DOI:
10.1039/D4PY01235F
(Paper)
Polym. Chem., 2025,
16, 475-483
Modulating the packing and photovoltaic performance of (bisthiophene)benzene-linked polymer acceptors through simple methylation engineering†
Received
1st November 2024
, Accepted 5th December 2024
First published on 11th December 2024
Abstract
Three polymerized small molecule acceptors (PSMAs), namely PY-TP, PY-TPMe2, and PY-TPMe4, were designed and synthesized by employing (bisthiophene)benzene linkers containing various methyl-substituted phenylene groups. All the PSMAs exhibit similar absorption maxima in films as well as LUMO energy levels. However, the increased number of methyl groups on the linkers induces steric hindrance and decreases the coplanarity of the polymer backbones, which, in turn, increases intermolecular π–π stacking distances. When the three acceptors are blended with a classical polymer donor PM6, PY-TPMe4 with a highly twisted backbone has a large π–π stacking distance and excessive phase separation, whereas PY-TP and PY-TPMe2 with moderately twisted backbones demonstrate more compact π–π stacking and suitable phase separation morphology. As a result, PY-TP and PY-TPMe2 exhibit better exciton dissociation and charge transport, leading to much higher photovoltaic performance compared to PY-TPMe4. Particularly, PY-TPMe2 effectively regulates the crystallinity and achieves a more suitable phase separation morphology in the blend films. The optimal PY-TPMe2-based photovoltaic device exhibits the best exciton dissociation and charge transport performance, achieving the highest power conversion efficiency (PCE) of 8.4% among the devices based on the three PSMAs, with a high open-circuit voltage (VOC) of 0.97 V, a short-circuit current density (JSC) of 14.74 mA cm−2 and a fill factor (FF) of 59.65%. These findings provide new insights into the regulation of the molecular packing and photovoltaic performance of polymer acceptors through simple methylation modification on linkers for designing novel PSMA materials.
Introduction
Over the past several years, all-polymer solar cells (all-PSCs) have garnered significant attention due to their exceptional flexibility, mechanical robustness, and large-area solution processability, making them promising candidates for the development of portable and wearable electronic devices.1–5 Recent advances in the field of all-PSCs have focused on the development of n-type conjugated polymers incorporating electron-deficient building blocks, such as aromatic diimides,6,7 dicyanobenzothiadiazole,8–10 and B–N bridged bipyridine moieties.11,12 However, the polymer acceptors based on these electron-deficient units usually suffer from some intrinsic flaws, including a narrow absorption range, low extinction coefficient and relatively deep energy levels, which limit the performance of the corresponding all-PSCs. To further improve the performance of polymer acceptor materials, researchers have recently adopted small molecule acceptors (SMAs) with a low bandgap A–D–A structure as the key building block connected by π-bridge linkers to create polymerized small molecule acceptors (PSMAs).13–20 This method utilizes small molecule acceptor units as polymerizable motifs, combining the advantages of polymers and small molecule receptors, like high absorption coefficients, broad light absorption ranges, and appropriate energy levels matched with efficient donors, which demonstrates significant potential in improving the power conversion efficiency (PCE) of all-PSCs.21 On the other hand, challenges remain in controlling the morphology and improving the performance of PSMAs. Further research efforts are needed to advance the development of all-polymer solar cells.
PSMAs are composed of small molecule acceptor fragments and linkers in their main chains, with the former playing a crucial role in determining their photovoltaic properties.22–25 The state-of-the-art Y-series SMAs as small molecule acceptor fragments in polymer acceptors have maintained narrow absorption spectra and high crystallinity, significantly contributing to improved PCE.26–34 However, researchers have primarily focused on optimizing the small molecule acceptor building fragments, while the π-bridge linkers have received relatively little attention. To date, representative PSMAs typically employ aromatic π-bridges, such as thiophene, bithiophene and selenophene, as linking units.27,29 In general, the linkers induce twisted conjugated backbones between the linker and end groups, which are considered detrimental to ordered molecular packing, charge transport, and photovoltaic performance in all-PSCs.35–37 Consequently, most of the research studies on the design of linkers have been aimed at improving the coplanarity of the linking units to achieve tighter interchain packing and better charge transport. On the other hand, moderately twisted conjugated backbones or partially conjugated unit configurations are helpful for mitigating excessive molecular aggregation as well as regulating the solubility and pre-aggregation capabilities of PSMAs.38–41 Utilizing steric hindrance to finely adjust the planarity of the polymer main chain is an effective strategy for modulating intermolecular interactions, packing morphology and crystallinity. However, the modulation of steric effects has primarily been achieved through the control of alkyl side chains in small molecule acceptor fragments to improve the performance of all-PSCs.26,42–45 There is still insufficient research on utilizing the steric effects of linkers to finely adjust polymer conformation, intermolecular packing and crystallinity to improve charge transport and photovoltaic performance. Li et al. employed thiophene–thiazolothiazole–thiophene as linking units to construct PSMAs, in which alkyl substitutions at various positions on the thiophene units play a crucial role in steric hindrance, self-aggregation, and crystallinity.27,42 Therefore, a systematic investigation into the steric effects of linker units on polymer acceptors is essential for elucidating structure–performance relationships and developing high-performance all-PSCs.
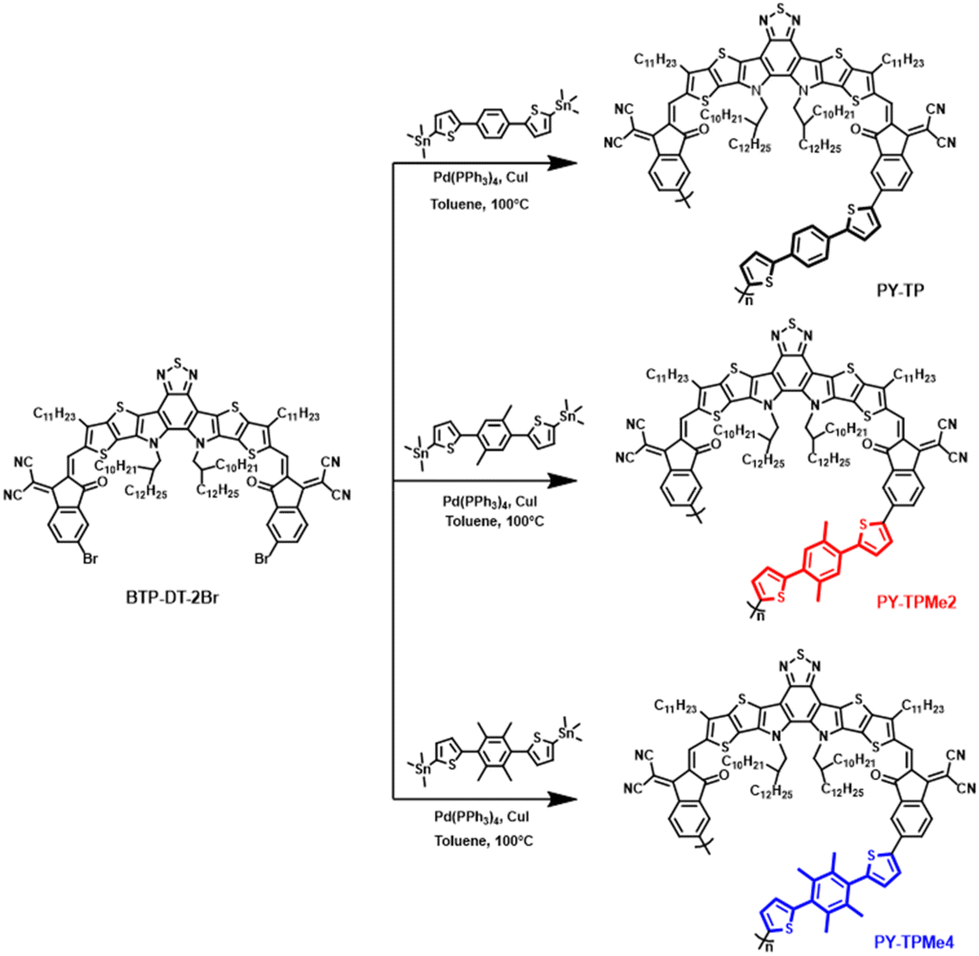
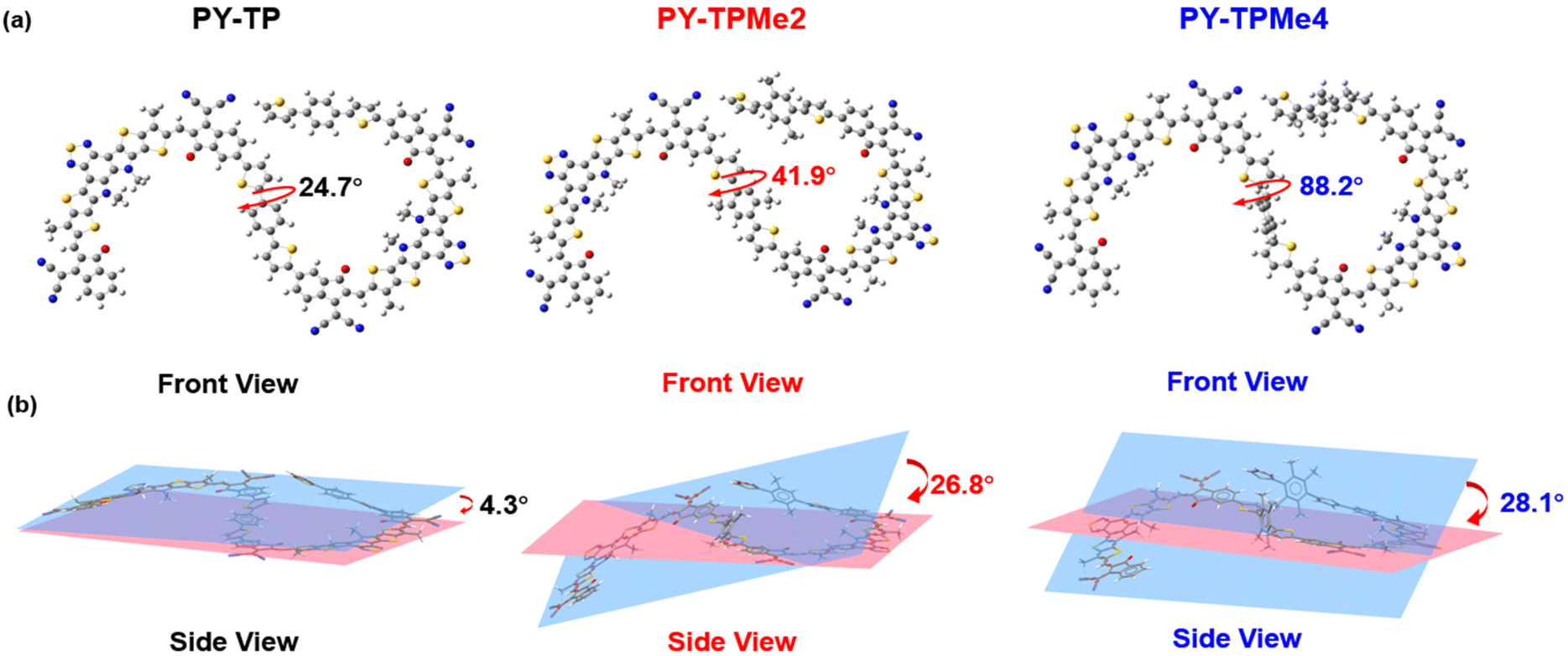
In this study, we designed and synthesized three polymers, namely PY-TP, PY-TPMe2, and PY-TPMe4, incorporating different numbers of methyl-substituted (bithiophene)benzene linkers, and explored their potential as PSMAs for all-PSCs. The methylation of benzene rings on the linkers induces different degrees of distortion compared to the pristine (bisthiophene)benzene linker, which in turn regulates the polymer backbone conformation and intermolecular stacking. As the number of methyl groups increases, the degree of distortion of (bithiophene)benzene also increases, and the dihedral angle between the benzene and thiophene increases from 24.7° to 41.9° and then to 88.2°, leading to a decrease in the planarity of polymer acceptors. Although the absorption in films and LUMO energy levels of the three polymer acceptors changed minimally, the intermolecular π–π stacking distance became larger with the increased distortion. When blended with PM6 to fabricate all-PSCs, the optimized binary device based on PY-TPMe2 exhibits the best PCE of 8.43% with a VOC of 0.97 V, a JSC of 14.74 mA cm−2, and a FF of 59.65% among all three PSMA-based devices. PY-TP with better planarity achieves a relatively lower PCE of 6.90% due to the decreased VOC, while PY-TPMe4 with the largest steric hindrance shows a poor photovoltaic response with a PCE of only 1.61% as a result of a large π–π stacking distance and excessive phase separation.
Results and discussion
Synthesis and characterization
The synthesis routes of polymer monomers BTP-DT-2Br, TP-Sn, TPMe2-Sn and TPMe4-Sn are depicted in Scheme S1.† Through the lithiation reaction with n-butyllithium followed by quenching with tributyltin chloride, TP-Sn, TPMe2-Sn and TPMe4-Sn were afforded in yields of about 80%. BTP-DT-2Br was obtained via BF3·OEt2-catalyzed Knoevenagel condensation at room temperature between BTP-DT and IC-γ-Br in a yield of 90%.46,47 Finally, we obtained the target polymer products PY-TP, PY-TPMe2 and PY-TPMe4 through the Stille polymerization reaction, as shown in Scheme 1. In the Stille polymerization reaction, a moderate amount of CuI was added as an additive to facilitate the transmetalation of the tin compound and enhance the yield.48,49 The chemical structures of the above-mentioned compounds were characterized by 1H NMR spectroscopy and mass spectrometry (MALDI-TOF-MS) (Fig. S14–S22, ESI†). The number-average molecular weights (Mn) are 9.2 kDa for PY-TP, 8.8 kDa for PY-TPMe2, and 9.1 kDa for PY-TPMe4, with the corresponding polydispersity index (PDI) of 1.72, 1.92 and 1.78, respectively. All polymers display good solubility in organic solvents such as chloroform and chlorobenzene, which ensures the preparation of all-PSC devices using the solution-processing method. The thermal decomposition temperature (5% weight loss) tested by thermogravimetric analysis (TGA) is 335 °C for all the polymers, which indicates good thermal stability of all three polymers (Fig. S2†).
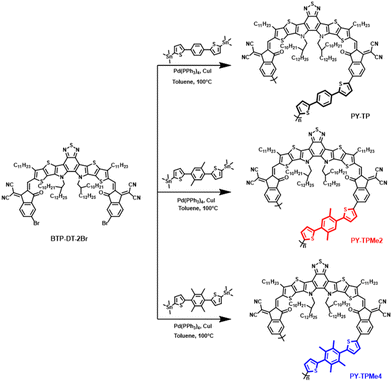 |
| | Scheme 1 Synthesis routes of PY-TP, PY-TPMe2 and PY-TPMe4. | |
Theoretical calculations and electrochemical properties
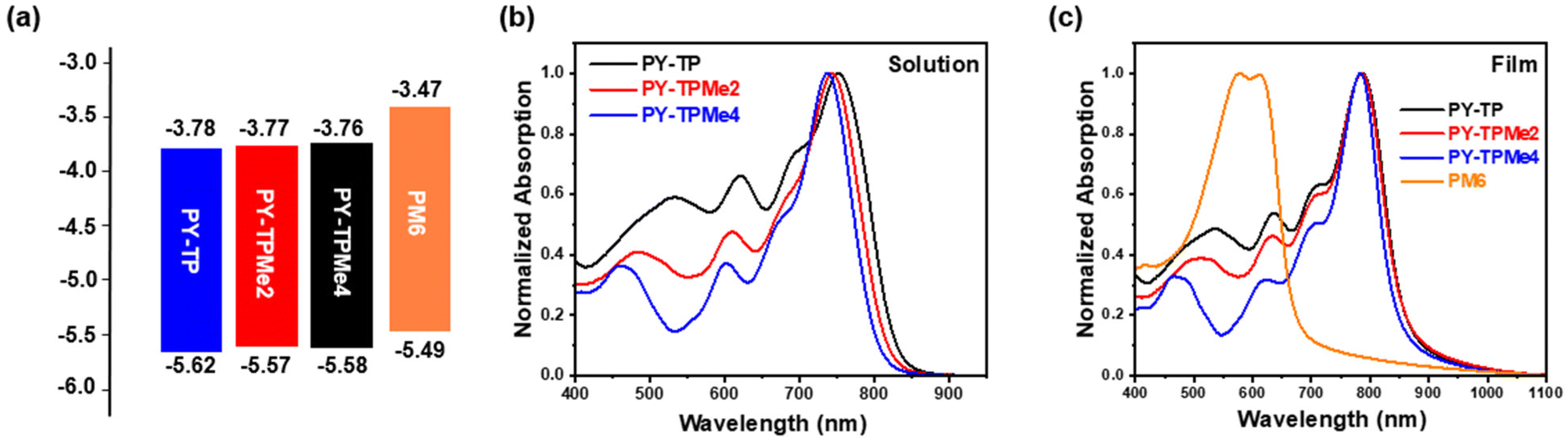
In order to better explain the influence of different linker units on the molecular geometry of the three polymer molecules, density functional theory (DFT) calculations at the B3LYP/6-31G (d,p) level are performed based on two repeated units of the polymer acceptors. It is clearly seen that compared with the (bithiophene)benzene linker without methyl groups, the introduction of methyl groups brings about different steric hindrance, which makes the dihedral angles between the thiophene ring and benzene ring increase from 24.7° for the benzene-containing linker to 41.9° for the dimethyl benzene-containing linker and 88.2° for the tetramethyl benzene-containing linker (Fig. 1a). Since the acceptor units (BTP and IC) almost maintain a coplanar structure, the distorted linkers induce the included angles of the adjacent acceptor planes to enlarge from 4° to over 28°, implying the decreased coplanar structures of polymer main chains (Fig. 1b).37 According to the calculated wave function distributions, the highest occupied molecular orbital (HOMO) energy levels and the lowest unoccupied molecular orbital (LUMO) energy levels of the three polymers are localized on the BTP-DT-IC units but not on (bithiophene)benzene linkers (Fig. S1 and Table S1, ESI†). Cyclic voltammetry (CV) measurements were used to test the energy levels of the three polymer acceptors using ferrocene/ferrocerium (−4.8 eV) as the reference (Fig. 2a and Fig. S3, ESI†). The HOMO/LUMO energy levels are −5.62/−3.78 eV for PY-TP, −5.57/−3.77 eV for PY-TPMe2, and −5.58/−3.76 eV for PY-TPMe4, which agree with the calculation results. The close HOMO/LUMO energy levels of the three polymer acceptors suggest that different methylated (bithiophene)benzene linkers have little influence on the energy levels of polymer acceptors.
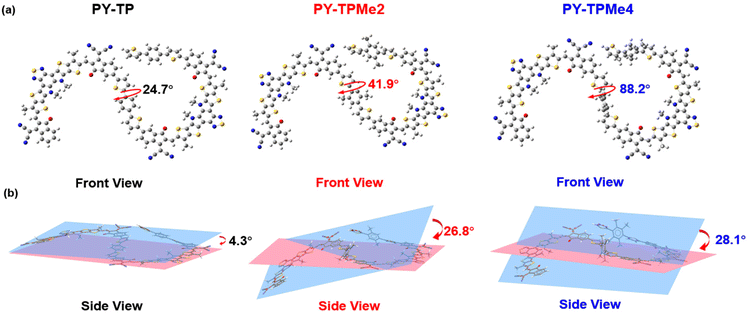 |
| | Fig. 1 The optimized dimer structures of PY-TP, PY-TPMe2 and PY-TPMe4 calculated by a DFT method at the B3LYP/6-31G(d,p) level. (a) front view and (b) side view. | |
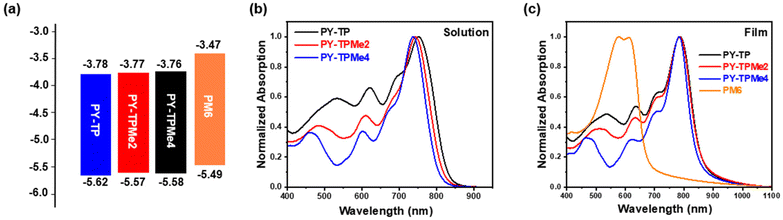 |
| | Fig. 2 (a) The energy level diagrams of PM6, PY-TP, PY-TPMe2 and PY-TPMe4. (b) Normalized absorption spectra of PY-TP, PY-TPMe2 and PY-TPMe4 in solution. (c) Normalized absorption spectra of PM6, PY-TP, PY-TPMe2 and PY-TPMe4 in films. | |
Photophysical properties
The ultraviolet-visible (UV-vis) absorption spectra of PY-TP, PY-TPMe2 and PY-TPMe4 in CF solutions and as thin films are shown in Fig. 2b and c. In solution, the maximum absorption peaks of PY-TP, PY-TPMe2, and PY-TPMe4 blue shift from 752 nm to 743 nm and 737 nm, possibly due to the reduced planarity of the polymer backbone by the twisted methyl-containing linkers (Fig. 2b). The maximum molar extinction coefficients (εsol.) are 7.47 × 104 M−1 cm−1 for PY-TP, 9.23 × 104 M−1 cm−1 for PY-TPMe2 and 8.38 × 104 M−1 cm−1 for PY-TPMe4 (Table 1). The polymer acceptor films exhibit absorption peaks at 786, 785 and 783 nm for PY-TP, PY-TPMe2 and PY-TPMe4, respectively, which red-shift by 34, 42 and 46 nm, respectively, as compared to their solution-state absorption, indicating strong aggregation in the solid state (Fig. 2c). The maximum extinction coefficients of PY-TP, PY-TPMe2 and PY-TPMe4 films are 8.97 × 104 cm−1, 1.02 × 105 cm−1 and 9.51 × 104 cm−1, respectively. According to the absorption onsets, the optical bandgaps of PY-TP, PY-TPMe2, and PY-TPMe4 films are calculated to be 1.42, 1.43, and 1.45 eV, respectively. The energy levels and absorption spectra of the three polymer acceptors match well with the wide bandgap polymer donor PM6, which is suitable for fabricating all-polymer solar cell devices. Temperature-dependent absorption spectra were recorded to further investigate the aggregation behaviors of the three polymer acceptors (Fig. S4, ESI†). Upon raising the solution temperature from 25 °C to 95 °C, the absorption maxima of PY-TP, PY-TPMe2 and PY-TPMe4 in solution are blue-shifted by 12, 9 and 6 nm, respectively. The most blue-shifted absorption peak of PY-TP indicates the strongest preaggregation in solution. Compared to PY-TP, PY-TPMe2 and PY-TPMe4 show gradually reduced blue shifts of absorption peaks at an elevated temperature, suggesting weakened intermolecular interactions, which are attributed to the twisted polymer backbones.42,50
Table 1 Optical and electrochemical properties of PY-TP, PY-TPMe2 and PY-TPMe4
| Acceptors |
M
n
|
PDI |
λ
Sol.max (nm) |
λ
filmmax (nm) |
ε
sol. (M−1 cm−1) |
ε
film (cm−1) |
E
opt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g
(eV) g
(eV) |
E
HOMOb (eV) |
E
LUMO
(eV) |
|
Calculated from the equation Eoptg = 1240/λfilmonset.
Calculated according to EHOMO = −e(Eox + 4.80) eV and ELUMO = −e(Ered + 4.80) eV.
|
| PY-TP |
9.2 kDa |
1.72 |
752 |
786 |
7.47 × 104 |
8.97 × 104 |
1.42 |
−5.62 |
−3.78 |
| PY-TPMe2 |
8.8 kDa |
1.92 |
743 |
785 |
9.23 × 104 |
1.02 × 105 |
1.43 |
−5.57 |
−3.77 |
| PY-TPMe4 |
9.1 kDa |
1.78 |
737 |
783 |
8.38 × 104 |
9.51 × 104 |
1.45 |
−5.58 |
−3.76 |
Photovoltaic performance
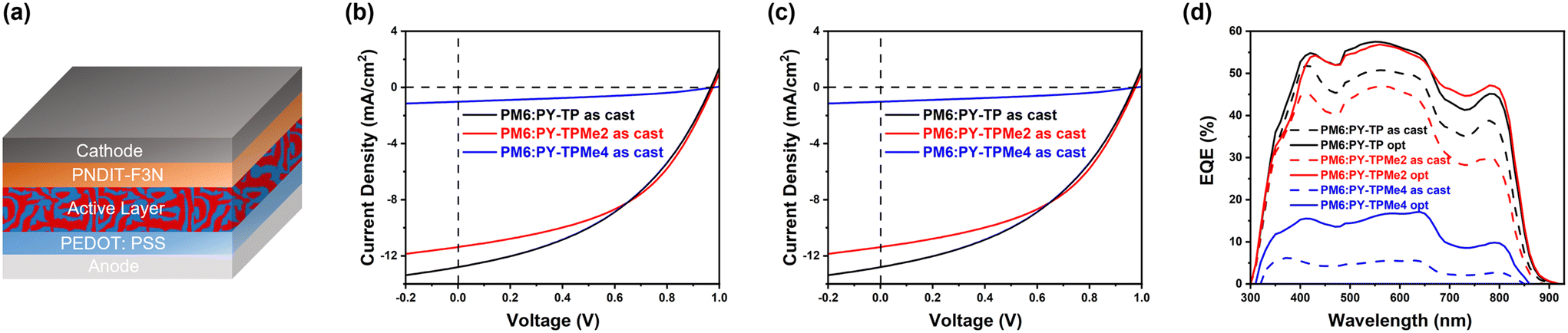
The photovoltaic performance of the three polymer acceptors was evaluated using bulk heterojunction organic solar cells with a conventional device structure: indium-doped tin oxide (ITO)/poly(3,4-ethylenedioxythiophene): polystyrene sulfonate (PEDOT:PSS)/PM6:PY-TP series/poly[(9,9-bis(3′-(N,N-dimethylamino)propyl)2,7-fluorene)-alt-5,5′-bis(2,2′-thiophene)-2,6-naphthalene-1,4,5,8-tetracaboxylic-N,N′-di(2-ethylhexyl)imide] (PNDIT-F3N)/Ag, as displayed in Fig. 3a. The current density–voltage (J–V) characteristics of the as-cast and optimized device performance measured under the illumination of AM 1.5 G at 100 mW cm2 are shown in Fig. 3b and c, respectively, and the corresponding photovoltaic parameters are summarized in Table 2. Chloroform was selected as the processing solvent. The optimal donor-to-acceptor (D:A) ratio was determined to be 1:1 by weight, with a total concentration of 12 mg mL−1. Owing to the similar LUMO energy levels, PY-TP-, PY-TPMe2- and PY-TPMe4-based devices show high VOC (0.97–0.98 V) with a low energy loss (Eloss) of 0.45–0.47 eV, according to the equation Eloss = Eg − eVOC. The as-cast devices based on PM6:PY-TP and PM6:PY-TPMe2 exhibit comparable PCE (5.34% and 5.33%) with similar VOC, short current density (JSC) and fill factor (FF). After incorporating 1-phenylnaphthalene (PN, 1% v/v) as an additive and applying thermal-annealing treatment, the optimized PY-TP-based device experienced significant voltage loss, which hindered effective improvement in PCE, yielding a PCE of 6.90%. In contrast, the optimized PY-TPMe2-based device maintains a high VOC of 0.97 V along with an improved JSC of 14.74 mA cm−2 and FF of 59.65%, resulting in a better PCE of 8.43%. Compared with the devices based on PM6:PY-TP and PM6:PY-TPMe2, the devices based on PM6:PY-TPMe4 maintain a high VOC of 0.98 V before and after applying the additive and thermal-annealing treatment, but the rather low JSC and unsatisfactory FF lead to the lowest PCE of 0.36% and 1.61% for the as-cast and optimized devices, respectively. The external quantum efficiency (EQE) spectra of the three polymer acceptor-based all-PSCs also reveal the difference in JSC, as shown in Fig. 3d. Compared with the as-cast acceptor-based devices, the EQE values of the three optimized devices increase remarkably, which contributes to the improved JSC values. The EQE responses of the PY-TP and PY-TPMe2-based all-PSCs range from 300 to 880 nm with a high EQE maximum of around 57%. In contrast, the PY-TPMe4-based device exhibits a slightly contracted photon response with the lowest EQE value of less than 20%, which seriously limits the photocurrent. The integrated current densities calculated from the corresponding EQE spectra (Jcal.) are consistent with the values obtained from the J–V characteristic curves.
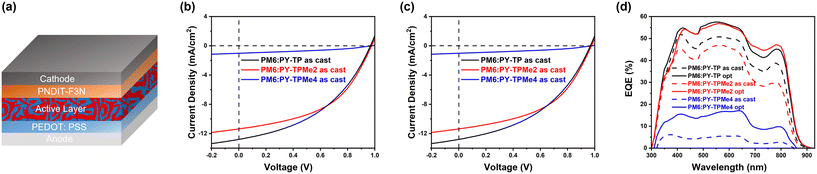 |
| | Fig. 3 (a) Device structure of the all-PSCs. (b) J–V curves of as-cast PM6:PY-TP, PM6:PY-TPMe2 and PM6:PY-TPMe4-based devices. (c) J–V curves of optimized PM6:PY-TP, PM6:PY-TPMe2 and PM6:PY-TPMe4-based devices. (d) EQE curves of ITO-based polymer acceptor-based devices. | |
Table 2 Detailed photovoltaic parameters based on PM6: acceptor-based devices
| Donor:acceptor |
V
oc (V) |
J
sc (mA cm−2) |
J
cal. (mA cm−2) |
FF (%) |
PCE (%) |
| PM6:PY-TP as-cast |
0.97 |
12.80 |
12.88 |
43.44 |
5.34 |
| PM6:PY-TP opt |
0.88 |
14.33 |
14.87 |
51.35 |
6.90 |
| PM6:PY-TPMe2 as-cast |
0.98 |
11.37 |
11.60 |
48.15 |
5.33 |
| PM6:PY-TPMe2 opt |
0.97 |
14.74 |
15.02 |
59.65 |
8.43 |
| PM6:PY-TPMe4 as-cast |
0.98 |
1.02 |
1.17 |
36.17 |
0.36 |
| PM6:PY-TPMe4 opt |
0.98 |
3.82 |
3.86 |
43.67 |
1.61 |
Exciton dissociation, charge transport and recombination
To investigate the charge-transfer and exciton dissociation process of the three all-PSCs, photoluminescence (PL) quenching testing was conducted (Fig. S13 and Table S5, ESI†). Upon selective excitation of the donor PM6 at 560 nm, the emissions from PM6:PY-TP and PM6:PY-TPMe2 blend films are quenched with high efficiencies of over 90%, suggesting efficient electron transfer from the excited polymer donor PM6 to PY-TP and PY-TPMe2 acceptors. In contrast, the PM6:PY-TPMe4 blend film exhibits less effective PL quenching than PM6:PY-TP and PM6:PY-TPMe2, and even after optimization, its PL quenching efficiency only increases from 49% to 81%. When excited at 560 nm, the PM6:PY-TPMe2 blend yields the highest quenching efficiency of 60%, compared to PM6:PY-TP (49%) and PM6:PY-TPMe4 (42%), indicating that the hole transfer from excited PY-TPMe2 to the donor polymer PM6 is the most effective among the three polymer acceptors. The highest quenching efficiency of the PM6:PY-TPMe2 blend is beneficial for the exciton dissociation to generate the photocurrent efficiently, while the lowest quenching efficiency of the PM6:PY-TPMe4 blend is one of the main reasons for its low JSC. Given the close LUMO–LUMO and HOMO–HOMO offsets between PM6 and the three acceptors, the differences in the PL quenching efficiencies of the three blend films may be related to their blend morphology, in which more D–A interfaces in the PM6:PY-TP and PM6:PY-TPMe2 blend films that are conducive to efficient charge transfer may be formed.
To understand the charge generation properties of the devices, the curves of photocurrent density (Jph) versus effective voltage (Veff) of all-PSC devices were obtained (Fig. S6, ESI†). Jph is defined as the difference between the photocurrent densities under illumination (JL) and dark conditions (JD), expressed as Jph = JL − JD. The effective voltage (Veff) is calculated as the difference between the voltage at which the photocurrent is zero (V0) and the applied external voltage (Vappl), given by Veff = V0 − Vappl. The charge dissociation and collection probability, denoted as P(E,T), can be estimated by calculating the ratio of Jph/Jsat. The calculated P(E,T) values for devices based on PY-TP and PY-TPMe2 are relatively higher compared to those of PY-TPMe4-based devices. Among all the devices, the optimized PY-TPMe2-based device achieves a P(E,T) value of 96.8%. This suggests that the optimized device based on the PM6:PY-TPMe2 blend exhibits the most efficient charge dissociation and charge collection. The charge recombination mechanism of all three devices is investigated by measuring the relationship between JSC and light intensity (Plight) (Fig. S7, ESI†). Typically, the correlation between JSC and Plight adheres to a power-law relationship, expressed as JSC ∝ (Plight)α, where the closer the α value is to 1, the less bimolecular recombination occurs. The α values of the optimized devices based on PY-TP, PY-TPMe2 and PY-TPMe4 are 0.94, 0.94 and 0.88, respectively, indicating the lower bimolecular recombination of the PY-TP and PY-TPMe2-based devices.
The charge carrier mobilities were evaluated using the space charge-limited current (SCLC) method to examine the charge transport characteristics of the blends, as shown in Fig. S8 and Table S2, ESI.† The hole- and electron-only devices were fabricated with the structure of ITO/PEDOT:PSS/active layer/MoO3/Ag and ITO/ZnO/active layer/PNDIT-F3N/Ag, respectively. The as-cast PM6:PY-TP and PM6:PY-TPMe2 blend films have similar hole mobility (μh) and electron mobility (μe), which are higher than those of the PM6:PY-TPMe4 blend film, accounting for the comparable JSC and FF of PM6:PY-TP- and PM6:PY-TPMe2-based devices and the lowest JSC and FF of the PM6:PY-TPMe4-based device. Under the optimal conditions, μh and μe were 6.42 × 10–5 and 4.36 × 10–4 cm2 v−1 s−1 for the PM6:PY-TP blend film, 1.26 × 10–4 cm2 v−1 s−1 and 5.82 × 10–4 cm2 v−1 s−1 for the PM6:PY-TPMe2 blend film and 1.28 × 10–5 cm2 v−1 s−1 and 2.96 × 10–4 cm2 v−1 s−1 for the PM6:PY-TPMe4 blend film. Among the three acceptor-based blend films, the PM6:PY-TPMe2 blend film shows the highest hole and electron mobilities and the most balanced charge transport, which are beneficial for suppressing space–charge accumulation and promoting charge extraction, resulting in the higher JSC and FF and thus the best device performance.
Morphology and molecular packing
We further investigated the effect of methylation on the morphological characteristics of blend films using atomic force microscopy (AFM) (Fig. 4). In the height images, both the as-cast PM6:PY-TP and PM6:PY-TPMe2 blend films show a relatively smooth surface with the root-mean-square (RMS) roughness values of 1.45 and 1.58 nm, respectively. After treating with 1% 1-phenylnaphthalene as an additive and thermal annealing, the RMS roughness values increase to 2.18 and 2.30 nm, and the phase separation becomes larger and more obvious in the phase images. Moreover, the PM6:PY-TPMe2 blend film exhibits a more distinct interpenetrating fibrous network with uniform phase separation morphology in the phase image, and these morphological features are thought to facilitate efficient exciton diffusion to the donor–acceptor interface and effective charge separation, as indicated by the improved PL quenching efficiencies in the optimized blend films. In contrast, the surface of PM6:PY-TPMe4 blend films is the roughest among all the blend films with large RMS roughness values (3.21 nm for the as-cast film and 5.22 nm for the optimized film). The overlarge phase separation of the PM6:PY-TPMe4 blend films leads to reduced D/A interfaces for exciton dissociation and thus seriously limits the JSC of PY-TPMe4-based devices. Additionally, the morphological characteristics of blend films are also consistent with those of the neat polymer acceptor films (Fig. S9, ESI†). These results suggest that the methylation of (bithiophene)benzene linkers in polymer acceptors plays a crucial role in finely regulating the film morphology.
 |
| | Fig. 4 (a–f) AFM height images of (a) as-cast PM6:PY-TP, (b) optimized PM6:PY-TP, (c) as-cast PM6:PY-TPMe2, (d) optimized PM6:PY-TPMe2, (e) as-cast PM6:PY-TPMe4 and (f) optimized PM6:PY-TPMe4. (g–l) AFM phase images of (g) as-cast PM6:PY-TP, (h) optimized PM6:PY-TP, (i) as-cast PM6:PY-TPMe2, (j) optimized PM6:PY-TPMe2, (k) as-cast PM6:PY-TPMe4 and (l) optimized PM6:PY-TPMe4. | |
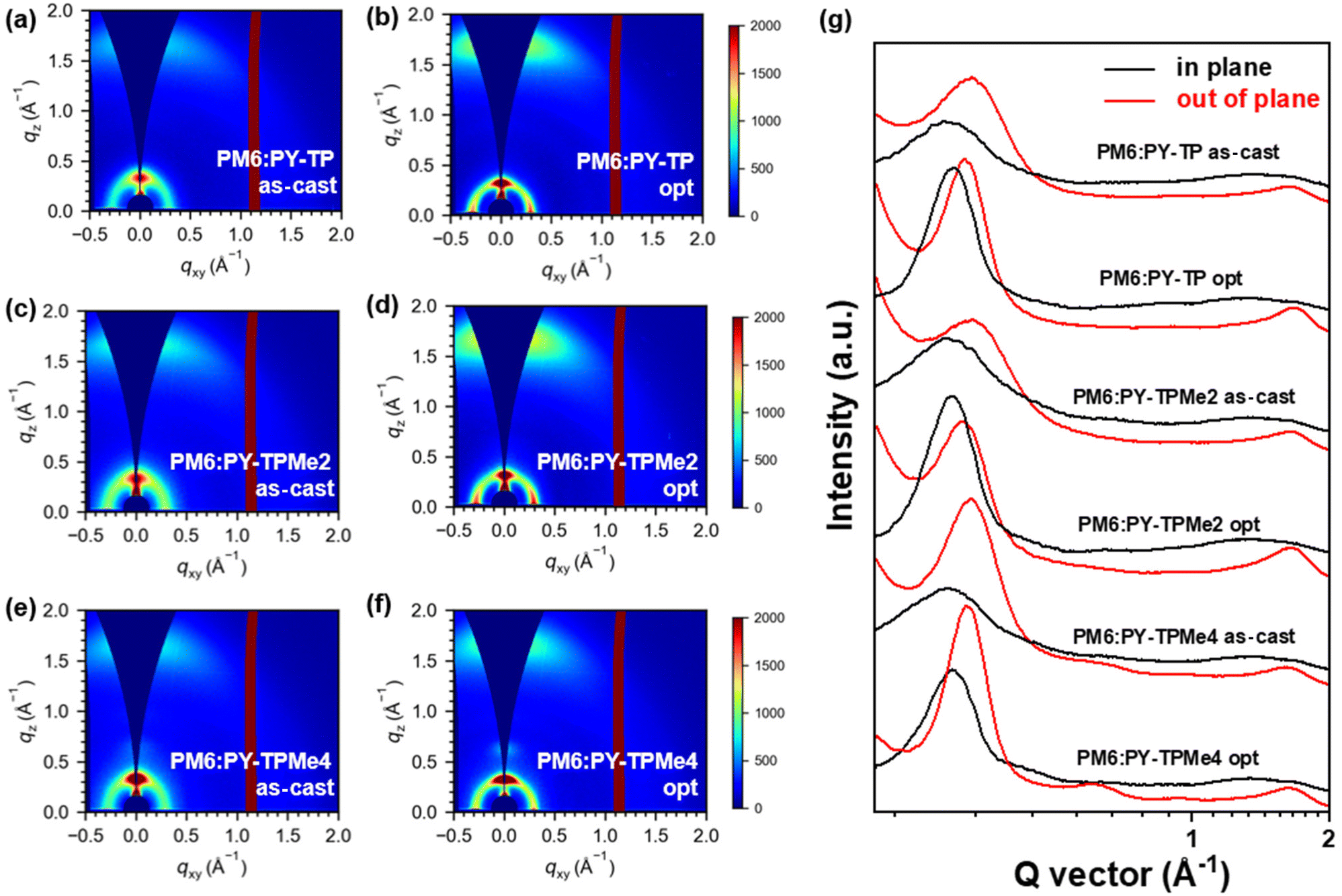
Grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were performed to evaluate the molecular packing and orientation of the neat and blend films based on PY-TP, PY-TPMe2 and PY-TPMe4.51 All the neat PY-TP, PY-TPMe2 and PY-TPMe4 films show a preferential face-on orientation, with a lamellar stacking peak (100) in the in-plane (IP) direction and a π–π stacking peak (010) in the out-of-plane (OOP) direction (Fig. S10 and 11, ESI†). The (010) peaks of the PY-TP, PY-TPMe2, and PY-TPMe4 neat films in the out-of-plane (OOP) direction appeared at 1.64 Å−1, 1.60 Å−1 and 1.59 Å−1 respectively, corresponding to the π–π stacking distances of 3.84 Å, 3.92 Å and 3.96 Å (Table S3, ESI†). The gradually increased π–π stacking distances in the PY-TP, PY-TPMe2, and PY-TPMe4 neat films demonstrate that the number of methyl groups on the benzene rings of the linkers weakens the cofacial interchain orientation and π–π interaction between the neighboring chains. When blended with the polymer donor PM6, both the face-on and edge-on orientations coexist in both as-cast and optimized blend films owing to the appearance of strong peaks at 0.31 Å−1 in OOP directions associated with the lamellar (100) stacking of PM6 (Fig. 5 and Fig. S12, ESI†). The trend of reduced π–π stacking distances from PY-TP, PY-TPMe2 to PY-TPMe4 is the same as that of the neat films. Treating with additives and thermal annealing leads to closer and more ordered intermolecular packing and higher crystallinity. Under the optimized conditions, the π–π stacking distances of the blend films from PY-TP, PY-TPMe2 to PY-TPMe4 increased from 3.76 Å, 3.80 Å to 3.84 Å, and the corresponding crystal coherence lengths (CCLs) of (010) orientation also reduced from 18.9 Å, 17.1 Å to 16.0 Å (Table 4, ESI†). These results indicate that the large steric hindrance and twisted backbone caused by methyl groups lead to decreased molecular packing order and crystallinity. Among all three blend films, the PY-TPMe4-based blend film exhibits both the largest π–π stacking distance and the smallest CCLs, which are unfavorable for charge transport, leading to the worst JSC and FF. Compared with the PY-TP-based blend film with the closest π–π stacking and strongest crystallinity, the moderate π–π stacking distance and CCL of the PY-TPMe2 based blend film contribute to more effective and balanced charge transport, which accounts for its higher FF. In addition, the rational crystallization intensity ratio between donor/acceptor components is crucial for achieving minimum energy loss as well as the best device performance.52–54 Before and after thermal annealing and the addition of the additive, the PM6:PY-TP blend films show a larger CCL variation from 16.6 to 18.9 Å as compared to PY-TPMe2- and PY-TPMe4-based blend films with the CCLs changing from 16.5 to 17.1 Å and 16.6 to 16.0 Å, respectively. The larger variation of CCLs in PY-TP-based blends before and after optimization suggests a larger crystallinity difference between PM6 and PY-TP, which may aggravate non-radiative recombination energy losses and lower the VOC of PY-TP-based devices.53
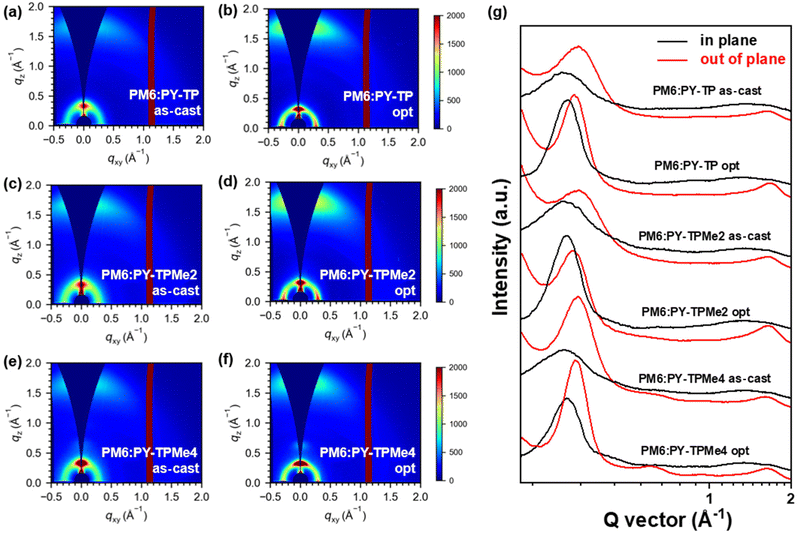 |
| | Fig. 5 2D-GIWAXS patterns (a–f) and line-cut profiles (g) of as-cast PM6:PY-TP, optimized PM6:PY-TP, as-cast PM6:PY-TPMe2, optimized PM6:PY-TPMe2, as-cast PM6:PY-TPMe4 and the optimized PM6:PY-TPMe4 blend film. | |
Conclusions
In summary, we have designed and synthesized three PSMAs constructed with benzene-, dimethylbenzene- and tetramethylbenzene-containing (bisthiophene)benzene linkers, respectively. Through changing the number of methyl groups, the degree of distortion of the (bisthiophene)benzene linkers can be effectively tuned, which in turn regulates the planarity of polymer backbones. Although the distortion of linker units has minimal impact on their light harvesting and frontier orbital energy levels, the intermolecular π–π stacking distance increases with the number of methyl groups. PY-TPMe4 with four methyl groups exhibits the largest π–π stacking distance and excessive phase separation morphology in the blend film, resulting in unfavorable exciton dissociation and charge transport, yielding an inferior PCE (1.61%) for its corresponding all-PSC device. In contrast, PY-TP and PY-TPMe2 have more suitable phase separation and better exciton dissociation and charge transport performance, and their photovoltaic performance is significantly better than that of PY-TPMe4. Particularly, the PY-TPMe2-based device not only exhibits the best exciton dissociation and charge transport performance, but also maintains a high VOC after thermal annealing and treatment with the additive PN. Consequently, the PY-TPMe2-based device achieves an optimal PCE of 8.4% with a high VOC of 0.97 V, an improved JSC of 14.74 mA cm−2 and a FF of 59.65%. This work demonstrates that designing linkers with suitable steric hindrance to modulate the intramolecular distortion and intermolecular aggregation behavior of PSMAs is valuable for improving the performance of all-PSC devices.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is financially supported by the Ministry of Science and Technology (National Key R&D Program of China, No. 2022YFB4200400), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB0520102), the National Natural Science Foundation of China (Grant No. 51973211, 22275182, 22075272, 51833009, 21674111 and 52261135541), and Sichuan Science and Technology Program (2023NSFSC0990). A portion of this work is based on the data obtained at 1W1A Diffuse X-ray Scattering Station, Beijing Synchrotron Radiation Facility (BSRF-1W1A). The authors gratefully acknowledge the cooperation of the beamline scientists at BSRF-1W1A beamline.
References
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS PubMed.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- Y. Tong, B. Xu and F. Ye, Adv. Funct. Mater., 2024, 34, 2310865 CrossRef CAS.
- Z.-G. Zhang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 4422–4433 CrossRef CAS.
- L. Zhang, Z. Yao, H. Wang, J. Zhang, X. Ma and F. Zhang, Sol. RRL, 2023, 7, 2300219 CrossRef CAS.
- B. He, L. Liu, Y. Liu, G. Chen, M. Xiao and C. Dai, RSC Adv., 2022, 12, 17898–17904 RSC.
- Y. Wu, Z. Du, X. Kang, J. Wang, X. Liang, P. Wang, X. Qu, Y. Li and X. Bao, Dyes Pigm., 2022, 199, 110081 CrossRef CAS.
- J. Shao, C. Liao, X. Xu, M. Deng, L. Yu, R. Li and Q. Peng, Chem. Mater., 2022, 34, 7971–7981 CrossRef CAS.
- S. Shi, P. Chen, Y. Chen, K. Feng, B. Liu, J. Chen, Q. Liao, B. Tu, J. Luo, M. Su, H. Guo, M.-G. Kim, A. Facchetti and X. Guo, Adv. Mater., 2019, 31, 1905161 CrossRef CAS.
- W. Wei, X. Yuan, J. Zhong, Z. Wang, X. Zhou, F. Zhao, D. Feng, Y. Zhang, W. Chen, M. Yang, W. Zhang, Z. Ma, Z. Tang, X. Lu, F. Huang, Y. Cao and C. Duan, Energy Environ. Sci., 2024, 17, 6627–6639 RSC.
- Y. Yu, L. Zhang, J. Liu and L. Wang, Polymer, 2022, 242, 124547 CrossRef CAS.
- Y. Wang, N. Wang, Q. Yang, J. Zhang, J. Liu and L. Wang, J. Mater. Chem. A, 2021, 9, 21071–21077 RSC.
- Y. Dou, J. Zhang, Z. Zhong, C. Zhu, Z. Chen, K. Zhang and F. Huang, Adv. Energy Mater., 2024, 2402268 CrossRef CAS.
- X.-J. Li, G.-P. Sun, Y.-F. Gong and Y.-F. Li, Chin. J. Polym. Sci., 2023, 41, 640–651 CrossRef CAS.
- H. Zhuo, X. Li, J. Zhang, C. Zhu, H. He, K. Ding, J. Li, L. Meng, H. Ade and Y. Li, Nat. Commun., 2023, 14, 7996 CrossRef CAS PubMed.
- Y. Liang, D. Zhang, Z. Wu, T. Jia, L. Lüer, H. Tang, L. Hong, J. Zhang, K. Zhang, C. J. Brabec, N. Li and F. Huang, Nat. Energy, 2022, 7, 1180–1190 CrossRef CAS.
- Y. Bai, Z. Zhang, Q. Zhou, H. Geng, Q. Chen, S. Kim, R. Zhang, C. Zhang, B. Chang, S. Li, H. Fu, L. Xue, H. Wang, W. Li, W. Chen, M. Gao, L. Ye, Y. Zhou, Y. Ouyang, C. Zhang, F. Gao, C. Yang, Y. Li and Z.-G. Zhang, Nat. Commun., 2023, 14, 2926 CrossRef CAS.
- Z.-G. Zhang, Y. Yang, J. Yao, L. Xue, S. Chen, X. Li, W. Morrison, C. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 13503–13507 CrossRef CAS PubMed.
- H. Yu, C. Zhao, H. Hu, S. Zhu, B. Zou, T. A. D. Peña, H. M. Ng, C. H. Kwok, J. Yi, W. Liu, M. Li, J. Wu, G. Zhang, Y. Chen and H. Yan, Energy Environ. Sci., 2024, 17, 5191–5199 RSC.
- S. Seo, C. Sun, J.-W. Lee, S. Lee, D. Lee, C. Wang, T. N.-L. Phan, G.-U. Kim, S. Cho, Y.-H. Kim and B. J. Kim, Adv. Funct. Mater., 2022, 32, 2108508 CrossRef CAS.
- Q. Bai, Q. Liang, Q. Liu, B. Liu, X. Guo, L. Niu and H. Sun, Chin. J. Chem., 2023, 41, 3714–3728 CrossRef CAS.
- T. Yang and C. Zhan, Sci. China:Chem., 2023, 66, 2513–2531 CrossRef CAS.
- J. Wang, T. Chen, W. Zhao, X. Tang, Y. Bai, W. Zhou, G. Long, X. Ji, G. Lu, W. Feng, X. Wan, B. Kan and Y. Chen, Adv. Funct. Mater., 2024, 2414941 CrossRef.
- Y. Ding, W. A. Memon, D. Zhang, Y. Zhu, S. Xiong, Z. Wang, J. Liu, H. Li, H. Lai, M. Shao and F. He, Angew. Chem., Int. Ed., 2024, 63, e202403139 CrossRef CAS PubMed.
- J.-W. Lee, J. S. Park, H. Jeon, S. Lee, D. Jeong, C. Lee, Y.-H. Kim and B. J. Kim, Chem. Soc. Rev., 2024, 53, 4674–4706 RSC.
- X. Li, L. Chen, L. Meng, C. Zhang, X. Duan, Y. Man, M. H. Jee, L. Han, Y. Pan, D. Wei, X. Wan, H. Y. Woo, Y. Chen and Y. Sun, Adv. Funct. Mater., 2024, 34, 2316090 CrossRef CAS.
- L. Zhou, X. Xia, L. Meng, J. Zhang, X. Lu and Y. Li, Chem. Mater., 2021, 33, 8212–8222 CrossRef CAS.
- M. Kataria, H. D. Chau, N. Y. Kwon, S. H. Park, M. J. Cho and D. H. Choi, ACS Energy Lett., 2022, 7, 3835–3854 CrossRef CAS.
- K. Ma, H. Liang, Y. Wang, Z. Xiao, C. Jiang, W. Feng, Z. Zhang, X. Si, J. Liu, X. Wan, B. Kan, C. Li, Z. Yao and Y. Chen, ACS Mater. Lett., 2023, 5, 884–892 CrossRef CAS.
- Y.-C. Lin, C.-H. Chen, B.-S. Tsai, T.-F. Hsueh, C.-S. Tsao, S. Tan, B. Chang, Y.-N. Chang, T.-Y. Chu, C.-E. Tsai, C.-S. Chen, Y. Yang and K.-H. Wei, Adv. Funct. Mater., 2023, 33, 2215095 CrossRef CAS.
- Y. Pan, L. Guo, M. H. Jee, G. Dai, Z. Ge, J. Zhang, X. Duan, J. Song, X. Li, H. Y. Woo and Y. Sun, Adv. Energy Mater., 2024, 2403747 CrossRef.
- B. Wu, Y. Li, K. Liu, S. Kim, X. Yuan, L. Pan, X. Zhou, S. Tian, C. Yang, F. Huang, Y. Cao and C. Duan, Nano Energy, 2024, 128, 109874 CrossRef CAS.
- J.-W. Lee, J. Kim, T. H.-Q. Nguyen, D. C. Lee, Z. Tan, J. Park, T. N.-L. Phan, S. Cho and B. J. Kim, Nano Energy, 2024, 122, 109338 CrossRef CAS.
- J.-W. Lee, C. Sun, S.-W. Lee, G.-U. Kim, S. Li, C. Wang, T.-S. Kim, Y.-H. Kim and B. J. Kim, Energy Environ. Sci., 2022, 15, 4672–4685 RSC.
- Q. Wang, M. Li, X. Zhang, Y. Qin, J. Wang, J. Zhang, J. Hou, R. A. J. Janssen and Y. Geng, Macromolecules, 2019, 52, 4464–4474 CrossRef CAS.
- X. Shen, X. Lai, H. Lai, Y. Wang, H. Li, M. Ou and F. He, Adv. Funct. Mater., 2024, 2404919 CrossRef CAS.
- H. Yu, Y. Wang, H. K. Kim, X. Wu, Y. Li, Z. Yao, M. Pan, X. Zou, J. Zhang, S. Chen, D. Zhao, F. Huang, X. Lu, Z. Zhu and H. Yan, Adv. Mater., 2022, 34, 2200361 CrossRef CAS.
- X. Bi, T. Zhang, C. An, P. Bi, K. Ma, S. Li, K. Xian, Q. Lv, S. Zhang, H. Yao, B. Xu, J. Zhang, S. Cao and J. Hou, J. Mater. Chem. A, 2020, 8, 14706–14712 RSC.
- C. Xue, T. Zhang, K. Ma, P. Wan, L. Hong, B. Xu and C. An, Macromol. Rapid Commun., 2019, 40, 1900246 CrossRef CAS PubMed.
- T. Zhang, C. An, K. Ma, K. Xian, C. Xue, S. Zhang, B. Xu and J. Hou, Org. Chem. Front., 2020, 7, 261–266 RSC.
- Z. Zhang, Z. Li, P. Wang, H. Chen, K. Ma, Y. Zhang, T. Duan, C. Li, Z. Yao, B. Kan, X. Wan and Y. Chen, Adv. Funct. Mater., 2023, 33, 2214248 CrossRef CAS.
- L. Zhou, L. Meng, J. Zhang, S. Qin, J. Guo, X. Li, X. Xia, X. Lu and Y. Li, Macromolecules, 2022, 55, 4420–4428 CrossRef CAS.
- Q. Wang, M. Li, Y. Sui, Z. Wang, Z. Liang and Y. Geng, J. Mater. Chem. C, 2019, 7, 9581–9590 RSC.
- Y. Guo, Z. Chen, J. Ge, J. Zhang, L. Xie, R. Peng, W. Ma and Z. Ge, Sci. China: Chem., 2023, 66, 500–507 CrossRef CAS.
- T. Hu, X. Zheng, T. Wang, A. Saparbaev, B. Gao, J. Wu, J. Xiong, M. Wan, T. Cong, Y. Li, E. Wang, X. Wang and R. Yang, Aggregate, 2024, 5, e632 CrossRef CAS.
- H. Fu, J. Yao, M. Zhang, L. Xue, Q. Zhou, S. Li, M. Lei, L. Meng, Z.-G. Zhang and Y. Li, Nat. Commun., 2022, 13, 3687 CrossRef CAS.
- H. Fu, M. Zhang, Y. Zhang, Q. Wang, Z. A. Xu, Q. Zhou, Z. Li, Y. Bai, Y. Li and Z.-G. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202306303 CrossRef CAS.
- V. Farina, S. Kapadia, B. Krishnan, C. Wang and L. S. Liebeskind, J. Org. Chem., 1994, 59, 5905–5911 CrossRef CAS.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- Z. Zhang, Z. Li, B. Kan, T. Chen, Y. Zhang, P. Wang, Z. Yao, C. Li, B. Zhao, M. Li, T. Duan, X. Wan and Y. Chen, Nano Energy, 2023, 116, 108766 CrossRef CAS.
- X. Cui, H. Li, H. Lu, Y. Liu, G. Ran, R. Liu, H. Zhang, X. Ma, D. Li, Y. Lin, J. Yu, W. Zhang, L. Cai, Y. Liu, P. Cheng, A. Zhang, Z. Ma, G. Lu and Z. Bo, Sci. China: Chem., 2024, 67, 890–897 CrossRef CAS.
- D. He, F. Zhao, C. Wang and Y. Lin, Adv. Funct. Mater., 2022, 32, 2111855 CrossRef CAS.
- Z.-H. Chen, P.-Q. Bi, X.-Y. Yang, M.-S. Niu, K.-N. Zhang, L. Feng and X.-T. Hao, J. Phys. Chem. C, 2019, 123, 12676–12683 CrossRef CAS.
- J.-H. Park, J. Kim, J. H. Kim, D. W. Kim, C. H. Ryoo, O. K. Kwon, C. Lee, J. E. Kwon and S. Y. Park, ACS Appl. Energy Mater., 2021, 4, 259–267 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hui
Tong
*a,
Hui
Tong