
Photoinitiated thermoset polymerization through controlled release of metathesis catalysts encapsulated in poly(phthalaldehyde)†
Oleg
Davydovich
 a,
Josephine
Lewis
a,
Mikayla
Romero
a,
Julia
Deitz
a,
Francesca
C'deBaca
a,
Jared M.
Schwartz
b,
Anthony C.
Engler
c,
Paul A.
Kohl
b,
Samuel C.
Leguizamon
a and
Brad H.
Jones
*a
a,
Josephine
Lewis
a,
Mikayla
Romero
a,
Julia
Deitz
a,
Francesca
C'deBaca
a,
Jared M.
Schwartz
b,
Anthony C.
Engler
c,
Paul A.
Kohl
b,
Samuel C.
Leguizamon
a and
Brad H.
Jones
*a
aSandia National Laboratories, P.O. Box 5800, MS 0888, Albuquerque, NM 87185, USA. E-mail: bhjones@sandia.gov
bChemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
cCain Department of Chemical Engineering, Louisiana State University, Baton Rouge, LA 70803, USA
First published on 27th November 2024
Abstract
Photoinitiated polymerization enables spatiotemporal control of reaction conditions and can thereby generate materials with high complexity while consuming minimal energy. Where ring opening metathesis polymerization (ROMP) is concerned, photo-activated processes are typically enabled by chemical inhibition of ruthenium carbenes via the careful design of complexed ligands such that photoactivation can proceed through an isomerization or ligand dissociation event. In this contribution, we have explored a new approach to photoinitiation of ROMP based on physical inhibition through microencapsulation and controlled release of metathesis catalysts. Micron-sized particles of poly(phthalaldehyde) (PPA), catalyst, and photoacid generator were fabricated by spray drying. The particles were dispersed in dicyclopentadiene monomer, after which polymerization was initiated through temperature or UV exposure, both inducing depolymerization of the PPA particles and in situ catalyst release. The monomer/particle dispersions were found to be stable and reproducibly polymerizable with 3 weeks of storage at room temperature. Furthermore, the dispersions can be used for both photo- and thermal-initiated frontal ROMP, yielding a polymerized thermoset of equivalent properties to conventional bulk- and frontally-polymerized analogues. This work will ultimately enable new manufacturing techniques for ROMP-based materials, due to the modular, easily tunable nature of the underlying initiating system and its unparalleled stability.
Introduction
Ring opening metathesis polymerization (ROMP) has become a mainstay of polymer chemistry, rivalling established techniques including free radical and condensation polymerizations. Early research in ROMP1,2 that led to the discovery of materials mimicking conventional polyolefins has since been expanded to encompass a plethora of building blocks and architectures, producing materials with advantageous features ranging from nanostructured morphologies3 to specific functional properties4–6 and even inherent recyclability.7 One example, polydicyclopentadiene (pDCPD), a tough thermosetting polymer with high impact strength and thermal and chemical stability, is created by the ROMP of the highly strained monomer dicyclopentadiene (DCPD).8 pDCPD has emerged as a potential alternative to epoxies, acrylonitrile-butadiene-styrene, and other comparable polymers in automotive, military, and consumer applications. There is also widespread interest currently in additive manufacturing (AM) of pDCPD and many other materials created by ROMP, due to the potential for rapid formation of complex structures whilst minimizing waste and energy consumption.9–15From a manufacturing perspective, particularly where AM is concerned, the initiation or modulation of ROMP by light is a powerful tool that enables spatiotemporal control over the polymerization with minimal energy input. Yet, photo-initiated ROMP systems currently lack the stability, versatility, and commercial availability of radical-mediated polymerizations, which has limited uptake of such systems in comparison to acrylic resins. For the most part, photo-initiation of ROMP has been achieved by the design and introduction of latent ruthenium carbene catalysts that are subsequently activated by ultraviolet (UV) or visible light. Eivgi and Lemcoff16 and, more recently, Greenlee, et al.17 have summarized the available strategies for photo-initiated ROMP in perspective articles. Direct photo-activation by isomerization or ligand dissociation is possible in several classes of metathesis catalysts, for example the S-chelated compounds pioneered by the Lemcoff group.18,19 Indirect photo-activation is similarly driven by chemical transformation of a latent catalysis; however, it is through the reaction of the catalyst with a complementary species generated by irradiation, such as an acid,20,21 oxidant,22,23 or sensitizer.10,12 In contrast, physical control over initiation, rather than chemical control, may be more accessible to engineers, designers, and formulators as it does not necessarily rely on catalyst latency, opening the door to light-driven manufacturing with the entire range of commercial metathesis catalysts. However, to the best of our knowledge, there is only one prior example of photophysical activation of ROMP where microcapsules containing 2nd generation Grubbs catalyst (GC2) and carbon nanotubes were photothermally ruptured to release the catalyst and effectively activate ROMP.24 While this work demonstrated long-term resin stability and successful ROMP of DCPD, the release mechanism – relying on the conversion of laser light to heat by the carbon nanotubes thereby rupturing a solvent-filled capsule – is likely incompatible with most UV- and visible-based AM modalities or, at a minimum, would give extraordinarily slow polymerizations. Moreover, it is unclear whether this approach would be viable for emerging polymerization techniques of interest, such as frontal polymerizations. Finally, the use of solvent-filled capsules is undesirable due to the subsequent incorporation of solvent into the polymerized product.
We have discovered the remarkable stability of encapsulated catalysts in frontal ROMP (FROMP) resins such as DCPD.25 FROMP involves the local initiation of polymerization, after which a polymerization front develops and propagates through a sample sustained by its own exothermic release of heat.26 The primary advantage of frontal polymerization is that energy input is minimized relative to bulk polymerization as the system efficiently utilizes its intrinsic energy density. The thermodynamic driving force for polymerization arises from cyclic olefin monomers that are highly strained like dicyclopentadiene and cyclooctadiene which enables the propagation of thermal front. Again, FROMP is typically performed with a latent or otherwise inhibited catalyst, one that is then thermally activated.27–30 Our previous work demonstrated that microencapsulation of non-latent, highly active catalysts, such as the common 2nd generation Hoveyda–Grubbs catalyst (HG2) is markedly superior to the conventional modus operandi where FROMP is concerned.25 Microencapsulated HG2 is stable in DCPD indefinitely (>1 year), in comparison to only several days for typical latent catalyst FROMP systems, yet gives high and tunable front rates whilst yielding pDCPD of comparable thermomechanical properties.
Building on this discovery, we sought to expand our approach to encapsulate and release similar (non-latent) catalysts triggered by light. Indeed, there are few prior examples of FROMP initiated by light. Stawiasz, et al. showed that DCPD containing GC2 inhibited by various phosphine species can undergo FROMP when irradiated with 375 nm light, likely due to dissociation of the inhibiting ligand.31 Dean, et al. found that the same resin system can be photothermally initiated with the inclusion of various nanoscopic forms of carbon.32 Finally, Stawiasz, et al. demonstrated that a bis-N-heterocyclic carbene catalyst in conjunction with a photoredox co-catalyst and a copper transmetallating agent to stimulate ligand exchange undergo FROMP with blue light.33 These examples all rely on ligand dissociation from a precatalyst (ROMP inactive species), which is inherently characterized by reduced and poorly tunable reactivity under frontal conditions.
In contrast to these prior examples, we hypothesized that physical inhibition of metathesis catalysts via microencapsulation would: (i) drastically improve long-term stability, and (ii) enable polymerization characteristics to be tuned rationally through particle design. Here, we utilize cyclic poly(phthalaldehyde) (cPPA) as an encapsulant. PPA is a metastable polymer with high glass transition temperature (Tg), but also a low ceiling temperature (Tc = −36 °C).34 In its cyclic form, it is rendered kinetically stable and spontaneously depolymerizes to its constituent monomer, ortho-phthalaldehyde, only when heated to temperatures in excess of 150 °C to cleave backbone bonds and trigger unzipping of the polymer chain.34–38 We have previously established that a PPA copolymer effectively inhibits encapsulated HG2, yet very efficiently releases the catalyst in situ when heated.39 PPA is also destabilized by catalytic quantities of acid and other bond-cleaving species. The light-triggered depolymerization of PPA is facile when the polymer is loaded with appropriate photoactive compounds, such as a photoacid generator (PAG).40–45 In this work, the co-encapsulation of metathesis catalysts and PAGs in cPPA was explored. Gratifyingly, we report here that these unique formulations enable photoinitiated ROMP and FROMP of DCPD with excellent resin stability and tunable polymerization characteristics.
Results and discussion
Our initial objective was to fabricate microparticles consisting primarily of cPPA encapsulant, with PAG and metathesis catalyst as co-encapsulated cargoes (Fig. 1). The synthetic procedure and relevant characteristics of the cPPA encapsulant used in our work are provided in the Experimental section. Various procedures for microencapsulation of cargo within cPPA or other polyaldehydes have been reported. These procedures rely on emulsification of a lipophilic solution of polymer and cargo in an aqueous matrix, after which the emulsified droplets are converted to core–shell microcapsules or dense microparticles.46–49 UV-triggered breakdown of core–shell microcapsules has even been claimed in one distinct case by Eriksson, et al., where direct irradiation of low molecular weight, linear PPA led to apparent release of the core contents despite the lack of a photoactive cargo (i.e., PAG).49 In our initial tests, however, we found it unnecessarily challenging to co-encapsulate PAG and metathesis catalyst in cPPA using emulsification strategies. Uncontrolled partitioning of the cargoes between phases led to low and unpredictable encapsulation efficiency.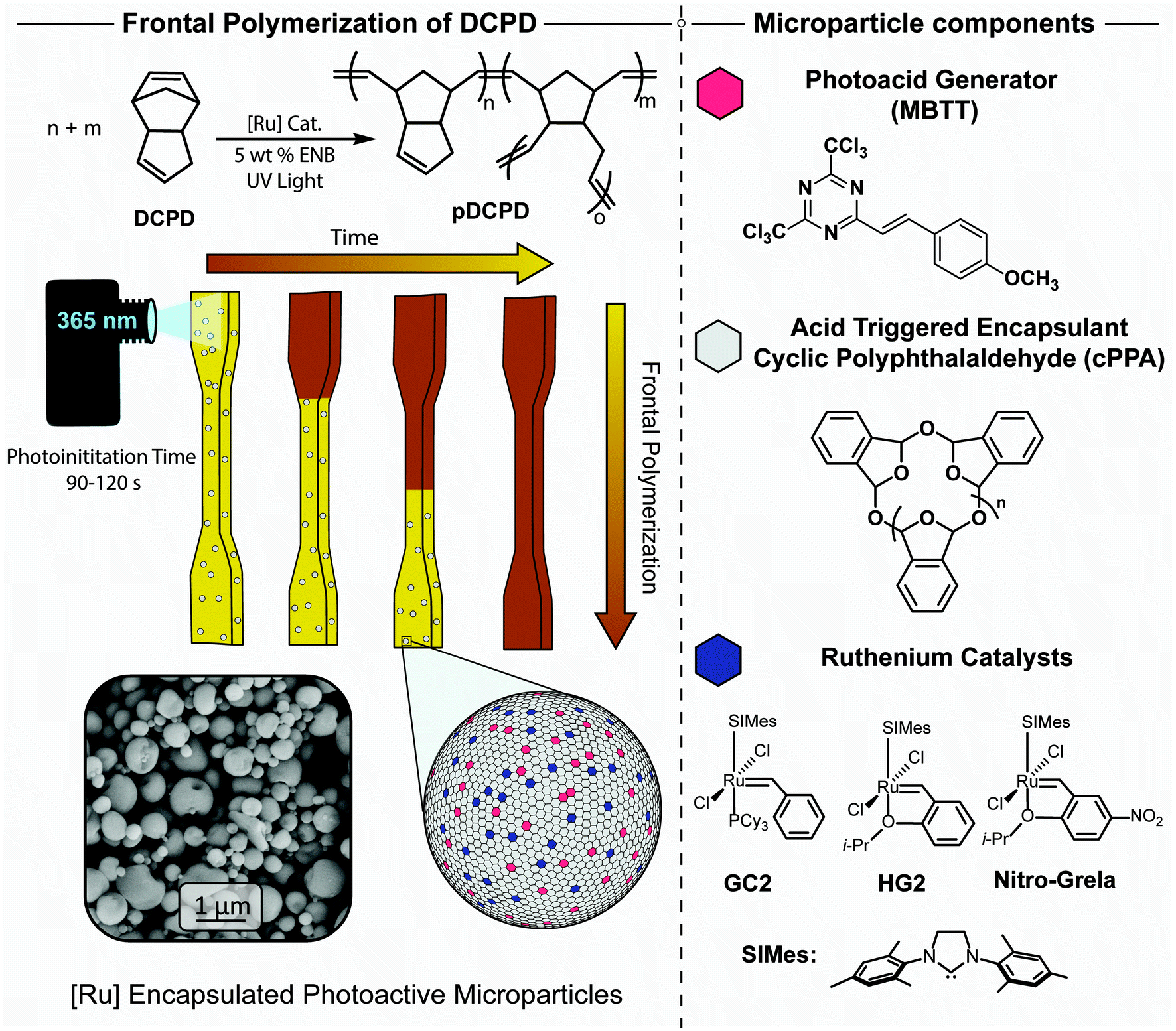 | ||
| Fig. 1 Photoinitiated thermoset polymerization through controlled release of metathesis catalysts encapsulated in poly(phthalaldehyde). PAG (MBTT) and catalysts such as GC2, HG2, and nitro-Grela are co-encapsulated in cPPA yielding photo and thermally triggered microparticles. These particles are stable in DCPD, but trigger FROMP to pDCPD when heated or when illuminated with UV light. The image shown at bottom left was acquired by scanning electron microscopy (SEM). | ||
Instead, we successfully generated the desired microparticles via spray drying, a widely used technique in the food,50 pharmaceutical,51 and coatings industries.52 Microencapsulation via spray drying involves the aerosolization of a solution containing a cargo (either homogenous or suspended mixtures) that is flowed into a heater via a carrier gas and collected as a dry powder (ESI, Fig. S10†).53 This method is particularly versatile since it removes the need for emulsification, typically requiring careful control of solution formulations. Further, spray drying can be run continuously which enables scalable manufacturing of microparticles at rates exceeding several tons per h. We spray dried a homogenous dichloromethane solution containing cPPA, catalyst, and PAG to generate photoactive microparticles (PMPs) ranging from 0.2 to 2 μm diameter. The microparticles displayed a combination of spherical, buckled, and irregular morphologies (Fig. 1).541H nuclear magnetic resonance (NMR) spectroscopy shows that 95% of HG2 and 75% of MBTT were successfully encapsulated (Experimental section and ESI, Fig. S1†). Thermogravimetric analysis (TGA) of the PMPs showed that the particles were thermally stable until 130 °C to 150 °C at which point cPPA began to depolymerize to monomer (ESI, Fig. S2†).
After successfully generating PMPs, we evaluated their ability to initiate ROMP of DCPD. To our satisfaction, we found that the PMPs could be dispersed in DCPD (e.g., by bath sonication) with no apparent DCPD polymerization, indicating that the encapsulated HG2 was well-sequestered by cPPA. Upon heating above ca. 80 °C, the dispersions rapidly polymerized, exhibiting a sharp exotherm and rapid increase in modulus (ESI, Fig. S3†). Given that the onset of DCPD polymerization occurred well below the depolymerization temperature of cPPA, we presume that thermal release of HG2 from PMPs is driven by increased solubility or miscibility of cPPA in DCPD, as in our previous work.25 In addition to thermal release, encapsulated PAGs are known to liberate acid upon UV irradiation which in turn triggers cPPA depolymerization. This results in microparticle breakdown, which releases the highly active HG2 into the surrounding DCPD solution, thereby initiating ROMP. Our goals were to (i) generate a viable PMP system capable of photoinitiating ROMP and (ii) understand the underlying factors that govern photoinitiation. Specifically, we evaluated PAGs with different absorption characteristics, PAG concentration, metathesis catalyst type, and temperature (ESI, Table S1†).
First, we characterized photoinitiated ROMP of DCPD/PMP dispersions in a rheometer equipped with a UV light source. We determined the onset of ROMP by evaluating the storage modulus from small amplitude oscillatory shear imposed under continuous illumination with broadband or 365 nm radiation. We screened two different PAGs, 4-isopropyl-4′-methyldiphenyliodonium tetrakis(pentafluorophenyl)borate (FABA) and 2-(4-methoxystyryl)-4,6-bis(trichloromethyl)-1,3,5-triazine (MBTT), which absorb primarily near 254 nm and 379 nm, respectively (ESI, Fig. S4†), and have been reported to depolymerize cPPA.43,55 PMPs containing FABA did not show any significant ability to polymerize DCPD when irradiated with a broadband source at intensities as high as 356 mW cm−2. Successful polymerization only occurred with a combination of high intensity broadband UV irradiation and heating; even still, the influence of UV radiation was rather limited (ESI, Fig. S5†). Presumably, given that the broadband source had little output below 300 nm,56 poor optical absorption limited the efficient generation of acid from the FABA. On the other hand, MBTT successfully caused polymerization at room temperature with 365 nm radiation (Fig. 2). Thus, subsequent work used MBTT as the PAG. We define photoinduction time (PIT) as the irradiation duration required to generate a measurable storage modulus value above the noise, roughly 10 Pa. Thus, a small PIT is associated with faster PMP breakdown.
 | ||
Fig. 2 Photorheology of DCPD/PMP dispersions with 365 nm irradiation at 27 mW cm−2. A. PMPs with varied PAG (MBTT) loadings. B. Varied sample temperature. C. Varied PMP concentration in DCPD. D. PMPs with different metathesis catalyst selection. Photoinduction time (PIT) is determined by the time at which the storage modulus (G′) reaches 10 Pa, as indicated by the arrows in panel A. Unless deliberately varied, the MBTT and metathesis catalyst are present in PMPs at 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 MBTT:cPPA and 12:88 catalyst:cPPA by wt, the PMPs are added to DCPD at 8 mg ml−1, the dispersions are irradiated at 25 °C, and the sample thickness is 0.5 mm. :100 MBTT:cPPA and 12:88 catalyst:cPPA by wt, the PMPs are added to DCPD at 8 mg ml−1, the dispersions are irradiated at 25 °C, and the sample thickness is 0.5 mm. | ||
We hypothesized that cPPA depolymerization is the rate determining step in the photo-ROMP process (i.e., faster particle breakdown accelerates catalyst release). To probe this effect, we varied the PAG loading and sample temperature (Fig. 2 and ESI, Table S1†). With no or low MBTT loading in the PMPs, UV irradiation did not result in polymerization over the course of 30 min (Fig. 2A). This indicates that cPPA particles containing little to no PAG are stable under prolonged UV irradiation. Increasing the PAG concentration to 5 and 10 wt% resulted in successful ROMP, with PITs measured to be 6.8 and 3.9 min, respectively. To further illustrate the significance of acid-catalyzed cPPA depolymerization as the rate determining step, we also studied DCPD/PMP dispersions where the MBTT was dissolved in the DCPD rather than incorporated into the PMPs. We presumed that cPPA would be exposed to much lower effective acid concentration in this ex situ approach, as compared to scenarios in which the acid is generated in situ within a PMP. Indeed, although photoinitiation was successful via external MBTT, it required 30 times higher MBTT to achieve a comparable PIT (ESI, Fig. S6†). Along similar lines, we suspected that particle size and polydispersity may significantly impact the catalyst release behavior and, consequently, polymerization behavior. For example, smaller particles may reduce PIT because the length scales for diffusion of catalyst into the surrounding DCPD are smaller. However, in this initial work, we found it difficult to control particle size and polydispersity with spray drying, thus improving control over the particle morphology remains a subject of our ongoing investigations. Instead, Fig. 2B further shows that temperature plays a substantial role in accelerating the rate of photoinitiation, approaching a PIT of 1.5 min at 50 °C. Higher temperature is expected to increase the diffusivity of the photogenerated acid and increase the cPPA depolymerization kinetics, with both factors potentially eliciting faster HG2 release.
We further examined the effect of total PMP loading (4 to 16 mg ml−1) in DCPD on photoinitiation. Fig. 2C shows that the PIT decreases with higher particle loadings. Given that solutions with higher particle loading have a higher catalyst concentration, a lower UV dose was required to release the critical amount of catalyst needed to initiate ROMP. Relatedly, the PIT decreased slightly with thinner samples (ESI, Fig. S7†), indicating more efficient absorption and concomitant photogeneration of acid. As a final test, we investigated several different metathesis catalysts in the PMPs (Fig. 2D). The chemical structures of GC2 and nitro-Grela are shown in Fig. 1. While GC2-PMPs seemingly show a significant increase in PIT when compared to HG2-PMPs, both catalysts were incorporated at an equivalent weight ratio, thus the relative molar concentration of HG2 (M = 626.6 Da) was 35% higher compared to that of GC2 (M = 849.0 Da). On the contrary, particles containing nitro-Grela showed a substantially lower PIT (4 min), yet the molar concentration of HG2 was only 7% higher than nitro-Grela (M = 671.6 Da) at equivalent weights. Notably, the physical inhibition afforded by microencapsulation, as well as its modular nature, enabled us to quickly assess different catalysts and easily discover species with improved performance.
Having established that the photoinitiation rates of the DCPD/PMP dispersions can be substantially tuned via PMP composition and environmental conditions, we next studied the long-term stability of the particles (Fig. 3). DCPD/PMP dispersions were stored at room temperature in the absence of light for more than one month. Photorheology was used to probe the difference in photoinitiation rates. After the first week, the PIT showed little to no change. During the second and third week, a significant decrease in PIT occurred. The decrease in PIT is likely caused by the de-aggregation of PMPs that results in a more homogenized dispersion of particles, an effect that was observed in previous work.25 We suspect that photoinitiation was accelerated by de-aggregation of PMPs, decreasing the diffusive length scale for effective initiation. Upon reaching the four-week mark, however, the PIT drastically increased. At this point, large masses of PMPs were visually apparent. Since cPPA slowly depolymerizes when stored at room temperature,43 incremental breakdown of PMPs likely released catalyst after extended storage, insufficient for bulk polymerization but sufficient for localized gelation. This effect reduced the amount of viable PMPs and thus reduced the overall concentration of active catalyst. Alternatively, the catalyst may be slowly leached from PMPs to give the same effect. DSC data similarly revealed that over the course of three weeks, the enthalpic peak contracts, resulting in more narrow heat flow distributions. This contraction is attributed to faster and more uniform catalyst release. The enthalpy of reaction remains roughly the same during the first three weeks and is reduced after four weeks of storage wherein a 23% reduction in enthalpy is observed (ESI, Table S2†). We also found that samples stored in ambient light for 7 days exhibited similar photorheological behavior (PIT ca. 6 min) to those stored in the absence of light.
 | ||
| Fig. 3 A. Photorheology (365 nm, 27 mW cm−2, 25 °C, 0.5 mm) and B. DSC (10 °C min−1) of DCPD/PMP dispersions after extended storage at room temperature in the absence of light. The MBTT and metathesis catalyst are present in PMPs at 5:100 MBTT:cPPA and 12:88 catalyst:cPPA by wt, and the PMPs are added to DCPD at 8 mg ml−1. | ||
Given that the DCPD/PMP dispersions possess the capacity for both photo- and thermal-initiated polymerization, we postulated that the dispersions were capable of photo-initiated, thermally-sustained FROMP. Indeed, our preferred formulation (ESI, Table S1,† sample 6, containing HG2 with a 16 mg mL−1 PMP loading in DCPD) was generated on a larger scale and transferred to ASTM D638 type-V dog bone silicone molds that were covered by a glass slide (more details in Experimental and ESI†). The DCPD/PMP dispersions were initiated locally by placing a UV light directly next to the glass. Initially, a low UV intensity (27 mW cm−2) was utilized but was unsuccessful in initiating FROMP. This is likely due to incomplete breakdown of the PMPs and catalyst release as the PMPs diffuse away from the localized UV-irradiated area. Additionally, the sample thickness was significantly greater in the dog bone molds as compared to the photorheology experiments. To compensate for these effects, we significantly increased the UV intensity to 330 mW cm−2 to address this issue. After roughly 90–120 s of irradiation (ca. 30 J cm−2 dose), FROMP was successfully initiated and sustained through the entire sample (Fig. 4A and ESI Movie†). Of particular note, temperature measurements collected in situ during photorheology under comparable irradiation conditions (by insertion of a thermocouple directly into the DCPD/PMP dispersion) showed minimal temperature rise (ESI, Fig. S8†), indicating that initiation can be achieved purely by light as opposed to a photothermal effect. Furthermore, insertion of a thermocouple into the FROMP mold configuration showed only a maximum temperature of 37 °C when DCPD was irradiated without PMPs.
 | ||
| Fig. 4 A. Time-lapse of FROMP of DCPD/PMP dispersion after initiation by high intensity 365 nm irradiation. The polymerization front is propagating downward from left to right. B. Average front velocity and maximum temperature of photo- and thermal-initiated DCPD/PMP dispersions compared to thermal-initiated DCPD containing tributylphosphite-inhibited GC2. Error bars indicate ± one standard deviation (n = 3). | ||
We monitored the maximum temperature and frontal velocity during photo-initiated and thermal-initiated FROMP of DCPD/PMP dispersions, which are common metrics used to compare different frontal polymerization systems (Fig. 4B). As a control, we subjected DCPD containing GC2 and tributylphosphite (TBP) inhibitor29 to the same thermal-initiation configuration. A higher frontal velocity was seen in the PMP/DCPD dispersions (8.6 cm min−1) compared to the control (6.5 cm min−1); however, the maximum temperature showed an opposite trend. The maximum temperature for the PMP system reached 93 °C whereas the control showed a maximum temperature of 108 °C. We believe the lower front temperature was due to the endothermic nature of cPPA depolymerization in the PMP, in addition to potential vaporization of the ortho-phthalaldehyde product generated therein. Gratifyingly, DCPD/PMP dispersions gave identical maximum temperature and frontal velocity values whether photo- or thermal-initiated. We note that alternative DCPD/PMP compositions – for example, with different PMP loadings or with different encapsulated catalysts (i.e., nitro-Grela) – may give different maximum temperatures and frontal velocities based simply on the observed differences in their photopolymerization characteristics (Fig. 2). However, we did not explore alternative compositions for FROMP extensively at present due to the significant sample sizes required.
Fig. 5 shows the thermomechanical properties of the resulting pDCPD materials, characterized using dynamic mechanical analysis and quasi-static deformation in uniaxial tension. There were negligible differences in properties depending on the initiation mode. Photo-initiation resulted a similar glass transition temperature compared to thermal-initiated samples, 157 °C and 158 °C, respectively. The rubbery storage modulus of photo-initiated pDCPD was similar to that of thermal-initiated pDCPD. The ultimate tensile strength and Young's modulus were also similar in both sets of materials with values at 40 ± 1 MPa and 1.8 ± 0.1 GPa, respectively. The ultimate strain also showed minimal differences between UV and thermally initiated reactions, 8.0 ± 4.4% and 6.6 ± 2.6%. These values are well within the range of pDCPD generated by conventional techniques.8 The complete set of stress–strain curves collected is provided in the ESI, Fig. S9.† Additionally, the monomer conversion was estimated by post-curing pDCPD materials generated from DCPD/PMP dispersions, with DSC analysis indicating consistent conversions of 91–92% (ESI, Table S3†).
 | ||
| Fig. 5 A. Temperature dependence of shear storage modulus and loss tangent, and B. uniaxial tensile response of pDCPD generated by photo- and thermal-initiation of DCPD/PMP dispersions. Curves are representative among several replicates (n = 3–5). Tg is determined via peak tan(δ). | ||
Building on our successful photo-initiation of DCPD/PMP dispersions, we concluded our studies with a preliminary investigation into their potential as precursors for thin film patterning. We achieved precise spatiotemporal control over polymerization using a 365 nm source and successfully formed a patterned thunderbird (Fig. 6). Post-exposure, the unpolymerized liquid resin in the unirradiated regions was readily washed away, leaving behind a well-defined pDCPD pattern. It is important to note the use of a thin bath of resin prevents FROMP as heat is quickly lost to the surroundings. This outcome underscores the promising future applications of this photoresponsive particle system in advanced additive manufacturing technologies.
 | ||
| Fig. 6 A. Cartoon schematic of simple configuration tested for patterning of pDCPD from DCPD/PMP dispersions, along with B. the Sandia National Laboratories logo used as a photomask and C. the resulting printed pDCPD object. | ||
Experimental
Materials
cPPA was synthesized according to previously published procedures.36,57 Briefly, o-phthalaldehyde (>99.7%, purchased from TCI and used as-received) was charged into a glass round bottom flask, which had been thoroughly cleaned and dried overnight in a 150 °C oven, and dissolved in anhydrous DCM (purchased from EMD Millipore) to a concentration of 0.746 M. A dilute catalyst solution was prepared in a separate, dry 20 mL vial with boron trifluoride diethyl etherate, (ca. 48% BF3, purchased from Acros Organics) and anhydrous DCM. A small volume of this catalyst solution was taken up in a syringe to ensure a molar ratio of monomer-to-catalyst of 500:1. These preparations took place in a N2-rich glovebox before sealing the flask with rubber septa. Polymerizations were chilled to −78 °C with the use of dry ice and acetone before injecting the catalyst solution into the flask. After >1 h a small volume of pyridine (99%, Alfa Aesar) in THF (ACS grade, BDH Chemicals) was injected to quench the catalyst and mix for >10 min. The polymerization solution was then precipitated dropwise into a volumetric excess of vigorously stirred MeOH (BDH Chemicals) and stirred for >1 h before separating the resulting white powder via vacuum filtration. The powder was dried under vacuum overnight and stored in a −80 °C freezer when not in use. The sample number-averaged molecular weight Mn = 75 kg mol−1 and polydispersity Đ = 2.48 were measured via size exclusion chromatography calibrated with polystyrene standards.
cPPA synthesized in this manner is cyclic in nature. The cPPA sample degraded at 150 °C and was stable under sonication conditions to generate microparticles, consistent with previous reports. The glass transition temperature of cPPA cannot be measured as it is greater than its degradation temperature.58,59 Depolymerization was rapid and spontaneous following the cleavage of the acetal bonds in the polymer backbone whether by thermal or chemical dissociation. The degradation kinetics of cPPA capsules with similar PAGs were discussed in Shin et al., where the depolymerization products were shown to be o-phthalaldehyde (i.e., cPPA monomer) with no observable byproducts.60o-Phthalaldehyde is not expected to be reactive with the metathesis polymerization catalyst due to lack of C–C π bonds, and positive experimental results indicate it was not prohibitive of the overall goal of frontal polymerizations.
DCPD, 5-ethylidene-2-norbornene (ENB), dichloromethane (DCM), d2-DCM, methanol (MeOH), acetonitrile, GC2, and nitro-Grela were obtained from Millipore Sigma. HG2 was obtained from Umicore. Tributylphosphite (TBP), MBTT, and FABA were obtained from TCI Chemicals. All of these materials were used as received without further purification.
DCPD/ENB mixtures were formulated by melting DCPD at 40–50 °C and mixing in a 95/5 wt/wt ratio with ENB. All references to DCPD in the Results and discussion refer to this 95:5 DCPD/ENB mixture.
Spray drying
PMPs were created using a Buchi B-290 Mini-Spray Dryer equipped with a B-295 Inert Loop and B-296 Dehumidifier. A schematic diagram showing the spray drying process is provided in the ESI, Fig. S10.† 0.5 g cPPA was dissolved in 20 mL DCM, after which the desired amount of PAG and metathesis catalyst were added and fully dissolved. This solution was pumped through the spray dryer at approximately 3.1 mL min−1, using an inlet temperature of 65 °C, aspirator setting of 95% (approximately 37 m3 h−1), and N2 flow rate of approximately 1.4 m3 h−1. The solution was pumped through a standard 0.7 mm nozzle and the resulting PMPs were collected in a high-performance cyclone. To increase the amount of PMPs recovered from the cyclone, the cyclone was rinsed with MeOH and the PMPs were separated from the MeOH by filtration through a 0.8 μm nylon membrane. Lastly, the PMPs were dried overnight under vacuum.SEM
The SEM image was acquired using a ThermoFisher Helios 5 Laser PFIB. The image was taken using the through-lens detector in backscatter mode at 5 kV and 0.40 nA.NMR spectroscopy
1H NMR spectroscopy of d2-DCM solutions containing HG2-encapsulated particles was performed on a Bruker Avance III 500 MHz instrument. For measuring composition, 5–20 mg of sample was dissolved in 700–1000 μL d2-DCM. Catalyst encapsulation efficiency was determined by comparing the ratio of the carbene proton from HG2 to the six protons in the cPPA backbone (shown in ESI, Fig. S1†).Encapsulation efficiency of HG2 was calculated using the following equation:
 | (1) |
Encapsulation efficiency of MBTT was calculated using the following equation:
 | (2) |
TGA
Thermogravimetric analysis (TGA) was performed using a TA Instruments 5500 TGA under a flow of nitrogen (25 mL min−1). 3–5 mg of HG2-PMP particles containing either FABA or MBTT (5 wt%) were loaded on a platinum pan. The samples were subjected to a 10 °C min−1 heating ramp starting at room temperature until 250 °C.DSC
DSC was performed using a TA Instruments Discovery X3 calorimeter. 7.5 μL of freshly sonicated DCPD/PMP dispersion was dispensed into and sealed within an aluminum hermetic pan. The pan was equilibrated at 0 °C and then heated at a rate of 10 °C min−1.UV-Visible spectroscopy
UV-visible spectra were collected using an Agilent Cary 60 spectrophotometer, scanning from 800 to 200 nm at 600 nm min−1. Analyzed compounds were dissolved in acetonitrile at 0.01 mg ml−1 and placed in a quartz cuvette with path length 1 cm.Rheology
Rheology was performed using a TA Instruments ARES-G2 strain-controlled rheometer equipped with a photocuring accessory. The photocuring accessory consisted of an OmniCure S2000 mercury lamp directed through a light guide containing either an in-line 365 nm filter or no filter. The light is directed into the sample confined between 20 mm parallel plates through the top via an acrylate plate, while the bottom plate is aluminum. The sample temperature is controlled via an advanced Peltier system that cools the bottom plate. Light intensity was measured using a SilverLine radiometer that covers a spectral range of 230–410 nm.Freshly sonicated DCPD/PMP dispersion was placed between the parallel plates and the gap set to the desired level, typically 0.5 mm. Small amplitude oscillatory shear was applied to the sample with a strain amplitude of 1% and oscillation frequency of 1 Hz, and the shear storage and loss moduli were measured over time with continuous illumination. For non-isothermal experiments, the sample was heated at a rate of 3 °C min−1.
FROMP
000 equiv.) was added TBP (2 μL, 7.6 μmol, 1 equiv.). The mixture was shaken and added directly to a scintillation vial containing GC2 (6.42 mg, 7.6 μmol, 1 equiv.). The resulting suspension was sonicated for 5 minutes prior to FROMP experiments. A 0.7–1 mL portion of the solution was then transferred to an RTV-630 silicone dog bone mold (ASTM D638 Type V) which was clamped with a glass slide. FROMP was triggered by the tip of a 70 W soldering iron (Weller, WE1010) to the surface of the glass slide until front propagation occurred.
Monomer conversion (%) was determined via the following equation:
 | (3) |
Additive manufacturing
Spacers (0.635 mm on each side) were clamped between two glass slides, and resin formulations were introduced between the slides via disposable pipette. The resin was irradiated for 3 min at ca. 50 mW cm−2 for 3 min using the 365 nm light engine from a Kudo3D Micro SLA printer. After, the slides were separated, rinsed with DCM, and allowed to dry in an oven at 100 °C.Conclusion
In summary, our work clearly demonstrates a novel microencapsulated catalyst platform for light-activated polymerization. cPPA is an excellent encapsulant for metathesis catalysts, enabling their facile dispersion in olefin monomers like DCPD with no significant interaction between catalyst and monomer over one month of storage. The encapsulated catalyst can be rapidly released to initiate polymerization in situ by depolymerizing the cPPA, either thermally or through photo-generation of acid via co-encapsulated PAG. Due to the ease and modular nature of microencapsulation via spray drying, we fabricated DCPD/PMP dispersions with different PAGs and catalysts in a wide range of compositions, including catalysts like HG2 and nitro-Grela that have not been used previously to photoinitiate FROMP. The PIT of these dispersions was strongly impacted by PMP composition. We are currently working to improve control over particle morphology, particularly particle size and dispersity, which we expect will provide additional means to control polymerization characteristics of these resin systems.Leveraging the dual photo- and thermal-initiated capabilities of our PMPs, we have shown one of the very few examples of photo-initiated FROMP, where localized UV irradiation of a DCPD/PMP dispersion initiates a polymerization front that is thermally sustained. Our approach yields pDCPD of comparable thermal and mechanical properties compared to conventional pDCPD, yet is distinguished from the prior examples of photo-initiated FROMP by the excellent stability of the resin, with reproducible polymerization characteristics. We expect that this work will enable new modes of spatiotemporal control for scalable additive manufacturing (AM) techniques, particularly due to the exceptional pot life of the resin systems. While we have provided a simple example of pDCPD patterning from DCPD/PMP dispersions, additional work is ongoing to further decrease PIT to levels suitable for typical stereolithographic AM tools. Furthermore, we hope to adapt this microencapsulated catalyst platform to include alternative catalysts and polymerization resins beyond the current scope of ROMP, so to access a wider variety of commercially-relevant thermoset materials.
Author contributions
O. D.: conceptualization, data curation, formal analysis, investigation, methodology, supervision, validation, visualization, writing – original draft, writing – review & editing; J. L.: investigation, methodology, validation; M. R.: investigation, validation; J. D.: investigation, writing – review & editing; F. C.: investigation; J. M. S.: conceptualization, resources, writing – review & editing; A. C. E.: conceptualization, resources, writing – review & editing; P. A. K.: project administration, resources, supervision, writing – review & editing; S. C. L.: conceptualization, investigation, resources, visualization, writing – original draft, writing – review & editing; B. H. J.: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, writing – original draft, writing – review & editing.Data availability
The U.S. Department of Energy (DOE) will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Ren Bean for their review of this manuscript. This work was supported by the Laboratory Directed Research Development program at Sandia National Laboratories (project # 225931). This article has been authored by an employee of National Technology & Engineering Solutions of Sandia, LLC under contract no. DE-NA0003525 with the U.S. Department of Energy (DOE). The employee owns all right, title and interest in and to the article and is solely responsible for its contents. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this article or allow others to do so, for United States Government purposes. The DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the DOE or the United States Government.References
- R. H. Grubbs and W. Tumas, Science, 1989, 243, 907–915 CrossRef CAS PubMed
.
- H. S. Eleuterio, J. Mol. Catal., 1991, 65, 55–61 CrossRef CAS
- S. Varlas, S. B. Lawrenson, L. A. Arkinstall, R. K. O'Reilly and J. C. Foster, Prog. Polym. Sci., 2020, 107, 101278 CrossRef CAS
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS
- Z. He, G. Wang, C. Wang, L. Guo, R. Wei, G. Song, D. Pan, R. Das, N. Naik, Z. Hu and Z. Guo, Polym. Rev., 2021, 61, 689–713 CAS
- J. McQuade, M. I. Serrano and F. Jäkle, Polymer, 2022, 246, 124739 CAS
- V. B. Purohit, M. Pięta, J. Pietrasik and C. M. Plummer, Eur. Polym. J., 2024, 208, 112847 CrossRef CAS
- S. Kovačič and C. Slugovc, Mater. Chem. Front., 2020, 4, 2235–2255 RSC
- I. D. Robertson, M. Yourdkhani, P. J. Centellas, J. E. Aw, D. G. Ivanoff, E. Goli, E. M. Lloyd, L. M. Dean, N. R. Sottos, P. H. Geubelle, J. S. Moore and S. R. White, Nature, 2018, 557, 223–227 CAS
- S. C. Leguizamon, A. W. Cook and L. N. Appelhans, Chem. Mater., 2021, 33, 9677–9689 CAS
- S. C. Leguizamon, K. Lyons, N. T. Monk, M. T. Hochrein, B. H. Jones and J. C. Foster, ACS Appl. Mater. Interfaces, 2022, 14, 51301–51306 CAS
- S. C. Leguizamon, N. T. Monk, M. T. Hochrein, E. M. Zapien, A. Yoon, J. C. Foster and L. N. Appelhans, Macromolecules, 2022, 55, 8273–8282 CAS
- Z. Chen, M. Ziaee, M. Yourdkhani and X. Zhang, Addit. Manuf., 2022, 59, 103182 CAS
- J. E. Aw, X. Zhang, A. Z. Nelson, L. M. Dean, M. Yourdkhani, R. H. Ewoldt, P. H. Geubelle and N. R. Sottos, Adv. Mater. Technol., 2022, 7, 2200230 CrossRef CAS
- M. Ziaee, I. Naseri, J. W. Johnson, K. A. Franklin and M. Yourdkhani, ACS Appl. Polym. Mater., 2023, 5, 1715–1724 CrossRef CAS
- O. Eivgi and N. G. Lemcoff, Synthesis, 2018, 49–63 CAS
- A. J. Greenlee, R. A. Weitekamp, J. C. Foster and S. C. Leguizamon, ACS Catal., 2024, 14, 6217–6227 CrossRef CAS PubMed
- A. Ben-Asuly, A. Aharoni, C. E. Diesendruck, Y. Vidavsky, I. Goldberg, B. F. Straub and N. G. Lemcoff, Organometallics, 2009, 28, 4652–4655 CrossRef CAS
- N. Alassad, N. B. Nechmad, R. S. Phatake, O. Reany and N. G. Lemcoff, Catal. Sci. Technol., 2023, 13, 321–328 RSC
- B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 2038–2039 Search PubMed
- W. Joo, C. H. Chen, J. P. Moerdyk, R. P. Deschner, C. W. Bielawski and C. G. Willson, J. Polym. Sci., Part A: Polym.
Chem., 2019, 57, 1791–1795 CAS
- C. Theunissen, M. A. Ashley and T. Rovis, J. Am. Chem. Soc., 2019, 141, 6791–6796 CAS
- J. V. Musso, P. Gebel, V. Gramm, W. Frey and M. R. Buchmeiser, Macromolecules, 2023, 56, 2878–2888 CrossRef CAS
- S. J. Pastine, D. Okawa, A. Zettl and J. M. J. Frechet, J. Am. Chem. Soc., 2009, 131, 13586–13587 CAS
- O. Davydovich, A. J. Greenlee, H. D. Root, A. L. Jansen, S. C. Gallegos, M. J. Warner, M. S. Kent, J. A. Cardenas, L. N. Appelhans, D. J. Roach, B. H. Jones and S. C. Leguizamon, Macromolecules, 2023, 56, 7543–7550 CAS
- B. A. Suslick, J. Hemmer, B. R. Groce, K. J. Stawiasz, P. H. Geubelle, G. Malucelli, A. Mariani, J. S. Moore, J. A. Pojman and N. R. Sottos, Chem. Rev., 2023, 123, 3237–3298 CAS
- A. Mariani, S. Fiori, Y. Chekanov and J. A. Pojman, Macromolecules, 2001, 34, 6539–6541 CAS
- A. Ruiu, D. Sanna, V. Alzari, D. Nuvoli and A. Mariani, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2776–2780 CAS
- I. D. Robertson, L. M. Dean, G. E. Rudebusch, N. R. Sottos, S. R. White and J. S. Moore, ACS Macro Lett., 2017, 6, 609–612 CrossRef CAS PubMed
- B. A. Suslick, A. N. Yazdani, M. M. Cencer, J. E. Paul, N. A. Parikh, K. J. Stawiasz, I. P. S. Qamar, N. R. Sottos and J. S. Moore, Macromolecules, 2022, 55, 5459–5473 CrossRef CAS
- K. J. Stawiasz, J. E. Paul, K. J. Schwarz, N. R. Sottos and J. S. Moore, ACS Macro Lett., 2020, 9, 1563–1568 CrossRef CAS PubMed
- L. M. Dean, A. Ravindra, A. X. Guo, M. Yourdkhani and N. R. Sottos, ACS Appl. Polym. Mater., 2020, 2, 4690–4696 CrossRef CAS
- K. J. Stawiasz, C. I. Wendell, B. A. Suslick and J. S. Moore, ACS Macro Lett., 2022, 11, 780–784 CAS
- J. P. Lutz, O. Davydovich, M. D. Hannigan, J. S. Moore, P. M. Zimmerman and A. J. McNeil, J. Am. Chem. Soc., 2019, 141, 14544–14548 CAS
- J. A. Kaitz, C. E. Diesendruck and J. S. Moore, J. Am. Chem. Soc., 2013, 135, 12755–12761 CAS
- J. M. Schwartz, O. Phillips, A. Engler, A. Sutlief, J. Lee and P. A. Kohl, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1166–1172 CAS
- F. Wang and C. E. Diesendruck, Macromol. Rapid Commun., 2018, 39, 1700519 CrossRef PubMed
- A. Engler and P. A. Kohl, Macromolecules, 2020, 53, 1543–1549 CrossRef CAS
- M. J. Warner, J. P. Lassa, H. Narcross, A. Commisso, K. Ghosh, M. Romero, J. M. Schwartz, A. C. Engler, P. A. Kohl, S. C. Leguizamon and B. H. Jones, ACS Sustainable Chem. Eng., 2023, 11, 14538–14548 CrossRef CAS
- K. M. Lee, O. Phillips, A. Engler, P. A. Kohl and B. P. Rand, ACS Appl. Mater. Interfaces, 2018, 10, 28062–28068 CAS
- C. Shi, A. Leonardi, Y. Zhang, P. Ohlendorf, A. Ruyack, A. Lal and C. K. Ober, ACS Appl. Mater. Interfaces, 2018, 10, 28928–28935 CAS
- J. Jiang, O. Phillips, A. Engler, M. H. Vong and P. A. Kohl, Polym. Adv. Technol., 2019, 30, 1656–1662 CAS
- J. Jiang, O. Phillips, A. Engler, M. H. Vong and P. A. Kohl, Polym. Adv. Technol., 2019, 30, 1198–1204 CAS
- C. Wu, J. Jiang, H. Guo, X. Pu, L. Liu, W. Ding, P. A. Kohl and Z. L. Wang, Adv. Electron. Mater., 2019, 5, 1900725 CrossRef CAS
- E. C. Feinberg, O. Davydovich, E. M. Lloyd, D. G. Ivanoff, B. Shiang, N. R. Sottos and J. S. Moore, ACS Cent. Sci., 2020, 6, 266–273 CrossRef CAS PubMed
- A. M. DiLauro, A. Abbaspourrad, D. A. Weitz and S. T. Phillips, Macromolecules, 2013, 46, 3309–3313 CrossRef CAS
- S. Tang, M. Yourdkhani, C. M. Possanza Casey, N. R. Sottos, S. R. White and J. S. Moore, ACS Appl. Mater. Interfaces, 2017, 9, 20115–20123 CrossRef CAS PubMed
- S. Tang, L. Tang, X. Lu, H. Liu and J. S. Moore, J. Am. Chem. Soc., 2018, 140, 94–97 CrossRef CAS PubMed
- V. Eriksson, M. Andersson Trojer, S. Vavra, M. Hulander and L. Nordstierna, J. Colloid Interface Sci., 2020, 579, 645–653 CAS
- A. Gharsallaoui, G. Roudaut, O. Chambin, A. Voilley and R. Saurel, Food Res. Int., 2007, 40, 1107–1121 CAS
- A. Ziaee, A. B. Albadarin, L. Padrela, T. Femmer, E. O'Reilly and G. Walker, Eur. J. Pharm. Sci., 2019, 127, 300–318 CAS
- E. A. Stefanescu and E. Soto-Cantu, Recent Pat. Mater. Sci., 2011, 4, 106–121 CAS
- E. Boel, R. Koekoekx, S. Dedroog, I. Babkin, M. R. Vetrano, C. Clasen and G. Van den Mooter, Pharmaceutics, 2020, 12, 625 CAS
- K. Park, A. Otte, F. Sharifi, J. Garner, S. Skidmore, H. Park, Y. K. Jhon, B. Qin and Y. Wang, J. Controlled Release, 2021, 329, 1150–1161 CrossRef CAS
- H. L. Hernandez, S.-K. Kang, O. P. Lee, S.-W. Hwang, J. A. Kaitz, B. Inci, C. W. Park, S. Chung, N. R. Sottos, J. S. Moore, J. A. Rogers and S. R. White, Adv. Mater., 2014, 26, 7637–7642 CrossRef CAS PubMed
- OmniCure S2000 UV Curing System, https://www.excelitas.com/product/omnicure-s2000-uv-curing-system, (accessed 25 July 2024).
- A. Engler, O. Phillips, R. C. Miller, C. Tobin and P. A. Kohl, Macromolecules, 2019, 52, 4020–4029 CrossRef CAS
- J. A. Kaitz and J. S. Moore, Macromolecules, 2014, 47, 5509–5513 CrossRef CAS
- M. Warner, A. Engler and P. A. Kohl, Polymer, 2020, 202, 122588 CrossRef CAS
- Y. Shin, J. M. Schwartz, A. C. Engler, B. Jones, O. Davydovich and P. A. Kohl, ACS Appl. Mater. Interfaces, 2024, 16, 43951–43960 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00882k |
| This journal is © The Royal Society of Chemistry 2025 |