
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
HFIP-mediated, regio- and stereoselective hydrosulfenylation of ynamides: a versatile strategy for accessing ketene N,S-acetals†
Appanapalli N. V.
Satyanarayana
,
Paramita
Pattanayak
 and
Tanmay
Chatterjee
*
and
Tanmay
Chatterjee
*
Department of Chemistry, Birla Institute of Technology and Science, Pilani, Hyderabad Campus, Jawahar Nagar, Hyderabad – 500078, India. E-mail: tanmay@hyderabad.bits-pilani.ac.in
First published on 23rd January 2025
Abstract
Herein, we report an HFIP-mediated, versatile, sustainable, atom-economical, and regio- and stereoselective hydro-functionalization of ynamides with various S-nucleophiles (1 equiv.) such as thiols, thiocarboxylic acids, carbamates, xanthates, and O,O-diethyl S-hydrogen phosphorothioate to access a wide variety of stereodefined trisubstituted ketene N,S-acetals under mild conditions. This protocol requires only HFIP, which plays multiple roles, such as acting as a Brønsted acid to protonate the ynamide regioselectively at the beta carbon to generate the reactive keteniminium intermediate, stabilizing the intermediate as solvent through H-bonding. After the nucleophilic attack of the S-nucleophile on the keteniminium intermediate and deprotonation, HFIP is regenerated in most of the cases and can be easily recovered and recycled, revealing the high sustainability of the protocol. Remarkably, all the reactions are highly efficient and furnish ketene N,S-acetals in excellent yields and in many cases pure products were obtained just by washing the crude reaction mixture with pentane. Significantly, the green chemistry metrics of the protocol are found to be excellent.
Introduction
Ketene N,S-acetals represent valuable entities in the realm of organic synthesis and exhibit a wide range of applications across multiple fields, including pharmaceuticals,1,2 agrochemicals3,4 and materials science. This class of molecules serves as valuable building blocks in numerous organic transformations, thereby facilitating the synthesis of diverse heterocycles.5–8 Furthermore, certain cyclic ketene N,S-acetals demonstrate enhanced efficacy in various applications. For instance, nithiazine A, a neonicotinoid insecticide,3,4,8 ranks among the most widely utilized insecticides globally. Additionally, compounds such as etozolin B and ralitoline C are employed in the treatment of neurological disorders and hypertension (Fig. 1).1,2 Thiazole Orange (TO) D functions as a probe for the early detection of cancer cell labelling,9,10 while SYBR Safe E is an effective probe for investigating interactions between G-quadruplexes and their ligands (Fig. 1).10,11 Although several strategies have been developed for the hydrosulfenylation of alkynes to synthesize functionalized vinyl sulphides, the synthesis of ketene N,S-acetals, a special class of vinyl sulphides, was found underdeveloped.12 Previous studies have documented various multi-step synthetic methodologies for the synthesis of a very specific type of ketene N,S-acetal bearing an electron-withdrawing carbonyl functional group.5,8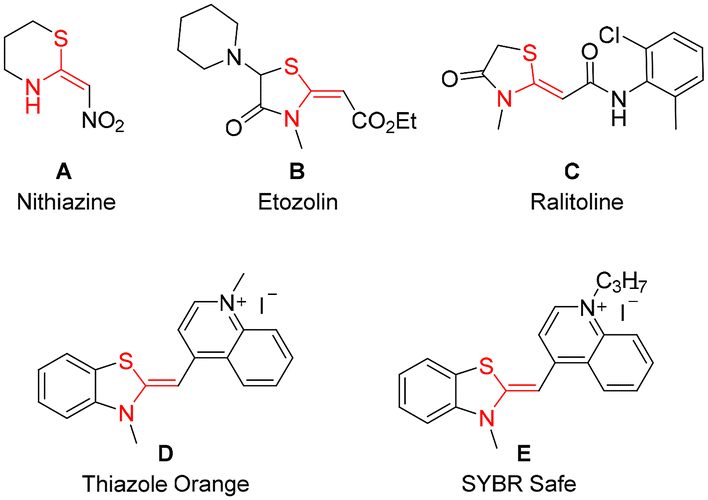 | ||
| Fig. 1 Some important ketene N,S-acetal-containing drugs, insecticides, and probes. | ||
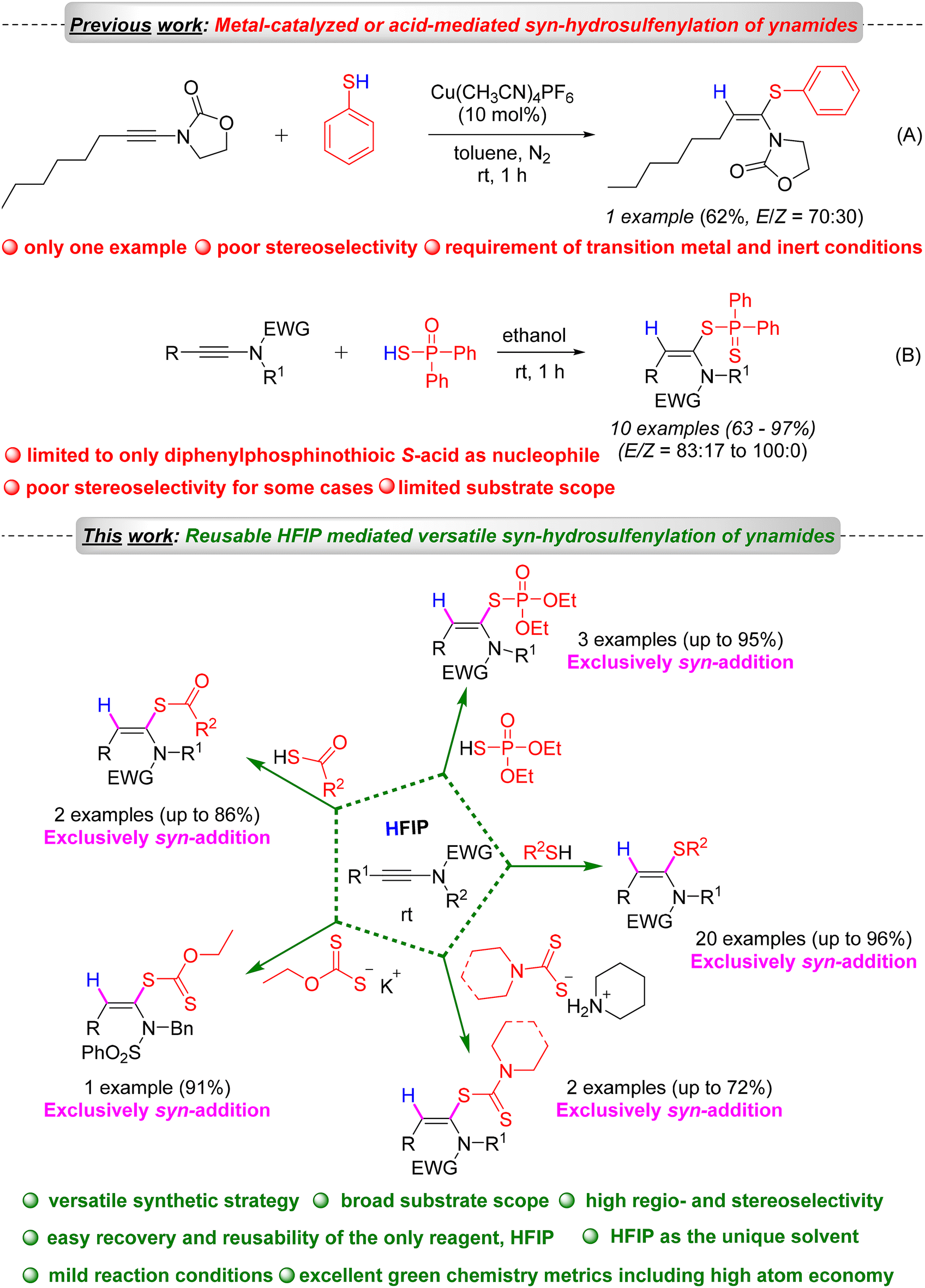
In 2023, the Taillefer group reported the synthesis of only one example of ketene N,S-acetals, with moderate yield (62%) and poor stereoselectivity (E/Z = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30) (Scheme 1).13 In 2008, Koichiro Oshima first reported the direct synthesis of ketene N,S-acetal derivatives (only 10 examples) from ynamides using diphenyldithiophosphinic acid in dimethoxyethane at ambient temperature (Scheme 1).14
:30) (Scheme 1).13 In 2008, Koichiro Oshima first reported the direct synthesis of ketene N,S-acetal derivatives (only 10 examples) from ynamides using diphenyldithiophosphinic acid in dimethoxyethane at ambient temperature (Scheme 1).14
 | ||
| Scheme 1 Synthetic strategies to access ketene N,S-acetals. | ||
However, these methodologies suffer from some serious limitations, such as limited substrate scope, use of only diphenyldithiophosphinic acid as the S-nucleophile (limited scope), poor stereoselectivity, and the need for a transition metal. A straightforward, one-pot, regio- and stereoselective synthesis of ketene N,S-acetals is underdeveloped. Recently, various regio- and stereoselective difunctionalization and hydrofunctionalization methods of ynamides have been developed by several research groups to access various functionalized enamides.15–17 As part of our continued interest in developing metal-free and sustainable synthetic methodologies,18–23 in particular, exploring hexafluoroisopropanol (HFIP) and ynamides in organic synthesis,19 and based on the importance of ketene N,S-acetals, we developed a metal-free, HFIP-mediated highly regio- and stereoselective hydrosulfenylation of ynamides for the sustainable and versatile synthesis of a wide variety of ketene N,S-acetals under mild reaction conditions such as room temperature and aerobic atmosphere.
Results and discussion
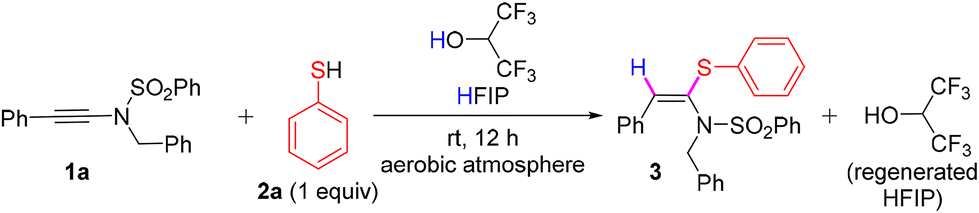
We commenced our investigation by reacting N-benzyl-N-(phenylethynyl)benzenesulfonamide (1a) with thiophenol 2a in the presence of HFIP (0.6 M) at ambient temperature under an aerobic atmosphere and gratifyingly, 1a underwent syn-hydrosulfenylation with 2a to furnish the desired product, (E)-N-benzyl-N-(2-phenyl-1-(phenylthio)vinyl)benzenesulfonamide 3 in an excellent yield of 92% and with exclusive (E)-stereoselectivity (entry 1, Table 1). When the loading of HFIP was decreased to 10 equiv., we observed that the solubility of 1a decreased, resulting in an unsuccessful reaction. To check the feasibility of the reaction using HFIP as only the reagent, not as the solvent, we treated 1a with HFIP (1 equiv.) in toluene (0.6 M) at ambient temperature and also at 70 °C; however, only trace amounts of 3 were formed in both cases (entries 3 and 4, Table 1). When another Brønsted acid, such as acetic acid, was used instead of HFIP, the acetic acid addition product of 1a was formed instead of 3 (entry 5, Table 1). Similarly, when TfOH was used instead of HFIP, the TfOH addition product of 1a was formed along with 3 (entry 6, Table 1). With the change of the solvent from HFIP to trifluoroethanol and ethanol, the reaction was unsuccessful, and 1a was recovered fully (entries 7 and 8, Table 1). When pentafluorophenol (0.6 M) was used as the solvent instead of HFIP, the hydro-pentafluorophenoxylation product of 1a, i.e., X (major), was formed along with the formation of 3 (minor amounts) in a 9:1 ratio with an overall quantitative yield (see Scheme S1 in the ESI†). Hence, for the synthesis of 3via the hydrosulfenylation of 1a, the use of only one equiv. of 2a in the presence of HFIP (0.6 M) was found to be optimum at room temperature under an aerobic atmosphere.
|
|
|||
|---|---|---|---|
| Entry | Variation in conditions from the scheme | Temperature (°C) | Yieldb (%) |
|
a All the reactions were conducted on a 0.1 mmol scale.
b Yield of the product was determined by 1H NMR of the crude reaction mixture using 1,3,5-trimethoxybenzene as the internal standard.
c Acetic acid addition product of 1a was formed exclusively.
d TfOH addition product of 1a was formed along with the formation of 3 (messy NMR).
e Pentafluorophenol addition product of 1a, i.e.X, was formed along with the formation of 3 (X:3 = 9:1) in a quantitative yield and the product X was isolated when the reaction was performed on a 0.3 mmol scale and characterized by NMR (see the ESI†).
|
|||
| 1 | None | rt | 92 |
| 2 | 10 equiv of HFIP was used | rt | Trace |
| 3 | HFIP (1 equiv) and toluene (0.6 M) were used | rt | Trace |
| 4 | HFIP (1 equiv) and toluene (0.6 M) were used | 70 | Trace |
| 5 | CH3COOH (2 equiv) and toluene (0.6 M) were used instead of HFIP | rt | 0c |
| 6 | TfOH (1 equiv) and toluene (0.6 M) were used instead of HFIP | rt | 20d |
| 7 | Trifluoroethanol was used instead of HFIP | rt | 0 |
| 8 | Ethanol was used instead of HFIP | rt | 0 |
| 9 | Pentafluorophenol was used instead of HFIP | rt | 10e |
With the optimized conditions in hand, we next explored the scope of syn-hydrosulfenylation of ynamides with thiols first (Table 2). When the reaction of 1a was performed with 2a on a 0.3 mmol scale, the desired product 3 was formed without any compromise in yield (92%). Both electron-withdrawing (4-Br, 4-F, and 4-CF3) and electron-donating (4-Me and 4-OMe) group substituted aryl thiols underwent the syn-hydrosulfenylation reaction with 1a smoothly to furnish the desired products (4–8) in excellent yields (85–96%, average yield: 91.8%). Next, we explored the scope of aliphatic thiols and, notably, both primary and secondary aliphatic thiols such as benzyl mercaptan, furan-2-ylmethanethiol, butane-1-thiol, and butane-2-thiol participated in the syn-hydrosulfenylation reaction of 1a to afford the corresponding desired products (9–12) in excellent yields (88–92%, average yield = 90%). The scope of ynamides was explored next, and both electron-withdrawing (4-Br, 4-F, and 2-CN) and electron-donating (4-Me) group substituted aryl ynamides underwent the syn-hydrosulfenylation reaction with 2a to afford the desired products (13–16) in good to excellent yields (80–93%). A heteroaryl, i.e., 2-thienyl-substituted ynamide, participated in the syn-hydrosulfenylation reaction with 2a and afforded the desired product 17 in 76% yield. Aliphatic ynamides, i.e., N-benzyl-N-(cyclohexylethynyl)benzenesulfonamide and N-benzyl-N-(cyclohexylethynyl)methanesulfonamide, also reacted with 2a to afford the desired products (18 and 19) in 94% and 87% yields, respectively. A variation in the sulfonamide part of ynamides did not affect the outcome of the syn-hydrosulfenylation reaction with 2a, as the desired products (20–22) were formed in 88–92% yields (average yield = 90%). When an N-alkynyl carbamate, i.e., methyl benzyl(phenylethynyl)carbamate 1m, was treated with 2a in HFIP, the desired product 23 was formed in 83% yield at room temperature; however, unfortunately, the ynamide 1-(2-phenylethynyl)-2-pyrrolidinone did not participate in the reaction with 2a and the desired product 24 was not formed.
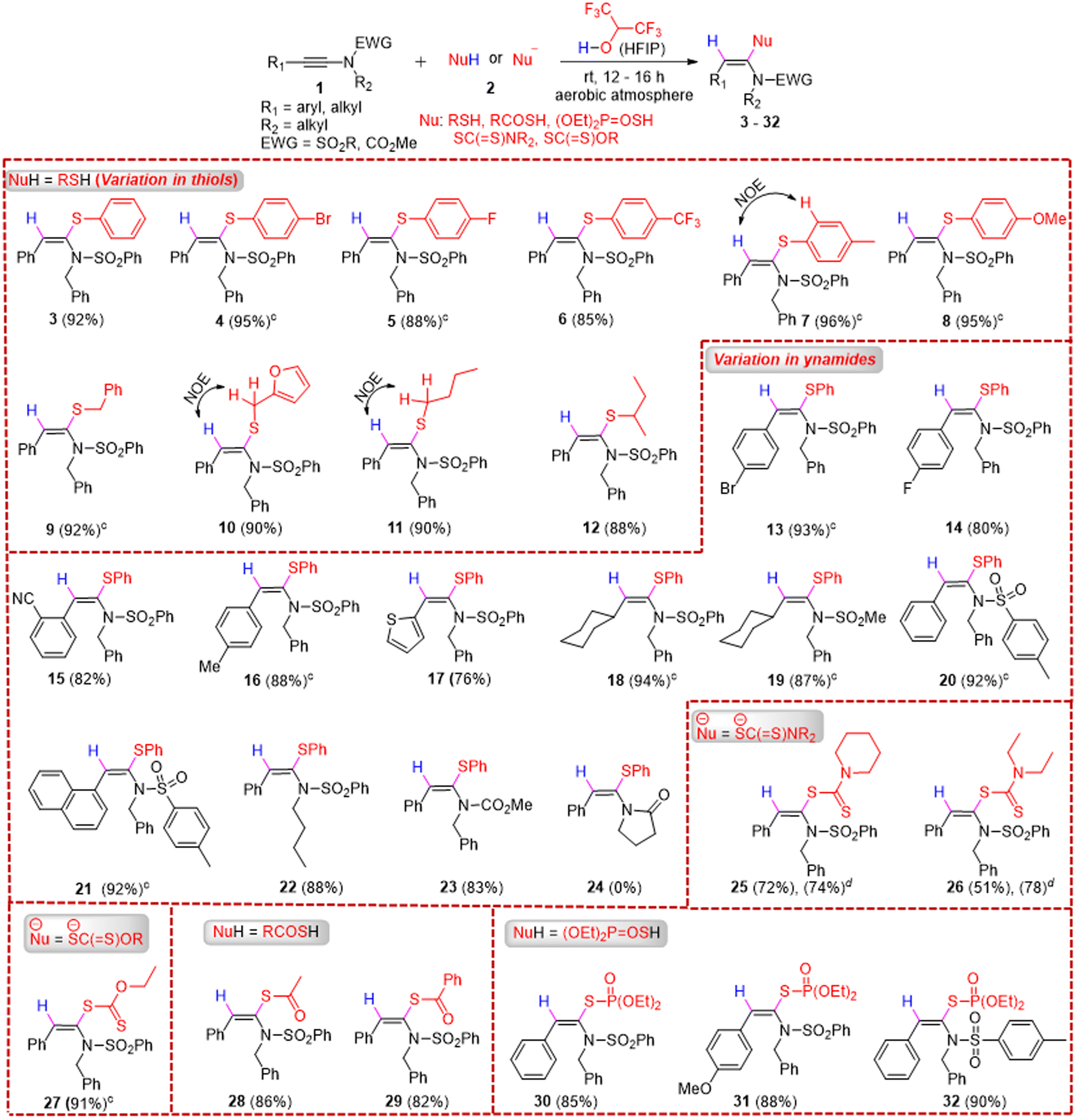
Next, we explored the scope of dithiocarbamates and xanthates for the hydrosulfenylation reaction with ynamides to access a new class of ketene N,S-acetals. When 1a was treated with in situ-generated dithiocarbamates, formed by the reaction of CS2 and amines, in the presence of HFIP, the desired syn-α-hydro-dithiocarbamation products of 1a, i.e., 25 and 26, were formed respectively in moderate to good yields. Both primary and secondary (cyclic and acyclic) amines (piperidine and diethyl amine) underwent the reaction smoothly. When potassium O-ethyl carbonodithioate was treated with 1a, the desired product 27 was formed in 91% yield. Other sulphur nucleophiles such as ethanethioic S-acid and benzothioic S-acid were employed for the hydrosulfenylation of 1a, and notably another new class of products, i.e., (E)-S-(1-(N-benzylphenylsulfonamido)-2-phenylvinyl) ethanethioate 28 and (E)-S-(1-(N-benzylphenylsulfonamido)-2-phenylvinyl) benzothioate 29, were formed in excellent yields (82–86%) under the optimized conditions. Finally, O,O-dimethyl S-hydrogen phosphorothioate was subjected to reaction with various ynamides, i.e., 1a, 1f, and 1j, under the optimized conditions, and the desired products 30–32 were also formed in excellent yields (85–90%).
The stereochemistry of various products, such as 7, 10, and 11, was confirmed by NOE difference spectroscopy (see the ESI†). Interestingly, most of the reactions were very clean, and several products such as -4, 5, 7, 8, 9, 13, 16, 18, 19, 20, 21, and 27 were obtained pure just by evaporating the solvent (HFIP) first after the reaction and then washing the crude product with a small amount of cold pentane or cold ethanol; no work-up or column chromatography was required. Thus, the requirement of huge amounts of organic solvents in purifying the product from the crude reaction mixture via work-up and column chromatographic techniques was avoided, which certainly revealed the high sustainability of our developed protocol. Moreover, the recovered HFIP could be reused for subsequent reactions (Scheme 2).19
 | ||
| Scheme 2 (A) Recovery and (B) reusability of HFIP. | ||
To demonstrate the recovery and reusability of HFIP, we performed the reaction between 1a and 2a on a 1 mmol scale, and after the completion of the reaction, 96% HFIP (1.6 mL) was recovered through simple distillation at 65 °C in an oil bath (Scheme 2A). The recovered HFIP was recycled for the synthesis of 23, and the result was reproducible (Scheme 2B).
To measure the greenness of our developed protocol quantitatively, we evaluated the green chemistry metrics17–19 of our protocol for the synthesis of 7 from 1a and 2e, and the results are summarized in Table 3.
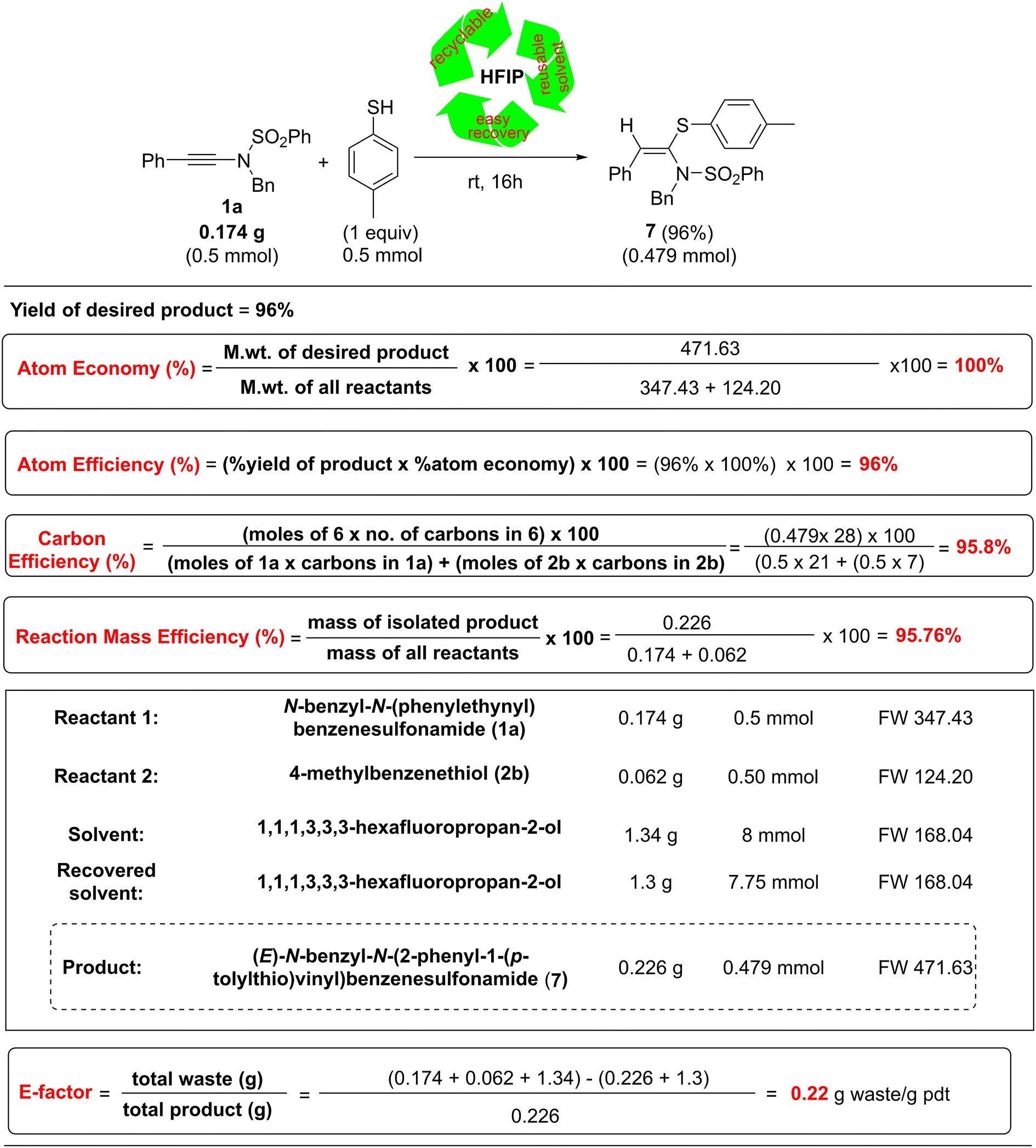
|
Notably, our protocol was found to possess 100% atom economy, 96% atom efficiency, 95.8% carbon efficiency, and 95.76% reaction mass efficiency (Table 3). Another important green parameter, i.e., the E-factor,18 which is based on the generated waste, is found to have a very low value of 1.22 g of waste generated per gram of product formed. These results demonstrated the high sustainability of our developed protocol.
Captopril (brand name: Capoten) is a marketed drug used for the treatment of hypertension. It possesses two functional groups, i.e., thiol (–SH) and carboxylic acid (–COOH), offering scope for bis-late-stage functionalization (bis-LSF) for the drug with ynesulfonamide (Scheme 3).
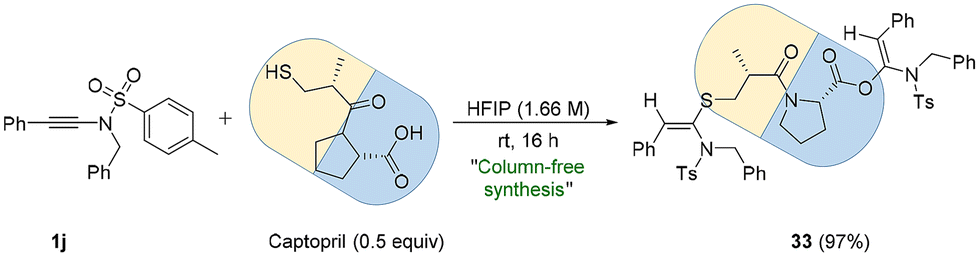 | ||
| Scheme 3 Bis-late-stage functionalization (bis-LSF) of captopril with ynamide 1j. | ||
When 1j was treated with Captopril (0.5 equiv.) in HFIP at room temperature, the desired bis-functionalized product 33 was formed in almost quantitative yield (97%), and notably, 33 was obtained pure without the requirement of column chromatography (Scheme 3).
Furthermore, to demonstrate the synthetic application of the products, we diversified one of the synthesized products, i.e., 7, into other useful classes of its derivatives, i.e., the corresponding sulfoxide 34 and sulfone 35, in 89% and 98% yields, respectively, via controlled oxidation of 7 using 0.6 and 3 equivalents of oxone in ethanol, respectively (Scheme 4).
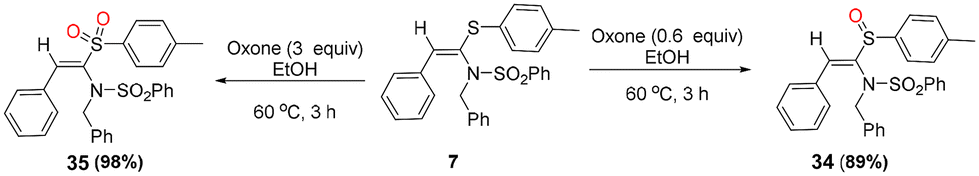 | ||
| Scheme 4 Oxone-mediated controlled oxidation of 7 to its corresponding sulfoxide (34) and sulfone (35). | ||
Based on the literature reports,16b,19 we proposed that at first, the keteniminium ion intermediate A was formed via the regioselective protonation of yensulfonamide 1a at the β-position by HFIP, which was also stabilized by HFIP. Next, A underwent a favoured syn-attack by the nucleophile to furnish the desired syn-hydrosulfenylation product of the ynamide (1a), as the anti-attack is disfavoured due to the steric hindrance from the R-group (Scheme 5).
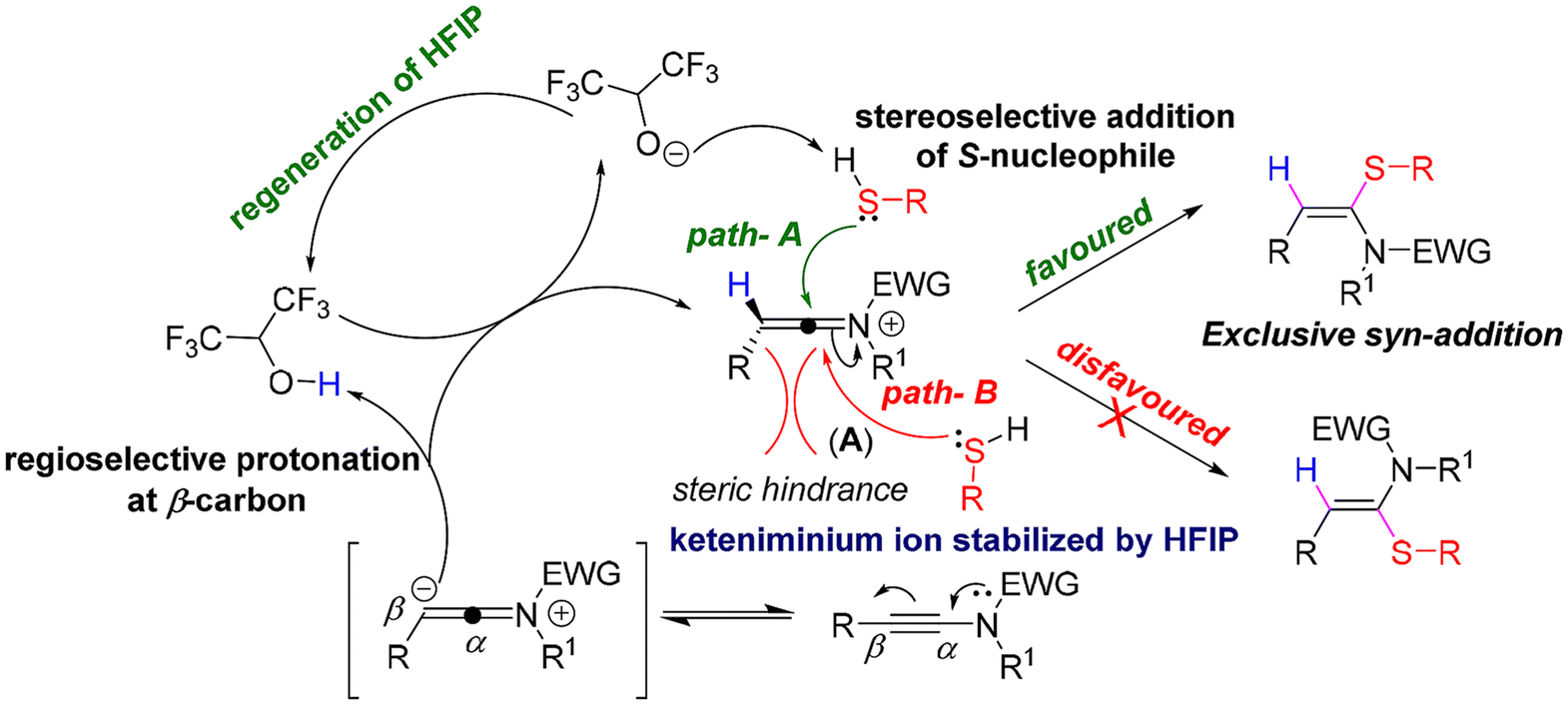 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusion
In conclusion, we developed an HFIP-mediated, highly regio- and stereoselective, and versatile protocol for the synthesis of a wide variety of stereodefined ketene N,S-acetals in good to excellent yields via the hydrosulfenylation of ynamides under mild reaction conditions such as room temperature and an aerobic atmosphere.Interestingly, a wide variety of S-nucleophiles such as thiols, thio S-acids, carbamates, xanthates, and O,O-diethyl S-hydrogen phosphorothioate were employed, and all underwent the reaction smoothly with ynesulfonamides to furnish a wide variety of N,S-acetals. The transformation required only HFIP, which played multiple roles such as Brønsted acid, keteniminium ion stabilizer, and solvent. HFIP was regenerated after the reactions when thiols, thio S-acids, and O,O-diethyl S-hydrogen phosphorothioate were used as S-nucleophiles, and it could easily be recovered and reused for subsequent reactions.19 Moreover, this protocol could be utilized for the late-stage functionalization of some drug molecules, such as captopril. Notably, this protocol features excellent green chemistry metrics including 100% atom economy, 96% atom efficiency, 95.8% carbon efficiency, and 95.76% reaction mass efficiency, and a very low value of E-factor, i.e., 1.22 g waste generated per gram of product (7) formed. We believe that this sustainable and versatile strategy will find useful applications in accessing useful N,S-acetals.
Experimental section
General experimental procedure for the synthesis of ketene N,S-acetals (3–22) via the hydrothiolation of ynesulfonamides
General experimental procedure for the synthesis of ketene N,S-acetals (25–26) via the hydro-dithiocarbamation of ynesulfonamides
General experimental procedure for the synthesis of ketene N,S-acetals (28–29) with thio S-acids
General experimental procedure for the synthesis of ketene N,S-acetals (30–32) with O,O-diethyl S-hydrogen phosphorothioate
Author contributions
A. N. V. Satyanarayana carried out most of the experiments, including optimization of the reaction conditions, synthesis of products, their synthetic diversifications, and mechanistic studies. P. Pattanayak also synthesized some of the ketene N,S-acetals. T. Chatterjee conceived and supervised the work and wrote and edited the manuscript. All the authors have given their final approval to the final version of the manuscript.Data availability
The data underlying this study are available in the published article, and its experimental and spectroscopic details are included as part of the online ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. Chatterjee gratefully acknowledges the financial support from the Science and Engineering Research Board (SERB), Govt. of India [File no. CRG/2023/003045]. The NMR facility at the BITS-Pilani, Hyderabad campus, and the HRMS facility, funded by DST-FIST (Grant number: SR/FST/CS-I/2020/158), at BITS-Pilani, Hyderabad campus, are acknowledged. A. N. V. Satyanarayana thanks CSIR [File No. 09/1026(17969)/2024-EMR-I] for the senior research fellowship (SRF Direct).References
- A. K. Jain, A. Vaidya, V. Ravichandran, S. K. Kashaw and R. K. Agrawal, Bioorg. Med. Chem., 2012, 20, 3378–3395 CrossRef CAS PubMed.
- W. Fischer, R. Bodewei and G. Satzinger, Arch. Pharmacol., 1992, 346(4), 442–452 CAS.
- S. Kagabu, J. Agric. Food Chem., 2011, 59, 2887–2896 CrossRef CAS PubMed.
- P. Jeschke, R. Nauen and M. E. Beck, Angew. Chem., Int. Ed., 2013, 52, 9464–9485 CrossRef CAS PubMed.
- H. Junjappa, H. Ii-A and C. V. Asokan, Tetrahedron, 1990, 16, 5423–5506 CrossRef.
- H. Ila and H. Junjappa, Chimia, 2013, 67, 17–22 CrossRef CAS PubMed.
- Saigal, S. Khan, H. Rahman, Shafiullah and M. M. Khan, RSC Adv., 2019, 9, 14477–14502 RSC.
- L. Zhang, J. Dong, X. Xu and Q. Liu, Chem. Rev., 2016, 116, 287–322 CrossRef CAS PubMed.
- X. Fei, Y. Gu, Y. Ban, Z. Liu and B. Zhang, Bioorg. Med. Chem., 2009, 17, 585–591 CrossRef CAS PubMed.
- W. E. Evenson, L. M. Boden, K. A. Muzikar and D. J. Oleary, J. Org. Chem., 2012, 77, 10967–10971 CrossRef CAS PubMed.
- P. Agarwala, S. Pandey and S. Maiti, Org. Biomol. Chem., 2015, 13, 5570–5585 RSC.
- (a) V. P. Ananikov, D. A. Malyshev, I. P. Beletskaya, G. G. Aleksandrov and I. L. Eremenko, Adv. Synth. Catal., 2005, 347, 1993–2001 CrossRef CAS; (b) A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente, M. Crucianelli, V. Polo, R. Sancho, F. J. Lahoz and L. A. Oro, J. Am. Chem. Soc., 2012, 134, 8171–8183 CrossRef CAS PubMed; (c) A. Dondoni and A. Marra, Eur. J. Org. Chem., 2014, 2014, 3955–3969 CrossRef CAS; (d) D. A. Malyshev, N. M. Scott, N. Marion, E. D. Stevens, V. P. Ananikov, I. P. Beletskaya and S. P. Nolan, Organometallics, 2006, 25, 4462–4470 CrossRef CAS; (e) A. Ogawa, T. Ikeda, K. Kimura and T. Hirao, J. Am. Chem. Soc., 1999, 121, 5108–5114 CrossRef CAS.
- L. Pagès, M. Bouquin, F. Jaroschik, F. Monnier and M. Taillefer, J. Org. Chem., 2023, 88, 1168–1176 CrossRef PubMed.
- H. Yasui, H. Yorimitsu and K. Oshima, Chem. Lett., 2008, 37, 40–41 CrossRef CAS.
- (a) A. Mishra, S. S. Pati and J. P. Das, Eur. J. Org. Chem., 2024, e202400637 CrossRef CAS; (b) S. Dutta, R. K. Mallick and A. K. Sahoo, Angew. Chem., Int. Ed., 2023, 62, e202300816 CrossRef CAS PubMed; (c) Y. B. Chen, P. C. Qian and L. W. Ye, Chem. Soc. Rev., 2020, 49, 8897–8909 RSC; (d) S. Shandilya, M. P. Gogoi, S. Dutta and A. K. Sahoo, Chem. Rec., 2021, 21, 4123–4149 CrossRef CAS PubMed.
- (a) V. Debrauwer, A. Turlik, L. Rummler, A. Prescimone, N. Blanchard, K. N. Houk and V. Bizet, J. Am. Chem. Soc., 2020, 142, 11153–11164 CrossRef CAS PubMed; (b) S. S. Pati, A. Mishra and J. P. Das, J. Org. Chem., 2024, 89, 1727–1735 CrossRef CAS PubMed; (c) R. Prasad, S. Kanikarapu, S. Dutta, S. Vangara and A. K. Sahoo, New J. Chem., 2022, 46, 13981–13986 RSC; (d) H. Wang, M. Hu, X. Wang and J. Chang, Org. Biomol. Chem., 2022, 20, 3408 RSC.
- (a) G. Compain, K. Jouvin, A. M. Mingot, G. Evano, J. Marrot and S. Thibaudeau, Chem. Commun., 2012, 48, 5196–5198 RSC; (b) W. Cao, P. Chen, L. Wang, H. Wen, Y. Liu, W. Wang and Y. Tang, Org. Lett., 2018, 20, 4507–4511 CrossRef CAS PubMed.
- N. Mukherjee and T. Chatterjee, Green Chem., 2023, 25, 8798–8807 RSC.
- A. N. V. Satyanarayana and T. Chatterjee, J. Org. Chem., 2024, 89, 12439–12451 CrossRef CAS PubMed.
- A. N. V. Satyanarayana, N. Mukherjee and T. Chatterjee, Green Chem., 2022, 25, 779–788 RSC.
- (a) A. N. V. Satyanarayana, P. Pattanayak and T. Chatterjee, J. Org. Chem., 2024, 89, 11455–11466 CrossRef CAS PubMed; (b) N. Mukherjee, A. N. V. Satyanarayana, P. Singh, M. Dixit and T. Chatterjee, Green Chem., 2022, 24, 7029–7038 RSC.
- (a) P. Pattanayak, A. N. V. Satyanarayana and T. Chatterjee, J. Org. Chem., 2024, 89, 13215–13223 CrossRef CAS PubMed; (b) A. Naresh, H. S. Keerthana, N. Mukherjee and T. Chatterjee, Chem. Commun., 2024, 60, 7057–7060 RSC; (c) N. Mukherjee and T. Chatterjee, Adv. Synth. Catal., 2023, 365, 2255–2263 CrossRef CAS.
- T. Chatterjee, P. Pattanayak, A. N. V. Satyanarayana and N. Mukherjee, Current Opinion in Green and Sustainable Chemistry., 2023, 41, 100826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data of the synthesized molecules, and 1H, 13C, and 19F spectra of the synthesized compounds. See DOI: https://doi.org/10.1039/d4ob01984a |
| This journal is © The Royal Society of Chemistry 2025 |