
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of counterions on the thermal and solution properties of strong polyelectrolytes†
Théophile
Pelras
*ab,
Julien
Es Sayed
 b,
Jin
Pierik
b,
Andrea
Giuntoli
c,
Anton H.
Hofman
b,
Katja
Loos
a and
Marleen
Kamperman
*b
b,
Jin
Pierik
b,
Andrea
Giuntoli
c,
Anton H.
Hofman
b,
Katja
Loos
a and
Marleen
Kamperman
*b
aMacromolecular Chemistry and New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: theophile.pelras@rug.nl; k.u.loos@rug.nl
bPolymer Science, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: j.s.es.sayed@rug.nl; a.h.hofman@rug.nl; marleen.kamperman@rug.nl
cMicromechanics, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: a.giuntoli@rug.nl
First published on 23rd November 2024
Abstract
Strong polyelectrolytes (i.e., macromolecules whose charge density is independent of the medium's pH) are invaluable assets in the soft matter toolbox, as they can readily disperse in aqueous media, complex to oppositely charged species – polymers and small molecules alike – and can be implemented in a plethora of applications, ranging from surface modification to chelating agents and lubricants. However, the direct synthesis of strong polyelectrolytes in a controlled fashion remains a challenging endeavour, and their in-depth characterisation is often limited. Additionally, producing a set of charged macromolecules with the same chain length but varying counterions would open doors towards a fine control of the polymer's chemistry and physical properties. Unfortunately, this either necessitates the direct polymerisation of several monomers with potentially varying reactivities, or a time-consuming ion exchange from a single batch. Herein we explore the facile and efficient production of strong polyanions through the deprotection of a poly(3-isobutoxysulphopropyl methacrylate) using a range of inorganic and organic iodide-containing salts. Owing to the contrasting nature of their counterions, the resulting polyanions exhibit a wide range of glass transition temperatures, which follow a non-monotonic trend with increasing counterion size. While all polymers readily dissolve in water, some can also be dissolved in non-aqueous media as well. This strategy, applied to block copolymers, permits the production of a library of amphiphilic macromolecules with consistent hydrophilic and hydrophobic blocks, yet varying nature of their polyanionic segments. All amphiphiles, regardless of their counterions, readily disperse in aqueous media and form well-defined micelles featuring a hydrophobic core and a charged hydrophilic shell, as evidenced by dynamic light scattering, ζ-potential and transmission electron microscopy. Additionally, a handful of block copolymers are capable of yielding polymer micelles in organic solvents, opening an avenue to the build-up of nanostructured soft matter in non-aqueous media.
Introduction
Polyelectrolytes (i.e., macromolecules possessing either positively or negatively charged repeating motifs throughout their chains) can readily be found in nature, where their charged character is required to fulfil specific tasks. In materials sciences, charged polymers play an important role, as they can be used in a variety of applications including energy storage and ion transport,1 surface lubrication2 and even for the stabilisation of micelles and other types of nanoparticles featuring a hydrophobic core.3 Additionally, when combined with an opposite-charged polymer, electrostatic interactions occur and permit the build-up of intricate nanostructures,4–7 the production of underwater adhesives,8,9 the formation of cargos for the pinpoint delivery of therapeutics10–12 or genetic material,13,14 as well as for the functionalisation of surfaces with anti-fouling properties.15,16The counterion, which is a small molecule compensating the charge of the polyionic repeating motif, is a key component of polyelectrolyte systems. Oftentimes, small-sized inorganic counterions (e.g., iodide anion in quaternised amino-based polycations17 or a proton in polystyrene sulphonate18) are present, but larger organic-based ones (e.g., tetra-substituted ammonium,19–21 oligo-ethylene glycol ammonium21 or imidazolium20,22 species) can also be featured, as their nature vastly influences the chemical and physical properties of the polymer chains.23
Research on surfactants has already shed light on the influence of the inorganic counterions on their surface excess,24–26 primarily explained by their difference in size, with smaller ions possessing a larger hydration layer and therefore looser packing.24 Furthermore, some organic counterions (e.g., bis(trifluoromethylsulfonyl)imide) act as plasticisers and hence reduce the glass transition temperature of polyelectrolytes. This effect is explained by the weaker ionic interactions between charged components, which cause fewer and weaker physical crosslinks within the polymer chains.27–29 However, while size is a key factor in determining the strength of these interactions, other intrinsic parameters of the counterion, such as symmetry, nucleophilicity, charge delocalisation and flexibility also play important roles.30 Interestingly, the effect of counterion size on the glass transition temperature has been postulated to be non-monotonic, due to competing effects of electrostatic and van der Waals interactions,31,32 an effect that has been clearly observed in simple ionic liquids but not distinctly in poly(ionic liquids).
Beyond the fundamental research aimed at understanding the behaviour of macromolecules, the ability to readily tailor their physical properties can be exploited in applications where polymer softness is of prime importance. For instance, the introduction of plasticising counterions within polymers and ionomers lowers their glass transition temperature, which vastly increases their electronic and/or ionic conductivity.27–29,33–35 Furthermore, self-assembled nanoparticles can respond to the exchange of either organic or inorganic counterions. Gröschel et al. demonstrated the transition of nanoparticles into worm-like micelles and superstructures by a simple exchange of iodide to triiodide anions, explained by a decrease in the hydrophilicity of the polymer chains.36 Another study demonstrated the transition from disk-like to worm-like micelles and later to spherical particles by introducing a variety of diamine-based counterions.37
However, few studies have so far systematically investigated the effect of the nature of the counterion on the physical properties of polyelectrolytes, mostly due to the challenges of producing a large set of nearly identical macromolecules. Whilst vast developments in controlled polymerisation techniques have facilitated the production of lab-made (strong) polyelectrolytes, their synthesis with tailored chemistries and functionalities, as well as their in-depth characterisation remain challenging endeavours. Strategies to produce positively charged macromolecules often involve the quaternisation of tertiary amines38,39 which, albeit not always driven to completion,40,41 remains efficient and permits thorough analyses of the hydrophobic precursor. This route is however not as straightforward for strong polyanions i.e., whose charge density is independent of the medium's pH, as they often rely on sulphonate groups that are soluble in a very limited number of media and can be incompatible with certain types of polymerisation methods. Furthermore, a systematic study of the influence of the counterion either necessitates the production of multiple polymers each requiring the same chain length, or the synthesis of one precursor whose counterion can be exchanged. The latter method is often favoured, but requires cumbersome ion exchange processes (e.g., passage through an ion exchange membrane and extensive dialysis21) or the use of a large excesses of salt to induce a selective precipitation.30
We have previously developed a mild yet effective strategy to produce sulphonate polymers through (i) controlled polymerisation of a protected sulphonate acrylate22,42 or methacrylate20,43 and (ii) nucleophilic deprotection using iodide-based salts. This methodology not only permits more straightforward characterisation of precursors and easier synthesis of (amphiphilic) block copolymers, but also allows the tailoring of the nature of the counterion through simple change in the nucleophile. For instance, sodium iodide typically used for the nucleophilic deprotection can be replaced by other inorganic20 or inorganic20,22 salts while maintaining similar reaction conditions and purification protocols. To the best of our knowledge and besides our preliminary work,20,22 no study has yet systematically screened a wide range of counterions and investigated their effect on the thermal and solution properties of polysulphonates with constant chain lengths.
Herein, we produced an extensive range of polyelectrolytes based on poly(sulphopropyl methacrylate) (PSPMA) with the same chain length and rigidity, but featuring a variety of inorganic and organic counterions. Our previously reported methodology was employed, starting with the synthesis of a hydrophobic poly(3-isobutoxysulphopropyl methacrylate) (PBSPMA) precursor through controlled radical polymerisation, and subsequent deprotection using iodide-based nucleophiles (Scheme 1). These polyanions not only exhibit different thermal properties (i.e., thermal stability and glass transition) but also solubilities. Their capacity to be dispersed in aqueous and sometimes non-aqueous media, coupled to a block copolymer synthesis strategy, was used to produce charged polymer micelles suspended in organic media.
 | ||
| Scheme 1 Tailoring the nature of the polyelectrolyte counterion through nucleophilic deprotection. (i) Controlled radical polymerisation is used to produce (ii) a protected poly(3-isobutoxysulphopropyl methacrylate) homopolymer. (iii) Subsequent nucleophilic deprotections using iodide-based salts are performed to remove the protective groups and yield (iv) a range of strong poly(sulphopropyl methacrylate) polyanions featuring various inorganic and organic counterions. | ||
Results and discussion
Synthesis of polyelectrolyte homopolymers
First, we employed reversible addition–fragmentation chain-transfer (RAFT) polymerisation, using a commercially available 4-cyano-4-(thiobenzoylthio)pentanoic acid chain-transfer agent (CTA) and tailor-made 3-isobutoxysulphopropyl methacrylate to produce a poly(3-isobutoxysulphopropyl methacrylate) homopolymer (PBSPMA). The isobutoxy moiety, installed on the monomer, acts as a protective group and renders the polymer hydrophobic, which greatly facilitates its synthesis as well as its characterisation prior to modification. Proton nuclear magnetic resonance (1H NMR) spectroscopy was invoked to determine the conversion and thereby the homopolymer chain length and molecular weight (DPPBSPMA = 114, Mn PBSPMA = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 Da). Further 1H NMR analyses of the purified polymer (Fig. 1A) revealed an excellent correlation between the isobutoxy protective group and alkyl spacer signals, confirming the absence of unwanted deprotection during the polymerisation. Additionally, size exclusion chromatography in N,N-dimethylformamide (DMF-SEC) was used to determine its dispersity (ĐPBSPMA = 1.10) and verify the absence of chain–chain termination (ESI Fig. S1-1†). Notably, poly(methyl methacrylate) (PMMA) was produced under similar conditions as a reference, yet yielded shorter chain length (DPPMMA = 68, Mn PMMA = 7100 Da), due to a lower conversion attained after 18 hours (conv.MMA = 60% vs. conv.BSPMA = 96%, Fig. S1-2 and Table S1†).
400 Da). Further 1H NMR analyses of the purified polymer (Fig. 1A) revealed an excellent correlation between the isobutoxy protective group and alkyl spacer signals, confirming the absence of unwanted deprotection during the polymerisation. Additionally, size exclusion chromatography in N,N-dimethylformamide (DMF-SEC) was used to determine its dispersity (ĐPBSPMA = 1.10) and verify the absence of chain–chain termination (ESI Fig. S1-1†). Notably, poly(methyl methacrylate) (PMMA) was produced under similar conditions as a reference, yet yielded shorter chain length (DPPMMA = 68, Mn PMMA = 7100 Da), due to a lower conversion attained after 18 hours (conv.MMA = 60% vs. conv.BSPMA = 96%, Fig. S1-2 and Table S1†).
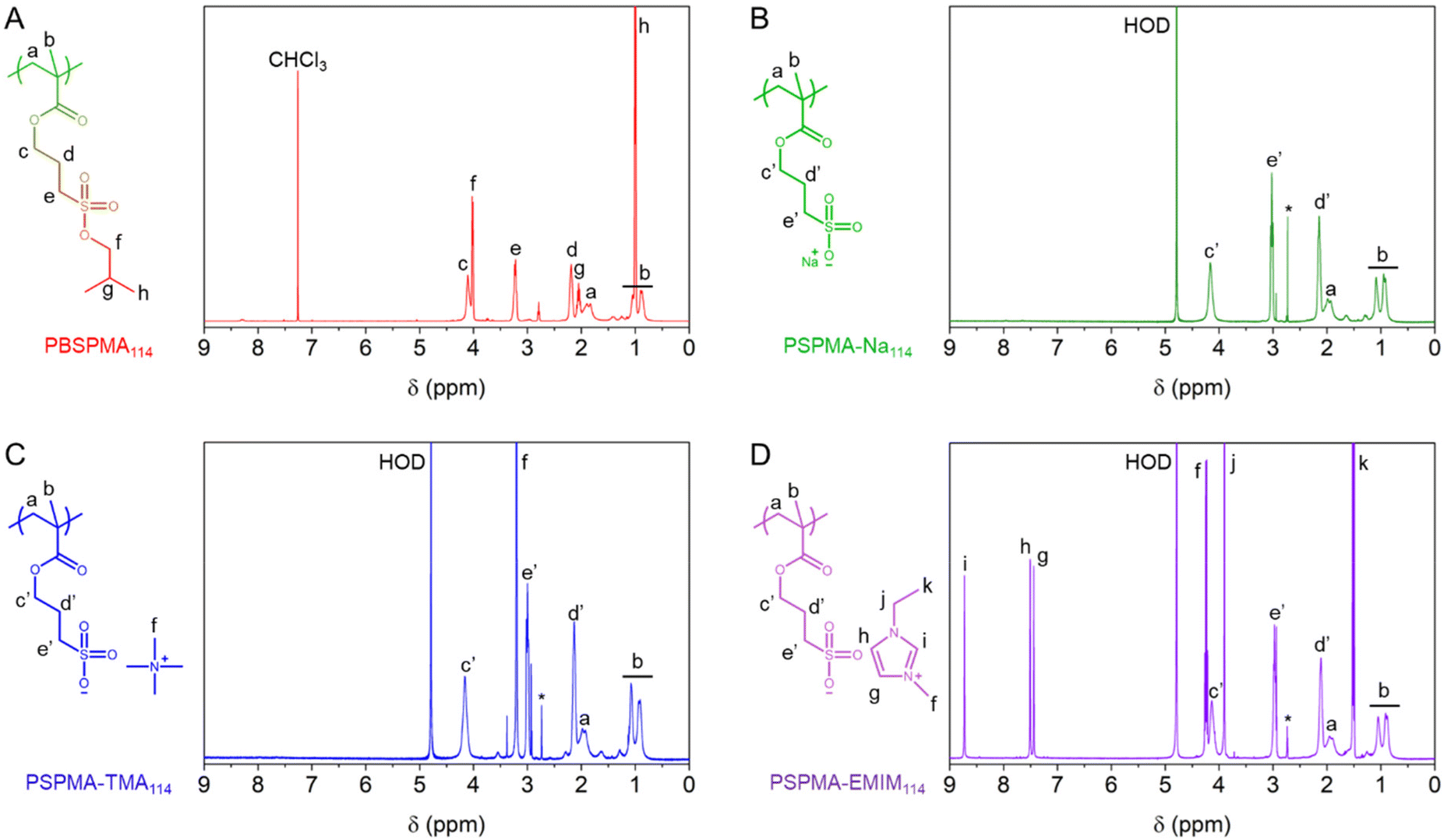 | ||
| Fig. 1 Comparative 1H NMR spectra (400 MHz) of (A) protected PBSPMA114 and polyelectrolytes featuring various counterions, including: (B) PSPMA-Na114, (C) PSPMA-TMA114 and (D) PSPMA-EMIM114. | ||
The PBSPMA114 homopolymer was then subjected to nucleophilic deprotection in dimethylsulphoxide (DMSO) at 70 °C for 24 hours using a variety of iodide-based salts. This enables (i) the exposure of the sulphonate groups, yielding strong polyanionic poly(sulphopropyl methacrylate) (PSPMA-R, R = counterion), and (ii) the inclusion of various inorganic and organic counterions. One advantage of this method over more time-consuming ion exchange protocols, is the straightforward purification step. Isolation of the polymer only involves a single precipitation in a non-solvent system (i.e., a mixture of n-hexane and ethanol, see the ESI for further details†), followed by a few washing cycles to remove the excess salt. The success of the deprotection reactions was confirmed by 1H NMR, as the polymers are now soluble in D2O, but also possess different proton signals. After reaction with sodium iodide, the spectrum of poly(sulphopropyl methacrylate) sodium salt (PSPMA-Na114, Fig. 1B) i.e., our benchmark polyanion, no longer features the characteristic signals from the isobutoxy protective groups (CH2, 2H, 4.1 ppm; CH, 1H, 2.1 ppm and CH3, 6H, 1.0 ppm) and only signals from the backbone and the C3 spacer (3 × CH2, 2H each, 4.2 ppm, 3.1 ppm and 2.2 ppm) are visible. Quantitative deprotection was also achieved when other iodide salts from the alkali metal group (i.e., lithium iodide, potassium iodide and caesium iodide) were used in lieu of sodium iodide, which enables the formation of polyelectrolytes with differently sized counterions. Although the efficacy of the deprotection can still be verified by 1H NMR (Fig. S2†), no further information about the nature of the counterion itself can be extracted using this technique.
The use of organic iodide salts however brings additional proton signals, based on the chemical nature of the counterion. A simple salt such as tetramethylammonium iodide (TMAI) only features one sharp proton signal at 3.2 ppm (originating from the four CH3 groups of the ammonium) in the spectrum of poly(sulphopropyl methacrylate) tetramethylammonium salt (PSPMA-TMA114, Fig. 1C). Increasing the length of the ammonium alkyl chains (i.e., using tetraethylammonium iodide (TEAI) or tetrabutylammonium iodide (TBAI) to produce poly(sulphopropyl methacrylate) tetramethylammonium salt and poly(sulphopropyl methacrylate) tetrabutylammonium salt, respectively; Fig. S2†) adds additional CH2 proton signals to the spectra. Here, 1H NMR can be used to verify that adequate amounts of SPMA units and organic counterions are present in the samples (i.e., no deficit or excess of salt is present). While a slight excess of TMA can be found in PSPMA-TMA114, despite extensive washing cycles (1.2:1 salt:polymer ratio), quantitative amounts of counterions were found for the longer alkyl chains.
Furthermore, a range of iodide salts with more complex chemical structures (i.e., 1-ethyl-3-methylimidazolium iodide (EMIMI), triethylphenylammonium iodide (PhTEAI), 3-(trifluoromethyl)phenyltrimethylammonium iodide (FPhTMAI) and butyrylthiocholine iodide (BTCI)) were used for nucleophilic deprotection. Again, the success of the deprotection can be assessed not only by the loss of the isobutoxy protective groups, but also by the presence of the organic counterions. For instance, poly(sulphopropyl methacrylate) 1-ethyl-3-methylimidazolium salt (PSPMA-EMIM114, Fig. 1D) features very distinct signals from the imidazolium ring (3 × CH, 1H each, 8.8 ppm, 7.5 ppm and 7.4 ppm), while poly(sulphopropyl methacrylate) 3-(trifluoromethyl)phenyltrimethylammonium salt (PSPMA-FPhTMA114, Fig. S2†) features aromatic (4H, 7.8–8.3 ppm) and methyl ammonium (CH3, 12H, 3.7 ppm) signals. Interestingly, fluorine nuclear magnetic resonance (19F NMR) spectroscopy was also conducted on PSPMA-FPhTMA114 and the pristine salt (Fig. S3†). Both species exhibited fluorine signals at ∼−62 ppm, further confirming the presence of the fluorinated counterion in the polyelectrolyte.
Albeit neither quantitative nor as precise as 1H NMR, Fourier transform infrared (FTIR) spectroscopy was also used to characterise the various polyelectrolytes (Fig. S4†). All the polymers feature typical S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1045 and 1192 cm−1 after deprotection, and some include signals that are specific to their counterions, such as the C–F stretching mode of PSPMA-FPhTMA114 at 1324 cm−1 or the CO ketone of poly(sulphopropyl methacrylate) butyrylthiocholine salt (PSPMA-BTC114) stretching at 1699 cm−1.
O stretching at 1045 and 1192 cm−1 after deprotection, and some include signals that are specific to their counterions, such as the C–F stretching mode of PSPMA-FPhTMA114 at 1324 cm−1 or the CO ketone of poly(sulphopropyl methacrylate) butyrylthiocholine salt (PSPMA-BTC114) stretching at 1699 cm−1.
Due to their charged nature and to the absence of large hydrophobic spacers, our polyelectrolytes can no longer be dissolved in DMF, so aqueous SEC (Fig. S5†) was used to verify that nucleophilic deprotections did not affect the integrity of the polymer chains. Except for of PSPMA-FPhTMA114 (possibly due to interaction with the column material30), all samples feature a monomodal peak, which confirms the absence of side-reactions during deprotection. The various polyelectrolytes also possess the same retention volume and similar molecular weights, as observed elsewhere,21 which suggests little influence of the nature and size of the counterion on the polymer coil size and polymer–solvent interaction.
Thermal and solution properties of polyelectrolyte homopolymers
As evidenced in previous studies,22,44 the nature of the counterion, particularly inorganic vs. organic, strongly affects the thermal properties of a polyelectrolyte. Therefore, the thermal stability and the presence of transition temperatures were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), respectively, and compared to those of the PBSPMA114 and PMMA68 reference materials. The protected homopolymer is typically stable up to a temperature of ∼180 °C, after which it decomposes with an initial loss of mass of ∼15–20%, attributed to the removal of the isobutoxy protective groups. Further loss of mass occurs at ∼200 °C through acid-catalysed hydrolysis of the acrylic ester,46 before the final degradation of the backbone around ∼400 °C (Fig. S6-1†).While all samples featuring inorganic counterions are now stable up to 200–250 °C (excluding the evaporation of trace amounts of solvent, Fig. 2A and ESI S7†), their decomposition still occurs through a multi-step process.44 Interestingly, PSPMA-K114 features an early loss of mass at 210 °C, while other polyanions only start to degrade at 250–270 °C; therefore, their thermal stability is not entirely dictated by the size of the counterion. While the protected precursor almost fully degrades before reaching the maximal temperature of 700 °C, polyelectrolytes with inorganic counterions retain a large weight fraction, and the heavier the cation is (i.e., lower in the alkali metal group), the greater the weight percentage remaining at the end of the ramp. The residues at the end of the heating ramp typically consist of complex salts/ceramics that are not volatile in the measured temperature range. Most notably however, no thermal events occurred for any of the inorganic counterions (Fig. 2B), while the PBSPMA114 precursor exhibited a glass transition temperature (Tg PBSMA = 16.7 °C, Fig. S6-2†). This absence of a Tg in the measured range, previously observed for both acrylic22,42 and methacrylic20,43 systems, is consistent with the brittle nature of polyelectrolytes, which can possess glass transition temperatures up to 228 °C,31 exceeding the capability of our instrument and the thermal stability of our polymers.
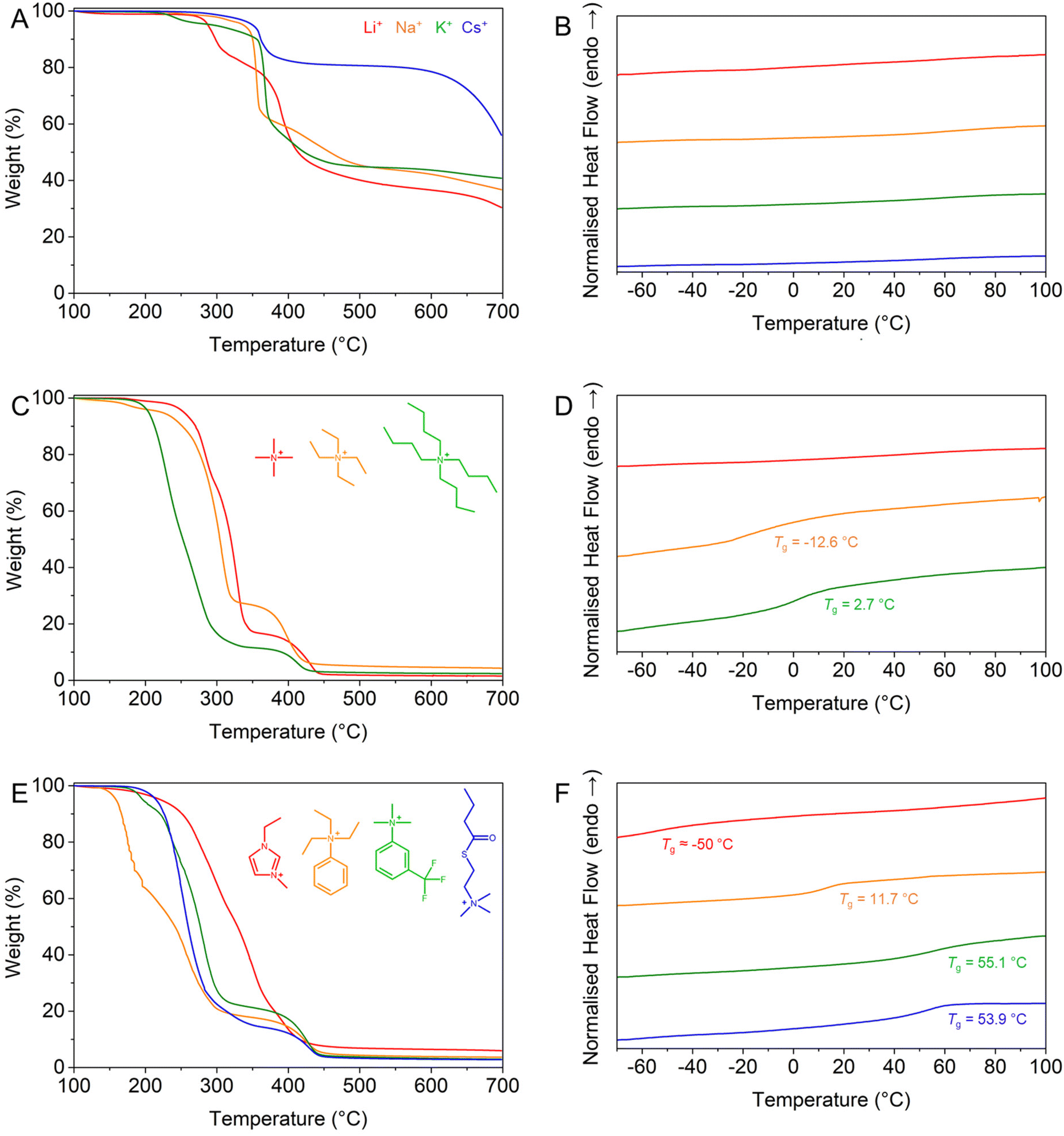 | ||
| Fig. 2 Thermogravimetric and differential scanning calorimetry analyses of the polyelectrolyte homopolymers. (A) TGA and (B) DSC of polyelectrolytes with inorganic counterions: PSPMA-Li114 (red), PSPMA-Na114 (orange), PSPMA-K114 (green) and PSPMA-Cs114 (blue). (C) TGA and (D) DSC of polyelectrolytes with simple quaternary ammonium counterions: PSPMA-TMA114 (red), PSPMA-TEA114 (orange) and PSPMA-TBA114 (green). (E) TGA and (F) DSC of polyelectrolytes with other organic counterions: PSPMA-EMIM114 (red), PSPMA-PhTMA114 (orange), PSPMA-FPhTEA114 (green) and PSPMA-BTC114 (blue). | ||
The presence of an organic counterion appears to greatly decrease the thermal stability of the polyelectrolyte to 150–170 °C, without any remarkable trend (Fig. 2C and E). However, the fully organic nature of the polyelectrolytes permits their complete decomposition with a remaining char weight ≤5 wt%, thus the salts that are formed upon heating are somewhat more volatile and do not yield complex salts/ceramics.44 Most importantly however, new glass transition temperatures, generally below room temperature, appear for most polyelectrolytes with organic counterions, akin to the plasticising effect observed in poly(ionic liquids).45 The only outlier, PSPMA-TMA114, remains brittle and does not exhibit any Tg in the measured temperature range, while longer alkyl analogues do possess one, also affected by the number of carbons (Tg PSPMA-TEA = −12.6 °C and Tg PSPMA-TBA = 2.7 °C, Fig. 2D). Other organic counterions also provide the polyelectrolyte with a Tg ranging from −50.2 °C for PSPMA-EMIM114 to 55.1 °C for poly(sulphopropyl methacrylate) phenyltriethyl ammonium salt (PSPMA-PhTEA114, Fig. 2F and Table S4†).
The exact relationship between the counterion and the observed glass transition temperature is however difficult to pinpoint, as several competing effects may be at stake. The size of the hydration layer around the counterion,24 a plasticising effect due to weaker ion/counterion interactions,27–29 or intrinsic parameters such as symmetry, nucleophilicity and the charge delocalisation30 may all play important and possibly contradictory roles. As a general observation, the glass transition temperature seems to be very high for small inorganic counterions, decreasing sharply for sufficiently large organic counterions but increasing again for even larger species. These findings are in line with the empirical model proposed by Bocharova et al.,31 which predicts a non-monotonic dependence of Tg on the molecular volume Vm (i.e., the volume of the monomer unit + counterion complex) based on the competing contributions of electrostatic (∼Vm−1/3) and van der Waals (∼Vm2/3) interactions. Interestingly, this non-monotonic behaviour is predicted by molecular dynamics simulations32,33 and has been clearly observed in simple ionic liquids, while the presence of a clear minimum is not obvious for polymeric ionic liquids. Admittedly, only TMA+, TEA+, and TBA+ are the three counterions with a clearly defined trend in molecular volume Vm, and the difference in Tg between TEA+ and TBA+ (∼15 °C) is significant but not larger than the fluctuations in data reported in previous literature.31 A larger difference in the Tg (∼100 °C) is observed for the large organic counterions EMIM, PhTMA and FPhTEA, but their complex structure makes it challenging to estimate and compare their molecular volumes.
A change in the counterion, while having large repercussions on the thermal behaviour of the polyelectrolytes, also greatly influences their ability to be dissolved in non-aqueous solvents. While apolar solvents such as n-hexane or cyclohexane cannot dissolve any of the polyelectrolytes featured in this study, a series of polar solvents were selected and solubility tests were conducted (Table 1 and Fig. S8-1†). Inorganic counterions do not provide solubility in organic solvents, except for ‘wet’ DMSO (i.e., DMSO with 10–20 vol% water, also used as a medium for the deprotection reaction) and only PSPMA-Li114 can be dissolved in ‘wet’ methanol and ethanol. Surprisingly, PSPMA-K114 did not dissolve at all in DMSO, as evidenced during deprotection, when the polymer precipitated over the course of the reaction. Note that this phenomenon does not hinder full deprotection, as shown previously using acetone i.e., another non-solvent for PSPMA-Na as medium.43
| Polyelectrolyte | Water | DMSO | Methanol | Ethanol | Acetone | THF | ACN | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Qualitative assessment of the solubility at 25 °C with concentration kept at 5 g L−1 across the whole series. o: fully soluble. p: partially soluble (i.e., particles and hair-like aggregates can be seen by eye). p*: partially soluble in neat solvent and soluble in ‘wet’ solvent (i.e., 10–20 vol% water). x: insoluble. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-Li114 | o | p* | p* | p* | x | x | x | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-Na114 | o | p* | x | X | x | x | x | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-K114 | o | x | x | x | x | x | x | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-Cs114 | o | p* | x | x | x | x | x | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-TMA114 | o | p* | p* | x | x | x | x | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-TEA114 | o | p* | p* | o | x | x | p | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-TBA114 | o | p* | p* | o | o | x | p | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-EMIM114 | o | o | o | o | x | x | p | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-PhTEA114 | o | o | o | o | x | x | p | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-FPhTMA114 | o | o | o | p* | x | x | x | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PSPMA-BTC114 | o | o | o | p* | x | x | p | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Polyelectrolytes with simple counterions (i.e., PSPMA-TMA114, PSPMA-TEA114 and PSPMA-TBA114) also dissolve in ‘wet’ DMSO, in ‘wet’ methanol and in dry ethanol for longer alkane chains, while larger cations (i.e., EMIM+, PhTEA+, FPhTMA+ and BTC+) permit dissolution in neat solvents. Aprotic polar organics (i.e., acetone, tetrahydrofuran and acetonitrile) provide little ability to dissolve the polyelectrolytes, except for PSPMA-TBA114, which disperses into acetone (see ESI†). To further confirm the capacity of some organic solvents to dissolve polyelectrolytes, 1H NMR spectra were recorded in DMSO-d6, methanol-d4, ethanol-d6 and acetone-d6 (Fig. S2†). For all polyelectrolytes, the relative integration of signals was maintained, and no peak broadening was observed, confirming their solubility in these organic solvents.
Synthesis and self-assembly of block copolymers
To expand our study, a series of amphiphilic block copolymers (BCPs) featuring a hydrophobic block and a hydrophilic PSPMA-R segment were produced via sequential RAFT polymerisation, followed by nucleophilic deprotection (Scheme 2). A poly(methyl methacrylate) macro-CTA was synthesised (PMMA114, Mn SEC = 11400 Da, Đ = 1.27) and chain extended with our protected sulphonate monomer, yielding an organo-soluble protected precursor (PMMA114-b-PBSPMA126, Mn SEC+NMR = 44800, Đ = 1.18) that is straightforward to characterise (Fig. S9-1†). 1H NMR analysis of the purified BCP confirmed the successful addition of BSPMA units onto the PMMA114 segment and enabled an accurate calculation of the chain length of the second block by comparing the PBSPMA CH2 signals (2H, 4.2 ppm, 2H, 3.2 ppm and 2H, 2.2 ppm) to the PMMA methyl signal (3H, 3.6 ppm). Note that a consistent ratio was found between the PBSPMA isobutoxy CH3 signal (6H, 1.0 ppm) and abovementioned CH2 signals, which confirms that the isobutoxy protective groups are preserved during chain extension. Furthermore, SEC corroborated the quantitative chain extension, with a full shift of the polymer signal towards a lower retention volume/higher molecular weight, as well as the absence of chain–chain termination.
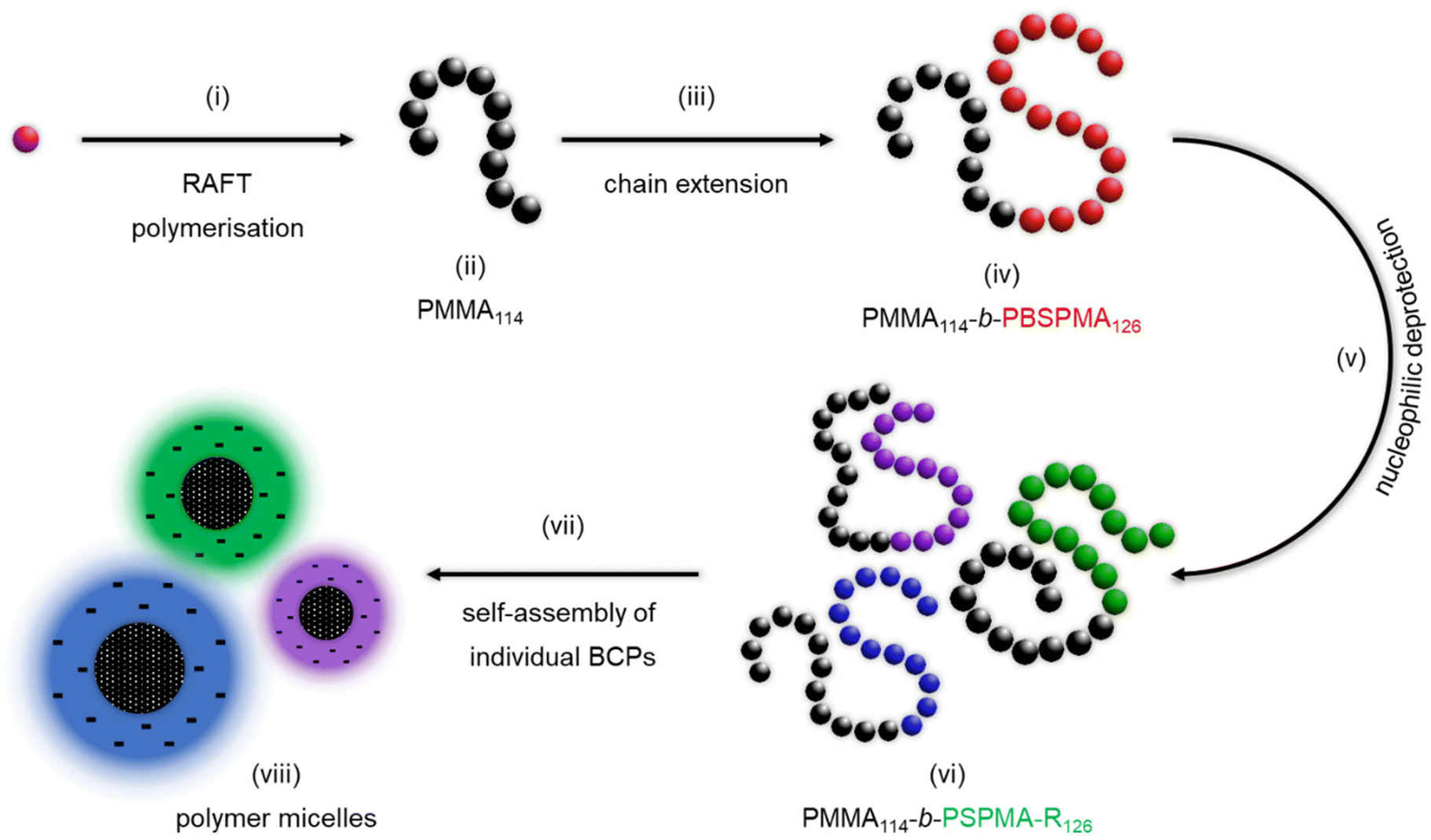 | ||
| Scheme 2 Synthesis of hydrophobic/strong anionic amphiphilic block copolymers capable of self-assembly in aqueous or organic media. (i) Controlled radical polymerisation is used to produce (ii) a poly(methyl methacrylate) segment, which is (iii) chain extended to yield (iv) a protected poly(methyl methacrylate)-block-poly(3-isobutoxysulphopropyl methacrylate) hydrophobic block copolymer. (v) Nucleophilic deprotection is then performed to produce (vi) a range of poly(methyl methacrylate)-block-poly(sulphopropyl methacrylate) amphiphiles, (vii) capable of self-assembly in various media to produce (viii) polymer micelles with consistent hydrophobic cores and varying natures of their charged shells. | ||
Then, a series of nucleophilic deprotections were conducted with most of the iodide-based salts, which enabled the production of BCPs with a constant PMMA molar fraction (xPMMA = 0.48) but varied the nature of the polyelectrolyte. The successful deprotections, albeit established on homopolymers, were verified using 1H NMR (Fig. S9†) by monitoring the disappearance of the isobutoxy protective groups and, if applicable, the presence of new proton signals from the organic counterions. Once the chemical nature of the deprotected block copolymers was verified, the effect of the various counterions on the overall thermal behaviour was investigated using DSC. While PBSPMA and PMMA possess vastly different glass temperatures (Tg PBSMA = 16.7 °C vs. Tg PMMA = 108 °C), a single Tg was observed for the protected block copolymer (Tg BCP = 18.1 °C, Fig. 3A). This could indicate a mixed phase, albeit not in agreement with the volume fractions of the blocks and the Flory–Fox equation. Previous studies on acrylic analogues22,42 also evidenced this behaviour, where two separate Tg were observed for block copolymers with a low protected sulphonate content, and a single one close to that of the sulphonate homopolymer when this ratio increased. Therefore, we suggest that both blocks are capable of phase separation, but with a short-range order only. Upon nucleophilic deprotection with NaI or TMAI, no Tg can be detected in the thermograms of the BCPs, despite the PMMA block remaining pristine during the deprotection, a behaviour that has also been evidenced before.22,42 However, the presence of larger counterions, introduces new glass transition temperatures, ranging from 18.6 to 73.1 °C (Fig. 3 and Table S5†). The values appear to be in part dictated by the nature of the counterion and to lie between those of a polyelectrolyte homopolymer and the PMMA block, in line with the Flory–Fox equation. For instance, the glass transition temperatures of the PMMA114 and PSPMA-PhTEA126 homopolymers are Tg PMMA = 108 °C and Tg PSPMA-PhTEA = 11.7 °C, respectively, while that of the PhTEA-based block copolymer is measured at 68.6 °C. This could be indicative of a single mixed phase between PMMA and any of the polyelectrolytes featuring organic counterions.47 Comparative DSC thermograms of the PMMA114 macro-CTA, polyelectrolytes and deprotected BCPs are available in Fig. S10.†
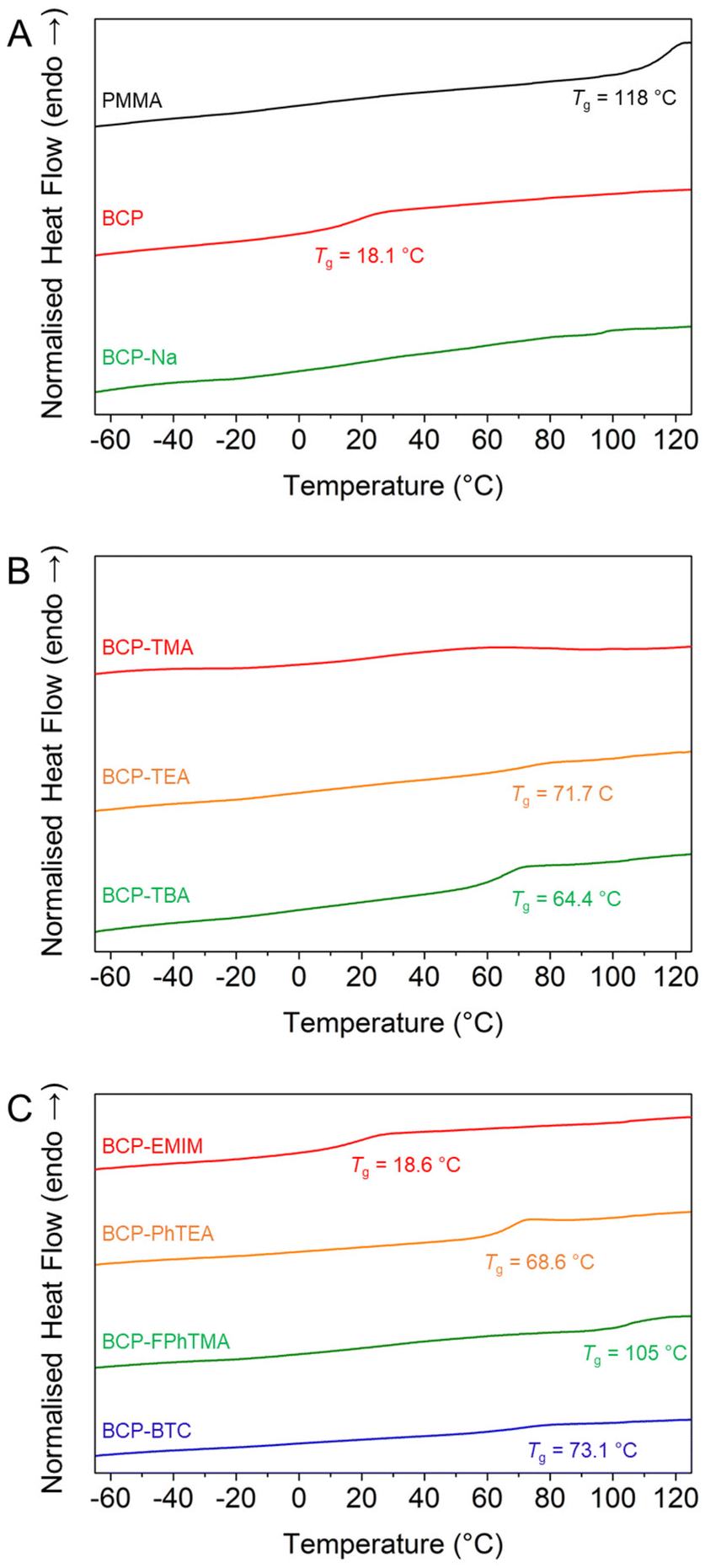 | ||
| Fig. 3 Influence of the type of counterion on the glass transition temperature of the block copolymers. (A) DSC thermograms of PMMA114 (black), PMMA114-b-PBSPMA126 (red) and PMMA114-b-PSPMA-Na126 (green). (B) DSC thermograms of PMMA114-b-PSPMA-TMA126 (red), PMMA114-b-PSPMA-TEA126 (orange) and PMMA114-b-PSPMA-TBA126 (green). (C) DSC thermograms of PMMA114-b-PSPMA-EMIM126 (red), PMMA114-b-PSPMA-PhTEA126 (orange), PMMA114-b-PSPMA-FPhTMA126 (green) and PMMA114-b-PSPMA-BTC126 (blue). | ||
The amphiphilic nature of these BCPs allows them to self-assemble in aqueous media. To enable the screening of the polyelectrolyte segments and hence reduce particle aggregation (i.e., slow diffusion mode),48 a small amount of salt was added. The ‘direct dissolution’ method was employed (i.e., an appropriate volume of 10 mM KNO3 was charged into a vial loaded with dried BCP to achieve a 1 g L−1 concentration), which readily enabled the formation of micelles with a hydrophobic core and a negatively charged hydrophilic shell, as evidenced by ζ-potential measurements (Table S6†). Dynamic light scattering (DLS, Fig. 4 and Fig. S11†) revealed the presence of polymer micelles with hydrodynamic diameters between Dh = 50–75 nm and relatively narrow polydispersity indices PDI ≈ 0.2 for all but one block copolymer. Only the FPhTMA-based BCP possesses a larger mean particle diameter and polydispersity index (Dh BCP-FPhTMA = 95 nm, PDIBCP-FPhTMA = 0.383), suggesting a slight inhomogeneity in the self-assembly process. Transmission electron microscopy (TEM, Fig. 4 and Fig. S12†) was used to visualise the particles, aided by the application of a negative stain. Here the PMMA core (i.e., low electron density) appears bright, while the uranyl acetate stain (i.e., high electron density) penetrates the negatively charged shell of the micelles, thus appearing dark. All amphiphilic BCPs form well-defined spherical micelles in aqueous media, once more suggesting that the counterions have little influence on the self-assembly mechanism. A few elongated or fused micelles within a sphere-dominated population were observed for the PMMA114-b-PSPMA-FPhTMA126 block copolymer, in accordance with the larger Dh and PDI values measured by dynamic light scattering.
 | ||
| Fig. 4 Self-assembly of selected amphiphilic block copolymers in aqueous media. (A1–C1) DLS size distribution plots (bars) and corresponding correlograms (solid lines) and (A2–C2) negatively stained TEM images of polymer micelles obtained from PMMA114-b-PSPMA-Na126 (green), PMMA114-b-PSPMA-TMA126 (blue) and PMMA114-b-PSPMA-EMIM126 (purple) at 1 g L−1 in 10 mM KNO3. | ||
With some of the polyelectrolytes possessing the ability to dissolve in non-aqueous solvents, attempts have been made to produce nanoparticles in organic media using the same ‘direct dissolution’ method. The key to this protocol lies in the choice of the medium, one that would enable the dissolution of the polyelectrolyte domain but not of the PMMA segment. As such, acetone, acetonitrile, DMSO and THF are incompatible, while methanol and ethanol remain potential candidates. Multiple populations, large dispersity indices and poor correlograms were measured by DLS for all samples dispersed in methanol (Fig. S13†) and for most dispersed in ethanol (Fig. S14†), suggesting non-homogeneous self-assembly in those media. Nonetheless, negatively stained specimens were prepared and imaged using TEM (Fig. S15 and Fig. S16†). Through a direct comparison with polymer micelles self-assembled in aqueous media (Fig. 5), it is evident that methanol and ethanol, albeit usable solvents for the formation of aggregates, do not permit the fabrication of particles with homogeneous shapes and narrow polydispersities. Often, very large and ill-formed particles are present within populations of small spherical micelles, and a few worm-like specimens were observed (i.e., PMMA114-b-PSPMA-EMIM126 in methanol and PMMA114-b-PSPMA-EMIM126 in ethanol). This might be explained by the limited solubility of PMMA in these solvents and/or by their low dielectric constant, resulting in limited charge repulsion between the charged coronas of the particles and therefore poor dispersion.49,50
 | ||
| Fig. 5 Comparative self-assembly of amphiphilic block copolymers in various types of media. Negatively stained TEM images of polymer micelles obtained from (A) PMMA114-b-PSPMA-EMIM126, (B) PMMA114-b-PSPMA-PhTEA126, (C) PMMA114-b-PSPMA-FPhTMA126 and (D) PMMA114-b-PSPMA-BTC126 in 10 mM KNO3, methanol or ethanol. | ||
Conclusion & outlook
Herein, we report a simple yet effective methodology to produce polyelectrolytes featuring a range of counterions, from simple alkali metals to more complex organic salts. The nature of the counterion has a strong effect on the thermal stability, on the presence of a glass transition temperature, as well as on the solubility of the polyelectrolytes. Polyelectrolytes including inorganic counterions tend to have a higher thermal stability, but exhibit no phase transition and lack solubility in organic solvents. Larger organic counterions on the other hand provide a wide range of glass transition temperatures, which appear to follow a non-monotonic trend with respect to counterion size, and provide moderate solubility in polar organics.Our strategy also permits the production of copolymers, including a hydrophobic block and a polyelectrolyte segment. The protected diblock precursors are fully hydrophobic, which facilitates their characterisation, yet tailoring the counterion relies on the simple choice of a nucleophile during deprotection. All the amphiphilic block copolymers readily self-assemble into spherical micelles in aqueous media, with similar hydrodynamic diameters and relatively narrow polydispersities. The dispersion of some copolymers in methanol and ethanol has been attempted, and while our system does not exhibit consistent formation of well-defined polymer micelles, it opens up avenues for the design of structured soft matter outside the boundaries of aqueous media.
Author contributions
The manuscript was written through contributions of all the authors. All the authors have approved the final version of the manuscript.Data availability
The data supporting this article (Materials and methods, as well as additional 1H NMR spectra, SEC elugrams, DSC, and TGA thermograms, DLS plots, and TEM images) have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors warmly thank Jur van Dijken, Léon Rohrbach and Dr Marc C. A. Stuart for their technical assistance with thermogravimetric analyses, aqueous size exclusion chromatography, and electron microscopy, respectively. This research received funding from the Dutch Research Council (NWO) in the framework of the ENW PPP Fund for the top sectors and from the Ministry of Economic Affairs in the framework of the ‘PPS-Toeslagregeling’. M. K. is the grateful recipient of a European Research Council grant (European Union's Horizon 2020 research and innovation program, consolidator grant agreement no. 864982). T.P. warmly thanks Uluru.References
- M. Armand and J. M. Tarascon, Building better batteries, Nature, 2008, 451(7179), 652–657 CAS.
- U. Raviv, S. Giasson, N. Kampf, J.-F. Gohy, R. Jérôme and J. Klein, Lubrication by charged polymers, Nature, 2003, 425(6954), 163–165 CAS.
- J. Kötz, S. Kosmella and T. Beitz, Self-assembled polyelectrolyte systems, Prog. Polym. Sci., 2001, 26(8), 1199–1232 Search PubMed.
- T. Pelras, Nonappa, C. S. Mahon and M. Müllner, Cylindrical Zwitterionic Particles via Interpolyelectrolyte Complexation on Molecular Polymer Brushes, Macromol. Rapid Commun., 2021, 42(8), 2000401 CrossRef CAS.
- T. Pelras, C. S. Mahon, Nonappa, O. Ikkala, A. H. Gröschel and M. Müllner, Polymer Nanowires with Highly Precise Internal Morphology and Topography, J. Am. Chem. Soc., 2018, 140(40), 12736–12740 CrossRef CAS.
- T. I. Löbling, J. S. Haataja, C. V. Synatschke, F. H. Schacher, M. Müller, A. Hanisch, A. H. Gröschel and A. H. E. Müller, Hidden Structural Features of Multicompartment Micelles Revealed by Cryogenic Transmission Electron Tomography, ACS Nano, 2014, 8(11), 11330–11340 CrossRef PubMed.
- J.-M. Malho, M. Morits, T. I. Löbling, Nonappa, J. Majoinen, F. H. Schacher, O. Ikkala and A. H. Gröschel, Rod-Like Nanoparticles with Striped and Helical Topography, ACS Macro Lett., 2016, 5(10), 1185–1190 CrossRef CAS PubMed.
- L. van Westerveld, J. Es Sayed, M. de Graaf, A. H. Hofman, M. Kamperman and D. Parisi, Hydrophobically modified complex coacervates for designing aqueous pressure-sensitive adhesives, Soft Matter, 2023, 19(45), 8832–8848 RSC.
- L. van Westerveld, T. Pelras, A. H. Hofman, K. Loos, M. Kamperman and J. Es Sayed, Effect of Polyelectrolyte Charge Density on the Linear Viscoelastic Behavior and Processing of Complex Coacervate Adhesives, Macromolecules, 2024, 57(2), 652–663 CrossRef CAS.
- M. Li, W. Song, Z. Tang, S. Lv, L. Lin, H. Sun, Q. Li, Y. Yang, H. Hong and X. Chen, Nanoscaled Poly(l-glutamic acid)/Doxorubicin-Amphiphile Complex as pH-responsive Drug Delivery System for Effective Treatment of Nonsmall Cell Lung Cancer, ACS Appl. Mater. Interfaces, 2013, 5(5), 1781–1792 CrossRef CAS.
- W. S. Cheow and K. Hadinoto, Self-assembled amorphous drug–polyelectrolyte nanoparticle complex with enhanced dissolution rate and saturation solubility, J. Colloid Interface Sci., 2012, 367(1), 518–526 CrossRef CAS.
- S. Kamble, P. Varamini, M. Müllner, T. Pelras and R. Rohanizadeh, Bisphosphonate-functionalized micelles for targeted delivery of curcumin to metastatic bone cancer, Pharm. Dev. Technol., 2020, 25(9), 1118–1126 CrossRef CAS PubMed.
- Y. Chen, D. Diaz-Dussan, Y.-Y. Peng and R. Narain, Hydroxyl-Rich PGMA-Based Cationic Glycopolymers for Intracellular siRNA Delivery: Biocompatibility and Effect of Sugar Decoration Degree, Biomacromolecules, 2019, 20(5), 2068–2074 CrossRef CAS PubMed.
- B. S. Kim, S. Chuanoi, T. Suma, Y. Anraku, K. Hayashi, M. Naito, H. J. Kim, I. C. Kwon, K. Miyata, A. Kishimura and K. Kataoka, Self-Assembly of siRNA/PEG-b-Catiomer at Integer Molar Ratio into 100 nm-Sized Vesicular Polyion Complexes (siRNAsomes) for RNAi and Codelivery of Cargo Macromolecules, J. Am. Chem. Soc., 2019, 141(8), 3699–3709 CrossRef CAS.
- A. M. C. Maan, A. H. Hofman, T. Pelras, I. M. Ruhof, M. Kamperman and W. M. de Vos, Toward Effective and Adsorption-Based Antifouling Zipper Brushes: Effect of pH, Salt, and Polymer Design, ACS Appl. Polym. Mater., 2023, 5(10), 7968–7981 Search PubMed.
- A. M. C. Maan, C. N. Graafsma, A. H. Hofman, T. Pelras, W. M. de Vos and M. Kamperman, Scalable Fabrication of Reversible Antifouling Block Copolymer Coatings via Adsorption Strategies, ACS Appl. Mater. Interfaces, 2023, 15(15), 19682–19694 CrossRef CAS PubMed.
- C. V. Synatschke, T. I. Löbling, M. Förtsch, A. Hanisch, F. H. Schacher and A. H. E. Müller, Micellar Interpolyelectrolyte Complexes with a Compartmentalized Shell, Macromolecules, 2013, 46(16), 6466–6474 CrossRef CAS.
- J. E. Coughlin, A. Reisch, M. Z. Markarian and J. B. Schlenoff, Sulfonation of polystyrene: Toward the “ideal” polyelectrolyte, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(11), 2416–2424 CAS.
- P. M. Reichstein, J. C. Brendel, M. Drechsler and M. Thelakkat, Poly(3-hexylthiophene)-block-poly(tetrabutylammonium-4-styrenesulfonate) Block Copolymer Micelles for the Synthesis of Polymer Semiconductor Nanocomposites, ACS Appl. Nano Mater., 2019, 2(4), 2133–2143 CrossRef CAS.
- A. H. Hofman, M. Pedone and M. Kamperman, Protected Poly(3-sulfopropyl methacrylate) Copolymers: Synthesis, Stability, and Orthogonal Deprotection, ACS Polym. Au, 2022, 2(3), 169–180 CAS.
- G. J. Tudryn, W. Liu, S.-W. Wang and R. H. Colby, Counterion Dynamics in Polyester−Sulfonate Ionomers with Ionic Liquid Counterions, Macromolecules, 2011, 44(9), 3572–3582 Search PubMed.
- T. Pelras, A. H. Hofman, L. M. H. Germain, A. M. C. Maan, K. Loos and M. Kamperman, Strong Anionic/Charge-Neutral Block Copolymers from Cu(0)-Mediated Reversible Deactivation Radical Polymerization, Macromolecules, 2022, 55(19), 8795–8807 CAS.
- A. Kisliuk, V. Bocharova, I. Popov, C. Gainaru and A. P. Sokolov, Fundamental parameters governing ion conductivity in polymer electrolytes, Electrochim. Acta, 2019, 299, 191–196 CAS.
- K. Lunkenheimer, D. Prescher and K. Geggel, Role of Counterions in the Adsorption and Micellization Behavior of 1:1 Ionic Surfactants at Fluid Interfaces–Demonstrated by the Standard Amphiphile System of Alkali Perfluoro-n-octanoates, Langmuir, 2022, 38(3), 891–902 CrossRef CAS.
- K. Lunkenheimer, K. Geggel and D. Prescher, Role of Counterion in the Adsorption Behavior of 1:1 Ionic Surfactants at Fluid Interfaces—Adsorption Properties of Alkali Perfluoro-n-octanoates at the Air/Water Interface, Langmuir, 2017, 33(39), 10216–10224 CrossRef CAS.
- K. Lunkenheimer, D. Prescher, R. Hirte and K. Geggel, Adsorption Properties of Surface Chemically Pure Sodium Perfluoro-n-alkanoates at the Air/Water Interface: Counterion Effects within Homologous Series of 1:1 Ionic Surfactants, Langmuir, 2015, 31(3), 970–981 CrossRef CAS.
- M. Lee, U. H. Choi, R. H. Colby and H. W. Gibson, Ion Conduction in Imidazolium Acrylate Ionic Liquids and their Polymers, Chem. Mater., 2010, 22(21), 5814–5822 CrossRef CAS.
- U. H. Choi, Y. Ye, D. Salas de la Cruz, W. Liu, K. I. Winey, Y. A. Elabd, J. Runt and R. H. Colby, Dielectric ansd Viscoelastic Responses of Imidazolium-Based Ionomers with Different Counterions and Side Chain Lengths, Macromolecules, 2014, 47(2), 777–790 Search PubMed.
- U. H. Choi, M. Lee, S. Wang, W. Liu, K. I. Winey, H. W. Gibson and R. H. Colby, Ionic Conduction and Dielectric Response of Poly(imidazolium acrylate) Ionomers, Macromolecules, 2012, 45(9), 3974–3985 CrossRef CAS.
- Y. Ye and Y. A. Elabd, Anion exchanged polymerized ionic liquids: High free volume single ion conductors, Polymer, 2011, 52(5), 1309–1317 Search PubMed.
- V. Bocharova, Z. Wojnarowska, P.-F. Cao, Y. Fu, R. Kumar, B. Li, V. N. Novikov, S. Zhao, A. Kisliuk, T. Saito, J. W. Mays, B. G. Sumpter and A. P. Sokolov, Influence of Chain Rigidity and Dielectric Constant on the Glass Transition Temperature in Polymerized Ionic Liquids, J. Phys. Chem. B, 2017, 121(51), 11511–11519 Search PubMed.
- Y. Fu, V. Bocharova, M. Ma, A. P. Sokolov, B. G. Sumpter and R. Kumar, Effects of counterion size and backbone rigidity on the dynamics of ionic polymer melts and glasses, Phys. Chem. Chem. Phys., 2017, 19(40), 27442–27451 Search PubMed.
- Y. Cheng, J. Yang, J.-H. Hung, T. K. Patra and D. S. Simmons, Design Rules for Highly Conductive Polymeric Ionic Liquids from Molecular Dynamics Simulations, Macromolecules, 2018, 51(17), 6630–6644 CAS.
- B. Doughty, A.-C. Genix, I. Popov, B. Li, S. Zhao, T. Saito, D. A. Lutterman, R. L. Sacci, B. G. Sumpter, Z. Wojnarowska and V. Bocharova, Structural correlations tailor conductive properties in polymerized ionic liquids, Phys. Chem. Chem. Phys., 2019, 21(27), 14775–14785 CAS.
- J. R. Keith, N. J. Rebello, B. J. Cowen and V. Ganesan, Influence of Counterion Structure on Conductivity of Polymerized Ionic Liquids, ACS Macro Lett., 2019, 8(4), 387–392 CrossRef CAS PubMed.
- A. Hanisch, A. H. Gröschel, M. Förtsch, M. Drechsler, H. Jinnai, T. M. Ruhland, F. H. Schacher and A. H. E. Müller, Counterion-Mediated Hierarchical Self-Assembly of an ABC Miktoarm Star Terpolymer, ACS Nano, 2013, 7(5), 4030–4041 CAS.
- H. Cui, Z. Chen, K. L. Wooley and D. J. Pochan, Controlling Micellar Structure of Amphiphilic Charged Triblock Copolymers in Dilute Solution via Coassembly with Organic Counterions of Different Spacer Lengths, Macromolecules, 2006, 39(19), 6599–6607 CAS.
- Z. Zhang, M. M. Rahman, C. Abetz, B. Bajer, J. Wang and V. Abetz, Quaternization of a Polystyrene-block-poly(4-vinylpyridine) Isoporous Membrane: An Approach to Tune the Pore Size and the Charge Density, Macromol. Rapid Commun., 2019, 40(3), 1800729 CrossRef.
- M. Billing, J. K. Elter and F. H. Schacher, Sulfo-and carboxybetaine-containing polyampholytes based on poly(2-vinyl pyridine)s: Synthesis and solution behavior, Polymer, 2016, 104, 40–48 CrossRef CAS.
- C. Facciotti, V. Saggiomo, A. Bunschoten, R. Fokkink, J. B. T. Hove, J. Wang and A. H. Velders, Cyclodextrin-based complex coacervate core micelles with tuneable supramolecular host–guest, metal-to-ligand and charge interactions, Soft Matter, 2018, 14(47), 9542–9549 RSC.
- H. M. van der Kooij, E. Spruijt, I. K. Voets, R. Fokkink, M. A. Cohen Stuart and J. van der Gucht, On the Stability and Morphology of Complex Coacervate Core Micelles: From Spherical to Wormlike Micelles, Langmuir, 2012, 28(40), 14180–14191 CrossRef CAS PubMed.
- T. Pelras, A. Eisenga, G. Érsek, A. Altomare, G. Portale, M. Kamperman and K. Loos, One-Pot Synthesis of Strong Anionic/Charge-Neutral Amphiphilic Block Copolymers, ACS Macro Lett., 2023, 12(8), 1071–1078 CrossRef CAS.
- A. H. Hofman, R. Fokkink and M. Kamperman, A mild and quantitative route towards well-defined strong anionic/hydrophobic diblock copolymers: synthesis and aqueous self-assembly, Polym. Chem., 2019, 10(45), 6109–6115 RSC.
- Y. S. Vygodskii, O. A. Mel'nik, A. S. Shaplov, E. I. Lozinskaya, I. A. Malyshkina and N. D. Gavrilova, Synthesis and ionic conductivity of polymer ionic liquids, Polym. Sci., Ser. A, 2007, 49(3), 256–261 CrossRef.
- J. Yuan, D. Mecerreyes and M. Antonietti, Poly(ionic liquid)s: An update, Prog. Polym. Sci., 2013, 38(7), 1009–1036 CrossRef CAS.
- J. Kolomanska, P. Johnston, A. Gregori, I. Fraga Domínguez, H.-J. Egelhaaf, S. Perrier, A. Rivaton, C. Dagron-Lartigau and P. D. Topham, Design, synthesis and thermal behaviour of a series of well-defined clickable and triggerable sulfonate polymers, RSC Adv., 2015, 5(82), 66554–66562 RSC.
- M. Aubin and R. E. Prud'homme, Analysis of the glass transition temperature of miscible polymer blends, Macromolecules, 1988, 21(10), 2945–2949 CAS.
- M. Sedlák, Generation of multimacroion domains in polyelectrolyte solutions by change of ionic strength or pH (macroion charge), J. Chem. Phys., 2002, 116(12), 5256–5262 Search PubMed.
- R. Hoogenboom, R. Becer, C. Guerrero-Sanchez, S. Hoeppener and U. S. Schubert, Solubility and Thermoresponsiveness of PMMA in Alcohol-Water Solvent Mixtures, Aust. J. Chem., 2010, 63(8), 1173–1178 CAS.
- R. Hoogenboom, S. Rogers, A. Can, R. Becer, C. Guerrero-Sanchez, D. Wouters, C. Hoeppener and U. S. Schubert, Self-assembly of double hydrophobic block copolymers in water–ethanol mixtures: from micelles to thermoresponsive micellar gels, Chem. Commun., 2009, 5582–5584 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01218f |
| This journal is © The Royal Society of Chemistry 2025 |