoscope IIIa, VEECO Instruments in tapping mode with a HQ:NSC19/ALBS probe (80 kHz; 0.6 N m−1) (MikroMasch). Samples were prepared by drop cast of the aqueous suspension on exfoliated mica substrates. The obtained AFM images were analysed in S3 Gwyddion 2.58.
TEM and STEM-EDX mapping images were recorded with a TEM – JEOL JEM – 2100F UHR transmission electron microscope equipped with a field emission electron beam FEG (field emission gun) with variable acceleration between 80 and 200 kV, a 4-megapixel CMOS camera (TVIPS TemCam-F216) and two STEM detectors (VF & HAADF) in combination with an EDX detector (Oxford UltimMax). Samples were prepared by dropcast of the aqueous suspensions on Lacey carbon 300 mesh copper grids.
X-ray photoelectron spectroscopy (XPS) spectra were registered with an XPS/UPS – SPECS SAGE HR-100 equipped with a 100 mm radius PHOIBOS analyser, where 421 Mg Kα X-ray source was used. The samples were prepared by depositing 0.5 mg on copper tape. The spectra were fitted using CasaXPS and calibration was performed using the major component of the C high-resolution spectrum as a reference at 284.8 eV, which is the value reported for adventitious and sp3 C–C carbon.
Thermogravimetric analysis (TGA) measurements were carried out with a TGA Discovery (TA Instruments), where 0.4 mg of material were weighed in a platinum HT pan and were heated in N2 atmosphere. First, an isotherm at 100 °C was applied 20 min, subsequently the samples were heated to 600 °C with a ramp of 10 °C min−1.
EIS was registered using an Autolab MSTAT204 potentiostat/galvanostat (Metrohm). A three-electrode configuration, composed by GCE as a working electrode (WE), Pt wire as a counter electrode (CE), and an Ag/AgCl reference electrode (RE), was employed. 10 μl of the materials suspensions (0.5 mg ml−1) were drop-casted onto the WE (electrode area 0.785 cm2). Measurements were performed in a 5 mM solution of K3[Fe(CN)6] using as an electrolyte 100 mM PBS buffer (containing 100 mM KCl, Ph 7.4), A potential of 0.21 V (vs. reference) was applied with a perturbation of 10 mV, in the frequency range between 100 kHz and 100 mHz.
1T-MoS2 chemical exfoliation
The exfoliated 1T-MoS2 was obtained following procedure reported in the literature.21 250 mg of MoS2 were dried at 100 °C in a 50 mL Schlenck tube for 16 h and then heated with a heat gun under vacuum to eliminate any trace of water. Under an inert atmosphere (Ar), 15 mL of dry hexane and 4 mL of butyllithium (2.5 M in hexane) were added, and the mixture was heated at 70 °C for 24 h. The mixture was then cooled with an ice bath and BuLi was quenched by adding water drop by drop until the disappearance of bubbling. The crude was sonicated to disperse the material, then the material was extracted from the organic phase with ultrapure water using a separating funnel. The aqueous phase was washed twice with hexane, was sonicated for 1 h, and subsequently centrifuged at 1000 rpm for 1.5 h at 5 °C. The supernatant was filtered on PTFE filters and washed with ultrapure water to recover the material, which was finally resuspended in ultrapure water.ζ-Potential titration
For the titration with ChNH2 and Et3NEtNH2, 7 aliquots of 1T-MoS2 aqueous suspension (0.5 mg mL−1) were incubated with different concentrations of ammonium salts (0.1, 0.2, 0.3, 0.4, 0.5, 0.75 and 1 equivalents with respect to the mol of Mo), and ζ-potential was measured after 10 minutes. For the titration with PDADMAC, 12 aliquots of 1T-MoS2 aqueous suspension (0.5 mg mL−1) were incubated with different concentrations of PDADMAC (0.015, 0.04, 0.06, 0.075, 0.1, 0.15, 0.2, 0.4, 0.75, 1.5, 3.75 and 5.6 equivalents with respect to the mol of Mo) and ζ-potential was measured after 10 minutes.Functionalization with alkylammonium salt
2 mL of 1T-MoS2 aqueous suspension (0.5 mg mL−1) was sonicated in ice bath for 15 min. 1 equivalent of alkylammonium salts (with respect to Mo) was solubilized in 100 μL of ultrapure water and added to the 1T-MoS2 suspension. The mixture was stirred for 30 min at room temperature, then sonicated for 5 min in ice bath, and finally stirred for other 30 min.The precipitate was separated by centrifugation (2000 rpm, 30 min). The solid was washed twice with 1 mL of milliQ water, each time sonicating for 5 min and centrifuging at 2000 rpm for 30 min. Finally, water was removed by lyophilization, obtain a dry powder.
Post-functionalization with p-fluorobenzaldehyde
The obtained 1T-MoS2-ChNH3 was suspended in methanol (1 mL), by adding 20 μL of triethylamine 0.72 mol L−1 (14.4 μmol, 4.6 equiv. relative to the starting ammonium salt), and sonicating for 10 min in ice bath. Then, p-fluorobenzoaldehyde (8.7 μL of a 10% solution in methanol, 6.25 μmol, 1 equiv. relative to the starting ammonium salt) was added and the mixture was sonicated for 5 min and then incubated for 1 h. Finally, the imine derivative was isolated by centrifugation (6000 rpm, 10 min), resuspending, and washing twice with methanol (1 mL).Author contributions
Giuseppe Misia: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft; Michele Cesco: data curation, formal analysis, methodology, writing – review & editing; Maurizio Prato: funding acquisition, resources, supervision, writing – review & editing; Alessandro Silvestri: data curation, funding acquisition, methodology, resources, supervision, writing – original draft, writing – review & editing.Data availability
The experimental data related to the paper have been uploaded on Datarepository Unive (https://datarepository.unive.it/). The data are identified by the following https://doi.org/10.71731/DATA_UNIVE/ML4R7D.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. P. is the AXA Chair for Bionanotechnology (2016–2026). The authors gratefully acknowledge the financial support from the Università degli Studi di Trieste, Università Ca’ Foscari di Venezia, INSTM, and the Italian Ministry of Education Ministero dell'Istruzione, dell'Università e della Ricerca (cofin Prot. 20228YFRNL).References
- D. Voiry, A. Mohite and M. Chhowalla, Chem. Soc. Rev., 2015, 44, 2702–2712 RSC.
- X. Yin, C. S. Tang, Y. Zheng, J. Gao, J. Wu, H. Zhang, M. Chhowalla, W. Chen and A. T. S. Wee, Chem. Soc. Rev., 2021, 50, 10087–10115 RSC.
- D. Escalera-López, C. Iffelsberger, M. Zlatar, K. Novčić, N. Maselj, C. Van Pham, P. Jovanovič, N. Hodnik, S. Thiele, M. Pumera and S. Cherevko, Nat. Commun., 2024, 15, 3601 CrossRef PubMed.
- E. Er, H.-L. Hou, A. Criado, J. Langer, M. Möller, N. Erk, L. M. Liz-Marzán and M. Prato, Chem. Mater., 2019, 31, 5725–5734 CrossRef CAS.
- E. Er, A. Sánchez-Iglesias, A. Silvestri, B. Arnaiz, L. M. Liz-Marzán, M. Prato and A. Criado, ACS Appl. Mater. Interfaces, 2021, 13, 8823–8831 CrossRef CAS PubMed.
- L. Chen, J. P. Merino, M. Torrent-Sucarrat, H.-L. Hou and M. Prato, Adv. Mater. Interfaces, 2024, 11, 2400272 CrossRef CAS.
- C. Wetzl, A. Silvestri, M. Garrido, H.-L. Hou, A. Criado and M. Prato, Angew. Chem., 2023, 135, e202212857 CrossRef.
- Q. Tang and D. Jiang, Chem. Mater., 2015, 27, 3743–3748 CrossRef CAS.
- L. Chen, H.-L. Hou and M. Prato, Chem. Mater., 2023, 35, 5032–5039 CrossRef CAS.
- X. Chen, N. C. Berner, C. Backes, G. S. Duesberg and A. R. McDonald, Angew. Chem., 2016, 128, 5897–5902 CrossRef.
- L. Daukiya, J. Teyssandier, S. Eyley, S. E. Kazzi, M. C. R. González, B. Pradhan, W. Thielemans, J. Hofkens and S. D. Feyter, Nanoscale, 2021, 13, 2972–2981 RSC.
- M. Vera-Hidalgo, E. Giovanelli, C. Navío and E. M. Pérez, J. Am. Chem. Soc., 2019, 141, 3767–3771 CrossRef CAS PubMed.
- M. Garrido, A. Criado and M. Prato, Nanoscale, 2024, 16, 13525–13533 RSC.
- J. Zheng, H. Zhang, S. Dong, Y. Liu, C. T. Nai, H. S. Shin, H. Y. Jeong, B. Liu and K. P. Loh, Nat. Commun., 2014, 5, 2995 CrossRef PubMed.
- T. H. M. Lau, S. Wu, R. Kato, T.-S. Wu, J. Kulhavý, J. Mo, J. Zheng, J. S. Foord, Y.-L. Soo, K. Suenaga, M. T. Darby and S. C. E. Tsang, ACS Catal., 2019, 9, 7527–7534 CrossRef CAS.
- Z. Chen, K. Leng, X. Zhao, S. Malkhandi, W. Tang, B. Tian, L. Dong, L. Zheng, M. Lin, B. S. Yeo and K. P. Loh, Nat. Commun., 2017, 8, 14548 CrossRef CAS PubMed.
- D. Voiry, A. Goswami, R. Kappera, C. de Carvalho Castro e Silva, D. Kaplan, T. Fujita, M. Chen, T. Asefa and M. Chhowalla, Nat. Chem., 2015, 7, 45–49 CrossRef CAS PubMed.
- S. Geng, W. Yang, Y. Liu and Y. Yu, J. Catal., 2020, 391, 91–97 CrossRef CAS.
- S. Presolski, L. Wang, A. H. Loo, A. Ambrosi, P. Lazar, V. Ranc, M. Otyepka, R. Zboril, O. Tomanec, J. Ugolotti, Z. Sofer and M. Pumera, Chem. Mater., 2017, 29, 2066–2073 CrossRef CAS.
- D. Iglesias, S. Ippolito, A. Ciesielski and P. Samorì, Chem. Commun., 2020, 56, 6878–6881 RSC.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- G. Gamer and H. Wolff, Spectrochimica Acta Part A, 1973, 29, 129–137 CrossRef CAS.
- H. Namli and O. Turhan, Spectrochim. Acta, Part A, 2006, 64, 93–100 CrossRef PubMed.
- D. Mahadevan, S. Periandy and S. Ramalingam, Spectrochim. Acta, Part A, 2011, 84, 86–98 CrossRef CAS PubMed.
- M. Garrido, A. Naranjo and E. M. Pérez, Chem. Sci., 2024, 15, 3428–3445 RSC.
- X. S. Chu, A. Yousaf, D. O. Li, A. A. Tang, A. Debnath, D. Ma, A. A. Green, E. J. G. Santos and Q. H. Wang, Chem. Mater., 2018, 30, 2112–2128 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05515b |
| This journal is © The Royal Society of Chemistry 2025 |