
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom burst to controlled release: using hydrogel crosslinking chemistry to tune release of micro-crystalline active pharmaceutical ingredients†
Purnima N.
Manghnani
 a,
Arif Z.
Nelson
a,
Kelvin
Wong
a,
Yi Wei
Lee
a,
Saif A.
Khan
*ad and
Patrick S.
Doyle
*abc
a,
Arif Z.
Nelson
a,
Kelvin
Wong
a,
Yi Wei
Lee
a,
Saif A.
Khan
*ad and
Patrick S.
Doyle
*abc
aSingapore-MIT Alliance for Research and Technology, 1 CREATE Way, #04-13/14 Enterprise Wing, Singapore 138602
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue Room E17-504F, Cambridge, MA, 02139 USA
cHarvard Medical School Initiative for RNA Medicine, Boston, MA, 02115 USA. E-mail: pdoyle@mit.edu
dDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: saifkhan@nus.edu.sg
First published on 5th November 2024
Abstract
Hydrogels have been widely studied as substrates for drug delivery and tissue engineering owing to their biocompatibility and ability to swell in aqueous media. Encapsulation of lipophilic active pharmaceutical ingredients (API) as crystalline micro-/nanoparticles within hydrogel formulations has shown promise for improving their bioavailability and achieving high drug load. Despite the size reduction of the API within the hydrogel mesh, the bioavailability of these formulations is largely governed by the inherent ability of the hydrogel polymer backbone to release the API. In this work, Michael addition-based Polyethylene glycol (PEG) hydrogels are developed for micro-crystalline fenofibrate (Fen) encapsulation. Using a parallelized step emulsification device, API nanoemulsion (NE) loaded micro-hydrogels are fabricated and subsequently subjected to anti-solvent extraction for API crystallization. The bi-molecular nature of the Michael addition reaction provides modular incorporation of crosslinking functional groups leading to precise temporal control over hydrogel degradation, thereby offering a sensitive handle on the release of micro-crystalline fenofibrate. By merely changing the chemical identity of the hydrogel cross-link, complete Fen release could be tuned from 4 hours to 10 days. Furthermore, the interaction of crystallizing Fen and PEG within the micro-hydrogel environment led to eutectic formation. This unique feature offered a second handle on the Fen release from the composite micro-hydrogels.
Introduction
Pharmaceutical formulations face significant challenges such as poor bioavailability of BCS class II and IV drugs, costly manufacture, and control of drug release.1 To address poor bioavailability, formulation of nano or microcrystalline API can be a potential strategy to improve aqueous solubility. However, most top-down processes used in industry for size reduction are energy-intensive. Recently, hydrogels have been explored as potential carriers for the co-processing of nano or microcrystalline API. Hydrogels typically comprise a biocompatible cross-linked polymer backbone of natural or synthetic origin capable of swelling in an aqueous environment. Hydrogels of varying compositions and cross-linking mechanisms allow for the tunability of bulk dimensions, internal mesh size, degradation, and mode of delivery. Crystalline lipophilic APIs have been incorporated into hydrogels by either introducing hydrophobic domains in the aqueous media such as oil-in-water NE2,3 or nanoparticles4,5 before cross-linking or by functionalizing the polymer backbone with hydrophobic groups6,7 or adding API containing solvent followed by solvent extraction8 in cross-linked hydrogels. While the nano or microcrystalline nature of the API embedded within hydrogel matrices may offer better solubility compared to bulk crystals, the drug release from hydrogel formulation is largely governed by the inherent nature of the hydrogel polymer backbone. While the hydrophilicity of the hydrogel network allows for aqueous media to be present in the vicinity of the API, release can be diffusion-controlled or chemically controlled based on the network response to stimuli present in the aqueous media such as pH or temperature. A fast-eroding formulation is preferred for bolus delivery and a sustained release is preferred for long-acting formulations.9Conventional hydrogel-based drug delivery systems like hydroxypropyl methylcellulose, Pluronics®, or alginate powders physically blended with API exhibit drug release governed by swelling of the polymer to create a diffusion barrier for migration of small molecules. Drug release from a specific polymer-API blend can be tailored to some extent by changing the concentration or the polymer grade, but the matrix erosion occurs fairly quickly once the polymer is completely hydrated.10,11 Cross-linked hydrogels encapsulating cargo larger than the gel mesh size, in this regard, offer release governed by the hydration, diffusion barrier, dissolution of the API, and erosion of the polymer network. However, hydrogels comprising a single cross-linked monomer are limited by their incipient erosion profiles.12 Bimolecular polymer hydrogels with covalent crosslinks, on the other hand, can provide a handle on the erosion of the polymer network as well.13
Polyethylene glycol (PEG) is one of the most widely explored synthetic polymers for hydrogels due to its versatility and biocompatibility.14 Highly structured PEG hydrogel networks with tunable degradation profiles have been fabricated through a Michael-type addition reaction which involves a step-growth reaction between thiol and an electron-deficient carbon–carbon double bond like an acrylate, alkyne, or vinyl sulfone. The bimolecular nature of the polymer network allows for rendering the hydrogel network degradable by insertion of degradable groups into the multi-arm PEG polymer backbone or the cross-linker molecule.15 This method allows for mild reaction conditions along with fast reaction time scales and no side-product formation. Michael addition-based PEG hydrogels have been primarily explored for biomolecule delivery via in situ cross-linking post-injection due to their rapid reaction at physiological conditions. So far the flexibility in terms of degradation afforded by these bimolecular networks has not been leveraged for controlling the release of lipophilic APIs. In the present work, we develop a system to maintain overall hydrogel-API composite properties and obtain different API release profiles by modifying the rate of polymer scaffold erosion. The vinyl sulfone–thiol reaction is a cross-linking strategy that leads to a hydrolytically stable thioether sulfone bond, whereas the acrylate–thiol reaction for cross-linking results in an acrylate thioether ester which is hydrolytically labile.16 By combining these two cross-linking reactions in different stoichiometry, we tune hydrogel erosion rates to coordinate API release.
Here, an oil-in-water NE of model BCS class II drug, fenofibrate (Fen), was cross-linked via a Michael-type addition reaction to yield micro-crystalline Fen-embedded micro-hydrogels with tunable degradation. More specifically, an aqueous phase containing polymer precursors of four-armed PEG-acrylate (PEG-Ac) or PEG-vinyl sulfone (PEG-Vs) or both, PEG-dithiol (PEG-DT) as cross-linker and Fen-containing NE was subjected to step emulsification in a microfluidic device to generate highly uniform droplets. The droplets were subsequently cross-linked by modulating the pH of the aqueous phase to generate micro-hydrogels.17 The Fen was subjected to crystallization by placing the NE-containing micro-hydrogel into an anti-solvent sink. The Fen-loaded micro-hydrogels were then characterized using powder X-ray diffraction (PXRD), differential scanning calorimetry (DSC), Fourier-transform infrared spectroscopy (FTIR), optical microscopy and electron microscopy to explain the drug release of different particle compositions. Changing the chemical structure of the micro-hydrogel crosslinks from acrylate–thiol to vinyl sulfone–thiol affected the hydrolytic degradation rate of the micro-hydrogels. As a result, a modular approach of varying the ratios of functional groups incorporated within the micro-hydrogels led to precise temporal control of drug release. In this manner, a complete Fen release could be tuned from 4 hours to 10 days. While several prior studies have explored the encapsulation of NEs within hydrogels, most have not focused on the crystallization of the payload.18–20 Of the studies where the API was crystallized, the drug release was tuned by changing the drug crystal size embedded within the matrix or changing the physical attributes of the micro-hydrogels.8,21–23 So far such a wide tunability of crystalline API release from a micro-hydrogel formulation has not been reported. Another interesting feature of this formulation was the eutectic formation between Fen and PEG. The concentration of Fen in the dispersed phase of the NE governed the eutectic formation of the PEG-Fen composites. This in turn offered another modality to tune the release of Fen from the micro-hydrogel matrix. Eutectic formation has been widely reported for composites prepared through the interaction of Fen-PEG melts.11,24 This, however, is the first report of Fen-PEG eutectic formation in a micro-hydrogel formulation.
Materials and methods
Materials
4-Arm poly (ethylene glycol) vinylsulfone (PEG-Vs, Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), 4-arm poly (ethylene glycol) acrylate (PEG-Ac, Mn = 10000 g mol−1), poly (ethylene glycol) dithiol (PEG-DT, Mn = 3400 g mol−1), pluronic F-127 (PF127), 1H,1H,2H,2H-perfluorooctyltriethoxysilane, fenofibrate (Fen), anisole, triethylamine (TEA), sodium phosphate monobasic, and sodium dodecyl sulfate (SDS) were purchased from Merck, Singapore. Fluorinated surfactant Pico-surf 1 was purchased from Sphere Fluidics Limited and fluorinated oil Novec HFE-7500 was purchased from 3M, Singapore. Ethanol, acetone, and hydrochloric acid (HCl) were purchased from VWR Singapore. All chemicals were used as received.
000 g mol−1), 4-arm poly (ethylene glycol) acrylate (PEG-Ac, Mn = 10000 g mol−1), poly (ethylene glycol) dithiol (PEG-DT, Mn = 3400 g mol−1), pluronic F-127 (PF127), 1H,1H,2H,2H-perfluorooctyltriethoxysilane, fenofibrate (Fen), anisole, triethylamine (TEA), sodium phosphate monobasic, and sodium dodecyl sulfate (SDS) were purchased from Merck, Singapore. Fluorinated surfactant Pico-surf 1 was purchased from Sphere Fluidics Limited and fluorinated oil Novec HFE-7500 was purchased from 3M, Singapore. Ethanol, acetone, and hydrochloric acid (HCl) were purchased from VWR Singapore. All chemicals were used as received.
Millipede device fabrication
Step emulsification devices were fabricated using soft lithography. Master molds were 3D printed with a co-polyester using an Ultimaker S3. A nozzle channel height of 500 μm was used. Devices were molded from the masters using the PDMS Sylgard 184 kit (Dow Corning). The base and cross-linker were mixed at a 10:1 mass ratio, poured over the mold, degassed, and cured at 70 °C for 2 hours. The PDMS devices and PDMS base were then activated via air plasma and bonded together. The bonded devices were silanized overnight with 1H,1H,2H, and 2H-Perfluorooctyltriethoxysilane to render the channels fluorophilic.
Fabrication of drug loaded PEG micro-hydrogels
The continuous phase of the NE solution was prepared by dissolving 10% (w/v) PEGVs (or PEGAc) and PEGDT (molar ratio 1:1) and 10% (w/v) F127 in ultrapure water. To the continuous phase (80 wt%), the surfactant blend of Tween 80 and Span 80 (HLB = 13.5) was added under continuous stirring (5 wt%). Anisole containing Fen (800–200 mg mL−1) was emulsified into nanodroplets in the continuous phase under vortex for 1 minute (15 wt%). The NE served as the dispersed phase for microdroplet generation in a millipede device. 0.03% (v/v) pico-surf 1 in HFE7500 was the continuous phase within which the millipede device was immersed. Droplets were then generated by flowing the NE through the millipede immersed in the HFE7500 at 1 mL min−1. The generated droplets were cross-linked into micro-hydrogels upon the addition of TEA to the HFE7500 bath (0.25% v/v) for 5 minutes and were immediately isolated in the anti-solvent sink. It was noted that the acrylate–dithiol reaction was particularly sensitive to the reaction time since a prolonged pH > 7 led to ester degradation in the polymer backbone. The anisole was extracted in anti-solvent sinks with varying ethanol composition – 5–80% in ultrapure water for 30 minutes. Afterward, the particles were soaked in DI water for another 30 minutes. The resulting micro-hydrogels were then dried under vacuum at room temperature for 24 h.
Formulation characterization
The polymorphism of crystalline Fen within the micro-hydrogels was characterized by powder X-ray diffraction (PXRD) raging from a 2θ angle of 4°–40° at a scan rate of 1° min−1. Particle size distribution was determined by ImageJ using optical upright microscopy images. Micro-hydrogel and Fen morphology were characterized using field emission scanning electron microscopy (FESEM, 7610, JEOL). Before FESEM observation at an operating voltage of 5 kV, dried samples were cut to expose their internal morphology and sputter coated with a platinum layer. The Fen-PEG interactions were characterized through a DSC thermogram obtained using DSC25 with Tzero pans from TA instruments. Each sample was heated from 25 to 125 °C at a rate of 10 °C min−1. The micro-hydrogels were further characterized using Fourier transform infrared spectroscopy (Bruker Tensor 27). Lastly, the Fen mass loading (w/w) in the micro-hydrogels was determined using a Cary 60 UV-visible spectrophotometer. Approximately 4 mg of the composite micro-hydrogels were placed in ethanol and allowed to dissolve for 2 hours. The Fen load was determined by comparing the measured UV-visible spectrum maxima at 290 nm to a calibration curve obtained in the concentration range of 1 to 25 μg mL−1 Fen dissolved in ethanol.Drug dissolution experiment
Fen mass load (w/w) of micro-hydrogels was determined before each dissolution experiment. Afterward, the particles were suspended in 40 mL of simulated gastric fluid (SGF) (50 × 10−3 M sodium phosphate monobasic, 25 × 10−3 M SDS, pH 1.2). SDS was added to ensure sink conditions by significantly increasing the saturation concentration of Fen in aqueous media, following previously established protocol. The mass of micro-hydrogels suspended in SGF was calculated to ensure that the final Fen concentration after drug release would remain at 30 μg mL−1, well below the saturation concentration (193 μg mL−1). The particles were stirred using an orbital shaker at 200 rpm at 37 °C throughout the dissolution study. At specified time points, 500 μL samples were taken out to measure the amount of Fen released using a Cary 60 UV–vis spectrophotometer. The concentration of Fen released was quantified using the UV absorbance at 290 nm.Results and discussion
Nanoemulsion synthesis and micro-hydrogel fabrication
An oil phase, anisole containing Fen, was emulsified in an aqueous continuous phase containing pre-polymers, surfactants and Pluronic® F127 (PF127) to obtain a nanoemulsion. The oil phase was stabilized using a non-ionic surfactant blend of Span 80 and Tween 80 with HLB = 13.5 (Fig. S1†). Similar to the low-energy method reported in Domenech et al.,3 the surfactants were dispersed uniformly in the continuous phase, after which the oil phase was added to the continuous phase under vigorous stirring (Fig. 1a). The dynamic light scattering (DLS) measurements (Fig. 1b) of the NE without PF127 showed a characteristic monomodal size distribution with an average hydrodynamic diameter (Dh) of 129 ± 11 nm. In the presence of PF127, the NE Dh shifted to 157 ± 12 nm, which was attributed to the adsorption of the polyoxypropylene (PPO) segments of PF127 on the oil interface. Furthermore, another peak appeared at 23 ± 2 nm due to the presence of pure PF127 micelles. | ||
| Fig. 1 Preparation and characterization of anisole NE and micro-hydrogels. (a) Scheme showing low-energy emulsion generation of anisole in aqueous phase containing pre-polymer and surfactants, (b) anisole droplet size measured through dynamic light scattering for emulsions prepared with 0 and 8 wt% F127, (c) scheme showing microfluidic generation of droplets containing PEG-DT, 4 arm PEG-Vs or PEG-Ac and Fen-anisole emulsion in the aqueous phase, and HFE-7500 as the oil phase. The droplets are subsequently cross-linked by subjecting the droplets to TEA, (d) micro-hydrogel size distribution measured through bright field optical imaging after solvent exchange, and (e) optical micrograph of Fen-anisole NE loaded PEGVs-DT micro-hydrogels after cross-linking and solvent exchange. | ||
Highly uniform NE-laden micro-droplets were generated in fluorinated oil, HFE-7500 using step emulsification in a millipede device. As shown in Fig. 1c, the NE solution flowed through multiple nozzles in the millipede, which opened into a tall reservoir containing fluorinated oil. The sudden increase in channel height led to a quasi-static instability causing the droplets to pinch off. As the droplets were generated, they rose to the surface of the oil due to the density gradient. We hypothesized that 10% PF127 in the NE continuous phase played a crucial role in micro-hydrogel fabrication in fluorinated oil by imparting stability to the NE micro-droplets at the fluorinated oil–air interface (Fig. S2†). PF127 is a non-ionic amphiphilic triblock copolymer that forms a highly ordered packing of micelles at high concentrations (10–25 wt%) and temperatures (25–60 °C). Apart from PF127 acting as a conventional surfactant at the fluorinated oil–NE interface, we suspect that the interaction between PF127 micelles led to improved micro-droplet stability due to enhanced shielding of the dispersed anisole phase (Video S1 and S2†). The aqueous continuous phase of the NE droplets contained cross-linkable polymer precursors of four-arm PEG-acrylate (PEG-Ac) or PEG-vinyl sulfone (PEG-Vs) or a mix of both and a cross-linker, PEG-dithiol (PEG-DT). Cross-linking of the polymers was achieved by modulating the pH of the dispersed droplets through the addition of an organic base, TEA to the fluorinated oil bath (0.25% v/v). Michael addition reaction is catalysed by the use of TEA. In the presence of the base, the thiol undergoes deprotonation to form a thiolate anion and a conjugate acid. The thiolate anion is a strong nucleophile that attacks the electron-deficient beta-carbon of the terminal alkene bond in the acrylate or vinyl sulfone, forming the thioether addition product. The micro-hydrogels were cross-linked for 5 minutes and collected by filtration. To induce crystallization of Fen by extraction of anisole, the NE-embedded micro-hydrogels were placed in an anti-solvent sink. The anti-solvent sink containing varying amounts of ethanol in DI water (5–80% v/v) was explored. The feasibility of extracting anisole from the hydrogel was dictated by the ternary phase diagram between water–ethanol–anisole in Fig. S3.† A single-phase formation between the three solvents outside of the two-phase composition is necessary for complete extraction. To ensure that all the anisole was completely extracted, no more than 200 micro-hydrogel particles were placed in 10 mL of anti-solvent. The calculation entailing the molar fraction of anisole, water, and ethanol in the anti-solvent sink post-extraction and its comparison with the theoretical miscibility limit can be found in the ESI S3.† The volume of anisole in 200 micro-hydrogels formed a single phase with 10 mL of 5% v/v ethanol. As the micro-hydrogel equilibrated in the anti-solvent, Fen crystallization occurred. The micro-hydrogels were filtered 30 minutes post-solvent extraction and placed in DI water for another 30 minutes. As seen in the particle size distribution (Fig. 1d) and optical micrograph (Fig. 1e), highly uniform micro-hydrogels of about 1.67 ± 0.06 mm were obtained.
Characterization of the canonical formulation – 800 mg mL−1 Fen in anisole NE in PEG-Vs cross-linked with PEG-DT (PEGVs-DT) micro-hydrogel is presented here in detail and is representative of all the polymer compositions. The analogous characterization for the PEG-Ac cross-linked with PEG-DT (PEGAc-DT) formulations can be found in the ESI.† Post solvent extraction, the micro-hydrogels were isolated and subjected to vacuum drying at 100 mbar. In Fig. 2, optical microscopy images of the micro-hydrogels in the anti-solvent and FESEM characterization of the dried micro-hydrogels are presented. The opacity of the micro-hydrogels visualized using optical microscopy in comparison to the blank formulation (Fig. 2a) was indicative of the presence of the Fen payload. The presence of crystalline Fen within the micro-hydrogels environment was confirmed through FESEM imaging. In addition to the microcrystals present in the cross-section, larger Fen crystals were observed on the particle surface. This was attributed to the diffusion of Fen out of the micro-hydrogel along with anisole during solvent exchange. Due to Fen's poor solubility in water, a region of supersaturation led to crystal formation around the particle surface.8 A distinct variation in Fen crystal morphology within the micro-hydrogels was observed as the ethanol concentration in the sink varied (Fig. 2b and c). This can be appreciated by comparing the Fen-loaded micro-hydrogels with blank micro-hydrogels formulated without Fen in anisole. The final drug load in the micro-hydrogels was also affected by the ethanol concentration in the anti-solvent. Table S2† shows the final Fen load in the dried micro-hydrogels measured through UV-visible spectrophotometry. The anti-solvent sink composition of 5% ethanol offered the highest drug load of 55.05 ± 3.50% by weight and was therefore maintained as the default condition for further studies.
 | ||
| Fig. 2 Characterization of Fen morphology in PEG micro-hydrogels. Optical micrographs, surface and cross-section morphology FESEM characterization of (a) blank anisole NE loaded PEGVs-DT micro-hydrogels post cross-linking and solvent exchange in 5% EtOH anti-solvent sink, (b) Fen-anisole NE loaded PEGVs-DT micro-hydrogels after cross-linking and solvent exchange in 5% EtOH anti-solvent sink and (c) Fen-anisole NE loaded PEGVs-DT micro-hydrogels after cross-linking and solvent exchange in 60% EtOH anti-solvent sink. | ||
Characterization of fenofibrate crystals in the micro-hydrogels
The crystallinity of the Fen in the PEGVs-DT and PEGAc-DT micro-hydrogels (PEGVs-DT-Fen and PEGAc-DT-Fen respectively) was compared with neat polymer micro-hydrogels and raw Fen powder in Fig. 3a. The Fen-loaded micro-hydrogels displayed identical solid-state characteristics irrespective of the functional groups used for cross-linking. The PXRD profiles of both Fen micro-hydrogel formulations showed characteristic peaks of both PEG and Fen form I indicating the presence of crystalline drug and polymer. In Fig. 3b, the FTIR curves for the PEGVs-DT-Fen and PEGAc-DT-Fen micro-hydrogels showed an overlap of the individual Fen and PEG curves. The FTIR spectrum of pure Fen displayed notable absorption peaks at 1725 cm−1 and 1649 cm−1 corresponding to carbonyl groups in the molecule. Additional peaks were observed at 2984 cm−1 and 2935 cm−1 due to the C–H stretching vibrations from the Benzene ring. The characteristic Fen peaks were observed in all the micro-hydrogel formulations indicating a lack of specific interactions like hydrogen bonding between the hydrogel matrix and Fen.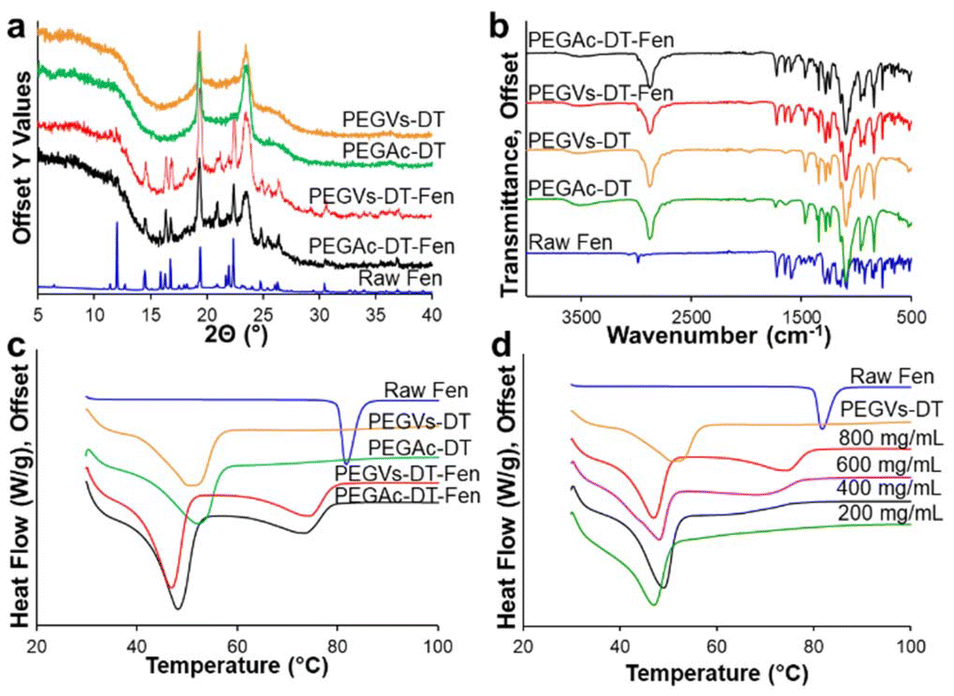 | ||
| Fig. 3 Characterization of Fen crystals in PEG hydrogel matrix. (a) PXRD curves of raw Fen crystalline form I, neat PEGVs-DT, PEGAc-DT hydrogel matrices, Fen PEGVs-DT and Fen PEGAc-DT composite micro-hydrogels prepared with 800 mg mL−1 Fen in anisole, (b) FTIR spectra of raw Fen crystalline form I, neat PEGVs-DT, PEGAc-DT hydrogel matrices, Fen PEGVs-DT and Fen PEGAc-DT composite micro-hydrogels prepared with 800 mg mL−1 Fen in anisole, (c) DSC thermograms of raw Fen crystalline form I, neat PEGVs-DT, PEGAc-DT hydrogel matrices, Fen PEGVs-DT and Fen PEGAc-DT composite micro-hydrogels prepared with 800 mg mL−1 Fen in anisole, (d) DSC thermograms of Fen PEGVs-DT micro-hydrogels prepared with varying Fen concentration in the anisole phase (800–200 mg mL−1). | ||
In Fig. 3c, the DSC curves for the Fen-loaded micro-hydrogels, irrespective of the polymer backbone, showed two endotherms at ∼46–48 °C and ∼74–76 °C distinct from the melting points of the individual Fen and PEG components. This outcome is characteristic of a eutectic system. A eutectic formation occurs when weak intermolecular interactions between two non-isomorphous materials lead to a disordered arrangement of their individual crystalline phases. Fen and PEG have been shown to form a eutectic when formulated through co-melting.25 This, however, is the first report of Fen NE extraction in a PEG hydrogel matrix for eutectic formation. A typical eutectic microstructure possesses alternating crystalline domains of one phase embedded in the other. Here, we suspect that upon anisole extraction, the nucleating Fen has the opportunity to interact with the semi-crystalline hydrated PEG in a convection-free gel environment26,27 (ESI S10†). As the micro-hydrogels are dried, the microcrystalline Fen is subsequently surrounded by a crystalline PEG microenvironment.
The Fen-PEG eutectic micro-hydrogels were further explored by changing their Fen load. It is typical of a eutectic mixture to show a single broad melting endotherm at the eutectic point, which occurs at a specific drug-polymer composition. For a Fen-PEG 8000 system, the eutectic point was recorded at 20–25% w/w Fen in PEG.24 NEs with different Fen loads were prepared by varying the Fen concentration in the anisole phase from 800–200 mg mL−1. The NE was formulated in PEGVs-DT micro-hydrogels and extracted 5% ethanol anti-solvent. The micro-hydrogels possessed the characteristic crystalline XRD peaks of Fen and PEG for all the Fen concentrations (Fig. S8†). Moreover, the DSC curves of the PEGVs-DT-Fen micro-hydrogels exhibited an identical low-temperature endotherm at ∼48 °C. The high-temperature endotherm, however, shifted to lower temperatures as the amount of Fen in the micro-hydrogels decreased as seen in Fig. 3d. At the 200 mg mL−1 Fen formulation, the drug load obtained was 23.45 ± 2.32% w/w in contrast to 55.05 ± 3.50% w/w obtained at 800 mg mL−1 Fen (Table S3†). The 200 mg mL−1 formulation appeared to be close to the eutectic composition of PEG and Fen since it presented a single melting endotherm. The lower Fen concentration in the micro-hydrogel environment may have led to higher Fen-PEG interactions since the PEG concentration remained constant. Moreover, a lower degree of supersaturation of Fen may lead to smaller crystal domains.
Drug release
Drug release from the micro-hydrogels was determined in simulated gastric fluid (SGF) at 37 °C following a previously published protocol.2 The number of micro-hydrogels added to the SGF was calculated based on a final dissolved Fen concentration of ∼30 μg mL−1. First, two distinctly different release profiles were observed based on the polymer backbone of the micro-hydrogels. As shown in Fig. 4a, at constant Fen concentration in anisole phase (800 mg mL−1) and anti-solvent condition of 5% ethanol, all the Fen crystals in the PEGAc-DT micro-hydrogels dissolved in SGF within 4 hours in contrast to those embedded in the PEGVs-DT micro-hydrogels which took significantly longer (∼10 days). This was attributed to the mechanism of release from the hydrogel matrix. The thiol-vinyl sulfone Michael addition catalysed through base, TEA, yields a thioether sulfone bond that is stable and resistant to hydrolytic degradation up to pH 7.16,28 Therefore, the hydrogel matrix, PEGVs-DT, forms a polymeric mesh that maintains its cross-links upon hydration in the release media. As the micro-hydrogels maintain structure, there is a diffusion barrier for the dissolution of Fen. A shrinking core of Fen and a hydrogel periphery free of Fen mark the dissolution front of Fen. In contrast, the thiol–acrylate Michael addition yields a thioether acrylate bond which renders the PEG-Ac ester hydrolytically labile. This leads to the cleavage of the modified ester bond in the PEGAc-DT micro-hydrogel in the release media and the overall degradation of the cross-linked network. Due to the hour time-scale of degradation, a burst release of Fen occurs from the PEGAc-DT micro-hydrogels. The controlled release from PEGVs-DT and burst release from PEGAc-DT micro-hydrogels were visualized in the optical micrographs presented in Fig. 4d and e. As a result of the primary dissolution studies, the polymer composition was identified as a possible lever for tuning drug release. To further explore the tunability of Fen release, micro-hydrogels with a mixed 4-arm polymer backbone comprising 75% PEGVs–25% PEGAc or 50% PEGVs–50% PEGAc cross-linked with PEG-DT were developed (Table S4†). As seen in Fig. 4b, at a constant Fen concentration in NE and ethanol extraction setting, the Fen dissolution could be systematically controlled by changing the polymer composition to obtain complete Fen dissolution at different time points. The ability to combine these two distinct polymer behaviors was a result of the modular tuning of functional groups incorporated in the polymer backbone. The Fen concentration in anisole affects the eutectic outcome of the micro-hydrogels which can also alter the dissolution profile of Fen due to higher crystal imperfections of Fen in the eutectic solid solution. The slow-releasing PEGVs-DT micro-hydrogels containing NE with different Fen loads (800, 400, and 200 mg mL−1 in anisole) were evaluated for Fen dissolution. As seen in Fig. 4c, PEGVs-DT micro-hydrogels formulated with 800 mg mL−1 of Fen showed a slower release relative to those formulated with 200 mg mL−1. As the crystalline PEG microstructure of the micro-hydrogels hydrated, the imperfect crystal structure of the embedded Fen was solubilized faster in formulations close to the eutectic composition. The release exponents obtained from a Korsmeyer-Peppas fit of the dissolution profiles are compared in Fig. S12.† The release exponents are 0.36, 0.85, and 0.89 for PEGVs-DT micro-hydrogels formulated with 200, 400, and 800 mg mL−1 of Fen respectively. An identical behavior was observed for PEGAc-DT micro-hydrogels, however not as obvious due to the hydrogel disintegration at competing timescales (Fig. S13†). The eutectic nature of the Fen-PEG composite therefore offers another route to tune drug release. The methodology used for formulating Fen in this study can be readily applied to several hydrophobic APIs like NSAIDS, polyphenols and alkaloids. Certain factors that need to be considered are:1. Solvent compatibility – Since the drug is dissolved in the dispersed phase of the nanoemulsion, the drug must possess a high solubility in relatively non-polar organic solvents like ethyl acetate, dimethyl carbonate, and anisole.
2. Crystallization tendency – For drugs that are slow crystallizers, there is a possibility of encapsulating the drug melt or amorphous drug solid. In this study, we have worked with Fen which has slow crystallization kinetics29 but still resulted in a crystalline outcome of the drug. This may not necessarily apply to all compounds.
3. Drug-PEG interactions – Fen exhibited no specific molecular interactions with the PEG hydrogel matrix in this study. This, however, may not apply to all drugs. For instance, drugs like flurbiprofen and ibuprofen11 show strong hydrogen bonding with PEG and it can affect the solid-state outcome of the drug when formulated through this method.
 | ||
| Fig. 4 Dissolution of Fen crystals in PEG micro-hydrogel matrix. (a) Dissolution profiles of raw Fen in SGF in comparison to the burst release formulation of PEGAc-DT-Fen and the controlled release formulation of PEGVs-DT-Fen, (b) dissolution in SGF of Fen embedded in varying composition of the 4-arm monomers cross-linked with PEG-DT of 100% PEGVs, 100% PEGAc, 50–50 PEG-Vs-Ac and 75–25 PEG-Vs-Ac, (c) dissolution in SGF of Fen embedded in PEGVs-DT micro-hydrogels prepared with varying Fen concentration in the anisole phase (800–200 mg mL−1), (d) optical micrographs of PEGVs-DT-Fen micro-hydrogels subjected to Fen dissolution in SGF from 0–24 h time points, (e) optical micrographs of PEGAc-DT-Fen micro-hydrogels subjected to Fen dissolution in SGF from 0–24 h time points. | ||
Conclusion
Michael addition-based PEG hydrogels possess fast-reacting (∼ms) bimolecular polymer networks which allow for modular incorporation of functional groups. Until now, the tunability of these hydrogel systems has not been explored in the context of the delivery of crystalline lipophilic APIs. Spherical co-processed API micro-hydrogels formulated through microfluidic platforms allow control over crystal domain size, powder flow properties, and overall product uniformity. In this work, we present fabrication of NE-loaded Michael addition-based PEG micro-hydrogels for encapsulation of model API, Fen. The fast reaction time of Michael addition and the unique chemical composition of the NE made step emulsification a feasible particle production strategy. The solidified micro-hydrogels presented several interesting formulation outcomes that affected the drug release profiles. The functional groups incorporated in the polymer backbone of the micro-hydrogels and their relative concentration directly affected the polymer degradation and therefore, offered a modular strategy for tuning drug release from the matrix. Fen dissolution from the micro-hydrogels was regulated from a fast degrading regime of the acrylate-dithiol crosslinks to a slow degrading regime of the vinylsulfone-dithiol crosslinks – tuning release from 4 hours to 10 days. Furthermore, the Fen-PEG micro-hydrogels presented a eutectic behavior, which offered Fen concentration as a second lever to tune drug dissolution.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Longfei Chen and Denise Z. L. Ng for meaningful suggestions and discussion. This research was supported by the Pharmaceutical Innovation Programme Singapore (grants number A19B3a0012 and M22B3a0051).References
- G. L. Amidon, H. Lennernäs, V. P. Shah and J. R. Crison, A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability, Pharm. Res., 1995, 12(3), 413–420 CrossRef CAS.
- L. H. Chen and P. S. Doyle, Design and Use of a Thermogelling Methylcellulose Nanoemulsion to Formulate Nanocrystalline Oral Dosage Forms, Adv. Mater., 2021, 33(29), 2008618 CrossRef CAS.
- T. Domenech and P. S. Doyle, High Loading Capacity Nanoencapsulation and Release of Hydrophobic Drug Nanocrystals from Microgel Particles, Chem. Mater., 2020, 32(1), 498–509 CrossRef CAS.
- M. A. Casadei, F. Cerreto, S. Cesa, M. Giannuzzo, M. Feeney and C. Marianecci, et al., Solid lipid nanoparticles incorporated in dextran hydrogels: A new drug delivery system for oral formulations, Int. J. Pharm., 2006, 325(1), 140–146 CrossRef CAS PubMed.
- C. Desfrançois, R. Auzély and I. Texier, Lipid Nanoparticles and Their Hydrogel Composites for Drug Delivery: A Review, Pharmaceuticals, 2018, 11(4), 118 CrossRef.
- T. T. H. Thi, Y. Lee, S. B. Ryu, H. J. Sung and K. D. Park, Oxidized cyclodextrin-functionalized injectable gelatin hydrogels as a new platform for tissue-adhesive hydrophobic drug delivery, RSC Adv., 2017, 7(54), 34053–34062 RSC.
- J. Cui, M. A. Lackey, G. N. Tew and A. J. Crosby, Mechanical Properties of End-Linked PEG/PDMS Hydrogels, Macromolecules, 2012, 45(15), 6104–6110 CrossRef CAS.
- M. Bora, M. N. Hsu, S. A. Khan and P. S. Doyle, Hydrogel Microparticle-Templated Anti-Solvent Crystallization of Small-Molecule Drugs, Adv. Healthc. Mater., 2022, 11(8), 2102252 CrossRef CAS PubMed.
- L. Macdougall, H. Culver, C. C. Lin, C. Bowman and K. Anseth, 1.3.2F - Degradable and Resorbable Polymers, ed. W. R. Wagner, S. E. Sakiyama-Elbert, G. Zhang and M. J. B. T. B. S. Yaszemski, Biomaterials Science, 4th edn, Academic Press, 2020, pp. 167–190 Search PubMed.
- S. Adepu and S. Ramakrishna, Controlled Drug Delivery Systems: Current Status and Future Directions, Molecules, 2021, 26, 5905 CrossRef CAS PubMed.
- S. R. Vippagunta, Z. Wang, S. Hornung and S. L. Krill, Factors Affecting the Formation of Eutectic Solid Dispersions and Their Dissolution Behavior, J. Pharm. Sci., 2007, 96(2), 294–304 CrossRef CAS.
- J. Li and D. J. Mooney, Designing hydrogels for controlled drug delivery, Nat. Rev. Mater., 2016, 1(12), 16071 CrossRef CAS PubMed.
- G. W. Ashley, J. Henise, R. Reid and D. V. Santi, Hydrogel drug delivery system with predictable and tunable drug release and degradation rates, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(6), 2318–2323 CrossRef CAS PubMed.
- S. Zalipsky and J. M. Harris, Introduction to Chemistry and Biological Applications of Poly(ethylene glycol), in Poly(ethylene glycol), American Chemical Society, ACS Symposium Series, 1997, vol. 680, pp. 1 Search PubMed.
- E. Jain, L. Hill, E. Canning, S. A. Sell and S. P. Zustiak, Control of gelation, degradation and physical properties of polyethylene glycol hydrogels through the chemical and physical identity of the crosslinker, J. Mater. Chem. B, 2017, 5(14), 2679–2691 RSC.
- A. Metters and J. Hubbell, Network Formation and Degradation Behavior of Hydrogels Formed by Michael-Type Addition Reactions, Biomacromolecules, 2005, 6(1), 290–301 CrossRef CAS PubMed.
- J. M. de Rutte, J. Koh and D. Di Carlo, Scalable High-Throughput Production of Modular Microgels for In Situ Assembly of Microporous Tissue Scaffolds, Adv. Funct. Mater., 2019, 29(25), 1900071 CrossRef.
- L. H. Chen, L. C. Cheng and P. S. Doyle, Nanoemulsion-Loaded Capsules for Controlled Delivery of Lipophilic Active Ingredients, Adv. Sci., 2020, 7(20), 2001677 CrossRef CAS.
- D. Jagadeesan, I. Nasimova, I. Gourevich, S. Starodubtsev and E. Kumacheva, Microgels for the Encapsulation and Stimulus-Responsive Release of Molecules with Distinct Polarities, Macromol. Biosci., 2011, 11(7), 889–896 CrossRef CAS PubMed.
- D. J. McClements, Emulsion Design to Improve the Delivery of Functional Lipophilic Components, Annu. Rev. Food Sci. Technol., 2010, 1(1), 241–269 CrossRef CAS PubMed.
- A. Z. M. Badruddoza, A. Gupta, A. S. Myerson, B. L. Trout and P. S. Doyle, Low Energy Nanoemulsions as Templates for the Formulation of Hydrophobic Drugs, Adv. Ther., 2018, 1(1), 1700020 Search PubMed.
- L. Attia, L. H. Chen and P. S. Doyle, Orthogonal Gelations to Synthesize Core–Shell Hydrogels Loaded with Nanoemulsion-Templated Drug Nanoparticles for Versatile Oral Drug Delivery, Adv. Healthc. Mater., 2023, 2301667 CAS.
- A. Z. M. Badruddoza, P. D. Godfrin, A. S. Myerson, B. L. Trout and P. S. Doyle, Core–Shell Composite Hydrogels for Controlled Nanocrystal Formation and Release of Hydrophobic Active Pharmaceutical Ingredients, Adv. Healthc. Mater., 2016, 5(15), 1960–1968 CAS.
- D. Law, W. Wang, E. A. Schmitt, Y. Qiu, S. L. Krill and J. J. Fort, Properties of Rapidly Dissolving Eutectic Mixtures of Poly(ethylene glycol) and Fenofibrate:The Eutectic Microstructure, J. Pharm. Sci., 2003, 92(3), 505–515 CAS.
- S. Cherukuvada and A. Nangia, Eutectics as improved pharmaceutical materials: design, properties and characterization, Chem. Commun., 2014, 50(8), 906–923 CAS.
- D. Eagland, N. J. Crowther and C. J. Butler, Interaction of poly(oxyethylene) with water as a function of molar mass, Polymer, 1993, 34(13), 2804–2808 CAS.
- V. V. Kuzmin, V. S. Novikov, L. Y. Ustynyuk, K. A. Prokhorov, E. A. Sagitova and G. Y. Nikolaeva, Raman spectra of polyethylene glycols: Comparative experimental and DFT study, J. Mol. Struct., 2020, 1217, 128331 CrossRef CAS.
- E. J. Weber and V. C. Stickney, Hydrolysis kinetics of Reactive Blue 19-Vinyl Sulfone, Water Res., 1993, 27(1), 63–67 CrossRef CAS.
- K. Kawakami, T. Usui and M. Hattori, Understanding the glass-forming ability of active pharmaceutical ingredients for designing supersaturating dosage forms, J. Pharm. Sci., 2012, 101(9), 3239–3248 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00186a |
| This journal is © The Royal Society of Chemistry 2025 |