
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe carbonate exchange reaction strategy for chemical recycling of poly(bisphenol A carbonate) into an epoxy-curing agent†
Zengping
Peng‡
abc,
Haoyue
Chen‡
abcd,
Maoqing
Chai
abc,
Rulin
Yang
*abc,
Guangqiang
Xu
 *abc,
Hongguang
Sun
d and
Qinggang
Wang
*abc
*abc,
Hongguang
Sun
d and
Qinggang
Wang
*abc
aKey Laboratory of Photoelectric Conversion and Utilization of Solar Energy, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao, 266101, China. E-mail: yangrl@qibebt.ac.cn; xu_gq@qibebt.ac.cn; wangqg@qibebt.ac.cn
bShandong Energy Institute, Qingdao, 266101, China
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing, 100049, China
dCollege of Polymer Science and Engineering, Qingdao University of Science and Technology, Qingdao, 266042, China
First published on 10th December 2024
Abstract
Using chemical methods to depolymerize or convert plastic materials for recycling is a useful process to achieve a sustainable and circular plastic economy. Herein, different from conventional chemical methodologies to convert waste plastics into monomers or high value-added chemicals, a “waste polymer to precursor of polymer to new polymer” protocol was proposed. For the chemical recycling of poly(bisphenol A carbonate) (BPA-PC), a novel carbonate exchange strategy was explored, in which BPA-PC was depolymerized into “DPC-PC” via a diphenyl carbonate (DPC)-mediated carbonate exchange reaction. The unique structure of DPC-PC enables it to serve as a curing agent for preparing high-performance and recyclable epoxy thermosets, thus blazing a new trail for the chemical recycling of BPA-PC.
Introduction
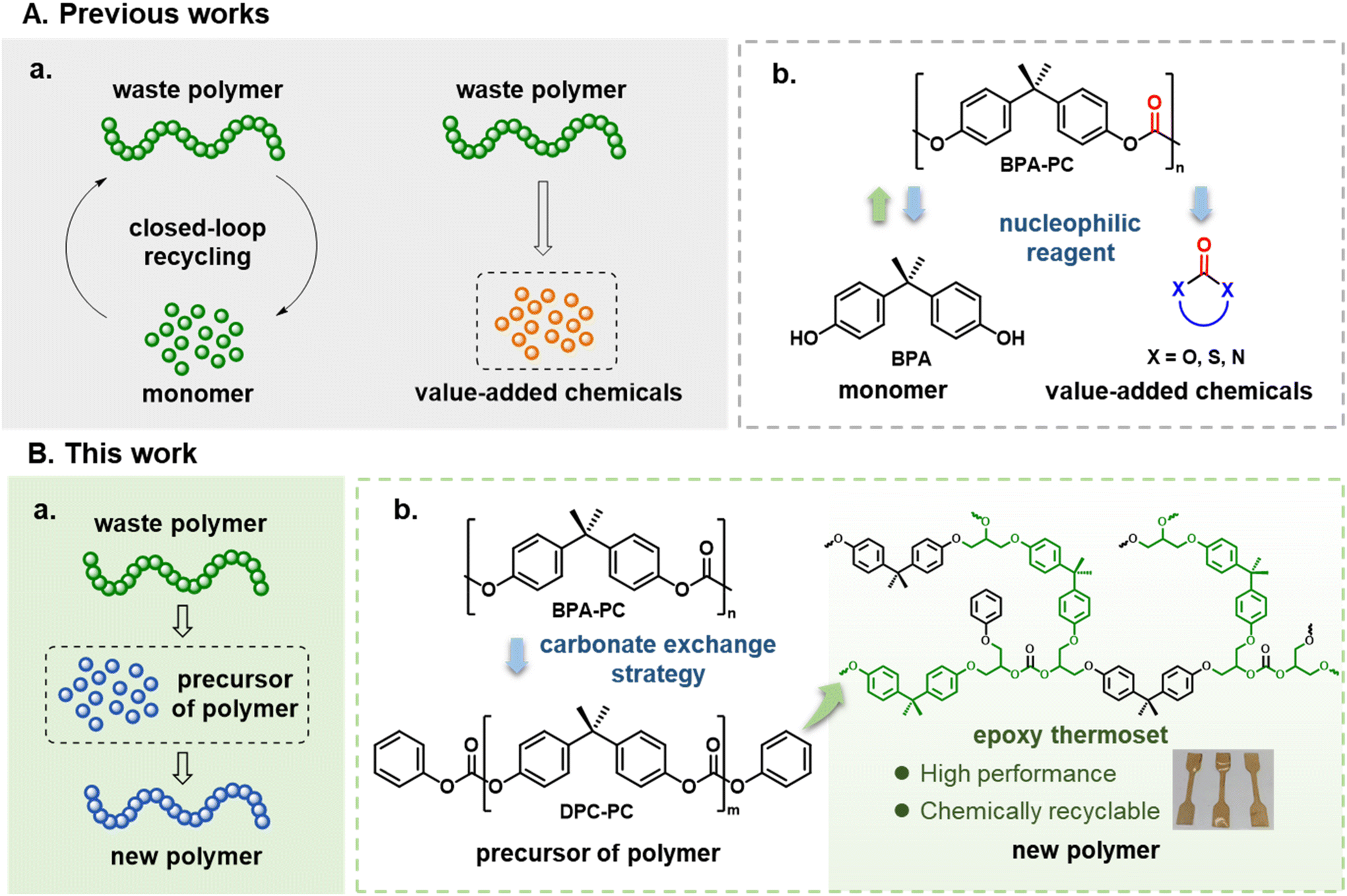
Plastics, due to their low cost and practicality, are playing an increasingly important and indispensable role in human society.1,2 However, with the vigorous development of the plastic industry and consumerism, the crises brought by plastics, such as environmental pollution, human disease, extreme climate, etc., are becoming irreversible global disasters.3–6 To improve this urgent situation, plastic recycling has been regarded as the ultimate choice to solve the plastic problem.7–9 In general, mechanical recycling is the first choice for plastic reutilization. However, mechanical recycling suffers from the inevitable limitations of restrictions on the source of raw materials and the decline in the quality of plastics during this downcycling process.10,11 To avoid these disadvantages caused by mechanical recycling, chemical recycling is considered a more ideal approach for achieving the depolymerization of waste plastics into commercially valuable chemicals.12–14The chemical recycling of plastics can be mainly classified into two pathways, according to the usefulness of depolymerization products (Scheme 1A(a)). One pathway is to depolymerize waste plastics into the initial monomers, which is suitable for polyester plastics, especially for obtaining polyesters by ring-opening polymerization.15–24 After purification, these recycled monomers can re-enter the polymerization process to produce pristine polymers, so as to achieve closed-loop recycling. The other way is to chemically convert waste plastics into other chemicals, which can be used for polymer materials such as polyolefins that are not easy to recycle into monomers.25–29 For polymers obtained by polycondensation of two monomers, such as poly(bisphenol A carbonate) (BPA-PC), polyethylene terephthalate (PET), etc., chemical recycling can recover one initial monomer and produce derivatives of the other monomer as by-products.30–36 For example, by attacking the carbonate bonds on BPA-PC with nucleophiles, a chemical containing carbonyl groups is synthesized and the monomer BPA is formed (Scheme 1A(b)).37–40 While there were numerous successful precedents, in these processes, the acquisition of BPA and additional products normally required a cumbersome post-processing. Besides, to promote the reaction efficiency and improve the yield of the product, an excess of nucleophiles is often required. While this approach enhances reaction outcomes, it can introduce additional steps in the separation and purification process, necessitating a balance between efficiency and operational simplicity. More importantly, although BPA could be reused in the repolymerization process, the utilization of carbonyl-containing chemicals also needed to be taken into account. For example, methanol is usually used as a depolymerizing agent, and the obtained carbonyl-containing chemical is dimethyl carbonate. Nevertheless, it is necessary to convert dimethyl carbonate into diphenyl carbonate before it can enter the synthesis process of BPA-PC.41 Meanwhile, other carbonyl-containing depolymerization products, which are referred to as “value-added” products, need further consideration for their real application market.42,43 Whether the recycling process is truly economical compared with the conventional production methods of these chemicals has not been tested. This is also a problem that needs to be contemplated in the current “chemical upcycling”. These challenges highlight the need for developing depolymerization strategies that minimize the reliance on complex separation processes and excess reaction reagents. By enabling the efficient reuse of depolymerization products, such strategies align more closely with the principles of green chemistry, promoting sustainability and operational simplicity.44
 | ||
| Scheme 1 Chemical recycling strategies of plastics. | ||
Facing the above issues, for polymers like BPA-PC that are difficult to chemically recycle into pristine monomers, converting them into precursors of polymers to directly prepare new polymers could provide a new solution for the recycling of waste polymers (Scheme 1B(a)). Based on this thread, they are then associated with epoxy thermosets, especially those based on bisphenol A (BPA). As indispensable materials, they are widely applied in rail transit, aerospace and other application scenarios.45–47 If the recycling of BPA-PC can be coupled with the synthesis of epoxy thermosets, it will be of great significance. To this end, a novel carbonate exchange reaction strategy is designed (Scheme 1B(b)). Using diphenyl carbonate (DPC) as a carbonate exchange reagent, the BPA-PC polymer chains can be transformed into a new chemical “DPC-PC”. The unique structure of DPC-PC enables its potential use as an epoxy-curing agent to prepare epoxy thermosets. Specifically, the carbonate groups at both ends act as active groups to react with epoxy groups,48 and the perfect double-capped structure increases the cross-linking density. The rigid skeleton of BPA brings mechanical strength to the resulting thermosets. In addition, the low molecular weight of DPC-PC also provides operational convenience for the curing process.
If this protocol is achieved, it will provide the following environmental benefits: (1) the efficient transformation of BPA-PC into epoxy thermosets will open a new path for the chemical recycling of end-of-life plastics. (2) In this process, the degradation products of BPA-PC could avoid isolation or purification as much as possible, with no excess reagents involved, thereby achieving 100% atomic utilization. (3) The synthesized epoxy thermosets will retain the carbonate group, which has the feasibility of achieving depolymerization and topological structure recombination, thus endowing the thermosets with recyclability. Therefore, in this work, this carbonate exchange reaction strategy was systematically explored in the chemical recycling of BPA-PC. Various end-of-life BPA-PC products were used to demonstrate the usefulness of this strategy. Besides, using DPC-PC as an epoxy-curing agent, the performance of the prepared epoxy thermosets was validated.
Experimental
Materials
The BPA-PC pellet (Mn = 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1, Sigma-Aldrich), diphenyl carbonate (DPC) (99%, Energy Chemical), bisphenol A (97%, Sigma-Aldrich), 2,2-bis(4-glycidyloxyphenyl)propane (DGEBA) (85%, TCI), and 4-dimethylaminopyridine (DMAP) (99%, Energy Chemical) were used as received without further purification. Sodium phenolate (98%, Macklin) was dissolved in anhydrous methanol to form a 10 mg mL−1 solution for easy weighing before use. The commercial BPA-PC materials, including veils, lampshades, goggles, discs, buckets, and tubes, were purchased from Taobao.com or amazon.cn and were cut into 5 mm square fragments before use. Chromatographic tetrahydrofuran (THF) was purchased from Honeywell Ltd for the analysis of SEC measurements.
400 g mol−1, Sigma-Aldrich), diphenyl carbonate (DPC) (99%, Energy Chemical), bisphenol A (97%, Sigma-Aldrich), 2,2-bis(4-glycidyloxyphenyl)propane (DGEBA) (85%, TCI), and 4-dimethylaminopyridine (DMAP) (99%, Energy Chemical) were used as received without further purification. Sodium phenolate (98%, Macklin) was dissolved in anhydrous methanol to form a 10 mg mL−1 solution for easy weighing before use. The commercial BPA-PC materials, including veils, lampshades, goggles, discs, buckets, and tubes, were purchased from Taobao.com or amazon.cn and were cut into 5 mm square fragments before use. Chromatographic tetrahydrofuran (THF) was purchased from Honeywell Ltd for the analysis of SEC measurements.
General procedure for the depolymerization of BPA-PC into DPC-PC
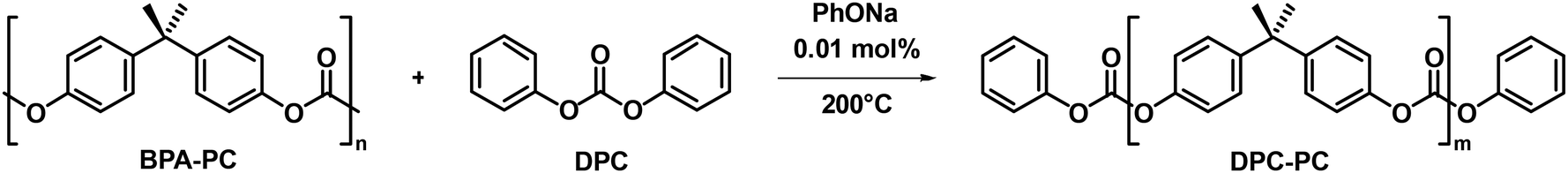
Taken from Table 1, entry 1 as an example, to a round-bottom flask equipped with a magnetic stir bar, sodium phenolate in methanol solvent (11.6 μL, 0.001 mmol) was added first, and then the methanol was removed by vacuum. The BPA-PC pellet (2.54 g, 10 mmol based on the BPA unit) and DPC (2.14 g, 10 mmol) were added into the flask. The reaction was carried out at 200 °C under sealed conditions. Initially, upon heating to 200 °C, BPA-PC did not dissolve in the molten DPC, but changed to a white opaque state, indicating that the reaction was ongoing. As the reaction proceeded, the opaque BPA-PC was gradually consumed. The reaction was stopped when the reaction mixture became clear and transparent. SEC and NMR measurements were performed to characterize the product. Then, the reaction mixture was directly subjected to column chromatography using petroleum ether (PE) and ethyl acetate (EA) as the eluent in a constant ratio of 5:1 to separate the products. The products of DPC-PC (m = 1) (2.87 g, 61%) and DPC-PC dimer (m = 2) (0.68 g, 19%) were confirmed through SEC and NMR measurements (Fig. S1–6†).
|
|
||||
|---|---|---|---|---|
| Entrya | DPCb | t (h) |
M
n
(g mol−1) |
Đ |
|
a All the reactions were carried out at 200 °C, adding 0.01% equivalent sodium phenolate as a catalyst and the BPA-PC material with Mn = 23400 g mol−1, Đ = 2.1, as determined by size-exclusion chromatography.
b Different equivalents of diphenyl carbonate (DPC) relative to polymer repeating units were used during the depolymerization of BPA-PC.
c Molecular weight and dispersity of DPC-PC were determined by size-exclusion chromatography (SEC) using THF as the eluent under 1 mL min−1 flow rate and using polystyrene standards for calibration.
d The reactions were carried out without a catalyst.
e The reaction was carried out at 150 °C.
|
||||
| 1d | 1 | 1 | 23400 |
2.1 |
| 2 | 1 | 1 | 800 | 1.4 |
| 3 | 0.5 | 5 | 1300 | 1.9 |
| 4 | 0.3 | 8 | 2200 | 2.5 |
| 5e | 1 | 24 | 720 | 1.4 |
DPC-PC (m = 1): 1H NMR (400 MHz, chloroform-d) δ 7.48–7.35 (m, 4H), 7.30–7.23 (m, 10H), 7.18 (d, J = 8.8 Hz, 4H), 1.69 (s, 6H).
13C NMR (100 MHz, chloroform-d) δ 152.2, 151.0, 149.0, 148.3, 129.6, 128.0, 126.3, 120.9, 120.4, 42.6, 31.0.
DPC-PC (m = 2):1H NMR (400 MHz, chloroform-d) δ 7.45–7.36 (m, 4H), 7.30–7.23 (m, 18H), 7.13–7.20 (m, 8H), 1.69 (s, 12H).
13C NMR (100 MHz, chloroform-d) δ 152.2, 152.2, 151.0, 149.0, 149.0, 148.4, 148.3, 129.6, 128.0, 128.0, 126.3, 120.9, 120.4, 120.4, 42.6, 31.0.
The reactions for the other entries in Table 1 were carried out using identical procedures, with only the amount of DPC or the reaction temperature changing.
General procedure for the depolymerization of commercial BPA-PC materials
To a Schlenk flask equipped with a magnetic stir bar, sodium phenolate in methanol solvent (4.6 μL, 0.394 μmol) was added first, and then the methanol was removed by vacuum. The fragmented commercial BPA-PC materials (1 g, 3.94 mmol based on BPA unit) and DPC (0.84 g, 3.94 mmol) were added into the flask. These reactions were carried out at 200 °C under sealed conditions. The reactions were stopped when the product became clear and transparent at ambient temperature. The products were characterized by SEC to determine the molecular weights after depolymerization.General procedure for preparing DPC-PC-based epoxy thermosets
2,2-Bis(4-glycidyloxyphenyl)propane (DGEBA) (2.9 g, 8.55 mmol) and DPC-PC (from Table 1, entry 1) (4 g, 8.55 mmol) were dissolved in anhydrous THF (20.7 g) to form a homogeneous solution, and then 4-dimethylaminopyridine (DMAP, 7.25 mg, 0.25 wt% of epoxy) was added. The solution was cast into a Teflon mold when DMAP dissolved completely, and then the sample was dried under vacuum overnight before curing. The epoxy resin was cured by periodically heating at 80 °C (4 h), 120 °C (2 h), 140 °C (2 h), 160 °C (2 h), 180 °C (2 h), 200 °C (2 h), and 220 °C (2 h) under an Ar atmosphere to obtain a yellow transparent film.General procedure for the depolymerization of DPC-PC-based epoxy thermosets
The cured epoxy resin (0.808 g, 1 mmol) was cut into 2 mm fragments before being added into a 5 mL round-bottle flask with N,N′-dimethyl-1,2-ethanediamine (0.186 g, 2.12 mmol), and then the flask was sealed and heated at 120 °C for 8 h. The depolymerization products 1,3-dimethyl-2-imidazolidinone (DMI) and phenoxy resin were separated by distillation. The crude DMI was purified by thin-layer chromatography (PE:EA = 1:1) to remove any remaining unreacted N,N′-dimethyl-1,2-ethanediamine. NMR and SEC analyses confirmed the structure of depolymerization products. After purification, the yield of DMI was calculated to be 52%, and the yield of phenoxy resin was calculated to be 99%.
Results and discussion
The carbonate exchange reaction between DPC and BPA-PC
To verify the feasibility of this strategy, the reaction between the carbonate exchange reagent DPC and BPA-PC was first investigated (Table 1). BPA-PC was first mixed with an equivalent amount of DPC relative to the carbonate groups, theoretically causing all carbonate groups to undergo the carbonate exchange reaction, thereby obtaining the target product DPC-PC (m = 1) (Scheme S2†). The reaction temperature was set at 200 °C to facilitate the carbonate exchange reaction, which was inspired by the conditions of polycondensation used to synthesize BPA-PC.49–51 The reaction was conducted without the use of solvents, as a solvent-free process reduces the complexity of separation, minimizes the generation of harmful by-products, and lowers the risk of volatile emissions. Firstly, the reaction results without the presence of a catalyst were explored (Table 1, entry 1). The almost unchanged molecular weight (Mn) and dispersity (Đ) of BPA-PC indicate that the addition of a catalyst is necessary. Therefore, 0.01 mol% sodium phenolate (PhONa) was added as a catalyst. As expected, in this case, BPA-PC was observed to melt rapidly in DPC, and a colorless transparent liquid was obtained after 1 h of reaction.Size-exclusion chromatography (SEC) was first employed to detect the reaction process. It was clearly observed that the peak with a molecular weight of 23400 g mol−1 for the initial BPA-PC completely disappeared, and a new group of peaks with a molecular weight of ca. 800 g mol−1 appeared (Table 1, entry 2, Fig. 1a), which confirmed that BPA-PC underwent a significant depolymerization under the action of DPC. It is worth noting that three groups of peaks could be clearly observed in the SEC trace (Fig. 1a, blue line). Therefore, in order to clarify each peak corresponding to the product in the SEC result, the reaction mixture was separated through column chromatography. By comparing the 1H NMR analysis and SEC trace (Fig. S1–3†), the product DPC-PC (m = 1) was clearly identified. Its molecular weight is 468 g mol−1, which is consistent with SEC results (SEC results: Mn = 434 g mol−1, Đ = 1.05). Similarly, the dimer (m = 2) and multimers (m = 3–5) were also found (Fig. S4–8†). That is to say, under these conditions, the main products were DPC-PC (m = 1 and 2), and there was also a small amount of multimers (m = 3–5) present due to the existence of equilibrium in the reversible reaction of carbonate exchange (Fig. 1b). In addition, the structure of depolymerization products was accurately characterized by high-resolution mass spectrometry (HRMS) (Fig. 1c). A phenyl-terminated carbonate structure containing bisphenol A was confirmed, with its molar mass corresponding to the quasi-molecular ion peak ([M + NH4]+, m/z = 486.19). The formation of this phenyl carbonate terminal structure revealed the occurrence of carbonate exchange reaction between the carbonate groups in DPC and BPA-PC.
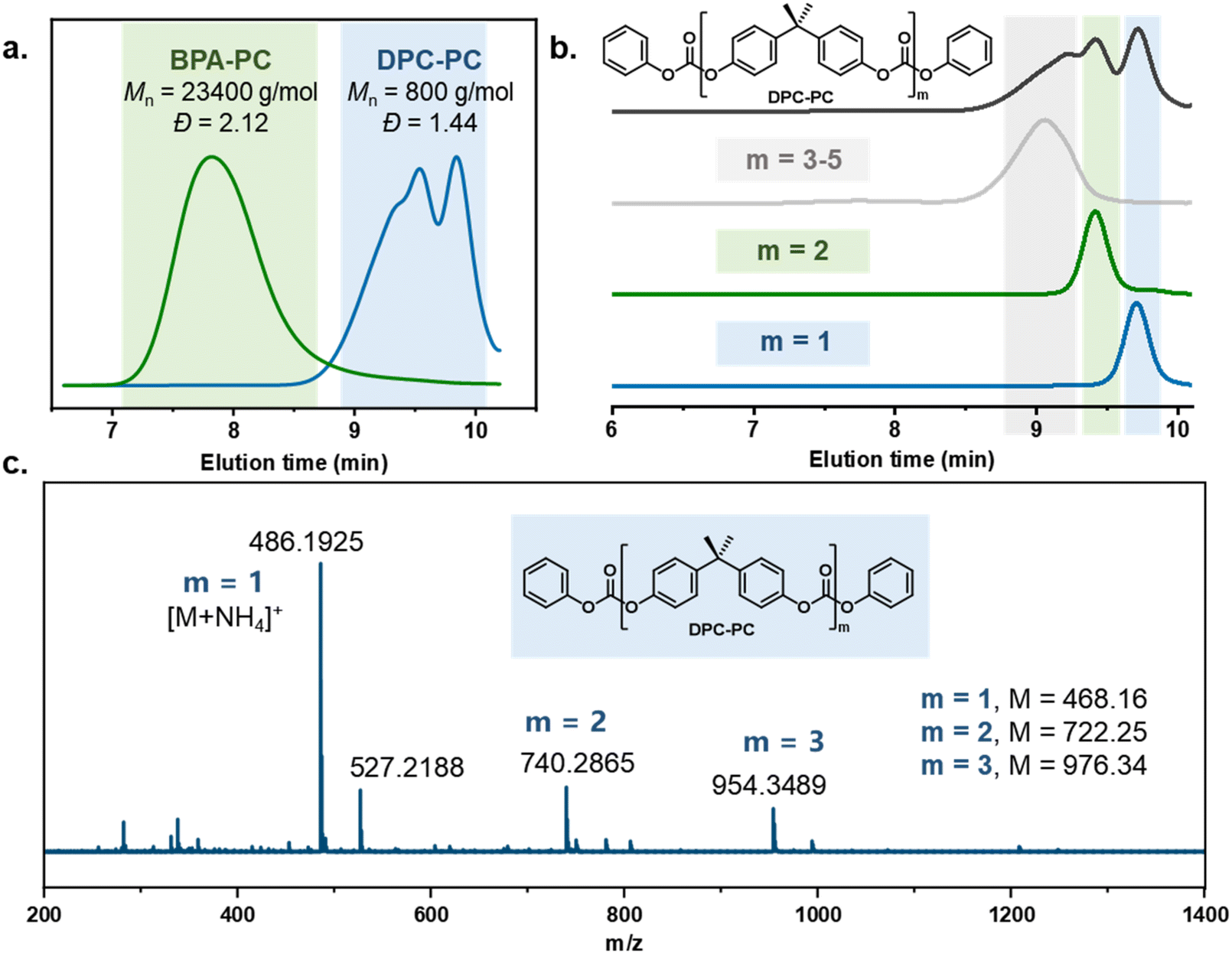 | ||
| Fig. 1 Characterization of the depolymerized product DPC-PC (Table 1, entry 2). (a) SEC analysis of the initial BPA-PC and the depolymerization product DPC-PC. (b) SEC analysis of DPC-PC of different molecular weights. (c) HRMS analysis of the depolymerization product DPC-PC. | ||
Meanwhile, the structure of depolymerization products was further explored by intensive NMR testing. The average repeated unit value (m) of the depolymerized products could be obtained through the integral area ratio of BPA repeating units and terminated phenyl carbonates (Fig. S9†). However, the characteristic peaks of the depolymerization products were highly coincident with those of the initial BPA-PC and DPC, making it difficult to accurately distinguish them from the raw material (Fig. S10†). Despite this, no other impurity peaks were observed in the NMR spectra, indicating that there were no other by-products generated during the process. To clearly identify the progress of the reaction, the depolymerization products were further analysed by 1H DOSY. Before the reaction started, DPC and BPA-PC showed obvious differences in diffusion coefficients due to discriminate molecular volumes (Fig. 2a). After depolymerization, a group of products with diffusion coefficients between that of BPA-PC and DPC appeared, which could be assigned to the products DPC-PC (Fig. 2b). The above characteristics fully reveal the structure of the obtained product.
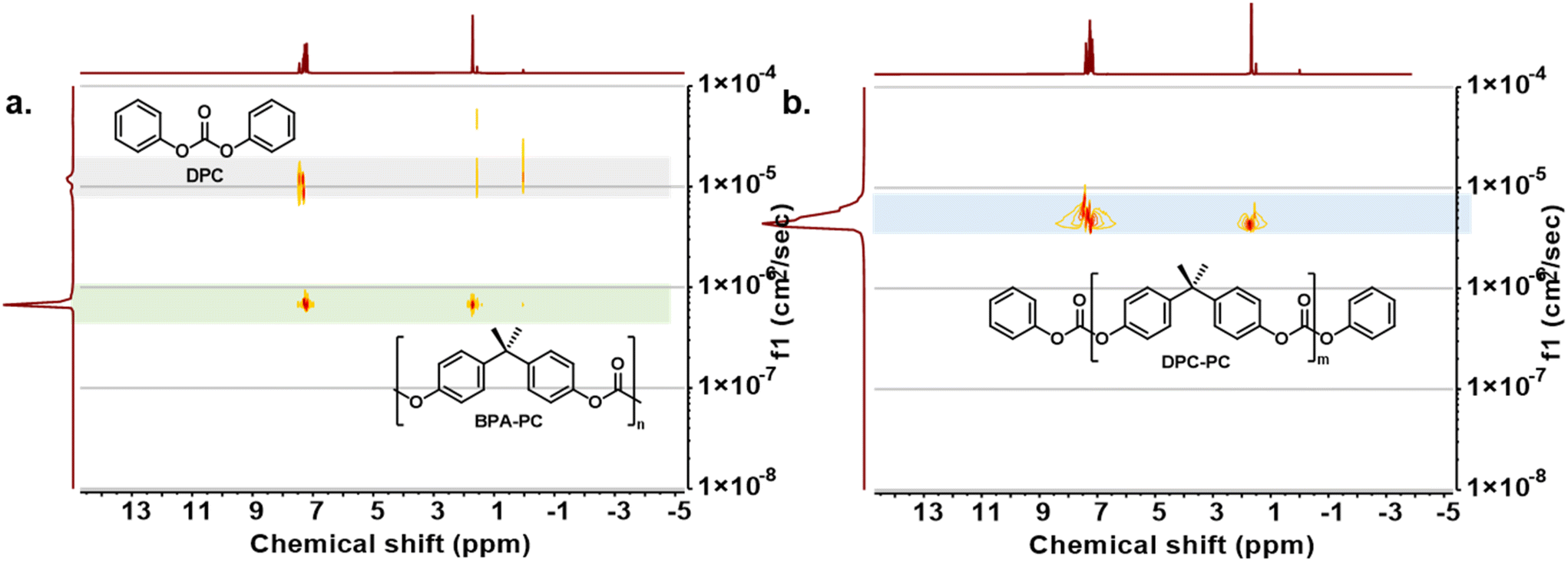 | ||
| Fig. 2 (a) 1H DOSY analysis of a BPA-PC and DPC equimolar mixture. (b) 1H DOSY analysis of the depolymerization product DPC-PC. | ||
The influence of the equivalent of carbonate exchange reagent and reaction temperature
Furthermore, it was suspected that if part of the carbonate groups reacted with the participation of a lower equivalent of DPC, longer chain structures might be obtained. To test this conjecture, the DPC addition was first reduced to 0.5 equivalents. After 5 h of reaction, a colorless liquid with a molecular weight of 1300 g mol−1 was obtained as well (Table 1, entry 3). Although small amounts of DPC-PC (m = 1) and DPC-PC dimer (m = 2) were observed, the multimers occupied a major position (Fig. 3, grey line). Further reducing the DPC equivalent to 0.3 equivalents, the depolymerization could still proceed smoothly within 8 h, yielding the DPC-PC products with a molecular weight of 2200 g mol−1 (Table 1, entry 4). At this point, higher molecular weight multimers dominated the depolymerization products, resulting in a more pronounced single peak trend (Fig. 3, orange line). As shown in Fig. 3, with the decrease of DPC addition, the elution time of the product in the SEC column shortened, which means that the average chain length of “DPC-PC” could be adjusted by regulating the amount of carbonate exchange reagent added. These results further demonstrated the feasibility of this strategy.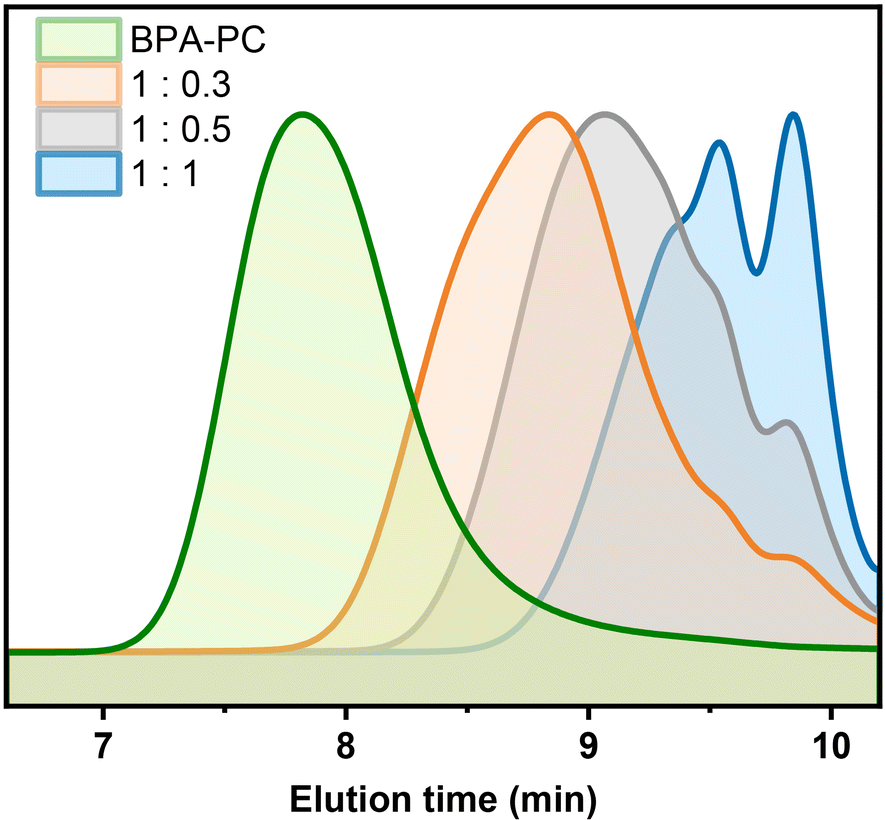 | ||
| Fig. 3 SEC analysis of depolymerization products with different DPC additions. | ||
Besides, when the temperature was further reduced to 150 °C, the reaction could still be carried out smoothly (Table 1, entry 5). The depolymerization products with the expected results were obtained after 24 h, which was consistent with the results at 200 °C (Mn = 800 vs. 720 g mol−1, Đ = 1.44 vs. 1.42). However, considering the efficiency and energy consumption of the reaction, the high reaction activity and shortened time at 200 °C were preferred. After comprehensive consideration, reactions at 200 °C were selected for further exploration.
Depolymerization of commercial BPA-PC materials
In practical BPA-PC products, additives are added to improve their performance.52 Therefore, it is necessary to explore the depolymerization process of BPA-PC used in real life to verify the practicability of this strategy. Hence, the common BPA-PC products in the market were collected, including veils, buckets, lampshades, discs, tubes, and goggles. The main components of these materials were proved to be BPA-PC by 1H NMR analysis (Fig. S11–16†). These end-of-life PC products exhibit different molecular weights, ranging from 12200 g mol−1 to 25500 g mol−1, as determined by SEC testing (Fig. S17–22†). Then, these plastic products were broken into about 5 mm square fragments and subjected to the carbonate exchange strategy. The reaction conditions were the same as before, involving 1 equiv. of DPC carbonate exchange reagent and 0.01 mol% PhONa catalyst at 200 °C. Satisfactorily, the rapid melting of these materials in DPC was observed. The reaction process was monitored by SEC testing of the samples taken during the reaction. As shown in Fig. 4, all the reactions could be completed within 2–4.5 h, yielding small molecule products with molecular weights from 700 g mol−1 to 1100 g mol−1, similar to those observed before. These results indicated that these commercial BPA-PC materials have also been completely depolymerized. It is worth noting that the addition of blue pigment was clearly observed in the BPA-PC bucket, but it did not affect the depolymerization process. In addition, there were other insoluble substances, such as polyacrylate and metallic silver, in the BPA-PC disc, which did not hinder the reaction as well and could be removed by filtration after the reaction. These successful processes fully demonstrate the tolerance of the catalytic strategy to additives in BPA-PC commodities.
 | ||
| Fig. 4 Depolymerization of commercial BPA-PC products. | ||
Synthesis of the DPC-PC-based epoxy thermoset
Next, further verification was conducted on whether the obtained DPC-PC could be employed as a curing agent for epoxy resins. Due to the absence of excessive reagents and other side reactions during the depolymerization process, DPC-PC (from Table 1, entry 2) was directly transferred to the next stage without cumbersome purification steps. 2,2-Bis(4-glycidyloxyphenyl)propane (DGEBA), as the most commonly used epoxy resin, was chosen as the epoxy resin. The epoxy thermoset was prepared using the casting method. DGEBA and DPC-PC were dissolved in tetrahydrofuran with a molar ratio of epoxy groups to carbonates equal to 1:1; then 4-dimethylaminopyridine (DMAP, 0.25 wt% of DGEBA) was added as a catalyst before casting into a Teflon mold. After removing the solvent and de-bubbling through vacuum, gradient heating to 220 °C ensured the curing process proceeded smoothly (Fig. 5a). The obtained epoxy thermoset displayed good solvent resistance and remained intact in various conventional organic solvents and aqueous solutions, revealing its highly cross-linked structure. Therefore, Fourier-transform infrared (FTIR) spectroscopy analysis was conducted to detect the structure of the cured epoxy resin. As shown in Fig. 5b, after curing, the peak of epoxy at 913 cm−1 disappeared, and the carbonate bond peak in the cured epoxy resin shifted to 1745 cm−1 compared to that in DPC-PC, 1769 cm−1. The above information proves that a chemical reaction occurred between the epoxy group and the carbonate group, indicating the successful preparation of the DPC-PC-based epoxy thermoset.
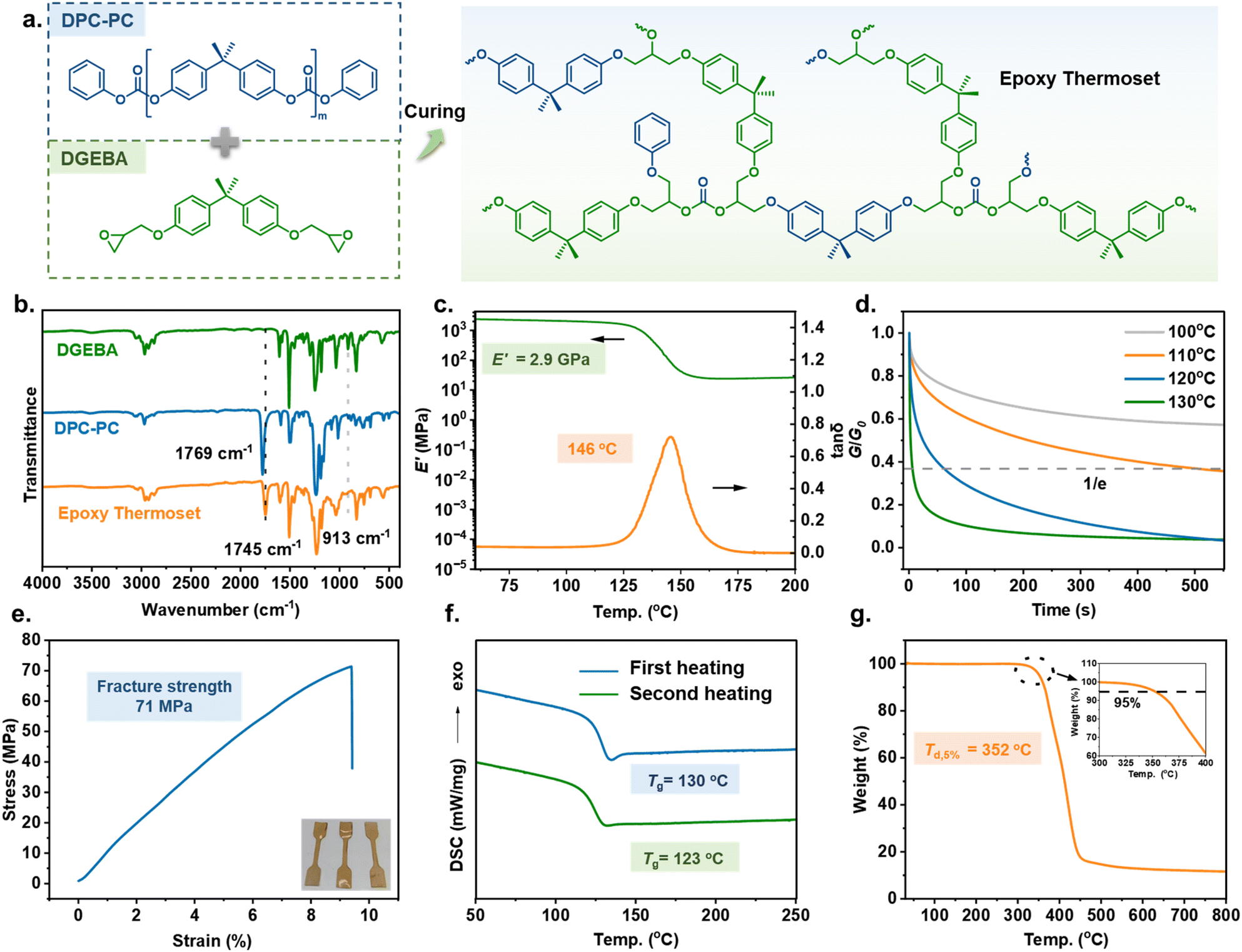 | ||
| Fig. 5 (a) Synthesis of the epoxy thermoset using DPC-PC as a curing agent. (b) FTIR spectra of DGEBA, DPC-PC and the obtained epoxy thermoset. (c) Dynamic mechanical analysis of DPC-PC-based epoxy thermoset; storage modulus E′ and loss angle tangent tanδ as a function of temperature from 60 to 200 °C with a heating rate of 3 °C min−1. (d) Stress relaxation curves of the epoxy thermoset in strain-control mode at temperatures from 100 to 130 °C. (e) Tensile stress–strain curves of the obtained epoxy thermoset with a stretching rate of 2 mm min−1 at room temperature. (f) DSC analysis of the obtained epoxy thermoset. (g) TGA analysis of the obtained epoxy thermoset. | ||
Performance of the DPC-PC-based epoxy thermoset
Investigations into the performance of this DPC-PC-based epoxy thermoset were conducted. Firstly, the dynamic mechanical properties of the curved sample were determined through dynamic mechanical analysis (DMA). As shown in Fig. 5c, a sharp loss factor (tan δ) peak was observed. The peak temperature of tan δ, that is, the glass transition temperature (Tg), reached 146 °C. The storage modulus (E′) of the epoxy thermoset reached 2.87 GPa in the glass state, and it still reached 24.3 MPa at Tg + 30 °C. Therefore, the cross-linking density (ve) was calculated to be 2170 mol m−3 by applying this value into the formula E′ = 3veRT (E′ is the storage modulus at Tg + 30 °C, R is the gas constant, and T is the absolute temperature). The Tg and modulus are positively correlated with cross-linking density and also depend on polymer structures. Therefore, the considerable Tg and E′ of this material are derived from its high cross-linking density and the rigidity of the BPA skeleton. In addition, a stable platform of rubber modulus was also detected, which confirmed the existence of cross-linked network structures from the side.The above data indicated that the synthesized thermosetting resin exhibited good viscoelastic characteristics. Furthermore, the dynamic performance of the DPC-PC-based epoxy thermoset was investigated by the stress relaxation experiment of DMA (Fig. 5d). The stress relaxation curves showed that this thermosetting material obeys typical Arrhenius temperature-dependent behaviour. The stress relaxed rapidly with increasing temperature. At 110 °C, the time required for the modulus to relax to 1/e of the initial one (the relaxation time τ*) was 505 s, whereas the τ* declined to 61 s at 120 °C, or even only 6 s at 120 °C. This phenomenon revealed the presence of rapid dynamic topological rearrangements in the DPC-PC-based epoxy networks, which is attributed to the carbonate exchange reaction of the dynamic carbonate bonds retained in the network.53,54
The mechanical properties were also taken into account by analyzing the tensile stress–strain curve. As shown in Fig. 5e, the stress increased continuously with strain until rupture, and no stress yield appeared. The fracture strength was up to 71 MPa, accompanied by the elongation at break of 9.5%. This material displayed the mechanical properties of a typical epoxy thermoset. The strong fracture strength is comparable to those of most epoxy thermosets reported at present.55,56
The thermal behavior of the polymer was investigated through differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). DSC analysis revealed that the DPC-PC-based epoxy thermoset displayed a Tg of 130 °C in the first heating scan, which decreased to 123 °C after the elimination of thermal history (Fig. 5f). Due to differences in testing principles, there was a slight difference in Tg obtained from DMA.57 Thermogravimetric analysis (TGA) showed that the decomposition temperature at 5% mass loss (Td,5%) reached 352 °C (Fig. 5g), demonstrating the excellent thermal stability of this material. The above performance characterization indicates that the DPC-PC-based epoxy thermoset possesses good comprehensive properties, such as dynamic performance, mechanical performance, and thermal stability, owing to the exquisite structures introduced into the thermoset by DPC-PC as a curing agent.
Depolymerization of the DPC-PC-based epoxy thermoset
Generally, the inherent high cross-linking characteristics of epoxy thermosets make their chemical recycling more challenging, which is also one of the environmental problems faced by such materials.58,59 The presence of easily chemically breakable carbonate bonds in DPC-PC-based epoxy thermosets has prompted us to consider whether this material can achieve efficient chemical recycling. For this purpose, N,N′-dimethyl-ethylenediamine (DMEDA) was considered as a depolymerization reagent, which could capture carbonyl to produce 1,3-dimethyl-2-imidazolidinone (DMI).60 Surprisingly, the depolymerization reaction of this DPC-PC-based epoxy thermoset could take place smoothly even at 120 °C without additional auxiliary solvents and catalysts (Fig. 6a). After depolymerization, DMI with a relatively low boiling point was obtained by distillation (Fig. 6b). The remaining high boiling point products were analyzed and confirmed to be phenoxy resin (Fig. 6c). DMI can be used as a green solvent, and phenoxy resin is widely used in most adhesives, coatings, prepregs, composites and molded parts,46,61 so as to realize the degradation of epoxy thermosetting substances into useful chemicals. Therefore, the depolymerization strategy of this work not only achieves the depolymerization of BPA-PC, but also couples the synthesis and recycling of the epoxy thermoset.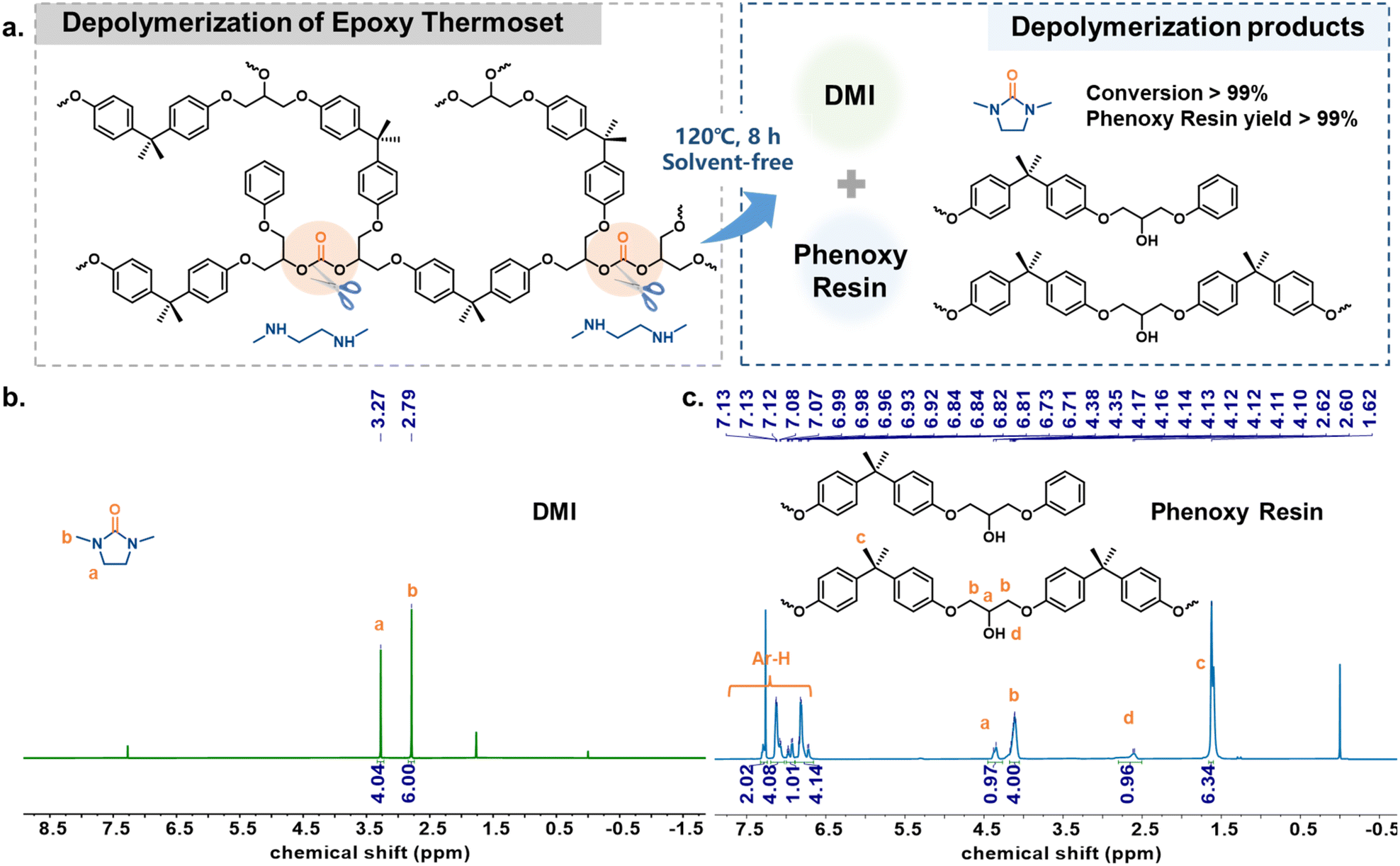 | ||
| Fig. 6 (a) Depolymerization of the epoxy thermoset using N,N′-dimethyl-ethylenediamine (DMEDA) as the depolymerizing agent. (b) 1H NMR spectrum of the depolymerization product DMI. (c) 1H NMR spectrum of the depolymerization product phenoxy resin. | ||
Conclusions
In summary, a carbonate exchange strategy was proposed to achieve chemical recycling of BPA-PC plastics into precursors of polymers to prepare new polymers. In this strategy, BPA-PC polymer chains were sheared by DPC, taking advantage of the carbonate exchange reaction, while transforming the polymer into a new chemical “DPC-PC”. The structure of DPC-PC was confirmed by SEC, HRMS analysis and 1H DOSY. The depolymerization strategy was tolerant to various impurities in commercial BPA-PC materials, demonstrating significant practical applications. More importantly, the unique structure of DPC-PC enables it to serve as a curing agent for epoxy resins, resulting in the preparation of a DPC-PC-based epoxy thermoset with excellent comprehensive properties. Meanwhile, the retention of the carbonate bond in the epoxy thermoset also gives it the feasibility of efficient chemical recycling, thus achieving the coupling of depolymerization of BPA-PC and the epoxy resin simultaneously. This strategy is innovative compared to the conventional approach of depolymerizing waste plastics into the initial monomers or high-value chemicals, and it will further inspire the development of plastic recycling.Author contributions
Zengping Peng: investigation, formal analysis, and writing – original draft. Haoyue Chen: investigation, formal analysis, and writing – original draft. Maoqing Chai: validation and formal analysis. Rulin Yang: methodology, formal analysis, visualization, and writing – original draft. Guangqiang Xu: conceptualization, supervision, writing – review & editing, and funding acquisition. Hongguang Sun: writing – review & editing. Qinggang Wang: conceptualization, writing – review & editing, supervision, project administration, and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province (ZR2023ME008), the National Natural Science Foundation of China (21901249), and the Taishan Scholars Program of Shandong Province (tstp20240520).References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- H. T. Pinheiro, C. MacDonald, R. G. Santos, R. Ali, A. Bobat, B. J. Cresswell, R. Francini-Filho, R. Freitas, G. F. Galbraith, P. Musembi, T. A. Phelps, J. P. Quimbayo, T. Quiros, B. Shepherd, P. V. Stefanoudis, S. Talma, J. B. Teixeira, L. C. Woodall and L. A. Rocha, Nature, 2023, 619, 311–316 CrossRef CAS PubMed.
- L. Cabernard, S. Pfister, C. Oberschelp and S. Hellweg, Nat. Sustain., 2021, 5, 139–148 Search PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- J. Guo, S. Ali and M. Xu, Green Carbon, 2023, 1, 150–153 Search PubMed.
- P. Stegmann, V. Daioglou, M. Londo, D. P. van Vuuren and M. Junginger, Nature, 2022, 612, 272–276 Search PubMed.
- W. W. Y. Lau, Y. Shiran, R. M. Bailey, E. Cook, M. R. Stuchtey, J. Koskella, C. A. Velis, L. Godfrey, J. Boucher, M. B. Murphy, R. C. Thompson, E. Jankowska, A. Castillo Castillo, T. D. Pilditch, B. Dixon, L. Koerselman, E. Kosior, E. Favoino, J. Gutberlet, S. Baulch, M. E. Atreya, D. Fischer, K. K. He, M. M. Petit, U. R. Sumaila, E. Neil, M. V. Bernhofen, K. Lawrence and J. E. Palardy, Science, 2020, 369, 1455–1461 CrossRef CAS PubMed.
- R. Meys, A. Kätelhön, M. Bachmann, B. Winter, C. Zibunas, S. Suh and A. Bardow, Science, 2021, 374, 71–76 CrossRef CAS PubMed.
- J. Zheng and S. Suh, Nat. Clim. Change, 2019, 9, 374–378 Search PubMed.
- Z. Ma, M. W. Ryberg, P. Wang, L. Tang and W.-Q. Chen, ACS Sustainable Chem. Eng., 2020, 8, 16861–16868 CrossRef CAS.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- R. Yang, G. Xu, W. Tao, Q. Wang and Y. Tang, Green Carbon, 2024, 2, 1–11 CrossRef.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- C. F. Gallin, W.-W. Lee and J. A. Byers, Angew. Chem., Int. Ed., 2023, 62, e202303762 CrossRef CAS PubMed.
- Y.-M. Tu, F.-L. Gong, Y.-C. Wu, Z. Cai and J.-B. Zhu, Nat. Commun., 2023, 14, 3198 CrossRef CAS PubMed.
- X.-L. Li, R. W. Clarke, H.-Y. An, R. R. Gowda, J.-Y. Jiang, T.-Q. Xu and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2023, 62, e202303791 CrossRef CAS PubMed.
- Z. Cai, Y. Liu, Y. Tao and J.-B. Zhu, Acta Chim. Sin., 2022, 80, 1165–1182 CrossRef CAS.
- T. M. McGuire, A. Buchard and C. Williams, J. Am. Chem. Soc., 2023, 145, 19840–19848 CrossRef CAS PubMed.
- Y. Wang, Y. Zhu, W. Lv, X. Wang and Y. Tao, J. Am. Chem. Soc., 2023, 145, 1877–1885 CrossRef CAS PubMed.
- L. Wursthorn, K. Beckett, J. O. Rothbaum, R. M. Cywar, C. Lincoln, Y. Kratish and T. J. Marks, Angew. Chem., Int. Ed., 2023, 62, e202212543 CrossRef CAS PubMed.
- Y. Xia, P. Yuan, Y. Zhang, Y. Sun and M. Hong, Angew. Chem., Int. Ed., 2023, 62, e202217812 CrossRef CAS PubMed.
- G. Xu and Q. Wang, Green Chem., 2022, 24, 2321–2346 RSC.
- R. Yang, G. Xu, C. Lv, B. Dong, L. Zhou and Q. Wang, ACS Sustainable Chem. Eng., 2020, 8, 18347–18353 CrossRef CAS.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- B. Sun, J. Zhang, M. Wang, S. Yu, Y. Xu, S. Tian, Z. Gao, D. Xiao, G. Liu, W. Zhou, M. Wang and D. Ma, Nat. Sustain., 2023, 6, 712–719 CrossRef.
- Z. Xu, N. E. Munyaneza, Q. Zhang, M. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. Liu, Science, 2023, 381, 666–671 CrossRef CAS PubMed.
- Z. Zhang, J. Wang, X. Ge, S. Wang, A. Li, R. Li, J. Shen, X. Liang, T. Gan, X. Han, X. Zheng, X. Duan, D. Wang, J. Jiang and Y. Li, J. Am. Chem. Soc., 2023, 145, 22836–22844 CrossRef CAS PubMed.
- S. Javed, D. Ropel and D. Vogt, Green Chem., 2023, 25, 1442–1452 RSC.
- M. Liu, J. Guo, Y. Gu, J. Gao, F. Liu and S. Yu, ACS Sustainable Chem. Eng., 2018, 6, 13114–13121 CrossRef CAS.
- P. McKeown, M. Kamran, M. G. Davidson, M. D. Jones, L. A. Román-Ramírez and J. Wood, Green Chem., 2020, 22, 3721–3726 RSC.
- K. Onida, M. Fayad, S. Norsic, O. Boyron and N. Duguet, Green Chem., 2023, 25, 4282–4291 RSC.
- E. Quaranta, A. Dibenedetto, F. Nocito and P. Fini, J. Hazard. Mater., 2021, 403, 123957 CrossRef CAS PubMed.
- S. Zhang, Q. Hu, Y.-X. Zhang, H. Guo, Y. Wu, M. Sun, X. Zhu, J. Zhang, S. Gong, P. Liu and Z. Niu, Nat. Sustain., 2023, 6, 965–973 CrossRef.
- S. Zhang, W. Xu, R. Du, X. Zhou, X. Liu, S. Xu and Y.-Z. Wang, Green Chem., 2022, 24, 3284–3292 RSC.
- T. Do, E. R. Baral and J. G. Kim, Polymer, 2018, 143, 106–114 CrossRef CAS.
- H. J. Jung, S. Park, H. S. Lee, H. G. Shin, Y. Yoo, E. R. Baral, J. H. Lee, J. Kwak and J. G. Kim, ChemSusChem, 2021, 14, 4301–4306 CrossRef CAS PubMed.
- K. Saito, C. Jehanno, L. Meabe, J. L. Olmedo-Martínez, D. Mecerreyes, K. Fukushima and H. Sardon, J. Mater. Chem. A, 2020, 8, 13921–13926 RSC.
- R. Singh, S. Shahi and Geetanjali, ChemistrySelect, 2018, 3, 11957–11962 CrossRef CAS.
- J. G. Kim, Polym. Chem., 2020, 11, 4830–4849 RSC.
- Z. Wang, R. Yang, G. Xu, T. Liu and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 4529–4537 CrossRef CAS.
- X. Zhou, M. Chai, G. Xu, R. Yang, H. Sun and Q. Wang, Green Chem., 2023, 25, 952–959 RSC.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- X. Wu, P. Hartmann, D. Berne, M. De Bruyn, F. Cuminet, Z. Wang, J. M. Zechner, A. D. Boese, V. Placet, S. Caillol and K. Barta, Science, 2024, 384, eadj9989 CrossRef CAS PubMed.
- T.-W. Jiang, K. S. K. Reddy, Y.-C. Chen, M.-W. Wang, H.-C. Chang, M. M. Abu-Omar and C.-H. Lin, ACS Sustainable Chem. Eng., 2022, 10, 2429–2440 CrossRef CAS.
- F.-L. Jin, X. Li and S.-J. Park, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- T. Yashiro, K. Matsushima, A. Kameyama and T. Nishikubo, Macromolecules, 2001, 34, 3205–3210 CrossRef CAS.
- X. Chang, T. Ding, H. Tian and T. Wang, J. Appl. Polym. Sci., 2016, 133, 43636 CrossRef.
- J. Tuominen, J. Kylmä and J. Seppälä, Polymer, 2002, 43, 3–10 CrossRef CAS.
- S. N. Vouyiouka, E. K. Karakatsani and C. D. Papaspyrides, Prog. Polym. Sci., 2005, 30, 10–37 CrossRef CAS.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruiperez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710–6717 CrossRef CAS PubMed.
- X. Jiao, Y. Ma, Z. Zhao, L. Gao, B. Zhang, J. Yang, M. H. Li and J. Hu, Adv. Funct. Mater., 2024, 2409223 CrossRef CAS.
- B. Zhang, T. Cui, X. Jiao, Y. Ma, L. Gao and J. Hu, Chem. Eng. J., 2023, 476, 146625 CrossRef CAS.
- S. You, S. Ma, J. Dai, Z. Jia, X. Liu and J. Zhu, ACS Sustainable Chem. Eng., 2017, 5, 4683–4689 CrossRef CAS.
- W. Yuan, S. Ma, S. Wang, Q. Li, B. Wang, X. Xu, K. Huang, J. Chen, S. You and J. Zhu, Eur. Polym. J., 2019, 117, 200–207 CrossRef CAS.
- M. Janvier, L. Hollande, A. S. Jaufurally, M. Pernes, R. Menard, M. Grimaldi, J. Beaugrand, P. Balaguer, P. H. Ducrot and F. Allais, ChemSusChem, 2017, 10, 738–746 CrossRef CAS PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Matter, 2019, 1, 1456–1493 CrossRef.
- M. Chai, G. Xu, R. Yang, H. Sun and Q. Wang, Molecules, 2024, 29, 640 CrossRef CAS PubMed.
- S. T. Nguyen, E. A. McLoughlin, J. H. Cox, B. P. Fors and R. R. Knowles, J. Am. Chem. Soc., 2012, 143, 12268–12277 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, 1H NMR spectra and GPC analysis for BPA-PC materials and DPA-PC products. See DOI: https://doi.org/10.1039/d4py01175a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |