
Ring-opening polymerization of a tri-substituted six-membered lactone derived from CO2/butadiene†
Jialin
Xu
,
Yuxuan
Niu
and
Bo-lin
Lin
 *
*
School of Physical Science and Technology, ShanghaiTech University, 393 Middle Huaxia Road, Pudong New Area, Shanghai, China. E-mail: linbl@shanghaitech.edu.cn
First published on 14th November 2024
Abstract
The selective ring-opening homo-polymerization of CO2/butadiene-derived lactone monomers has emerged as an appealing approach for synthesizing chemically recyclable polyesters from CO2. Previous research has only concentrated on CO2-derived di-substituted six-membered lactones. In this study, a newly designed tri-substituted six-membered CO2/butadiene-derived lactone monomer, 3,3,6-triethyltetrahydro-2H-pyran-2-one (Et-HL), was polymerized successfully through selective ring-opening polymerization (ROP) using NaOMe, tBu-P4/BnOH, or tBu-P4. tBu-P4/BnOH affords linear-poly(Et-HL) with typical living polymerization behaviors, while a maximum number-average molecular weight (Mn) of 1050 kg mol−1 and a dispersity (Đ) of 1.52 were achieved for cyclic-poly(Et-HL) using only tBu-P4. Catalytic methods were developed for monomer recycling of both linear- and cyclic-poly(Et-HL). Direct observation of key intermediates by Nuclear Magnetic Resonance (NMR) reveals the mechanistic differences between Et-HL and DEtP. A tail-to-head strain-releasing mechanism was proposed to rationalize the selective formation of cyclic polymers for both DEtP and Et-HL using only tBu-P4. In the case of linear polymers, DEtP and Et-HL share a similar mechanism involving the initiator anion attacking the monomer ester bond. This work represents the first example of the ROP of six-membered lactones bearing more than 2 substituents, simultaneously offering a fundamental understanding of the Thorpe–Ingold effect on the ROP of CO2/butadiene-derived six-membered lactones for the first time.
Introduction
The synthesis of CO2-derived polymers with a closed-loop lifecycle has been pursued for decades.1–3 The activation of CO2 presents a key significant challenge,4 necessitating the introduction of high-energy co-monomers.5,6 Simple olefins, as low-cost bulk chemicals, have been considered among the ideal co-monomers for the co-polymerization of CO2 due to their reactive double bonds.7–9 However, previous studies have demonstrated that the direct alternate co-polymerization of CO2 and olefins is thermodynamically and kinetically inaccessible.10,11 Consequently, considerable attention has been devoted to the polymerization of CO2/butadiene-derived lactone intermediates, especially di-substituted six-membered lactones starting from EVP (Fig. 1a).12–14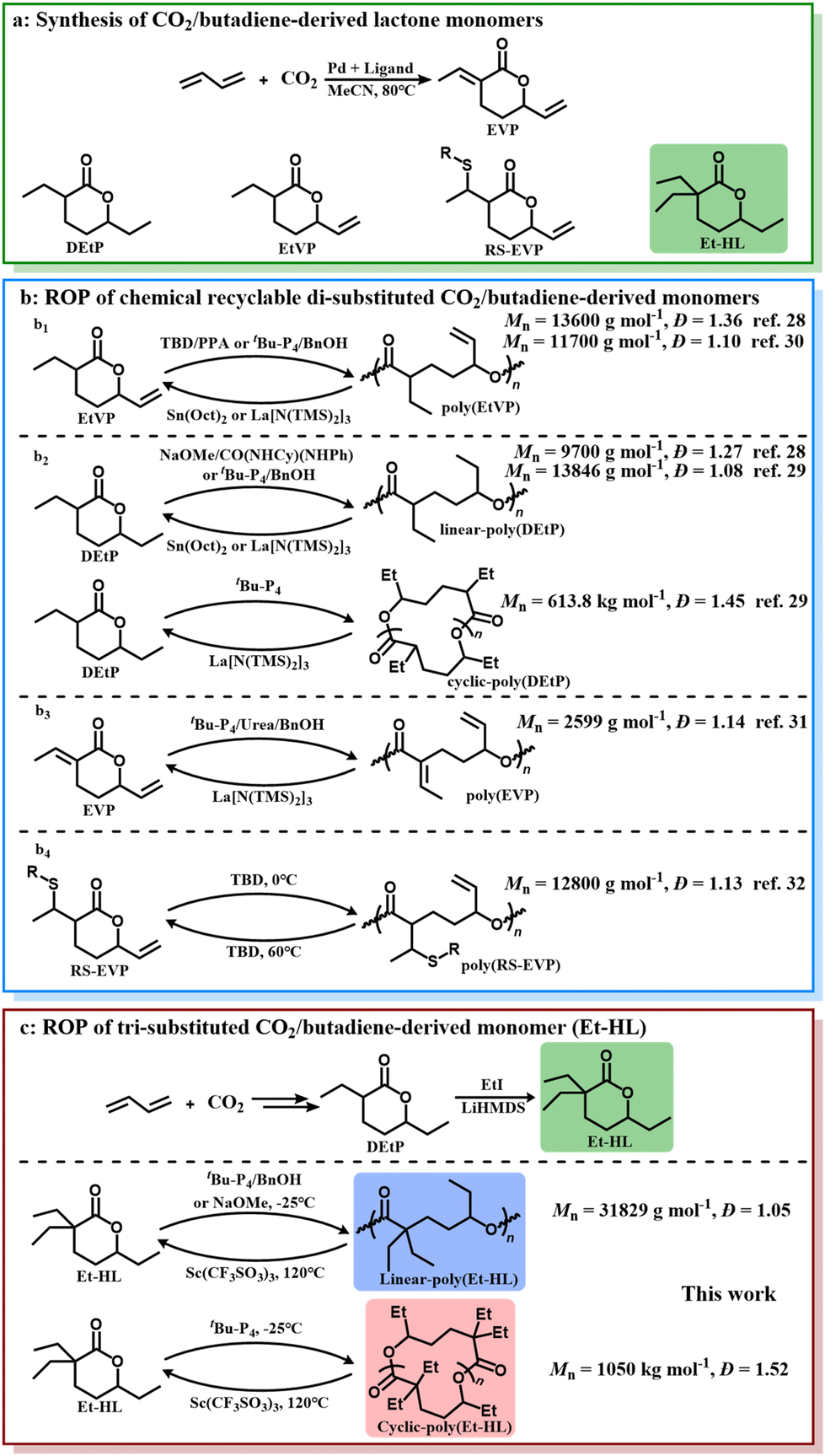 | ||
| Fig. 1 (a) Synthesis of CO2-derived lactones, EVP, EtVP, DEtP and Et-HL. (b) ROP of chemical recyclable CO2-derived lactones in previous works: b1 for EtVP, b2 for DEtP and b3 for EVP. (c) ROP of Et-HL. | ||
Various strategies, including radical homo-polymerization,11,15–19 co-polymerization20–26 and ring-opening homo-polymerization (ROP)27–32 of lactones, have been attempted, while only four approaches have successfully produced polyesters with chemical recyclability through ROP28–32 (Fig. 1b). Thermodynamic and kinetic analyses consistently pointed towards an enhanced difficulty in polymerizing these di-substituted six-membered lactones in comparison with their zero- and mono-substituted counterparts.33 Nonetheless, the polymerizability of more-substituted six-membered lactones remains unknown.
Herein, we present the ROP of a tri-substituted six-membered lactone monomer, 3,3,6-triethyltetrahydro-2H-pyran-2-one (Et-HL, Fig. 1c), derived from EVP. The monomer also provides the first example of examining the geminal di-substituted effect on the ROP characteristics of CO2/butadiene-derived six-membered lactones. Selective synthesis of linear- and cyclic-poly(Et-HL) was achieved using NaOMe, tBu-P4/BnOH, and tBu-P4, respectively. Both linear- and cyclic-poly(Et-HL) were depolymerized back to monomers under catalytic conditions at 120 °C. The impact of the third substituent on the ROP compared to other CO2/butadiene-derived di-substituted lactones was systematically studied. In particular, the identification of different intermediates highlights the distinct initiation mechanisms arising from the additional substituent.
Results and discussion
As shown in Fig. 1c, a two-step procedure was applied to synthesize DEtP from CO2, H2, and 1,3-butadiene.29 The subsequent reaction between DEtP and EtI, using LiHDMS as a base, was conducted to yield Et-HL (Fig. 1c) via a procedure modified from the literature.34 The successful installation of gem-disubstituted ethyl groups at the α-position of the carbonyl group of the six-membered lactone was confirmed via NMR spectroscopy (Fig. S1–5†).To achieve ROP of Et-HL, we initially tested organic bases such as 1,8-diazabicyclo[5.4.0]undec-7-ene and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), in conjunction with benzyl alcohol (BnOH) as the initiator, but observed no reaction (Table 1, runs 1 and 2). Subsequently, 1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)-phosphoranylide-namino]-2λ5,4λ5-catenadi(phos-phazene) (tBu-P4) was evaluated at a feeding ratio of [Et-HL]/[tBu-P4]/[BnOH] = 100/1/2 at −25 °C for 72 h in tetrahydrofuran (THF), resulting in a monomer conversion rate of 88% with a molecular mass Mn = 8120 g mol−1 and a polydispersity Đ = 1.24 (poly(Et-HL)) (Table 1, run 3). A feeding ratio of [Et-HL]/[tBu-P4]/[BnOH] = 50/1/1 led to a 91% monomer conversion rate with Mn = 9397 g mol−1 and Đ = 1.08 (Table 1, run 4). By reducing the ratio of [Et-HL]/[BnOH] to 200/1 and 300/1, we managed to increase the molecular mass to Mn = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 846 g mol−1 (Đ = 1.07) and Mn = 31712 g mol−1 (Đ = 1.12), respectively (Table 1, runs 5 and 6). The ROP reactivity of Et-HL decreased significantly when the reaction temperature was elevated from −25 °C to room temperature (Table 1, run 7), suggesting a rather low ceiling temperature (Tc).
846 g mol−1 (Đ = 1.07) and Mn = 31712 g mol−1 (Đ = 1.12), respectively (Table 1, runs 5 and 6). The ROP reactivity of Et-HL decreased significantly when the reaction temperature was elevated from −25 °C to room temperature (Table 1, run 7), suggesting a rather low ceiling temperature (Tc).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Run | Et-HL/cat./BnOH | Cat. | T (°C) | t (h) | [Et-HL]0 (mol L−1) | Conv.a (%) |
M
nb (kg mol−1) |
Đ |
| Reaction conditions: Et-HL = 0.1000 g (0.54 mmol) in THF; Et-HL was added to a tBu-P4/BnOH solution.a Monomer conversions were measured by 1H NMR; n.d. means not detected.b Mn and Đ were determined by GPC at 40 °C in THF relative to PMMA standards.c 1 equiv. of 15-crown-5 was added.d For run 12, the quenching reagent was replaced with dehydrated hexane. Detailed information of all the experiments is provided in the ESI.† | ||||||||
| 1 | 50/1/1 | TBD | −25 | 72 | 4.5 | n.d. | — | — |
| 2 | 50/1/0 | TBD | −25 | 72 | Neat | n.d. | — | — |
| 3 | 100/1/2 | t Bu-P4 | −25 | 72 | 4.5 | 88 | 8.12 | 1.24 |
| 4 | 50/1/1 | t Bu-P4 | −25 | 48 | 4.5 | 91 | 9.40 | 1.08 |
| 5 | 100/1/1 | t Bu-P4 | −25 | 48 | 4.5 | 98 | 19.85 | 1.07 |
| 6 | 200/1/1 | t Bu-P4 | −25 | 48 | 4.5 | 86 | 31.71 | 1.12 |
| 7 | 100/1/1 | t Bu-P4 | R.T. | 72 | 4.5 | 27 | 2.78 | 1.61 |
| 8 | 100/1/0 | NaOMe | −25 | 168 | 4.5 | 29 | 12.60 | 1.06 |
| 9c | 100/1/0 | NaOMe | −25 | 72 | Neat | 94 | 17.32 | 1.16 |
| 10c | 200/1/0 | NaOMe | −25 | 72 | Neat | 86 | 31.83 | 1.05 |
| 11 | 20/1/0 | t Bu-P4 | −25 | 2 | 3.0 | 75 | 36.78 | 1.24 |
| 12d | 20/1/0 | t Bu-P4 | −25 | 2 | 3.0 | 73 | 34.83 | 1.27 |
| 13 | 100/1/0 | t Bu-P4 | −25 | 24 | 4.5 | 84 | 513.9 | 1.16 |
| 14 | 100/1/0 | t Bu-P4 | −25 | 24 | Neat | 88 | 905.2 | 1.34 |
| 15 | 100/1/0 | t Bu-P4 | −25 | 72 | Neat | 96 | 1050 | 1.52 |
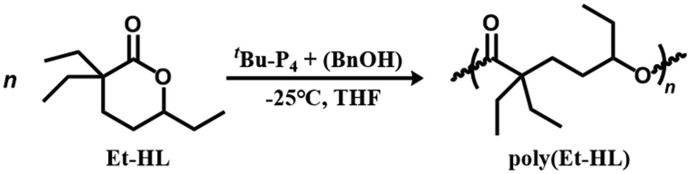
The ROP of Et-HL exhibits the characteristics of living polymerization under suitable conditions. The conversion of Et-HL over time was measured at a feeding ratio of [Et-HL]/[tBu-P4]/[BnOH] = 200/1/1 at −25 °C (Fig. 2a). The plot of ln([M]0/[M]) increases linearly with reaction time (Fig. 2b), and a linear correlation was observed between Mn and monomer conversion with a consistently low Đ (Đ < 1.2, Fig. 2c). The GPC curves obtained were all confirmed to be unimodal (Fig. 2d).
 | ||
| Fig. 2 ROP of Et-HL at a feeding ratio of Et-HL/tBu-P4/BnOH = 200/1/1 (a) plot of Et-HL conversion versus time. (b) Plot of ln([M]0/[M]t) versus time. (c) Plot of molecular weight (Mn) and Đ versus Et-HL conversion. (d) GPC traces of poly(Et-HL) obtained at different polymerization times. [Et-HL]0 = 4.5 M in THF and T = −25 °C. | ||
NaOMe, another common ROP agent, was also tested as a potential initiator. The poor solubility of NaOMe in both THF and Et-HL resulted in a very low monomer conversion rate (29%), even after extending the reaction time to 7 days (Table 1, run 8). Thus, we replaced THF with 15-crown-5 and successfully achieved a series of poly(Et-HL) with high conversion rates, and the molecular mass increased with the feeding ratio of Et-HL/NaOMe (Table 1, runs 9 and 10). The NMR spectra of both purified products initiated by [tBu-P4]/[BnOH] and NaOMe showed the presence of end-group signals, suggesting that linear structures were obtained (Fig. 3a and S6–14†). The signals of MALDI-TOF MS for poly(Et-HL) (Fig. 3b and S15†) showed only one set of peaks with a constant spacing of 184.2 g mol−1 plus corresponding end-group masses, confirming the linear topology of poly(Et-HL) initiated by [tBu-P4]/[BnOH] and NaOMe.
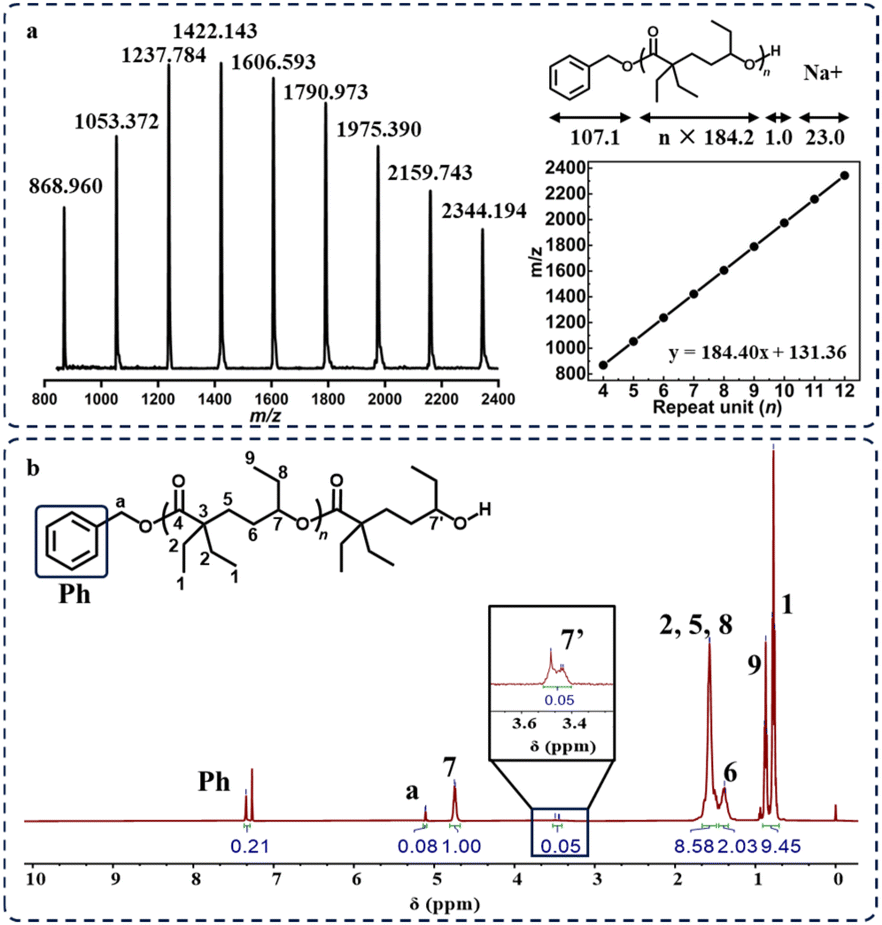 | ||
| Fig. 3 (a) MALDI-TOF mass spectrum of linear-poly(Et-HL) initiated by tBu-P4/BnOH. (b) 1H NMR spectrum of purified linear-poly(Et-HL) end-capped with BnOH. | ||
In contrast, pure cyclic-poly(DEtP) was observed previously in the ROP of DEtP, the di-substituted precursor of Et-HL, using tBu-P4 as the catalyst in the absence of any initiator (Mn,max = 613.8 kg mol−1 and Đ = 1.45, Fig. 1b2).29 In the present work, ROP of Et-HL was conducted under similar conditions (Table 1, run 11). A 75% monomer conversion to a polymer with Mn = 36781 g mol−1 and Đ = 1.24 was achieved. No end group signal was observed in NMR (Fig. S16 and S17†) and the MALDI-TOF MS data (Fig. S18†) for the purified product, consistent with the formation of cyclic-poly(Et-HL). To further probe the topology of the product, both linear- and cyclic-poly(Et-HL) were prepared independently to measure their respective dn/dc values. Consistent with previous literature,35 the dn/dc of linear-poly(Et-HL) (0.0668 ml g−1, Fig. S36†) was higher than that of cyclic-poly(Et-HL) (0.0664 ml g−1, Fig. S37†). The ratio of [η]cyclic/[η]linear was 0.73 according to the Mark–Houwink plot (Fig. S38†), which was very close to the theoretical ratio (∼0.7) for cyclic/linear.36 Also, the Mark–Houwink exponent α values of cyclic- and linear-poly(Et-HL) were nearly identical (0.85 and 0.88, respectively, random coils in THF). Based on this evidence, we identified this product as cyclic-poly(Et-HL).
Notably, an interesting dependence of polymer topology on the quenching conditions was observed previously for the ROP of a geminal di-substituted four-membered thiolactone, where dehydrated hexane and methanol as quenching agents led to zwitterionic linear polymers with a covalently-bonded tBu-P4 head and cyclic polymers, respectively.34 However, both quenching methods only resulted in cyclic polymers for Et-HL (Table 1, runs 11 and 12, Fig. S33 and S34†). Remarkably, the molecular weight can be enhanced to be over 1 million with further increases in the Et-HL/tBu-P4 ratio, monomer concentration and reaction time (Table 1, runs 13, 14 and 15).
Next, we investigated the thermostability of both linear- and cyclic-poly(Et-HL) using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The TGA and derivative thermogravimetry (DTG) curves revealed Td,5% = 322.6 °C and Tmax = 352.5 °C for cyclic-poly(Et-HL) (Mn = 36.8 kg mol−1 and Đ = 1.24, Fig. S20†) as well as Td,5% = 318.9 °C and Tmax = 343.3 °C for linear-poly(Et-HL) (Mn = 31.7 kg mol−1 and Đ = 1.12, Fig. S21†). Cyclic-poly(DEtP) also showed slightly higher thermostability than linear-poly(DEtP).29 The glass-transition temperatures (Tg) for linear- and cyclic-poly(Et-HL) derived from DSC curves (Fig. S22†) were −15.8 °C and −11.8 °C, respectively, which were higher than those observed for poly(DEtP) (Tg,linear = −30.6 °C, Mn = 19.6 kg mol−1, and Đ = 1.08; Tg,cyclic = −29.7 °C, Mn = 21.7 kg mol−1, and Đ = 1.13).29 The higher molecular weight and the increased main-chain rigidity of poly(Et-HL) might account for such a difference. Similar to poly(DEtP), no obvious crystalline peak was observed for poly(Et-HL).
Van't Hoff analysis was performed to study the thermodynamic parameters for the ROP of Et-HL. The corresponding plot was a line with a slope of −2.52 and an intercept of 9.50. The R2 value of the fitting line was 0.9990 (Fig. S35 and Table S1†). According to Dainton's equation, the thermodynamic parameters were calculated to be ΔHp = −20.9 kJ mol−1 and ΔSp = −79.0 J mol−1 K−1, and the ceiling temperature (Tc) was calculated to be 265 K (−8 °C) at [Et-HL]0 = 1.0 mol L−1 in THF. From a thermodynamic standpoint, the ROP reactivity of Et-HL was only slightly lower than that of DEtP (ΔHp = −13.1 kJ mol−1, ΔSp = −49.1 J mol−1 K−1, Tc = −6 °C, and [DEtP]0 = 1.0 mol L−1).
The impact of substituents on ring-opening polymerization of six-membered lactones has been studied previously.33,34 The length of the side chain has been found to impact both ring strain (ΔHp) and entropic losses (ΔSp), while the variations of Tc are primarily influenced by the entropic term.33 Additionally, the substituent on the α-position of δ-valerolactone was considered as the least entropically unfavorable substitution, which would maximize the thermodynamic polymerizability.33 Furthermore, the polymerization of various α,α-dialkyl δ-valerolactones (VLR2) exhibits much lower thermodynamic polymerizability compared to δ-valerolactone (δVL).34 The thermodynamic trend for the present ring-opening polymerizations of Et-HL and DEtP is consistent with the previous observations.
To test the chemical recyclability of poly(Et-HL), linear and cyclic polymers with Mn in the range of 9–200 kg mol−1 were studied. Various catalysts, including Sn(Oct)2, ZnCl2, Fe(acac)2, Sc(CF3SO3)3 and La[N(SiMe3)2]3 (La), were employed to catalyze the recycling of poly(Et-HL) in toluene ([Et-HL]0 = 0.5 M) at 80 °C for 24 h, but monomer recovery was only observed for Sc(CF3SO3)3, where 47% and 11% recoveries of Et-HL were obtained for linear and cyclic polymers, respectively. The monomer recovery of poly(Et-HL) is kinetically much less favorable than that of poly(DEtP), which may be attributed to the increased steric hindrance due to the additional ethyl group of Et-HL. By increasing the reaction time and temperature to 72 h and 120 °C, the monomer recovery was improved to 81% for the linear polymer and 62% for the cyclic polymer, respectively. Further prolonging the reaction time reached full monomer recovery in 5 days for linear-poly(Et-HL). The monomer before and after chemical recycling is shown in Fig. S23.†
Notably, the molecular weights of cyclic-poly(Et-HL) were inconsistent with the feeding ratio of [Et-HL]/tBu-P4. Three potential reasons may be invoked to rationalize this phenomenon. Firstly, the aggregation and/or decomposition of tBu-P4 before initiation should result in an unexpectedly large molecular weight. However, no corresponding evidence in the crude 1H NMR spectra (Fig. S39†) or phenomena in the reaction process were observed. Secondly, late-stage intermolecular chain transfer is also a possible pathway for the large molecular weights in many anionic ROPs. Linear polymer chains with concomitant smaller molecular weights would be produced as side products in this process. However, in our system, the GPC curve of the crude product (Fig. S40†) only showed a single peak with no smaller molecules detected. This partially proved that intermolecular chain extension was at least not the main reason for high molecular weight cyclic products. Thirdly, the amount of the initiating species in the system may be significantly less than the feeding ratio due to the low initiation reactivity, which will be further investigated later on.
To further investigate the polymerization of cyclic-poly(Et-HL), the mechanism of the formation of cyclic polyester from Et-HL was studied and compared to the corresponding mechanism of DEtP. Four possible pathways for the initiation step were considered, as shown in Fig. 4 at first. For DEtP, Path I is susceptible due to the strong acidity of the hydrogen at the α-position. α-Deuterium-labelled DEtP was successfully captured and identified in the 2H NMR spectrum using CF3COOD as a quenching reagent in a reaction with a 2:1 ratio of DEtP and tBu-P4 at −25 °C without solvent (Fig. S24†). Fig. S24a† shows a clear α-deuterium signal at δ 1.75 ppm, exactly matching the α-hydrogen in DEtP (δ 1.75 ppm in 1H NMR, Fig. S25†). Also, no deuterium signal corresponding to any other hydrogen in DEtP, including the hydrogen at the 7-position (Fig. S25†), was observed, suggesting that Path II is inaccessible in the ROP of DEtP. This evidence strongly suggested that Path I was the dominant initiating mechanism for the ROP of DEtP.
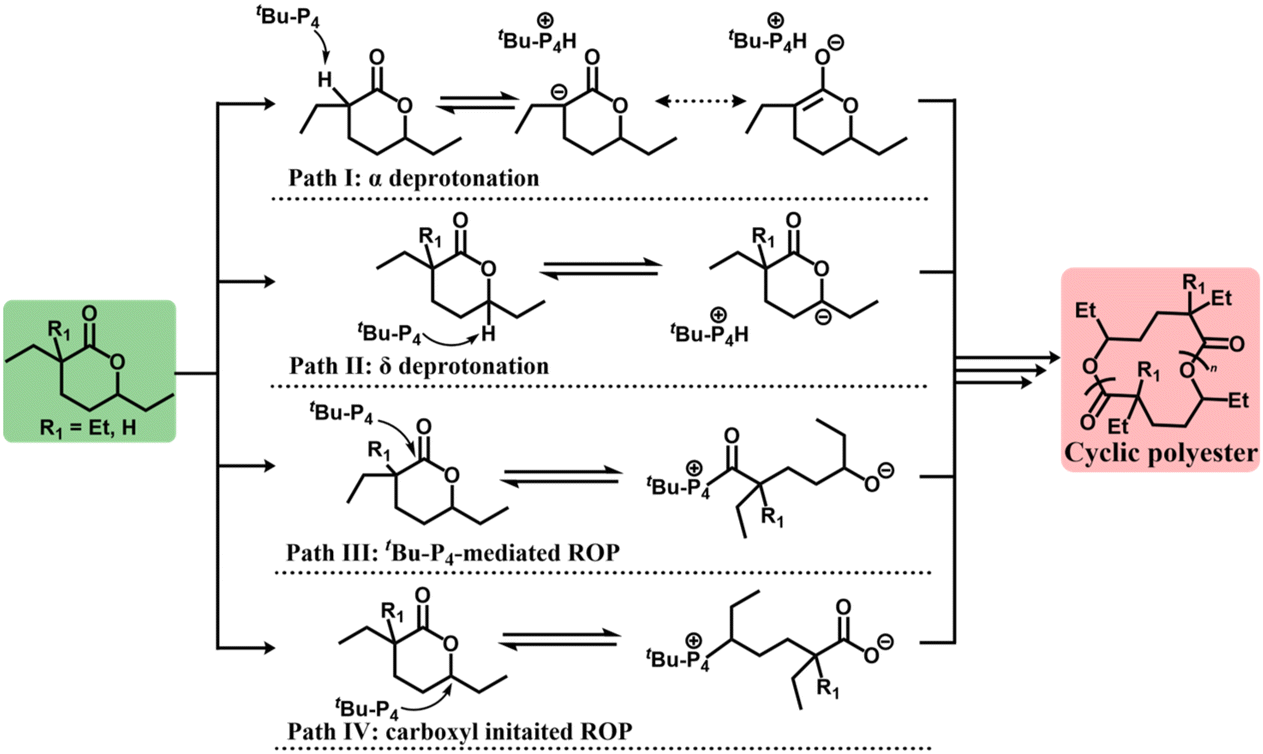 | ||
| Fig. 4 ROP initiation of DEtP and Et-HL. Path I: tBu-P4 abstracts R1, if R1 is hydrogen, generating an α-anion intermediate that initiates ROP (α deprotonation ROP); Path II: similar initiation with Path I occurring at the δ-position (δ deprotonation ROP); Path III: tBu-P4 attacks the ester bond directly at the carbonyl group to initiate ROP (tBu-P4-mediated ROP), leading to cyclic polymers; and Path IV: tBu-P4 attacks the δ-position resulting in the anionic carboxyl group (carboxyl initiated ROP). | ||
For Et-HL, the α-deprotonation initiated ROP (Path I) was prohibited due to the presence of the gem-diethyl substitution. Only one deuterium signal, other than the solvent, was detected at δ 2.29 ppm in the reaction quenched by CF3COOD (Et-HL:tBu-P4 = 2:1, Fig. S24c†). Such a signal matches the 2H NMR peak of the tBu-P4 and CF3COOD mixture closely (δ 2.35 ppm, Fig. S24d†), which means that Et-HL produced a signal consistent with a deuterated phosphazene tBu-P4-D+, with no evidence for activation of any hydrogen in Et-HL. These results suggested that Paths I and II (Fig. 4) can be ruled out for the ROP of Et-HL.
Next, three parallel reactions with different reaction times were attempted to probe potential initiating species for Et-HL. With a feeding ratio of [Et-HL]/[tBu-P4] = 10/1 at −25 °C, reaction times of 5 min, 15 min and 20 min were employed, respectively (1H NMR spectra in toluene-d8, Fig. S26 and 27†). The broad characteristic signals of poly(Et-HL), particularly at δ 4.91 ppm, increased noticeably with the reaction time. Reactions with the feeding ratio of [Et-HL]/[tBu-P4] = 10/1 and a reaction time of 10 min at −25 °C were conducted for both DEtP and Et-HL. Comparing their 1H NMR spectra, different novel signals arising from tBu-P4 in Et-HL (δ 2.52 ppm) and DEtP (δ 2.55, 2.56 ppm) were observed (Fig. S28 and S29†), suggesting a distinct mechanism for Et-HL derived from tBu-P4.
Diffusion Ordered Spectroscopy (DOSY) was applied to the same 10 min reaction for both Et-HL and DEtP (Fig. S30 and S31†) to identify the different tBu-P4 signals in DEtP and Et-HL. For Et-HL (Fig. S30†), two independent peaks were obtained on the Y-axis (f1) related to tBu-P4 signals. The first signal (Fig. S30†) correlating to δ 2.70 and 2.72 ppm, which was consistent with the 1H NMR of tBu-P4 (Fig. S29c†), represented the unreacted tBu-P4. The second one (Fig. S30†) correlating to δ 2.52 ppm, which was excluded for the monomer, BnOH or cyclic-poly(Et-HL), was considered as the novel catalytic species derived from tBu-P4. Several signals correlated with the second peak were observed at δ 1.00–1.50 ppm along with δ 2.52 ppm apart from the tBu- signal, indicating a covalent attachment between tBu-P4 and CH2- in this novel intermediate (Fig. S30†) since the Et-HL monomer was the only CH2- source in this reaction. A similar correlation was not observed at δ 1.00–1.50 ppm for DEtP (Fig. S31†). Thus, a mechanism involving covalent bonding between tBu-P4 and Et-HL should occur in the initiating state of Et-HL.
As shown in Fig. 4, both Path III and Path IV are tBu-P4-involved covalent pathways. The carboxyl group is the active species in Path IV for chain propagation. However, zero monomer conversion was observed in 72 hours for Et-HL ([Et-HL]/[CH3COOK]/[18-crown-6] = 50/1/2, [Et-HL]0 = 4.5 mol L−1, and T = −25 °C), which showed that the carboxyl group cannot open the ring of Et-HL and ruled out Path IV. Path III became the only option for the initiation of Et-HL by tBu-P4.
A detailed mechanism for the initiation and formation of cyclic-poly(Et-HL) is proposed in Fig. 5. Considering the significant steric hindrance from both tBu-P4 and the groups surrounding the carboxyl group on Et-HL, the initiating step may be an equilibrium significantly favoring Et-HL. This postulation is consistent with the weak NMR signals of the tBu-P4-involved intermediate observed above (Fig. S26–S30,†δ 2.52 ppm). Thus, the relative amount of the initiating intermediate would be much less than the feeding ratios of Et-HL to tBu-P4, which partially rationalizes the repeating units in the resultant polymers and the molecular weight over 1 million obtained in the tBu-P4-only system. Selective backbiting from a propagating chain tail to the chain head was proposed. The backbiting occurring at internal sites would produce linear-poly(Et-HL) as a side product, which is not supported by experimental data as we discussed above (Fig. S40†). We tentatively attribute this selectivity to the unusually strong steric strain between bulky tBu-P4 and the geminal carbon at the chain head. The strong tendency to release the chain-head strain presumably favored the selective tail-to-head backbiting that only formed cyclic-poly(Et-HL).
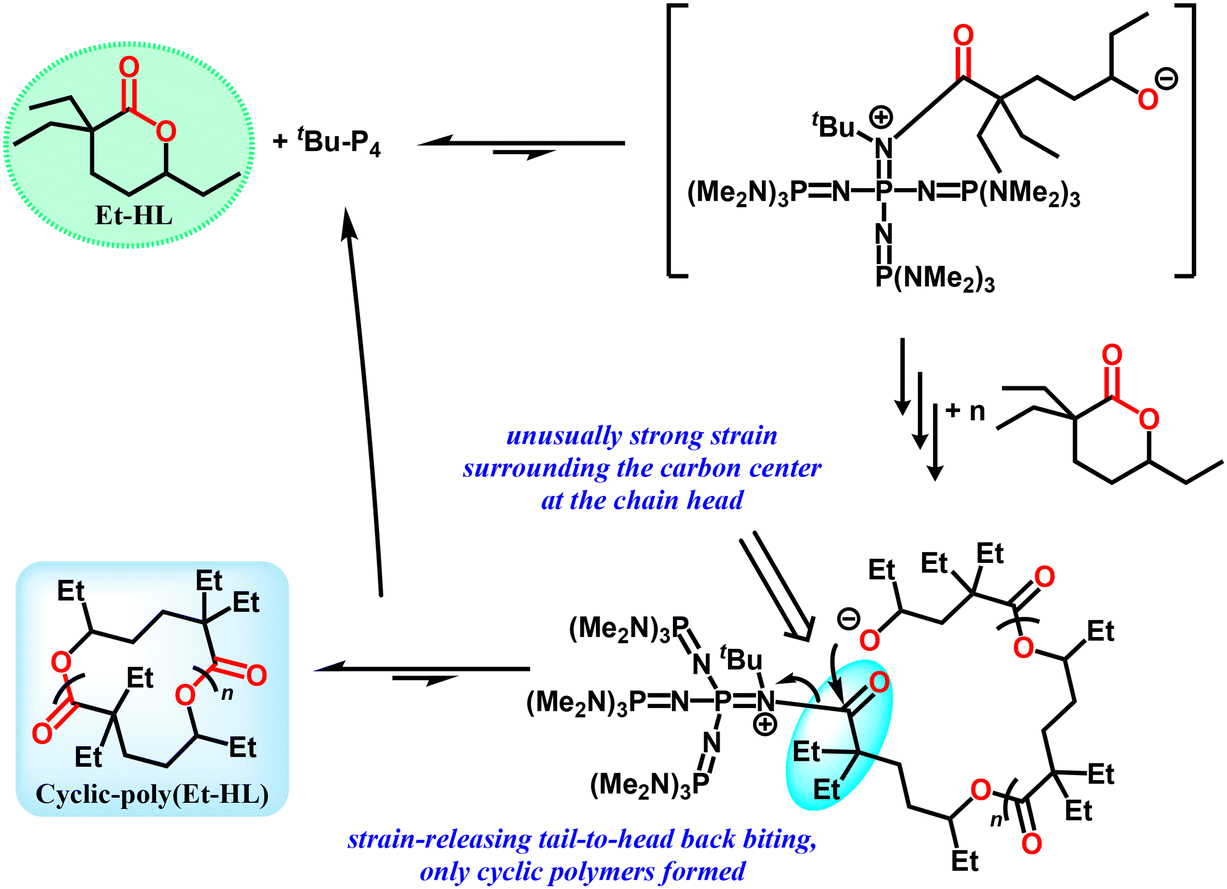 | ||
| Fig. 5 (a) Proposed mechanism for the formation of cyclic-poly(Et-HL). | ||
Although sharing similar cyclic polymer products using only tBu-P4 as a catalyst, Et-HL and DEtP exhibit different topological transition sensitivities from cyclic to linear products in their ROPs. The topological transition sensitivity here stands for the lowest feeding ratio of BnOH to tBu-P4 that can achieve the selective synthesis of a linear polymer. Linear-poly(Et-HL) can be produced selectively at a feeding ratio of BnOH/tBu-P4 = 1, while a ratio of 10 is needed for poly(DEtP). One equivalent of BnOH was not enough for the complete inhibition of α-deprotonation initiated cyclization by tBu-P4 in DEtP, but it was enough for the complete inhibition of tBu-P4-mediated cyclization in Et-HL. Such a difference might be tentatively attributed to the mechanistic difference between DEtP and Et-HL due to the presence of Path I for the formation of a cyclic polymer from DEtP.
Notably, a related, but different, mechanism has been reported in the ROP of a geminal disubstituted four-membered thiolactone (2,2-dimethyl-3-thiopropiolactone, (Me)2TPL).34 The linear and cyclic polymers of (Me)2TPL could be selectively synthesized by using hexane and methanol as the quenching reagent, respectively. Although the ROPs of Et-HL and (Me)2TPL share similar initiation and propagation mechanisms, their termination step appears quite different. The absence of topological dependence on the quenching agent for Et-HL was confirmed by using hexane (Table 1, run 12). Apparently, the zwitterionic linear-poly(Et-HL) can spontaneously backbite to form the cyclic product as we proposed in the last paragraph. This key reactivity difference might be attributed to the increased main chain flexibility resulting from the longer repeating unit of poly(Et-HL) relative to P3T(Me)2P, presumably allowing the facile formation of a close ion pair between the positive chain head and negative chain end.
The mechanism of producing a linear polymer is also proposed in Fig. S32.† The introduction of BnOH into the ROP provides an active hydroxyl group to neutralize tBu-P4 and affords benzoxide to initiate the polymerization. The resultant propagating secondary alkoxide should be less reactive than the initiating benzoxide, a primary alkoxide, for subsequent ring-opening reactions due to steric hindrance. Thus, a fast-initiation-slow-propagation kinetic situation occurs. In addition, due to the stronger basicity of the secondary alkoxide relative to the primary alkoxide, the former can be reversibly converted to a dormant alcohol chain end in the presence of excess benzyl alcohol. All these factors may jointly rationalize the observed living ROP of Et-HL to form linear-poly(Et-HL) (vide supra).
Conclusion
In summary, we presented the first study of ROP of Et-HL, a tri-substituted CO2/butadiene-derived six-membered lactone. The selective synthesis of linear-poly(Et-HL) and cyclic-poly(Et-HL) was precisely controlled and the polymer properties were characterized, establishing a new strategy for synthesizing CO2/butadiene-derived monomer-recyclable polyesters. Additionally, the initiation mechanism of both Et-HL and DEtP was elucidated through the identification of key intermediates. While they share similar mechanisms in controlling the selective formation of either cyclic or linear polymers, the introduction of a third substituent at the alpha position of the monomer carbonyl group does lead to mechanistic differences in the initiation step. This work represents the first example of the ROP of six-membered lactones bearing more than 2 substituents, simultaneously offering a fundamental understanding of the Thorpe–Ingold effect on the ROP of CO2/butadiene-derived six-membered lactones for the first time.Data availability
The data supporting the article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. U2032132) and the Cross-disciplinary Research Fund of Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine (JYJC202218). The authors appreciate the support from the Analytical Instrumentation Center (AIC) of the School of Physical Science and Technology, ShanghaiTech University.References
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Advances in the use of CO2 as a renewable feedstock for the synthesis of polymers, Chem. Soc. Rev., 2019, 48, 4466–4514 CAS.
- Y. H. Xua, L. M. Lin, M. Xiao, S. J. Wang, A. T. Smith, L. Y. Sun and Y. Z. Meng, Synthesis and properties of CO2-based plastics: Environmentally friendly, energy-saving and biomedical polymeric materials, Prog. Polym. Sci., 2018, 80, 163–182 Search PubMed.
- R. Meys, A. Katelhön, M. Bachmann, B. Winter, C. Zibunas, S. Suh and A. Bardow, Achieving net-zero greenhouse gas emission plastics by a circular carbon economy, Science, 2021, 374, 71–76 CrossRef CAS.
- J. Wang, C. S. Jia, C. J. Li, X.-L. Peng, L. H. Zhang and J. Y. Liu, Thermodynamic Properties for Carbon Dioxide, ACS Omega, 2019, 4, 19193–19198 CrossRef CAS PubMed.
- Y. Liu and X. B. Lu, Current Challenges and Perspectives in CO2-Based Polymers, Macromolecules, 2023, 56, 1759–1777 CrossRef CAS.
- R. Muthuraj and T. Mekonnen, Recent progress in carbon dioxide (CO2) as feedstock for sustainable materials development: co-polymers and polymer blends, Polymer, 2018, 145, 348–373 CAS.
- S. Tang and K. Nozaki, Advances in the Synthesis of Copolymers from Carbon Dioxide, Dienes, and Olefins, Acc. Chem. Res., 2022, 55, 1524–1532 CAS.
- K. Soga, S. Hosoda, Y. Tazuke and S. Ikeda, Copolymerization of Carbon Dioxide and Ethyl Vinyl Ether, J. Polym. Sci., Polym. Lett. Ed., 1975, 13, 265–268 CAS.
- F. Zhou, H. Dai, S. Tang and Y. Zhou, Synthesis of Polyethylenes with in-Chain Isolated Carbonyls from CO2 and Ethylene Via a Tandem Photoreduction/Polymerization Protocol, CCS Chem., 2023, 6, 1591–1599 Search PubMed.
- C. J. Price, B. J. E. Reich and S. A. Miller, Thermodynamic and Kinetic Considerations in the Copolymerization of Ethylene and Carbon Dioxide, Macromolecules, 2006, 39, 2751–2756 CAS.
- R. Nakano, S. Ito and K. Nozaki, Copolymerization of carbon dioxide and butadiene via a lactone intermediate, Nat. Chem., 2014, 6, 325–331 CrossRef CAS PubMed.
- S. Tang, B.-L. Lin, I. Tonks, J. M. Eagan, X. F. Ni and K. Nozaki, Sustainable Copolymer Synthesis from Carbon Dioxide and Butadiene, Chem. Rev., 2024, 124, 3590–3607 CrossRef CAS PubMed.
- J. M. Eagan, The Divergent Reactivity of Lactones Derived from Butadiene and Carbon Dioxide in Macromolecular Synthesis, Macromol. Rapid Commun., 2023, 44, 2200348 CrossRef CAS.
- R. M. Rapagnani and I. A. Tonks, 3-Ethyl-6-vinyltetrahydro-2H-pyran-2-one (EVP): A Versatile CO2-Derived Lactone Platform for Polymer Synthesis, Chem. Commun., 2022, 58, 9586–9593 CAS.
- S. Moon, K. Masada and K. Nozaki, Reversible Polymer-Chain Modification: Ring-opening and Closing of Polylactone, J. Am. Chem. Soc., 2019, 141, 10938–10942 CAS.
- V. Haack, E. Dinjus and S. Pitter, Synthesis of Polymers with an Intact Lactone Ring Structure in the Main Chain, Angew. Makromol. Chem., 1998, 257, 19–22 CAS.
- M. Kamigaito and K. Satoh, Stereoregulation in Living Radical Polymerization, Macromolecules, 2008, 41, 269–276 CAS.
- L. F. Chen, Y. Li, S. C. Yue, J. Ling, X. F. Ni and Z. Q. Shen, Chemoselective RAFT Polymerization of a Trivinyl Monomer Derived from Carbon Dioxide and 1,3-Butadiene: From Linear to Hyperbranched, Macromolecules, 2017, 50, 9598–9606 CAS.
- M. H. Liu, Y. Y. Sun, Y. Q. Liang and B.-L. Lin, Highly Efficient Synthesis of Functionalizable Polymers from a CO2/1,3-Butadiene-Derived Lactone, ACS Macro Lett., 2017, 6, 1373–1378 CAS.
- M. R. Hill, S. Tang, K. Masada, Y. Hirooka and K. Nozaki, Incorporation of CO2-Derived Bicyclic Lactone into Conventional Vinyl Polymers, Macromolecules, 2022, 55, 3311–3316 CrossRef CAS.
- S. Tang and K. Nozaki, Advances in the Synthesis of Copolymers from Carbon Dioxide, Dienes, and Olefins, Acc. Chem. Res., 2022, 55, 1524–1532 CrossRef CAS PubMed.
- Y. J. Zhao, X. H. Zhang, Z. Li, Z. K. Li and S. Tang, Functional and Degradable Polyester-co-polyethers from CO2, Butadiene, and Epoxides, ACS Macro Lett., 2024, 13, 315–321 CrossRef CAS.
- S. Tang, Y. Zhao and K. Nozaki, Accessing Divergent Main Chain-Functionalized Polyethylenes Via Copolymerization of Ethylene with a CO2/Butadiene-Derived Lactone, J. Am. Chem. Soc., 2021, 143, 17953–17957 CrossRef CAS PubMed.
- K. H. Chen, T. W. Bai, Y. X. Mei, T. L. Shen, J. Ling and X. F. Ni, A Topology-Defined Polyester Elastomer from CO2, and 1,3-Butadiene: A One-Pot-One-Step “Scrambling Polymerizations” Strategy, Angew. Chem., Int. Ed., 2022, 61, e202213028 CrossRef CAS PubMed.
- S. C. Yue, T. W. Bai, S. Y. Xu, T. Shen, J. Ling and X. F. Ni, Ring-Opening Polymerization of CO2-Based Disubstituted δ-Valerolactone toward Sustainable Functional Polyesters, ACS Macro Lett., 2021, 10, 1055–1060 CrossRef CAS.
- R. J. Anderson, R. L. Fine, R. M. Rapagnani and I. A. Tonks, Ring-Opening Copolymerizations of a CO2-Derived δ-Valerolactone with ε-Caprolactone and L-Lactide, Macromolecules, 2024, 57, 6248–6254 CrossRef CAS.
- L. D. G. Espinosa, K. W. Pavlantos, K. M. Turney, C. Wesdemiotis and J. M. Eagan, Degradable Polymer Structures from Carbon Dioxide and Butadiene, ACS Macro Lett., 2021, 10, 1254–1259 CrossRef.
- R. M. Rapagnani, R. J. Dunscomb, A. A. Fresh and I. A. Tonks, Tunable and recyclable polyesters from CO2 and butadiene, Nat. Chem., 2022, 14, 877–883 CrossRef CAS PubMed.
- Y. J. Lou, L. Y. Xu, N. L. Gan, Y. Y. Sun and B.-L. Lin, Chemically recyclable polyesters from CO2, H2, and 1,3-butadiene, Innovation, 2022, 3, 100216 CAS.
- Y. J. Lou, J. L. Xu, L. Y. Xu, Z. Chen and B.-L. Lin, Chemically Recyclable CO2-Based Solid Polyesters with Facile Property Tunability, Macromol. Rapid Commun., 2022, 43, 2200341 CrossRef CAS.
- J. L. Xu, Y. X. Niu and B.-L. Lin, Monomer-Recyclable Polyester from CO2 and 1,3-Butadiene, Macromol. Rapid Commun., 2024, 2400163 CrossRef CAS.
- Z. R. Zhang, T. Shen, K. H. Chen, J. J. Zeng, Y. X. Mei, J. Ling and X. F. Ni, Polyester Platform with High Refractive Indices and Closed-Loop Recyclability from CO2, 1,3-Butadiene, and Thiols, ACS Macro Lett., 2024, 13, 741–746 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Aliphatic Polyester Block Polymer Design, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- L. Zhou, L. T. Reilly, C. X. Shi, E. C. Quinn and E. Y.-X. Chen, Proton-triggered topological transformation in superbase-mediated selective polymerization enables access to ultrahigh-molar-mass cyclic polymers, Nat. Chem., 2024, 16, 1357–1365 CrossRef CAS PubMed.
- Y. Song, J. He, Y. Zhang, R. A. Gilsdorf and E. Y.-X. Chen, Recyclable cyclic bio-based acrylic polymer via pairwise monomer enchainment by a trifunctional Lewis pair, Nat. Chem., 2024, 15, 366–376 CrossRef.
- J. Roovers, Cyclic Polymers, ed. J. A. Semlyen, Kluwer Academic, 2nd edn, 2000, pp. 347–384 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01213e |
| This journal is © The Royal Society of Chemistry 2025 |