
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Foldameric receptors with domain-swapping cavities capable of selectively binding and transporting monosaccharides†
Geunmoo
Song
,
Seungwon
Lee
and
Kyu-Sung
Jeong
 *
*
Department of Chemistry, Yonsei University, Seoul 03722, South Korea. E-mail: ksjeong@yonsei.ac.kr
First published on 17th February 2025
Abstract
The development of synthetic receptors capable of selectively binding and transporting saccharides is crucial but highly challenging. In this study, two foldameric receptors 1 and 2, consisting of two repeating monomers, indolocarbazole and naphthyridine units, with different aromatic spacers in the middle of their sequences, have been synthesised. These receptors fold into helical conformations, and the two strands of each receptor are assembled to create domain-swapping cavities for binding monosaccharides by multiple hydrogen bonds. According to 1H NMR, CD spectroscopy, mass spectrometry, and ITC experiments, receptor 1 forms two distinct 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complexes with methyl β-D-galactopyranoside and methyl β-D-glucopyranoside: (1-MM)2⊃(methyl β-D-galactopyranoside·2H2O)2 and (1-MP)2⊃(methyl β-D-glucopyranoside)2. Despite being composed of identical foldamer strands, these two complexes exhibit notably different folding and assembly modes to achieve optimal stability. The binding affinities of 1 for methyl β-D-galactopyranoside and methyl β-D-glucopyranoside are estimated to be logK = 12.7 and 13.3, respectively, in 5% (v/v) DMSO/CH2Cl2. On the other hand, receptor 2 forms a stable 2:2 receptor/guest complex with methyl β-D-glucopyranoside, (2-MP)2⊃(methyl β-D-glucopyranoside)2, with an association constant of logK = 13.9, which is significantly higher than that of methyl β-D-galactopyranoside (logK = 11.1) and methyl α-D-glucopyranoside (logK = 10.6). Furthermore, receptor 2 facilitates the selective transport of methyl β-D-glucopyranoside over other glycosides across an organic phase (CH2Cl2) in U-tube experiments.
:2 complexes with methyl β-D-galactopyranoside and methyl β-D-glucopyranoside: (1-MM)2⊃(methyl β-D-galactopyranoside·2H2O)2 and (1-MP)2⊃(methyl β-D-glucopyranoside)2. Despite being composed of identical foldamer strands, these two complexes exhibit notably different folding and assembly modes to achieve optimal stability. The binding affinities of 1 for methyl β-D-galactopyranoside and methyl β-D-glucopyranoside are estimated to be logK = 12.7 and 13.3, respectively, in 5% (v/v) DMSO/CH2Cl2. On the other hand, receptor 2 forms a stable 2:2 receptor/guest complex with methyl β-D-glucopyranoside, (2-MP)2⊃(methyl β-D-glucopyranoside)2, with an association constant of logK = 13.9, which is significantly higher than that of methyl β-D-galactopyranoside (logK = 11.1) and methyl α-D-glucopyranoside (logK = 10.6). Furthermore, receptor 2 facilitates the selective transport of methyl β-D-glucopyranoside over other glycosides across an organic phase (CH2Cl2) in U-tube experiments.
Introduction
Saccharides play crucial roles in various biological processes, including cell recognition, signaling, and immune responses.1 Over the past several decades, significant efforts have been dedicated to the development of synthetic receptors capable of selectively binding saccharides in supramolecular chemistry.2,3 However, the rational design of such synthetic receptors remains highly challenging owing to the structural and functional group similarities among saccharides.4 In addition, multiple equilibria between constitutional isomers (e.g., furanose vs. pyranose, α vs. β, and cyclic vs. acyclic forms) of simple monosaccharides provide further difficulties in the design of synthetic receptors.Recently, we described a novel approach for developing synthetic receptors capable of selectively binding monosaccharides utilising two key principles: dynamic covalent chemistry and complexation-induced equilibrium shift.5,6 This method enabled us to quantitatively assemble synthetic receptors from its precursors through imine linkages, when a specific monosaccharide guest with strong affinity was present in solution. Specifically, foldameric receptor 3 was quantitatively assembled through imine bond formation between the aldehyde and amine precursors in the presence of methyl β-D-galactopyranoside (me-β-D-gal) or methyl β-D-glucopyranoside (me-β-D-glc) (Fig. 1). Both monosaccharides formed 2:2 receptor/guest complexes. However, the folding and assembly modes of receptor 3 in these complexes were quite different, featuring domain-swapping7 cavities and guest-adaptive folding.8 In the me-β-D-gal complex, the strand folded into a helical conformation with two identical half cavities in a transoid manner, adopting the same left-handed orientation (M,M). The two strands dimerised by stacking one half-cavity of a strand on top or below the other, generating two identical domain-swapping cavities, each encapsulating one me-β-D-gal with two water molecules (Fig. 1a). In contrast, in the me-β-D-glc complex, the strand adopted a folding structure with the two partial helices of opposite orientation: one left-handed (M) and one right-handed (P). In the M-helix, all four repeating monomers are fully folded. However, the P-helix is partially unfolded, with the naphthyridine monomer next to the imino linkage undergoing a 180° rotation. These two partial helices were positioned in a cisoid mode around a central fluorene plane. The two strands were dimerised by face-to-face stacking in an antiparallel manner, which resulted in two binding cavities, each accommodating one molecule of me-β-D-glc (Fig. 1b).
 | ||
| Fig. 1 X-ray crystal structures of (a) (3-MM)2⊃(me-β-D-gal·2H2O)2 and (b) (3-MP)2⊃(me-β-D-glc)2. me-β-D-gal: methyl β-D-galactopyranoside. me-β-D-glc: methyl β-D-glucopyranoside. Two separate strands are shown in green and orange tubes, and two guests and H2O are shown in CPK views as grey and red, respectively. CH hydrogen atoms and t-butyls in 3 are omitted for clarity.5 | ||
Despite the selective binding of specific monosaccharides, receptor 3 has limitations for potential applications in the transport, purification, and separation of monosaccharides in aqueous environments due to the chemical instability of the imine bond.9 To overcome these limitations, we herein synthesised analogous foldameric receptors 1 and 2 by replacing the imine linkage with chemically stable ethenyl and ethynyl bonds, respectively (Scheme 1). These receptors were found to provide nearly identical folding and binding properties to 3, forming stable 2:2 receptor/guest complexes with methyl glycosides: (1-MM)2⊃(me-β-D-gal·2H2O)2 and (1-MP)2⊃(me-β-D-glc)2 for receptor 1, and (2-MP)2⊃(me-β-D-glc)2 for receptor 2. Furthermore, the more soluble receptor 2 in chlorinated solvents was used in U-tube transport experiments, showing the selective transport of me-β-D-glc over other glycosides.
 | ||
| Scheme 1 Chemical structures of foldameric receptors 1, 2, and 3. | ||
Results and discussion
The syntheses of receptors 1 and 2 are outlined in Scheme 2. The synthesis of compound 5 was described in ESI,† and the syntheses of compounds 6,108,1110,12 and 1113 were previously reported. Compound 7 was synthesised by a PPh3/NaH-mediated coupling reaction14 between 5 (2.0 equiv.) and 6 (1.0 equiv.) in 44% yield. After protodesilylation with tetrabutylammonium fluoride (TBAF), compound 7 was directly coupled with excess 8 (8 equiv.) to yield compound 9 in 62% yield. Subsequently, compound 9 was coupled with 10 (2.2 equiv.) to afford receptor 1 in 52% yield. On the other hand, a Pd(PPh3)2Cl2/CuI-catalysed coupling reaction15 between 10 and 11, followed by protodesilylation with TBAF, gave compound 12 in 81% yield. Finally, compound 12 (2.1 equiv.) was coupled with 2,7-diiodo-9H-fluorene (1.0 equiv.) to yield receptor 2 in 54% yield.
 | ||
| Scheme 2 Syntheses of foldameric receptors 1 and 2: (a) PPh3, NaH, DMF, 40 °C; (b) CuI, Pd(PPh3)2Cl2, TBAF (1.0 M in THF), DMF/TEA, 45 °C; (c) CuI, Pd(PPh3)2Cl2, DMF/TEA, 45 °C; (d) TBAF (1.0 M in THF), THF, 25 °C. | ||
As reported previously,12,16 indolocarbazole-naphthyridine foldamers have a strong propensity to adopt helical conformations due to dipole–dipole interactions and π-stacking between aromatic planes in CD2Cl2 and toluene-d8. As a result, a tubular cavity is formed inside the aromatic helical backbone, where the indolocarbazole NHs and naphthyridine nitrogen atoms are located, allowing the binding of polyhydroxy guest molecules by the formation of multiple hydrogen bonds. Both receptors 1 and 2 contain two tetrameric units at the 2,7-positions of fluorene, which are spatially separated to form two independent helices. Each helical cavity is too small to fully encapsulate a monosaccharide, and therefore these receptors may assemble into dimers, forming larger cavities capable of encapsulating monosaccharides more effectively, as observed previously with receptor 3.
The binding properties of receptor 1 with monosaccharides were investigated using 1H NMR spectroscopy in 5% (v/v) DMSO-d6/CD2Cl2, containing water (ca. 0.05%) (Fig. S1†). Upon the addition of me-β-D-gal to receptor 1, a new separate set of 1H NMR signals appeared (Fig. 2c and Fig. S17†), due to slow exchange between free 1 and its complex under the given conditions. As the amount of guest was gradually increased, this new set of signals intensified at the expense of free signals, without appearing any other sets of 1H NMR signals for other possible complexes. Under these titration conditions, the signal intensities were saturated at an approximately 1:1 molar ratio of receptor 1 to me-β-D-gal. Upon complex formation, the CH signals of indolocarbazole (H11–H14 and H11*–H15*) were characteristically upfield-shifted (Δδ = 0.4–1.7 ppm) compared to those in free 1. In addition, the naphthyridine CH signals (H9*, H17, H18*, and H19*) were upfield-shifted by Δδ = 0.7–1.2 ppm due to stacking between the aryl planes. Indolocarbazole NH signals split from four to eight peaks, and the OH signals of me-β-D-gal appeared in the downfield region between 10.5 and 6.1 ppm due to the formation of hydrogen bonds. Furthermore, 1H–1H ROESY experiment exhibited characteristic NOE cross-peaks between two different strands (e.g., t-Bu4*-17, t-Bu4-16*, t-Bu3*-6, and t-Bu3-21*) (Fig. 2a, e and Fig. S6†). All these observations are consistent with an energy-minimised structure of a 2:2 stoichiometric assembly, (1-MM)2⊃(me-β-D-gal·2H2O)2 (Fig. 2b and Fig. S5†).17 It should be noted that the trends in splitting patterns and chemical shifts described above are nearly identical to those observed in the formation of complex (3-MM)2⊃(me-β-D-gal·2H2O)2, shown in Fig. 1. Additional evidence for the formation of a 2:2 complex between 1 and me-β-D-gal was obtained from diffusion ordered spectroscopy (DOSY). The diffusion coefficient for a 1:1 mixture of 1 and me-β-D-gal was estimated to be 3.0 × 10−10 m2 s−1, while that of 1 alone was 3.6 × 10−10 m2 s−1 (Fig. 2d). Finally, ESI-MS also supported the formation of a 2:2 complex, showing characteristic peaks at m/z = 1867.9 ([(1-MM)2⊃(me-β-D-gal·2H2O)2 + 2H + Na]3+) and at m/z = 1869.2 ([(1-MM)2⊃(me-β-D-gal·2H2O)2 + H + 2Na − H2O]3+) (Fig. 2f and Fig. S26†).
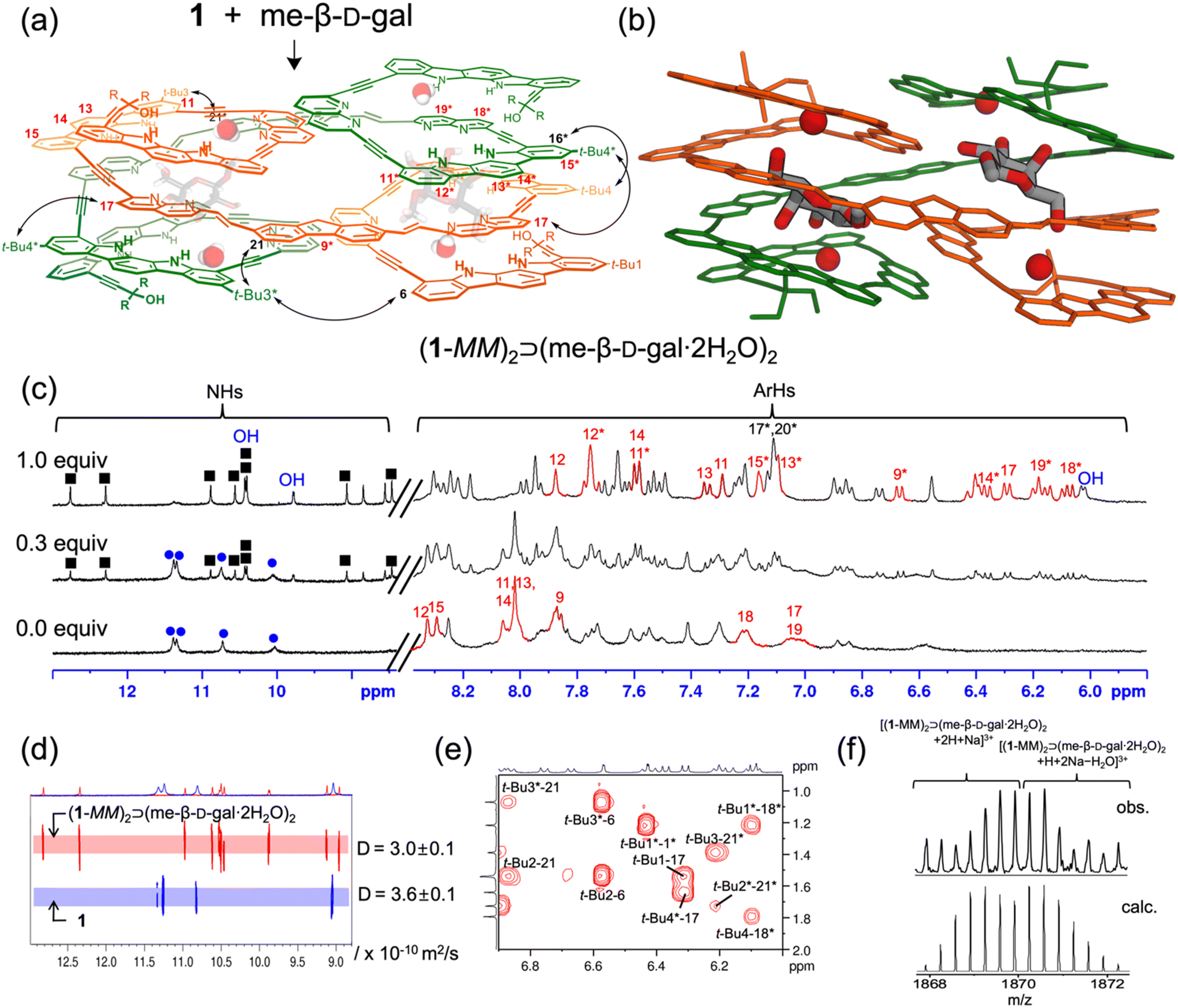 | ||
| Fig. 2 (a) Possible molecular structure of complex (1-MM)2⊃(me-β-D-gal·2H2O)2 with NOE correlations (R = Et). (b) Energy-minimised structure of (1-MM)2⊃(me-β-D-gal·2H2O)2 (MacroModel 9.1, MMFFs force field, CHCl3 phase). (c) Partial 1H NMR spectra (400 MHz, 25 °C) of 1 (1.5 mM) with increasing me-β-D-gal in 5% (v/v) DMSO-d6/CD2Cl2 at 25 °C. NH signals of 1 and its 2:2 complex are marked with blue circles and black squares, respectively. Helically stacked aromatic CH signals are depicted in red, and OH signals of me-β-D-gal are in blue. (d) Partial DOSY spectra (400 MHz, 25 °C) of 1 in the absence and presence of me-β-D-gal (2 mM) in 2% (v/v) DMSO-d6/CD2Cl2. (e) Partial 1H–1H ROESY spectrum (400 MHz, 25 °C, mixing time 400 ms) of (1-MM)2⊃(me-β-D-gal·2H2O)2 (4 mM) in 2% (v/v) DMSO-d6/CD2Cl2. (f) ESI-MS of a 2:2 complex: observed distribution (top) and calculated isotopic distribution (bottom) as [(1-MM)2⊃(me-β-D-gal·2H2O)2 + 2H + Na]3+ and [(1-MM)2⊃(me-β-D-gal·2H2O)2 + H + 2Na − H2O]3+. | ||
Similarly, the binding properties of receptor 1 with me-β-D-glc were examined using 1H NMR spectroscopy in 5% (v/v) DMSO-d6/CD2Cl2, containing water (ca. 0.05%). Upon the addition of me-β-D-glc at room temperature, a new separate set of 1H NMR signals appeared. The intensities of these new signals were gradually increased at the expense of the original free signals, reaching saturation at a 1:1 molar ratio of receptor 1 to me-β-D-glc (Fig. 3c and Fig. S18†). It is noted that no 1H NMR signals for other possible complexes were observed during the titration. Upon complex formation, significant upfield shifts (Δδ = 0.3–2.5 ppm) were observed in the CH signals of naphthyridine (H7*–H10* and H17–H20), fluorene (H21, H21*, and H22), and indolocarbazole (H2–H5) relative to those in free 1. The OH signals of me-β-D-glc appeared between 10.9 to 8.1 ppm, indicative of hydrogen-bonding formation. Furthermore, 1H–1H ROESY experiment showed NOE correlations between the remote aromatic protons (e.g., t-Bu2-21, t-Bu1-17, and 20-5) (Fig. S9†). The formation of a 2:2 complex between receptor 1 and me-β-D-glc was supported by DOSY experiments, which estimated a diffusion coefficient of 3.0 × 10−10 m2 s−1 for a 1:1 mixture of 1 and me-β-D-glc, compared to 3.6 × 10−10 m2 s−1 for 1 alone (Fig. S10†). Finally, ESI-MS spectra provided additional evidence, showing characteristic peaks at m/z = 1836.6 ([(1-MP)2⊃(me-β-D-glc)2 + 3H]3+) and at m/z = 2754.3 ([(1-MP)2⊃(me-β-D-glc)2 + 2H]2+) (Fig. 3b and Fig. S26†). All these observations confirm the formation of a 2:2 complex between 1 and me-β-D-glc.
 | ||
| Fig. 3 (a) Possible molecular structure of (1-MP)2⊃(me-β-D-glc)2 with NOE correlations (R = Et). (b) ESI-MS of the 2:2 complex: observed distribution (top) and calculated isotopic distribution (bottom) as [(1-MP)2⊃(me-β-D-glc)2 + 3H]3+. (c) Partial 1H NMR spectrum (400 MHz, 25 °C) of 1 (2.0 mM) after adding me-β-D-glc (1 equiv.) in 5% (v/v) DMSO-d6/CD2Cl2 at 25 °C. NH signals of complex are marked with black squares. Helically stacked aromatic CH signals are depicted in red, and OH signals of me-β-D-glc are in blue. | ||
However, 1H NMR spectra showed clear differences between the two complexes, me-β-D-glc and me-β-D-gal complexes. In the me-β-D-glc complex, the CH signals of fluorene (H24, H24*) and naphthyridine (H7*, H10*, and H17–H20) were more upfield-shifted by 0.7–2.4 ppm compared to the me-β-D-gal complex. Conversely, the indolocarbazole (H11–H13, H12*–H15*) were downfield-shifted by Δδ = 0.3–1.2 ppm in the me-β-D-glc complex. In addition, 1H–1H ROESY experiment revealed characteristic NOE cross-peaks (t-Bu4*-21, t-Bu3*-17, and t-Bu2*-8) that are absent in the me-β-D-gal complex (Fig. 3a and Fig. S9†). Furthermore, circular dichroism (CD) intensities between the two complexes were considerably different from each other. Receptor 1 was CD-inactive in 5% (v/v) DMSO/CH2Cl2 (Fig. 4 and Fig. S16†). Upon the addition of me-β-D-gal and me-β-D-glc, induced CD signals were observed, indicating the bias of helical handedness in the resulting complexes. Specifically, the CD intensity of the me-β-D-gal complex was Δε(475 nm) = −134.7 M−1 cm−1, while that of the me-β-D-glc complex was much smaller, only Δε(446 nm) = −51.5 M−1 cm−1 and Δε(478 nm) = −47.7 M−1 cm−1. These results are nearly identical to those observed with receptor 3, strongly suggesting that receptor 1 adopts (M,M)-helices in the me-β-D-gal complex but (M,P)-helices of a pseudo-meso type in the me-β-D-glc complex (for an energy-minimised structure, see Fig. S8†)17
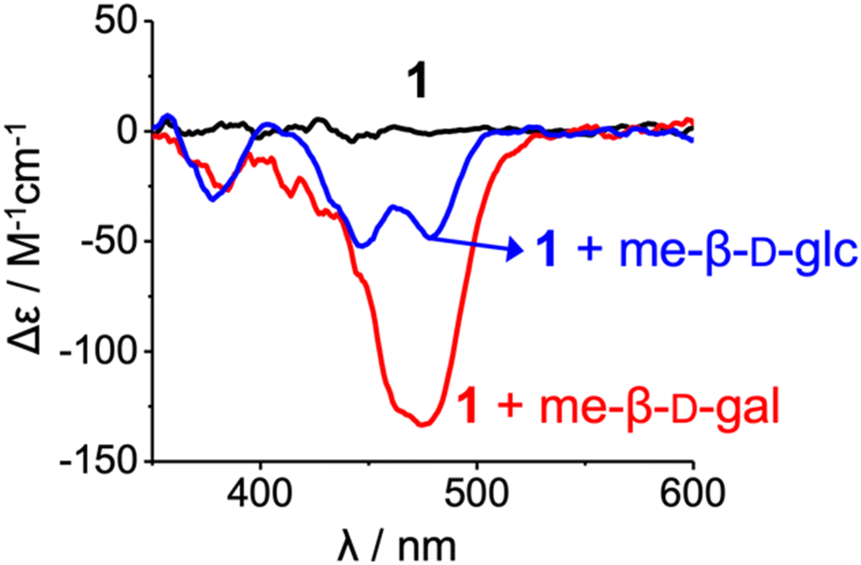 | ||
| Fig. 4 CD spectra of 1 (2.00 × 10−5 M, 25 ± 1 °C) and their complexes with me-β-D-gal and me-β-D-glc (ca. 200 equiv.) in 5% (v/v) DMSO/CH2Cl2. | ||
The quantitative binding properties of receptor 1 with me-β-D-gal and me-β-D-glc were investigated using isothermal titration calorimetry (ITC) in 5% (v/v) DMSO/CH2Cl2 (containing ca. 0.05% water, 22 °C) (Fig. S20, S21 and Tables S4, S5†). As mentioned in the 1H NMR studies, only signals corresponding to 2:2 complexes between 1 and these guests were observed throughout the titrations. No signals for possible 1:1, 1:2, and 2:1 complexes were detected. Therefore, the binding constants were calculated under the assumption that 2:2 complexes formed through a single step, as shown in eqn (1).
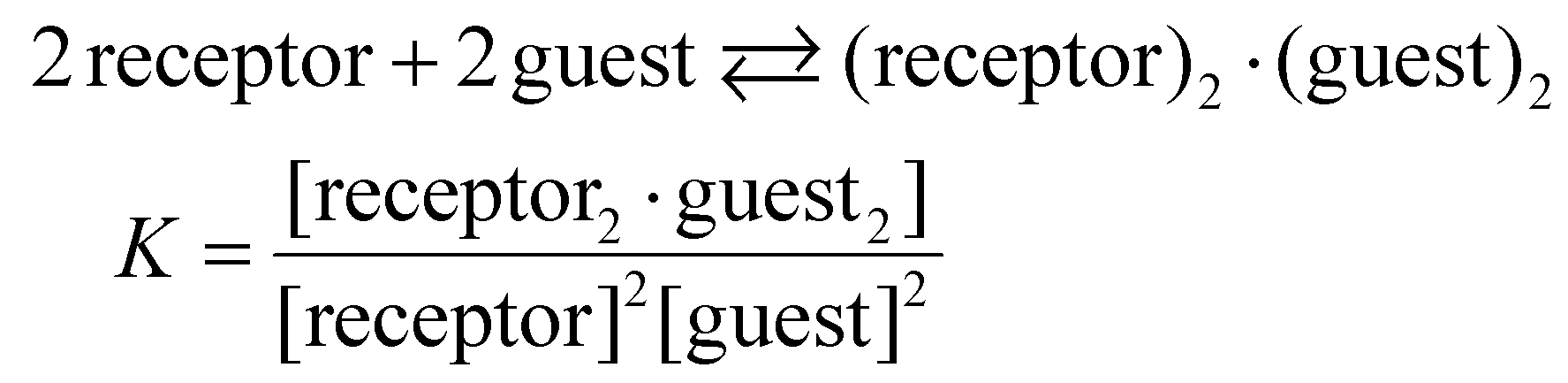 | (1) |
The ITC experiments also support this assumption, showing sigmoidal curves with single inflection points at molar ratios of approximately 1 (1/guest). The binding curves were analysed using HypCal software,18 and the association constants (logK) for the 2:2 complexes were estimated to be 12.7 for me-β-D-gal and 13.3 for me-β-D-glc. Although their comparable binding affinities, the two complexes exhibited very different thermodynamic parameters. For the me-β-D-gal complex, the enthalpy (ΔH°) and entropy (TΔS°) values were −117.0 kJ mol−1 and −45.4 kJ mol−1, respectively. In contrast, the values for the me-β-D-glc complex were ΔH° = −55.0 kJ mol−1 and TΔS° = +20.3 kJ mol−1 (Table 1). These differences are possibly attributed to the presence of water molecules contained in the 2:2 complexes, as observed in the complexes between receptor 3 and these guests (Fig. 1). The me-β-D-gal complex may include four water molecules but no water molecules are involved in the me-β-D-glc complex, as supported by their ESI-mass spectra. Under the titration conditions containing approximately 0.05% water, several water molecules are present in the internal cavities of free receptor 1 through hydrogen-bonding interactions, as seen in the X-ray structures of an indolocarbazole-naphthyridine foldamer.16 Some or all of these water molecules are likely released upon guest binding. The thermodynamic parameters indicate that the me-β-D-gal complex is enthalpically more favourable while the me-β-D-glc complex is entropically more favourable. This result is consistent with the fact that water molecules are partially liberated in the former complex but completely liberated in the latter complex.
| Receptor | Guest | LogK |
ΔG° | ΔH° | TΔS° |
|---|---|---|---|---|---|
| (2:2 complex) |
(kJ mol−1) | ||||
| 1 | me-β-D-gal | 12.7 ± 0.1 | −71.6 | −117.0 | −45.4 |
| me-β-D-glc | 13.3 ± 0.1 | −75.3 | −55.0 | +20.3 | |
| me-α-D-glc | 10.9 ± 0.1 | −61.4 | −36.4 | +25.0 | |
| 2 | me-β-D-gal | 11.1 ± 0.1 | −62.6 | −80.3 | −17.7 |
| me-β-D-glc | 13.9 ± 0.1 | −78.3 | −51.1 | +27.2 | |
| me-α-D-glc | 10.6 ± 0.1 | −60.1 | −48.6 | +11.5 | |
Next, the binding properties of receptor 2 with monosaccharides were investigated using 1H NMR spectroscopy in 5% (v/v) DMSO-d6/CD2Cl2 containing water (ca. 0.05%) (Fig. S2†). Interestingly, receptor 2 showed a well-resolved 1H NMR spectrum only when complexed with me-β-D-glc, while broad, unresolved spectra were observed in the presence of other guests (me-β-D-gal, me-α-D-glc, me-α-D-gal, and me-α-D-man). The addition of me-β-D-glc to 2 resulted in the splitting of NH signals, from four to eight (Fig. 5c and Fig. S19†). Moreover, the CH signals of indolocarbazole (H1–H6), naphthyridine (H7*–H10*, H17–H20), and fluorene (H24, H24*) are noticeably upfield-shifted (Δδ = 0.3–1.8 ppm) due to aromatic stacking interactions between the aryl planes. All OH signals of me-β-D-glc were largely downfield-shifted (δ = 10.9–7.5 ppm) as a result of the formation of strong hydrogen bonds. Furthermore, 1H–1H ROESY experiments revealed NOE correlations between the two different strands (t-Bu4*-21, t-Bu3*-17, t-Bu3-7*, and t-Bu2*-10) (Fig. 5a, e and Fig. S14†). All these results were consistent with an energy-minimised structure of (2-MP)2⊃(me-β-D-glc)2 (Fig. 5b and Fig. S13†).17 As anticipated, CD spectroscopy of 2 exhibited a low induced negative Cotton effects (Δε(439 nm) = −66.8 M−1 cm−1) upon the addition of me-β-D-glc, further supporting pseudo-meso conformation of (2-MP)2⊃(me-β-D-glc)2 (Fig. 5d and Fig. S16†). Finally, ESI-MS spectra also showed 2:2 complex peaks at m/z = 1833.9 ([(2-MP)2⊃(me-β-D-glc)2 + 3H]3+) and at m/z = 2750.3 ([(2-MP)2⊃(me-β-D-glc)2 + 2H]2+) (Fig. 5f and Fig. S27†).
 | ||
| Fig. 5 (a) Possible molecular structure of (2-MP)2⊃(me-β-D-glc)2 with NOE correlations (R = Et). (b) Energy-minimised structure of (2-MP)2⊃(me-β-D-glc)2 (MacroModel 9.1, MMFFs force field, CHCl3 phase). (c) Partial 1H NMR spectra (400 MHz, 25 °C) of 2 (1.5 mM) in the absence and presence of me-β-D-glc (1.0 equiv.) in 5% (v/v) DMSO-d6/CD2Cl2. Helically stacked CH signals are depicted in red, and me-β-D-glc signals are in blue. The blue circles and black squares correspond to the NH signals of free 2 and its 2:2 complex. (d) CD spectra of 2 (2.00 × 10−5 M, 25 ± 1 °C) and its complex with me-β-D-glc in 5% (v/v) DMSO/CH2Cl2. (e) Partial 1H–1H ROESY spectrum (400 MHz, 25 °C, mixing time 400 ms) of (2-MP)2⊃(me-β-D-glc)2 (4 mM) in CD2Cl2. (f) ESI-MS of the 2:2 complex: observed distribution (top) and calculated isotopic distribution (bottom) as [(2-MP)2⊃(me-β-D-glc)2 + 3H]3+. | ||
The binding affinities of receptor 2 for monosaccharides were measured and compared using isothermal titration calorimetry (ITC) in 5% (v/v) DMSO/CH2Cl2 (containing ca. 0.05% water, 22 °C) (Fig. S23–S25 and Tables S7–S9†). For the formation of a 2:2 complex between 2 and me-β-D-glc, the association constant (logK) was estimated to be 13.9, with ΔH° = −51.1 kJ mol−1 and TΔS° = +27.2 kJ mol−1 (Table 1). These values were nearly identical to the 2:2 complex formation between 1 and me-β-D-glc, further supporting the formation of (2-MP)2⊃(me-β-D-glc)2. Under the same conditions, the association constants (logK) were calculated to be 11.1 for me-β-D-gal and 10.6 for me-α-D-glc, both of which are two to three orders of magnitudes weaker than that for me-β-D-glc. These weaker affinities are likely responsible for the unresolved and broad 1H NMR spectra of their complexes under the given conditions.
Finally, we performed liquid–liquid transport experiments using a U-tube apparatus to investigate whether receptors 1 and 2 could facilitate the transport of monosaccharides across an organic solvent layer.19 However, receptor 1 was not suitable for the transport experiment due to the low solubility in chlorinated solvents, and the experiments were conducted with the more soluble receptor 2. As depicted in Fig. 6a, a source phase of D2O containing a mixture of me-β-D-glc, me-β-D-gal, and me-α-D-glc (2.0 M of each) and a receiving D2O phase were placed on opposite sides of a CH2Cl2 liquid membrane with or without receptor 2 (3.0 mM). A stirring bar was positioned at the bottom of the CH2Cl2 phase, and the system was stirred at 10 °C. The amount of transported guests was quantified by integrating the 1H NMR signals relative to DMSO (2 mM in D2O) used as an external standard (Fig. S15†). As a result, the presence of 2 in the CH2Cl2 layer significantly enhanced the transport of monosaccharides from the source phase to the receiving phase. Specifically, the concentration of me-β-D-glc in the receiving phase reached 17.8 mM in the presence of 2 after 96 h, compared to only 1.8 mM in its absence under the same conditions. Furthermore, the selective transport of me-β-D-glc over me-β-D-gal and me-α-D-glc was shown (Fig. 6b). The observed trend in transport efficiency is in parallel with their binding affinities.
 | ||
| Fig. 6 (a) Cartoon representation of the U-tube experiment. (b) Time-dependent concentration profiles of me-β-D-glc (red), of me-α-D-glc (green), and me-β-D-gal (blue) in receiving phase, in the absence and presence of receptor 2 in liquid membrane. Source phase (0.7 mL D2O, [me-β-D-glc] = [me-α-D-glc] = [me-β-D-gal] = 2.0 M); liquid membrane (2 mL, CH2Cl2, [2] = 3.0 mM); receiving phase (0.7 mL D2O), 10 °C. | ||
Conclusions
In this study, we present foldameric receptors 1 and 2 which adopt helical conformations and assemble to generate domain-swapping cavities for monosaccharides. Comprehensive analysis using 1H NMR, ESI-mass, CD spectroscopy, and ITC experiments revealed that receptor 1 forms two distinct 2:2 receptor/guest complexes: (1-MM)2⊃(me-β-D-gal·2H2O)2 and (1-MP)2⊃(me-β-D-glc)2, exhibiting different folding and assembly modes for optimal stability. In contrast, receptor 2 forms a single stable 2:2 complex, (2-MP)2⊃(me-β-D-glc)2, with significantly stronger binding affinity for me-β-D-glc compared to other methyl glycoside guests. U-tube transport experiments showed that receptor 2 selectively transports me-β-D-glc across an organic phase (CH2Cl2). This study clearly demonstrates that aromatic foldamers can serve as versatile synthetic receptors, with the right balance between rigidity and flexibility, enabling adaptive folding and assembly, thereby optimising the binding and transport of specific guests. Furthermore, the modification of the repeating units and sequences could enable the development of foldameric receptors capable of binding and transporting other saccharides.
Experimental section
Receptor 1
A yellow solid; mp > 275 °C (dec); 1H NMR (400 MHz, DMSO-d6, 25 °C, ppm) δ 11.63 (s, 1H), 11.45 (s, 1H), 11.38 (s, 1H), 10.74 (s, 1H), 8.57 (d, J = 8.4 Hz, 1H), 8.45 (s, 1H), 8.43 (s, 1H), 8.35 (d, J = 1.4 Hz, 1H), 8.35 (d, J = 8.3 Hz, 1H), 8.18 (d, J = 1.4 Hz, 1H), 8.14 (s, 4H), 8.01 (d, J = 8.3 Hz, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 8.3 Hz, 1H), 7.94 (d, J = 1.5 Hz, 1H), 7.82 (s, 1H), 7.80 (d, J = 7.5 Hz, 1H), 7.79 (d, J = 8.3 Hz, 1H), 7.76 (d, J = 8.2 Hz, 1H), 7.68 (d, J = 16.1 Hz, 1H), 7.66 (d, J = 1.4 Hz, 1H), 7.63 (s, 1H), 7.45 (d, J = 7.5 Hz, 1H), 7.43 (d, J = 1.6 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.21 (d, J = 16.2 Hz, 1H), 5.18 (s, OH), 3.70 (s, 1H), 1.75–1.62 (m, 4H), 1.50 (s, 18H, t-Bu), 1.44 (s, 9H, t-Bu), 1.38 (s, 9H, t-Bu), 0.98 (t, J = 7.3 Hz, 6H); 13C NMR (100 MHz, DMSO-d6, 25 °C, ppm) δ 158.9, 155.3, 155.2, 146.7, 145.7, 143.9, 142.2, 142.1, 141.8, 141.7, 141.5, 138.0, 137.9, 137.8, 137.1, 134.7, 127.1, 126.3, 126.0, 125.9, 125.9, 125.8, 125.7, 124.2, 124.1, 123.7, 121.2, 120.8, 120.7, 120.6, 120.4, 119.1, 118.8, 116.7, 112.5, 112.3, 112.1, 105.0, 103.2, 102.9, 97.5, 93.2, 93.1, 92.9, 89.4, 89.0, 87.9, 79.7, 70.9, 34.5, 34.4, 34.3, 33.8, 31.7, 31.6, 29.0, 20.7, 8.7; ESI-HRMS, m/z calcd for C179H154N16O2 [M + 2H]2+ 1280.6293 found 1280.6277.Receptor 2
A yellow solid; mp > 265 °C (dec); 1H NMR (400 MHz, DMSO-d6, 25 °C, ppm) δ 11.56 (s, 1H), 11.44 (s, 2H), 10.78 (s, 1H), 8.61 (d, J = 8.4 Hz, 1H), 8.47 (d, J = 8.4 Hz, 1H), 8.43 (s, 1H), 8.42 (s, 1H), 8.38 (d, J = 1.5 Hz, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 1.6 Hz, 1H), 8.11 (d, J = 8.4 Hz, 1H), 8.47 (s, 2H), 8.04 (d, J = 8.3 Hz, 1H), 7.98 (d, J = 8.3 Hz, 1H), 7.98 (d, J = 8.3 Hz, 1H), 7.89 (d, J = 8.3 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.83 (d, J = 7.5 Hz, 1H), 7.83 (d, J = 1.5 Hz, 1H), 7.81 (d, J = 1.6 Hz, 1H), 7.72 (d, J = 1.6 Hz, 1H), 7.66 (s, 1H), 7.44 (d, J = 1.6 Hz, 1H), 7.42 (d, J = 7.5 Hz, 1H), 7.29 (d, J = 8.3 Hz, 1H), 5.21 (s, OH), 3.78 (s, 1H), 1.76–1.64 (m, 4H), 1.49 (s, 18H, t-Bu), 1.43 (s, 9H, t-Bu), 1.37 (s, 9H, t-Bu), 1.00 (t, J = 7.3 Hz, 6H); 13C NMR (100 MHz, DMSO-d6, 25 °C, ppm) δ 155.8, 155.5, 147.3, 147.2, 146.9, 146.7, 144.2, 142.7, 142.6, 142.4, 142.2, 142.0, 138.6, 138.4, 138.4, 138.3, 138.2, 137.9, 137.6, 131.4, 129.0, 126.9, 126.5, 126.4, 126.4, 126.2, 125.5, 124.7, 124.6, 124.6, 124.2, 121.8, 121.4, 121.3, 121.2, 121.1, 120.9, 120.0, 119.5, 119.4, 117.2, 113.1, 112.8, 112.6, 105.4, 103.4, 103.4, 103.3, 98.0, 93.6, 93.4, 92.1, 90.1, 90.0, 89.5, 89.3, 80.1, 71.4, 35.0, 34.9, 34.8, 34.3, 33.7, 32.2, 32.1, 23.1, 15.2, 9.2; ESI-HRMS, m/z calcd for C179H150N16O2 [M + 2H]2+ 1278.6137 found 1278.6130.NMR titrations
A solvent mixture of 5% (v/v) DMSO-d6/CD2Cl2 with water content of ca. 0.05% was prepared using dried DMSO-d6 and a 10:1 (v/v) mixture of anhydrous and water-saturated CH2Cl2. Organic solvents were dried over molecular sieves (4 Å). Water-saturated CD2Cl2 was prepared by sonicating the CD2Cl2 with a few drops of distilled water for 20 min, followed by carefully separating the organic layer. Stock solution of 1 and 2 (1.50 mM), and each guest (6.00 mM) were prepared separately using this solvent mixture. A 400 μL of the 1 and 2 solution was taken in a NMR tube and an initial spectrum was taken to determine the chemical shifts of free receptors. Aliquots of the guest solution were added to the NMR tube and the spectrum was recorded after each addition.
ITC experiments
Stock solutions of receptors 1 and 2 (0.09–0.30 mM) and each guest (4.00–7.00 mM) were prepared separately in 5% (v/v) DMSO/CH2Cl2 (containing ca. 0.05% water). ITC experiments were conducted by adding the solution of receptor to the ITC sample cell, followed by adding each solution of a guest using a syringe. Heats of dilution which was obtained by titrating each guest into the ITC sample cell in the absence of receptors were subtracted. ITC experiments were recorded using MicroCal VP-ITC (spacing time: 240 s, temperature: 22 °C, injection volume: 3 or 4 μL). Thermodynamic values were determined using HypCal software18 based on eqn (1).U-tube transport experiments
Stock solutions of a source phase containing a mixture of me-β-D-glc, me-β-D-gal, and me-α-D-glc (2.0 M of each) in D2O and receptor 2 (3.0 mM) in CH2Cl2 were prepared separately. A U-tube apparatus with an internal diameter of 0.4 cm, a height of 7.8 cm, and a 3.0 cm distance between the two arms was used. For the experiment, 0.7 mL of the D2O source phase and 0.7 mL of the receiving D2O phase were placed on opposite sides of the U-tube, with 2.0 mL of the receptor 2 solution in CH2Cl2 placed in the middle. A stirring bar was positioned at the bottom of the CH2Cl2 phase, and the system was stirred at 500 rpm at 10 °C. The background experiment was performed under identical conditions without receptor 2. The amount of transported guests was quantified by integrating the 1H NMR signals relative to DMSO (2.0 mM in D2O) used as an external reference.Author contributions
G. S. performed all experiments and data analysis. K.-S. J. conceived and supervised the project. All authors discussed the results and write, read, and reviewed the manuscript.Data availability
The data supporting this article are included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (2021R1A2C1093591 to K.-S. J.).References
- (a) H. G. Garg, M. K. Cowman and C. A. Hales, Carbohydrate Chemistry, Biology and Medical Applications, Elsevier, Amsterdam, 2008 Search PubMed; (b) H. M. Asif, M. Akram, T. Saeed, M. I. Khan, N. Akhtar, R. ur Rehman, S. M. A. Shah, K. Ahmed and G. Shaheen, Int. Res. J. Biochem. Bioinform., 2011, 1, 1–5 Search PubMed; (c) G. A. Rabinovich, Y. van Kooyk and B. A. Cobb, Ann. N. Y. Acad. Sci., 2012, 1253, 1–15 CrossRef CAS PubMed; (d) M. He, X. Zhou and X. Wang, Sig. Transduct. Target. Ther., 2024, 9, 194 CrossRef PubMed.
- (a) M. Mazik, Chem. Soc. Rev., 2009, 38, 935–956 RSC; (b) X. Sun and T. D. James, Chem. Rev., 2015, 115, 8001–8037 CrossRef CAS PubMed; (c) A. P. Davis, Chem. Soc. Rev., 2020, 49, 2531–2545 RSC; (d) R. Ahamed, J. Venkatesh, R. Srithar, S. Gaikwad and S. Pramanik, Org. Biomol. Chem., 2023, 21, 5492–5505 RSC; (e) X. Sun, B. M. Chapin, P. Metola, B. Collins, B. Wang, T. D. James and E. V. Anslyn, Nat. Chem., 2019, 11, 768–778 CrossRef CAS PubMed; (f) B. Wu, R. Tang and Y. Tan, Nat. Rev. Chem., 2025, 9, 10–27 CrossRef CAS PubMed.
- (a) Y. Ferrand, M. P. Crump and A. P. Davis, Science, 2007, 318, 619–622 CrossRef CAS PubMed; (b) N. Chandramouli, Y. Ferrand, G. Lautrette, B. Kauffmann, C. D. Mackereth, M. Laguerre, D. Dubreuil and I. Huc, Nat. Chem., 2015, 7, 334–341 CrossRef CAS PubMed; (c) R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, Nat. Chem., 2019, 11, 52–56 CrossRef CAS PubMed; (d) B. J. J. Timmer, A. Kooijman, X. Schaapkens and T. J. Mooibroek, Angew. Chem., Int. Ed., 2021, 60, 16178–16183 CrossRef CAS PubMed; (e) J. K. Awino, R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 9759–9762 CrossRef CAS PubMed; (f) R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 829–835 CrossRef CAS PubMed; (g) X. Li and Y. Zhao, Chem. Sci., 2021, 12, 374–383 RSC.
- (a) A. P. Davis and R. S. Wareham, Angew. Chem., Int. Ed., 1999, 38, 2978–2996 CrossRef; (b) S. Tommasone, F. Allabush, Y. K. Tagger, J. Norman, M. Köpf, J. H. R. Tucker and P. M. Mendes, Chem. Soc. Rev., 2019, 48, 5488–5505 RSC.
- G. Song, S. Lee and K.-S. Jeong, Nat. Commun., 2024, 15, 1501 CrossRef CAS PubMed.
- (a) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed; (b) C. G. Pappas, P. K. Mandal, B. Liu, B. Kauffmann, X. Miao, D. Komáromy, W. Hoffmann, C. Manz, R. Chang, K. Liu, K. Pagel, I. Huc and S. Otto, Nat. Chem., 2020, 12, 1180–1186 CrossRef CAS PubMed; (c) D. Yang, L. K. S. von Krbek, L. Yu, T. K. Ronson, J. D. Thoburn, J. P. Carpenter, J. L. Greenfield, D. J. Howe, B. Wu and J. R. Nitschke, Angew. Chem., Int. Ed., 2021, 60, 4485–4490 CrossRef CAS PubMed; (d) N. M. A. Speakman, A. W. Heard and J. R. Nitschke, J. Am. Chem. Soc., 2024, 146, 10234–10239 CrossRef CAS PubMed.
- (a) N. Nandwani, P. Surana, H. Negi, N. M. Mascarenhas, J. B. Udgaonkar, R. Das and S. Gosavi, Nat. Commun., 2019, 10, 452 CrossRef CAS PubMed; (b) S. Wang, B. Wicher, C. Douat, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2024, 63, e202405091 CrossRef CAS PubMed.
- (a) L.-P. Yang, F. Jia, Q.-H. Zhou, F. Pan, J.-N. Sun, K. Rissanen, L. W. Chung and W. Jiang, Chem. – Eur. J., 2017, 23, 1516–1520 CrossRef CAS PubMed; (b) S. H. Yoo, J. Buratto, A. Roy, E. Morvane, M. Pasco, K. Pulka-Ziach, C. M. Lombardo, F. Rosu, V. Gabelica, C. D. Mackereth, G. W. Collie and G. Guichard, J. Am. Chem. Soc., 2022, 144, 15988–15998 CrossRef CAS PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- D. Wang, M. V. Ivanov, D. Kokkin, J. Loman, J.-Z. Cai, S. A. Reid and R. Rathore, Angew. Chem., Int. Ed., 2018, 57, 8189–8193 CrossRef CAS PubMed.
- K.-J. Chang, D. Moon, M. S. Lah and K.-S. Jeong, Angew. Chem., Int. Ed., 2005, 44, 7926–7929 CrossRef CAS PubMed.
- K. M. Kim, G. Song, S. Lee, H.-G. Jeon, W. Chae and K.-S. Jeong, Angew. Chem., Int. Ed., 2020, 59, 22475–22479 CrossRef CAS PubMed.
- G. Song and K.-S. Jeong, ChemPlusChem, 2020, 85, 2475–2481 CrossRef CAS PubMed.
- (a) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS; (b) P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670–6696 RSC.
- (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef; (b) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- (a) J. Y. Hwang, H.-G. Jeon, Y. R. Choi, J. Kim, P. Kang, S. Lee and K.-S. Jeong, Org. Lett., 2017, 19, 5625–5628 CrossRef CAS PubMed; (b) G. Song, K. M. Kim, S. Lee and K.-S. Joeng, Chem. – Asian J., 2021, 16, 2958–2966 CrossRef CAS PubMed.
- (a) F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440–467 CrossRef CAS; (b) T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- (a) HypCal program in Hyperquad; ; (b) G. Arena, P. Gans and C. Sgarlata, Anal. Bioanal. Chem., 2016, 408, 6413–6422 CrossRef CAS PubMed.
- (a) T. Kida, T. Ohe, H. Higashimoto, H. Harada, Y. Nakatsuji, I. Ikeda and M. Akashi, Chem. Lett., 2004, 33, 258–259 CrossRef CAS; (b) H. Abe, T. Yoneda, Y. Ohishi and M. Inouye, Chem. – Eur. J., 2016, 22, 18944–18952 CrossRef CAS PubMed; (c) Q. He, G. M. Peters, V. M. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2017, 56, 13396–13400 CrossRef CAS PubMed; (d) A. Docker, I. Marques, H. Kuhn, Z. Zhang, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2022, 144, 14778–14789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols and characterisation new compounds and NMR spectra. See DOI: https://doi.org/10.1039/d4ob02061h |
| This journal is © The Royal Society of Chemistry 2025 |