
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of polymer-coated, drug-eluting self-expandable metal stents for the potential treatment of gastrointestinal cancers†
Mohammad
Arafat
 a,
Anthony
Wignall
b,
Kyle
Brewer
c,
Yunmei
Song
a,
Hugo
Albrecht
d,
Clive A.
Prestidge
b,
Sanjay
Garg
*a and
Anton
Blencowe
*c
a,
Anthony
Wignall
b,
Kyle
Brewer
c,
Yunmei
Song
a,
Hugo
Albrecht
d,
Clive A.
Prestidge
b,
Sanjay
Garg
*a and
Anton
Blencowe
*c
aPharmaceutical Innovation and Development Group (PIDG), Centre for Pharmaceutical Innovation (CPI), UniSA Clinical and Health Sciences, University of South Australia, Adelaide, SA 5000, Australia. E-mail: sanjay.garg@unisa.edu.au
bNanostructure and Drug Delivery Group, Centre for Pharmaceutical Innovation (CPI), UniSA Clinical and Health Sciences, University of South Australia, Adelaide, SA 5000, Australia
cApplied Chemistry and Translational Biomaterials (ACTB) Group, Centre for Pharmaceutical Innovation (CPI), UniSA Clinical and Health Sciences, University of South Australia, Adelaide, SA 5000, Australia. E-mail: anton.blencowe@unisa.edu.au
dCentre for Pharmaceutical Innovation (CPI), UniSA Clinical and Health Sciences, University of South Australia, Adelaide, SA 5000, Australia
First published on 19th February 2025
Abstract
Self-expandable metal stents (SEMS) represent the gold standard for the clinical management of malignant obstructions in the gastrointestinal tract. Gastrointestinal stent blockage (restenosis) caused by tumour growth is a common problem. The incorporation of anticancer drugs into SEMS for localised delivery could potentially address restenosis, although further studies are required to better understand the influence of the stent structure in combination with different drug-eluting polymer formulations and chemotherapeutics. Therefore, in this work, we investigated for the first time the suitability of a polyurethane-silicone (PUS) elastomer for the controlled encapsulation and release of 5-fluorouracil (5FU) from membrane-covered oesophageal stents (OS) and bare enteral colonic stents (CS). The stents were coated with a bilayer structure consisting of a 5FU-loaded (7.0% w/w) PUS basecoat and poly(ethylene-co-vinyl acetate) (PEVA) diffusion regulating topcoat. Physicochemical characterisation of the coatings revealed that 5FU is uniformly distributed and semi-crystalline in the PUS layer, and that 5FU did not leach into the topcoat during coating. Interestingly, drug release from the coated stents revealed a significant difference, with 5FU release from CS plateauing after ∼12 d, while a much more gradual release was observed with the OS over 150 d. Imaging revealed that defects in the coatings due to the underlying stent structure are likely contributors to these differences. The coated stents were found to be stable to gamma sterilisation and in accelerated stability tests. In vitro cytotoxicity, cell cycle and apoptosis assays revealed that 5FU released from the stents had comparable anticancer efficacy to free 5FU against human colon carcinoma cells. This research demonstrates the potential of polymer-coated SEMS for controlled drug-release and highlights the importance of the underlying stent structure on performance.
Introduction
Gastrointestinal cancers are the most common form of malignancy and currently account for 26% of the world's cancer incidence and 33% of all cancer-related deaths.1 Gastrointestinal cancers frequently cause obstructions (partial or complete) in the oesophageal region, biliary tract, duodenum, and colon. Obstruction in the gastrointestinal tract (GIT) is a serious and life-threatening complication, and requires emergency surgical intervention to restore or improve the luminal patency of the GIT, restoring the normal digestive functions.2,3 In this clinical setting, gastrointestinal SEMS are placed as a nonsurgical alternative in obstructive gastrointestinal cancer patients, either as a bridge to elective one-stage surgery in patients with potentially curable malignant gastrointestinal strictures or with palliative intent for inoperable malignant gastrointestinal strictures.2 In both scenarios, gastrointestinal stenting has been reported with high technical and clinical success, and SEMS currently represent the gold standard treatment for the local (non-surgical) management and palliation of obstructing gastrointestinal cancers.2,4 Despite a proven record of clinical safety and effectiveness, conventional gastrointestinal SEMS work as simple endoluminal scaffolds that only provide mechanical palliation of obstructions, and are therefore prone to in-stent restenosis due to malignant tumour growth.5,6 This limitation can result in a significant decrease in the working duration of gastrointestinal stents, and necessitate emergency surgical interventions and/or further placement of a second stent.2To address this limitation, there has been growing interest in drug-eluting gastrointestinal stents,7–10 following the remarkable success (both technical and clinical) of drug-stent combinations in the field of vascular coronary disease. Thus, gastrointestinal stents (e.g., oesophageal, biliary, or colorectal) have been studied in conjunction with contemporary anticancer drugs to serve both as typical gastrointestinal stenting devices and as drug reservoirs in the gastrointestinal tract.2 These functional gastrointestinal stents are intended to deliver anticancer drugs locally at the site of stent placement in a controlled manner to maximise the drug distribution and bioavailability within local intestinal cancerous tissue, and minimise potential systemic and non-target organ toxicities. Chemotherapeutics released from drug-stent combinations over a prolonged period of time (weeks to several months) are more likely to result in maintained high cytotoxic drug concentrations locally within the surrounding tumour microenvironments. Therefore, drug-eluting gastrointestinal stents could inhibit the proliferation of tumour cells covering the stent more potently than the standard administration of the anticancer drug and a non-drug-eluting stent.8,9
Drug-eluting stents are typically composed of three basic components: the active pharmaceutical ingredient, a polymeric coating and the metallic stent platform. A variety of anticancer drugs, including 5FU, oxaliplatin, irinotecan, capecitabine, and monoclonal antibodies, are used for the clinical treatment of gastrointestinal cancers.11 5FU is one of the most potent and broadly effective chemotherapeutics, however, due to its very low oral absorption and first-pass clearance, intravenous administration is currently used clinically, which is known to cause severe side effects. Furthermore, 5FU is metabolised quickly resulting in a very short circulation half-life (10–20 min) and low concentrations at tumour sites.8,11 Thus, 5FU is considered an excellent chemotherapeutic candidate for local stent-based delivery with reduced systemic side effects.4,6–8 For example, in porcine models, Wang et al.12 and Liu et al.10 have demonstrated that 5FU-loaded oesophageal stents result in significantly higher 5FU concentrations in the oesophageal tissue (cf. other organs) without any obvious signs of local tissue damage. In addition, Li et al. have shown that the release of 5FU from stents reduced in-stent restenosis and provided superior anti-tumour activity in a mouse model.6
As acknowledged by the United States Food and Drug Administration,13 the development of drug-eluting stents is a complex process and presents numerous safety and efficacy challenges, many of which are linked to the underlying stent platform, drug–polymer coatings, and the manufacturing approach. While previous preclinical/clinical studies support the potential benefits of local stent-based 5FU delivery with reduced systemic toxicities for the treatment of gastrointestinal malignancies, little consideration has been given to address the critical quality attributes of novel gastrointestinal drug-eluting stents. Importantly, a comprehensive evaluation of the coating process and/or polymer coating system, and their applicability to different stent formats, are crucial for the development of drug-eluting stents from both a product quality and regulatory perspective.14
Therefore, in this study, we comprehensively evaluated a novel 5FU-polymer coating formulation across two different clinical gastrointestinal SEMS formats, including membrane-covered OS with a fixed braided cell structure and bare enteral CS with a flexible woven cell structure. The successful performance of drug-eluting stents largely depends on the delivery of an effective drug dose with controlled kinetics. To create a tailored platform with controlled drug release kinetics, the drug-carrying polymer must possess adequate capacity to encapsulate the required amount of drug and deliver the drug with an effective dose regime. While biodurable (non-erodible) polymers such as polyurethanes (PUs), PEVA, and polysiloxanes have been commonly used in pharmaceutical applications, PUS elastomers (e.g., ChronoSil AL) are yet to be assessed for encapsulation and release of hydrophilic drugs (e.g., 5FU) from stents. Nevertheless, the high aqueous solubility of 5FU can lead to significant burst release, and therefore, the PUS reservoir was used in combination with PEVA to slow the drug diffusion and release. As the 5FU-loaded stents are intended primarily for treating obstructions in two different parts of the gastrointestinal tract (oesophagus and colon), this study represents a novel aspect of drug-eluting stent design, fabrication and assessment that is more broadly applicable to other types of gastrointestinal stents.
Results and discussion
Fabrication and characterisation of drug-loaded stents
Drug-loaded stents were fabricated by sequential dip-coating of commercial SEMS to deposit a 5FU-loaded (7.0% w/w) PUS basecoat (PUSFU) followed by a PEVA topcoat. The PEVA topcoat was included to control the diffusion and release of 5FU from the underlying PUS reservoir.15,16 Interestingly, the use of PUS allowed for a slight improvement in the loading of 5FU as compared to other related polyurethanes (ChronoFlex AL),4 possibly because of the improved flexibility of the polymer matrix due to the inclusion of silicone segments. Membrane-covered OS with a fixed braided cell structure and bare enteral CS with an unfixed woven cell structure were compared (Fig. 1). Initially, PUSFU-OS and -CS were prepared and presented with excellent coating uniformity and reproducibility, with the average deposition of the PUSFU coating being 90.9 ± 10.0 and 28.2 ± 1.9 μg mm−2 (n = 6), respectively (ESI, Fig. S1 and 2†). The significantly lower amount of PUSFU deposited on PUSFU-CS resulted from their open-cell structure and lack of a supporting membrane. It should be noted that due to the membrane, the PUSFU coatings were deposited across both the abluminal and luminal sides of PUSFU-OS. Subsequently, the PEVA topcoat was applied on both the abluminal and luminal surfaces resulting in PEVA-PUSFU-OS and -CS with deposited PEVA coatings of 110.7 ± 9.2 and 89.4 ± 8.1 μg mm−2 (n = 6), respectively. To allow detailed characterisation, the coatings were also prepared on planar substrates using identical dip-coating parameters. | ||
| Fig. 1 (A) Membrane-covered OS with fixed braided cell structure and its (B) PUSFU and (C) PEVA-PUSFU coated derivatives. (D) Bare CS with unfixed woven cell structure and its (E) PUSFU and (F) PEVA-PUSFU coated derivatives. | ||
To assess the uniformity of the coatings and the incorporation of 5FU, Fourier transform infrared photoacoustic (FTIR-PA) spectroscopy was conducted on drug-loaded and drug-free polymer coatings, and then compared to pure 5FU (Fig. 2A). Comparison of the spectra between the PUSFU coating and pure PUS revealed very few distinctive spectral features resulting from 5FU due its relatively low concentration, with the exception of peak broadening at 1743 and 3110–3280 cm−1. Following coating with PEVA, all peaks from the underlying PUS and 5FU were absent from PEVA-PUSFU indicating the formation of a homogeneous PEVA topcoat with no leaching or dissolution of the PUS basecoat or 5FU during the second dip-coating cycle. These results were further confirmed by X-ray photoelectron spectroscopy (XPS) (Fig. 2B), which revealed the presence of characteristic structural elements (e.g., F 1s and Si 2s/2p peaks) from both PUS and 5FU in the PUSFU coating. In comparison, spectra of the PEVA-PUSFU coatings were identical to PEVA, corroborating the FTIR-PA results.
 | ||
| Fig. 2 (A) Fourier transform infrared photoacoustic spectra of pure 5FU and polymer coatings as indicated; the dashed boxes highlight peak broadening resulting from the inclusion of 5FU within the PUS matrix. (B) X-ray photoelectron spectroscopy survey spectra recorded for pure 5FU and polymer coatings as indicated. | ||
X-ray diffraction (XRD) was used to investigate the crystallinity of 5FU within the PUS basecoat (Fig. 3). The diffractogram of pure 5FU powder displayed a series of characteristic 2θ peaks consistent with previous literature reports17 with an intense peak at ∼28.6° corresponding to an interlayer distance (d-spacing) of 3.123 Å (ESI, Table S1†).4 The diffractogram obtained for the PUS coating revealed a semi-crystalline/amorphous structure with a broad peak centered at a 2θ of 19.7° and a low intensity but sharp peak at a 2θ of 29.1°, corresponding to the polycarbonate segments of PUS.18 In comparison, the diffractogram of the PUSFU coating revealed peaks characteristic of PUS, and a sharp peak at a 2θ of 28.4° (d-spacing = 3.145 Å) consistent with 5FU (ESI, Table S1†). These results indicate that a proportion of 5FU within the PUS matrix is in a crystalline state, which may result from the more hydrophobic silicone segments influencing the microphase separation and crystallisation of 5FU during solvent evaporation.
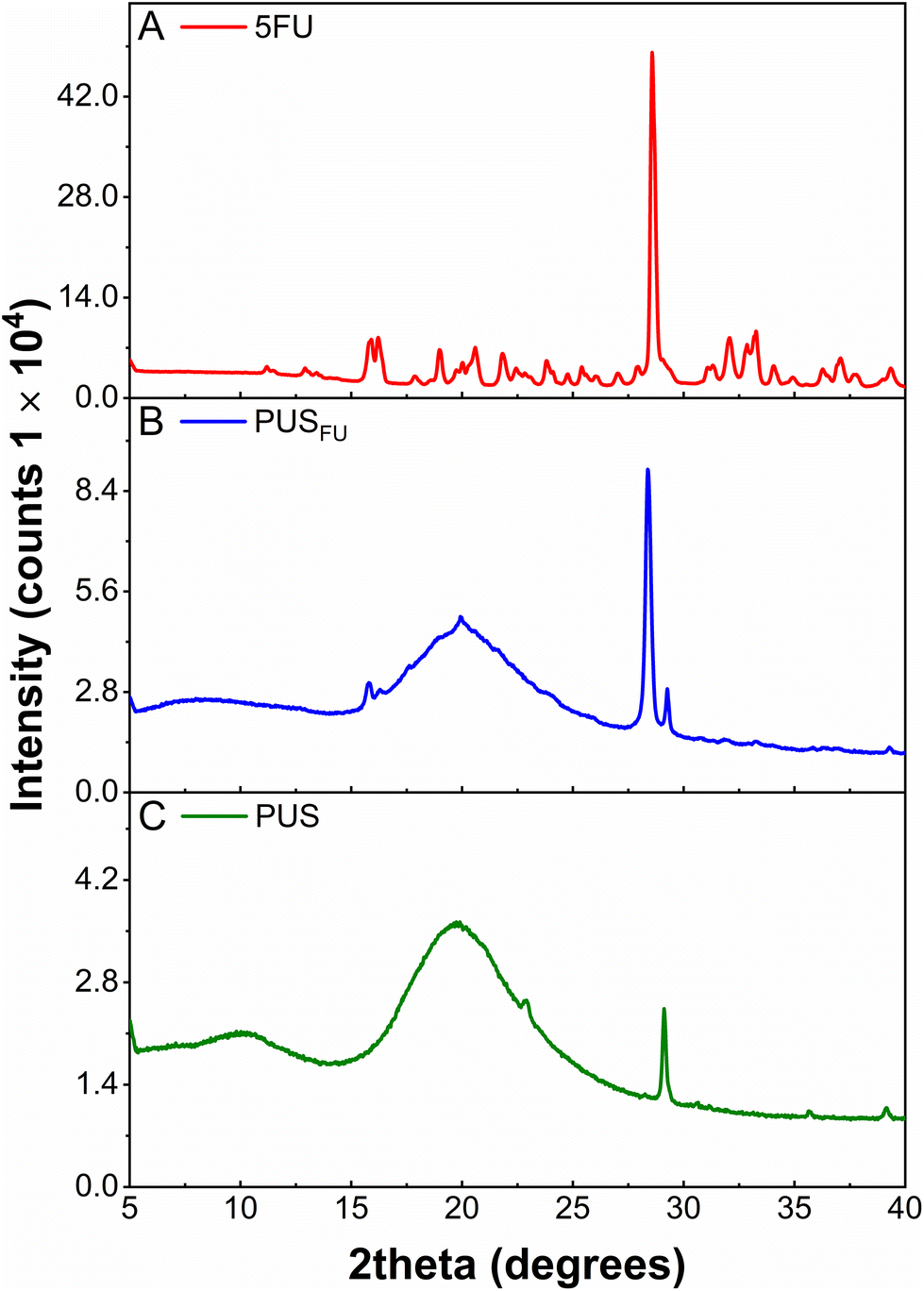 | ||
| Fig. 3 X-ray diffractograms of (A) pure 5FU, and (B) PUSFU and (C) PUS coatings. | ||
Modulated differential scanning calorimetry (MDSC) was used to determine any change in the thermal properties of the PUS upon incorporation of 5FU, as well as the existence of drug crystallites in the PUS matrix (Fig. 4A). The thermogram of pure 5FU revealed a sharp endothermic peak with a maximum at 283 °C, corresponding to its melting temperature (Tm),17 and an underlying endothermic feature from ∼250 °C upwards consistent with degradation. The thermogram of the PUS coating exhibited two melting peaks at ∼67 and 109 °C that correspond to soft and hard segments, respectively, as reported previously.19 In contrast, the thermogram of the PUSFU coating revealed two Tm peaks at ∼67 and 114 °C corresponding to the soft and hard PUS segments, respectively, and a broad endothermic peak between ∼250 and 280 °C ascribed to the simultaneous melting and degradation of the crystallites of 5FU in the PUS matrix. These results corroborate the XRD results (Fig. 3), indicating the presence of crystalline 5FU, but suggest that the crystallites are less well ordered and likely exist in combination with amorphous 5FU within the PUS matrix.
 | ||
| Fig. 4 (A) Modulated differential scanning calorimetry thermograms recorded at a heating rate of 2 °C min−1 and (B) derivative thermogravimetric curves recorded at a heating rate of 10 °C min−1 for pure 5FU, and PUSFU and PUS coatings. | ||
The thermal stability of 5FU within the PUS matrix was assessed by thermogravimetric analysis (TGA) (ESI, Fig. S3†), and the results were plotted as the corresponding derivative thermogravimetric (DTG) curves (Fig. 4B). The results for pure 5FU revealed a peak maximum at 294 °C, consistent with the previously reported degradation temperature (Td).20 The PUS coating displayed an onset degradation temperature of ∼270 °C and a peak maximum of 331 °C.4,18 In comparison, the PUSFU coating displayed a moderate weight loss from 206 to 294 °C, followed by complete decomposition of the PUS matrix with a peak maximum of 340 °C.4,21 Considering the slight degradation observed from the PUS matrix from ∼270 °C onwards, it was estimated from the DTG curves that the weight loss due to 5FU is ∼7.0%, which is consistent with the theoretical 5FU loading.
Stent coating surface topography and thickness
The polymer coating thickness needs to be well controlled to avoid stent-related complications and differences in performance between individual stents, as well as to allow for easy crimping and release of stents from their delivery system catheters.2,22 Therefore, scanning electron microscopy (SEM) was employed for topographical and cross-sectional imaging of the stents, allowing the thickness of each dip-coated layer to be determined (Fig. 5 and ESI, Fig. S4–6†). Prior to coating, the commercial OS had a smooth silicone membrane layer with a mean cross-sectional thickness of 15.7 ± 1.5 μm (Fig. 5A and ESI, Fig. S4†). | ||
| Fig. 5 Representative abluminal top-view (left (i); insets show high magnifications) and cross-section (right (ii)) SEM images of surface morphologies of the (A) silicone membrane-covered OS and (B) PUSFU-OS and (C) PEVA-PUSFU-OS. The arrows in B(i) indicate 5FU crystallites in the PUS matrix. Scale bars for A–C(i) = 500 μm, A–C(i) inset = 10 μm; and for A(ii), B(ii) and C(ii) are 20, 100 and 500 μm, respectively. Cross-sectional thickness values were determined from n = 50 individual measurements. | ||
After application of the PUSFU basecoat, the PUSFU-OS showed a textured and porous surface structure, consistent with the surface morphology of aliphatic poly(carbonate-co-urethane)s reported by others.23 Higher magnification images revealed structured aggregates (<5 μm in diameter) on the PUSFU-OS surfaces, which were attributed to crystallites of 5FU formed during solvent evaporation (Fig. 5B), which was consistent with the XRD and MDSC results. This was further confirmed by comparison to the surface topography images of PUS-OS, which showed no evidence of such aggregates (ESI, Fig. S7†). Cross-sectional imaging of the base-coated PUSFU-OS stents provided similar mean thickness values for the abluminal (47.7 ± 10.5 μm) and luminal coatings (55.2 ± 8.7 μm). Subsequent coating with PEVA afforded PEVA-PUSFU-OS having relatively flat and smooth surfaces without any pores and abluminal and luminal thicknesses of 113.2 ± 36.8 and 76.0 ± 20.9 μm, respectively (Fig. 5C).
Dip-coating of the CS with the PUSFU basecoat resulted in PUSFU-CS with surface topographical features (ESI, Fig. S5B†) similar to PUSFU-OS (Fig. 5B), however, the former had only a single PUSFU basecoat layer with a thickness of 65.0 ± 4.0 μm due to the absence of a supporting substrate (ESI, Fig. S5–6 and 8†). Subsequent dip-coating with PEVA afforded PEVA-PUSFU-CS with seemingly smooth surface morphologies, although higher magnification images revealed ring-like wrinkles ∼5 μm in diameter (ESI, Fig. S5C†). While the emergence of these features may be due to bubble formation during solvent evaporation from the PEVA topcoat, their absence in PEVA-PUSFU-OS indicated an insufficient thickness of the underlying layers and the increased vulnerability of the single PUSFU layer to solvent-induced swelling, which may affect the PEVA top layer. As previously reported, wrinkles often appear in double- or multiple-coating processes involving the utilisation of organic solvent(s) in the second or latter coating cycles.24 Therefore, the differences observed in the PEVA coatings between PEVA-PUSFU-CS and -OS may potentially result from swelling and deformation of the PUS basecoat layer on the former due to a single and unsupported PUS coating. Nevertheless, the thickness of both the PEVA abluminal (111.4 ± 29.0 μm) and luminal coatings (82.7 ± 12.8 μm) for the PEVA-PUSFU-CS (ESI, Fig. S5, 6 and 8†) were found to be close to the values measured for the PEVA-PUSFU-OS.
Drug loading and release from stents
The 5FU content and its homogeneity of distribution within the PUS basecoat of the stents was determined by cutting the stents into sections (n = 6 from two separate coated stents for each stent type), extracting and analysing the 5FU by high-performance liquid chromatography (HPLC) following dissolution and separation of the polymeric component. For both stents, the experimentally determined loadings were statistically equivalent to the theoretical loadings (ESI, Table S2†), and the low standard deviation values confirmed that the 5FU was uniformly distributed within the PUS basecoat across the different stents, regardless of the stent type.In vitro drug release from complete PEVA-PUSFU-OS and -CS was investigated in phosphate buffered saline (10 mM PBS, pH 7.4) at 37 °C under sink conditions, by measuring the 5FU concentration in the release media via HPLC at regular intervals. The release of 5FU from both stents displayed asymptotic profiles with significantly different release durations (Fig. 6). PEVA-PUSFU-OS exhibited a relatively rapid 5FU release in the first 14 d (∼30%) followed by a sustained and slow release from 14 to 150 d (∼62%). In comparison, 5FU was released rapidly from PEVA-PUSFU-CS, reaching a plateau around day 12 (∼90%). Compared to previous studies with related polyurethanes,4 the release of 5FU from PEVA-PUSFU-OS and -CS was accelerated, which was attributed to the silicone segments and a more flexible polymer matrix promoting water diffusion.
 | ||
| Fig. 6 In vitro drug release profiles of 5FU from (A) PEVA-PUSFU-OS and (B) PEVA-PUSFU-CS. All data expressed as mean (n = 3) ± SD. | ||
The difference in the 5FU release durations was attributed to the influence of the silicone membrane. For PEVA-PUSFU-OS the membrane allowed the uniform deposition of the PUSFU basecoat across the open cell structure of the stent, whereas for PEVA-PUSFU-CS, the absence of a membrane resulted in the PUSFU basecoat pooling at the mesh edges of the cell (ESI, Fig. S9†). This appeared to lead to thicker PUSFU coatings on the wire mesh, hindering the formation of an inclusive and uniform PEVA topcoat and resulting in quicker water and 5FU diffusion. The presence of wrinkles, as noted in the SEM images of PEVA-PUSFU-CS, may also be an indicator of defects in the PEVA topcoat that contribute to quicker drug release. In addition, the silicone membrane may act as an additional barrier to water diffusion in PEVA-PUSFU-OS, retarding 5FU release, which is absent for PEVA-PUSFU-CS.
Overall, the drug release studies demonstrated that PEVA-PUSFU-OS and PEVA-PUSFU-CS are capable of providing a controlled and sustained release of 5FU across two different time frames of 150 and 14 d, respectively. The safety and efficacy of drug-stent combinations is largely based on their ability to deliver appropriate amounts of drugs with controlled release kinetics.25 Cytotoxic anticancer drugs released gradually from coated stents over several weeks to months are potentially more likely to be delivered locally to gastrointestinal tumours over multiple cycles of cell division, which may lead to better control over cell growth and cancer progression.2,26 Furthermore, the European Society of Gastrointestinal Endoscopy recommends the insertion of gastrointestinal SEMS only when a cancer patient's survival is predicted to be more than 4 months;9 thus, the prolonged drug release (5 months) provided by PEVA-PUSFU-OS may make it a potential chemotherapeutic candidate for the treatment of obstructing gastrointestinal cancers. On the other hand, the comparatively rapid and much shorter duration of release (2 weeks) from PEVA-PUSFU-CS may be preferred for the palliative treatment of patients with advanced gastrointestinal cancers, in which the majority of patients are likely to reach end of life with the stent in place in their GIT.
To investigate the mechanism of drug release from the stents, the release data were fitted to various kinetic models (ESI, Table S3†)27 and the goodness-of-fit were assessed using a combination of adjusted coefficient of determination (R2 adjusted), Akaike Information Criterion (AIC) and root mean square error (RMSE) goodness-of-fit tests. The most suitable model was considered the one with the highest R2 adjusted and the smaller ACI and RMSE values.27,28 For PEVA-PUSFU-OS and -CS, the release data fitted best to the Weibull and Peppas–Sahlin kinetic models, respectively (ESI, Table S3†), both of which are consistent with previous studies with polyurethanes.29 The β (shape factor) parameter of the Weibull model was estimated to be <1, consistent with the pronounced parabolic shape (with higher initial slope) of the drug release profile,27,30 which corresponded to a Fickian diffusion mechanism.31 Comparatively, the Peppas–Sahlin kinetic model is consistent with Fickian diffusion and polymer chain relaxation, although the higher k1 value (k1 = 37.831 and k2 = −4.267) indicated the predominance of Fickian diffusion over polymer chain relaxation.27,28
Stent leachables, stability and sterilisation
During stent fabrication, several organic solvents (e.g., tetrahydrofuran (THF) and N,N-dimethylformaide (DMF)) were employed. Although most of these solvents are likely to evaporate during the drying process, it is plausible that trace amounts may remain trapped. Given the possible health risks associated with these organic chemicals, the amounts of residual THF and DMF in the stents were determined by gas chromatography (GC). Residual THF was absent in both PEVA-PUSFU-OS and -CS (ESI, Fig. S10†), while 1685 ± 443 and 1715 ± 133 ppm of DMF, respectively, was found to be present (ESI, Fig. S11†), both of which are above the permitted daily exposure (PDE) limit for DMF (8.8 mg d−1) by the FDA (ESI, Table S4†).32 The relatively high residual amounts of DMF may have resulted from insufficient drying, and further optimisation of the drying process would likely decrease the level of residual DMF. Nevertheless, unlike conventional drugs or dosage forms, the gradual release of payloads (and solvents)14 from the stents was postulated to limit the daily release of DMF. Hence, the release of DMF from the stents was measured,29 which revealed significantly lower daily amounts compared to the total extracted amounts and PDE limits (Fig. 7 and ESI, Fig. S12 and 13†). Thus, the maximum amount of residual DMF that would leach from the stents in a single day is highly unlikely to cause localised or systemic DMF toxicity. This was further supported by in vitro MTT assays with leachables from drug-free PEVA-PUS-OS and -CS, which showed similar cell viability of HCT 116 cells (∼97 and 100%, respectively) as compared to a negative control (vide infra).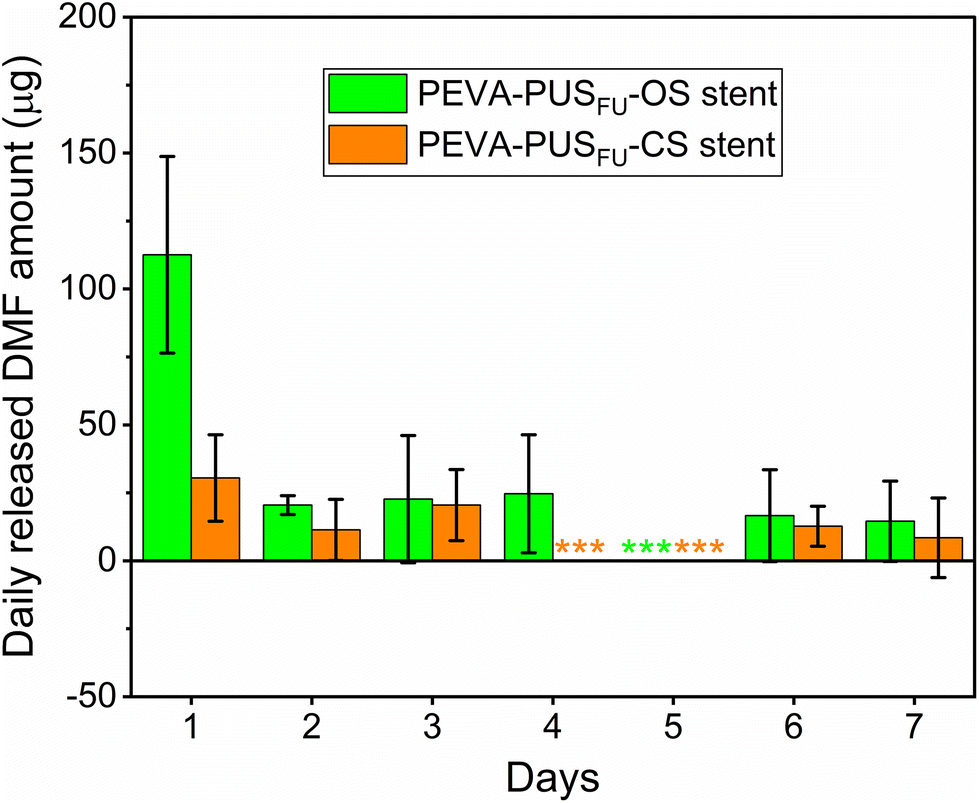 | ||
| Fig. 7 Amount of residual dimethylformamide (DMF) released from PEVA-PUSFU-OS and -CS as a function of time. Three asterisks (***) indicate the amount of DMF that was below the quantitation limit (5 ppm). Data are expressed as mean (n = 3) ± SD. | ||
The stability of the stents when stored at 25 °C/60% relative humidity (RH) and 40 °C/75% RH over 3 months was assessed according to the International Council for Harmonisation (ICH) guidelines (ESI, Table S5†).14,33 Mass balances (ESI, Table S5†) and microscopy revealed no changes in weight or appearance (ESI, Fig. S14–17†), indicating that the stents were physically stable. Similarly, no variations in the 5FU content (ESI, Table S5†) or release profiles (ESI, Fig. S18 and Table S5†) were observed, indicating that the drug and stents are stable under the accelerated temperature and humidity conditions. Gamma irradiation is commonly used as a standard sterilisation method for pharmaceutical products and many medical devices.14,34 To assess the influence of gamma sterilisation (25 kGy) on the stents, in vitro release of 5FU was assessed (ESI, Fig. S18†). For both stent types, there were no significant differences in 5FU release profiles observed between the non-irradiated (control) and gamma irradiated stents, indicating their stability to gamma sterilisation.
In vitro anticancer activity of stents
The anticancer cell activity of the stent-released 5FU was assessed using a human-derived colon carcinoma cell line, HCT 116.6,35–37 Initially, the release of 5FU from stent sections in cell medium (sterile 10% v/v foetal bovine serum supplemented Roswell Park Memorial Institute-1640 (RPMI-FBS)) was measured over 18 d to determine the daily dosing for subsequent cell studies (ESI, Fig. S19†). A noncumulative cytotoxicity test was considered suitable to simulate the best possible in vivo scenario of time-dependent release of 5FU from the stents.38 In addition, the cytotoxic activity of pure 5FU at different concentrations ranging from 0.05 to 97.6 μg mL−1 was initially determined via a MTT cell viability assay after treating the cells for 72 h (ESI, Fig. S20†), which resulted in a concentration-dependent decrease in HCT 116 cell viability with an IC50 value of 0.95 μg mL−1. Importantly, the measured IC50 value of pure 5FU was significantly lower than the concentrations of 5FU released from the stent sections (ESI, Fig. S19†). It is therefore conceivable that the stents would provide a sustained and localised therapeutic concentration of the drug in vivo over an extended period of time.The cytotoxic activity of the stent released 5FU at day 1 and 7 from PEVA-PUSFU-OS, and day 1 and 14 from PEVA-PUSFU-CS was compared with leachables obtained on day 1 from the blank stents (Fig. 8) and pure 5FU (positive control) (ESI, Fig. S21†).39 As anticipated, the stent-released 5FU resulted in a statistically significant concentration-dependent decrease in cell viability following incubation for 72 h, as compared to the media-treated negative control (Fig. 8). In comparison, cells exposed to drug-free PEVA-PUS-OS and -CS leachables displayed cell viabilities >98%, confirming the absence of other leachables at cytotoxic levels. The results demonstrate that the proliferation of HCT 116 cells was significantly inhibited by the stent-released 5FU, and therefore the stents have the potential of providing sustained anticancer activity.
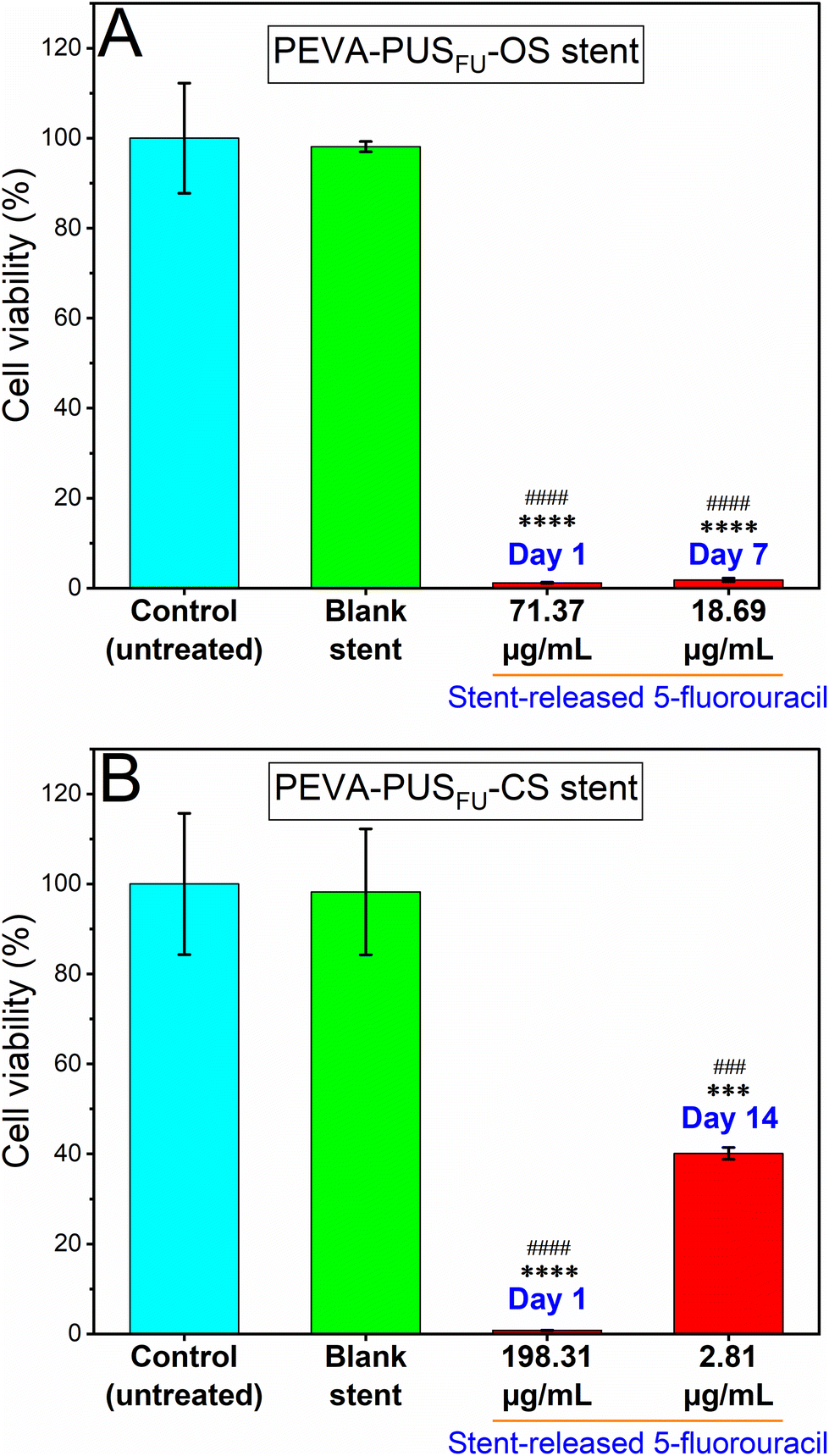 | ||
| Fig. 8 Growth inhibition effects of different concentrations of 5FU released (A) on day 1 and 7 from the PEVA-PUSFU-OS sections, and (B) at day 1 and 14 from the PEVA-PUSFU-CS sections, on HCT 116 cells treated for 72 h, analysed by MTT assay. Results are expressed as mean ± SD of at least triplicate measurements, and p-values were obtained using one-way analysis of variance (ANOVA). **** p < 0.0001 and *** p < 0.001 compared to untreated control (cells grown in culture media only); and #### p < 0.0001 and ### p < 0.001 compared to drug-free (blank) stent sections, respectively. | ||
Cell cycle distribution analysis is a valuable tool for obtaining information on different phases (G1, S, G2 and M) of eukaryotic cell cycle based on the DNA content.40 Previous reports suggest that 5FU exerts cytotoxicity through interference with DNA synthesis during the S phase of the cell cycle.6,35 Therefore, we assessed whether the stent-released 5FU affected the cell cycle profile of HCT 116 cells and induced an S phase accumulation, in the same manner as pure 5FU (Fig. 9A and B; and ESI, Fig. S22 and 23†).
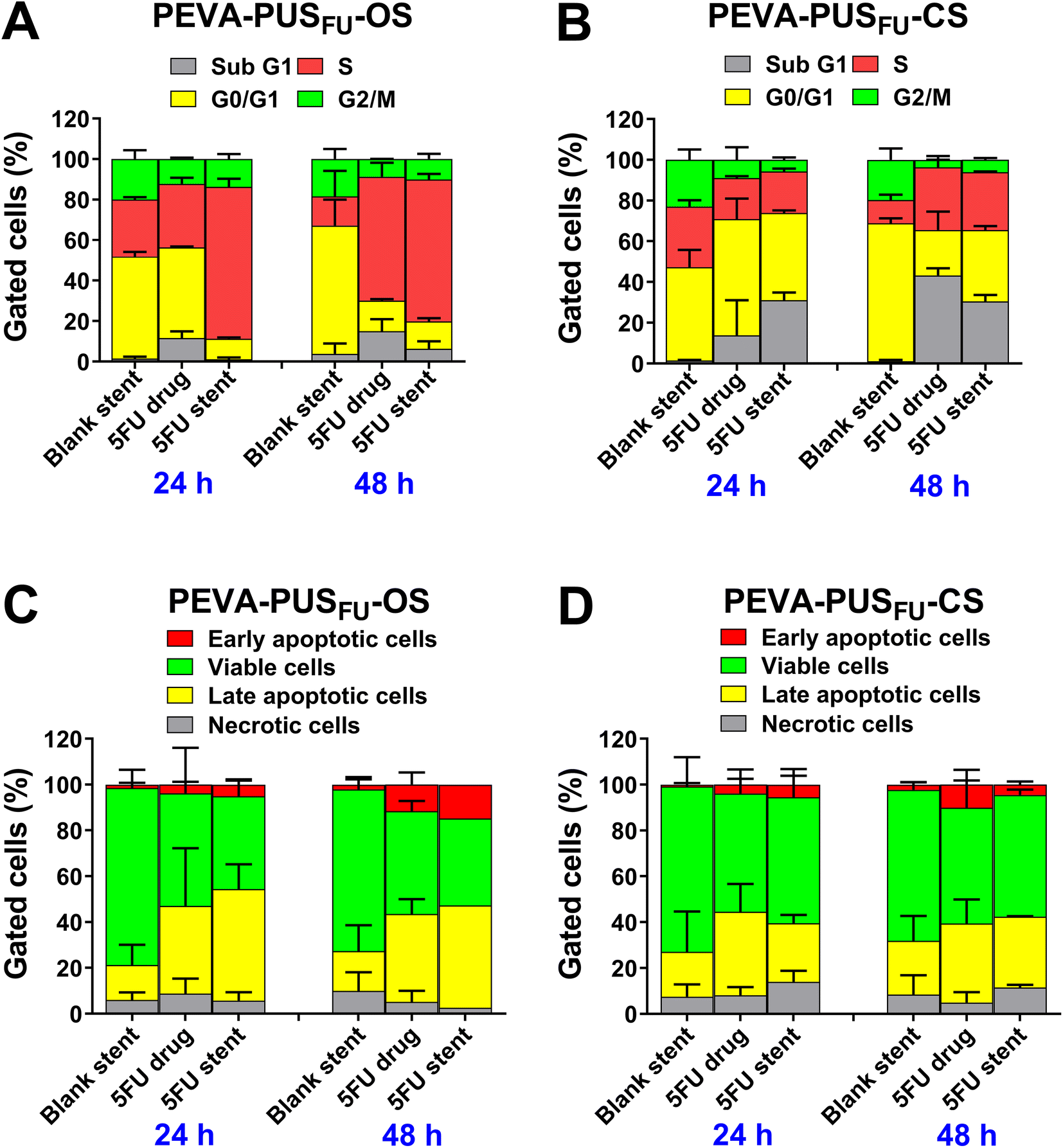 | ||
| Fig. 9 In vitro evaluation of the anticancer effect of 5FU released from (A and C) PEVA-PUSFU-OC and (B and D) -CS stents against HCT 116 cells following 24 and 48 h treatment, as determined by a flow cytometry-based fluorescein isothiocyanate (FITC) Annexin V and propidium iodide (PI) double staining assay. Graphs showing the effects on different phases of cells (A and B) or the distribution of cells at different cell death stages for cells (C and D) treated with leachables from blank (drug-free) stent controls, 5FU (positive control) and leachables from the 5FU-loaded stents. Mean ± SD of data (without subtracting media-treated negative control) pooled from two independent experiments (A and B) or at least two replicate measurements (C and D) are presented. | ||
No significant accumulation of cells in any particular phase of the cycle was observed following treatment with drug-free PEVA-PUS-OS and -CS leachables for up to 48 h (Fig. 9A and B, respectively). In contrast, treatment with 5FU released from PEVA-PUSFU-OS (equivalent to 5.36 μg mL−1) and -CS (equivalent to 17.02 μg mL−1) sections for 48 h caused a significant increase in the number of cells undergoing S phase arrest compared to the respective drug-free stent controls (ESI, Fig. S22†). Simultaneously, a substantial accumulation of sub G1 phase cells was observed for both the stent-released 5FU samples, which indicated the occurrence of cell death. Arrest in the cell cycle phase allows cells to repair DNA damage prior to replication followed by mitosis; however, if the damage is beyond repair, then cells undergo apoptotic programmed cell death.6,41 Thus, a significant increase in cell death may be attributed to the successful anticancer effect of 5FU released from the stents. Pure 5FU (positive control) at approximately similar concentrations (6.10 and 18.30 μg mL−1) exhibited similar cell cycle distribution profiles (statistically non-significant) to stent-released 5FU samples under the same experimental concentrations. These cell cycle analysis results confirmed that the 5FU released from the stents exhibited a similar cytotoxic effect as pure 5FU, which is consistent with its intrinsic cytotoxicity.4,35
As observed in the cell cycle analysis, the treatment with the stent-released 5FU caused an increase in cell death. The death of eukaryotic cells typically occurs either by necrosis or apoptosis,42 with 5FU43 being reported to cause apoptosis in HCT 116 cells in a concentration- and time-dependent manner.4,6,36,37 Hence, the mechanism of cancer cell death caused by the stent-released 5FU was assessed using the Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) double-staining assay after 24 and 48 h incubation (Fig. 9C and D; and ESI, Fig. S24 and 25†). As expected, there were no significant changes in the proportion of apoptotic, necrotic or viable cells following treatment with drug-free PEVA-PUS-OS and -CS leachables for up to 48 h. In comparison, treatment with 5FU released from the stent sections for up to 48 h induced a concentration- and time-dependent increase in the proportion of total apoptotic cells, as well as a decrease in necrotic or viable cells (Fig. 9C and D). These results imply that both the stents had a clear 5FU-induced cytotoxic anticancer effect on HCT 116 cells, predominantly via the apoptotic pathway, consistent with previously reported literature.4,6,35,37
Experimental
Procedures
Release studies in PBS. The stents (n = 3 for each stent type) were placed inside fabric mesh bags and submerged in separate Falcon tubes with 20–25 mL of PBS (10 mM, pH 7.4), which was calculated to be sufficient to maintain sink conditions based on the saturation solubility of pure 5FU (7.65 ± 1.50 mg mL−1).29 The Falcon tubes were then sealed and incubated in an orbital mixer incubator (Ratek Instruments) at 37 °C and 175 rpm. At predetermined time points, aliquots (1 mL) of the release medium were removed from each tube for further analysis by HPLC and replenished immediately with the same volume of fresh PBS.
Release studies in RPMI media. The stents were cut into small sections weighing ∼100 mg with titanium-coated stainless-steel scissors. The stent sections (n = 3 for each stent type) were placed in sterile containers and immersed in 4 and 3 mL, respectively, of RPMI-FBS medium. Each container was sealed and placed on a shaking orbital incubator (Ratek Instruments) at 37 °C and 175 rpm for 18 d. The whole release medium was collected daily from each container and subsequently replaced with an equal volume of fresh medium. Subsequently, the concentrations of 5FU in the daily collected medium were measured by HPLC.
Accelerated stability studies were conducted according to the ICH Q1A(R2) guidelines.33PEVA-PUSFU-OS and -CS sections were sealed individually in zip-lock bags and stored at either 40 ± 2 °C/75 ± 5% RH, 25 ± 2 °C/60 ± 5% RH or 4 °C. After 1 and 3 months of storage, stents were examined microscopically, and the drug content was determined by HPLC as previously described.
Human colon carcinoma HCT 116 cells (ATCC CCL-247) between 15 and 25 passages were grown in RPMI-FBS medium (refreshed every 2 d) and incubated at 37 °C/5% CO2 (Thermo Electron LED). At 80% confluency, the cells were washed with sterile PBS and then passaged using 0.25% trypsin and 0.02% ethylenediaminetetraacetic acid (EDTA) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 dilution.
:10 dilution.
Colorimetric MTT assay. HCT 116 cells were plated in 96-well plates at a density of 5 × 104 cells per well and incubated for 24 h at 37 °C/5% CO2. Stent-release media or pure 5FU solution (100 μL) was added to each well and the cells were incubated for a further 72 h. The medium in each well was replaced with MTT reagent, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (0.5 mg mL−1) in PBS (100 μL) and the plates were incubated for 4 h at ambient temperature in the dark. The medium in each well was replaced with dimethyl sulfoxide (DMSO; 100 μL) and the plates were shaken gently for 10 min. The absorbance of formazan was measured using a multi-well plate reader (Model 1420-012, PerkinElmer) at a wavelength of 540 nm. The results were presented as the percentage of relative viability of HCT 116 cells treated with stent-release media or pure 5FU solution as compared to a negative control.
Cell cycle distribution and apoptosis analyses by flow cytometry. Cell cycle distribution and apoptosis were analysed by flow cytometry as previously reported.4 A detailed description is provided in the ESI.†
Conclusions
Two novel drug-eluting gastrointestinal stents were successfully fabricated by dip-coating of either fully covered oesophageal or uncovered colonic self-expandable nitinol stents sequentially with a 5FU-incorporated polyurethane-silicone (PUS) basecoat and a 5FU-free (blank) poly(ethylene-co-vinyl acetate) (PEVA) topcoat. Physicochemical characterisation of the drug-eluting stents and coatings revealed that the 5FU is homogenously distributed throughout the PUS matrix, and is likely to be present in both crystalline and amorphous states. Determination of the drug loading content in the stents confirmed that the 5FU was dispersed uniformly throughout the PUS coating matrix across individual stents, regardless of the stent type. In vitro release studies demonstrated controlled and sustained release of 5FU across two different time scales of 150 and 14 d for the oesophageal and colonic stents, respectively, with mathematical modelling indicating a Fickian diffusion mechanism. The more rapid release appeared to correlate with defects in the uniformity of both the base and topcoats resulting from the design of the colonic stents, which highlighted the importance of a supporting membrane for consistent coating. Analysis of residual solvents in the stents remaining form the manufacturing process revealed that the amount of dimethylformamide (DMF) leached daily from the stents was far below daily exposure limits. The drug-loaded stents were shown to be stable to gamma irradiation sterilisation and accelerated storage conditions. Cell viability assay, and cell cycle and apoptosis analyses confirmed that the coated oesophageal and colonic stent-released 5FU had comparable anticancer activity to pure 5FU against human colon carcinoma HCT 116 cells. Overall, the results of the physicochemical characterisation, quality assessment, and in vitro biological evaluation are encouraging and suggest that the newly developed 5FU-eluting stents may serve as potential chemotherapeutic candidates for the treatment of obstructing oesophageal/colon cancers.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
All authors declare that they have no conflicts of interest. While Taewoong Medical Company gifted the nitinol stents used in this work to MF, he has no other financial or non-financial conflicts of interest with this organisation.Acknowledgements
The authors would like to extend their thanks to Taewoong Medical Co. Ltd (Gimpo-si, Gyeonggi-do, South Korea) for kindly providing commercial Niti-S oesophageal covered and Niti-S enteral colonic uncovered stents, Prof. Shudong Wang (Drug Discovery and Development, University of South Australia, Adelaide) for gifting HCT 116 human colon cancer cell line, and Dr Khandokar Sadique Faisal for his help with nuclear magnetic resonance spectroscopy and data processing.References
- M. Arnold, et al., Global Burden of 5 Major Types of Gastrointestinal Cancer, Gastroenterology, 2020, 159(1), 335–349 CrossRef PubMed.
- M. Arafat, et al., Drug-eluting non-vascular stents for localised drug targeting in obstructive gastrointestinal cancers, J. Controlled Release, 2019, 308, 209–231 CrossRef CAS PubMed.
- R. E. Sexton, et al., Gastric Cancer: A Comprehensive Review of Current and Future Treatment Strategies, Cancer Metastasis Rev., 2020, 39(4), 1179–1203 CrossRef PubMed.
- M. Arafat, et al., Development and In Vitro Evaluation of 5-Fluorouracil-Eluting Stents for the Treatment of Colorectal Cancer and Cancer-Related Obstruction, Pharmaceutics, 2021, 13(1), 17 CrossRef CAS PubMed.
- J. W. Lee, S.-G. Yang and K. Na, Gemcitabine-releasing polymeric films for covered self-expandable metallic stent in treatment of gastrointestinal cancer, Int. J. Pharm., 2012, 427(2), 276–283 CrossRef CAS PubMed.
- G. Li, et al., A 5-fluorouracil-loaded polydioxanone weft-knitted stent for the treatment of colorectal cancer, Biomaterials, 2013, 34(37), 9451–9461 CrossRef CAS PubMed.
- Q. Guo, S. Guo and Z. Wang, A type of esophageal stent coating composed of one 5-fluorouracil-containing EVA layer and one drug-free protective layer: In vitro release, permeation and mechanical properties, J. Controlled Release, 2007, 118(3), 318–324 CrossRef CAS PubMed.
- S.-R. Guo, et al., In Vivo Evaluation of 5-Fluorouracil-Containing Self-Expandable Nitinol Stent in Rabbits: Efficiency in Long-Term Local Drug Delivery, J. Pharm. Sci., 2010, 99(7), 3009–3018 CrossRef CAS PubMed.
- S.-Y. Kim, et al., Paclitaxel-eluting nanofiber-covered self-expanding nonvascular stent for palliative chemotherapy of gastrointestinal cancer and its related stenosis, Biomed. Microdevices, 2014, 16(6), 897–904 CrossRef CAS PubMed.
- J. Liu, et al., Paclitaxel or 5-fluorouracil/esophageal stent combinations as a novel approach for the treatment of esophageal cancer, Biomaterials, 2015, 53(Supplement C), 592–599 CrossRef CAS PubMed.
- B. M. Wolpin, et al., Systemic Treatment of Colorectal Cancer, Gastroenterology, 2008, 134(5), 1296–1310 CrossRef CAS PubMed.
- Z. Wang, et al., Nitinol stents loaded with a high dose of antitumor 5-fluorouracil or paclitaxel: esophageal tissue responses in a porcine model, Gastrointest. Endos., 2015, 82(1), 153–160 CrossRef PubMed.
- A. B. Boam, Regulatory issues facing the development of drug-eluting stents: a US FDA perspective, Expert Rev. Med. Devices, 2006, 3(3), 297–300 CrossRef PubMed.
- Food and Drug Administration Center for Devices and Radiological Health (CDRH), Guidance for Industry: Coronary Drug-Eluting Stents-Nonclinical and Clinical Studies (Draft), Food and Drug Administration (FDA), Rockville, MD, USA, 2008, p. 89 Search PubMed.
- R. A. Chappa, et al., Coating systems for the controlled delivery of hydrophilic bioactive agents, 2013, Google Patents.
- L. Lei, et al., 5-Fluorouracil-loaded multilayered films for drug controlled releasing stent application: Drug release, microstructure, and ex vivo permeation behaviors, J. Controlled Release, 2010, 146(1), 45–53 CrossRef CAS PubMed.
- P. Singh, et al., Thermal stability studies of 5-fluorouracil using diffuse reflectance infrared spectroscopy, Drug Test. Anal., 2009, 1(5), 240–244 CrossRef CAS PubMed.
- M. Rogulska and A. Kultys, Aliphatic polycarbonate-based thermoplastic polyurethane elastomers containing diphenyl sulfide units, J. Therm. Anal. Calorim., 2016, 126(1), 225–243 CrossRef CAS.
- E. L. Robertson, D. M. Hoffman and P. F. Pagoria, New Polycarbonate-Based Thermoplastic Polyurethane Binder for HMX Based Explosives, Conference: Portland, OR, United States, 2018; Lawrence Livermore National Lab, (LLNL), Livermore, CA (United States), Medium, ED.
- A. Gupta, et al., Enteric coated HPMC capsules plugged with 5-FU loaded microsponges: a potential approach for treatment of colon cancer, Braz. J. Pharm. Sci., 2015, 51, 591–605 CrossRef CAS.
- C. Zhang, et al., Preparation and characterization of 5-fluorouracil-loaded PLLA–PEG/PEG nanoparticles by a novel supercritical CO2 technique, Int. J. Pharm., 2012, 436(1), 272–281 CrossRef CAS PubMed.
- W. Schmidt and P. Lanzer, Instrumentation, in Catheter-Based Cardiovascular Interventions: A Knowledge-Based Approach, ed. P. Lanzer, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 445–472 Search PubMed.
- R. Araujo Borges, D. Choudhury and M. Zou, 3D printed PCU/UHMWPE polymeric blend for artificial knee meniscus, Tribol. Int., 2018, 122, 1–7 CrossRef CAS.
- M. Shimokawa, et al., Emergence of Wrinkles during the Curing of Coatings, Gels, 2018, 4(2), 41 CrossRef PubMed.
- R. Thipparaboina, W. Khan and A. J. Domb, Eluting combination drugs from stents, Int. J. Pharm., 2013, 454(1), 4–10 CrossRef CAS PubMed.
- J. B. Wolinsky, Y. L. Colson and M. W. Grinstaff, Local drug delivery strategies for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers, J. Controlled Release, 2012, 159(1), 14–26 CrossRef CAS PubMed.
- Y. Zhang, et al., DDSolver: an add-in program for modeling and comparison of drug dissolution profiles, AAPS J., 2010, 12(3), 263–271 CrossRef CAS PubMed.
- M. C. L. C. Freire, et al., Understanding Drug Release Data through Thermodynamic Analysis, Materials, 2017, 10(6), 651 CrossRef PubMed.
- M. Arafat, et al., Pharmaceutical Development of 5-Fluorouracil-Eluting Stents for the Potential Treatment of Gastrointestinal Cancers and Related Obstructions, Drug Des., Dev. Ther., 2021, 15, 1495–1507 CrossRef PubMed.
- N. Yuksel, A. E. Kanık and T. Baykara, Comparison of in vitro dissolution profiles by ANOVA-based, model-dependent and -independent methods, Int. J. Pharm., 2000, 209(1), 57–67 CrossRef CAS PubMed.
- J. KobryD, et al., Influence of Hydrophilic Polymers on the Factor in Weibull Equation Applied to the Release Kinetics of a Biologically Active Complex of Aesculus hippocastanum, Int. J. Polym. Sci., 2017, 2017, 1–8 CrossRef.
- The United States Pharmacopeia-National formulary (USP-NF), Residual Solvents, United States Pharmacopeial Convention, Rockville, MD, USA, 2019, p. 6639 Search PubMed.
- ICH HARMONISED TRIPARTITE GUIDELINE. STABILITY TESTING OF NEW DRUG SUBSTANCES AND PRODUCTS Q1A(R2). 2003. Accessed 22nd Janurary 2025: https://database.ich.org/sites/default/files/Q1A%28R2%29%20Guideline.pdf.
- C. R. Harrell, et al., Risks of Using Sterilization by Gamma Radiation: The Other Side of the Coin, Int. J. Med. Sci., 2018, 15(3), 274–279 CrossRef CAS PubMed.
- V. R. Silva, et al., A ruthenium-based 5-fluorouracil complex with enhanced cytotoxicity and apoptosis induction action in HCT116 cells, Sci. Rep., 2018, 8(1), 288 CrossRef PubMed.
- E. Tawfik, et al., Prolonged exposure of colon cancer cells to 5-fluorouracil nanoparticles improves its anticancer activity, Saudi Pharm. J., 2017, 25(2), 206–213 CrossRef PubMed.
- R. Yoshikawa, et al., Dual antitumor effects of 5-fluorouracil on the cell cycle in colorectal carcinoma cells: a novel target mechanism concept for pharmacokinetic modulating chemotherapy, Cancer Res., 2001, 61(3), 1029–1037 CAS.
- M. Shaikh, et al., In Vitro and In Vivo Assessment of Docetaxel Formulation Developed for Esophageal Stents, AAPS PharmSciTech, 2017, 18(1), 130–137 CrossRef CAS PubMed.
- S. R. Gadagkar and G. B. Call, Computational tools for fitting the Hill equation to dose–response curves, J. Pharmacol. Toxicol. Methods, 2015, 71, 68–76 CrossRef CAS PubMed.
- S. Biswas, et al., Evaluation of Novel 3-Hydroxyflavone Analogues as HDAC Inhibitors against Colorectal Cancer, Adv. Pharmacol. Sci., 2018, 2018, 4751806 Search PubMed.
- R. Singh, J. George and Y. Shukla, Role of senescence and mitotic catastrophe in cancer therapy, Cell Div., 2010, 5, 4–4 CrossRef PubMed.
- S. L. Fink and B. T. Cookson, Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells, Infect. Immun., 2005, 73(4), 1907–1916 CrossRef CAS PubMed.
- W. Leowattana, et al., Systemic treatment for metastatic colorectal cancer, World J. Gastroenterol., 2023, 29(10), 1569–1588 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00312h |
| This journal is © The Royal Society of Chemistry 2025 |