DOI:
10.1039/D4PY00759J
(Paper)
Polym. Chem., 2025,
16, 37-44
A new recyclable polymer based on Diels–Alder crosslinking networks derived from anisaldehyde†
Received
9th July 2024
, Accepted 17th November 2024
First published on 19th November 2024
Abstract
The preparation of recyclable cross-linked polymers by combining biomass-based resources with dynamic covalent bonds is a promising strategy for the preparation of sustainable and recyclable thermosetting materials. In this paper, a biomass-based epoxy resin was synthesized from anisaldehyde, and then a biomass-based polymeric material was prepared using Diels–Alder chemistry. The biomass-based material exhibits high mechanical strength and light transparency. The tensile strength and elongation of the biomass-based material at a temperature of 315 °C with a weight loss of 5% (Td5) were 79.7 MPa and 12.2%, respectively, which are higher than those of material synthesized from petroleum-based (bisphenol A) epoxy resin. The biomass-based film material with a thickness of 0.14 mm has a transmittance of up to 89% in the light wavelength range of 550–1200 nm. Moreover, the biomass-based material showed good recyclability.
Introduction
Traditional polymers were often constructed through stable irreversible chemical bonds, which ensure the good thermal and mechanical properties of the polymers. Researchers have strived to prevent the breaking of covalent bonds.1 Once damaged or discarded, these polymers can only be disposed of by landfilling or incineration, which causes great pressure on the environment.2,3 In recent years, scientists have tried to introduce dynamic covalent bonds into polymers, which can be broken and reorganized under specific conditions. When exposed to external stimuli, polymers can exhibit macroscopic responses.1,4–6 It is worth mentioning that the polymers designed based on dynamic covalent bonds have good stability, robustness and mechanical strength as compared with traditional non-covalent cross-linking networks7 (hydrogen bonds,8 ionic bonds,9 and supramolecular interactions10).
Cycloaddition reactions (such as the Diels–Alder reaction, hereinafter referred to as the DA reaction) have a higher specificity than the popular ionic or radical based reactions, because their intermediates will not be quenched by water, oxygen, and so on, which ensures their reversibility under ambient conditions.11 Most of the previous works focused on the reaction mechanism, for example, Sukhishvili12 studied the influence of crosslinking networks on the thermally reversible Diels–Alder reaction between furan and maleimide groups and analyzed the effect of different crosslinking agents and crosslinking densities on the DA reaction, which was instructive for the construction of polymer networks using the DA reaction. In addition, some researchers7,11 used DA networks to prepare elastomers that can be used in wearable electronics. Tang's group13 used 3-substituted furans instead of 2-substituted furans to react with maleimide, thereby stabilizing the double bonds in DA adducts by using the conjugated structure, thus increasing the retro-DA reaction temperature to 240 °C. However, 3-substituted furans are expensive and not suitable for industrial production. Meanwhile, the high inverse reaction temperature can cause damage to the material, which limits its application in electronic devices and other fields.
In recent years, a large number of reports have applied DA reactions to flexible electronic devices14 and the modification of polytriazole resins and epoxy resins.15–17 Taking the study of Huang's group as an example, furfuryl glycidyl ether and maleimides were used to prepare recyclable polymers. Although the polymers exhibited recycling performance, their thermal stability was not good, and the mechanical properties decreased significantly after three cycles of recycling. Brande's group18 designed a self-healing biomass-based polymer with a lignin content of up to 29 wt% based on a DA reaction. Although the toughness and creep resistance of the polymer were improved, the healing efficiency decreased with increasing mechanical stability, and the tensile strength was less than 20 MPa, exhibiting ductile fracture with yield. Zhu's group19 synthesized a biomass-based epoxy BOF using 2,5-furandimethanol as a raw material and toughened it through its DA reaction adduct DA-BOF, which contained a flexible pendant chain. With the increase of the DA-BOF content, the tensile strength decreased significantly, and the maximum elongation at break was only 6.3%. Therefore, it is of great interest to combine the DA reaction with traditional thermosetting resins to develop new thermosetting materials with excellent processability, suitable mechanical properties, high thermal stability, and recyclability.
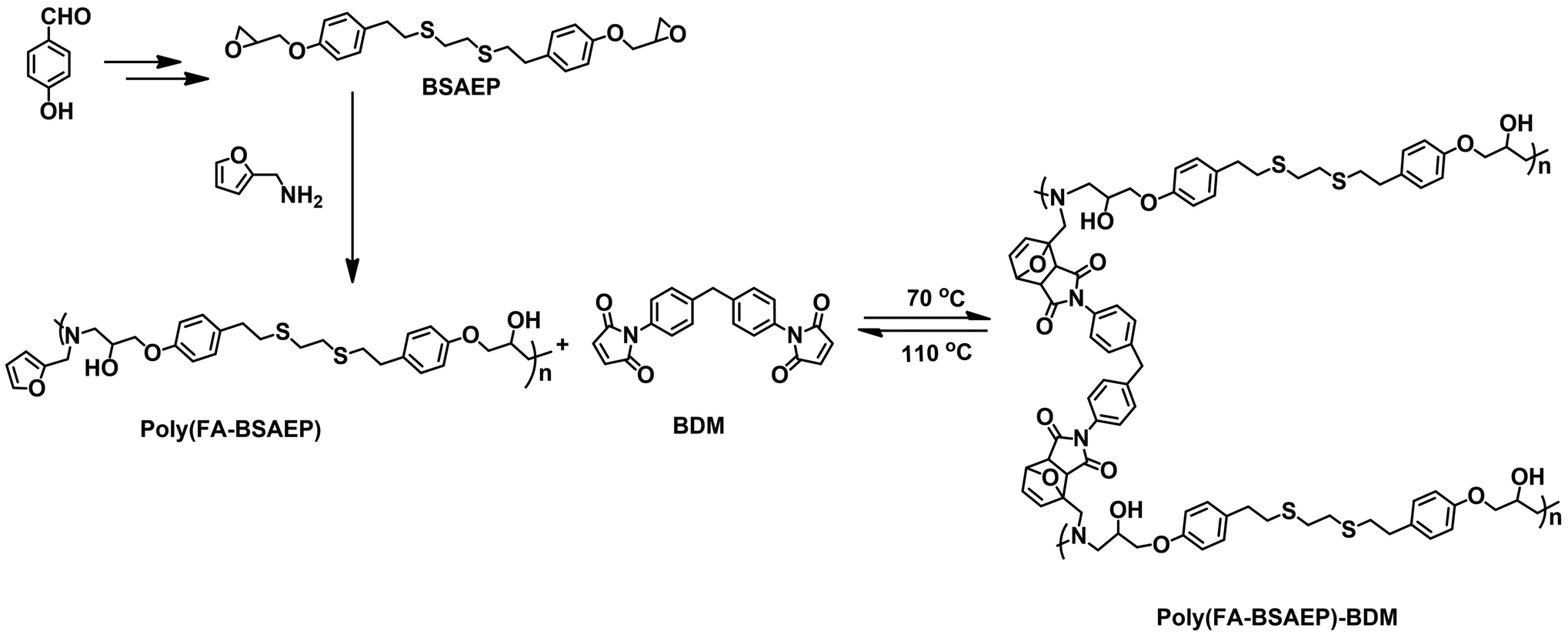
With increasing sustainability requirements, a large number of resins made from eugenol, vanillin, anethole, resveratrol and anisaldehyde as raw materials have been reported in recent years. These resins have comparable or even higher properties than petroleum-based resins.20–23 The introduction of dynamic covalent bonds into biomass-based polymer materials can make the materials more environmentally friendly, which is very promising. At present, there have been a large number of studies on recyclable biomass materials based on imine bonds,3,23 disulfide bonds,24–26 boronic ester bonds27 and ester bonds,28–31 but there have been few reports on biomass-based materials with DA bonds as dynamic covalent bonds. In this study, anisaldehyde was used as the raw material to prepare a biomass-based epoxy resin through a simple reaction, and a DA adduct system was constructed with 2-furfurylamine (FA) and 4,4′-bismaleimidodiphenylmethane (BDM) to prepare a reprocessable cross-linking system. It is worth noting that FA is also a biomass compound that can be obtained from lignin.18,19 Most previous reports13,30,32 on the preparation of recycled epoxy resin by DA addition reaction with an amino curing agent have been presented. Because the amino group is easily oxidized during the preparation process, the material is usually black and opaque. Instead, we first reacted FA with an epoxy group to prepare a linear polymer (poly(FA-BSAEP)) and then reacted BDM with a furan ring to prepare a crosslinked polymer (poly(FA-BSAEP)-BDM). This method effectively avoided the oxidation of the amino group in the preparation of DA adducts and made the prepared material more transparent. Meanwhile, the petroleum-based recyclable cross-linked polymer (poly(FA-BPAEP)-BDM) was prepared using bisphenol A (BPA) epoxy resin as the raw material in the same way. The performances of poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM were investigated in detail.
Experimental
Materials and measurements
Materials and characterization methods used in the experiments are described in the ESI.†
Synthesis of 4-vinylphenol and 2-((4-vinylphenoxy)methyl)oxirane
The detailed synthesis steps are given in the ESI,† while the 1H NMR and 13C NMR spectra of 4-vinylphenol and 2-((4-vinylphenoxy)methyl)oxirane are given in S1 and S2, respectively.
Synthesis of 1,2-bis((4-(oxiran-2-ylmethoxy)phenethyl)thio)ethane (BSAEP)
1,2-Ethanedithiol (3.20 g, 0.03 mol) and 2,2′-azobis(2-methylpropionitrile) (AIBN) (3.35 g, 0.02 mol) were dissolved in THF and heated to 60 °C in a 100 mL three-mouth flask. After 10 minutes, 2-((4-vinylphenoxy)methyl)oxirane (12.00 g, 0.06 mol) was added dropwise to the flask and allowed to react for 1 h. The THF was removed under vacuum at 45 °C and the residue was washed repeatedly with hot ethanol. The crude product was recrystallized in ethyl acetate, filtered and dried to obtain a white solid product BSAEP with a yield of about 91%. 1H NMR (400 MHz, CDCl3) δ: 7.11 (d, J = 8.6 Hz, 1H), 6.85 (d, J = 8.6 Hz, 1H), 4.18 (dd, J = 19.5, 9.7 Hz, 1H), 4.01–3.88 (m, 1H), 3.34 (dddd, J = 5.8, 3.1 Hz, 1H), 2.96–2.64 (m, 4H). 13C NMR (101 MHz, CDCl3) δ: 157.15 (s), 133.07 (s), 129.52 (s), 114.70 (s), 68.84 (s), 50.19 (s), 44.72 (s), 35.46 (s), 34.01 (s), 32.35 (s).
Preparation of linear epoxy polymers containing the furan group (poly(FA-BSAEP) and poly(FA-BPAEP))
BSAEP and 2-furfurylamine were dissolved in DMF and stirred at 120 °C for 4 h. The obtained mixture solution was settled in ethanol, filtered and dried under vacuum at 50 °C to yield a light yellow solid polymer poly(FA-BSAEP). Poly(FA-BPAEP) was prepared using BPA epoxy resin instead of BSAEP as the raw material by the same method.
Fabrication of a crosslinked polymer from poly(FA-BSAEP) through reaction with BDM
The preparation temperature of the crosslinked polymer was determined from the DSC results. Poly(FA-BSAEP) (240 mg) and BDM (80 mg) were dissolved in DMF (3 mL) and reacted at 60 °C for 15 minutes. Then, the solution was cast on the surface of smooth molds made of stainless steel and evaporated at 50 °C to obtain a pre-polymer. Poly(FA-BSAEP)-BDM was obtained by treating the pre-polymer at 70 °C for 6 h under air. After the removal of the molds, fully cured free-standing poly(FA-BSAEP)-BDM was obtained. Similarly, poly(FA-BPAEP)-BDM was obtained by treating the pre-polymer at 80 °C for 6 h under air. The structure is shown in Scheme 1.
 |
| | Scheme 1 Structural design of cross-linked biomass-based recyclable resins. | |
Recovery experiments of crosslinked polymer films by hot-pressing
The crosslinked polymer films of poly(FA-BSAEP)-BDM were cut into small pieces and placed between two steel plates. The plates were first put on a plate vulcanizer and preheated for 1 h at 110 °C under a pressure of 1 MPa, and then the temperature was reduced to 70 °C and held for 6 hours. After cooling to room temperature, uniform and smooth films were obtained. Poly(FA-BPAEP)-BDM was recovered through a similar process, but the hot-pressing process required holding at 5 MPa for 1 hour and then holding at 80 °C for 6 hours to obtain a uniform polymer film.
Recovery experiments of crosslinked polymer films by dissolving in DMF
1.0 g of crosslinked polymer films were cut into pieces and put into 10 mL of DMF, and stirred for about 1 h until completely dissolved. The solution was then poured into a stainless steel mold and heated to 50 °C to remove the solvent. Finally, the pre-polymer was heated to suitable curing temperatures (70 °C for poly(FA-BSAEP)-BDM and 80 °C for poly(FA-BPAEP)-BDM) and maintained at those temperatures for 12 hours to obtain polymer films.
Results and discussion
Synthesis and characterization
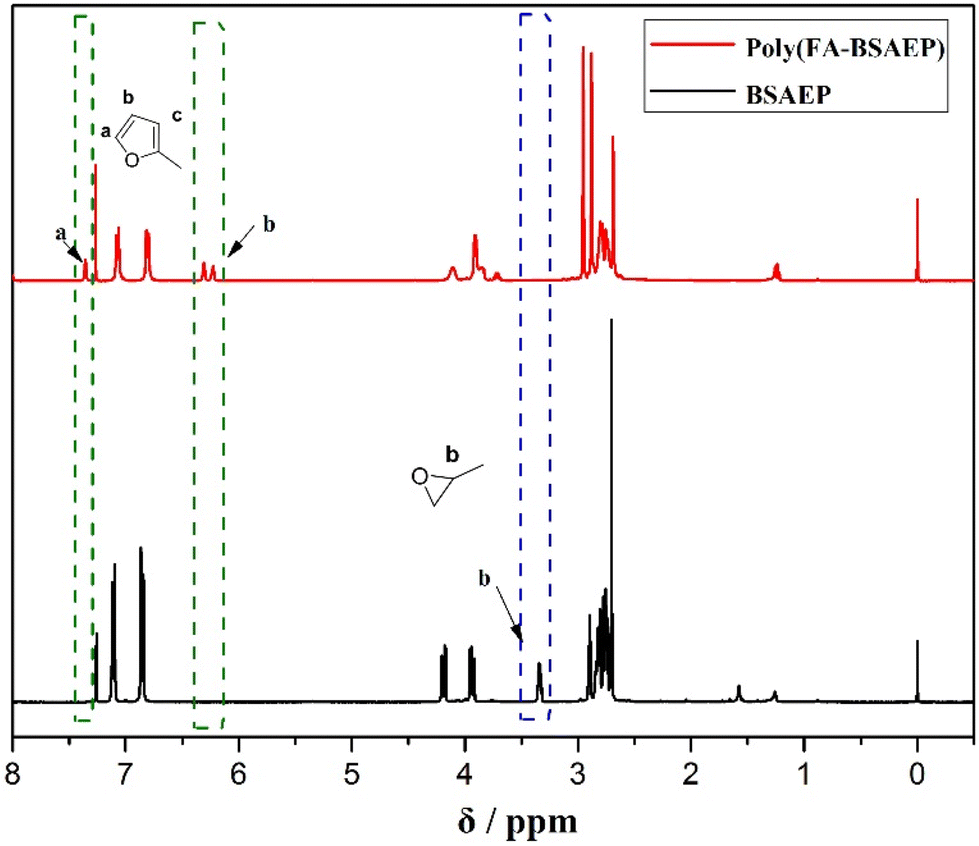
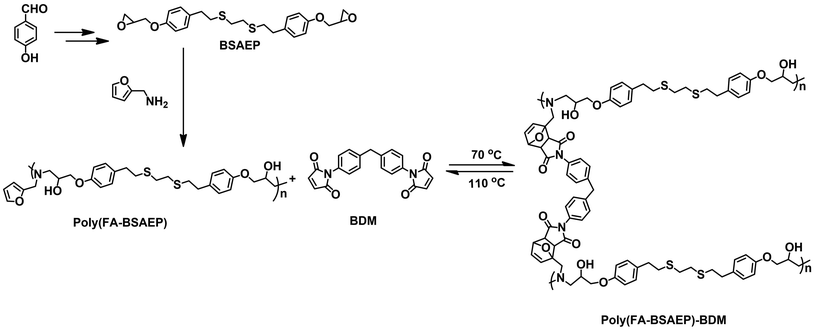
In this paper, a new difunctional biomass-based epoxy monomer (BSAEP) containing flexible chain segments was designed and synthesized using anisaldehyde as the raw material. The structure of BSAEP was characterized by 1H NMR (Fig. 1) and 13C NMR (Fig. S3†) spectroscopy. Linear polymers containing furan rings were prepared by utilizing the fact that the epoxy group can react with FA at a certain temperature. The linear polymer poly(FA-BSAEP) was characterized by 1H NMR spectroscopy (Fig. 1) and GPC. As shown in Fig. 1, the characteristic peak of epoxy at δ 3.34 ppm disappeared, which indicated that the epoxy groups completely reacted. The peaks at δ 7.36, 6.28 and 6.19 ppm are attributed to the furan ring, and the peaks at δ 7.04 and 6.39 ppm are attributed to the benzene ring. It is found that the ratio of the furan ring to the benzene ring is about 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, indicating that the content of FA and BSAEP in the addition polymer is about 1:1. The GPC results show that the number average molecular weight (Mn) is about 9000. Similarly, the linear polymer poly(FA-BPAEP) prepared using BPA epoxy resin was characterized by 1H NMR spectroscopy (Fig. S4†).
:8, indicating that the content of FA and BSAEP in the addition polymer is about 1:1. The GPC results show that the number average molecular weight (Mn) is about 9000. Similarly, the linear polymer poly(FA-BPAEP) prepared using BPA epoxy resin was characterized by 1H NMR spectroscopy (Fig. S4†).
 |
| | Fig. 1
1H NMR spectra of BSAEP and poly(FA-BSAEP) (CDCl3). | |
Thermo-crosslinking reaction of poly(FA-BSAEP) and BDM
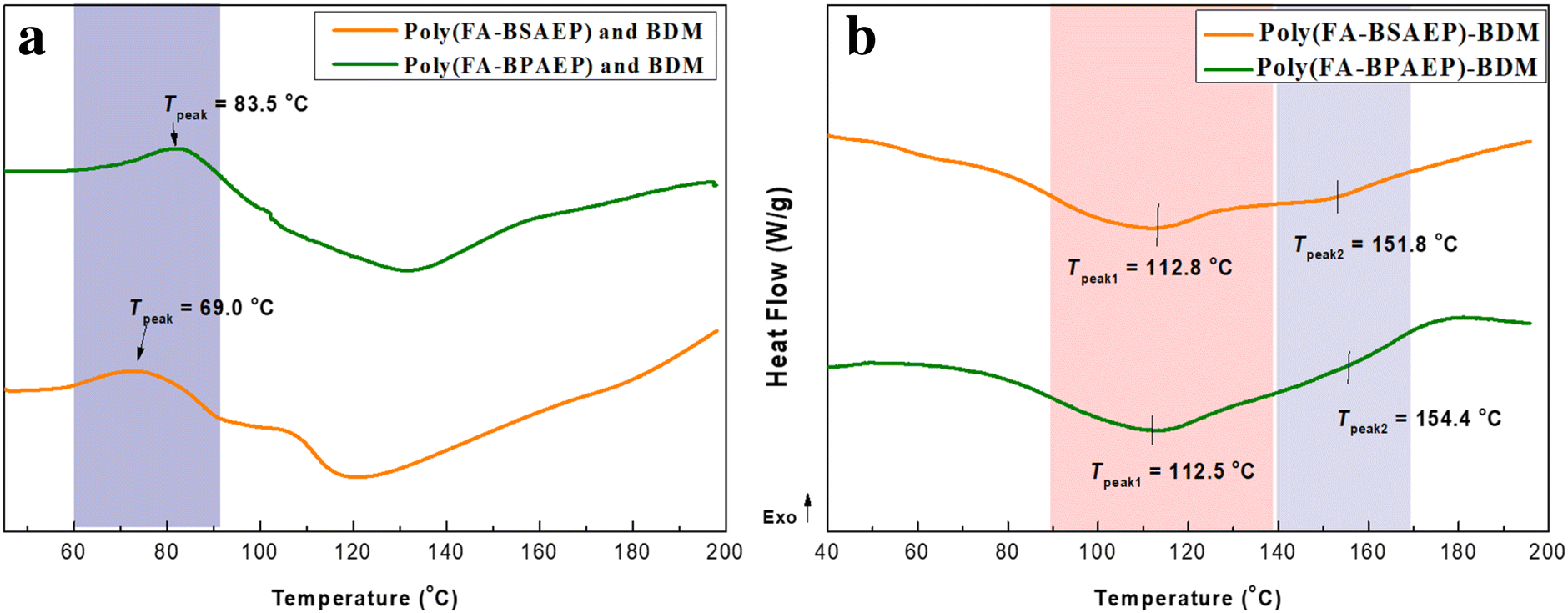
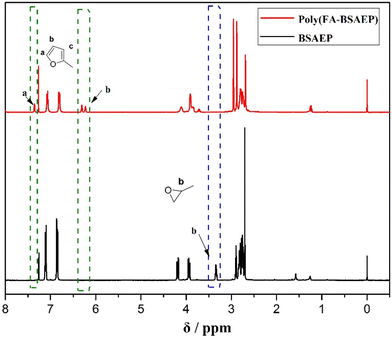
The furan ring can undergo a DA addition reaction with BDM at a certain temperature. Interestingly, the DA addition reaction is reversible at higher temperatures. This reversible property is the key to the preparation of recyclable materials. In this paper, we used BDM as a cross-linking agent and evaluated the polymerization process (Fig. 2a) and depolymerization process (Fig. 2b) by differential scanning calorimetry (DSC). It can be seen from Fig. 2a that due to the similar structure and the same curing group of the two monomers, their curing temperatures were close. The temperature of the DA reaction took place between 60 and 90 °C, and the peak temperatures of poly(FA-BSAEP) and BDM were about 69.0 °C, while the peak temperatures of poly(FA-BPAEP) and BDM were about 83.5 °C. The network dissociation of the retro-Diels–Alder (rDA) reaction can be detected by the appearance of endothermic peaks in DSC (Fig. 2b). After deconvolution of the endothermic peaks, the peak temperature was determined by analyzing the second derivatives of the heat flow; two distinct endothermic peaks were identified in the DSC scans: 112.8 °C and 151.8 °C, which correspond to the dissociation of endo adducts (Tendo) and the dissociation of exo adducts (Texo), respectively. This result is consistent with what has been previously reported in the literature.12 It is worth mentioning that although poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM have different structures, there is only a slight difference in the dissociation temperatures, because they are made of the same reversible units.
 |
| | Fig. 2 DSC curves of (a) the polymerization process (poly(FA-BSAEP) or poly(FA-BPAEP) with BDM) and (b) the depolymerization process (cross-linked polymers poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM). | |
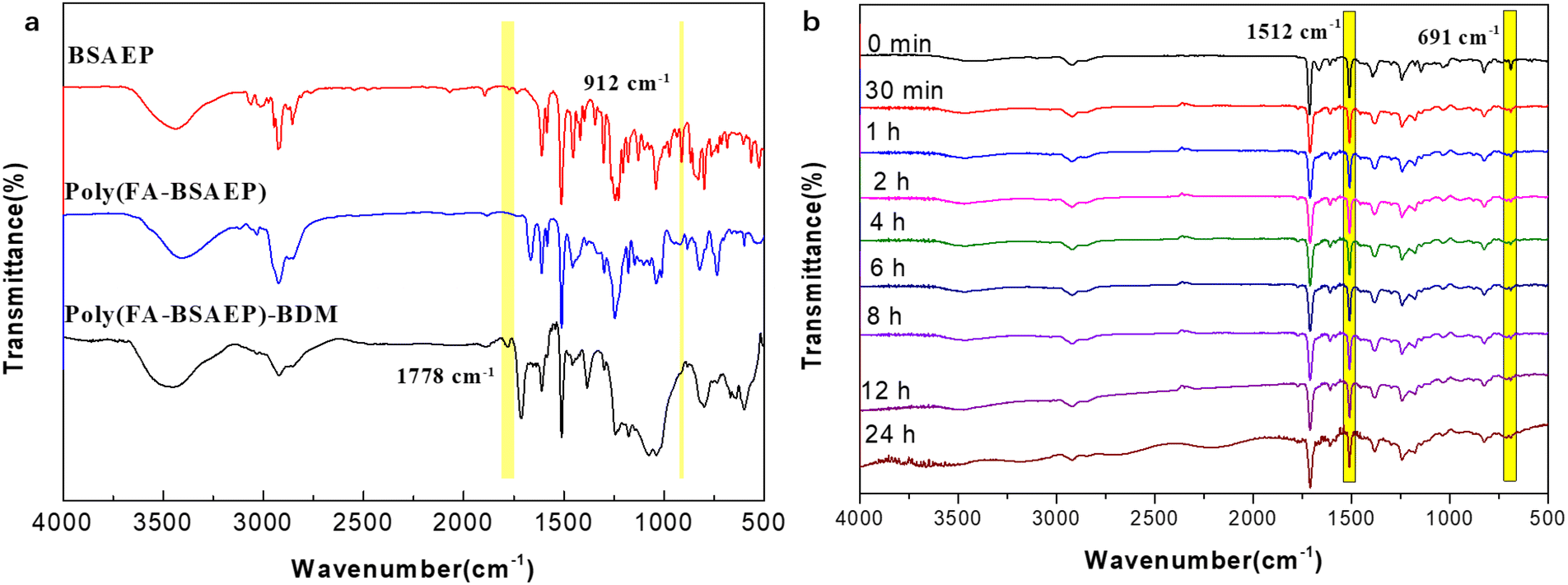
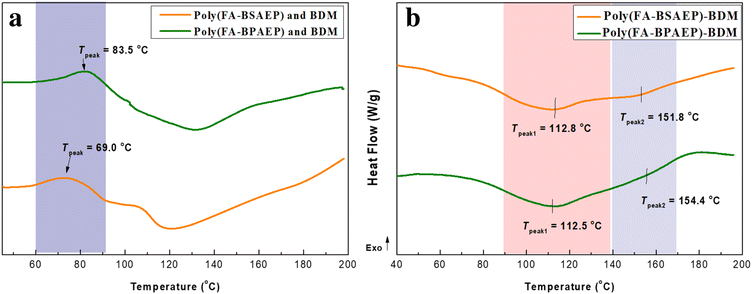
The thermo-curing behavior of the polymers was characterized by FT-IR spectroscopy, as shown in Fig. 3a. The absorption peak at 912 cm−1 attributed to the epoxy group disappeared in poly(FA-BSAEP) and poly(FA-BSAEP)-BDM, indicating that the reaction between the epoxy group and the amino group was complete. The absorption peak at 1778 cm−1 attributed to the succinimide ring generated through the DA reaction appeared in poly(FA-BSAEP)-BDM, indicating the occurrence of the DA reaction. In order to further confirm the optimal reaction temperature, poly(FA-BSAEP) was mixed with BDM and in situ infrared detection was carried out at 60, 70, and 80 °C, respectively. As shown in Fig. 3b for the variation of FT-IR spectroscopies with the time at 70 °C, the absorption peak at 691 cm−1 attributed to the characteristic absorption of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– of BDM gradually decreased and did not change anymore after about 12 h. We chose the absorption peak of the benzene ring (1512 cm−1), which was not involved in the reaction, as the reference peak and characterized the degree of reaction by calculating the relative change of the absorption peak at 691 cm−1. The results of the calculation showed that the degree of reaction was highest at 70 °C, reaching about 80% (Fig. S6†). Similarly, the degree of reaction of poly(FA-BPAEP) with BDM at different temperatures was characterized (Fig. S5 and S6†) and the optimum reaction temperature was 80 °C, which corresponded to the peak temperature of the exothermic peak in the DSC curve (Fig. 2a). It is worth noting that when the curing temperature was higher than 80 °C, the reaction degree had a significant decline, which was due to the lower temperature of the retro-DA reaction.
C– of BDM gradually decreased and did not change anymore after about 12 h. We chose the absorption peak of the benzene ring (1512 cm−1), which was not involved in the reaction, as the reference peak and characterized the degree of reaction by calculating the relative change of the absorption peak at 691 cm−1. The results of the calculation showed that the degree of reaction was highest at 70 °C, reaching about 80% (Fig. S6†). Similarly, the degree of reaction of poly(FA-BPAEP) with BDM at different temperatures was characterized (Fig. S5 and S6†) and the optimum reaction temperature was 80 °C, which corresponded to the peak temperature of the exothermic peak in the DSC curve (Fig. 2a). It is worth noting that when the curing temperature was higher than 80 °C, the reaction degree had a significant decline, which was due to the lower temperature of the retro-DA reaction.
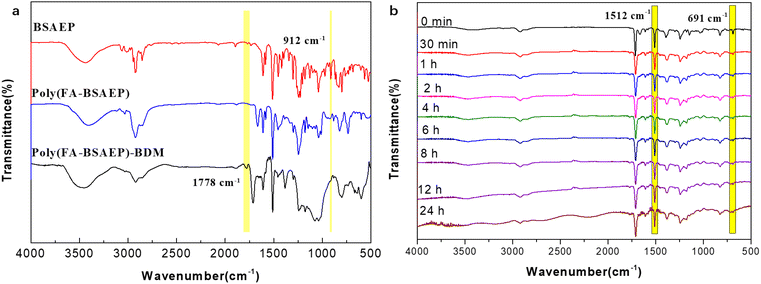 |
| | Fig. 3 FT-IR curves of BSAEP, poly(FA-BSAEP) and poly(FA-BSAEP)-BDM (a), and FT-IR curves (b) of poly(FA-BSAEP) reacted with BDM at 70 °C for different holding times. | |
Mechanical and thermal properties of the cross-linked polymers
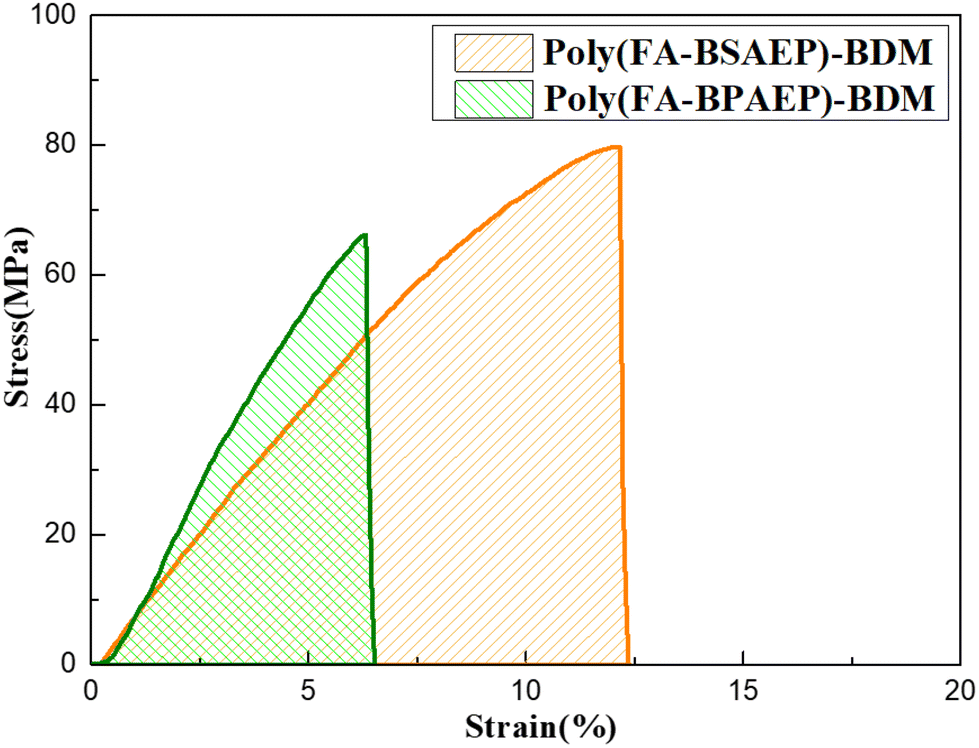
Tensile tests were used to characterize the mechanical properties of both films and the results are shown in Fig. 4. The stress–strain curves showed that poly(FA-BSAEP)-BDM had a tensile strength of 79.7 MPa, while poly(FA-BPAEP)-BDM had a tensile strength of 66.1 MPa. Additionally, poly(FA-BSAEP)-BDM had a high elongation at break of 12.2% compared to poly(FA-BPAEP)-BDM (6.3%), which was excellent compared to other commercially available epoxy resins (1–5%).33,34 This was all because poly(FA-BSAEP)-BDM had more flexible molecular chain segments than poly(FA-BPAEP)-BDM. The tensile modulus of poly(FA-BPAEP)-BDM was 1.38 GPa, which was higher than that of poly(FA-BSAEP)-BDM (0.86 GPa), indicating that poly(FA-BPAEP)-BDM was more rigid. It is worth mentioning that by calculating the area of the stress–strain curve, we found that the area of poly(FA-BSAEP)-BDM was about 3 times that of poly(FA-BPAEP)-BDM, which indicated that poly(FA-BSAEP)-BDM had better toughness than BPA epoxy resin.
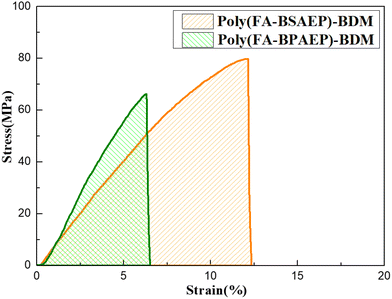 |
| | Fig. 4 The tensile tests of poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM. | |
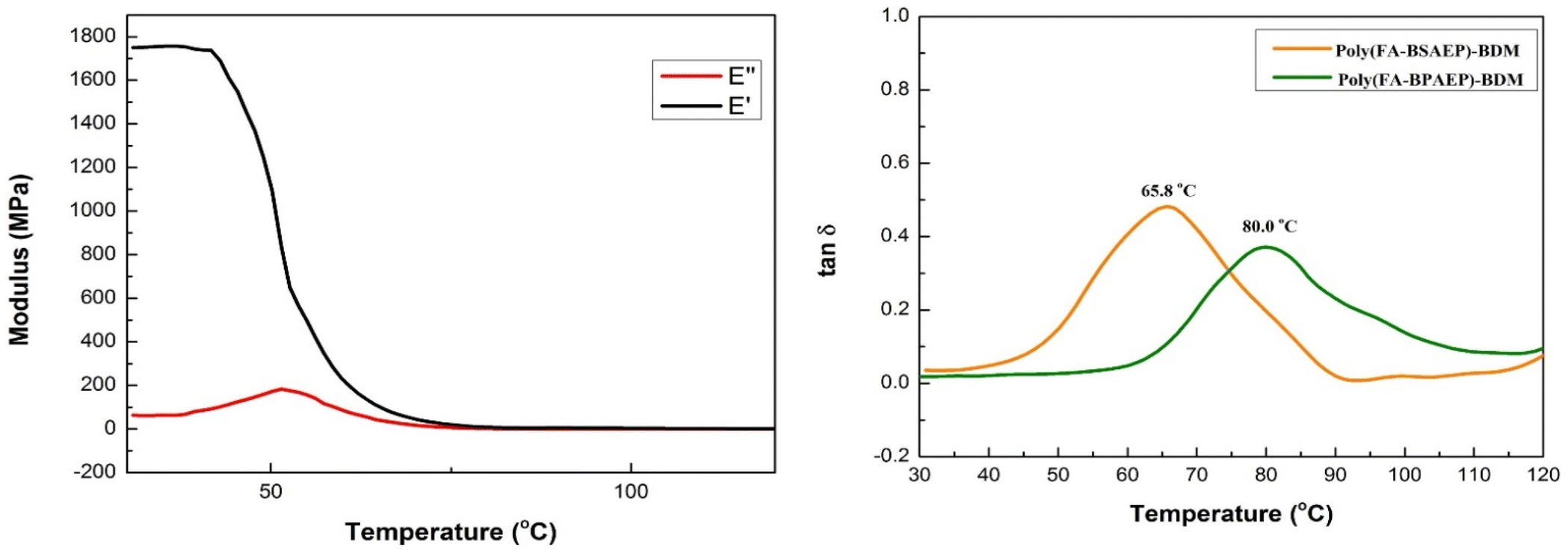
In order to further study the thermomechanical properties of these resins, dynamic mechanical analysis (DMA) was carried out. It can be seen from the DMA curves (Fig. 5 and Fig. S8†) that at room temperature, the storage modulus (E′) values of poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM were 1.7 GPa and 2.8 GPa, respectively. This gap was caused by poly(FA-BSAEP)-BDM having a more flexible molecular chain than poly(FA-BPAEP)-BDM. With the increase of temperature, the E′ of poly(FA-BSAEP)-BDM decreased rapidly and eventually approached 0 MPa; especially when the temperature was higher than 90 °C, both E′ and E′′ were almost 0 MPa. This process was accompanied by the fracture of some DA bonds, after which the polymer can be reprocessed. The Tg can be characterized by the change of tanδ. It can be concluded from the curve that the Tg of poly(FA-BSAEP)-BDM was 65.8 °C, and the Tg of poly(FA-BPAEP)-BDM was up to about 80.0 °C, indicating that both poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM have good thermal properties.
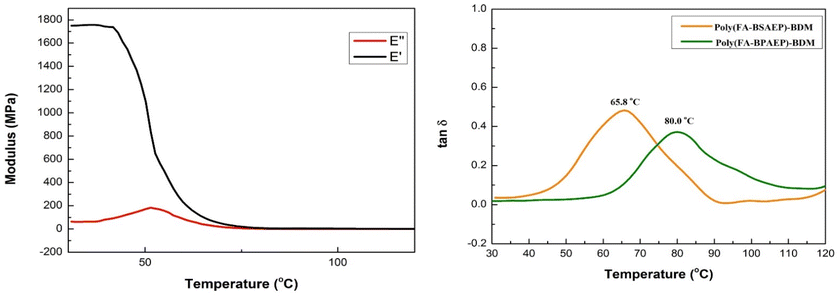 |
| | Fig. 5 DMA curves of poly(FA-BSAEP)-BDM (left) and tanδ of poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM (right). | |
Thermogravimetric analysis (TGA) was used to study the thermal stability of the two resins. As shown in Fig. 6, the heat resistance of poly(FA-BSAEP)-BDM was almost close to that of poly(FA-BPAEP)-BDM. Under a nitrogen atmosphere, the 5% weight loss temperature (Td5) of both poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM was 316.0 °C. This proves that the synthesized biomass-based resins have heat resistance comparable to that of commercially available resins.
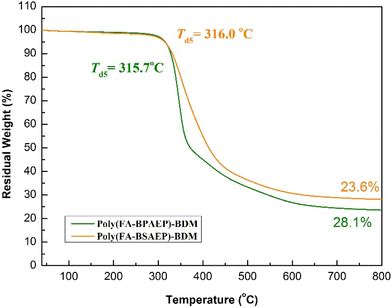 |
| | Fig. 6 Thermogravimetric analysis of poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM. | |
To further compare the thermal stability and mechanical properties of poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM, the detailed data are listed in Table 1. It can be observed that the thermal stability of poly(FA-BSAEP)-BDM is comparable to that of poly(FA-BPAEP)-BDM. However, due to the presence of flexible connecting segments in its structure, poly(FA-BSAEP)-BDM has a relatively lower energy storage modulus and Young's modulus of elasticity compared to poly(FA-BPAEP)-BDM, but the tensile strength and elongation at break of poly(FA-BSAEP)-BDM are higher. Because their DA adducts are composed of 2-furfurylamine and BDM, they exhibit similar reaction temperatures. This is consistent with the “furfurylamine-BDM” reversible system reported in the previous literature.12
Table 1 The mechanical and thermal properties of poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM
| Resin |
T
rDA/°C |
T
d5/°C |
T
g/°C |
E′/GPa |
E/GPa |
σ
t/MPa |
ε/% |
|
E′: storage modulus, E: Young's modulus, σt: tensile strength, and ε: elongation. |
|
Poly(FA-BSAEP)-BDM
|
112.5 & 154.4 |
316.0 |
65.8 |
1.8 |
0.86 |
79.7 |
12.2 |
|
Poly(FA-BPAEP)-BDM
|
113.2 & 151.8 |
316.0 |
80 |
2.8 |
1.38 |
66.1 |
6.3 |
Recyclability of the crosslinked polymer films (poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM)
From the DSC curves, we can see that the TrDA of poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM is about 112 °C. DMA curves show that E′ and E′′ of the material are almost 0 MPa at this temperature, and the materials are processable. This property makes it possible for the films to be physically recycled through hot-pressing. Fig. 7 shows the recycling diagram of poly(FA-BSAEP)-BDM. Due to its flexible molecular structure, the film can be recreated by applying a pressure of only 1 MPa to the film fragments. In the process of hot-pressing recovery of poly(FA-BPAEP)-BDM, we found that its relatively rigid structure limited the movement of molecules, so that higher pressure was needed to ensure adequate contact of molecules on the fracture surface during recovery. At the same temperature and time, a pressure of 5 MPa was required to obtain a complete polymer film.
 |
| | Fig. 7 Physical recycling of poly(FA-BSAEP)-BDM through hot-pressing at 110 °C under 1 MPa pressure. | |
In addition, we also attempted to recycle the polymer films by degradation in solution, taking into account the nature of DA bond breaking (Fig. S9†). We have found that these polymer films can be dissolved in DMF at 110 °C, and after removing the solvent, the polymer film can be obtained again. We attempted to reisolate the precursor polymer (poly(FA-BSAEP)) after degradation, but the results were not satisfactory. Although the film can be completely dissolved in solution and the solution was uniformly transparent at room temperature, because 2-furfurylamine has low steric hindrance and high electron cloud density, the DA reaction can occur rapidly in a short time to form crosslinked polymers, which makes it difficult to recover raw materials to a certain extent.7
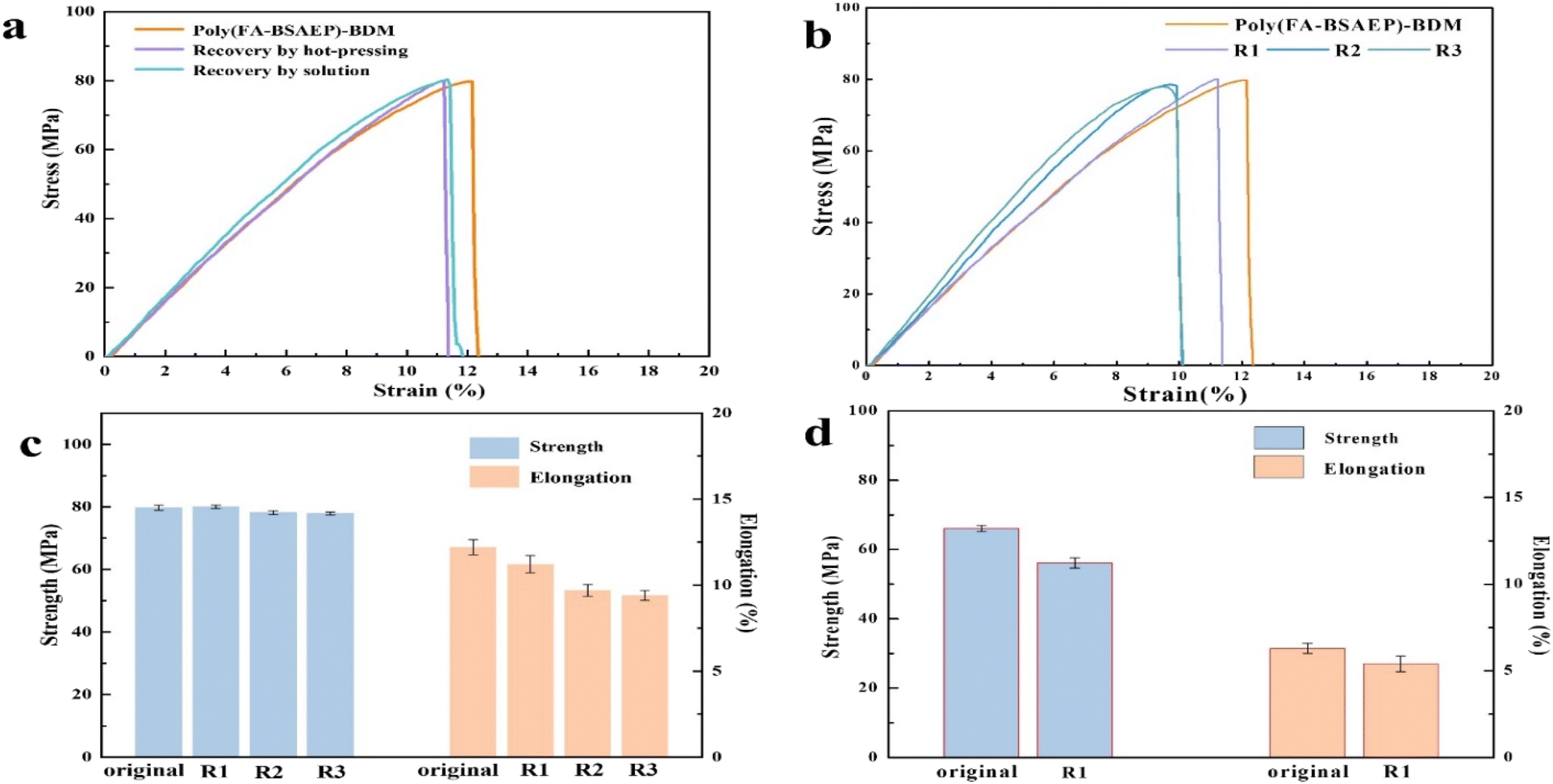
To assess the impact of recycling, we compared the tensile properties of the films before and after the process. Fig. 8a and b show the changes of the tensile properties of the two films before and after reprocessing by hot-pressing and solvent methods. No matter which method was used for reprocessing, the properties of poly(FA-BSAEP)-BDM changed very little, while the properties of poly(FA-BPAEP)-BDM were significantly reduced (Fig. S7†), which may be due to its rigid skeleton structure limiting the movement of the molecular chain segments, so that the contact surface cannot adequately react. Fig. 8c shows the performance changes of poly(FA-BSAEP)-BDM before and after three cycles of hot-pressing recycling; the data are presented in Table S1.† After three cycles of recovery, the tensile strength and elongation of the film decreased slightly, but the changes were not significant. In contrast, the recovered poly(FA-BPAEP)-BDM showed poor performance. When the film was recycled two times, the resulting film was very brittle and could not be cut into the shape required for the tensile test, probably due to the rigidity of its own molecular structure.
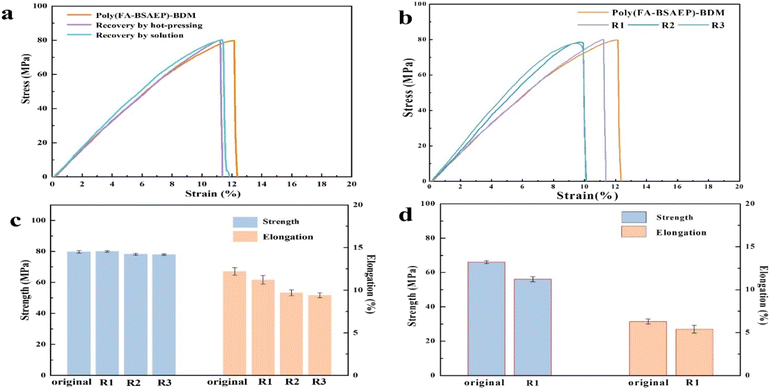 |
| | Fig. 8 The tensile tests of reprocessed poly(FA-BSAEP)-BDM ((a) processing using different methods and (b) and (c) recycling three times by hot-pressing) and poly(FA-BPAEP)-BDM ((d) recycling three times by hot-pressing). | |
Optical properties of poly(FA-BSAEP)-BDM
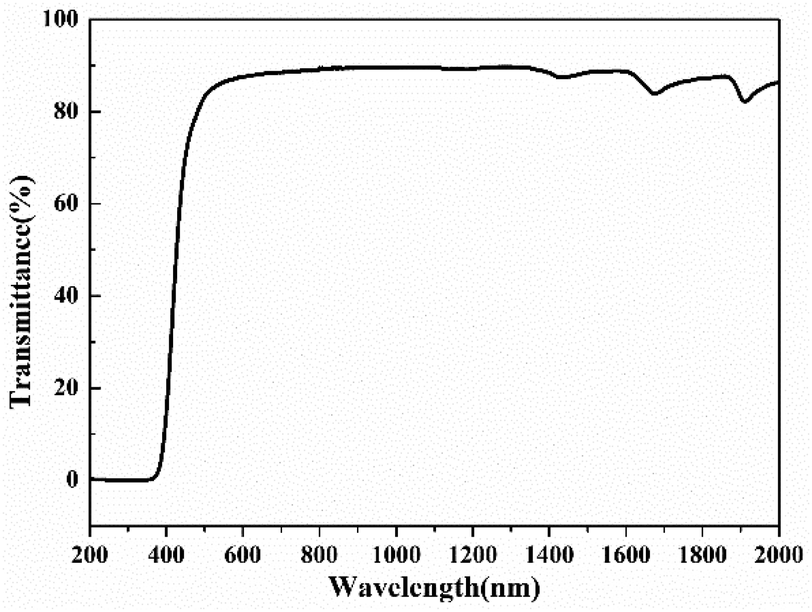
The transmittance of the self-supporting film was measured by UV-vis-NIR spectroscopy. As shown in Fig. 9, poly(FA-BSAEP)-BDM films (the thickness is 0.14 mm) have a transmittance of up to 89% in the wavelength range of 550–1200 nm. Its good transmittance is expected to provide a reference for the application of optical devices.
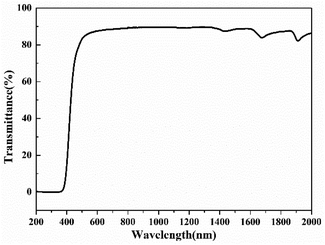 |
| | Fig. 9 UV-vis-NIR spectra of poly(FA-BSAEP)-BDM. | |
Conclusions
Using anisaldehyde as the raw material, a new epoxy resin containing flexible organic chain segments was synthesized by several steps of reactions. The furan ring was successfully introduced into a polymer by the reaction of furfurylamine with the epoxy resin, and a recyclable thermosetting polymer poly(FA-BSAEP)-BDM was prepared through a unique dynamic covalent crosslinking strategy of the DA reaction of the furfuryl ring with the maleimide ring. Similarly, a recyclable thermosetting polymer poly(FA-BPAEP)-BDM was prepared from BPA epoxy resin by the same method for comparison. Both poly(FA-BSAEP)-BDM and poly(FA-BPAEP)-BDM have high storage moduli at room temperature, which are 1.8 GPa and 2.8 Gpa, respectively. Poly(FA-BSAEP) and BDM can undergo the DA reaction at 70 °C, and the DA bond breaking at 110 °C allows the materials to be reprocessed either by hot-pressing or in solution. It is worth mentioning that the presence of flexible organic chain segments makes the intermolecular movement of poly(FA-BSAEP)-BDM more flexible, which gives it good toughness and allows it to be reprocessed under moderate conditions. Although after three cycles of recycling, the elongation of poly(FA-BSAEP)-BDM was greater than 10%, which was superior to that of BPA epoxy resin (<6.5%). Moreover, poly(FA-BSAEP)-BDM exhibits high transmittance in the visible and infrared regions, showing potential value as an infrared transmitting material. In summary, the recyclable thermosets prepared from anisaldehyde have excellent thermal and mechanical properties, and the more flexible structure than BPA epoxy resin gives them better fracture toughness and recyclability. This makes it a promising candidate for practical applications and provides an effective solution for the recovery of thermosetting resins.
Data availability
Data for this article, including all structural representations and performance representations, are available in the manuscript and ESI.†
Conflicts of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (JKD01241701).
References
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- L. Imbernon and S. Norvez, Eur. Polym. J., 2016, 82, 347–376 CrossRef CAS.
- Y. Tao, L. Fang, J. Zhou, C. Wang, J. Sun and Q. Fang, ACS Appl. Polym. Mater., 2019, 2, 295–303 CrossRef.
- S. J. Rowan, S. J. Cantrill, G. R. Cousins, J. K. Sanders and J. F. Stoddart, Chem. Commun., 2022, 58, 11103–11106 RSC.
- J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- T. P. Corbett, J. Leclaire, L. Vial, K. R. West, J. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 Search PubMed.
- Y. Guo, L. Yang, L. Zhang, S. Chen, L. Sun, S. Gu and Z. You, Adv. Funct. Mater., 2021, 31, 2106281 CAS.
- J. Hou, M. Liu, H. Zhang, Y. Song, X. Jiang, A. Yu, L. Jiang and B. Su, J. Mater. Chem. A, 2017, 5, 13138–13144 RSC.
- J. S. Kim, J. H. Kim, Y. Cho and T. S. Shim, Polymers, 2019, 11, 875 CrossRef CAS PubMed.
- H. Wang, J. Lu, H. Huang, S. Fang, M. Zubair and Z. Peng, J. Colloid Interface Sci., 2021, 588, 295–304 CrossRef CAS PubMed.
- Y. Guo, S. Chen, L. Sun, L. Yang, L. Zhang, J. Lou and Z. You, Adv. Funct. Mater., 2020, 31, 2009799 Search PubMed.
- Q. Zhou, Z. Sang, K. K. Rajagopalan, Y. Sliozberg, F. Gardea and S. A. Sukhishvili, Macromolecules, 2021, 54, 10510–10519 CAS.
- J. Wang, D. Chen, S. Xing, B. Ji, J. Yang and J. Tang, ACS Appl. Polym. Mater., 2024, 6, 466–474 CAS.
- Y. S. Ryu, K. W. Oh and S. H. Kim, Macromol. Res., 2016, 24, 874–880 CAS.
- K. Heng, J. Zhang, C. Wang, K. Wang, L. Wan and F. Huang, e-Polym., 2023, 23, 20230119 CAS.
- J. Wang, J. Li, J. Zhang, S. Liu, L. Wan, Z. Liu and F. Huang, Polymers, 2023, 15, 466–474 CrossRef PubMed.
- J. Yu, N. Gao, X. Xie, X. Xin, Z. Li, S. Chen and J. Xu, J. Polym. Sci., 2021, 60, 794–802 CrossRef.
- M. Thys, J. Brancart, G. Assche, R. Vendamme and N. Brande, Macromolecules, 2021, 54, 9750–9760 CrossRef CAS.
- X. Shen, X. Liu, J. Wang, J. Dai and J. Zhu, Ind. Eng. Chem. Res., 2017, 56, 8508–8516 CAS.
- S. Kumar, S. Krishnan, S. Mohanty and S. K. Nayak, Polym. Int., 2018, 67, 815–839 CrossRef CAS.
- L. Fang, J. Zhou, Y. Tao, Y. Wang, X. Chen, X. Chen, J. Hou, J. Sun and Q. Fang, ACS Sustainable Chem. Eng., 2019, 7, 4078–4086 Search PubMed.
- X. Chen, L. Fang, X. Chen, J. Zhou, J. Wang, J. Sun and Q. Fang, ACS Sustainable Chem. Eng., 2018, 6, 13518–13523 CAS.
- J. Ma, G. Li, X. Hua, N. Liu, Z. Liu, F. Zhang, L. Yu, X. Chen, L. Shang and Y. Ao, Polym. Degrad. Stab., 2022, 201, 109989 CAS.
- Y.-Y. Liu, J. He, Y.-D. Li, X.-L. Zhao and J.-B. Zeng, Ind. Crops Prod., 2020, 153, 112576 CrossRef CAS.
- C. Ocando, Y. Ecochard, M. Decostanzi, S. Caillol and L. Avérous, Eur. Polym. J., 2020, 135, 109860 CAS.
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 8, 255–260 CAS.
- W. Yang, Q. Yi, F. Liu, X. Pan and Y. Zeng, Eur. Polym. J., 2023, 196, 112278 CAS.
- S. Zhang, T. Liu, C. Hao, L. Wang, J. Han, H. Liu and J. Zhang, Green Chem., 2018, 20, 2995–3000 CAS.
- C. Hao, T. Liu, S. Zhang, L. Brown, R. Li, J. Xin, T. Zhong, L. Jiang and J. Zhang, ChemSusChem, 2019, 12, 1049–1058 CrossRef CAS PubMed.
- Y. Tao, L. Fang, M. Dai, C. Wang, J. Sun and Q. Fang, Polym. Chem., 2020, 11, 4500–4506 RSC.
- L. Zhong, Y. Hao, J. Zhang, F. Wei, T. Li, M. Miao and D. Zhang, Macromolecules, 2022, 55, 595–607 CrossRef CAS.
- P. Wu, L. Liu and Z. Wu, Macromol. Mater. Eng., 2020, 305, 2000359 CrossRef CAS.
- B. De and N. Karak, J. Mater. Chem. A, 2013, 1, 348–353 RSC.
- J. M. González-Domínguez, A. M. Díez-Pascual, A. Ansón-Casaos, M. A. Gómez-Fatou and M. T. Martínez, J. Mater. Chem., 2011, 21, 14948–14958 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, experimental details and supplemental figures. See DOI: https://doi.org/10.1039/d4py00759j |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Weibo
Liu
,
Keying
Wang
,
Kaili
Wang
,
Bowen
Deng
,
Liqiang
Wan
,
Qiaolong
Yuan
and
Farong
Huang
,
Weibo
Liu
,
Keying
Wang
,
Kaili
Wang
,
Bowen
Deng
,
Liqiang
Wan
,
Qiaolong
Yuan
and
Farong
Huang