
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinitiated thiol–ene mediated functionalisation of 4,5-enoses†
Alejandro
Prieto-Castañeda
 ac,
Harlei
Martin
a,
Tapasi
Manna
b,
Laura
Beswick
b,
Joshua T.
McLean
a,
Imlirenla
Pongener
b,
Inés Rabadán
González
a,
Brendan
Twamley
a,
Gavin J.
Miller
*b and
Eoin M.
Scanlan
*a
ac,
Harlei
Martin
a,
Tapasi
Manna
b,
Laura
Beswick
b,
Joshua T.
McLean
a,
Imlirenla
Pongener
b,
Inés Rabadán
González
a,
Brendan
Twamley
a,
Gavin J.
Miller
*b and
Eoin M.
Scanlan
*a
aSchool of Chemistry & Trinity Biomedical Sciences Institute (TBSI), Trinity College Dublin, 152-160 Pearse Street, Dublin D02 R590, Ireland. E-mail: eoin.scanlan@tcd.ie
bSchool of Chemical and Physical Sciences & Centre for Glycoscience, Keele University, Keele, Staffordshire, ST5 5BG, UK. E-mail: g.j.miller@keele.ac.uk
cChemical and Environmental Technology Department, ESCET, Universidad Rey Juan Carlos, 28933, Móstoles, Spain
First published on 16th May 2025
Abstract
The photoinitiated thiol–ene reaction is emerging as a highly efficient methodology for thioglycoside synthesis. Herein, the radical-mediated hydrothiolation reaction of 4,5-unsaturated saccharides was extended, offering efficient access to C4-position, S-linked glycosides. A diverse range of 4,5-unsaturated saccharides were investigated with high-yields achieved for the thioether products with complete regioselectivity and good diastereoselectivity. 1,2-Ethanedithiol products furnished a thiol-residue suitable for tagging and fluorescent labelling of a disaccharide.
Introduction
Thio-linked oligosaccharides are valuable glycomimetics with inherent resistance to glycosidase activity, offering significant potential both as probes for glycobiology and as therapeutics.1–4 Consequently, methods for the chemical synthesis of thioglycosides via the regio- and stereoselective introduction of thioether groups at specific sites on the carbohydrate backbone are of considerable interest.5–14 Thiol–ene ligation, between a thiol and an unsaturated sugar, offers an efficient and selective approach for thioglycoside synthesis (Fig. 1A). Borbás, Somsák and co-workers have extensively investigated the thiol–ene reaction of both endo- and exo-enoses to furnish anomeric S-glycans in high yields, often with high stereoselectivity.9,10 Furthermore, the thiol–ene reaction of 2,3-unsaturated glycosides was applied to the introduction of a thio-linkage at either C2- or C3-position of pyranoses, with regioselectivity highly influenced by the ring substituents. The methodology was further extended to furanose exomethylene derivatives to furnish thiol-analogues of nucleosides.9 The radical-mediated approach enabled access to novel thiosugars that would be challenging to synthesise through traditional ionic methods.15 4,5-Unsaturated sugars are of particular interest in glycobiology owing to their formation as byproducts of polysaccharide lyase activity.16,17 These enzymes operate through an elimination mechanism resulting in oligosaccharide products displaying a terminal 4,5-unsaturated uronic acid residue. Lyase enzymes that process human glycosaminoglycans (GAGs) include heparin lyase and chondroitin lyase, critical carbohydrate processing enzymes, often overexpressed in disease states.18–22 The thia-Michael addition and thiol–ene reaction have been used to investigate labelling or modification of the non-reducing end of low molecular weight heparin23–25 or electron-deficient enoses.26,27 | ||
| Fig. 1 (A) Photoinitiated thiol–ene in 1,2-enoses. (B) Prior thia-Michael addition study. (C) Photoinitiated thiol–ene in 4,5-enoses. | ||
Covalent modification of 4,5-unsaturated sugars with thiols remains relatively unexplored, with only one prior study reported to our knowledge. In this study, Liu and coworkers described a base-mediated thia-Michael addition in an electron-deficient 4,5-enose that required heating and prolonged reaction times, using thiophenol and benzyl mercaptan as model thiol reagents (Fig. 1B), and attempted the radical-mediated hydrothiolation reaction, concluding its inefficiency.28 We set out to explore if the radical-mediated thiol–ene reaction could be expanded to other classes of thiols and sugar derivatives, being a milder and more efficient reaction compared to the thia-Michael process (Fig. 1C).
Results and discussion
Reaction conditions and thiol scope
Given the unique steric challenges associated with 4,5-unsaturated systems, in particular protected uronic acid derivatives, we first sought to identify optimised reaction conditions. Prior to investigating 4,5-unsaturated sugars, we conducted our own extensive studies of commercially available 1,2-enoses to investigate if quantitative conversion could be achieved under photochemical thiol–ene conditions (see ESI†). A key finding emerging from these preliminary studies was that solvent-free reaction conditions could be employed to furnish thioether products with full conversion and simple work-up. These conditions were subsequently employed as the starting point for the 4,5-unsaturated systems.To commence our study, we first prepared 4,5-unsaturated saccharide 1,29 derived from D-glucuronic acid, using a modified protocol based on previously reported procedures (Scheme 1).29,30 The synthesis began with the methylation of the acetylated β-D-glucuronic acid 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 using methyl iodide and K2CO3 to furnish intermediate 3,32 followed by β-elimination of the acetyl group at C4 under basic conditions yielding the desired 4,5-enose 1.
31 using methyl iodide and K2CO3 to furnish intermediate 3,32 followed by β-elimination of the acetyl group at C4 under basic conditions yielding the desired 4,5-enose 1.
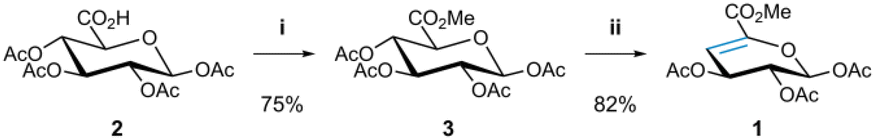 | ||
| Scheme 1 Synthesis of 4,5-unsaturated saccharide 1. Reaction conditions: (i) CH3I, K2CO3, DMF, rt, 5 h; (ii) DBU, CH2Cl2, 0 °C to rt, 19 h. | ||
Using 1 as a model substrate we carried out the photoinitiated reaction with 1,2-ethanedithiol 4a, employing the latter as the solvent, under irradiation at λmax 352 nm for 1 hour, at room temperature in the presence of 2,2-dimethoxy-2-phenylacetophenone (DPAP) and 4′-methoxyacetophenone (MAP), as a photoinitiator and a photosensitizer respectively. The ‘neat’ reaction conditions afforded the diastereomers 5a and 6a in a 26:74 ratio and a combined isolated yield of 97% (Table 1, entry 1).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Photo-initiator | Light source (λmax) | T (°C) | Time (h) | Solvent | Yielda (%) (5a/6a)b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Scale: 0.24 mmol in 0.06 mL of thiol, neat.a Combined yield.b Isolated compounds.c 0.5 mL solution.d 0.5 mL added for photoinitiator solubility.e Recovered 96% of unreacted 1.f MeOH/crushed ice cooling bath.g Thia-Michael addition using N,N-diethylamine (2 equiv.) as base led to a complex mixture of degradation products. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | DPAP/MAP | UV-A (352 nm) | 25 | 1 | Neat | 97 (26:74) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | DPAP/MAP | UV-A (352 nm) | 25 | 2 | EtOAcc | 26 (29:71) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Eosin Y | Blue LED (450 nm) | 25 | 2 | EtOAc:THF (1:1, v:v)d |
48 (22:78) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Thioxanthen-9-one | Blue LED (450 nm) | 25 | 2 | EtOAc:THF (1:1, v:v)d |
10 (n/a) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | DPAP/MAP | In dark | 25 | 2 | Neat | 0e (n/a) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | DPAP/MAP | UV-A (352 nm) | −18f | 1 | Neat | 97 (37:63) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | No catalystg | In dark | 50 | 1 | CHCl3 | — (n/a) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

NMR characterization and X-ray crystallography revealed the structures of the isolated diastereomers (Fig. 2A and ESI†). The observed coupling constants for 5a exhibited small values (3J ∼ 2–4 Hz), consistent with a trans-diequatorial arrangement of the coupled protons, in addition to long-range coupling constants (4J ∼ 0.8 Hz) between diequatorial protons in a W disposition (H1–H3 or H2–H4), hence supporting an α-L-idouronate in a 1C4 chair conformation.33 In contrast, 6a showed high values (3J ∼ 8–10 Hz) for the trans-diaxial arranged protons (H1, H2, and H3) and small values for the couplings with one of the pyranose ring protons (H4, J ∼ 2–4 Hz) in an equatorial disposition, characteristic of the 4C1 chair of a β-D-galacturonate. The assigned conformations of the uronates were confirmed by 2D-NOESY spectra. Thus, NOE contacts were observed between the equatorial-oriented H4 and the axial-oriented protons (H1, H3 and H5), justifying the 4C1 conformation of 6a. These axial NOE contacts were absent in 5a, as expected for the 1C4 chair, showing instead the spatial interactions of each proton with its immediate vicinal protons in an all-equatorial arrangement, except for the axial-oriented H5. The structure of α-L-idouronate 5a, was confirmed by single-crystal X-ray diffraction analysis, showing the absolute configuration of the substituents along the pyranose ring, albeit, the sugar crystalized in its 4C1 conformation. Suitable crystals of 6a could not be obtained, despite extensive crystallization attempts (see ESI†). However, suitable crystals of structurally related product 6b were successfully obtained, and the X-ray single-crystal diffraction analysis showed the absolute configuration of the substituents, confirming a β-D-galacturonate structure (see ESI†).
 | ||
| Fig. 2 (A) J values and NOE signals for 5a/6a structural determination and X-ray crystal structure of 5a. (B) Proposed mechanism and diastereoselectivity considerations for thiol–ene reaction on 1. | ||
The solved crystal structures and initial diastereoselectivity outcomes led us to propose a reaction mechanism, illustrated in Fig. 2B. The minor product 5a may arise from an α-side axial attack of the thiyl radical onto the 1H2 conformation of 1, followed by ring flip to the 1C4 conformation and axial hydrogen abstraction from the thiol, affecting an overall trans-diaxial addition across the enose. Comparatively, for the formation of the major product 6a, a β-side axial attack can occur on the ring flipped 2H1 conformation of 1, leading to a similar overall trans-diaxial addition but affording a 4C1 galacturonate-configured product.34–36 Furthermore, only C4 addition products were observed and this complete regioselectivity may be explained by enhanced captodative stability of the intermediate tertiary radical, formed upon addition to the less substituted olefinic carbon.
Since clearly not all thiol substrates are amenable to solvent-free reaction conditions, further optimisation studies were carried out in organic solvents, varying reaction parameters including photoinitiator, light source and temperature. Optimisation studies quickly revealed however that use of neat conditions furnished the highest yields (Table 1, entries 1 and 6), when paired with the optimised photoinitiation system of DPAP/MAP under UV light irradiation. A significant drop in yield to 26% was observed when using EtOAc as a solvent (Table 1, entry 2). However this improved to 48% upon using a mixture of EtOAc:THF (1:1) and using Eosin Y as an initiator under Blue LED (450 nm) conditions (Table 1, entry 3). A drop in yield to 10% was observed when a less efficient photoinitiator, thioxanthen-9-one, was used highlighting the importance of an efficient photoinitiator system for achieving high yields (Table 1, entry 4). Indeed, the dark reaction furnished no thioether products (as expected), demonstrating the radical nature of the process and the requirement for a photochemical initiation of the thiol–ene radical chain process (Table 1, entry 5). Reduced temperature furnished a 97% conversion, showing a slight variation in the diastereomeric ratio from 26:74 at room temperature to 37:63 at −18 °C (Table 1, entry 6), providing further evidence that the major product originates from thermodynamically favoured axial addition of the thiyl radical onto the alkene. The addition of a base to promote the competing thia-Michael addition was also investigated (Table 1, entry 7), however, this failed to furnish any thioether products. Finally, to demonstrate the ability to scale up, this solvent-free method was successfully completed on 1 mmol scale to produce 5a and 6a in 87% yield and 16:84 ratio (see ESI†). Interestingly, upon heating of compound 6a during crystallization studies, a novel bicyclic, dithioether derivative was formed and its X-ray structure was solved (6a′, see ESI†).
With optimised conditions in hand and some insight into the expected regio- and stereochemical outcomes of the thiol–ene reaction, we next sought to investigate the scope of the reaction using a variety of thiols, including alkyl, aromatic and thioacid derivatives (Table 2). Since all of the thiols screened were either oils or liquids, solvent-free conditions were used in each case. Thus, the reaction between 1 and 1,3-propanedithiol (4b) furnished a 92% yield of the corresponding diastereomers (5b/6b) in a 2:8 ratio (Table 2, entry 2). Similarly, the reaction with 3-chloropropanethiol (4c), 3-mercapto-1,2-propanediol (4d), 3-mercaptopropionate (4e) and 2-mercaptoacetate (4f) each afforded the corresponding diastereomers 5c–f and 6c–f in good to excellent yields (50–99%, Table 2, entries 3–6), with slightly lower yields observed for the more bulky thiol substrates (Table 2, entries 4 and 6). The ratios of the isolated isomers were ca. 2:8, in favour of the axial addition products, with a slightly reduced ratio of 3:7 observed for 4f (Table 2, entry 6), potentially arising from the proximity of the bulky carbonyl group to the thiol residue which may interfere with axial attack of the corresponding thiyl radical. Interestingly, reaction of 1 with thioacetic acid (4g), a well-known and highly utilised substrate for thiol–ene reactions with enoses, furnished a poor yield of only 21% and a 5g/6g ratio of ca. 1:1 even after 6 hours (Table 2, entry 7), suggesting that steric bulk around the thiyl radical is detrimental to efficient thiol–ene reaction with 1. Recent studies by Kelemen and co-workers suggest that degradation of the thioacid may also be a factor in the low yield for this substrate.37 The reaction with benzyl mercaptan (4h) only furnished diastereomer 6h, albeit in a low yield of 26% (Table 2, entry 8), again due to steric interference arising from the bulky benzyl moiety. Overall, these findings demonstrate that while the reaction does appear to be tolerant of a diverse range of thiols, having a bulky residue in the proximity of the reactive heteroatom radical hampers product formation, increases reaction time, lowers the reaction yields and/or varies the diastereomeric product ratios. Furthermore, in the series evaluated here the product ratio appears to follow a pattern whereby the 2H1 enose conformer reacts preferentially, perhaps avoiding unwanted steric (axial substituent) penalties via the competing 1H2 pathway (see Fig. 2B).

Synthesis and thiol–ene scope on 4,5-enoses
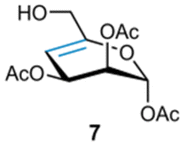
To further investigate the scope of the thiol–ene reaction, we next prepared a range of 4,5-unsaturated enoses (7–10, Scheme 2) including both protected and fully unprotected sugars. We also included non-uronate derivatives to investigate the impact of having an alkyl group at C5 since this should lessen the steric constraints for the thiol–ene addition. | ||
| Scheme 2 Synthesis of 4,5-unsaturated monosaccharides 7–10. Reaction conditions: (i) (a) DMP, CH2Cl2, rt, 6 h; (b) Et3N, CH2Cl2, rt, 1–2 h; (ii) NaBH4, THF, 0 °C to rt, 1–2 h; (iii) Ac2O, DMAP, pyridine, CH2Cl2, rt, 3 h; (iv) (a) CH3I, K2CO3, DMF, rt, 14 h; (b) BzCl, pyridine, 0 °C to rt, 12 h; (v) DBU, CH2Cl2, 0 °C to rt, 2 h; (vi) NaOH, EtOH/H2O (1:1), rt, 6 h. | ||
The synthesis of compounds 7 and 8 began with the preparation of intermediate 11 from D-mannose (see ESI†).38,39 Next, oxidation of the primary alcohol via Dess–Martin periodinane and subsequent β-elimination of the acetyl group at C4 under basic conditions furnished the 4,5-enose 12 as a separable mixture of α/β anomers. The partially protected mannosyl 4,5-unsaturated derivative 7 was obtained upon reduction of the aldehyde 12α in the presence of sodium borohydride, whilst the fully protected mannosyl 4,5-enose 8 was isolated following reduction of 12β to the respective primary alcohol 13, followed by acetylation. Synthesis of benzoyl-protected 4,5-unsaturated methyl uronate 9 involved conversion of methyl α-D-mannosiduronic acid 1440 into the fully protected derivative 1541via a two-step process involving methyl iodide mediated esterification under basic conditions, followed by perbenzoylation upon treatment with BzCl in pyridine. DBU-mediated β-elimination of the benzoyl group at C4 furnished the desired 4,5-enose 9. The fully unprotected 4,5-enose 10 was synthesised from the acetyl-protected methyl uronate 16,42 a derivative of α-D-mannosiduronic acid (see ESI†), whose acetyl group at C4 was subjected to base-promoted β-elimination to furnish 4,5-enose 17, which was subsequently deprotected upon treatment with NaOH in EtOH/H2O (1:1).
Next, we proceeded to investigate the thiol–ene reaction of these alternate 4,5-enoses using the previously optimised conditions with 1,2-ethanedithiol 4a employed as the thiol in each case (Table 3). For compound 7, a C1,2,3-protected mannosyl 4,5-enose, and compound 8, a fully acetylated form of 7, only a single diastereomer was isolated in each case, identified as 4C1 mannosyl derivatives 18 or 19, respectively. Indicatively, a combination of changing the C6 uronate oxidation level to hexose, together with a C2-axially-oriented substituent, promoted an alternate overall cis addition across the enose (Table 3, entries 1 and 2), although the yields were low in both cases. By contrast, return to a benzoyl protected 4,5-unsaturated mannose uronate 9 formed diastereomers 20a and 20b in 87% yield and in a 32:68 ratio (Table 3, entry 3). This pair were identified as derivatives of α-D-taluronate (C2 and C4 epimer of glucose) and β-L-guluronate respectively and supports the finding that the presence of an ester at C5 is optimum for a trans-diaxial thiol–ene addition onto 4,5-enoses (here using thiol 4a and considering D-manno and D-gluco configurations only). Finally, for a fully unprotected uronic acid enose 10, only guluronate-configured addition product 21 was isolated in 45% yield (Table 3, entry 4) demonstrating potential for compatibility of the methodology in functionalising free sugars.







Thiol–ene reaction on 4,5-unsaturated disaccharide
As outlined in the introduction, a key motivation for this research was the development of a methodology that would enable facile tagging of the 4,5-unsaturated terminal residues of di- and oligosaccharide products from lyase activity in biological samples. As further proof of concept towards this objective, we selected the disaccharide 22, a synthetic heparan sulfate building block, to test with the thiol–ene conditions (Scheme 3). | ||
| Scheme 3 (A) Synthesis of 4,5-unsaturated disaccharide 22. (B) Application of the photoinitiated thiol–ene methodology. Reaction conditions: (i) (a) Ac2O, pyridine, rt, 2 h; (ii) DBU, CH2Cl2, 0 °C to rt, 2 h; (iii) thioacetic acid, 2,6-lutidine, CHCl3, 60 °C, 36 h. | ||
The 4,5-unsaturated disaccharide 22 was synthesised from the commercially available building block 23 (Scheme 3A). Acetylation of the free hydroxyl group at the non-reducing terminus under standard conditions (Ac2O, pyridine) afforded intermediate 24, which was subsequently converted into the 4,5-enose derivative 25via β-elimination of the acetyl group at C4. To prevent unwanted side reactions, such as thiol-mediated azide reduction during the photoinitiated hydrothiolation, the azide group at C2 of the reducing end of 25 was modified into an acetamide group using thioacetic acid in the presence of 2,6-lutidine, following a previously reported method.43
Gratifyingly, hydrothiolation of 22 using the optimised thiol–ene conditions furnished the thioether product 26 in 40% yield (Scheme 3B). A trace quantity of a second product was detected by TLC but the quantity proved insufficient for isolation and analysis. The colourless, plate-shaped crystals, obtained during the isolation step (column chromatography, hexane/acetone 60:40 solvent system), allowed the determination of the absolute configuration of the monosaccharide units in disaccharide 26 by X-ray analysis. Thus, the modification of the 4,5-unsaturated terminal residue furnished the β-D-galacturonate (4C1 conformation), as observed previously with monosaccharides, and the methyl α-D-glucopyranoside unit remained unaltered, highlighting the potential for derivatisation of complex oligosaccharides.
Conclusions
The thiol–ene reaction has been demonstrated on sterically challenging 4,5-unsaturated derivatives of uronic acids together with complete regioselectivity and good diastereoselectivity. The thermodynamically favoured axial attack of the photoinitiated thiyl radical appears to be the dominant process and is tolerant of a wide range of conditions and substrates. Of particular relevance is the finding that solvent-free conditions are optimal for thiol–ene reactions on 4,5-unsaturated sugars, since these conditions are scalable and have proven to be operationally simple in terms of work-up. The removal of solvents from organic reactions is important in the context of sustainable synthesis. The approach is compatible with fully protected, partially protected and fully unprotected monosaccharides. Finally, we have demonstrated how the methodology can be applied to a relevant disaccharide, which may allow fluorescent labelling, utilising a tag and modify approach, to ensue.44 Further work on modification of oligosaccharide structures via thiol–ene ligation is ongoing in our laboratories.Author contributions
Conceptualisation: G. J. M. and E. M. S.; methodology and investigation: A. P.-C., H. M., T. M., L. B., J. T. M., I. P. and I. R. G.; data curation: A. P.-C., H. M., T. M., I. P., I. R. G. and B. T.; visualization: A. P.-C. and E. M. S.; writing – original draft: A. P.-C., H. M. and E. M. S.; writing – review and editing: A. P.-C., H. M., T. M., I. P., G. J. M. and E. M. S.; supervision: G. J. M. and E. M. S.; funding acquisition: G. J. M. and E. M. S.All authors have given approval to the final version of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI:†General methods, synthetic procedures and characterisation data for all compounds.
NMR spectra for all new compounds and X-ray crystallographic data for 5a, 6a′, 6b, and 26.
Crystallographic data for 5a, 6a′, 6b, and 26 has been deposited at The Cambridge Crystallographic Data Centre under CCDC accession codes 2389449 (5a), 2389448 (6a′), 2389447 (6b) and 2389446 (26).†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Irish Research Council (Grant GOIPD/2021/186) and Science Foundation Ireland (19/FFP/6667) and the EPSRC (19/EPSRC/3634). We are grateful to Dr John O'Brien and Dr Manuel Ruether for assistance with NMR spectroscopy, Dr Gary Hessman for assistance with mass spectrometry, and Dr Brendan Twamley for assistance with X-ray diffractometry and crystallographic analysis.References
- Z. J. Witczak and J. M. Culhane, Appl. Microbiol. Biotechnol., 2005, 69, 237–244 CrossRef CAS PubMed.
- A. Czubatka-Bienkowska, J. Sarnik and T. Poplawski, Carbohydr. Res., 2023, 533, 108934 CrossRef CAS PubMed.
- T. Morikawa, K. Nimomiya, G. Tanabe, G. Matsuda, M. Yoshikawa and O. A. Muraoka, J. Nat. Med., 2021, 75, 449–466 CrossRef CAS PubMed.
- J. Porter, A. R. Noble, N. Signoret, M. A. Fascione and G. J. Miller, ACS Omega, 2024, 9, 31703–31713 CrossRef CAS PubMed.
- M. Fiore, A. Marra and A. Dondoni, J. Org. Chem., 2009, 74, 4422–4425 CrossRef CAS PubMed.
- S. Staderini, A. Chambery, A. Marra and A. Dondoni, Tetrahedron Lett., 2012, 53, 702–704 CrossRef CAS.
- A. Dondoni and A. Marra, Chem. Soc. Rev., 2012, 41, 573–586 RSC and references cited therein.
- T. Xiong, R. Xie, C. Huang, X. Lan, N. Huang and H. Yao, J. Carbohydr. Chem., 2021, 40, 401–439 CrossRef CAS.
- A. Borbás, Chem. – Eur. J., 2020, 26, 6090–6101 CrossRef PubMed and references cited therein.
- M. Csávás, D. Eszenyi, E. Mező, L. Lázár, N. Debreczeni, M. Tóth, L. Somsák and A. Borbás, Int. J. Mol. Sci., 2020, 21, 573 CrossRef PubMed.
- A. Malone and E. M. Scanlan, Org. Lett., 2013, 15, 504–507 CrossRef CAS PubMed.
- L. McSweeney, F. Dénès and E. M. Scanlan, Eur. J. Org. Chem., 2016, 2080–2095 CrossRef CAS.
- J. Porter, D. Parisi, T. Miller, A. N. Cheallaigh and G. J. Miller, Carbohydr. Res., 2023, 530, 108854 CrossRef CAS PubMed.
- J. Porter, M. A. Lima, I. Pongener and G. J. Miller, Carbohydr. Res., 2023, 524, 108759 CrossRef CAS PubMed.
- G. Goti, ChemCatChem, 2022, 14, e202200290 CrossRef CAS.
- D. W. Abbott, H. J. Gilbert and A. B. Boraston, J. Biol. Chem., 2010, 285, 39029–39038 CrossRef CAS PubMed.
- D. Ndeh, A. Rogowski, A. Cartmell, A. S. Luis, A. Baslé, J. Gray, I. Venditto, J. Briggs, X. Zhang, A. Labourel, N. Terrapon, F. Buffetto, S. Nepogodiev, Y. Xiao, R. A. Field, Y. Zhu, M. A. O'Neill, B. R. Urbanowicz, W. S. York, G. J. Davies, D. W. Abbott, M.-C. Ralet, E. C. Martens, B. Henrissat and H. J. Gilbert, Nature, 2017, 544, 65–70 CrossRef CAS PubMed.
- N. Afratis, C. Gialeli, D. Nikitovic, T. Tsegenidis, E. Karousou, A. D. Theocharis, M. S. Pavão, G. N. Tzanakakis and N. K. Karamanos, FEBS J., 2012, 279, 1177–1197 CrossRef CAS PubMed.
- A. Joshi, P. Chopra, A. Venot and G.-J. Boons, Org. Lett., 2024, 26, 2462–2466 CrossRef CAS PubMed.
- S. Zhu, J. Li, R. S. Loka, Z. Song, I. Vlodavsky, K. Zhang and H. M. Nguyen, J. Med. Chem., 2020, 63, 4227–4255 CrossRef CAS PubMed.
- P. Chopra, T. Yadavalli, F. Palmieri, S. A. K. Jongkees, L. Unione, D. Shukla and G.-J. Boons, Angew. Chem., Int. Ed., 2023, 62, e202309838 CrossRef CAS PubMed.
- T. K. Kandola, C. J. Mycroft-West, M. L. Andrade De Lima, A. Turner, C. Gardini, E. Urso, G. J. Miller, A. Bisio, E. A. Yates, M. Guerrini and L. Wu, bioRxiv, 2024, preprint, DOI:10.1101/2024.09.26.615151.
- Z. Wang, C. Shi, X. Wu and Y. Chen, Chem. Commun., 2014, 50, 7004–7006 RSC.
- C. Przybylski, V. Bonnet and R. R. A. Vivès, Chem. Commun., 2019, 55, 4182–4185 RSC.
- R. T. Taylor and D. P. Poudel, Reactions, 2022, 3, 442–450 CrossRef CAS.
- L. Lázár, M. Csávás, Á. Hadházi, M. Herczeg, M. Tóth, L. Somsák, T. Barna, P. Herczegh and A. Borbás, Org. Biomol. Chem., 2013, 11, 5339–5350 RSC.
- L. Lázár, L. Juhász, G. Batta, A. Borbás and L. Somsák, New J. Chem., 2017, 41, 1284–1292 RSC.
- S.-T. Ma, C.-W. Lee and W.-M. Liu, RSC Adv., 2021, 11, 18409 RSC.
- D. Combaud, M. Thomas, S. Papot and J.-P. Gesson, Lett. Drug Des. Discovery, 2005, 2, 631–637 CrossRef CAS.
- L. Beswick, S. Ahmadipour, J. P. Dolan, M. Rejzek, R. A. Field and G. J. Miller, Carbohydr. Res., 2019, 485, 107819 CrossRef CAS PubMed.
- J. P. Malkinson, R. A. Falconer and I. Toth, J. Org. Chem., 2000, 65, 5249–5252 CrossRef CAS PubMed.
- W. Pilgrim and P. V. Murphy, J. Org. Chem., 2010, 75, 6747–6755 CrossRef CAS PubMed.
- B. M. Sattelle, B. Bose-Basu, M. Tessier, R. J. Woods, A. S. Serianni and A. Almond, J. Phys. Chem. B, 2012, 116, 6380–6386 CrossRef CAS PubMed.
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- Y. Araki, K. Matsuura, Y. Ishido and K. Kushida, Chem. Lett., 1973, 2, 383–386 CrossRef.
- B. H. Northrop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef CAS PubMed.
- V. Kelemen, M. Bege, N. Debreczeni, I. Bereczki, A. C. Bényei, P. Herczegh and A. Borbás, Chem. – Eur. J., 2025, 31, e202500104 CrossRef CAS PubMed.
- H. Yu and X. Chen, Org. Lett., 2006, 8, 2393–2396 CrossRef CAS PubMed.
- M. Lopez, J. Trajkovic, L. F. Bornaghi, A. Innocenti, D. Vullo, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2011, 54, 1481–1489 CrossRef CAS PubMed.
- M. M. Muthana, J. Qu, M. Xue, T. Klyuchnik, A. Siu, Y. Li, L. Zhang, H. Yu, L. Li, P. G. Wang and X. Chen, Chem. Commun., 2015, 51, 4595–4598 RSC.
- R. A. Edington, E. L. Hirst and E. E. Percival, J. Chem. Soc., 1955, 2281–2288 RSC.
- R. N. Rej, J. N. Glushka, W. Chewand and A. S. Perlin, Carbohydr. Res., 1989, 189, 135–148 CrossRef CAS.
- N. Shangguan, S. Katukojvala, R. Greenberg and L. J. Williams, J. Am. Chem. Soc., 2003, 125, 7754–7755 CrossRef CAS PubMed.
- M. Singh, M. Watkinson, E. M. Scanlan and G. J. Miller, RSC Chem. Biol., 2020, 1, 352–368 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2389449, 2389448, 2389447 and 2389446. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ob00507h |
| This journal is © The Royal Society of Chemistry 2025 |