
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoenzymatic synthesis of Tamsulosin†
Enol
de Prado
 ,
Juan
Mangas-Sánchez
* and
Vicente
Gotor-Fernández
*
,
Juan
Mangas-Sánchez
* and
Vicente
Gotor-Fernández
*
Organic and Inorganic Chemistry Department, Instituto Universitario de Química Organometálica “Enrique Moles”, University of Oviedo. Avenida Julián Clavería 8, 33006 Oviedo, Asturias, Spain. E-mail: mangasjuan@uniovi.es; vicgotfer@uniovi.es
First published on 24th January 2025
Abstract
Several chemoenzymatic pathways have been developed for the stereoselective production of the drug tamsulosin. The interest in the exclusive synthesis of its (R)-enantiomer lies in the greater activity compared to that displayed by its (S)-counterpart for the treatment of kidney stones and benign prostatic hyperplasia disease. Using different types of biocatalysts such as lipases, alcohol dehydrogenases and transaminases, three complementary strategies have been studied to introduce chirality into a key synthetic precursor. The first approach involved the lipase-catalyzed kinetic resolution of a racemic amine precursor, although low conversions and selectivities were found. A second strategy consisted in the synthesis of a chiral alcohol intermediate through a bioreduction proccess catalyzed by ADHs, with the identification of stereocomplementary redox enzymes capable of producing both enantiomers. The (S)-alcohol, obtained with ADH-A from Rhodococcus ruber, was subsequently converted into the corresponding amine through a telescoped approach. Alternatively, transaminases were also employed for the biotransamination of the previously studied intermediate ketone, which led directly to the enantiopure (R)-amine in high yield. Finally, the active pharmaceutical ingredient was prepared in enantiopure form and in 49% overall yield from the ketone precursor by a two-step sequential transformation of the chiral amine building block. These findings highlight the importance and versatility of enzyme catalysis for the stereoselective synthesis of drugs.
Introduction
Nowadays, it is estimated that around 60% of marketed drugs are chiral molecules and Biocatalysis represents a suitable and sustainable synthetic methodology for the production of valuable drugs and synthetic precursors.1 The capability of enzymes to produce optically active products, such as chiral amines, highlights their potential as ideal catalysts for the development of sustainable synthetic routes under mild conditions.2 Their remarkable capacity to discriminate between the enantiomers in a racemate, as well as their ability to carry out desymmetrization processes, has significantly expanded the implementation of biotransformations in the healthcare industry in recent years, providing practical and elegant alternative methods to existing chemical routes.3 This progress has allowed the design of efficient chemoenzymatic routes for producing single enantiomers, which display enormous differences in terms of pharmacological properties within the human body, such as absorption, distribution, excretion and toxicity, among others.4 Particularly, we were interested in designing a novel chemoenzymatic strategy towards enantiopure tamsulosin [(R)-1, Scheme 1] currently commercialized under the name Flomax®, and also known as (R)-5-{2-[(2-(2-ethoxyphenoxy)ethyl)amino]propyl}-2-methoxybenzenesulfonamide. Tamsulosin is a selective α1A adrenergic blocker currently used to treat lower urinary tract symptoms usually associated with benign prostatic hyperplasia, including enlargement, obstruction, and bladder outlet obstruction.5 Notably, the (R)-enantiomer of tamsulosin exhibits approximately 320 times greater activity than its (S)-homologue,6 stressing the importance of developing efficient and stereoselective methods in drug synthesis.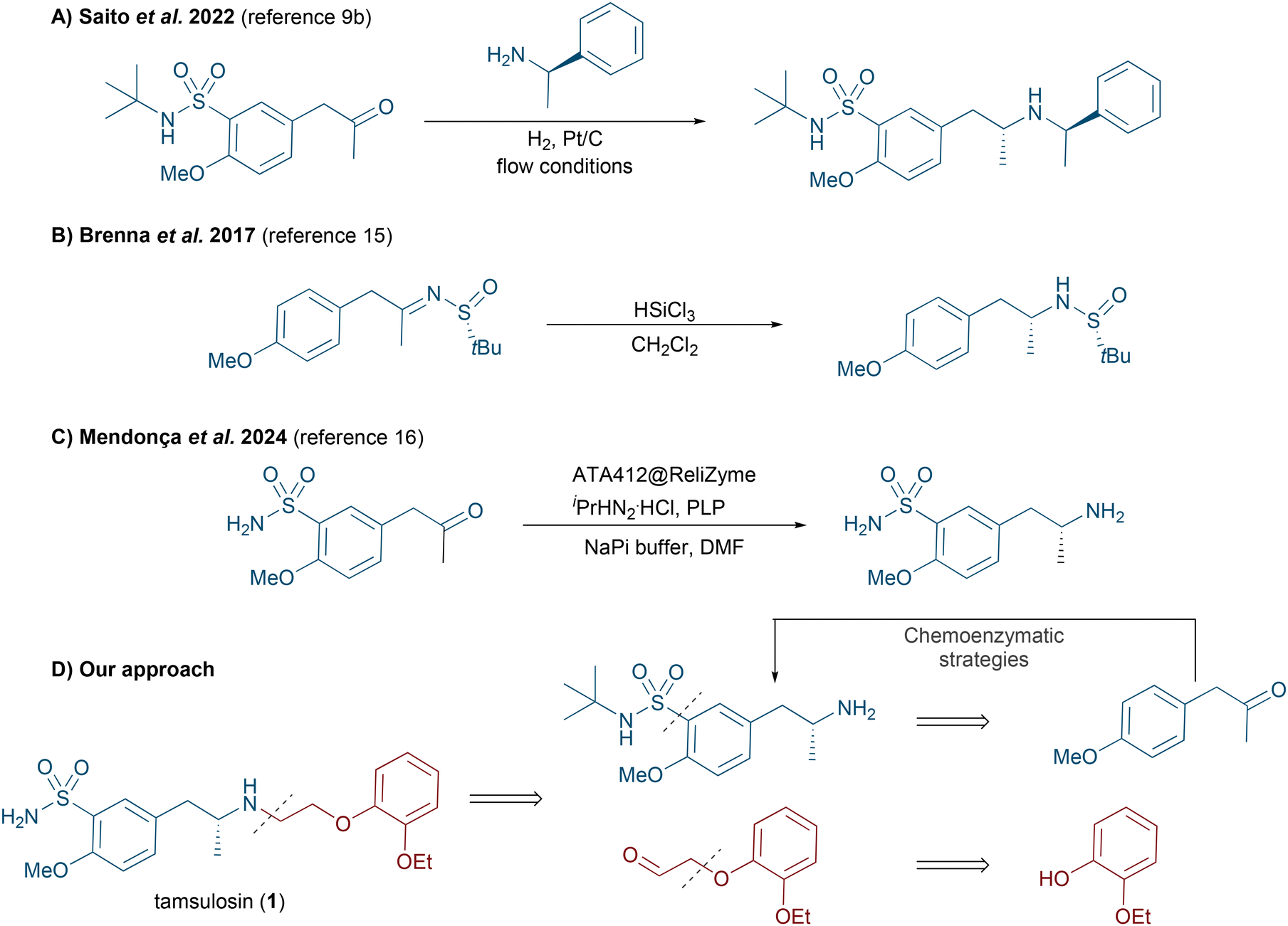 | ||
| Scheme 1 (Dia)stereoselective routes towards tamsulosin and retrosynthetic analysis for its stereoselective chemoenzymatic synthesis. | ||
Traditional synthetic routes towards tamsulosin are based on the use of chiral reagents, while others rely on the resolution or asymmetrization of key fragments. The first stereoselective approaches towards tamsulosin employed as chiral starting material (R)-1-(4-methoxyphenyl)propan-2-amine7 or its corresponding acetamide.8 However, more recently, Saito et al. employed a diastereoselective synthesis based on a reductive amination process using (R)-α-methylbenzylamine (Scheme 1A).9 Additionally, several strategies took advantage of resolution processes involving suitable amine fragments and optically active organic acids such as D-tartaric acid,10 (R,R)-dibenzoyl-D-tartaric acid,11L- or D-10-camphorsulfonic acid,6 and (–)- or (+)-N-(3,5-dinitrobenzoyl acid)-α-phenylglycine enantiomers.6 Alternatively, various asymmetric chemoenzymatic routes have been reported for the synthesis of tamsulosin and its precursors. These include: (a) the kinetic resolution of (±)-1-(4-methoxyphenyl)propan-2-amine catalyzed by lipases (CAL-B, RL IM and TL IM) using different ethyl haloacetates (X = Cl, Br or I);12 (b) the stereoselective alkene bioreduction of 1-methoxy-4-(2-nitroprop-1-en-1-yl)benzene using the ene-reductase from Gluconobacter oxydans;13 (c) the biotransamination of 2-methoxy-5-(2-oxopropyl)benzenesulfonamide;14 (d) the synthesis of a chiral alcohol intermediate through Baker's yeast fermentation processes (Scheme 1B);15 (e) after the submission of this manuscript, Mendonça and co-workers reported an elegant chemoenzymatic synthesis of tamsulosin combining continuous and batch reactions starting from commercially available 4-methoxyacetophenone. In this case, an immobilized transaminase preparation was identified as a very efficient biocatalyst for producing a key chiral intermediate via the stereoselective biotransamination of 2-methoxy-5-(2-oxopropyl)benzenesulfonamide (Scheme 1C).16
In order to further improve the existing routes towards enantiopure tamsulosin, we were encouraged to explore and optimize the action of different biocatalyst classes such as lipases, alcohol dehydrogenases (ADHs) and transaminases (TAs) in asymmetric processes including acylation, bioreduction and biotransamination, respectively (Scheme 1D).
Results and discussion
The stereoselective synthesis of tamsulosin [(R)-1] was initially designed to involve the independent preparation of two precursor fragments, the chirality being induced through the formation of an enantiopure (R)-amine (Scheme 1 bottom), which would then be reacted with a suitable aldehyde or derivative, and subsequently modified to obtain the desired drug. To achieve this, a comprehensive study for the production of the key (R)-amine was considered focusing on selective and high-yielding enzymatic steps, while for the synthesis of the aldehyde fragment, previously established protocols were adapted and optimized starting from commercially available 2-ethoxyphenol.Chemoenzymatic approaches for the synthesis of enantiopure amine (R)-5
Initially, three different approaches were considered for the synthesis of (R)-5-(2-aminopropyl)-N-(tert-butyl)-2-methoxybenzenesulfonamide [(R)-5] as the chiral amine fragment (Scheme 2), all of them involving the preparation of the common intermediate N-isopropyl-2-methoxy-5-(2-oxopropyl)benzenesulfonamide (4). In this context, the use of complementary biocatalyst types such as lipases, ADHs or ATAs, represents an elegant and sustainable approach for the introduction of chirality into a molecule. Ketone 4 was obtained from 4-methoxyphenylacetone (2) through the two-step process that involved an initial chlorosulfonation reaction with chlorosulfonic acid (HSO3Cl) in CH2Cl2, affording chlorosulfonamide 3 in 46% yield.17 In the second step, the chlorine atom of 3 was replaced with tert-butylamine (tBuNH2) in the presence of triethylamine (Et3N) as a base in CH2Cl2, producing 4 in 47% yield after 28 h reaction followed by column chromatography purification.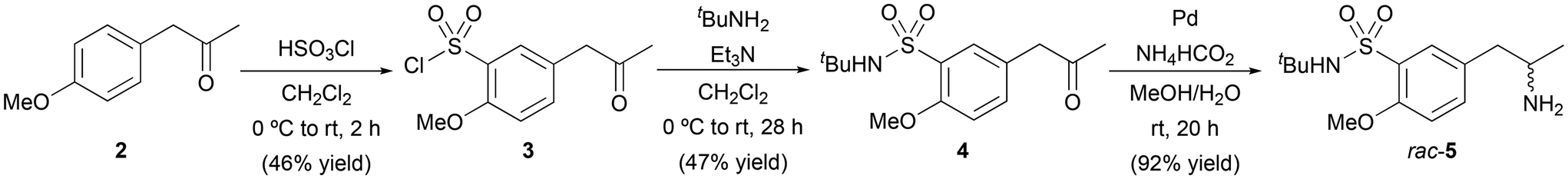 | ||
| Scheme 2 Chemical synthesis of racemic amine 5. | ||
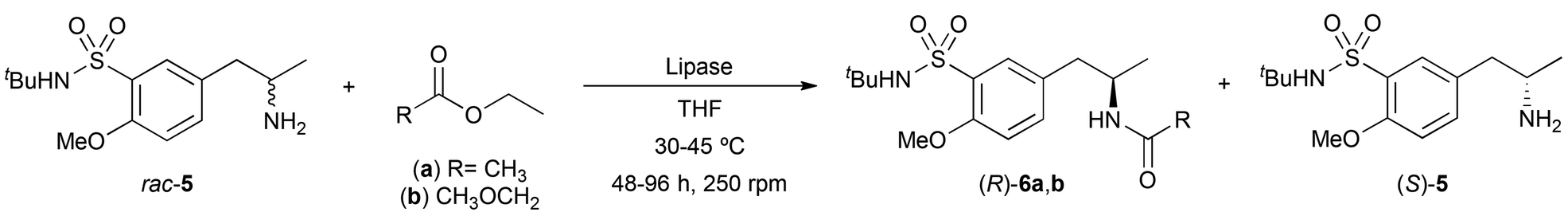
To prepare the racemic amine 5, the reductive amination of ketone 4 was carried out using ammonium formate (NH4HCO2) as a nitrogen source, Pd as a catalyst in a solvent mixture of MeOH and deoxygenated H2O (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) (Scheme 2), attaining (±)-5 in 92% yield and high purity after a simple filtration process over Celite® and liquid–liquid extraction. At this point, a classical lipase-catalyzed kinetic resolution of amine 5 was studied through acylation reactions with non activated esters such as ethyl acetate (3.0 equiv., Table 1). The initial screening of different biocatalysts was performed using an enzyme:substrate ratio (1:1 w/w) with 100 mM (±)-5 in tetrahydrofuran (THF) at 30 °C (entries 1–4). Unfortunately, none or low to moderate conversions and selectivities were found towards the formation of the corresponding (R)-amide 6a. On one hand, Pseudomonas cepacia lipase (PSL), Thermomyces lanuginosus lipase (TLL) and Rhizomucor miehei lipase did not react with the amine 5. On the other hand, Candida rugosa lipase (CRL, entry 1) and Pseudomonas fluorescens (PFL, entry 2) led to very low conversions, while both the Candida antarctica lipase type A (CAL-A, entry 3) and its isoenzyme CAL-B (entry 4) led to moderate enantioselectivities. Regarding the stereochemistry of the products obtained, all these lipases follow the so-called Kazlauskas rule,18 favoring the formation of the corresponding (R)-amides. Although structurally similar substrates have been reported to undergo efficient acylation with CAL-B,19 the use of higher temperatures (45 °C, entry 5) or even other acyl donor such as ethyl methoxyacetate (entry 6) did not provide any significant improvement.
:1 v/v) (Scheme 2), attaining (±)-5 in 92% yield and high purity after a simple filtration process over Celite® and liquid–liquid extraction. At this point, a classical lipase-catalyzed kinetic resolution of amine 5 was studied through acylation reactions with non activated esters such as ethyl acetate (3.0 equiv., Table 1). The initial screening of different biocatalysts was performed using an enzyme:substrate ratio (1:1 w/w) with 100 mM (±)-5 in tetrahydrofuran (THF) at 30 °C (entries 1–4). Unfortunately, none or low to moderate conversions and selectivities were found towards the formation of the corresponding (R)-amide 6a. On one hand, Pseudomonas cepacia lipase (PSL), Thermomyces lanuginosus lipase (TLL) and Rhizomucor miehei lipase did not react with the amine 5. On the other hand, Candida rugosa lipase (CRL, entry 1) and Pseudomonas fluorescens (PFL, entry 2) led to very low conversions, while both the Candida antarctica lipase type A (CAL-A, entry 3) and its isoenzyme CAL-B (entry 4) led to moderate enantioselectivities. Regarding the stereochemistry of the products obtained, all these lipases follow the so-called Kazlauskas rule,18 favoring the formation of the corresponding (R)-amides. Although structurally similar substrates have been reported to undergo efficient acylation with CAL-B,19 the use of higher temperatures (45 °C, entry 5) or even other acyl donor such as ethyl methoxyacetate (entry 6) did not provide any significant improvement.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Lipase | t (h) | T (°C) | Acyl donor | eePa (%) |
eeSa (%) |
c (%) | E |
| a Enantiomeric excess values of product and substrate measured by HPLC analyses (see Section IV in the ESI†). For the substrate, it was necessary performing the derivatization of the amine into a suitable amide (6a or 6b). b Conversion values: c = ees/(ees + eep). c Enantioselectivity values: E = ln[(1 − c)(1 − eep)]/ln[(1 − c)(1 + eep)].20 | ||||||||
| 1 | CRL | 48 | 30 | EtOAc | 91 | 1 | 1 | 21 |
| 2 | PFL | 48 | 30 | EtOAc | 70 | 3 | 4 | 6 |
| 3 | CAL-A | 48 | 30 | EtOAc | 91 | 13 | 12 | 24 |
| 4 | CAL-B | 72 | 30 | EtOAc | 70 | 25 | 26 | 7 |
| 5 | CAL-B | 72 | 45 | EtOAc | 74 | 42 | 36 | 10 |
| 6 | CAL-B | 96 | 30 | MeOCH2CO2Et | 84 | 33 | 28 | 16 |
To explore a more efficient transformation, the asymmetric synthesis of amine 5 was studied through biotransamination experiments of ketone 4. Thus, a screening of 37 TAs was undertaken at 30 °C and 250 rpm, including 9 enzymes overexpressed in E. coli from our in-house collection and 28 from a commercial kit (Table S1 in the ESI†). Isopropylamine (2-propylamine, 2-PrNH2) was used as an amine donor in large excess (50 equiv.) to drive the chemical equilibrium towards amine synthesis and forming acetone as by-product, which is easily removed and do not lead to competitive reactions in the reaction medium. Biobased 2-methyl tetrahydrofuran (2-MeTHF) was initially selected as a cosolvent. The best results in terms of selectivity for the formation of the chiral amine 5 have been highlighted in Table 2. Among this set of TAs, only the commercial enzymes displayed complete selectivity. Two enzymes yielded (S)-5 (entries 1 and 2) in enantiopure form while six TAs produced the opposite and desired enantiomer (R)-5 (entries 3–8).
|
|
|||
|---|---|---|---|
| Entry | TA | c (%) | eeb (%) |
| a Conversion values calculated by reverse phase HPLC using calibration curves (see Section IV in the ESI†). b Enantiomeric excess values of amine 5 were measured by HPLC after derivatization of the reaction crude with methoxyacetyl chloride using pyridine as a base and dichloromethane as a solvent (see Section IV in the ESI†). | |||
| 1 | ATA-260 | 10 | >99 (S) |
| 2 | ATA-P1-G05 | 14 | >99 (S) |
| 3 | ATA-412 | 30 | >99 (R) |
| 4 | ATA-P2-B01 | 5 | >99 (R) |
| 5 | ATA-007 | 5 | >99 (R) |
| 6 | ATA-113 | 16 | >99 (R) |
| 7 | ATA-301 | 7 | >99 (R) |
| 8 | ATA-P2-A07 | 4 | >99 (R) |
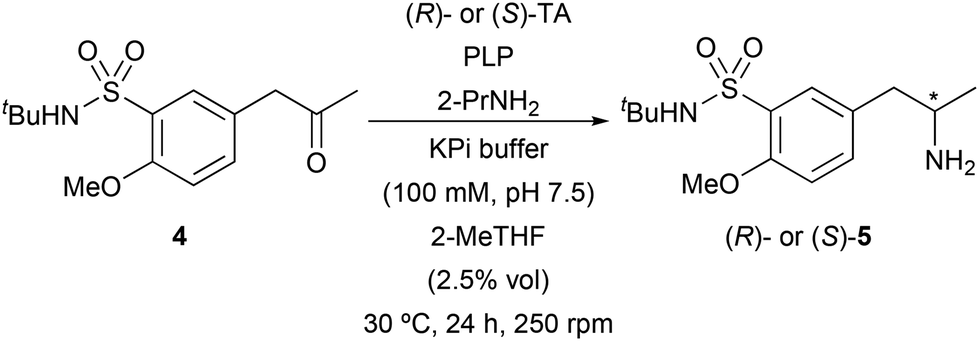
Since the (R)-amine was required for the synthesis of tamsulosin, TAs that showed complete stereoselectivity towards this enantiomer were selected for biotransamination optimization studies: ATA-412, ATA-P2-B01, ATA-007, ATA-113, ATA-301 and ATA-P2-A07. Firstly, to improve the ketone solubility, dimethyl sulfoxide (DMSO) was used as a cosolvent (5% v/v, Fig. 1) instead of 2-MeTHF. Notably, in all cases a significant increase in conversion was observed (from 4–30% to 18–94%), finding ATA-412 as the best enzyme for the formation of enantiopure amine (R)-7 with a remarkable 94% conversion. This finding is consistent with the recent study from Mendonça and co-workers, in that case using the unprotected sulfonamide as starting material, which also identified the same transaminase as the most effective catalyst for their process.16
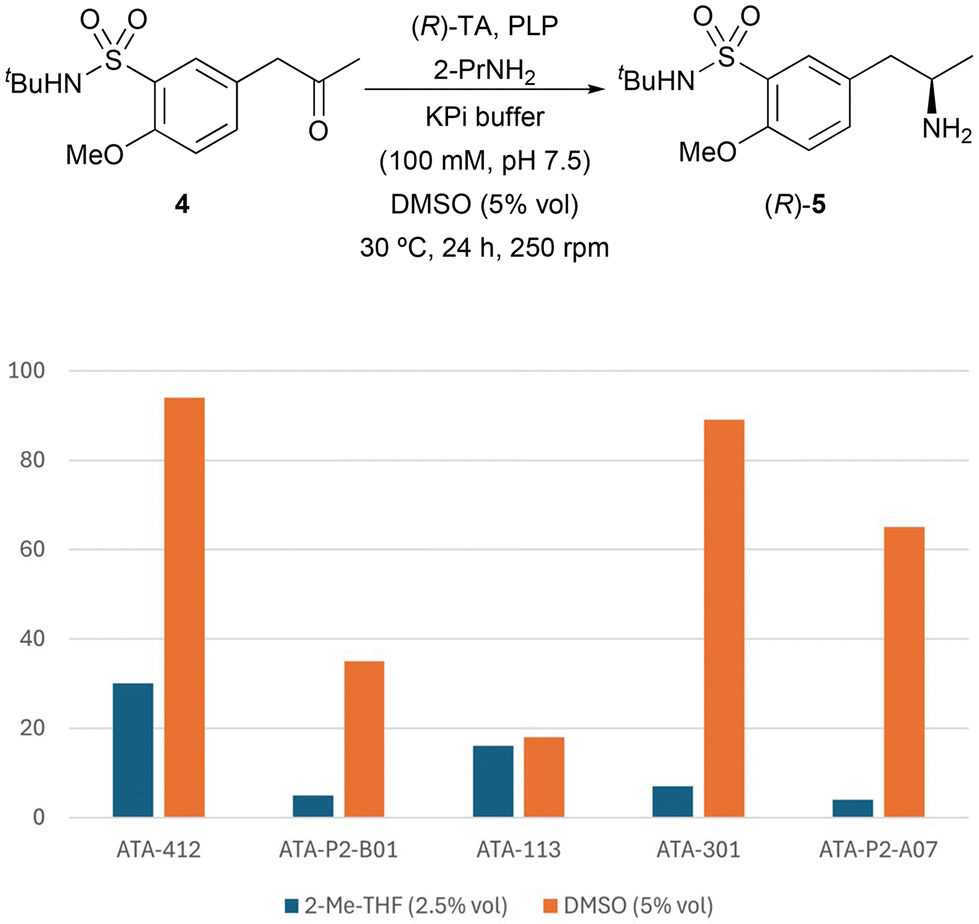 | ||
| Fig. 1 Influence of the organic cosolvent in the TA-catalyzed biotransamination of ketone 4 (25 mM) using 2-PrNH2 (50 equiv.) in KPi buffer (100 mM, pH 7.5) after 24 h at 30 °C and 250 rpm. | ||
At this point, the reaction was scale-up 20-fold (74 mg of substrate) while maintaining the same parameters for substrate, reagent concentrations, ratios and reaction time. With this, (R)-5 was obtained in 87% isolated yield after a liquid–liquid extraction and purification by column chromatography on silica gel.
We next considered an alternative chemoenzymatic route to access amine (R)-5 consisting of the initial bioreduction of ketone 4 followed by a functionalization-nucleophilic substitution strategy for the incorporation of the amine group, as shown in Scheme 3. Initially, a screening of ADHs was performed, including 6 enzymes from our in-house collection overexpressed in E. coli, and 16 obtained from commercial sources (Table S2 in the ESI†). The best results were found with the ADH from Rhodococcus ruber (E. coli ADH-A) and commercially available evo.1.1.200, which led to the enantiopure alcohols (S) and (R)-7, respectively, with complete conversion. These results agree with the exquisite activity and selectivity displayed by these two enzymes in the enantiocomplementary bioreduction of 1-(4-methoxyphenyl)propan-2-one (2).21 Activation of (S)-7 with mesyl chloride in the presence of Et3N and DMAP in CH2Cl2, followed by bimolecular nucleophilic substitution with sodium azide (NaN3) in dimethylsulfoxide (DMSO) and final reduction of the resulting azide through the Staudinger reaction, gave the desired (R)-5 in 78% isolated yield.
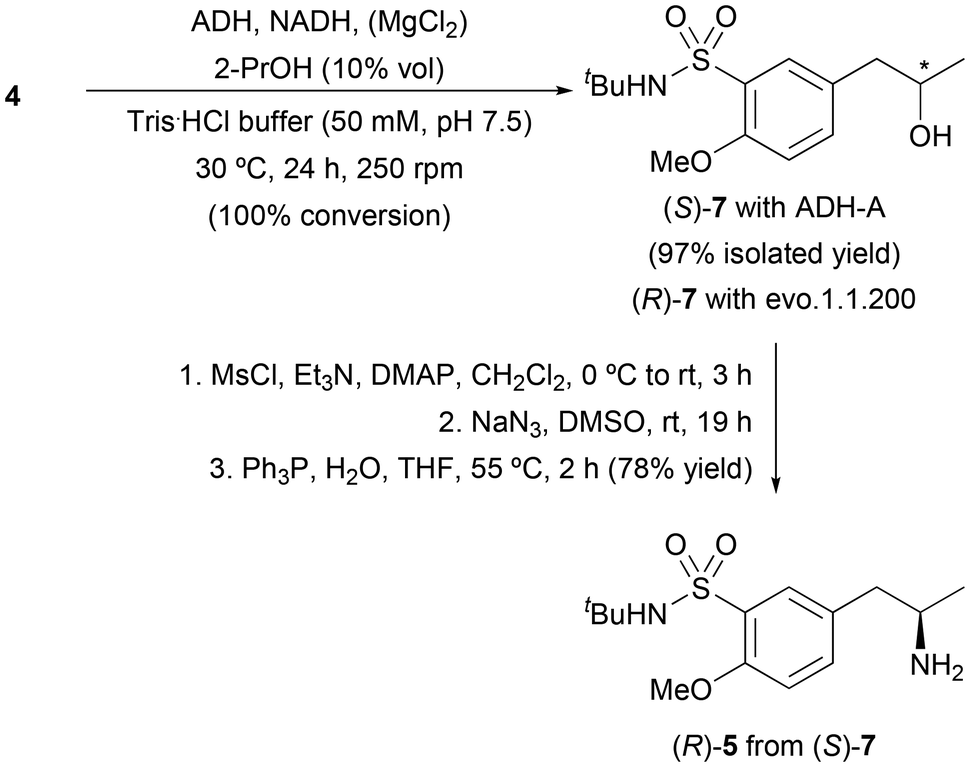 | ||
| Scheme 3 Chemoenzymatic route towards amine (R)-5 involving bioreduction of ketone 4 catalyzed by ADH-A and subsequent functionalization-nucleophilic substitution-reduction steps. | ||
Full chemoenzymatic routes towards the synthesis of tamsulosin
Once the stereoselective synthesis of the amine intermediate (R)-5 was successfully achieved using ADH- or ATA-catalyzed processes, the chemical synthesis of the aldehyde fragment 10 was explored. Initially, a two-step route was designed consisting of the alkylation of 2-ethoxyphenol (8) and subsequent alcohol oxidation (Scheme 4). The alkylation of 8 was performed with ethylene carbonate in the presence of K2CO3 as the base and (N,N)-dimethylformamide (DMF) as the solvent, achieving complete conversion to the 2-(2-ethoxyphenoxy)ethane-1-ol (9, 91% yield) after 24 h at 140 °C. At this stage, different methods were studied for the oxidation of the primary alcohol in compound 9, such as the use of a laccase mediator system, Swern oxidation or the use of 2-iodoxybenzoic acid (IBX) as the oxidizing agent. The reaction with IBX provided the best results, obtaining 2-(2-ethoxyphenoxy)acetaldehyde (10) with 90% isolated yield after 8 h at 77 °C in EtOAc as the solvent.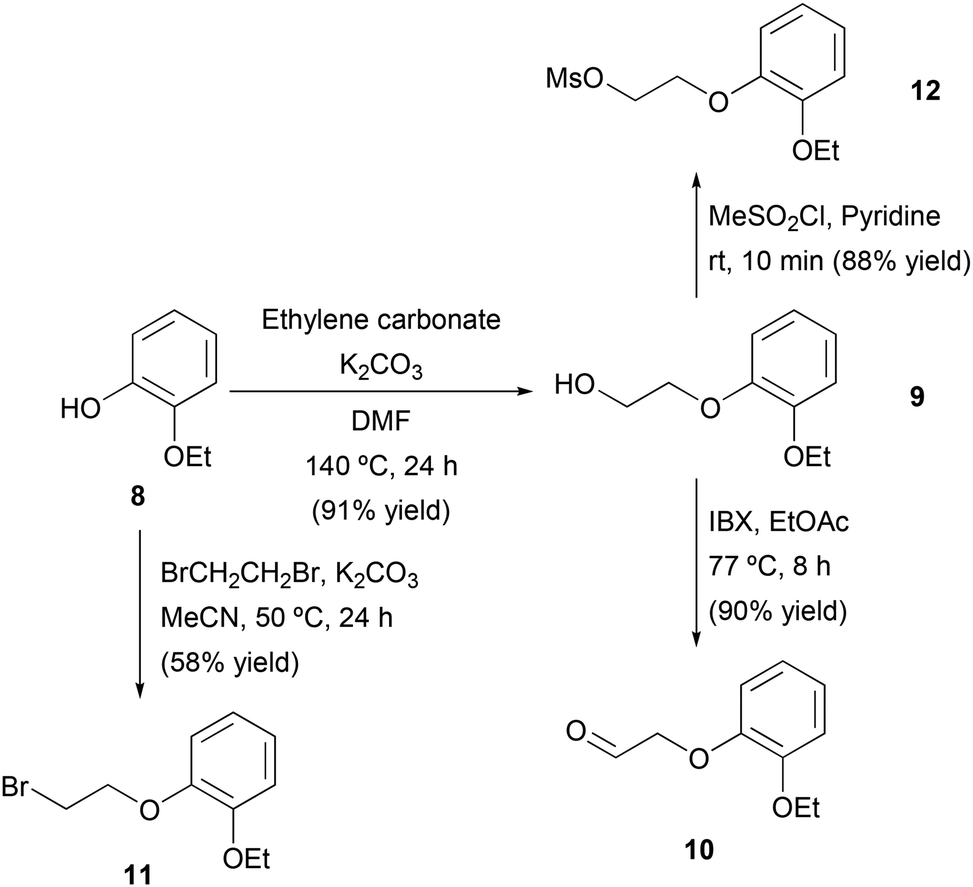 | ||
| Scheme 4 Chemical synthesis of fragments 10, 11 and 12. | ||
Attempts to purify 10 by column chromatography led to lower yields due to the decomposition of the product caused by the acidity of the silica gel; therefore, and considering the high conversion obtained, we decided to use 10 directly after liquid–liquid extraction without further purification. Unfortunately, the reductive amination between aldehyde 10 and amine 5 using sodium triacetoxyborohydride and acetic acid in THF was unsuccessful. To solve this problem, an alternative route was envisioned to access the final drug through an SN2 reaction between (R)-5 and fragments containing leaving groups such as bromo (11) or mesyl (12). On one hand, 2-ethoxyphenol (8) was reacted with 1,2-dibromoethane in the presence of K2CO3 and MeCN as a solvent, affording bromide 11 in 58% isolated yield after column chromatography purification. On the other hand, activation of alcohol 9 with the mesyl group using methanesulfonyl chloride (MeSO2Cl) and pyridine led to the protected alcohol 12 in short reaction time and with 88% isolated yield after filtration and washing with water.
Finally, both approaches were separately explored by reacting racemic or enantiopure amine (R)-5 with 11 and 12 (Scheme 5). Unfortunately, the reaction of (R)-5 with 11 in DMF at 85 °C for 4 h, proceeded in low yield (13%). However, and to our delight, the reaction between (R)-5 and 12 in the presence of sodium bicarbonate (NaHCO3) and sodium iodide (NaI) in MeCN, afforded (R)-13 in 60% isolated yield. Finally, the resulting sulfonamide was deprotected using scandium(III) trifluoromethanesulfonate and trifluoromethanesulfonic acid in MeCN, yielding enantiopure tamsulosin [(R)-1] in 93% yield after column chromatography.9
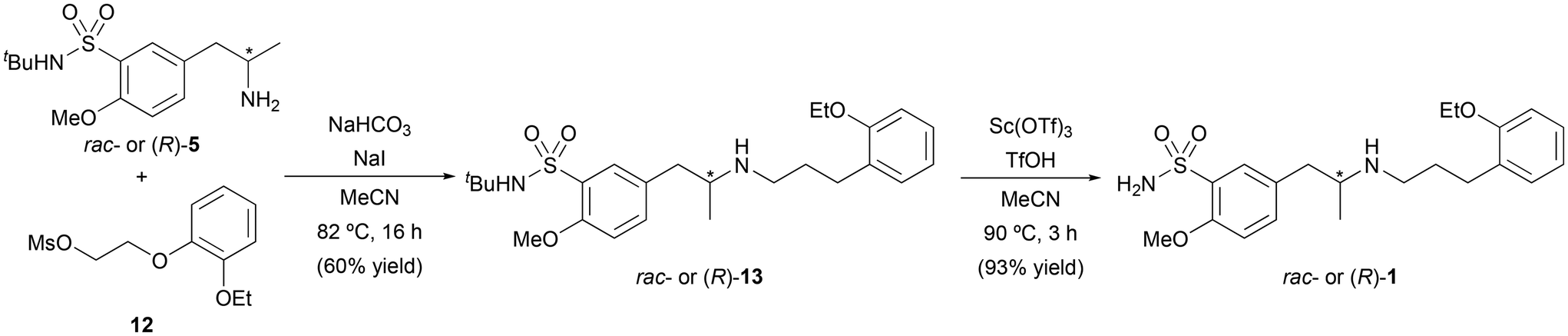 | ||
| Scheme 5 Reaction between fragments 5 and 12, followed by sulfonamide deprotection for the synthesis of chiral and racemic tamsulosin. | ||
Conclusions
In this report, different chemoenzymatic routes towards the stereoselective synthesis of the α1A blocker tamsulosin have been explored. After initial retrosynthetic analysis, a chiral amine fragment was identified as a key precursor, with three alternative strategies considered for its synthesis. Initially, the lipase-mediated kinetic resolution of the bulky amine intermediate was attempted; unfortunately, all the enzymes tested resulted in low conversion and selectivity values. Therefore, other biotransformations were explored finding suitable amine transaminases for the biotransamination of the ketone precursor. Particularly, ATA-412 was demonstrated to be an excellent and selective catalyst for the semipreparative transformation of the enantiopure (R)-5-(2-aminopropyl)-N-(tert-butyl)-2-methoxybenzenesulfonamide with high yield (87%). Alternatively, bioreduction of the ketone intermediate catalyzed by ADHs led to the formation of the (S)-alcohol in quantitative conversion and complete selectivity. A telescoped route to the corresponding (R)-amine also proved to be an efficient synthetic method, obtaining tamsulosin key precursor un 78% yield from the corresponding (S)-alcohol.Finally, after exploring different approaches, reaction between the chiral amine 5 and the mesylated intermediate 12 led to the formation of the corresponding protected sulfonamide, which was successfully deprotected to afford tamsulosin without any loss of the optical purity in 49% overall yield from the ketone precursor 4.
Experimental
General experimental procedures
4-Ethoxyphenol (8) and 1-(4-methoxyphenyl)propan-2-one (2), as well as the different chemical reagents and solvents used in this work, have been purchased from commercial sources such as BLDpharm, Scharlau, Sigma-Aldrich and VWR. Organic solvents were used as obtained, except those used under an inert atmosphere that were previously dried using the appropriate desiccant (CH2Cl2, MeCN and THF). Regarding the enzymes, ADHs and TAs overexpressed in E. coli and commercial ones from Codexis Inc. were used, while the NAD(P)+ and PLP cofactors were purchased from Sigma-Aldrich. The lipases used were obtained from Codexis Inc. and Sigma Aldrich, respectively. Immobilized Candida antarctica lipase B (CAL-B), marketed under the name Novozym 435, Thermomyces lanuginosus lipase and Rhozomucor miehei lipase were obtained from Novozymes; Pseudomonas cepacia (PSL IM) was a gift from Amano; Immobilized Candida antarctica lipase A (CAL-A), lyophilized lipase AK from Pseudomonas fluorescens (PFL) and lyophilized Candida rugosa lipase (CRL) were purchased from Sigma-Aldrich. When lyophilized E. coli cells heterologously overexpressing ADHs were used as biocatalysts, a control reaction with a model substrate was performed, and activity was measured via gas chromatography. For the ADH-A preparation overexpressed in E. coli BL21 cells the following procedure was performed: cells were grown in LB-amp medium (1 L, 100 mg L−1) at 30 °C and 250 rpm for 24 h. Then, an additional amount of ampicillin (50 mg L−1), ZnCl2 (100 mg L−1) and isopropyl thiogalactoside (IPTG, 450 mg L−1) were added and shaken for 24 h at 20 °C. The cells were centrifuged, and the pellet was washed and suspended in phosphate buffer (pH 7.5, 50 mm), frozen with liquid nitrogen and stored at 4 °C. The activity was determined to be approximately 0.5–1 U mg−1 of lyophilized catalyst ADH-A towards the reduction of 2-octanone.22Monitorization of the chemical reactions and analysis of the fractions collected in the chromatographic column purifications were carried out by thin layer chromatography analyses using silica gel chromatographs (230–400 mesh, Merck Silica Gel 60 F254). The monitorization of enzymatic reactions was performed using HPLC analyses as specified in the ESI.†1H, 13C and DEPT NMR experiments were carried out on a Bruker DPX-300 spectrometer (300.13 MHz for 1H and 75.5 MHz for 13C and DEPT), using deuterated chloroform (CDCl3) as a solvent. The chemical shifts (δ) have been expressed in parts per million (ppm) and the spectra have been calibrated taking as reference the residual signal of the protonated solvent at 7.26 ppm. Coupling constants (J) are given in hertz (Hz). The multiplicity of the signals has been indicated as follows: singlet (s), doublet (d), doublet (dd), doublet of doublets (ddd), and doublet of doublet of triplets (dddt), triplet of doublets (td), triplet (t), double triplet (dt), quadruplet (q), quintuplet (quint), quintuplet of doublets (quintd) and multiplets (m). All NMR spectra can be found in the ESI.†
Infrared (IR) experiments have been carried out using a Jasco FT/IR-4700 spectrophotometer using neat samples. Selected tension bands are given in cm−1, highlighting the most significant signals such as those of carbonyl, amine or alcohol groups. Melting points (Mp) were recorded using a Gallenkamp apparatus in open capillary tubes and they are uncorrected. High resolution mass spectra (HRMS) were obtained in a Bruker Daltonics spectrometer using the ESI-TOF positive mode. Measurements of the optical rotation values were carried out using a PerkinElmer 241 polarimeter at 590 nm.
Synthetic procedures
:7 ratio 1:1 w/w) in THF (0.80 mL) inside an Erlenmeyer flask. The reaction was stirred on an orbital shaker at 250 rpm at 30 or 45 °C for 48–96 h, following the reaction course by HPLC analysis. At a certain time, the reaction was stopped by filtering the enzyme and washing it with CH2Cl2 (3 × 1.5 mL). The solvent was distilled under reduced pressure and the reaction crude was analyzed by 1H NMR analysis to measure the conversion. Selected reactions were purified by column chromatography on silica gel (5% MeOH/CH2Cl2), while the other reaction crudes were conveniently derivatised for the measurement of the enantiomeric excess of the amine and amide resulting from the lipase-catalyzed process.
Author contributions
E. P. performed the experimental work (chemical synthesis, biotransformations and analytical data management), while J. M. S. and V. G.-F. were responsible of funding acquisition, project conceptualization, and manuscript writing (initial draft, revision and edition).Data availability
The data supporting this article have been included as part of the ESI.† Additional data are available upon request from the authors.The data that support the findings of this study are available from the corresponding author, Vicente Gotor-Fernández, upon reasonable request.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Agencia Estatal de Investigación (AEI), Spanish Ministry of Science and Innovation (MCI) and the EU is gratefully acknowledged through grants PID2022-137893OB-I00, PID2020-113351RA-I00/AEI/10.13039/501100011033 and TED2021-130803B-I00 MCIN/AEI/10.13039/501100011033 NextGenerationEU/PRTR. J. M.–S. also thanks the AEI for a Ramón y Cajal Fellowship (RYC2021-032021-I). Technical support from the Scientific–Technical Services (University of Oviedo) is acknowledged. Prof. Wolfgang Kroutil (University of Graz, Austria) is also acknowledged for providing us with ADHs and TAs overexpressed in E. coli cells.References
- (a) W. Jiang and B. Fang, Appl. Biochem. Biotechnol., 2020, 192, 146 CrossRef CAS PubMed; (b) W. Zawodny and S. L. Montgomery, Catalysts, 2022, 12, 595 CrossRef CAS; (c) S. P. France, R. D. Lewis and C. A. Martinez, JACS Au, 2023, 3, 715 CrossRef CAS PubMed.
- (a) E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, Ss. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS; (b) U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594 RSC; (c) R. D. Lewis, S. P. France and C. A. Martinez, ACS Catal., 2023, 13, 5571 CrossRef CAS; (d) A. O'Connell, A. Barry, A. J. Burke, A. E. Hutton, E. L. Bell, A. P. Green and E. O'Reilly, Chem. Soc. Rev., 2024, 53, 2828 RSC.
- (a) A. Fryszkowska and P. N. Devine, Curr. Opin. Chem. Biol., 2020, 55, 151 CrossRef CAS PubMed; (b) A. R. Alcántara, P. Domínguez de María, J. A. Littlechild, M. Schürmann, R. A. Sheldon and R. Wohlgemuth, ChemSusChem, 2022, 15, e202102709 CrossRef PubMed; (c) R. Buller, S. Lutz, R. J. Kazlauskas, R. Snajdrova, J. C. Moore and U. T. Bornscheuer, Science, 2023, 382, ead8615 CrossRef PubMed.
- H. Sun, H. Zhang, E. L. Ang and H. Zhao, Bioorg. Med. Chem., 2018, 26, 1275 CrossRef CAS PubMed.
- (a) H. L. Holtgrewe, Urology, 1998, 51, 1 CrossRef CAS PubMed; (b) G. Sagratini, P. Angeli, M. Buccioni, U. Gulini, G. Marucci, C. Melchiorre, E. Poggesi and D. Giardinà, Eur. J. Med. Chem., 2010, 45, 5800 CrossRef CAS PubMed.
- F. C. Blanco, B.-L. A. Lorente, J. J. Martin and G. L. O. Silva, WO2004006829A3, 2004.
- (a) T. Tanenaka, T. Fujikura, K. Honda, M. Asano and K. Niigata, Yakugaku Zasshi, 1995, 115, 773 CrossRef PubMed; (b) D. P. Konakanchi, R. Sambasiva and N. V. Chowdary, WO2004016582A1, 2004; (c) K. Niigata and T. Fujikura, WO2004087623, 2004; (d) D. Brenna, M. Pirola, L. Raimondi, A. J. Burke and M. Benaglia, Bioorg. Med. Chem., 2017, 25, 6242 CrossRef CAS PubMed.
- R. H. Jih, B. Magendran, S. K. Chakraborty, A. R. Das, S. C. Tsay, K. W. Sheu, C. M. Shu, C. K. Lu and W. M. Chang, EP 1734036A1, 2006.
- (a) J. Hajicek and M. Slavikova, WO2005075415A1, 2005; (b) Y. Saito, K. Nishizawa, B. Laroche, H. Ishitani and S. Kobayashi, Angew. Chem., Int. Ed., 2022, 61, e202115643 CrossRef CAS PubMed.
- S. Dubey, D. A. Patel, D. J. Patel, M. L. Rupapara, V. K. Agargawal, K. Pandita and P. R. Patel, WO2004058694A1, 2004.
- T. Gizur, E. Fogassy, J. Bálint, G. Egri, J. Törley, Á. Demeter and I. Greiner, Chirality, 2008, 20, 790 CrossRef CAS PubMed.
- J. Villasante-Prieto and F. Palomo-Nicolau, US2006018046A1, 2006.
- E. Burda, T. Reβ, T. Winkler, C. Giese, X. Krostov, T. Huber, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2013, 52, 9323 CrossRef CAS PubMed.
- W. Jingli, Z. Pentgao, S. Junchao and Y. Kai, CN111876449A, 2020.
- D. Acetti, E. Brenna and C. Fuganti, Tetrahedron: Asymmetry, 2007, 18, 488 CrossRef CAS.
- Á. Maltá-Lakó, R. M. Durão, L. Poppe and R. F. Mendonça, Org. Process Res. Dev., 2024, 28, 4281 CrossRef.
- E. P. Keaney, M. Connolly, M. Dobler, R. Karki, A. Honda, S. Sokup, S. Karur, S. Britt, A. Patnaik, P. Raman, L. G. Hamann, B. Wiedmann and M. J. LaMarche, Bioorg. Med. Chem. Lett., 2014, 24, 3714 CrossRef CAS PubMed.
- R. J. Kazlauskas, A. N. E. Weissfloch, A. T. Rappaport and L. A. Cuccia, J. Org. Chem., 1991, 56, 2656 CrossRef CAS.
- (a) J. González-Sabín, V. Gotor and F. Rebolledo, Tetrahedron: Asymmetry, 2002, 13, 1315 CrossRef; (b) L. Muñoz, A. M. Rodríguez, G. Rosell, M. P. Bosch and A. Guerrero, Org. Biomol. Chem., 2011, 9, 8171 RSC.
- C. S. Chen, Y. Fujimoto, G. Girdauskas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294 CrossRef CAS.
- J. Albarrán-Velo, V. Gotor-Fernández and I. Lavandera, Adv. Synth. Catal., 2021, 363, 4096 CrossRef.
- G. de Gonzalo, I. Lavandera, K. Faber and W. Kroutil, Org. Lett., 2007, 9, 2163 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of novel organic compounds, enzymatic screenings, analytical data, and NMR spectra. See DOI: https://doi.org/10.1039/d4ob02047b |
| This journal is © The Royal Society of Chemistry 2025 |