
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and characterization of efavirenz cocrystal-encapsulated pronanoliposomes for antiretroviral therapy with improved bioavailability†
Sucheta
Sarkar
 ,
Arijit Prosad
Roy
,
Sanchita
Mitra
,
Gouranga
Nandi
,
Ranabir
Sahu
,
Tarun Kumar
Dua
and
Paramita
Paul
*
,
Arijit Prosad
Roy
,
Sanchita
Mitra
,
Gouranga
Nandi
,
Ranabir
Sahu
,
Tarun Kumar
Dua
and
Paramita
Paul
*
Department of Pharmaceutical Technology, University of North Bengal, Darjeeling, West Bengal-734013, India. E-mail: paramita37@gmail.com; paramita37@nbu.ac.in; Tel: +919831424214
First published on 6th December 2024
Abstract
The present research work aims to improve the bioavailability of the antiretroviral drug efavirenz (EFV) using pharmaceutical cocrystallization technique. EFV is a potential antiretroviral drug that exhibits extremely poor water solubility and poor oral bioavailability and falls under the BCS-II category. EFV and L-proline were selected in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equimolar ratio to formulate efavirenz proline co-crystals, and a facile method was adopted to prepare co-crystals of EFV. The formation of a new solid phase was confirmed through advanced techniques such as Fourier-transform infrared (FTIR) spectroscopy, differential scanning calorimetry (DSC) and powder X-ray diffraction (pXRD) analysis, and solubility study was conducted utilising UV visible spectroscopy. Proliposomal vesicles containing EFV or EFV cocrystals were prepared using thin film hydration methods with few modifications. The vesicle size in dispersion, zeta potential, surface morphology, drug loading and in vitro drug release were assessed. Co-crystallization increased the solubility of EFV up to 3 fold, and the liposomes were found to release the drug in a sustained manner. The optimized formulation was found to have a substantial amount of EFV loading (32.70%) and entrapment efficiency (99.28%) with a narrow range of size distribution. The liposomes containing the pure drug showed 72% release of the drug in 72 h, whereas the liposomes containing co-crystals showed 99.98% release of the drug in 72 h. This was due to the presence of L-proline in association with EFV, which led to an enhancement in the polarity of hydrophobic EFV, thus increasing its dissolution in drug release media. The present work reports a cost-effective method for the enhancement of drug solubility, providing sustained drug release from liposome and thereby improving the oral bioavailability of the antiviral agent EFV.
:1 equimolar ratio to formulate efavirenz proline co-crystals, and a facile method was adopted to prepare co-crystals of EFV. The formation of a new solid phase was confirmed through advanced techniques such as Fourier-transform infrared (FTIR) spectroscopy, differential scanning calorimetry (DSC) and powder X-ray diffraction (pXRD) analysis, and solubility study was conducted utilising UV visible spectroscopy. Proliposomal vesicles containing EFV or EFV cocrystals were prepared using thin film hydration methods with few modifications. The vesicle size in dispersion, zeta potential, surface morphology, drug loading and in vitro drug release were assessed. Co-crystallization increased the solubility of EFV up to 3 fold, and the liposomes were found to release the drug in a sustained manner. The optimized formulation was found to have a substantial amount of EFV loading (32.70%) and entrapment efficiency (99.28%) with a narrow range of size distribution. The liposomes containing the pure drug showed 72% release of the drug in 72 h, whereas the liposomes containing co-crystals showed 99.98% release of the drug in 72 h. This was due to the presence of L-proline in association with EFV, which led to an enhancement in the polarity of hydrophobic EFV, thus increasing its dissolution in drug release media. The present work reports a cost-effective method for the enhancement of drug solubility, providing sustained drug release from liposome and thereby improving the oral bioavailability of the antiviral agent EFV.
Introduction
Acquired immunodeficiency syndrome (AIDS) caused by the human immunodeficiency virus (HIV) is a life-threatening infectious disease and is a challenge to public health by virtue of its easy transmittability.1 According to the United Nations, over 36.9 million people in the world are living with HIV/AIDS, including 1.8 million children.2 HIV is transmitted through contact of infected body fluids with mucosal tissue, blood or broken skin. Factors that increase the infectiousness of a person infected with HIV include high levels of the virus in plasma3 or genital secretions4 and other sexually transmitted infections.5 HIV attacks and destroys CD4+ T cells, which are the essential component of the immune system, hence leading to knockdown of the immune system.6 HIV patients are mostly treated with highly active antiretroviral therapy (HAART), which entails frequent administration of multiple antiretroviral (ARV) drugs such as abacavir, efavirenz, zidovudine, lamivudine, and doravirine.7 Unfortunately, this therapy only improves the quality of the affected person's life by slowing down the replication of the virus within infected cells.8 Moreover, owing to the lengthy drug administration regimen in HAART and severe adverse effects associated with ARV drugs, patients often do not adhere to it, which leads to the development of drug resistance and, ultimately, therapeutic failure.9 ARV drugs exhibit poor oral bioavailability due to hepatic first pass metabolism and short biological half-life, and this in fact raises further concerns as it necessitates frequent administration of high doses, which leads to low patient compliance.10 Since ARVs remain pivotal to the HAART regimen, there is an urgent need to improve the pharmacological profiles of existing ARVs.11The U.S. Food and Drug Administration (USFDA) has authorized efavirenz (EFV) as a non-nucleoside reverse transcriptase inhibitor (NNRTI) for the treatment of HIV. However, the limited water solubility of EFV necessitates high dose (maximum 800 mg daily) administration to achieve therapeutic activity.12,13 As a result, several adverse effects associated with EFV, including insomnia, liver failure, severe rash, nausea, diarrhoea, confusion, depression, hallucination and serious psychiatric effects such as suicidal ideation, are often observed.14,15 In recent times, pharmaceutical co-crystals have attracted much attention of the formulation scientists. Co-crystals are multiple component crystals or crystalline complexes stabilized by different types of interaction, including hydrogen bonding, π-stacking, and van der Waals forces. Cocrystallization of a drug with a suitable coformer is one of the many approaches for improving the pharmaceutical performances of a drug, including solubility, dissolution profile, pharmacokinetics, and stability.16 A variety of molecules can act as coformers, such as tartaric acid, adipic acid,17DL-alanine, oxalic acid, maleic acid, nicotinamide,18L-proline,19etc. Also proline is found to be an effective coformer with other drugs such as diclofenac,20 ezetimibe21 and many others. Among all coformers, L-proline was available in our lab, and is a water soluble molecule. Studies showed that L-proline effectively enhanced the solubility of many BCS class II drugs. Hence, it was selected as a coformer.
Pronanoliposomes are a free-flowing solid dispersion formulation of phospholipids, which form nanoliposomal vesicular dispersion in contact with an aqueous phase. These pronanoliposomes could be a promising approach to avoid fusion, aggregation, and sedimentation types of physicochemical instability generally found with liposome formulations and can be used to improve the stability as well as bioavailability of many drugs with poor oral bioavailability performance.22 Liposomes prepared with dimyristoylphosphatidylcholine and distearoyl-phosphatidylcholine containing EFV and glutathione have been found to enhance the uptake and reduce the cytotoxicity of EFV in the presence of glutathione.23 Liposomes have been adopted as an approach for many poorly water-soluble drugs.24,25 Here, we prepared co-crystals of EFV using L-proline to improve the solubility and dissolution rate of the pure drug without affecting its intact structure.19 This type of work has not been reported earlier and its novelty relies on designing a nanocarrier, such as liposomes, to increase the solubility of EFV and enable targeted and controlled distribution of the ARV drug, which could enhance the molecule's bioavailability and lessen its adverse effects. Liposomes are small artificial vesicles of spherical shape prepared using phospholipids and cholesterol (CHL), and have been extensively studied since the 1960s as delivery systems.26 By altering the absorption and metabolism, liposomes have increased the therapeutic index of known or newly discovered ARV drugs, leading to a longer plasma half-life and less toxicity.27 In this study, we also compare the co-crystal loaded liposomes with the liposomes containing pure EFV.
Materials and methods
Materials
Sisco Research Laboratories Private Limited, Mumbai, Maharashtra, India, provided soya lecithin (SLC) (30%) [Batch no. 9175679], CHL [Batch no. 4544907], butylated hydroxytoluene (BHT) [Batch no. 3925242], and L-proline [Batch no. 1979570]. Pristine EFV was gifted by Aurobindo Pharma Limited, Hyderabad, India. Avantor Performance Materials India Ltd, Maharashtra, India, provided disodium hydrogen phosphate. Emplura®, India, provided potassium dihydrogen phosphate, and sodium chloride was provided by Merck Life Science Pvt. Ltd, Mumbai, India. The other chemicals and solvents used in this experiment were all of high analytical grade. Double distilled water was used throughout the experiment and deionised water was used in the DLS study.Methods
:water in a ratio of 7:3 (v/v) and in phosphate buffer saline (PBS) (pH 7.4) for the purpose of drug loading and drug release, respectively. To detect the interaction between the excipients and the pure form of drug at the functional group level, Fourier transform infrared spectroscopy (FTIR) study was adopted.29
:water (7:3 (v/v)). PBS, pH 7.4 was used as a medium for the drug release study, and acetonitrile:water (7:3) as the medium for the drug loading study. 1 μg ml−1 concentration of the drug solution was prepared in two different solvents and scanned in a UV-visible spectrophotometer (Shimadzu, Japan) with a wavelength range from 200 nm to 400 nm using a fresh solvent (PBS 7.4 and acetonitrile-water mixture in a ratio of 7:3) as the blank solvent. Different drug concentrations were prepared for both solvents to prepare the standard curve, and absorbance was determined using a UV-visible spectrophotometer at a wavelength of maximum absorbance (λmax) using a fresh solvent as a blank reference. Then, the absorbance data were plotted against the concentration to get the standard curve. The same process was utilized to prepare a standard curve of EFV in phosphate buffer pH 7.4.
:50 (w/w) by using a clean mortar and pestle. The mixture was taken in sufficient amount in a die hole and the punch was placed above it and the set was pressed in a hydraulic press to prepare a transparent pellet. The pellet was then placed in a sample holder and scanned in OPUS software.
:1) of EFV and L-proline were taken in a petri dish. A mixture of two solvents viz. acetonitrile and diethyl ether in 1:1 (v/v) ratio was added to this. The whole mixture was heated slightly in a water bath to obtain a clear solution and then kept at room temperature for slow evaporation. After evaporation, the co-crystal of EFV was obtained in dry form.19,31
Characterization of the co-crystals
:4); lastly, the sample's absorbance was measured at its corresponding λmax using a UV-visible spectrometer.32
000 rpm for 45 min and then lyophilized using a lyophilizer (CoolSafe 4-15L Freeze Dryers, Labogene™, Denmark) to get a dried liposomal product.35,36
| Formulation code | Form of drug used | EFVa | SLC (mg)b | CHL (mg)c |
|---|---|---|---|---|
| BHT: 1% W/V used for all formulations.a EFV: efavirenz;b SLC: soya lecithin;c CHL: cholesterol. | ||||
| F1 | Pure EFV | 100 mg | 75 | 25 |
| F2 | Co-crystals of EFV | 100 mg | 75 | 25 |
| F3 | Pure EFV | 100 mg | 100 | 50 |
| F4 | Co-crystals of EFV | 100 mg | 100 | 50 |
| F5 | Pure EFV | 100 mg | 100 | 100 |
| F6 | Co-crystals of EFV | 100 mg | 100 | 100 |
Characterization of experimental liposome batches
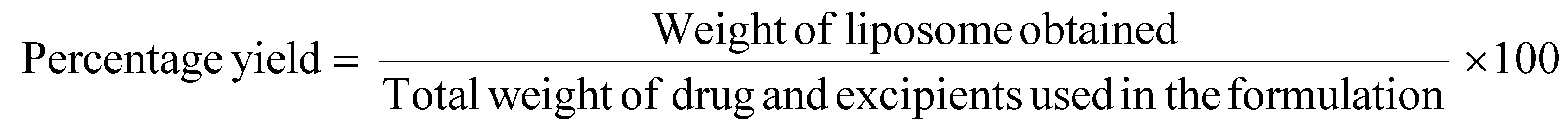 :3 v/v) solvent mixture and mixing with a vortex mixer. The dispersion was then sonicated for 2 hours in a bath sonicator before being centrifuged for 10 min at 15000 rpm in a cold centrifuge. The absorbance of the supernatant solution was measured in a UV-visible spectrophotometer against the acetonitrile–water (7:3) mixture as the blank solution at the λmax of EFV. The % loading of EFV was calculated using the equation below:40
:3 v/v) solvent mixture and mixing with a vortex mixer. The dispersion was then sonicated for 2 hours in a bath sonicator before being centrifuged for 10 min at 15000 rpm in a cold centrifuge. The absorbance of the supernatant solution was measured in a UV-visible spectrophotometer against the acetonitrile–water (7:3) mixture as the blank solution at the λmax of EFV. The % loading of EFV was calculated using the equation below:40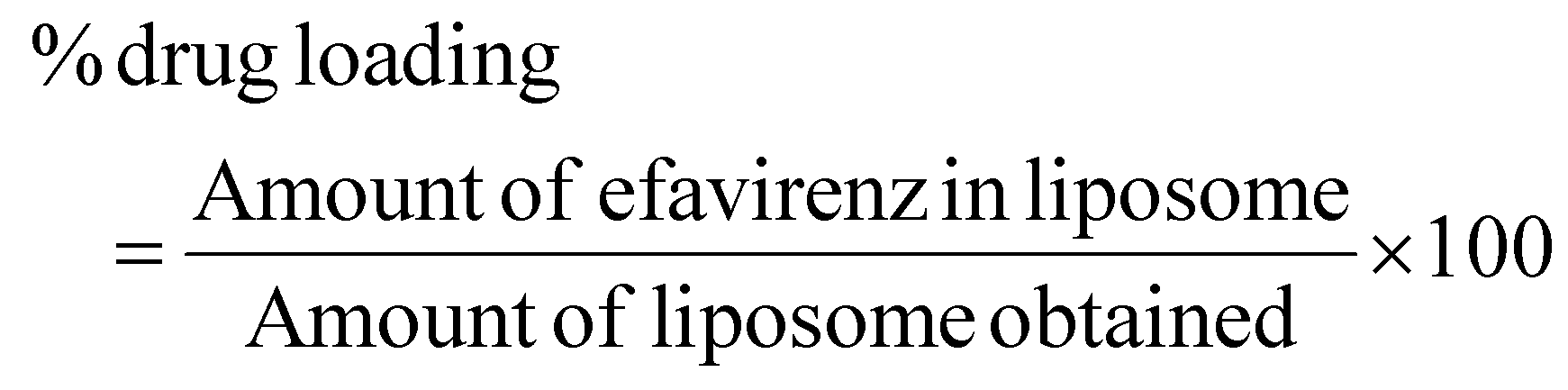
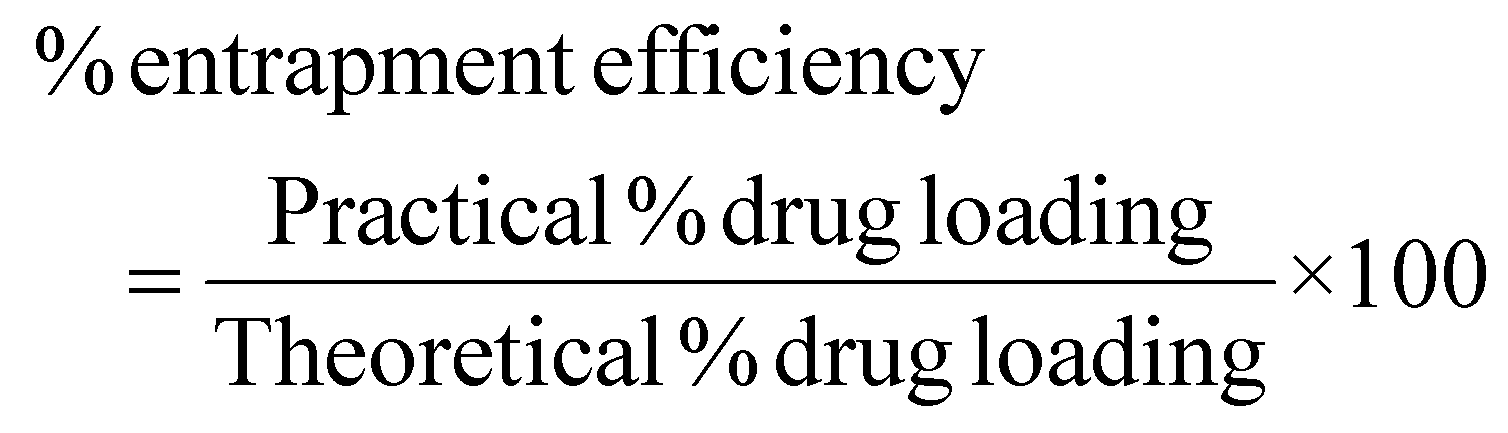
Results and discussion
FTIR spectroscopy
FTIR spectroscopy is a useful method for identifying the functional groups present in chemicals used in formulations.29 Hence, any changes in the functional group may indicate the presence of interaction. In this study, FTIR spectra of pure drug EFV, L-proline, and EFV co-crystals were recorded and are presented in Fig. 1.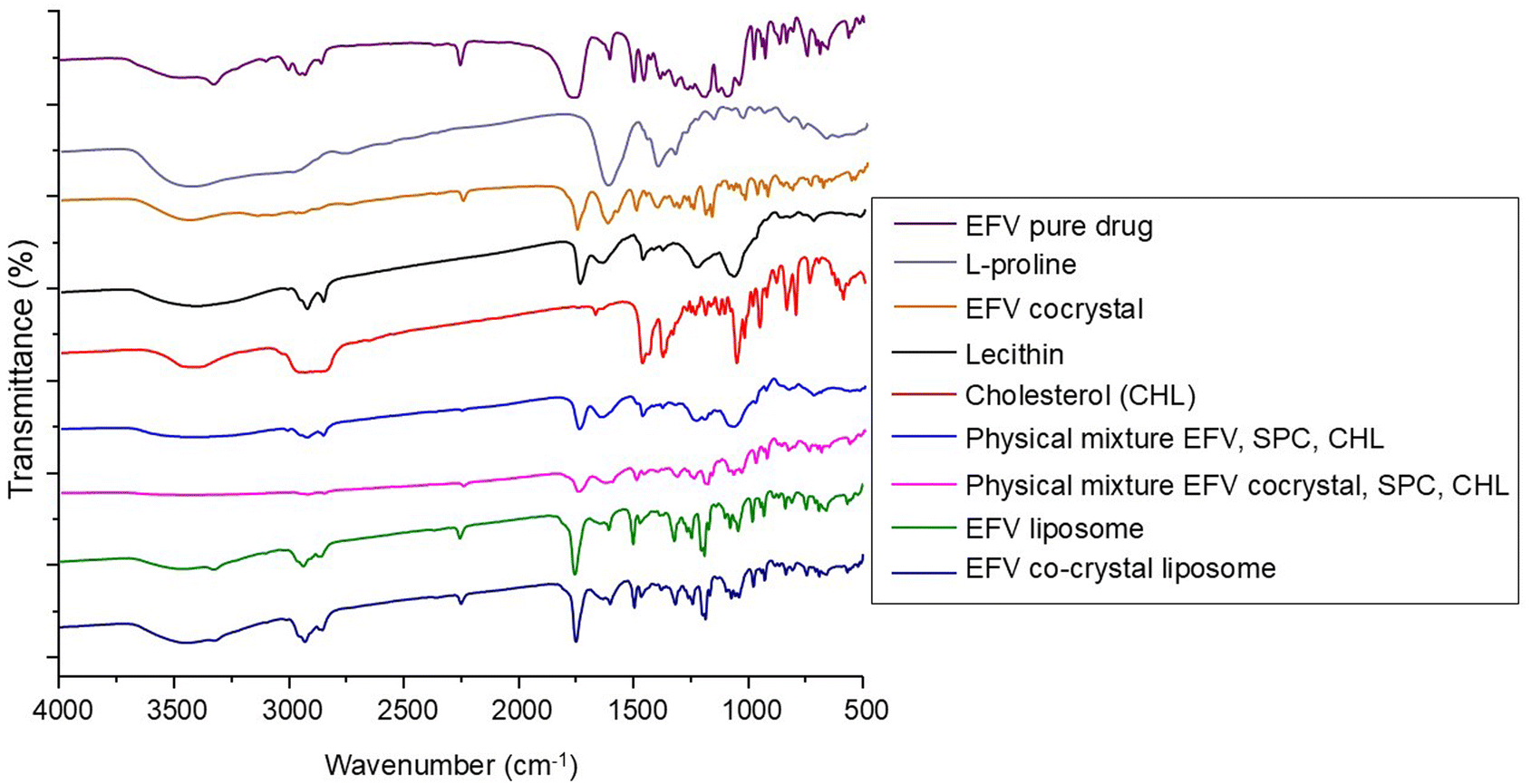 | ||
| Fig. 1 FTIR spectra of pure efavirenz (EFV); L-proline; EFV cocrystal; lecithin; cholesterol (CHL); physical mixture of EFV, lecithin and CHL; physical mixture of EFV co-crystal, lecithin, and CHL; EFV-loaded liposomes; and EFV cocrystal-loaded liposomes. | ||
The pure drug EFV showed a strong N–H stretching at 3320 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1749 cm−1, C–C stretching due to the alkyne group at 2250 cm−1, C–F stretching at 1317 cm−1 and 1242 cm−1, C–Cl stretching at 1039 cm−1, and C–O–C stretching at 1074 cm−1 and 1098 cm−1. The characteristic peaks of EFV for CC stretching of the aromatic ring were observed at 1602 cm−1 and 1496 cm−1, and the peak for C–H stretching was also observed at 2925 cm−1.12 The distinctive peaks of L-proline were observed at 1623, 2348, and 3486 cm−1, representing CO stretching of the carboxylic acid, N–H stretching of the amine heterocyclic ring, and O–H stretching of the carboxylic acid, respectively.48 FTIR spectra of EFV cocrystals showed characteristics peaks of L-proline and pure EFV with some changes due to the formation of hydrogen bonds between the components. It showed peaks at 1621 cm−1 and 1753 cm−1 for CO and 2363 cm−1 and 3433 cm−1 for N–H and O–H groups, respectively. It showed strong intensity at 2248 cm−1 for the C–C group. For the C–F group, it showed peaks at 1311 and 1242 cm−1. Peaks at 1025 and 1497 cm−1 confirmed the presence of C–Cl and CC groups.18,19
O stretching at 1749 cm−1, C–C stretching due to the alkyne group at 2250 cm−1, C–F stretching at 1317 cm−1 and 1242 cm−1, C–Cl stretching at 1039 cm−1, and C–O–C stretching at 1074 cm−1 and 1098 cm−1. The characteristic peaks of EFV for CC stretching of the aromatic ring were observed at 1602 cm−1 and 1496 cm−1, and the peak for C–H stretching was also observed at 2925 cm−1.12 The distinctive peaks of L-proline were observed at 1623, 2348, and 3486 cm−1, representing CO stretching of the carboxylic acid, N–H stretching of the amine heterocyclic ring, and O–H stretching of the carboxylic acid, respectively.48 FTIR spectra of EFV cocrystals showed characteristics peaks of L-proline and pure EFV with some changes due to the formation of hydrogen bonds between the components. It showed peaks at 1621 cm−1 and 1753 cm−1 for CO and 2363 cm−1 and 3433 cm−1 for N–H and O–H groups, respectively. It showed strong intensity at 2248 cm−1 for the C–C group. For the C–F group, it showed peaks at 1311 and 1242 cm−1. Peaks at 1025 and 1497 cm−1 confirmed the presence of C–Cl and CC groups.18,19
FTIR spectra of SLC, CHL, the physical mixture of EFV, CHL, and SLC, and the physical mixture of EFV cocrystals, CHL, and SLC are reported in Fig. 1. The FTIR spectra of SLC revealed characteristic peaks at 2925 cm−1 for C–H stretching, at 1725 cm−1 for strong intensity of CO stretching vibration, at 1350 cm−1 for medium intensity of C–O stretching vibration, and at 1050 cm−1 for C–O groups (glycerol) and C–N groups (choline) present in lecithin.34 CHL showed a peak at 3590 cm−1 due to the presence of the –OH group and a strong intensity band at 2930 cm−1 due to C–H stretching as well as at 2850 cm−1 due to C–OH stretching. CHL showed its characteristic strong intensity stretching vibration of C–O at 1054 cm−1.49 FTIR spectra of the physical mixture containing the drug, SLC, and CHL showed the individual peaks of components, which confirmed that the drug did not undergo any chemical interaction with the excipients present. Again, the FTIR spectra of the physical mixture containing co-crystals, SLC and CHL showed the individual peaks of components, which confirmed that the co-crystals did not undergo any chemical interaction with the excipients present.
The FTIR spectra of liposome formulations containing pure EFV and EFV cocrystals are shown in Fig. 1. The spectrum of EFV-loaded liposomes showed some characteristic peaks of EFV in the same spectral regions, in addition to peaks due to the lipid components that appeared at 2931 cm−1 and 1466 cm−1 for the alkane C–H stretching, at 1242 cm−1 for the ester sigma bonds and at 1107 cm−1 for the ether region.50
The spectrum of co-crystal loaded liposomes contains the characteristic peaks of EFV co-crystals, L-proline, and other components. It showed a peak at 2929 cm−1 for the lipid component, at 1242 cm−1 and 1317 cm−1 for C–F stretching, at 1632 cm−1 and 1750 cm−1 for CO stretching, at 2250 cm−1 for C–C stretching, and at 1496 cm−1 for CC stretching.
pXRD analysis
The p-XRD data for pristine EFV, L-proline, and EFV co-crystals with L-proline are represented in Fig. 2. Fig. 2A exhibits some sharp characteristic peaks (2θ) at 6.24°, 10.94°, 12.74°, 13.59°, 14.69°, 16.92°, 20.69°, 21.74°, 25.39, 28.59 and 39.19°, which may be attributed to the presence of crystalline character of EFV in its pure form.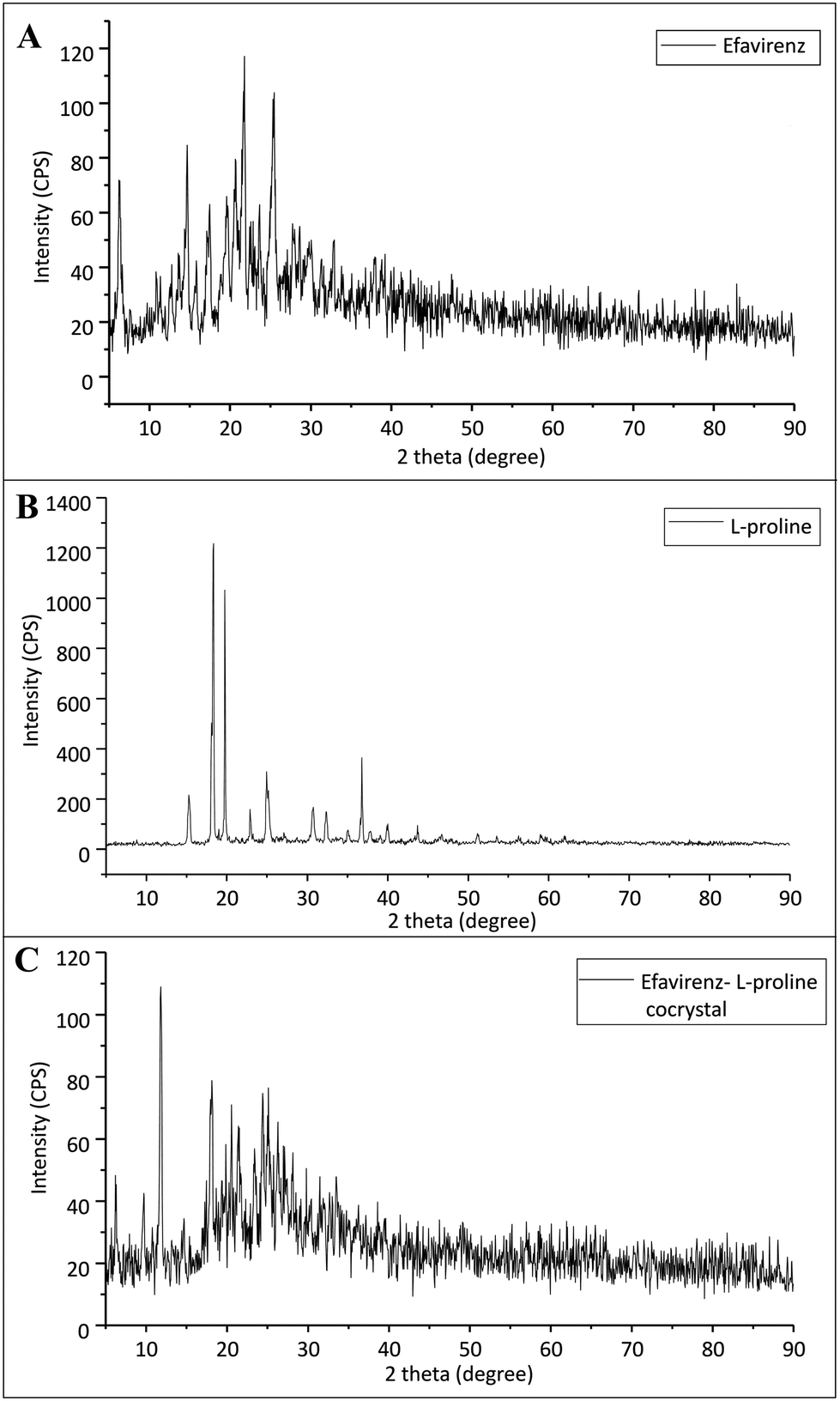 | ||
| Fig. 2 X ray diffraction pattern of (A) pristine efavirenz, (B) L-proline and (C) cocrystals of efavirenz with L-proline. | ||
Pure L-proline shows characteristic peaks at 15.39°, 18.29°, 19.74°, 22.89°, 24.89°, 30.69°, 32.34°, 35.14°, 36.79°, 39.99° and 43.69°. However, the p-XRD pattern of co-crystals exhibits a significant difference as compared to the characteristic peak of pristine EFV and L-proline. It suggests the formation of a new crystalline phase.
Solubility study of co-crystals
The λmax of efavirenz was determined and found to be 247.5 nm. In the aqueous solubility study of EFV and its co-crystals, the result revealed that the solubility of pure EFV was 5.46 μg ml−1 and that of EFV co-crystals with L-proline (equimolar) was 15.01 μg ml−1, which suggests a 3 fold improvement in the solubility of co-crystals compared to pure drug. This is because of the salification where a hydrogen bond donor group leads to an association with an acceptor group. A hydrogen bond was formed between the –N–H and –CO functional groups of L-proline.19 The –N–H band of EFV has broadened, with a considerable shift towards higher wavenumber from 1749 cm−1 to 1753 cm−1 (redshift) in the case of cocrystals, which actually occurred due to the involvement of intermolecular interactions such as hydrogen bonding. Due to the enhancement of solubility as a result of co-crystal formation, the dissolution profile of the drug can also be increased.17
DSC analysis
The DSC patterns are given in Fig. 3. The two endothermic peaks observed for pure L-proline that were close to one another were assigned to the melting of L-proline accompanied by decomposition, and it was similar to an observation by another study.51 The onset and peak temperatures of L-proline were 232 and 234 °C, respectively. The DSC of pure EFV showed a sharp endothermic peak at 138 °C. EFV co-crystals with L-proline showed an endothermic peak at 266 °C and 126 °C, which exhibits a unique and distinct melting endotherm compared to the EFV and L-proline, which confirms the formation of a new crystalline phase. Earlier studies reported that the formation of co-crystals with lower melting points and lower enthalpy value implies an increased dissolution rate compared to the parent compound.18 The liposomes encapsulated with co-crystals showed none of the endothermic peaks, suggesting the complete encapsulation of the co-crystal within the liposome core.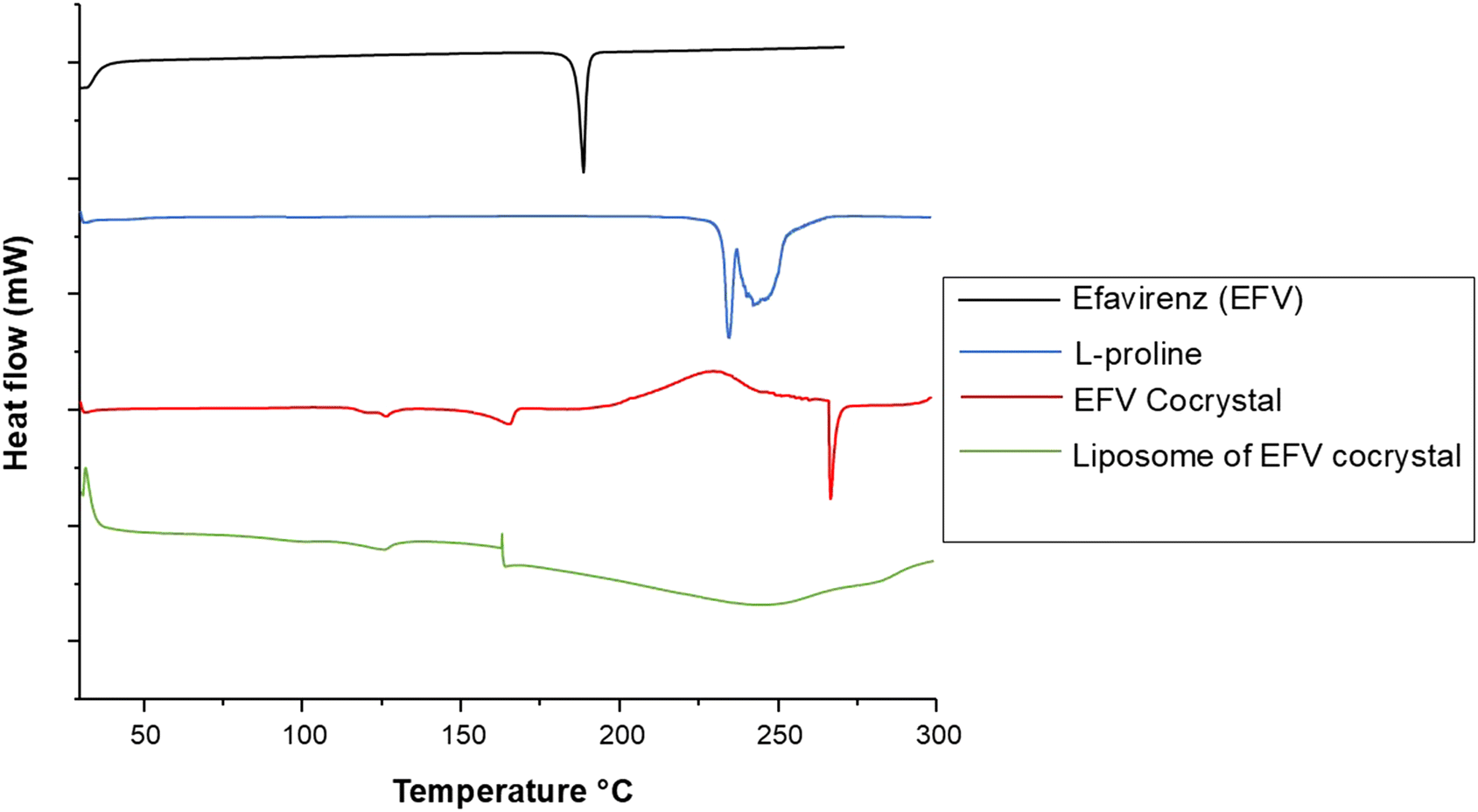 | ||
| Fig. 3 DSC thermogram of pristine efavirenz, L-proline, cocrystals of efavirenz with L-proline, and liposomal formulation containing cocrystals of efavirenz. | ||
Percentage yield of liposomes
The yield percentage of different batches was determined after the formulation of different batches to know the amount of liposomes recovered in each batch.52,53 The percentage yield of liposomes containing co-crystals is more than that of the liposomes containing pure drugs (Table 2). The maximum yield was observed in the F6 batch.| Formulation code | % yield | Practical drug loading (%) | Theoretical drug loading (%) | Entrapment efficiency (%) |
|---|---|---|---|---|
| F1 | 23.67 ± 2.77 | 28.41 | 50.00 | 56.82 ± 3.30 |
| F2 | 51.20 ± 1.04 | 31.00 | 49.14 | 63.10 ± 1.51 |
| F3 | 44.25 ± 1.54 | 24.50 | 40.00 | 61.26 ± 3.72 |
| F4 | 67.78 ± 6.74 | 36.62 | 39.44 | 92.86 ± 2.08 |
| F5 | 53.98 ± 1.02 | 29.98 | 33.33 | 89.97 ± 2.16 |
| F6 | 72.28 ± 0.80 | 32.70 | 32.94 | 99.28 ± 1.05 |
Drug loading and entrapment efficiency study
The λmax value of EFV in the acetonitrile water mixture was found to be 247 nm. Drug loading and entrapment efficiency of the formulation batches were determined, and the drug loading values of the F1, F2, F3, F4, F5, and F6 batches were found to be 28.41%, 31.00%, 24.40%, 36.62%, 29.98%, and 32.70%, respectively. The entrapment efficiency of the F1, F2, F3, F4, F5, and F6 formulations was found to be 56.82%, 63.10%, 61.26%, 92.86%, 89.97% and 99.28%, respectively (Table 2). The encapsulation efficiency augmented as the amount of CHL incorporated into the liposomes increased.54 The formulation with the highest entrapment efficiency contained 100 mg of SLC and CHL individually.Particle size, PDI and zeta potential of liposomes
Particle size, PDI and zeta potential were measured with the particle size analyzer instrument using the software KALLIOPEPM, which used the principle of dynamic light scattering. Particle size and zeta potential of the experimental formulation batches (F1–F6) are given in Table 3.| Formulation code | Particle size (nm) ± SD | PDI (%) | Zeta potential (mV) ± SD |
|---|---|---|---|
| F1 | 333 ± 0.12 | 22.5 | −48.82 ± 1.21 |
| F2 | 342 ± 12.26 | 19.9 | −53.85 ± 0.21 |
| F3 | 408 ± 13.86 | 36.0 | −36.80 ± 0.90 |
| F4 | 425 ± 4.89 | 37.1 | −46.30 ± 1.00 |
| F5 | 411 ± 7.40 | 29.1 | −41.10 ± 1.00 |
| F6 | 437 ± 84.80 | 21.3 | −43.53 ± 0.86 |
The particle size of the liposomes was in a range of 300–440 nm. Fig. 4 shows the narrow size range of the experimentally developed liposomes for both F5 and F6. Here, we can see that with an increase in the amount of CHL, the particle size of the liposomal vesicles increased. F5 and F6 are considered optimal batches because the particle size of these batches is larger but in the range of standard size of liposomal vesicles (usually 50–500 nm in diameter).26 PDI values of all formulations were found to be below 38% or 0.38. A PDI value less than 0.3 is considered to be satisfactory and indicates a homogeneous distribution of phospholipid vesicles.55 The optimal formulations F5 and F6 had PDI values of 29.1 and 21.3, respectively.
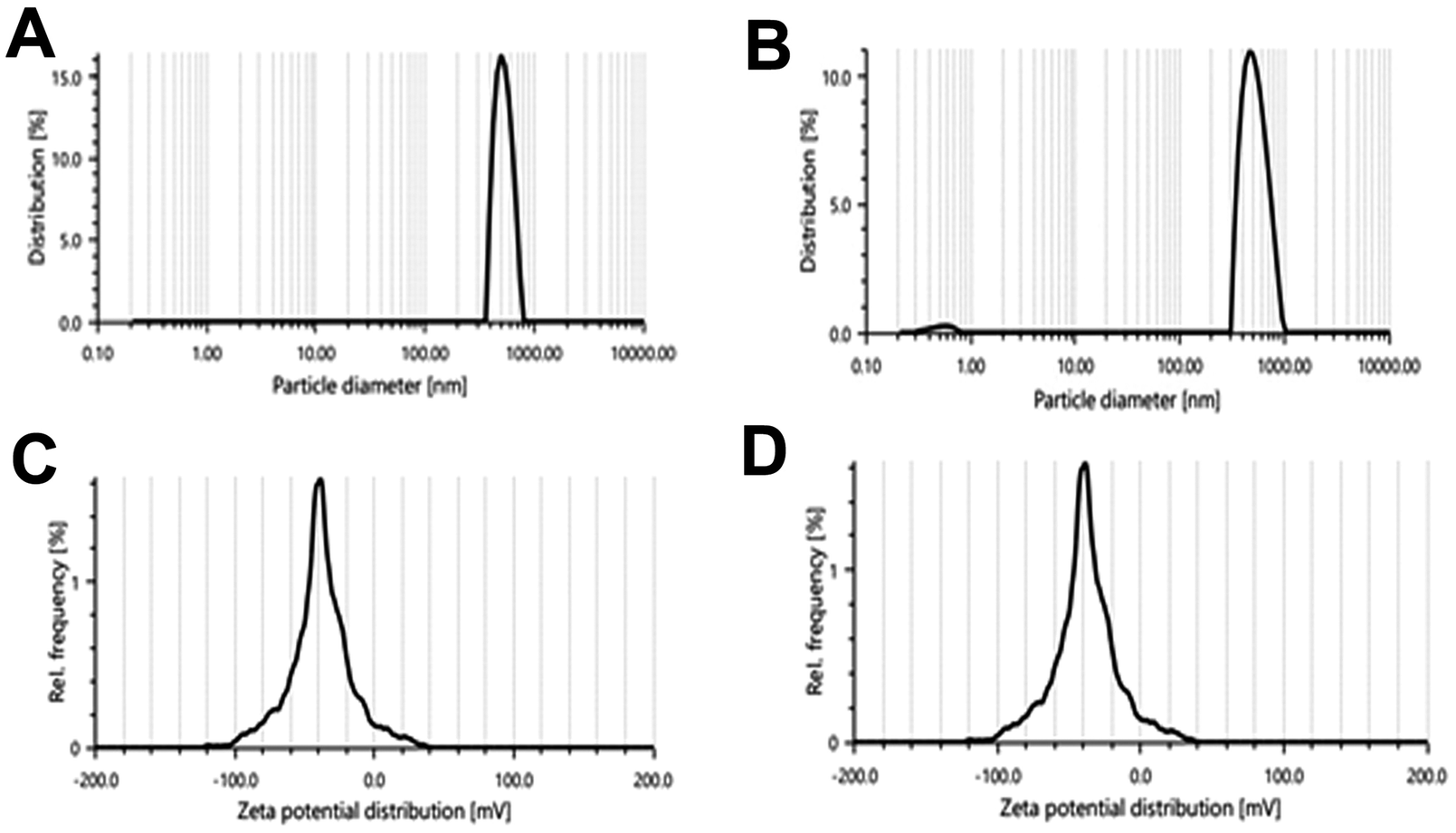 | ||
| Fig. 4 Particle size distribution of (A) the F5 batch and (B) F6 batch; zeta potential of (C) the F5 batch and (D) F6 batch. | ||
Depending on the composition, the zeta potential of the liposomes can be positive, neutral, or negative. It affects the physical stability (aggregation) and in vivo behavior of the formulation. Generally, the zeta potential of liposomes is negative due to the presence of carboxylic groups in lipids.36 A zeta potential less than −30 mV or greater than +30 mV is considered to be stable in colloidal form for a prolonged period and prevents settling down while in suspension.36,56 Here we can see that all formulations had zeta potential less than −30 mV (Fig. 4C and D), which means that the surface charge of the prepared liposomes was negative and the vesicles were stable in colloidal form.56 Based on the particle size, zeta potential, drug loading, and entrapment efficiency data, formulation batches F5 and F6 were found to be optimal batches and selected for further studies.
Study of surface morphology of liposomes by SEM
SEM images of the freshly prepared optimized formulations (F5 and F6) revealed that the prepared liposomes had a smooth surface and spherical shape and average sizes of the liposomes were found to be around 300–400 nm with a thick dense distribution. The SEM images of formulation batches F5 and F6 are given in Fig. 5.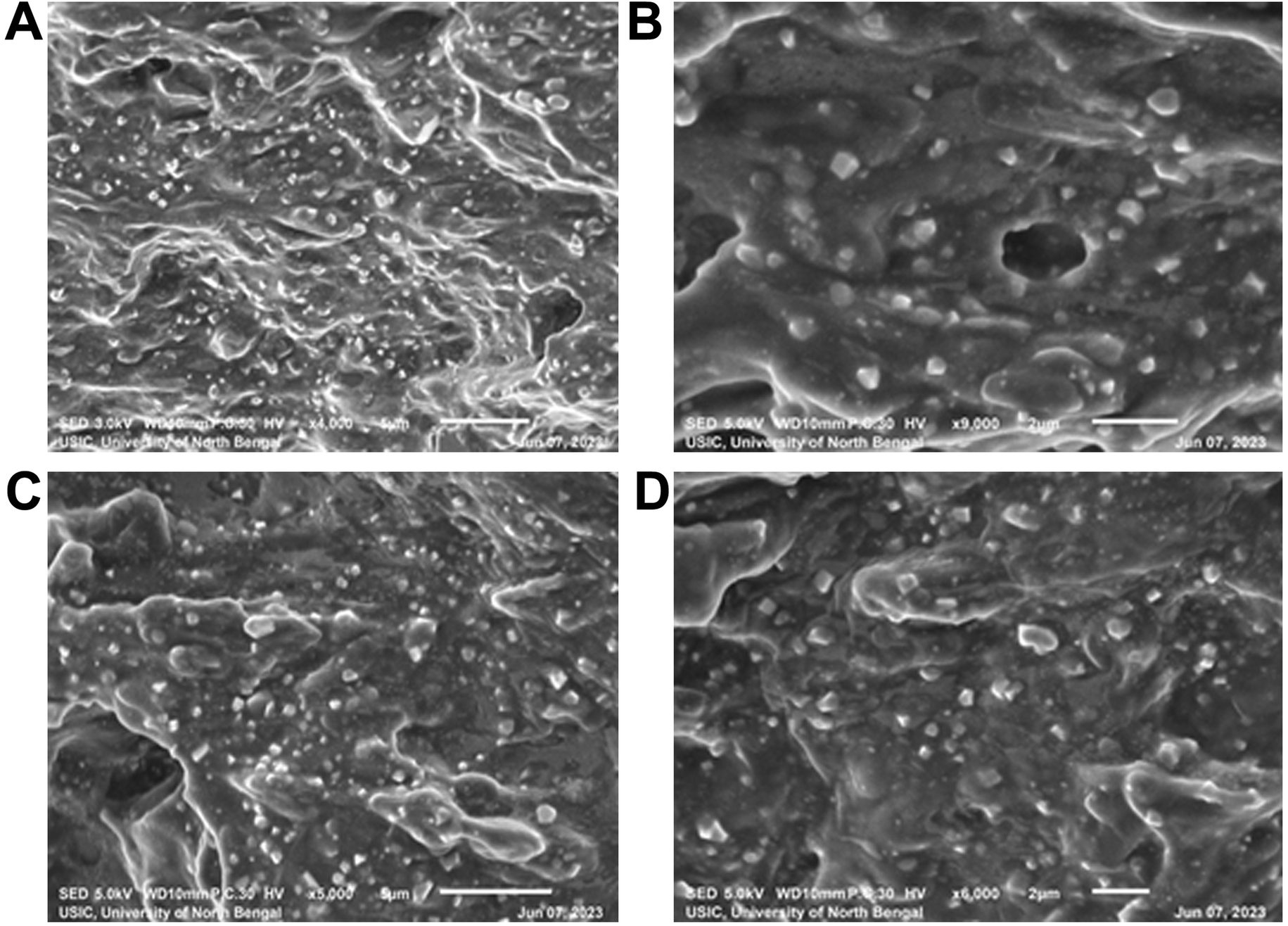 | ||
| Fig. 5 SEM images of pure EFV loaded liposomes (F5) measured at (A) 1700× and (B) 9000× magnification and EFV cocrystal-loaded liposomes (F6) measured at (C) 5000× and (D) 6000× magnification. | ||
The formulation F5 containing the pure drug (Fig. 5A and B) had distinct spherical particles with uniform sizes compared to other formulations, and most particles were below 500 nm. The formulation F6 containing EFV-cocrystals (Fig. 5C and D) showed spherical particles of the range 300–450 nm, obtained from visual estimation.
SEM images of the freshly prepared optimized formulations (F5 and F6) revealed more homogeneous distribution of the nanoliposomes, and the size of the liposomal vesicles was found to be within 400 nm, which is smaller than those detected in DLS. The difference in size may be due to the DLS method measuring the hydrodynamic diameter, and while preparing the sample in double distilled water, the liposomal formulation may swell, increasing the size.57
In vitro drug release study
On the basis of drug loading, particle size and SEM study, F5 and F6 formulation batches were found to be the best based on the parameters investigated and considered for the release study experiment. This study was performed in PBS pH 7.4 at 247 nm maximum wavelength. The linear graph had a regression coefficient value of 0.998 with a slope of 0.026. A distinct enhancement in EFV release was obtained in the in vitro drug release testing of plain EFV loaded liposomes as compared to liposomes containing EFV co-crystals as shown in Fig. 6. The increase in drug release can be attributed to the formation of hydrogen bonding between the –N–H and –CO functional groups of L-proline.19 Similar data were seen in liposomes prepared with EFV.50 The release data of EFV showed that the drug had limited solubility in its pure form and in the liposomal state, the release was modified in the presence of pluronic F127 in addition to carbopol in biofilm.58 In both batches, F5 and F6, the drug was released in two clear phases: the first phase with burst release of the drug and the 2nd phase with sustained drug release. The liposomes containing the pure drug showed burst release of the drug until 2 h and sustained release up to 168 h, while the liposomes containing co-crystals showed burst release until 1 h and sustained release up to 72 h. The drug release of liposomes containing the co-crystals of EFV (F6) was found to be 99.98% in 72 hours, whereas the drug release of liposomes containing plain EFV (F5) was found to be 72%. The presence of L-proline in association with EFV leads to an enhancement in the polarity of hydrophobic EFV, thus increasing its dissolution in dissolution media. Thus, the cumulative drug release is more in the case of F6 than the batch F5.
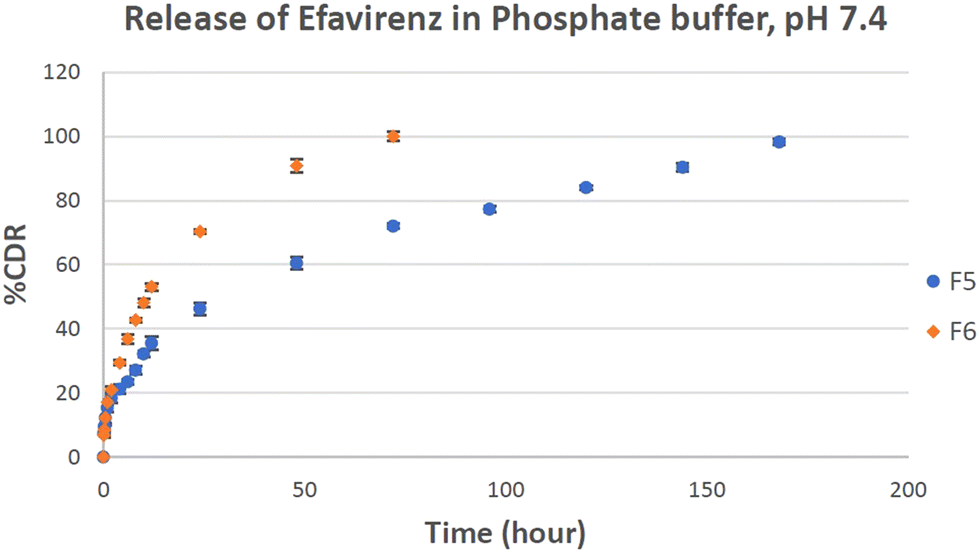 | ||
| Fig. 6 Percentage cumulative release of efavirenz in PBS (pH 7.4) medium from optimized F5 and F6 batches with respect to time (data represented as n = 3, mean ± SD). | ||
Release kinetics
The drug release data have been tested by plotting them in different kinetic models (zero order, first order, Higuchi model, Hixson–Crowell Model, and Korsmeyer–Peppas model), and the data are given in Table 4. Zero order and first order kinetics only depict whether the drug release is concentration independent or dependent. Zero order release kinetics of a drug is only a function of time. First-order reaction kinetics showed that the drug release mechanism is concentration dependent. Hence, the amount of drug release lessened with decreasing concentration gradient over time.59 The Higuchi kinetic model describes the drug release from a matrix type system. This model is followed when the matrix is swelled by the solvent and the concentration gradient becomes linear and is decreasing from the saturated concentration at the interface with the core.60 The Korsmeyer–Peppas kinetic model is used to describe drug release following non-Fickian mechanisms from a polymeric matrix. The mechanism depends on the Korsmeyer–Peppas release exponent (or “n” value). When n < 0.5, the drug diffuses by the Fickian diffusion mechanism. When n is equal to 0.89, the case II transport mechanism prevails and an n value > 0.89 describes the super case II transport mechanism. When the n value lies between 0.5 and 0.9, a non-Fickian release mechanism with anomalous diffusion is considered.59,61 The Hixson–Crowell cube root law specifically correlates the drug release rate from the systems made of polymers which erode over time from the exposed surface. This model depends on the change in surface area and diameter of the particles.62| Sample | Kinetic model | Equation | R 2 value |
|---|---|---|---|
| F5 batch | Zero order | y = 0.540x + 19.120 | R 2 = 0.900 |
| First order | y = −0.008x + 1.955 | R 2 = 0.928 | |
| Higuchi model | y = 7.203x + 7.052 | R 2 = 0.992 | |
| Korsmeyer–Peppas | y = 0.399x + 1.101 | R 2 = 0.993 | |
| Hixson–Crowell | y = −0.017x + 4.383 | R 2 = 0.975 | |
| F6 batch | Zero order | y = 1.338x + 20.398 | R 2 = 0.829 |
| First order | y = −0.021x + 1.947 | R 2 = 0.993 | |
| Higuchi model | y = 12.037x + 6.007 | R 2 = 0.984 | |
| Korsmeyer–Peppas | y = 0.454x + 1.214 | R 2 = 0.994 | |
| Hixson–Crowell | y = −0.056x + 4.428 | R 2 = 0.985 |
The maximum linearity (as assumed by R2 values) was observed for the Korsmeyer Peppas model (Table 4). For the F5 batch, the regression coefficient (R2 value) is 0.993, and for the F6 batch, it is 0.994. The release exponent (n) is used to characterize the release mechanism of the drug. For both batches (F5 and F6), the n value is less than 0.5, which indicates the Fickian release mechanism (Fig. S1 and S2 of ESI†).
Stability studies
The stability of selected batches F5 and F6 was assessed over a period of 3 months. No significant color changes were observed in the liposomes. The mean values of particle size, zeta potential, and drug loading of fresh formulations were compared to those of formulations stored for 1 month and 3 months (Table 5). The particle size and zeta potential remained almost constant over the storage period. The zeta potential results indicated the good stability of the experimental formulations as only a negative charge can prevent the emulsion from cracking and phase separation. The percentage drug loading also remained almost constant after 3 months, indicating the stability of the stored formula. The unchanged physical appearance, size, and zeta potential of the liposome vesicles are supported indicators of kinetically stable formulation redispersed in water in suspended form.| Time period | Particle size (nm) | Zeta potential (mV) | %Drug loading | |
|---|---|---|---|---|
| F5 batch | Fresh formulation | 411.10 ± 7.40 | −41.1 ± 1.0 | 29.98 ± 0.31 |
| After 1 month | 415.23 ± 0.7 | −39.9 ± 0.7 | 29.97 ± 0.40 | |
| After 3 months | 413.11 ± 1.0 | −38.0 ± 0.4 | 29.97 ± 0.83 | |
| F6 batch | Fresh formulation | 437.4 ± 84.80 | −43.53 ± 0.86 | 32.72 ± 0.30 |
| After 1 month | 441.3 ± 22.4 | −40.43 ± 2.53 | 32.70 ± 0.48 | |
| After 3 months | 440.42 ± 38.1 | −30.79 ± 30 | 32.71 ± 1.14 |
Conclusion
In the present study, liposomes encapsulating EFV co-crystals were prepared using L-proline as a coformer, particularly focusing on the anti-HIV action of the drug. The drug EFV belongs to BCS class II, having poor water solubility and high permeability. Due to the solubility problem, the bioavailability of the drug is very less. Here, we developed co-crystals to increase the solubility up to 3 fold and co-crystal entrapped liposomes to release the drug in a sustained manner. The formation of cocrystals of EFV was confirmed by XRD and DSC analysis. The optimized formulation was found to have a substantial amount of EFV loading (32.70%) and entrapment efficiency (99.28%) with a standard range of size distribution. The drug release study was performed for the pure drug containing liposomes and co-crystal containing liposomes, and both formulations were released in two clear phases: the first phase with burst release and the 2nd phase with sustained release. The liposomes containing the pure drug showed 72% release of the drug in 72 h, while the liposomes containing co-crystals showed 99.98% release of the drug in 72 h. This is due to the presence of L-proline in association with EFV, which leads to an enhancement in the polarity of hydrophobic EFV, thus increasing its dissolution in dissolution media. The sustained release minimizes the frequency of dosing. Other studies, including zeta potential, SEM, and FTIR studies, were done to confirm the stability of the formulations, spherical shape of liposomes, and compatibility of the components, respectively. To date, co-crystal-loaded liposomes have not been reported, and this is the first time we are following this approach to deliver EFV for antiretroviral therapy with much better therapeutic efficacy in markedly reduced doses.Data availability
The study's original contributions are accessible in the article, and additional inquiries can be directed to the corresponding author.Conflicts of interest
The authors declare to have no financial or non-financial conflicts of interest.Acknowledgements
This work reports no funding. The authors sincerely acknowledged the indebted support, e-resources, and infrastructural facilities provided by the University of North Bengal, Darjeeling. The authors further acknowledge Central instrumentation facility of BIT Mesra, Ranchi for performing DSC analysis.References
- E. Ojewole, I. Mackraj, P. Naidoo and T. Govender, Eur. J. Pharm. Biopharm., 2008, 70, 697–710 CrossRef CAS.
- T. Melis, T. Sahle, K. Haile, A. Timerga, A. Zewdie, Y. Wegu, K. Zepire and J. Bedewi, J.Pharm. Policy Pract., 2024, 17, 2290672 CrossRef PubMed.
- T. C. Quinn, M. J. Wawer, N. Sewankambo, D. Serwadda, C. Li, F. Wabwire-Mangen, M. O. Meehan, T. Lutalo and R. H. Gray, N. Engl. J. Med., 2000, 342, 921–929 CrossRef CAS PubMed.
- J. M. Baeten, E. Kahle, J. R. Lingappa, R. W. Coombs, S. Delany-Moretlwe, E. Nakku-Joloba, N. R. Mugo, A. Wald, L. Corey and D. Donnell, Sci. Transl. Med., 2011, 3, 77ra29–77ra29 Search PubMed.
- J.-A. Røttingen, D. W. Cameron and G. P. Garnett, Sex. Transm. Dis., 2001, 579–597 CrossRef.
- Q. Le Hingrat, I. Sereti, A. L. Landay, I. Pandrea and C. Apetrei, Front. Immunol., 2021, 12, 695674 CrossRef CAS.
- F. E. T. Foka and H. T. Mufhandu, Viruses, 2023, 15, 1732 CrossRef CAS PubMed.
- M. Ullah Nayan, B. Sillman, M. Hasan, S. Deodhar, S. Das, A. Sultana, N. Thai Hoang Le, V. Soriano, B. Edagwa and H. E. Gendelman, Adv. Drug Delivery Rev., 2023, 200, 115009 CrossRef CAS.
- S. SeyedAlinaghi, A. M. Afsahi, A. Moradi, Z. Parmoon, P. Habibi, P. Mirzapour, M. Dashti, A. Ghasemzadeh, E. Karimi, F. Sanaati, Z. Hamedi, A. Molla, E. Mehraeen and O. Dadras, AIDS Res. Ther., 2023, 20, 74 CrossRef PubMed.
- B. Yavuz, J. L. Morgan, L. Showalter, K. R. Horng, S. Dandekar, C. Herrera, P. LiWang and D. L. Kaplan, Adv. Ther., 2018, 1, 1800054 CrossRef.
- M. H. Dinh, M. R. Anderson, M. D. McRaven, G. C. Cianci, S. G. McCoombe, Z. Kelley, C. J. Gioia, A. J. Fought, A. W. Rademaker and R. S. Veazey, PLoS Pathog., 2015, 11, e1004729 CrossRef PubMed.
- P. K. Gaur, S. Mishra, M. Bajpai and A. Mishra, BioMed Res. Int., 2014, 2014, 363404 Search PubMed.
- S. Velmurugan, M. A. Ali and P. Kumar, Int. J. Pharm. Pharm. Sci., 2014, 6, 31–39 CAS.
- A. Carr and D. A. Cooper, Lancet, 2000, 356, 1423–1430 CrossRef CAS.
- L. N. Ramana, A. R. Anand, S. Sethuraman and U. M. Krishnan, J. Controlled Release, 2014, 192, 271–283 CrossRef CAS PubMed.
- M. Guo, X. Sun, J. Chen and T. Cai, Acta Pharm. Sin. B, 2021, 11, 2537–2564 CrossRef CAS.
- B. J. Gowda, S. K. Nechipadappu, S. Shankar, M. Chavali, K. Paul, M. G. Ahmed, A. Sanjana and H. Shanthala, Mater. Today: Proc., 2022, 51, 394–402 CAS.
- B. J. Gowda, M. G. Ahmed, S. Shankar, K. Paul, R. Chandan, A. Sanjana, S. Narayana, A. Nasrine, N. Noushida and M. Thriveni, Mater. Today: Proc., 2022, 57, 878–886 CAS.
- P. M. Szell, J. R. Lewandowski, H. Blade, L. P. Hughes, S. O. N. Lill and S. P. Brown, CrystEngComm, 2021, 23, 6859–6870 RSC.
- I. Nugrahani, D. Utami, S. Ibrahim, Y. P. Nugraha and H. Uekusa, Eur. J. Pharm. Sci., 2018, 117, 168–176 CrossRef CAS PubMed.
- P. Prajapati, J. Pandey, P. Tandon, K. Sinha and M. R. Shimpi, Front. Chem., 2022, 10, 848014 CrossRef CAS.
- E. Parhizkar, M. Rashedinia, M. Karimi and S. Alipour, J. Microencapsulation, 2018, 35, 301–311 CrossRef CAS PubMed.
- V. Kenchappa, R. Cao, V. Venketaraman and G. V. Betageri, Appl. Sci., 2022, 12, 1468 CrossRef CAS.
- A. R. Mohammed, N. Weston, A. G. A. Coombes, M. Fitzgerald and Y. Perrie, Int. J. Pharm., 2004, 285, 23–34 CrossRef CAS PubMed.
- H. Liang, F. Zou, Q. Liu, B. Wang, L. Fu, X. Liang, J. Liu and Q. Liu, Int. J. Pharm., 2021, 599, 120418 CrossRef CAS PubMed.
- H. Nsairat, D. Khater, U. Sayed, F. Odeh, A. Al Bawab and W. Alshaer, Heliyon, 2022, 8, e09394 CrossRef CAS PubMed.
- M. D. Fulton and W. Najahi-Missaoui, Int. J. Mol. Sci., 2023, 24, 6615 CrossRef CAS PubMed.
- J. Douda, L. G. Miranda Calderón, T. Kryshtab, J. S. Arias Cerón and A. Kryvko, J. Mater. Sci.: Mater. Electron., 2018, 29, 15570–15578 CrossRef CAS.
- B. Han, Y. Yang, J. Chen, H. Tang, Y. Sun, Z. Zhang, Z. Wang, Y. Li, Y. Li and X. Luan, Int. J. Nanomed., 2020, 553–571 CrossRef CAS.
- F. H. Blindheim and J. Ruwoldt, Polymers, 2023, 15, 2901 CrossRef CAS.
- V. Rajurkar, N. Sunil and V. Ghawate, Med. Chem., 2015, 2, 2161–2444 Search PubMed.
- T. A. Ahmed, A. O. Bawazir, W. S. Alharbi and M. K. Safo, Int. J. Nanomed., 2020, 15, 4001–4020 CrossRef CAS.
- L. Dutta, B. Mukherjee, T. Chakraborty, M. K. Das, L. Mondal, S. Bhattacharya, R. H. Gaonkar and M. C. Debnath, Drug Delivery, 2018, 25, 504–516 CrossRef CAS.
- A. Mallick, R. Sahu, G. Nandi, T. K. Dua, T. K. Shaw, A. Dhar, A. Kanu and P. Paul, J. Pharm. Innovation, 2023, 18, 1020–1029 CrossRef.
- S. Sengupta, P. Paul, B. Mukherjee, R. H. Gaonkar, M. C. Debnath, R. Chakraborty, N. Khatun and S. Roy, Nanomedicine, 2018, 13, 3009–3023 CrossRef CAS PubMed.
- S. Satapathy and A. Naik, Complex Intell. Syst., 2016, 2, 173–203 CrossRef.
- N. S. Dey, B. Mukherjee, R. Maji and B. S. Satapathy, Curr. Cancer Drug Targets, 2016, 16, 357–372 CrossRef CAS.
- D. E. Large, R. G. Abdelmessih, E. A. Fink and D. T. Auguste, Adv. Drug Delivery Rev., 2021, 176, 113851 CrossRef CAS PubMed.
- F. Weber, L. Rahnfeld and P. Luciani, Talanta, 2020, 220, 121320 CrossRef CAS.
- P. Paul, S. Sengupta, B. Mukherjee, T. K. Shaw, R. H. Gaonkar and M. C. Debnath, Nanomedicine, 2018, 13, 501–520 CrossRef CAS PubMed.
- S. Kakad and S. Kshirsagar, Heliyon, 2021, 7, e08368 CrossRef CAS.
- P. K. Gaur, S. Mishra, M. Bajpai and A. Mishra, BioMed Res. Int., 2014, 2014, 363404 Search PubMed.
- R. Kumar and V. Sinha, AAPS PharmSciTech, 2017, 18, 884–894 CrossRef CAS PubMed.
- C. Cheng, Z. Wu, D. J. McClements, L. Zou, S. Peng, W. Zhou and W. Liu, Colloids Surf., B, 2019, 183, 110460 CrossRef CAS PubMed.
- S. Das, A. Samanta, S. Mondal, D. Roy and A. K. Nayak, Sens. Int., 2021, 2, 100077 CrossRef.
- K. Karami, N. Jamshidian, A. Hajiaghasi and Z. Amirghofran, New J. Chem., 2020, 44, 4394–4405 RSC.
- S. Ugwu, A. Zhang, M. Parmar, B. Miller, T. Sardone, V. Peikov and I. Ahmad, Drug Dev. Ind. Pharm., 2005, 31, 223–229 CrossRef CAS PubMed.
- I. Nugrahani, R. A. Kumalasari, W. N. Auli, A. Horikawa and H. Uekusa, Pharmaceutics, 2020, 12, 690 CrossRef CAS PubMed.
- A. M. Siddiquee, A. Houri, K. A. Messalea, J. Lin, T. Daeneke, B. Abbey, A. Mechler and S. Kou, J. Phys. Chem. Lett., 2020, 11, 9476–9484 CrossRef CAS PubMed.
- N. I. Okafor, C. I. Nkanga, R. B. Walker, X. S. Noundou and R. W. M. Krause, J. Pharm. Invest., 2020, 50, 201–208 CrossRef CAS.
- A. Neacsu, D. Gheorghe, C. Marinescu, E. Stancu, V. Tecuceanu and C. Ciuculescu, Radiat. Phys. Chem., 2019, 156, 115–127 CrossRef.
- M. K. Das and B. Kalita, J. Appl. Pharm. Sci., 2014, 4, 051–057 CrossRef.
- J. Zhang, Q. Tang, X. Xu and N. Li, Int. J. Pharm., 2013, 448, 168–174 CrossRef CAS PubMed.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
- K. Ogurtsova, J. da Rocha Fernandes, Y. Huang, U. Linnenkamp, L. Guariguata, N. H. Cho, D. Cavan, J. Shaw and L. Makaroff, Diabetes Res. Clin. Pract., 2017, 128, 40–50 CrossRef CAS.
- M. R. Rao and L. S. Babrekar, Indian J. Pharm. Sci., 2018, 80, 1115–1124 CAS.
- N. I. Okafor, M. Ngoepe, X. S. Noundou and R. W. Maçedo Krause, J. Drug Delivery Sci. Technol., 2019, 54, 101312 CrossRef CAS.
- F. Shafiei, M. Ghavami-Lahiji, T. S. Jafarzadeh Kashi and F. Najafi, Dent. Res. J., 2021, 18, 94 CrossRef PubMed.
- C. Mircioiu, V. Voicu, V. Anuta, A. Tudose, C. Celia, D. Paolino, M. Fresta, R. Sandulovici and I. Mircioiu, Pharmaceutics, 2019, 11, 140 CrossRef CAS.
- N. S. Heredia, K. Vizuete, M. Flores-Calero, V. K. Pazmiño, F. Pilaquinga, B. Kumar and A. Debut, PLoS One, 2022, 17, e0264825 CrossRef CAS PubMed.
- M. A. Malana and R. Zohra, Daru, J. Pharm. Sci., 2013, 21, 10 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00215f |
| This journal is © The Royal Society of Chemistry 2025 |