
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall far-red cationic benzoquinone diimine dyes†
Tatiana
Munteanu
 a,
Carmelo
Naim
b,
Gabriel
Canard
a,
Denis
Jacquemin
*bc,
Olivier
Siri
*a and
Simon
Pascal
*ab
a,
Carmelo
Naim
b,
Gabriel
Canard
a,
Denis
Jacquemin
*bc,
Olivier
Siri
*a and
Simon
Pascal
*ab
aAix Marseille Univ, CNRS UMR 7325, Centre Interdisciplinaire de Nanoscience de Marseille (CINaM), Campus de Luminy, case 913, Marseille cedex 09 13288, France. E-mail: simon.pascal@cnrs.fr
bNantes Université, CEISAM UMR 6230, CNRS, Nantes F-44000, France
cInstitut Universitaire de France (IUF), Paris F-75005, France
First published on 14th February 2025
Abstract
Two compact far-red cationic benzoquinone diimine dyes were synthesized, having molecular weights lower than 400 or 300 Da and featuring light absorption properties centered around 700 nm. Their redox and optical properties were investigated experimentally, alongside theoretical studies of their structural and excited-state characteristics.
Introduction
The development of far-red and near-infrared (NIR) dyes has been a key focus over recent decades, driven by their potential in cutting-edge applications. These wavelengths are particularly significant due to their compatibility with the biological transparency window, making red-NIR dyes valuable for fluorescence and photoacoustic bioimaging, as well as therapeutic applications.1 Additionally, since a substantial portion of solar radiation falls within this spectral range, red-NIR dyes are crucial for enhancing the efficiency of solar cells, and enabling the development of transparent panels.2,3 However, designing red-NIR dyes is often challenging, as it typically requires large chromophores with extensive π-delocalization to achieve the low HOMO–LUMO gaps needed for long-wavelength absorption.4–6 In the actual context of decrease in natural resources and with the aim of atom economy, reducing the size of chromophores is a pressing objective. To address this challenge, different emerging strategies focus on the design of compact red-NIR cationic dyes with low molecular weights (MW < 400 Da), such as antiaromatic systems like aminofluorenes (e.g., A+, Fig. 1),7–9 or coupled polymethine structures like benzoquinone imines (e.g., B+, Fig. 1).10,11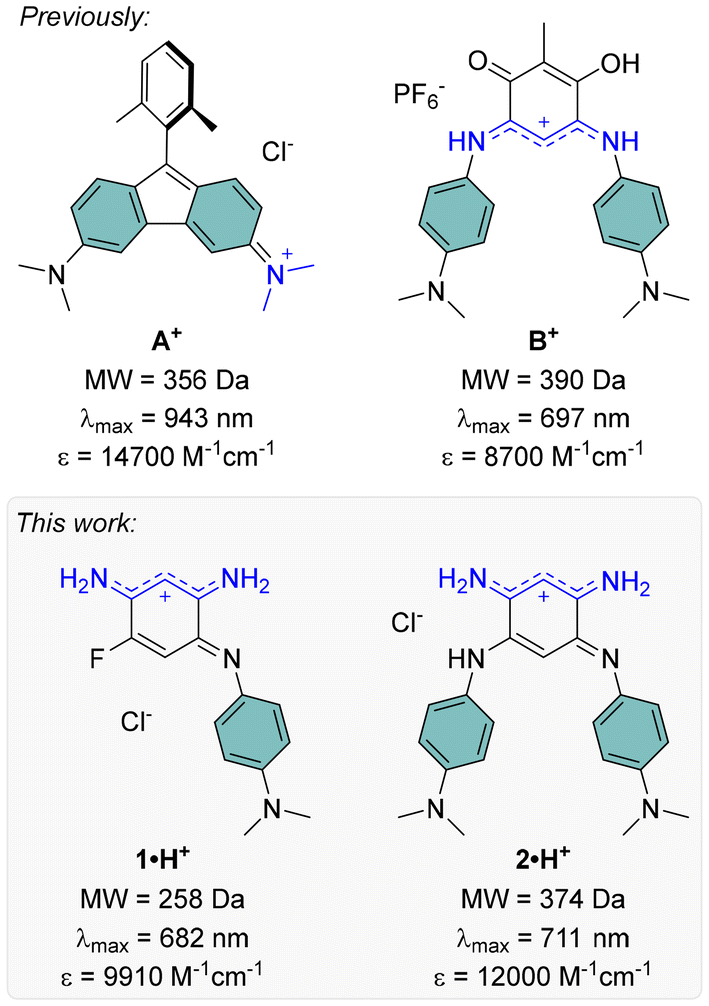 | ||
| Fig. 1 Low molecular weight red-NIR cationic dyes previously reported8,10 (top) and presented in this work (bottom), with their absorption maxima and corresponding molar extinction coefficients in solution (in water–DMSO for A+, in DCM for B+, 1·H+ and 2·H+). | ||
The design of such small quinoidal coupled polymethines relies on the combination of two 6π-electron subunits, typically forming six-membered rings linked by two single bonds. This arrangement induces electronic coupling between the subunits, reducing the HOMO–LUMO gap and redshifting the absorption (and possibly emission) of the resulting chromophore.12 We have been actively investigating the synthesis of 2,5-diaminobenzoquinone diimine coupled polymethines and their structure–property relationships to understand how N-substituents influence their electronic characteristics.13 Since the early 2000s, it has been recognized that protonation of these quinones to form mono- or dicationic species induces pronounced bathochromic shifts, extending absorption into the visible range.14 For example, introducing two weakly electron-donating trimethoxyphenyl N-substituents, as in quinone C (Fig. 2), enables reaching the red domain upon protonation in acidic solution.15 Conversely, the incorporation of strongly electron-withdrawing N-substituents, as in quinone D (Fig. 2), stabilizes a zwitterionic ground state with a lower-energy absorption band in the red range, driven by intramolecular charge transfer (ICT); a feature however vanishing upon protonation.16–18 In contrast, quinones bearing both types of aryl N-substituents, such as quinone E (Fig. 2), do not exhibit visible absorption for the neutral form and undergo noticeable red shifts upon generation of a cationic trimethine subunit in the protonated form.19
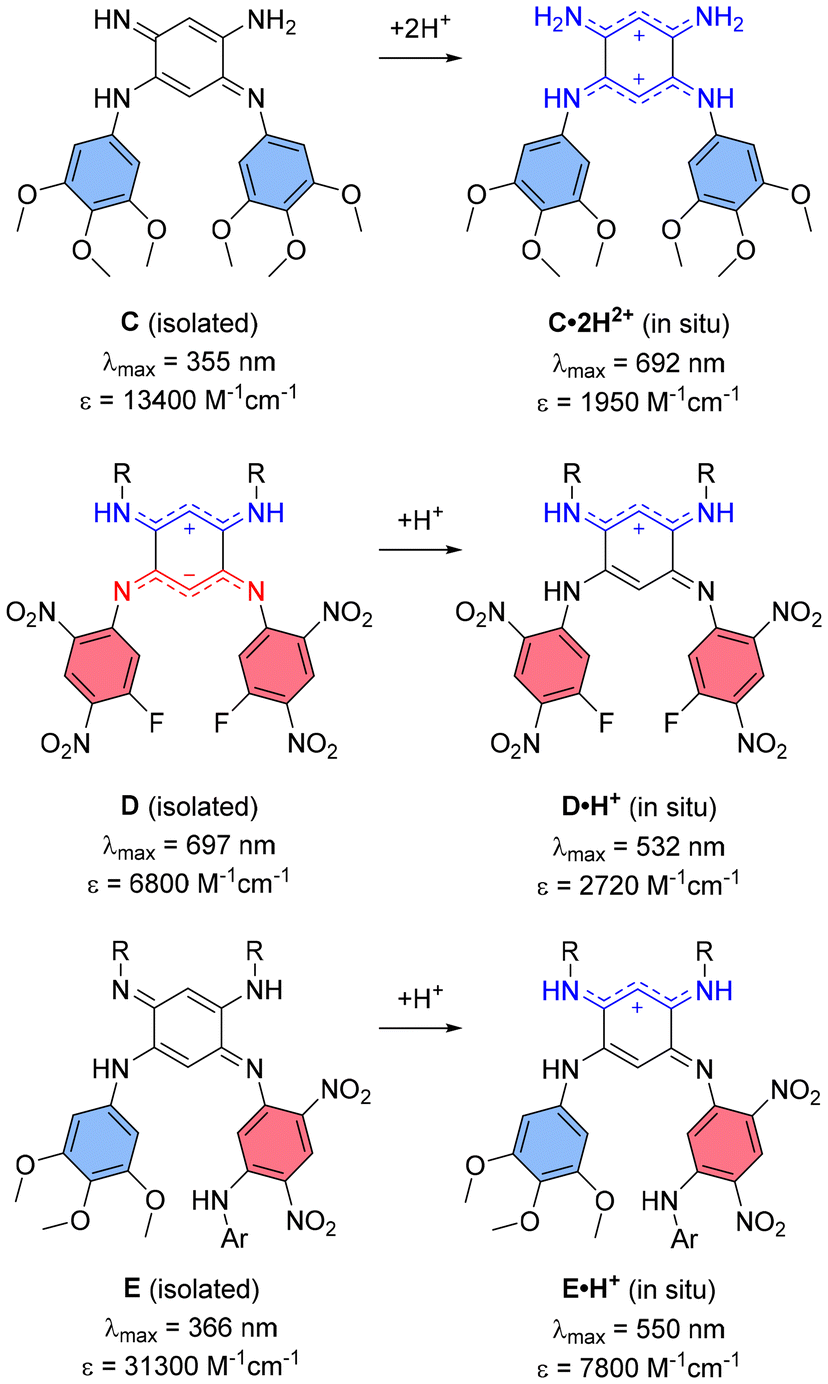 | ||
| Fig. 2 Previous works on diaminobenzoquinone diimines (R = C8H17; Ar = C6H2(OCH3)3) with their absorption maxima and corresponding molar extinction coefficients in solution (in methanol–water for C, in DCM for D and E). | ||
A notable drawback of small 2,5-diaminobenzoquinone diimines with far-red absorption is their particularly low molar extinction coefficients (ε), often attributed to nearly forbidden low-energy transitions sometimes of ICT character. While we recently succeeded in enhancing the ε of coupled heptamethine-oxonol dyes, this improvement came at the cost of significantly increasing the molecular weights of these more extended dyes, undermining efforts to maintain their compactness.20 In the present work, we report the preparation of two new benzoquinone diimines incorporating strong electron-donating dimethylaniline substituents, 1·H+ and 2·H+, which were isolated as stable cationic salts and exhibit strong absorption in the far-red range, while having molecular weight lower than 400 Da (Fig. 1). The optical properties and electronic structures of these compounds, along with their various protonation states, were explored through both experimental and theoretical investigations.
Results and discussion
Synthesis
The cationic dyes 1·H+ and 2·H+ were synthesized via a straightforward two-step route starting from commercially available 1,5-difluoro-2,4-dinitrobenzene (DFDNB), a highly reactive electrophile, and N,N-dimethyl-p-phenylenediamine, which acts as the nucleophile (Scheme 1). At room temperature, the reaction selectively yields the mono-substituted product 3 in 97% yield. In contrast, heating the reaction to 145 °C facilitates the substitution of both fluorine atoms in DFDNB, providing compound 4 in 92% yield.17 The subsequent reduction of the nitro groups is performed by catalytic hydrogenation with Pd/C in acidified tetrahydrofuran or methanol. This step is immediately followed by air oxidation of the resulting aromatic tri- or tetra-amino intermediates (which are not isolated) and a basic treatment with triethylamine to neutralize the excess hydrochloric acid until the apparition of the characteristic blue coloration of the cationic dyes. The salts 1·H+ and 2·H+ were obtained as blue-purple solids in 26% and 21% yield, respectively. The striking coloration of these compounds strongly suggests their charged nature, consistent with prior observations of similar benzoquinone diimine cations. Additionally, the cationic structure of 2·H+ was further confirmed by performing anion metathesis with potassium hexafluorophosphate, and the 19F NMR analysis demonstrated the presence of the PF6− counterion (Fig. S11, ESI†).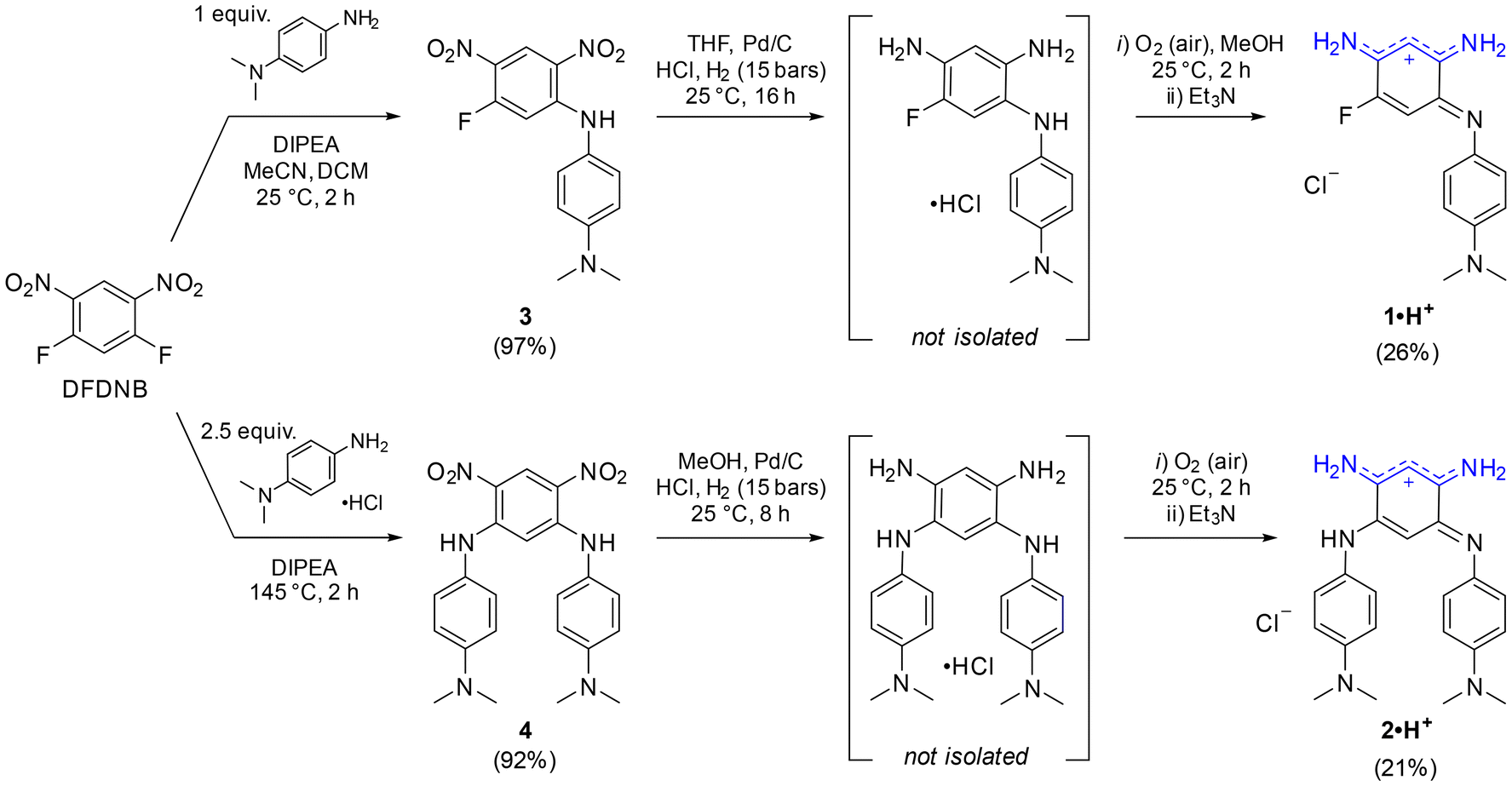 | ||
| Scheme 1 Synthesis of benzoquinone diimines 1·H+ and 2·H+. | ||
To the best of our knowledge, aminobenzoquinone diimine structures such as 1·H+ have not been reported in the literature. In an effort to expand the scope of analogs of 1·H+, we replaced N,N-dimethyl-p-phenylenediamine with 4-methoxyaniline. The transient blue coloration that was observed during this reaction suggested the formation of the desired product, unfortunately, it decomposed during purification (Scheme S1, ESI†). Attempts to reduce an analog of compound 3, introducing a single o-phenylenediamine N-substituent, resulted in a mixture of unidentified fractions. Similarly, efforts to isolate a congener of 1·H+ with a hydrogen atom instead of the fluorine atom failed due to product decomposition during the purification steps. Collectively, these results highlight the critical roles played by both the strongly electron-donating N,N-dimethyl-p-phenylenediamine group and the presence of the fluorine atom at the position 5 in stabilizing and enabling the successful isolation of aminobenzoquinone diimine 1·H+.
Electrochemical and photophysical properties
The electrochemical and spectroscopic properties of the isolated benzoquinone diimines were investigated experimentally and theoretically. Cyclic voltammetry studies provided insight into how structural modifications influence the redox behavior of the dyes (Fig. 3). Both derivatives exhibited a single irreversible one-electron reduction, occurring at −0.30 V and −0.45 V vs. Fc/Fc+ for 1·H+ and 2·H+, respectively. At anodic potentials, two one-electron oxidation processes were observed for each dye, with oxidation potentials at 0.89 V and 0.99 V for 1·H+, and at 0.38 V and 0.83 V for 2·H+. The shifts toward more cathodic potentials for the redox processes of 2·H+ indicate its greater electron-rich nature relative to 1·H+.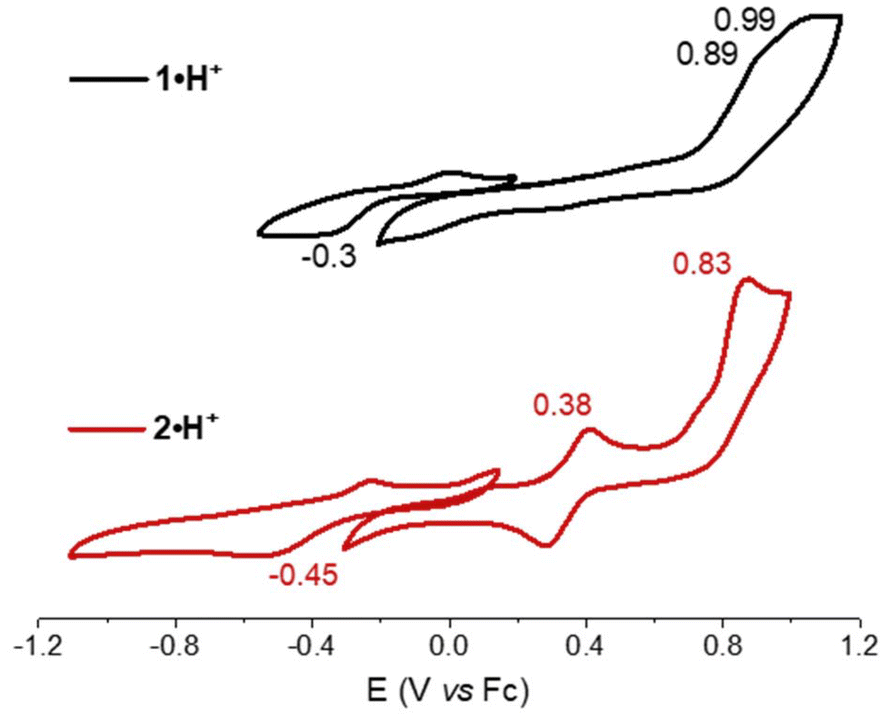 | ||
| Fig. 3 Cyclic voltammograms of 1·H+ (black) and 2·H+ (red) recorded in N,N-dimethylformamide (5 × 10−4 M) in the presence of tetra-n-butylammonium hexafluorophosphate as supporting electrolyte (10−1 M), with a scan rate of 100 mV s−1. | ||
The electronic absorption properties of the dyes were measured in dichloromethane (DCM) at a concentration of ca. 10−5 M, with the resulting spectra presented in Fig. 4 (top row) and key values compiled in Table 1. Theoretical calculations (vide infra) enabled the identification of the stable tautomers and isomers responsible for the observed spectra; these calculations, which also include conformational analysis and excited states computations, successfully reproduce the experimental absorption spectra, as illustrated in Fig. 4 (bottom row). Starting with the cationic species 1·H+ or 2·H+, different protonation states were systematically accessed: neutral compounds were obtained by the addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), while higher protonation states were achieved using trifluoroacetic acid (TFA). It is noteworthy that no emission was observed for any of the investigated species, which aligns with the non-emissive character of previously reported diaminobenzoquinone diimines.21,22
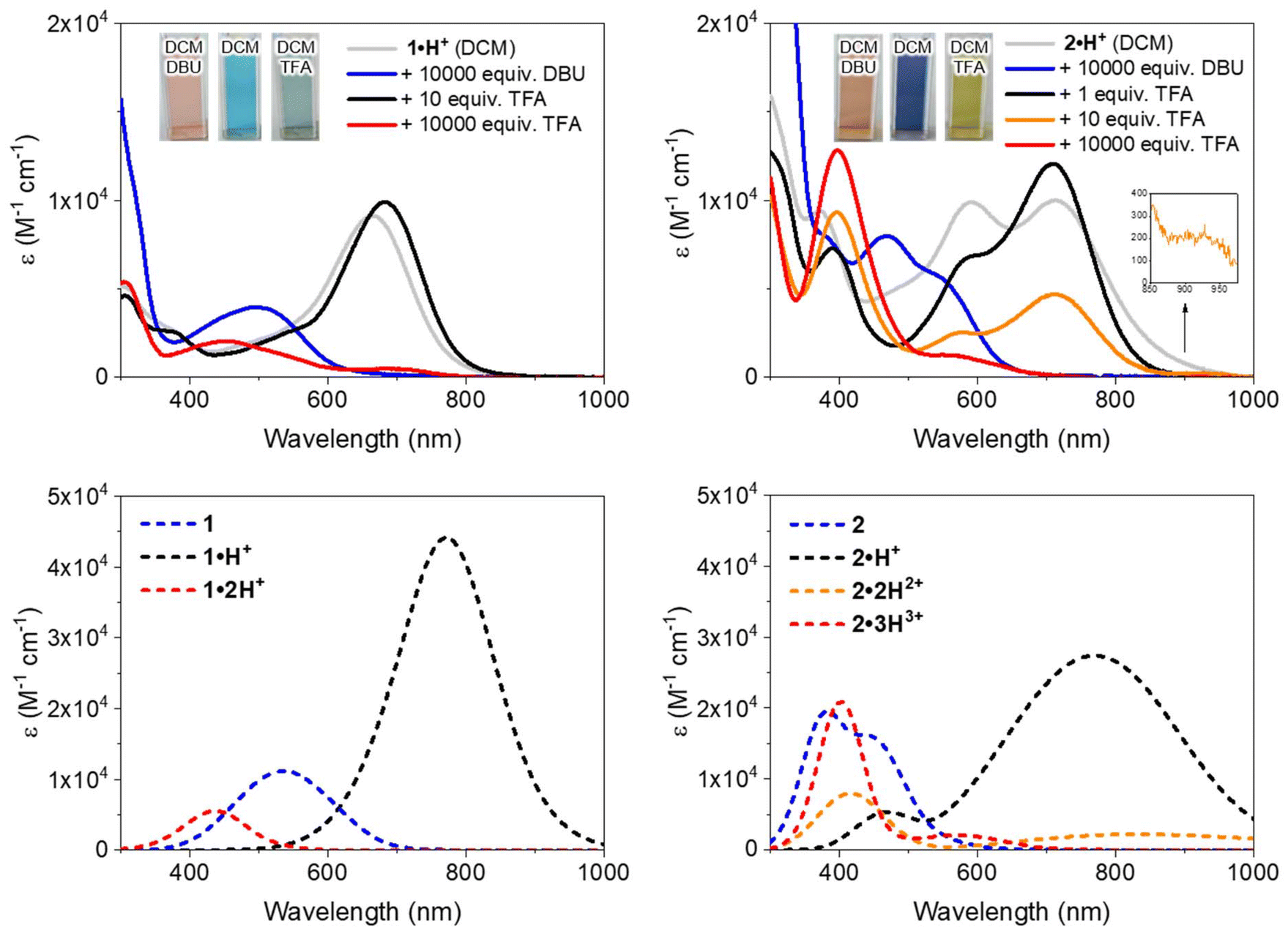 | ||
| Fig. 4 Top: electronic absorption spectra of 1·H+ (left) and 2·H+ (right) in DCM (ca. 10−5 M) without or with the presence of DBU (1,8-diazabicyclo(5.4.0)undec-7-ene) or TFA (trifluoroacetic acid). Bottom: theoretically determined electronic absorption spectra of the protonation states of 1 (left) and 2 (right) in DCM. | ||
| Species | Conditions in DCM | λ expabs [nm] (εexp [M−1 cm−1])a | λ theoabs [nm] (εtheo [M−1 cm−1])b |
|---|---|---|---|
| a Absorption maxima determined in diluted DCM solution (10−5 M). b Theoretical data extracted from Fig. 4. c Shoulder. | |||
| 1 | 1·H+ + 0.1 M DBU | 497 (3950) | 535 (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 140) 140) |
| 1·H+ | 1·H+ + 10 equiv. TFA | 682 (9910) | 771 (44160) |
| 1·2H2+ | 1·H+ + 0.1 M TFA | 454 (2040) | 437 (5510) |
| 2 | 2·H+ + 0.1 M DBU | 540c (2780) | 435 (16210) |
| 471 (7970) | 382 (19570) |
||
| 2·H+ | 2·H+ + 1 equiv. TFA | 712 (12060) |
771 (27400) |
| 600c (6920) | 470 (5340) | ||
| 390 (7290) | |||
| 2·2H2+ | 2·H+ + 10 equiv. TFA | 928c (262) | 833 (2210) |
| 713 (4680) | 415 (7970) | ||
| 578 (2550) | |||
| 396 (9320) | |||
| 2·3H3+ | 2·H+ + 0.1 M TFA | 548 (1280) | 569 (2060) |
| 397 (12830) |
402 (20900) |
||
When dissolved in DCM, 1·H+ exhibits a broad absorption band in the red region, with a maximum at 663 nm. Upon addition of DBU, the neutral species 1 is formed (Scheme 2), displaying an absorption maximum around 500 nm, which aligns well with the theoretically predicted maximum at 535 nm. Conversely, the addition of TFA induces a redshift, moving the maximum to 682 nm and increasing the molar extinction coefficient to ε682 = 9910 M−1 cm−1. This suggests a complete conversion to the cationic species 1·H+ (see titration, Fig. S29, ESI†). The observed peak is fitting with theoretical calculations, which predict a strongly-allowed transition redshifted to 771 nm for that species. This corresponds to an 89 nm shift from the experimental value, consistent with the expected error of the selected level of theory. However, the molar extinction coefficient is significantly overestimated in the theoretical model, calculated as ε = 44160 M−1 cm−1. Further addition of more than 10 equivalents of TFA causes a hypochromic shift of the red-region band and the emergence of a new transition at 454 nm with ε ∼ 2000 M−1 cm−1, corresponding to the formation of the dicationic species 1·2H2+ (red spectrum, Fig. 4). This new band is well-supported by theoretical calculations, which predict an absorption at approximately 440 nm. The shift can be attributed to the protonation of the dimethylamine group, disrupting the ICT mechanism (see below).
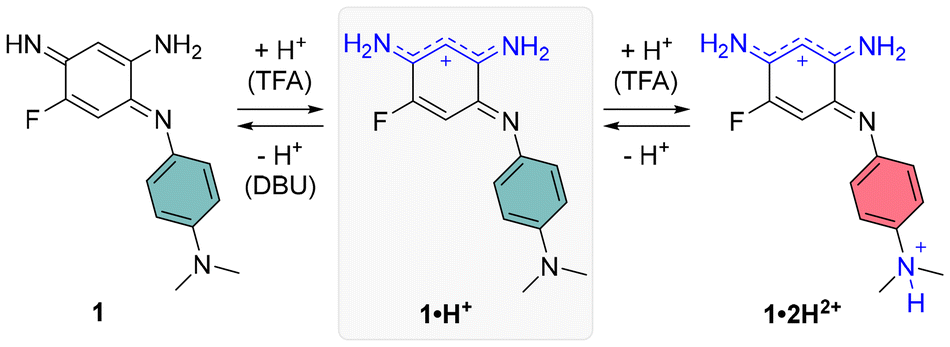 | ||
| Scheme 2 Protonation states of compound 1. | ||
Regarding the symmetrical compound 2·H+ in DCM, it presents a broad absorption spectrum with two main bands centered around 700 and 600 nm, which appear to originate from a mixture of the neutral and mono-protonated forms, 2 and 2·H+ (see grey spectrum in Fig. 4). Indeed, the theoretical absorption of 2·H+ predicts a lower-energy transition at 771 nm, hinting that the experimental spectrum in DCM likely represents a mixture of two species. Upon addition of DBU to the solution, the absorption undergoes a significant blueshift to the 400–550 nm region (blue spectrum in Fig. 4), corresponding to the formation of the neutral quinone 2 (Scheme 3), which is also predicted by theory to absorb in this range. This absorption range is consistent with those of similar electron-rich quinones bearing aniline groups, such as indamine.23 To achieve full conversion to the cationic species 2·H+, an acidic titration was performed (Fig. S30, ESI†), which revealed that upon adding one equivalent of TFA, the mixture of 2 and 2·H+ is fully converted to the mono-cationic species 2·H+. This species is characterized by an absorption maximum centered at 712 nm with a high ε of 12000 M−1 cm−1 (black spectrum, Fig. 4). The absorption signature of the mono-protonated form again fits the theoretically calculated values, although the experimental absorption is slightly shifted to a higher energy (711 nm vs. 770 nm).
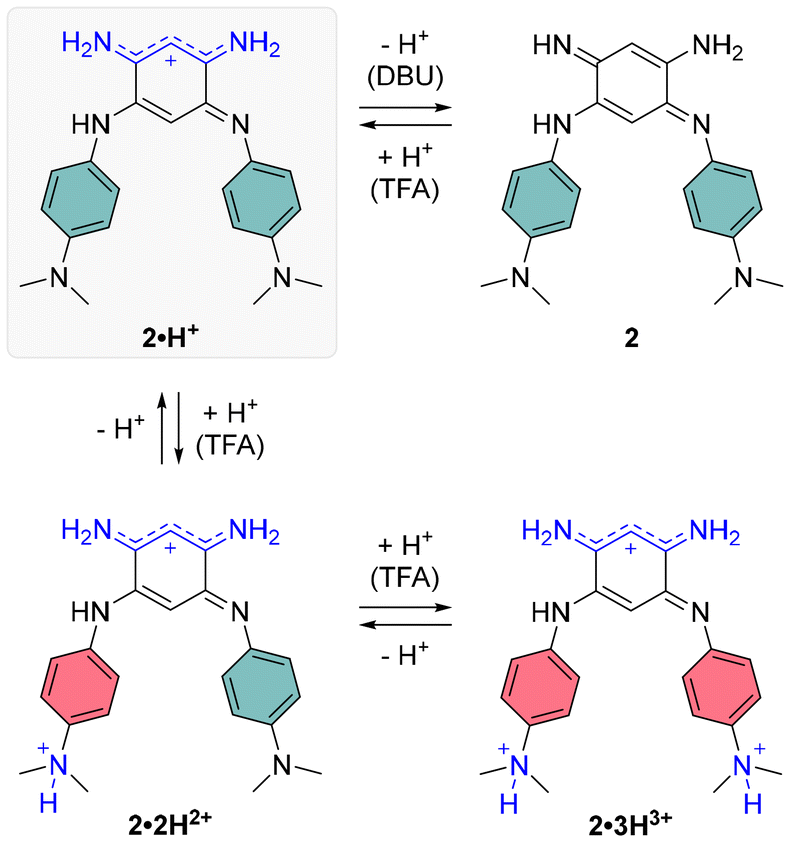 | ||
| Scheme 3 Protonation states of compound 2. | ||
The addition of ten equivalents of TFA results in a noticeable hypochromic shift in the far-red absorption band, attributed to the second protonation of the compound, progressively forming the di-cationic species 2·2H2+ (orange spectrum, Fig. 4). The experimental spectrum reveals a new band at 396 nm and a very weak transition in the NIR (800–1000 nm) with ε928 = 262 M−1 cm−1, corresponding to a strong ICT from the non-protonated dimethylamine towards the core of the dye. With a large excess of TFA (10000 equivalents), protonation of the second dimethylamine moiety diminishes the electron-donating effects of the aryl substituents, resulting in a blueshifted absorption maximum at ca. 400 nm, with a shoulder around 550 nm.
The absorption solvatochromism of 1·H+ and 2·H+ recorded in various solvents (Fig. S31, ESI†) reveals that the spectral variations primarily arise from the coexistence of protonated and neutral dye species in solution. Notably, slightly basic solvents (such as DMF or DMSO) favor deprotonation, increasing the proportion of neutral dyes, whereas acidic (e.g., DCM) or protic solvents (e.g., MeOH, H2O) stabilize the cationic forms.
Theoretical analysis
To gain deeper insights into the structure–property relationships for the two chromophores and their various protonation states, we studied their ground-state geometries and low-lying excited states using (TD-)DFT calculations. In the neutral species 1, conformational analysis of the tautomers reveals that the most stable geometry exhibits distinct structures for the two nitrogen atoms in the unsubstituted trimethine chain: one adopts an amine-like configuration, while the other forms an imine group (see Scheme 2 and Table S1, ESI†). This asymmetry imparts a quinone-like character to the aromatic ring. Upon protonation to form the cationic species 1·H+, the proton preferentially binds to the imine group, localizing the positive charge on the upper trimethine subunit and stabilizing the overall structure. For the dicationic species 1·2H2+, the second proton attaches to the dimethylamino group, suppressing its electron-donating character and leaving the aromatic ring largely unchanged. Excited-state calculations attribute the observed absorption bands (Fig. 4, upper left) primarily to HOMO → LUMO transitions, characterized by high oscillator strengths (see Tables S3 and S4, ESI†). The electron density difference maps between the S0 and S1 states (EDD1) for 1, 1·H+, and 1·2H2+ further elucidate the nature of these transitions (Fig. 5). In both 1 and 1·H+, EDD1 indicates significant ICT across the molecule, with this effect being more pronounced in the neutral species. Conversely, in 1·2H2+, excitation is localized within the two trimethine subunits, consistent with the diminished electron-donating character of the protonated dimethylamino group.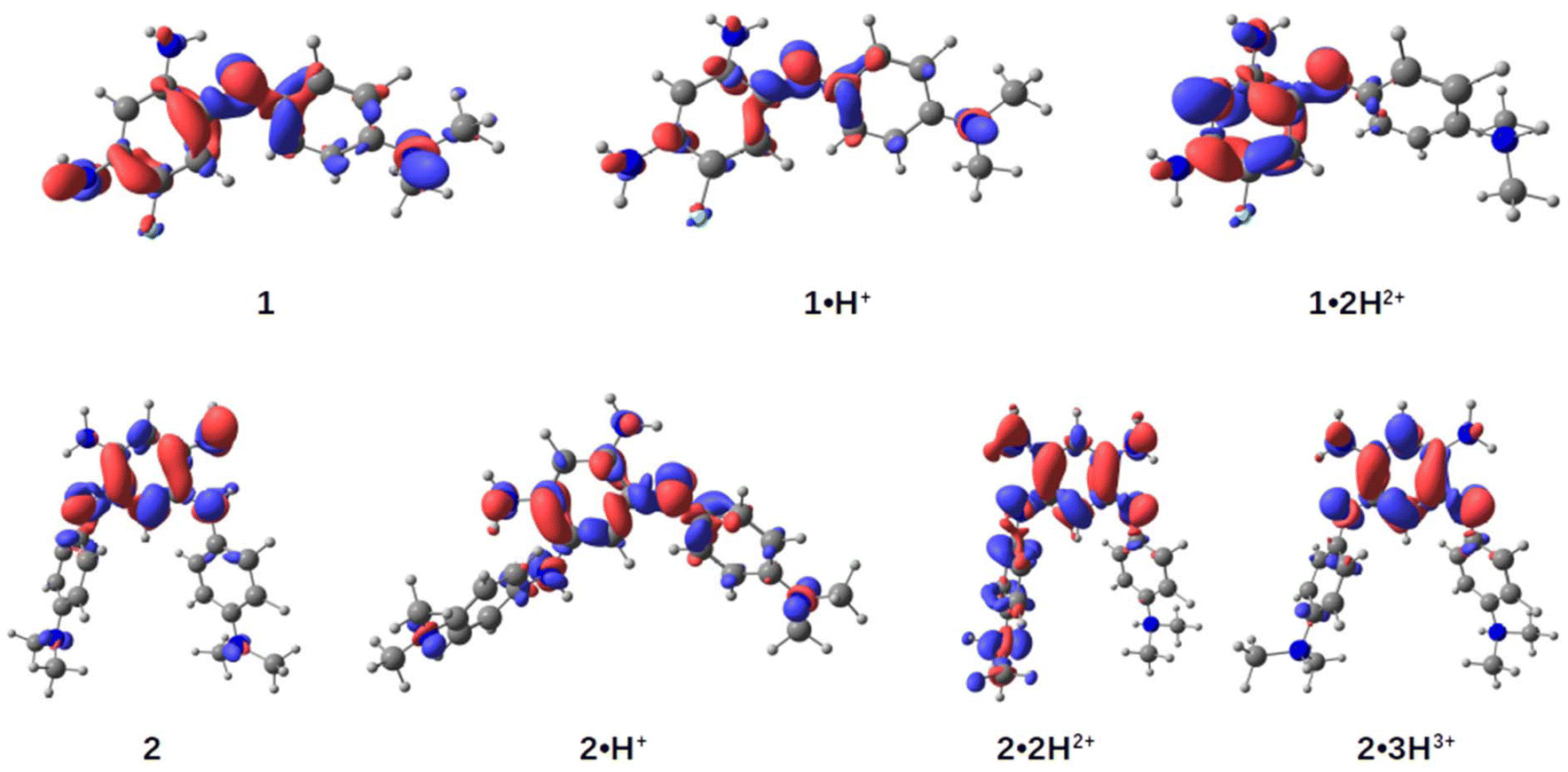 | ||
| Fig. 5 Electronic density differences (EDD) between the ground (S0) and the first excited state (S1) for molecules 1, 2 (most stable isomers) and their corresponding cationic forms. Regions in red indicate an increase in electronic density from S0 to S1, while regions in blue indicate a decrease. The isosurfaces are visualized with an isovalue of 0.003. | ||
For the neutral compound 2, tautomer and conformer analyses indicate that the canonical quinone structure is the most stable form, characterized by an almost coplanar arrangement of its branches (see ESI†). Similar to 1, the cationic form 2·H+ exhibits charge delocalization along the unsubstituted trimethine chain. Protonation in 2·H+ occurs at one of the nitrogen atoms within the unsubstituted trimethine chain (Scheme 3), resulting in two distinct isomers with a minimal free energy difference of less than 0.6 kcal mol−1 at room temperature (see Table S2, ESI†). The most stable isomer adopts a twisted conformation, with one branch oriented approximately 120° out of the molecular plane, whereas the less stable isomer retains coplanarity between both branches. Further protonation states, 2·2H2+ and 2·3H3+, predominantly involve the sequential protonation of the two dimethylamino groups rather than the second trimethine subunit.
Regarding excited-state calculations, the absorption spectra of 2 and its protonated forms (Fig. 4) arise from contributions of the first lowest three excited states. In the neutral compound 2, which adopts a quinone structure, the S1 band is situated closer to other transitions in the 350–450 nm range, all exhibiting relatively low oscillator strengths. For 2, the EDD1 is predominantly localized on the core of the dye, with only minor contributions from the dimethylaniline moieties. A gain in electron density is observed at the single bonds within the core ring, which aligns with findings from related compounds previously studied by some of us.15
In the monocation 2·H+, the strong NIR absorption originates from the S1 state, dominated by a HOMO → LUMO transition with high oscillator strength (see Table S3, ESI†). Protonation of the nitrogen atom in the upper ring induces a pronounced charge-transfer character, leading to delocalization of the EDD1 across the entire molecule. We highlight that the experimental spectra likely represent a mixture of both isomers; however, as the spectral differences between these two conformers are not significant (see Fig. S34, ESI†), only the most stable isomer is reported here.
For the dication 2·2H2+, protonation of one dimethylamino group restricts delocalization to the remaining moieties of the molecule. In the trication 2·3H3+, where both dimethylamino groups are protonated, the excited state becomes confined to the core ring. In these protonation states, the S1 band is well-separated but exhibits a reduced molar absorption coefficient. In contrast, the intense maxima in their spectra arise from the S2 and S3 states, which involve transitions such as HOMO−1 → LUMO and HOMO−2 → LUMO, respectively (see Table S3, ESI†).
Conclusions
In summary, we have synthesized and characterized two novel small cationic benzoquinone diimine dyes, 1·H+ and 2·H+, which demonstrate strong absorption in the far-red region. Through a combination of experimental studies and theoretical calculations, we established how structural modifications, protonation states, and electronic properties influence their optical behavior. Notably, the dyes exhibit higher molar extinction coefficients compared to previously reported small coupled polymethines based on quinoidal structures. These findings provide valuable insights into the design of compact red-to-NIR dyes, paving the way for further exploration of small and efficient chromophores for applications in bioimaging and optoelectronics.Experimental section
General remarks and analysis conditions
To improve the accuracy of vertical excitation energies, CC2 corrections30 were applied to the gas-phase TD-DFT results. Such corrections consist in shifting the TD-DFT values computed in solution (with cLR2) using the difference between CC2 and TD-DFT values in the gas phase. CC2 calculations employed the aug-cc-pVDZ with the associated auxiliary (RI) basis set31 and were performed using the Turbomole software applying the frozen-core approximation.32
Vibronic calculations were performed with the FCClasses 3 software33 using the Vertical Hessian (VH) model within the time-dependent (TD) formalism.34 The Franck–Condon approximation was applied for dipole expansion, i.e., Herzberg–Teller effects were neglected. Geometries, gradients, and Hessians were calculated with the (TD-)DFT level described above, with transition energies refined using the cLR2 combined with CC2 protocol. Internal coordinates were employed. To ensure easy comparisons with experimental observations, the absorption spectra were convoluted with a Gaussian broadening of 0.10 eV. For compound 1 and relative cations, absorption spectra included the first excited state, while for compound 2 and relative cations, the first three excited states were considered.
Synthetic protocols and characterizations
:1 to 85:15) as eluent and the product was finally precipitated in diethyl ether to afford 1·H+ as a blue-violet solid (26% yield, 48 mg, 0.16 mmol). Rf: 0.5 (Al2O3, dichloromethane/methanol, 9:1). 1H NMR (CD3OD, 400 MHz): δ = 7.31 (d, 3J = 8.6 Hz, 2H, CH), 7.01 (d, 3JH–F = 12.4 Hz, 1H, CH), 6.91 (d, 3J = 8.91 Hz, 2H, CH), 6.09 (d, 4JH–F = 8.5 Hz, 1H, CH), 3.18 (s, 6H, CH3). 13C NMR (CD3OD, 101 MHz): δ = 159.5 (d, 1JC–F = 163.1 Hz, C), 156.1 (d, 2JC–F = 19.3 Hz, C), 155.2 (CH), 154.0 (C), 141.5 (d, 2JC–F = 5.7 Hz, C), 139.0 (C), 130.1 (CH), 114.1 (CH), 107.5 (d, 2JC–F = 17.7 Hz, CH), 92.5 (CH), 40.5 (CH3). 19F (CD3OD, 376 MHz): δ = −127.19. HRMS (ESI+) calculated for (C+): 259.1354 (C14H16FN4+), found: 259.1357.
:5 to 9:1) as eluent to afford 2·H+ as a blue-violet solid (21% yield, 40 mg, 0.1 mmol). Rf: 0.20 (Al2O3, dichloromethane/methanol, 95:5). 1H NMR (CD3OD, 400 MHz): δ = 7.03 (d, 3J = 8.7 Hz, 4H, CH), 6.75 (d, 3J = 8.9 Hz, 4H, CH), 6.15 (s, 1H, CH), 6.00 (s, 1H, CH), 2.94 (s, 12H, CH3). 1H NMR (DMSO-d6, 400 MHz): δ = 9.36 (br s, 2H, NH), 8.71 (br s, 3H, NH, NH2), 7.03 (br s, 4H, CH), 6.75 (d, 3J = 8.6 Hz, 4H, CH), 5.98 (s, 1H, CH), 5.96 (s, 1H, CH), 2.91 (s, 12H, CH3). 13C NMR did not show any signals in DMSO-d6 and only partially resolved signals in CD3OD due to the limited solubility of the compound. 13C {DEPT135} NMR (CD3OD, 101 MHz): δ = 125.8 (CH), 113.5 (CH), 96.8 (CH), 92.3 (CH), 40.1 (CH3). HRMS (ESI+) calculated for (C+): 375.2292 (C22H27N6+), found: 375.2287.
Anion exchange with KPF6
2·H+ was dissolved in dichloromethane and washed with an aqueous solution of KPF6 (0.1 M), then with water. The organic layer was dried, evaporated and the product 2·H+ PF6− was analyzed by 1H and 19F NMR. 1H NMR (CD3OD, 400 MHz): δ = 6.99 (br d, 3J = 7.6 Hz, 4H, CH), 6.72 (br d, 3J = 6.4 Hz, 4H, CH), 6.12 (s, 1H, CH), 6.01 (s, 1H, CH), 2.91 (s, 12H, CH3). 19F NMR (CD3OD, 376 MHz): δ = −73.78 (d, 1JP–F = 720 Hz).Author contributions
Conceptualization: S. P., D. J., O. S.; investigations: T. M., S. P., G. C., C. N.; supervision: S. P., D. J., O. S.; writing – original draft: T. M., S. P., C. N., D. J.; writing – review & editing: T. M., C. N., G. C., D. J., O. S., S. P.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agence Nationale de la Recherche, in the frame of the CONDOR (ANR-21-CE07-0058) and SOCOOL (ANR-20-CE07-0024) projects. We thank the CNRS and the Ministère de l'Enseignement Supérieur et de la Recherche (PhD grant to TM). We thank the Spectropole (Aix-Marseille Univ.) for HRMS analyses. This research used resources of the GLiCID Computing Facility (Ligerien Group for Intensive Distributed Computing, 10.60487/glicid, Pays de la Loire, France).References
- H. Li, Y. Kim, H. Jung, J. Y. Hyun and I. Shin, Near-Infrared (NIR) Fluorescence-Emitting Small Organic Molecules for Cancer Imaging and Therapy, Chem. Soc. Rev., 2022, 51, 8957–9008 RSC.
- F. Grifoni, M. Bonomo, W. Naim, N. Barbero, T. Alnasser, I. Dzeba, M. Giordano, A. Tsaturyan, M. Urbani, T. Torres, C. Barolo and F. Sauvage, Toward Sustainable, Colorless, and Transparent Photovoltaics: State of the Art and Perspectives for the Development of Selective Near-Infrared Dye-Sensitized Solar Cells, Adv. Energy Mater., 2021, 11, 2101598 CrossRef CAS.
- D. Meng, R. Zheng, Y. Zhao, E. Zhang, L. Dou and Y. Yang, Near-Infrared Materials: The Turning Point of Organic Photovoltaics, Adv. Mater., 2022, 34, 2107330 CrossRef CAS PubMed.
- J. Fabian, H. Nakazumi and M. Matsuoka, Near-Infrared Absorbing Dyes, Chem. Rev., 1992, 92, 1197–1226 CrossRef CAS.
- G. Qian and Z. Y. Wang, Near-Infrared Organic Compounds and Emerging Applications, Chem. – Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed.
- S. Pascal, S. David, C. Andraud and O. Maury, Near-Infrared Dyes for Two-Photon Absorption in the Short-Wavelength Infrared: Strategies towards Optical Power Limiting, Chem. Soc. Rev., 2021, 50, 6613–6658 RSC.
- A. Barker and C. C. Barker, 3:6-Disubstituted Fluorenes. Part III. Fluorene Analogues of Michler's Hydrol, Malachite-Green, and Crystal-Violet, J. Chem. Soc., 1954, 1307–1309 RSC.
- M. Grzybowski, O. Morawski, K. Nowak and P. Garbacz, Fluorene Analogues of Xanthenes – Low Molecular Weight near-Infrared Dyes, Chem. Commun., 2022, 58, 5455–5458 RSC.
- K. Yan, Z. Hu, P. Yu, Z. He, Y. Chen, J. Chen, H. Sun, S. Wang and F. Zhang, Ultra-Photostable Small-Molecule Dyes Facilitate near-Infrared Biophotonics, Nat. Commun., 2024, 15, 2593 CrossRef CAS PubMed.
- A. Torres Ruiz, M. H. E. Bousquet, S. Pascal, G. Canard, V. Mazan, M. Elhabiri, D. Jacquemin and O. Siri, Small Panchromatic and NIR Absorbers from Quinoid Zwitterions, Org. Lett., 2020, 22, 7997–8001 CrossRef PubMed.
- T. Munteanu, V. Mazan, M. Elhabiri, C. Benbouziyane, G. Canard, D. Jacquemin, O. Siri and S. Pascal, A Strategy to Design Substituted Tetraamino-Phenazine Dyes and Access to an NIR-Absorbing Benzoquinonediimine-Fused Quinoxaline, Org. Lett., 2023, 25, 3886–3891 CrossRef CAS PubMed.
- S. Dähne and D. Leupold, Coupling Principles in Organic Dyes, Angew. Chem., Int. Ed. Engl., 1966, 5, 984–993 CrossRef.
- B. Mourot, D. Jacquemin, O. Siri and S. Pascal, Coupled Polymethine Dyes: Six Decades of Discoveries, Chem. Rec., 2024, 24, e202400183 CrossRef CAS PubMed.
- O. Siri, P. Braunstein, M.-M. Rohmer, M. Bénard and R. Welter, Novel “Potentially Antiaromatic”, Acidichromic Quinonediimines with Tunable Delocalization of Their 6π-Electron Subunits, J. Am. Chem. Soc., 2003, 125, 13793–13803 CrossRef CAS PubMed.
- L. Lavaud, Z. Chen, M. Elhabiri, D. Jacquemin, G. Canard and O. Siri, Di- vs. Tetra-Substituted Quinonediimines: A Drastic Effect on Coordination Chemistry, Dalton Trans., 2017, 46, 12794–12803 RSC.
- S. Pascal, L. Lavaud, C. Azarias, G. Canard, M. Giorgi, D. Jacquemin and O. Siri, Controlling the Canonical/Zwitterionic Balance through Intramolecular Proton Transfer: A Strategy for Vapochromism, Mater. Chem. Front., 2018, 2, 1618–1625 RSC.
- S. Pascal, L. Lavaud, C. Azarias, A. Varlot, G. Canard, M. Giorgi, D. Jacquemin and O. Siri, Azacalixquinarenes: From Canonical to (Poly-)Zwitterionic Macrocycles, J. Org. Chem., 2019, 84, 1387–1397 CrossRef CAS PubMed.
- T. Horáčková, M. H. E. Bousquet, A. Morice, U. Triballier, G. Canard, P. Lhoták, D. Jacquemin, S. Pascal and O. Siri, Fully Zwitterionic Diaminobenzoquinonediimines Promoted by Cyanoaromatic N-Substituents, Dyes Pigm., 2022, 206, 110681 CrossRef.
- J.-F. Longevial, Z. Chen, S. Pascal, G. Canard, D. Jacquemin and O. Siri, Stabilization of a 12-π Electrons Diamino-Benzoquinonediimine Tautomer, Chem. Commun., 2021, 57, 548–551 RSC.
- B. Mourot, V. Mazan, M. Elhabiri, R. Sarkar, D. Jacquemin, O. Siri and S. Pascal, Insights into Extended Coupled Polymethines through the Investigation of Dual UV-to-NIR Acidochromic Switches Based on Heptamethine–Oxonol Dyes, Chem. Sci., 2024, 15, 1248–1259 RSC.
- S. Pascal and O. Siri, Benzoquinonediimine Ligands: Synthesis, Coordination Chemistry and Properties, Coord. Chem. Rev., 2017, 350, 178–195 CrossRef CAS.
- H. Lei, S. M. Aly, P.-L. Karsenti, D. Fortin and P. D. Harvey, Luminescent Organometallic Complexes Built upon the Nonemissive Azophenine, Organometallics, 2017, 36, 572–581 CrossRef CAS.
- J. F. Corbett and E. P. Gamson, Benzoquinone Imines. Part XI. Mechanism and Kinetics of the Reaction of p-Benzoquinone Di-Imines with Aniline and Its Derivatives, J. Chem. Soc., Perkin Trans. 2, 1972, 1531 RSC.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 16A.03, Gaussian Inc., Wallingford, USA, 2016 Search PubMed.
- Y. Zhao and D. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- C. A. Guido, A. Chrayteh, G. Scalmani, B. Mennucci and D. Jacquemin, Simple Protocol for Capturing Both Linear-Response and State-Specific Effects in Excited-State Calculations with Continuum Solvation Models, J. Chem. Theory Comput., 2021, 17, 5155–5164 CrossRef CAS PubMed.
- Chemcraft – Graphical Software for Visualization of Quantum Chemistry Computations (version 1.8) https://www.chemcraftprog.com.
- O. Christiansen, H. Koch and P. Jørgensen, The Second-Order Approximate Coupled Cluster Singles and Doubles Model CC2, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef CAS.
- C. Hättig and F. Weigend, CC2 Excitation Energy Calculations on Large Molecules Using the Resolution of the Identity Approximation, J. Chem. Phys., 2000, 113, 5154–5161 CrossRef.
- TURBOMOLE, V7.4.1 2019, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007. https://www.turbomole.com.
- F. Santoro and J. Cerezo, FCclasses3 (version 1.0 (v3-0.1-177-g4b1514a)), 2021. https://www.iccom.cnr.it/en/fcclasses/.
- F. J. Avila Ferrer, J. Cerezo, J. Soto, R. Improta and F. Santoro, First-Principle Computation of Absorption and Fluorescence Spectra in Solution Accounting for Vibronic Structure, Temperature Effects and Solvent Inhomogenous Broadening, Comput. Theor. Chem., 2014, 1040–1041, 328–337 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic protocols and characterizations, 1H, 13C and 19F NMR spectra, HRMS spectra, additional electronic absorption spectra, additional theoretical data. See DOI: https://doi.org/10.1039/d5ob00082c |
| This journal is © The Royal Society of Chemistry 2025 |