
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the effect of side chain on RAFT depolymerization; identifying the rate determining step†
Francesco
Felician
a,
Maria-Nefeli
Antonopoulou
a,
Nghia P.
Truong
 a,
Asja A.
Kroeger
b,
Michelle L.
Coote
b,
Glen R.
Jones
*a and
Athina
Anastasaki
*a
a,
Asja A.
Kroeger
b,
Michelle L.
Coote
b,
Glen R.
Jones
*a and
Athina
Anastasaki
*a
aLaboratory of Sustainable Polymers, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zürich, Switzerland. E-mail: glen.jones@mat.ethz.ch; athina.anastasaki@mat.ethz.ch
bInstitute for Nanoscale Science and Technology, College of Science and Engineering, Flinders University, Bedford Park, South Australia 5042, Australia
First published on 19th March 2025
Abstract
Reversible addition–fragmentation chain-transfer (RAFT) depolymerization represents an attractive and low-temperature chemical recycling methodology enabling the near-quantitative regeneration of pristine monomer. Yet, several mechanistic aspects of the process remain elusive. Herein, we shine a light on the RAFT depolymerization mechanism by elucidating the effect of pendant side chains on the depolymerization kinetics. A systematic increase of the number of carbons on the side chain, or the number of ethylene glycol units, revealed a significant rate acceleration. Notably, radical initiator addition during the depolymerization of poly(methyl methacrylate) and poly(hexyl methacrylate) resulted in rate equilibration, indicating that chain activation is the rate-determining step in RAFT depolymerization. Moreover, incorporation of a low DP of hexyl monomer as the second block of poly(methyl methacrylate) led to comparable rates with poly(hexyl methacrylate) homopolymer, confirming the rate determining step. Computational investigations further corroborate this finding, revealing that chain-end fragmentation is energetically more favorable in longer-side-chain methacrylates, which accounts for the experimentally observed rate acceleration. These insights not only deepen our understanding of depolymerization but also pave the way for developing more efficient and customizable depolymerization systems.
Introduction
Reversible deactivation radical polymerization (RDRP), also known as controlled radical polymerization (CRP), allows for the synthesis of polymers with tailored properties such as molecular weight, dispersity, and architectures.1–6 The fundamental concept responsible for this high degree of control is a deactivation step which renders the propagating radical in a dormant state and yields polymers with functional end groups.7,8 These end groups are highly desirable as they allow for facile chain extensions to afford block copolymers, and modifications to yield polymers with ω-end-group functionality.7,9–11 Notably, the end groups inherent in RDRP-synthesized polymers can also be utilized to facilitate depolymerization, a topic which is of increasing interest due to the need to improve the sustainability of polymeric materials.12–14 Through the reactivation of the chain end, polymeric radicals are formed, which can depropagate to yield monomer under thermodynamically favorable conditions.15 Importantly, this can be achieved at temperatures at which analogous polymers synthesized by traditional radical polymerization are thermodynamically stable, as high energy is required to break back-bone bonds and generate depropagating radicals.16Ouchi and coworkers first reported that poly(methyl methacrylate) (PMMA) with chlorine end groups synthesized by atom transfer radical polymerization (ATRP) could undergo depolymerization to a limited extent.17 Subsequent studies by Matyjaszewski and coworkers demonstrated high-yielding depolymerizations by utilizing chlorine capped polymers with both copper18,19 and iron catalysts.20 Our group later reported a photothermal depolymerization system21 and expanded the scope of ATRP-based depolymerization to include bromine-terminated polymers.22 The first example of depolymerization of polymers synthesized by RAFT polymerization was reported in 2018, where brush polymers of bulky methacrylates produced monomer (∼30%) when heated under dilute conditions.23 In 2022, our group reported that depolymerization of both bulky and non-bulky polymethacrylates synthesized by RAFT proceeded to up to 92% depolymerization conversion by heating at 120 °C in 1,4-dioxane.24 Further advancements demonstrated that activation of thiocarbonylthio moieties by direct photolysis25 or using photocatalysts26,27 could lower the depolymerization temperature to 100 °C, and enable temporal control.28 More recently, a controlled depolymerization was achieved by enhancing deactivation through high-activity chain transfer agents, giving a linear decrease in molecular weight over time.29 Additionally, RDRP-synthesized polymers have also significantly lowered depolymerization temperatures under bulk conditions, when compared to analogous polymers synthesized by free radical polymerization.30–33
The work described above demonstrates that the depolymerization of RDRP-synthesized materials is a rapidly evolving field, with significant advancements further expanding the scope of depolymerizable materials and enhancing control over the process. However, as a relatively new area of research, many aspects of these processes remain largely unexplored.12,13 Although the thermodynamic parameters that drive the depolymerization have been adequately investigated and understood, detailed and thorough kinetic analysis for the depolymerization of various polymethacrylates has not received considerable attention. For example, a kinetic comparison between the depolymerization of various polymethacrylates with different ester side chains remains elusive. Inspired by seminal work demonstrating an increase in the kp with increasing ester side chain length during polymerization,34–36 we envisioned that studying their respective depolymerizations would also uncover interesting trends and potentially reveal the rate determining step of a depolymerization reaction.
Results and discussion
We first synthesized a PMMA polymer via RAFT polymerization in toluene using 2-cyano-2-propyl dithiobenzoate as the chain transfer agent at 70 °C, yielding a well-defined polymer with a narrow molar mass distribution (Đ = 1.11, Fig. S1, 2 and Table S1†). PMMA is a commodity polymer, and is the most commonly studied polymethacrylate in terms of depolymerization, thus it serves as a benchmark for comparing the depolymerization of other methacrylate polymers.22 The PMMA-DTB polymer was depolymerized at 120 °C in 1,4-dioxane, with a repeat unit concentration (RUC) of 20 mM (Fig. 1a). Aliquots were taken at regular intervals to determine depolymerization conversion by nuclear magnetic resonance (NMR) spectroscopy. In line with previous reports, depolymerization proceeded to 71% conversion after 7 hours (see Fig. S3 and Table S2†). From the detailed kinetics, an apparent depolymerization rate constant of 0.41 h−1 was obtained by plotting the logarithm of the depolymerization conversion for the first hour of the reaction (Fig. 1b, black trace), similar to the pseudo first-order kinetic plots commonly utilized in the analysis of polymerization reactions.37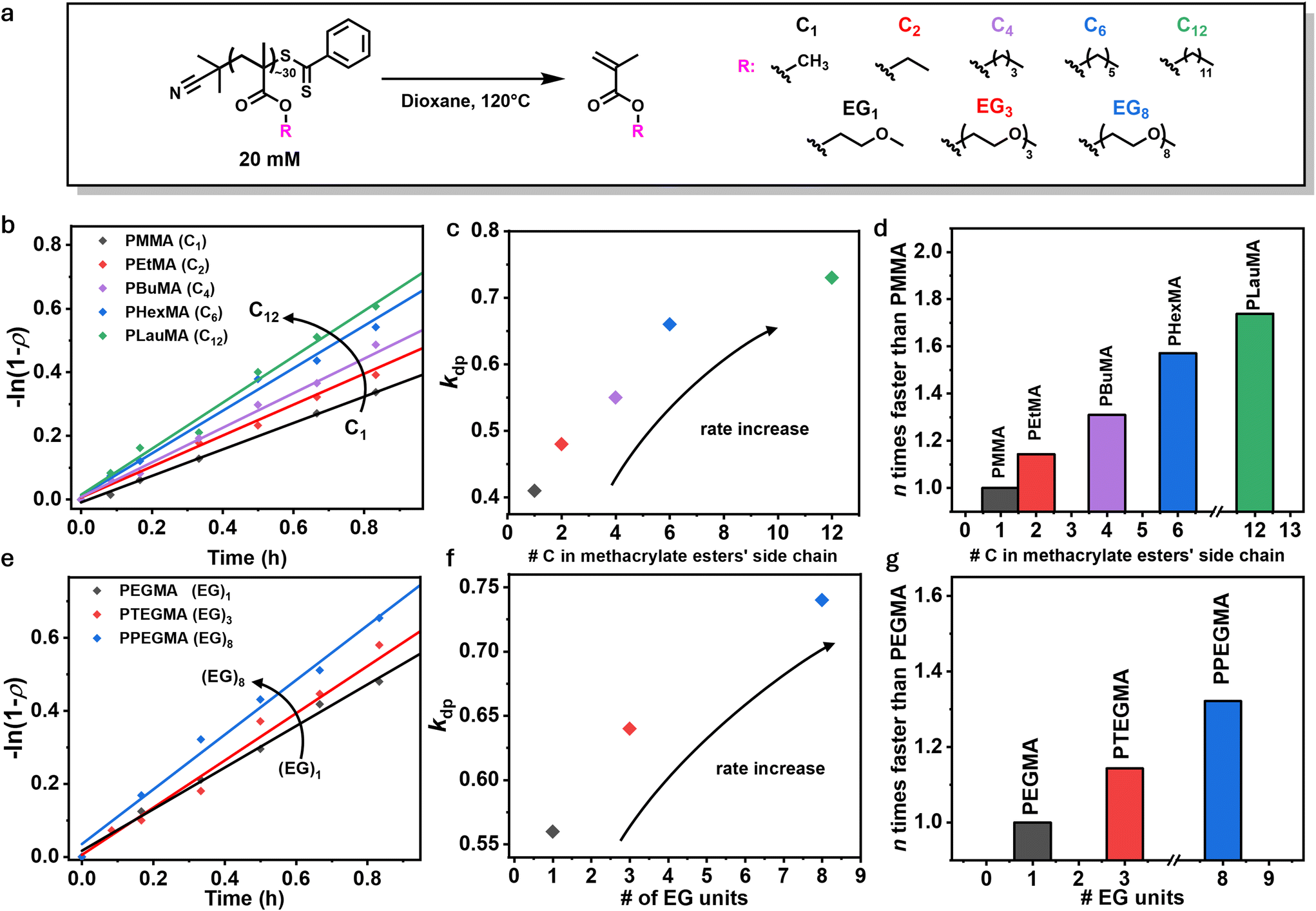 | ||
| Fig. 1 (a) Reaction scheme of depolymerization reaction showing the different side chains used. (b) Pseudo-first order plot for the depolymerization kinetics and (c) extracted of kinetic parameters of polymethacrylates characterized by different alkyl chain lengths. (d) Comparison between depolymerization rate constants normalized to PMMA. (e) Pseudo-first order plot for the depolymerization kinetics and (f) extracted of kinetic parameters of polymethacrylates characterized by different numbers of ethylene glycol units. (g) Comparison between depolymerization rate constants normalized to PEGMA. | ||
To investigate the effect of the pendant alkyl side chain on depolymerization rate, a series of homopolymers with variable side chains was then synthesized consisting of ethyl (Et), butyl (Bu), hexyl (Hex), and lauryl (Lau) methacrylate using similar methodology to PMMA as described above (see Fig. S4–10†). In the depolymerization of RAFT-synthesized polymers, depolymerizability has previously been shown to be highly dependent on the degree of polymerization (DP), and thus a DP of 30 was targeted for each polymer used in this study.38,39 Polymethacrylates are known to have large zip lengths,40 meaning that once activation occurs, full depolymerization is likely to proceed.41 Therefore, selecting this relatively low DP ensures that the majority of chains will depolymerize completely upon activation. The purified polymers were subsequently subjected to identical depolymerization conditions (120 °C, 20 mM RUC, in 1,4-dioxane, Fig. 1a) and the kinetic profiles in the first hour were analyzed (Fig. 1b). As shown in Fig. 1c, the depolymerization rate clearly increases with increasing alkyl chain length (see Table S3†). For example, while in the case of PMMA approximately 24% monomer was successfully regenerated in 40 minutes, one additional carbon on the side chain led to 28% conversion for PEtMA within the same timeframe. Following a similar trend, the depolymerizations of PBuMA, PHexMA, and PLauMA resulted in 31%, 35%, and 40% regenerated monomer, respectively. This relationship appears to be linear for methacrylate monomers ranging from methyl to hexyl, but the effect diminishes beyond this point, likely because the incremental impact of additional carbons becomes less significant. By normalizing these rate constants to the 0.41 h−1 value obtained for PMMA, it can be seen that PLauMA depolymerizes ∼1.7 times faster than PMMA, clearly indicating that longer alkyl chains play a significant role in increasing depolymerization rate. Interestingly, previous studies on polymerization kinetics have shown that the propagation rate constant (kp) increases with increasing alkyl chain length.34–36 Given that polymerization and depolymerization are equilibrium-driven, one might expect the reverse trend for depolymerization. However, our results suggest that both processes follow a similar trend, highlighting an unexpected relationship between side-chain length and depolymerization efficiency.
Systematic studies of propagation rate constants for n-alkyl methacrylates have suggested that interaction or ‘pre-organization’ between the side-chains of adjacent monomer units42 could play a role in altering rate constants by bringing methacrylic groups into closer proximity and facilitating faster propagation.35 To investigate whether a similar effect influences depolymerization rates, we expanded our study beyond n-alkyl methacrylates to include polymethacrylates with ethylene glycol (EG) units in the side chain. Since the hydrophilic EG units are less likely to undergo pre-organization, this allowed us to test whether the previously observed trend in depolymerization rates was due to side-chain interactions or an inherent effect of chain length. Poly(ethylene glycol methyl ether methacrylate) (PEGMA), poly(triethylene glycol methyl ether methacrylate) (PTEGMA), and poly(polyethylene glycol methyl ether methacrylate) (PPEGMA), bearing 1, 3, or 8 EG units per side-chain were therefore synthesized (see Fig. S11–16†) and subjected to the previously optimized depolymerization conditions (Fig. 1a and e). Comparing the rates of depolymerization, as shown in Fig. 1e and f, longer side-chains increase the rate of depolymerization, with PPEGMA depolymerizing 1.34 times faster than PEGMA (Fig. 1g). Specifically, the apparent depolymerization rate constants for PEGMA, PTEGMA, and PPEGMA, were found to be 0.56, 0.64, and 0.74 h−1 respectively, indicating a similar trend to the n-alkyl methacrylate series (see Table S4†). These results suggest that side-chain interactions are not a necessary factor in determining depolymerization rates, and instead, the increase in rate is likely an intrinsic effect of side-chain length, irrespective of specific side-chain interactions.
To better understand why longer side chains lead to faster depolymerization, we next examined the underlying mechanisms governing this process. In conventional radical polymerization, under steady-state conditions, the rate of polymerization is often expressed as a product of the propagation constant (kp), the monomer concentration, and the radical concentration (determined by the initiator decomposition and termination rates).37 Therefore, if the polymerization rates of two different monomers are compared experimentally using the same conditions (i.e. same temperature, initiator, and concentrations), any difference observed is likely to highly depend on the difference in the respective propagation constants, as the radical concentration will be the same. However, in the depolymerization of RAFT-synthesized polymers, the rate-determining step has not yet been fully clarified. Depolymerization is a complex process, but can be summarized in two main steps: the activation of the polymer chain (i.e. the C–S bond cleavage to form a radical at the chain end) and the subsequent depropagation (i.e. the removal of monomer units from the polymer chain). A recent study from our group indicated that activation is driven by solvent-derived (i.e. 1,4-dioxane) radical species which react at the chain end, undergoing fragmentation to yield a depropagating polymeric radical and a solvent-derived thiocarbonylthio compound.43 Given that depropagation has been reported to be a fast process16 (albeit at higher temperatures than 120 °C), we hypothesized that the difference in rate of depolymerization that we observe could potentially originate from differences in the rate of the activation step alone.44
To test whether activation of the polymer chain is the rate determining step, we designed and performed depolymerizations of PMMA and PHexMA in the presence of a radical initiator, 1,1′-azobis(cyclohexanecarbonitrile) (ABCN, 2 eq. with respect to chain end). Our hypothesis here is that when a sufficient number of radicals is provided, any potential kinetic barrier caused by fragmentation differences at the chain end will be overcome and the corresponding depolymerization rates should become comparable. Fig. 2a shows kinetic plots of these reactions in comparison to depolymerizations in the absence of initiator (see Table S5† for detailed information). Notably, ABCN significantly accelerates the depolymerization, with 64% conversion being attained in just 10 minutes for PMMA, compared to only 6% that was previously obtained in the absence of radical initiator. Adding initiator also effectively eliminated the rate difference between PMMA and PHexMA, making their depolymerization rates nearly identical. At 120 °C, ABCN will decompose quickly, providing an abundant source of radicals to add to the chain ends and fragment to form a depropagating polymer chain, in a much more efficient manner than chain activation by the solvent-derived species. The highly similar rates of depolymerization in the presence of ABCN indicate that depropagation is a fast process at 120 °C, and chain activation is governed by the decomposition of the radical initiator. The rate-determining step of depolymerization of RAFT-synthesized polymers is therefore likely to be activation of the chain end, rather than the depropagation step itself. Importantly, these experiments also highlight that a conventional radical initiator can significantly improve the depolymerization of RAFT-synthesized polymers for a range of different polymethacrylates.
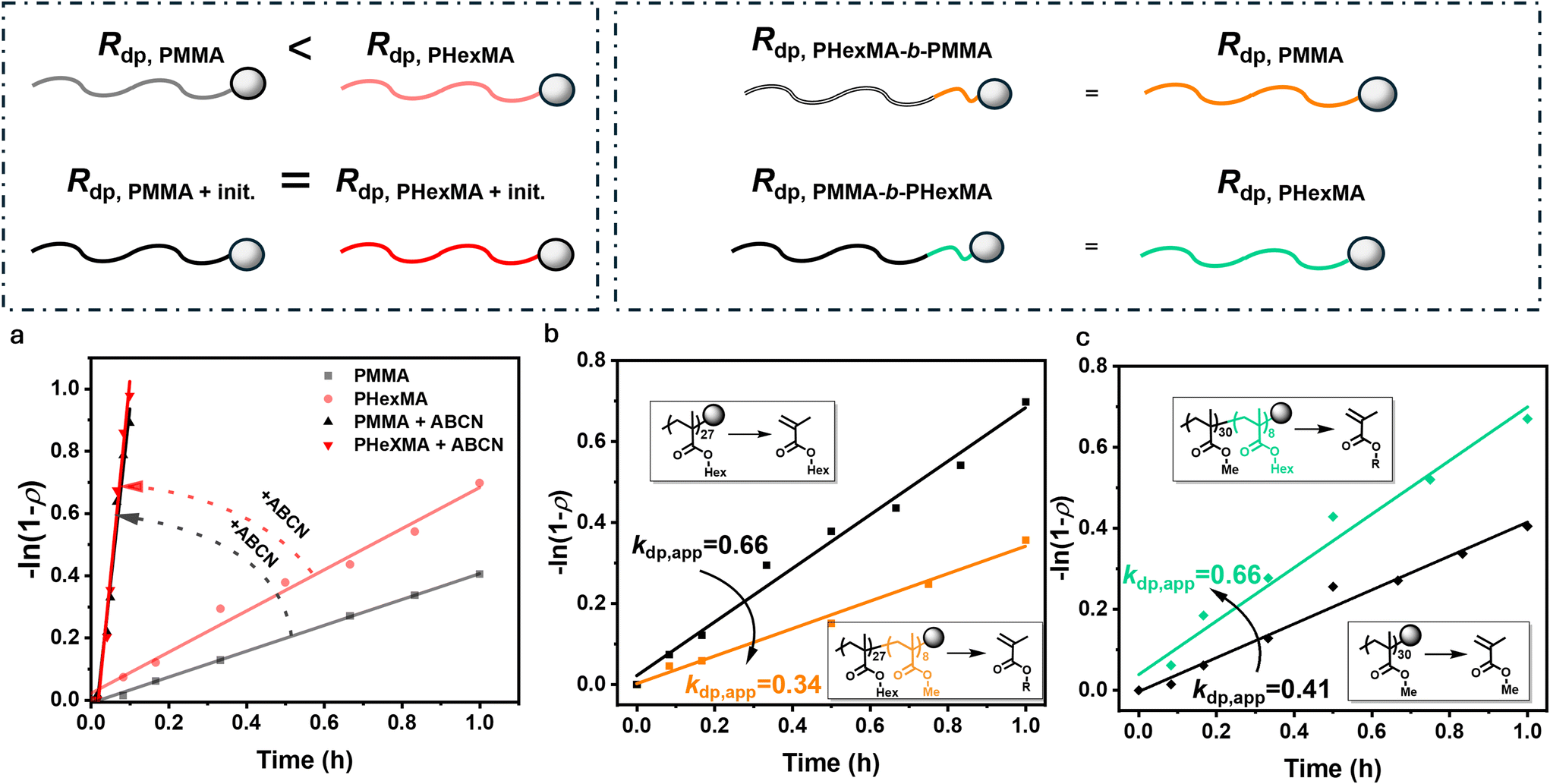 | ||
| Fig. 2 (a) Pseudo-first order plot of depolymerization kinetics of PMMA and PHexMA, compared with the analogues in the presence of 2 eq. of ABCN. (b and c) First order plot of depolymerization kinetics of block-co-polymers PHexMA-(b)-PMMA (b) and PMMA-(b)-PHexMA (c) compared to the homopolymers PHexMA and PMMA, respectively. | ||
To further explore whether chain end activation is indeed the rate-determining step, we hypothesized that the very last monomer unit, would play a significant role in the activation of the C–S bond. Comparing the steric hindrance of the side chains, a hexyl moiety is much bulkier than a methyl side-chain, and could potentially modify the energy of the C–S bond. It is widely known that sterically hindered bonds are generally characterized by lower bond energy, owing to the less effective orbital overlap, increased repulsion, and torsional strain.45,46 Since end-group modification in polymethacrylates is rather challenging, we envisioned that the incorporation of a small amount of hexyl monomer as the second block of PMMA would potentially alter the depolymerization kinetics. To realize this, we synthesized a block copolymer of PMMA-(b)-PHexMA by chain extending the PMMA-DTB species with HexMA in the presence of AIBN (see Fig. S17 and 18†). The target DP of the HexMA block was selected to be 12, and the polymerization was stopped at 68% conversion, thus corresponding to a DP of ∼8. This value was chosen according to the guidelines set out by Harrisson and coworkers who calculated the minimum block lengths required to ensure that the vast majority of chains will contain at least one PHexMA unit (see Table S8†).47 Moreover, this low DP minimizes molecular weight variations between the homopolymer and block copolymer. Interestingly, depolymerization of this block copolymer resulted in an apparent rate constant of 0.66 h−1, which is exactly the same value as the PHexMA homopolymer (Fig. 2b). This demonstrates that the terminal monomer unit of a polymer chain dictates the overall rate of depolymerization. In a similar vein, chain extending PHexMA with a short block of PMMA (Fig. S19 and 20†) lowers the overall rate of depolymerization from 0.66 to 0.34 h−1, thus nearly matching the rate of a PMMA homopolymer (Fig. 2c). Taken altogether, this work shows that the length of the side-chain significantly affects the depolymerization rate with longer side chains being responsible for faster reactions. Importantly, a wide range of polymethacrylates can be rigorously depolymerized in the presence of a radical initiator irrespective of the length of the side chain.
To support our experimental findings, we conducted computational investigations to evaluate the Gibbs free energies and reaction energies associated with the activation and depropagation steps of depolymerization. Specifically, we examined PMMA and PHexMA, which experimentally exhibited a 1.6-fold acceleration in depolymerization rate in favor of PHexMA. The initiation process involves the addition of a dioxane radical to the RAFT agent, followed by fragmentation to generate a depropagating carbon-centered radical.43 Our calculations revealed that while dioxane radical addition is similarly exergonic for both PMMA and PHexMA (Fig. S21†), fragmentation of the resulting intermediate occurs at a significantly slower rate, highlighting this step as the primary contributor to the initiation kinetics. To examine depolymerization as a whole, we compared the free energy barriers for fragmentation and depropagation in both systems (Fig. 3), and found that depropagation proceeds with nearly identical energetics for both methyl and hexyl chains, with differences falling within the error of the calculations. In contrast, fragmentation of PHexMA was found to have a considerably lower Gibbs free energy barrier (7.1 kJ mol−1 lower) and a more exergonic reaction energy (5.8 kJ mol−1 lower) than PMMA. Further insights from non-covalent interaction (NCI) analyses of the model RAFT adduct radicals (Fig. S22†) revealed a hydrogen-bonding interaction that stabilizes the PMMA-derived adduct radical but is significantly weakened in PHexMA due to steric crowding. These findings rationalize the experimentally observed acceleration in depolymerization of polymethacrylates with longer pendant groups, identify sterics as a key factor, and reinforce our conclusion that chain activation, rather than depropagation, plays a more significant role in dictating the rates of depolymerizations. While our study focuses on thermally induced RAFT depolymerization, alternative activation pathways, such as photoiniferter or photoinduced electron/energy transfer (PET) RAFT mechanisms,48 could alter the relationship between side-chain length and depolymerization rate. Under photolytic conditions, chain activation would not involve a fragmentation step but rather homolytic cleavage of the C–S bond, shifting the dependence on side-chain effects to the stability of the resultant polymeric radicals rather than fragmentation energetics.49,50 Further exploration of photochemical RAFT depolymerization will be the subject of future work.
 | ||
| Fig. 3 Gibbs free energy barriers and reaction energies (kJ mol−1, 120 °C) as calculated at the wB97X-D/aug-cc-pVTZ//M062X/6-31G(d) level of theory using SMD to model the 1,4-dioxane solvent environment. | ||
Conclusions
In conclusion, we have demonstrated that the rate-determining step in the depolymerization of RAFT-synthesized polymers is the initial fragmentation of chain ends forming propagating radicals. Crucially, we present both experimental and computational evidence to support this finding and confirm that varying depolymerization rate is not due to differences in the depropagation step itself. As a direct consequence of this, radical initiators which rapidly decompose at elevated temperatures are able to significantly enhance the rate of depolymerization as they provide an abundant and efficient source of activating radicals. Chain-extension experiments further supported our findings by showing that the depolymerization rate of a block copolymer is determined by the rate of the block which is directly attached to the thiocarbonylthio moiety. NCI analyses reveal that steric factors during the fragmentation step could be highly influential in rate determination. This work significantly enhances our understanding of the depolymerization of RAFT-synthesized polymers, and will inform the further development and optimization of depolymerization systems by tuning rates through initiator addition or chain-end modifications.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
A. A. gratefully acknowledges the ETH Zurich for financial support. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (DEPO: Grant Agreement No. 949219). M. L. C. gratefully acknowledges financial support from the Australian Research Council Centre of Excellence in Quantum Biotechnology (CE230100021) and generous allocations on the National Facility of the Australian National Computational Infrastructure.References
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- N. P. Truong, G. R. Jones, K. G. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS PubMed.
- D. A. Shipp, Polym. Rev., 2011, 51, 99–103 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- M.-N. Antonopoulou, N. P. Truong and A. Anastasaki, Chem. Sci., 2024, 15, 5019–5026 RSC.
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef CAS PubMed.
- G. R. Jones, H. S. Wang, K. Parkatzidis, R. Whitfield, N. P. Truong and A. Anastasaki, J. Am. Chem. Soc., 2023, 145, 9898–9915 CrossRef CAS PubMed.
- H. Tang, Y. Luan, L. Yang and H. Sun, Molecules, 2018, 23, 2870 CrossRef PubMed.
- B. Qin and X. Zhang, CCS Chem., 2024, 6, 297–312 CrossRef CAS.
- M. R. Martinez and K. Matyjaszewski, CCS Chem., 2022, 4, 2176–2211 CrossRef CAS.
- V. Lohmann, G. R. Jones, N. P. Truong and A. Anastasaki, Chem. Sci., 2024, 15, 832–853 RSC.
- Y. Sano, T. Konishi, M. Sawamoto and M. Ouchi, Eur. Polym. J., 2019, 120, 109181 CrossRef CAS.
- M. R. Martinez, F. De Luca Bossa, M. Olszewski and K. Matyjaszewski, Macromolecules, 2021, 55, 78–87 CrossRef.
- M. R. Martinez, S. Dadashi-Silab, F. Lorandi, Y. Zhao and K. Matyjaszewski, Macromolecules, 2021, 54, 5526–5538 CrossRef CAS.
- M. R. Martinez, D. Schild, F. De Luca Bossa and K. Matyjaszewski, Macromolecules, 2022, 55, 10590–10599 CrossRef CAS.
- K. Parkatzidis, N. P. Truong, K. Matyjaszewski and A. Anastasaki, J. Am. Chem. Soc., 2023, 145, 21146–21151 CAS.
- S. A. Mountaki, R. Whitfield, K. Parkatzidis, M.-N. Antonopoulou, N. P. Truong and A. Anastasaki, RSC Appl. Polym., 2024, 2, 275–283 CAS.
- M. J. Flanders and W. M. Gramlich, Polym. Chem., 2018, 9, 2328–2335 CAS.
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CAS.
- J. B. Young, J. I. Bowman, C. B. Eades, A. J. Wong and B. S. Sumerlin, ACS Macro Lett., 2022, 11, 1390–1395 CrossRef CAS PubMed.
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. D. A. Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Polym. Chem., 2023, 14, 253–258 RSC.
- G. Ng, S. W. Prescott, A. Postma, G. Moad, C. J. Hawker, A. Anastasaki and C. Boyer, J. Polym. Sci., 2024, 62, 3920–3928 CrossRef CAS.
- V. Bellotti, H. S. Wang, N. P. Truong, R. Simonutti and A. Anastasaki, Angew. Chem., Int. Ed., 2023, 62, e202313232 CrossRef CAS PubMed.
- H. S. Wang, K. Parkatzidis, T. Junkers, N. P. Truong and A. Anastasaki, Chem, 2024, 10, 388–401 CAS.
- F. De Luca Bossa, G. Yilmaz and K. Matyjaszewski, ACS Macro Lett., 2023, 12, 1173–1178 CrossRef CAS PubMed.
- J. B. Young, R. W. Hughes, A. M. Tamura, L. S. Bailey, K. A. Stewart and B. S. Sumerlin, Chem, 2023, 9, 2669–2682 CAS.
- R. Whitfield, G. R. Jones, N. P. Truong, L. E. Manring and A. Anastasaki, Angew. Chem., 2023, 135, e202309116 CrossRef.
- F. D. L. Bossa, G. Yilmaz, C. Gericke and K. Matyjaszewski, Eur. Polym. J., 2025, 223, 113646 CrossRef.
- M. D. Zammit, M. L. Coote, T. P. Davis and G. D. Willett, Macromolecules, 1998, 31, 955–963 CrossRef CAS.
- A. P. Haehnel, M. Schneider-Baumann, K. U. Hiltebrandt, A. M. Misske and C. Barner-Kowollik, Macromolecules, 2013, 46, 15–28 CrossRef CAS.
- R. A. Hutchinson, S. Beuermann, D. Paquet and J. McMinn, Macromolecules, 1997, 30, 3490–3493 CrossRef CAS.
- C. Barner-Kowollik, P. Vana and T. P. Davis, Handbook of radical polymerization, 2002, pp. 187–261 Search PubMed.
- N. De Alwis Watuthanthrige, R. Whitfield, S. Harrisson, N. P. Truong and A. Anastasaki, ACS Macro Lett., 2024, 13, 806–811 CrossRef CAS PubMed.
- H. S. Wang, N. P. Truong, G. R. Jones and A. Anastasaki, ACS Macro Lett., 2022, 11, 1212–1216 CrossRef CAS PubMed.
- I. Mita, K. Obata and K. Horie, Polym. J., 1990, 22, 397–410 CrossRef CAS.
- M. T. Chin, T. Yang, K. P. Quirion, C. Lian, P. Liu, J. He and T. Diao, J. Am. Chem. Soc., 2024, 146, 5786–5792 CrossRef CAS PubMed.
- M. Xiang, D. Lyu, Y. Qin, R. Chen, L. Liu and Y. Men, Polymer, 2020, 210, 123034 CrossRef CAS.
- F. Häfliger, N. P. Truong, H. S. Wang and A. Anastasaki, ACS Macro Lett., 2023, 12, 1207–1212 CrossRef PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- S. W. Benson, J. Chem. Educ., 1965, 42, 502 CrossRef CAS.
- P. Vermeeren, T. A. Hamlin and F. M. Bickelhaupt, Chem. Commun., 2021, 57, 5880–5896 RSC.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS PubMed.
- Y. Lee, C. Boyer and M. S. Kwon, Chem. Soc. Rev., 2023, 52, 3035–3097 RSC.
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS PubMed.
- M. A. Bereś, B. Zhang, T. Junkers and S. Perrier, Polym. Chem., 2024, 15, 3166–3175 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00212e |
| This journal is © The Royal Society of Chemistry 2025 |