
Increasingly threaded polypseudorotaxanes with reduced enthalpies of melting†
He
Sun
 ,
Sean R.
Gitter
,
Kiana A.
Treaster
,
Joshua D.
Marquez
,
Brent S.
Sumerlin
,
Kenneth B.
Wagener
and
Austin M.
Evans
*
,
Sean R.
Gitter
,
Kiana A.
Treaster
,
Joshua D.
Marquez
,
Brent S.
Sumerlin
,
Kenneth B.
Wagener
and
Austin M.
Evans
*
George & Josephine Butler Polymer Research Laboratory, Department of Chemistry, Center for Macromolecular Science & Engineering, University of Florida, Gainesville, FL 32611, USA. E-mail: AustinEvans@ufl.edu
First published on 24th October 2024
Abstract
Polymers that contain mechanical bonds have unique and useful properties. It is difficult to prepare linear polyrotaxanes that contain well-defined mechanical bonds, which has made it challenging to establish the influence of mechanical bonding on linear polyrotaxane properties. We disclose the synthesis of 1,9-decadiene-pseudorotaxane linear copolymers with variable densities of threaded macrocycles using acyclic diene metathesis (ADMET) polymerization. This enabled our investigation into how macrocyclic threading impacts the thermomechanical properties of linear polypseudorotaxanes. Specifically, we observe that the melting enthalpy decreased from 67 to 5 J g−1 as we increased the molar incorporation of a pseudorotaxane unit. Going forward, we expect ADMET to be a useful strategy to prepare linear mechanically interlocked macromolecules at scales relevant for characterizing their thermomechanical properties.
 Austin M. Evans | Austin Evans was born and raised in Oklahoma. Austin completed his Ph.D. in Chemistry (2022) at Northwestern University as an NSF Graduate Research Fellow. Austin was a postdoctoral Schmidt Science Fellow studies at Columbia University working with Professors Latha Venkataraman and Colin Nuckolls. Austin is currently an assistant professor at the University of Florida, where he is a member of the Butler Polymer Laboratory. His research focuses on the precise structural control of macromolecular materials. In his free time, Austin enjoys competing in triathlons. |
Introduction
Mechanical interactions between polymer chains govern macromolecular material properties.1–5 Well-defined mechanical bonds represent a new method to install these interactions into macromolecular materials in a systematic way.6–8 For example, creating a (pseudo-)rotaxane by threading a macrocycle onto a polymer backbone is reminiscent of loop structures in entangled thermoplastics.9 Many researchers have investigated mechanical bonds in network materials, such as slide-ring networks held together by mechanical cross-links.10–12 The mobility of these cross-linked units imbues these materials with unique material properties that have been demonstrated as useful in electrode binders,13–16 biomedical drug delivery,17–20 and self-healing elastomers.21–23 However, the influence of mechanical cross-links on linear polyrotaxane properties is not as well-established.The limited understanding of mechanical bonding's influence on linear polymer properties primarily results from the synthetic difficulty of generating linear polyrotaxanes or polycatenanes at large-scale. Recently, strategies have emerged to either scale or control the number of macrocycles on a polymer backbone.24–30 These recent synthetic breakthroughs inspire our renewed interest in understanding how the incorporation of mechanical bonds influences the properties of linear poly[n]rotaxanes. Here, we synthesize a linear pseudo-rotaxane diene that we then randomly copolymerize with 1,9-decadiene by acyclic diene metathesis (ADMET). By varying the molar ratio of the monomeric precursors, we systematically vary the density of mechanical bonds and probe their impact on linear polyrotaxane material properties. Finally, thermal characterization revealed threading-dependent thermal stability and melting enthalpies.
Results and discussion
Pseudorotaxane diene synthesis
Secondary dialkylammonium salts complex dibenzo-24-crown-8 (DB24C8) macrocycles strongly (Ka of 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 in CDCl3) to form threaded pseudorotaxanes.31–33 This understanding inspired us to prepare copolymers of a pseudorotaxane diene and a linear alkyl diene to study the effect of macrocyclic threading on thermomechanical properties. We began our investigation by preparing a pseudorotaxane monomer in a 3-step sequence at large-scale (3 g, Fig. 1).30 Briefly, we alkylated tBu-carbamate with 6-bromo-1-hexene to yield the boc-protected amino diene. Subsequent deprotection and ion exchange yielded an ammonium-containing diene with a hexafluorophosphate counterion. Finally, treatment of this salt with DB24C8 furnished the desired pseudorotaxane diene monomer, as confirmed by 1H NMR spectroscopy (Fig. S7†). This pseudorotaxane diene was then isolated and polymerized without further purification.
000 M−1 in CDCl3) to form threaded pseudorotaxanes.31–33 This understanding inspired us to prepare copolymers of a pseudorotaxane diene and a linear alkyl diene to study the effect of macrocyclic threading on thermomechanical properties. We began our investigation by preparing a pseudorotaxane monomer in a 3-step sequence at large-scale (3 g, Fig. 1).30 Briefly, we alkylated tBu-carbamate with 6-bromo-1-hexene to yield the boc-protected amino diene. Subsequent deprotection and ion exchange yielded an ammonium-containing diene with a hexafluorophosphate counterion. Finally, treatment of this salt with DB24C8 furnished the desired pseudorotaxane diene monomer, as confirmed by 1H NMR spectroscopy (Fig. S7†). This pseudorotaxane diene was then isolated and polymerized without further purification.
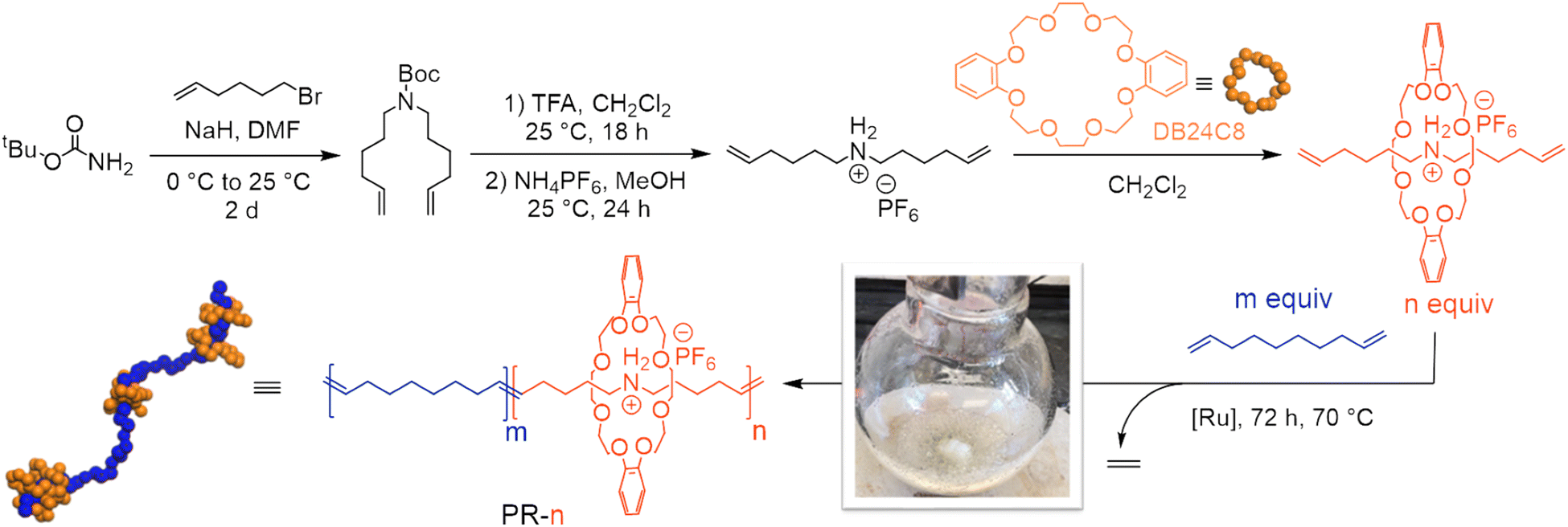 | ||
| Fig. 1 Synthesis of pseudorotaxane diene (orange) and subsequent copolymerization with 1,9-decadiene (blue) via acyclic diene metathesis polymerization to yield variably threaded polypseudorotaxanes (PR-n), where n indicates the mole fraction of threaded monomer. The embedded photograph shows ethylene being liberated and removed during polymerization. TFA = trifluoroacetic acid. | ||
Linear polyrotaxanes by ADMET polymerization
We prepared variably threaded polypseudorotaxanes (PRs) by copolymerizing the pseudorotaxane diene with 1,9-decadiene via ADMET polymerization (Table 1). In this case, the central axle is the resultant polyalkeneamer. We define our materials as PR-n, where n represents the molar fraction of the pseudorotaxane monomer used to produce these polyrotaxanes. We used acyclic diene metathesis (ADMET) polymerization with Ru-based catalysts to prepare PRs at a moderate scale (>300 mg of total monomer). ADMET polymerizations were performed at 70 °C and under a constant vacuum to remove liberated ethylene gas, which increases the monomer conversion. We began by copolymerizing 96 mol% 1,9-decadiene with 4 mol% of the pseudorotaxane monomer (Table 1, entry 2). Analysis of the isolated material with 1H NMR spectroscopy revealed a 5 mol% incorporation of the pseudorotaxane monomer by comparing the relative integrations of the macrocycle (3.80, 3.90, 4.16, and 6.90 ppm, Fig. 2) and backbone olefins (5.38 ppm, Fig. 2). Notably, no resonances associated with terminal olefins (4.98 and 5.78 ppm) were observed in the crude post-polymerization NMR spectrum, suggesting near-quantitative monomer conversion (>95%). Additionally, gel permeation chromatography (GPC) with inline differential refractive index (dRI) detection revealed a single monomodal peak consistent with Mn = 8.2 kg mol−1. We further confirmed the incorporation of the pseudorotaxane monomer with Fourier-transform infrared (FTIR) spectroscopy. Notably, we observed a signal at 1000–1500 cm−1 in the FTIR spectrum of PR-04 and no signal at the same frequency in PR-00, consistent with incorporating the pseudorotaxane monomer in the linear PR.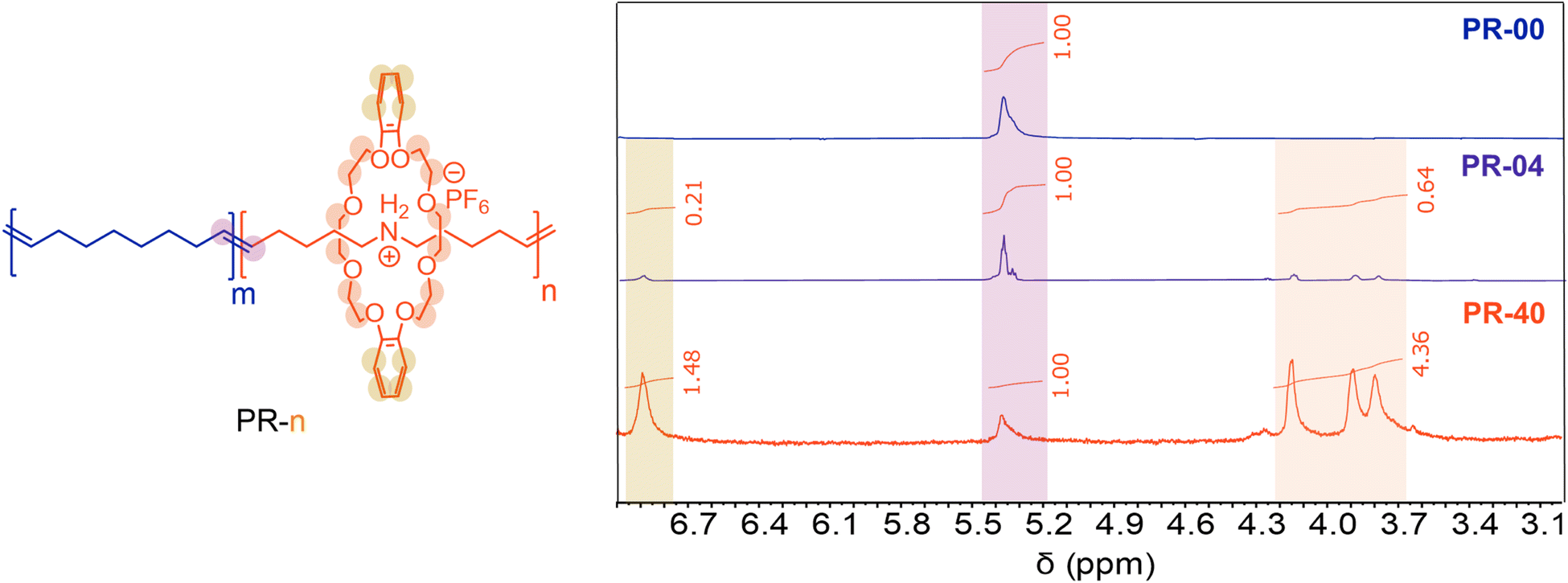 | ||
| Fig. 2 Peak shading indicates assignment. 1H NMR spectra of variably threaded copolymers in CDCl3. | ||
| 1 mol% of catalyst was used in each case.a Amount of pseudorotaxane in reaction feed.b Determined by relative integrations of 1H NMR spectra.c Determined by gel permeation chromatography with inline differential refractive index detection in THF. Mn calculated by conventional calibration using polystyrene standards. See ESI for full synthetic procedures and structures of G1 and G2.† |
|---|

|
Copolymerizing higher molar loadings of the pseudorotaxane monomer led to PRs with systematically higher numbers of mechanical bonds. We found that the resulting materials displayed an increase in pseudorotaxane incorporation (Table 1, entries 3–6) as we increased the molar loading of the pseudorotaxane monomer from 4% to 40%. Using ADMET polymerization under constant vacuum, we incorporated up to 37 mol% of the pseudorotaxane monomer (Table 1, entry 6). GPC dRI chromatograms of each sample lacked significant low MW impurities (Fig. 3A) and the resulting Mn values were consistently ≥8.2 kg mol−1. These molecular weights are analogous to those achieved in other reported ADMET polymerizations.30 Similar to the PR-04, the other PR systems had increased 1H NMR intensities (3.80, 3.90, 4.16, and 6.90 ppm) consistent with the increased pseudorotaxane monomer incorporation. Additionally, we found increased FTIR signal intensity (Fig. 3B) associated with aryl C–H stretches (3175 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretches (1592 cm−1), C–O stretches (1252 and 1209 cm−1), N–H stretches (1121 and 1049 cm−1), and aryl C–H out-of-plane bends (831 cm−1) as the molar incorporation of the pseudorotaxane in the material increased. In summary, ADMET polymerization produces variably threaded PRs at sufficient scales (>300 mg) for thermal characterization.
C stretches (1592 cm−1), C–O stretches (1252 and 1209 cm−1), N–H stretches (1121 and 1049 cm−1), and aryl C–H out-of-plane bends (831 cm−1) as the molar incorporation of the pseudorotaxane in the material increased. In summary, ADMET polymerization produces variably threaded PRs at sufficient scales (>300 mg) for thermal characterization.
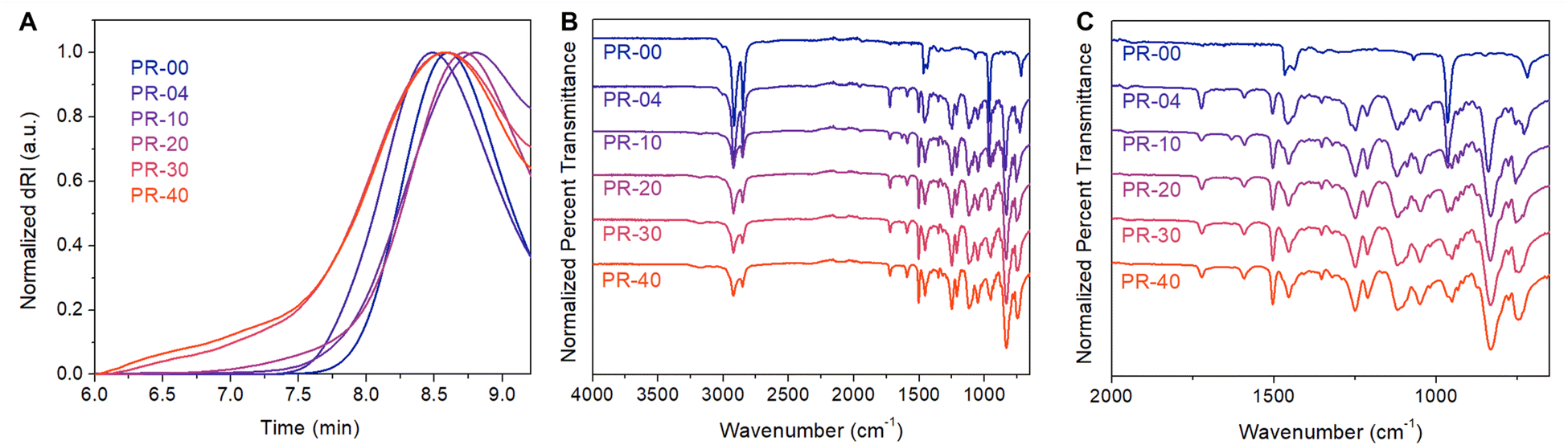 | ||
| Fig. 3 (A) Gel permeation chromatograms (GPC) of the variably threaded copolymers. GPC was conducted using differential refractive index detection and a tetrahydrofuran mobile phase. (B) Offset Fourier-transform infrared spectra of the variably threaded polyrotaxanes and a (C) zoomed in view of this data to more clearly show the low wavenumber region of this data. | ||
Thermal properties of linear polypseudorotaxanes
Having established polymerization conditions that generate materials with tunable pseudorotaxane content, we turned our attention to thermally characterizing these materials. Thermogravimetric analysis (TGA) reveals that incorporating the pseudorotaxane units decreases the temperature of the primary mass loss event from 400 to 300 °C (Fig. 4A). This finding is consistent with the presence of the C–O and C–N bonds in the pseudorotaxane components and the TGA profiles of the bare macrocycle and amine-containing diene monomer (Fig. S24†). As the incorporation of pseudorotaxane is increased from 0 to 40 mol% the magnitude of this lower temperature mass loss event increases, consistent with an increased threading density. Moreover, as more macrocycles are incorporated along the polymer backbone, the char yield of these materials increases from 0 to 10%. It is conceivable that the variable thermal stabilities of the macrocycle and polymer backbone could be leveraged to create responsive materials that evolve on thermal curing due to selective cleavage of the macrocyclic structure.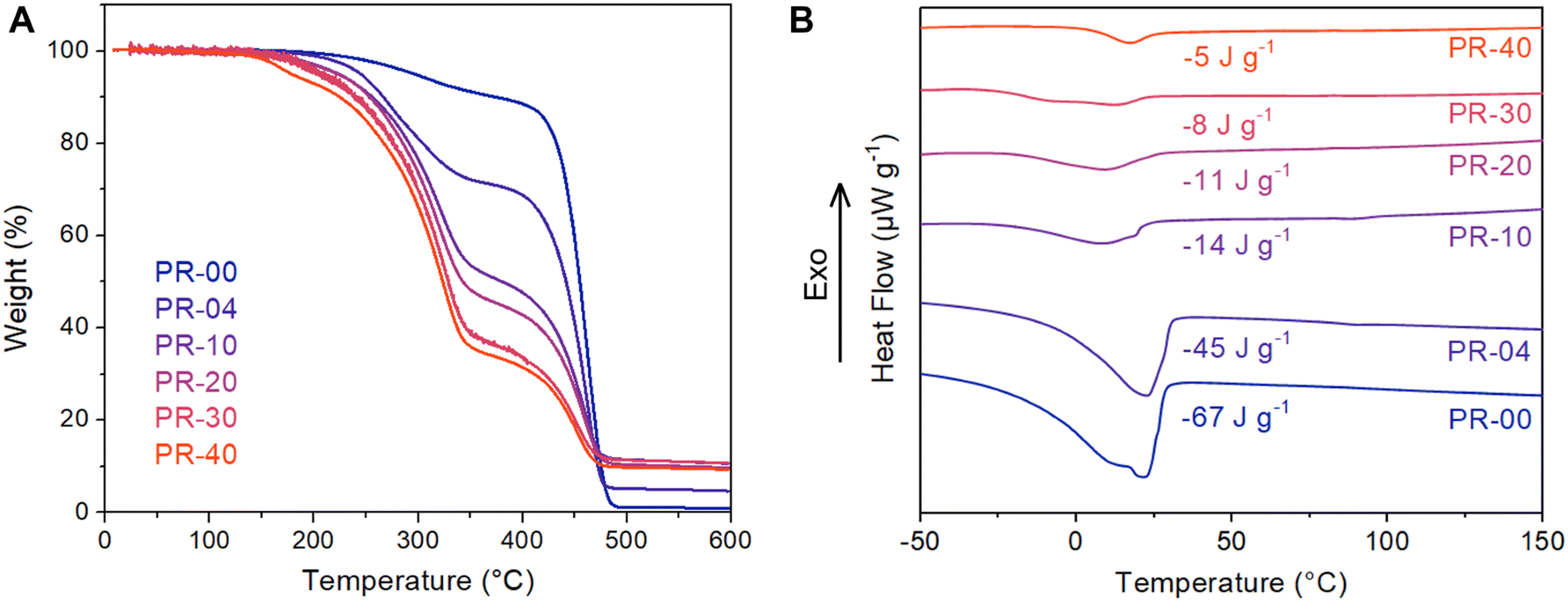 | ||
| Fig. 4 (A) Thermogravimetric analysis (TGA) of the variably threaded polypseudorotaxanes. TGA was conducted with a heating rate of 10 °C min−1 under an N2 atmosphere. (B) Differential scanning calorimetry (DSC) thermograms of variably threaded copolymers under heating flow with a heating rate of 10 °C min−1 under an N2 atmosphere. | ||
Differential scanning calorimetry (DSC) revealed that threading a larger number of macrocycles onto the polymer backbone leads to gradual changes in the melting behavior of the polyoctenamer (Fig. 4B). In the pure polyoctenamer system, we observe a melting transition at approximately 22 °C that is consistent with other reports of polycyclooctene with equimolar backbone cis-/trans-olefins produced by ring-opening metathesis polymerization.34 The melting enthalpy drops by over an order of magnitude, from 67 to 5 J g−1, as a macrocyclic threading density of 40% is achieved. Notably, we do not observe the melting enthalpy associated with pure DB24C8 or the pseudorotaxane monomer, indicating that the macrocycles and polymer axles are intimately mixed and not phase-separated (Fig. S25–27†). Taken together, these DSC observations indicate that the interchain packing of the bare polyoctenamer is disrupted as macrocycles are threaded onto the backbone. Intriguingly, these findings are counter to previous work by Tonelli and coworkers on a poly(ε-caprolactone) cyclodextrin system (CD) that showed increasing crystallinity with increasing macrocyclic threading.35 We note that many interchain interactions (macrocyclic threading, H-bonding, etc.) are present in these CD-based systems, complicating understanding of structure–property relationships in these materials.
Conclusions
We have prepared linear polyalkeneamers with predictable and variable degrees of macrocyclic threading. We investigated the thermal characteristics of these materials with TGA and DSC. TGA showed that the materials became less thermally stable as the percentage of macrocyclic threading increased. It is conceivable that the disparate thermal stabilities of macrocycles and polymer rotors may unlock unique temperature-responsiveness in linear PRs. DSC revealed that threading macrocycles onto polyalkeneamers lowers their melting enthalpy, which we attribute to disrupted interchain interactions in polyrotaxanes. DSC measurements also reveal that the macrocycle and polyalkeneamer do not phase-separate and indicate that mechanical bonding may be one method to compatibilize otherwise immiscible macromolecular functionalities. For example, macrocyclic threading could be used to compatibilize polyethylene and polypropylene waste streams, simplifying current recycling methods. Moreover, mechanical bonding may find value in plasticizing thermoplastics with molecules that would otherwise phase-separate from the thermoplastic host. This would enhance the processability of unique polymer blends enabling access to otherwise inaccessible materials. As methods to prepare mechanically interlocked macromolecules mature, we expect that these areas will be of valuable fundamental investigation.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors have no competing financial interests.Acknowledgements
This work was supported by funding from the Defense Advanced Research Projects Agency (DARPA) under award number HR00112430352 and the donors of ACS Petroleum Research Fund with a Doctoral New Investigator (DNI) award (PRF #67035-DNI7). We thank Umicore AG & Co. KG for providing the metathesis catalysts used in this work.References
- L. F. Hart, J. E. Hertzog, P. M. Rauscher, B. W. Rawe, M. M. Tranquilli and S. J. Rowan, Nat. Rev. Mater., 2021, 6, 508–530 CAS.
- L. Chen, X. Sheng, G. Li and F. Huang, Chem. Soc. Rev., 2022, 51, 7046–7065 RSC.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- J. F. Stoddart and C. J. Bruns, The Nature of the Mechanical Bond: From Molecules to Machines, John Wiley & Sons: Hoboken, NJ, 2017 Search PubMed.
- R. Brighenti, Y. Li and F. J. Vernerey, Front. Mater., 2020, 7, 176 CrossRef.
- L. Fang, M. A. Olson, D. Benitez, E. Tkatchouk, W. A. Goddard 3rd and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17–29 RSC.
- S. Mena-Hernando and E. M. Perez, Chem. Soc. Rev., 2019, 48, 5016–5032 CAS.
- N. H. Perez and J. E. M. Lewis, Org. Biomol. Chem., 2020, 18, 6757–6780 Search PubMed.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022–5049 CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CAS.
- K. Ito, Polym. J., 2007, 39, 489–499 CrossRef CAS.
- K. Kato, Y. Ikeda and K. Ito, ACS Macro Lett., 2019, 8, 700–704 CrossRef CAS PubMed.
- S. Choi, T.-W. Kwon, A. Coskun and J. W. Choi, Science, 2017, 357, 279–283 CrossRef CAS PubMed.
- D. J. Yoo, A. Elabd, S. Choi, Y. Cho, J. Kim, S. J. Lee, S. H. Choi, T. W. Kwon, K. Char, K. J. Kim, A. Coskun and J. W. Choi, Adv. Mater., 2019, 31, e1901645 CrossRef.
- H. Yang, R. M. Gao, X. D. Zhang, J. Y. Liang, X. H. Meng, Z. Y. Lu, F. F. Cao and H. Ye, Adv. Mater., 2022, 34, e2204835 CrossRef.
- Y. Cai, C. Liu, Z. Yu, W. Ma, Q. Jin, R. Du, B. Qian, X. Jin, H. Wu, Q. Zhang and X. Jia, Adv. Sci., 2023, 10, e2205590 CrossRef PubMed.
- T. Hirotsu, T. Higashi, K. Motoyama and H. Arima, Carbohydr. Polym., 2017, 164, 42–48 CrossRef CAS PubMed.
- Z. Liu, G. A. Simchick, J. Qiao, M. M. Ashcraft, S. Cui, T. Nagy, Q. Zhao and M. P. Xiong, ACS Nano, 2021, 15, 419–433 CrossRef CAS PubMed.
- G. Yu, Z. Yang, X. Fu, B. C. Yung, J. Yang, Z. Mao, L. Shao, B. Hua, Y. Liu, F. Zhang, Q. Fan, S. Wang, O. Jacobson, A. Jin, C. Gao, X. Tang, F. Huang and X. Chen, Nat. Commun., 2018, 9, 766 Search PubMed.
- T. K. Paira, S. Banerjee and T. K. Mandal, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2130–2141 CAS.
- M. Nakahata, S. Mori, Y. Takashima, H. Yamaguchi and A. Harada, Chem, 2016, 1, 766–775 CAS.
- S. Choi, B. Kim, S. Park, J. H. Seo and S. K. Ahn, ACS Appl. Mater. Interfaces, 2022, 14, 32486–32496 CAS.
- Y. Wang, R. Shu and X. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303446 CAS.
- Q. Wu, P. M. Rauscher, X. Lang, R. J. Wojtecki, M. J. A. H. Juan, J. de Pablo and S. J. Rowan, Science, 2017, 358, 1434–1439 CrossRef CAS PubMed.
- N. D. Colley, M. A. Nosiglia, S. L. Tran, G. H. Harlan, C. Chang, R. Li, A. O. Delawder, Y. Zhang and J. C. Barnes, ACS Cent. Sci., 2022, 8, 1672–1682 CrossRef CAS.
- G. H. Harlan, S. L. Tran, N. D. Colley, M. A. Nosiglia, Y. Zhang and J. C. Barnes, Cell Rep. Phys. Sci., 2024, 5, 101767 CrossRef CAS.
- H. Han, J. S. W. Seale, L. Feng, Y. Qiu and J. F. Stoddart, J. Polym. Sci., 2023, 61, 881–902 CAS.
- J. S. W. Seale, B. Song, Y. Qiu and J. F. Stoddart, J. Am. Chem. Soc., 2022, 144, 16898–16904 CrossRef CAS PubMed.
- M. E. Belowich, C. Valente, R. A. Smaldone, D. C. Friedman, J. Thiel, L. Cronin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 5243–5261 CAS.
- N. Momcilovic, P. G. Clark, A. J. Boydston and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 19087–19089 CAS.
- P. R. Ashton, E. J. T. Chrystal, P. T. Glink, S. Menzer, C. Schiavo, N. Spencer, J. F. Stoddart, P. A. Tasker, A. J. P. White and D. J. Williams, Chem. – Eur. J., 1996, 2, 709–728 CAS.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS.
- S. D. P. Fielden, D. A. Leigh, C. T. McTernan, B. Perez-Saavedra and I. J. Vitorica-Yrezabal, J. Am. Chem. Soc., 2018, 140, 6049–6052 CrossRef CAS PubMed.
- R. Walker, R. M. Conrad and R. H. Grubbs, Macromolecules, 2009, 42, 599–605 CrossRef CAS.
- G. Narayanan, Y. Caydamli, H. Tekinalp, I. Matai, R. Boy, C. C. Chung, J. Shen, B. S. Gupta and A. E. Tonelli, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 1529–1537 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01006j |
| This journal is © The Royal Society of Chemistry 2025 |