
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular polymerization and self-assembly of transition metal complexes
Merlin R.
Stühler
ab and
Alex J.
Plajer
 *ac
*ac
aMakromolekulare Chemie, Universität Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: alex.plajer@uni-bayreuth.de
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34-36, 14195 Berlin, Germany
cBayrisches Polymer Institut (BPI), Universität Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany
First published on 21st May 2025
Abstract
Metallo-supramolecular polymers (MSPs) have emerged as a dynamic field in soft materials and nanomaterials over the past three decades. The unique interplay of ligands and metal ions plays a crucial role in defining their properties, such as binding strength and topology, making these systems distinct from their all-organic counterparts. Furthermore, the incorporation of metal centers introduces redox activity, unique photophysical properties, and electronic functionality. As a result, these systems can exhibit stimuli-responsive behavior, essential for applications in optoelectronics and biology. However, challenges remain in achieving precise control over self-assembly, morphology, and spatiotemporal responsiveness, as well as a clear rationale between coordination and self-assembly behavior. Our review, therefore, summarizes recent advances in ligand design, coordination modulation, and polymerization techniques, offering new strategies to rationally develop MSPs with tailored functionalities.
Introduction
Inspired by biological systems and building on the fundamental concepts of supramolecular chemistry, as established by Lehn, synthetic supramolecular polymers (SPs) have become a flourishing area of research over the past decades.1–10 SPs are based on directional and reversible interactions between small monomeric building blocks, such as hydrogen bonds, π–π and metallophilic interactions.11,12 One-dimensional assemblies driven by unidirectional interactions have been of particular interest for studying controlled self-assembly and structure–activity relationships, as they allow for the strategic design of monomers and the precise positioning of linking points, enabling the construction of highly defined supramolecular structures.13 Among the most studied monomers are peptides, benzene-1,3,5-tricarboxamides (BTAs), and perylene bisimide (PBI) dyes.14–19 In contrast to these purely organic systems, metallosupramolecular polymers (MSPs) have been shown to impart enhanced stability through additional metal-specific interactions, stimuli-responsiveness (e.g., redox, temperature), and structural versatility.20–22 The inclusion and variation of counterions within charged systems further affect the assembly behavior, contributing to dynamicity and increased variability in interactions.23 A number of metal ions, coordination motifs, and ligand classes have been explored, and prior reviews have discussed these with regard to aggregation, the assembly pathway, and morphology control.24–29 The following sections highlight recent developments in MSPs with particular focus on how coordination chemistry, i.e the roles of the ligand and metal ion and their interaction between monomers influences and controls assembly behavior, morphology, and stimuli responsiveness. The discussion is sectioned by ligand classes as presented in Fig. 1. | ||
| Fig. 1 Schematic representation of the ligand classes discussed in this mini-review. | ||
Monodentate ligands (L2MX2)
Current examples of metal-containing monomers comprising monodentate ligands are mostly based on square-planar complexes of heavy d8 metals, namely Pt(II) and Pd(II), with pyridine-derived N-donor ligands, as these form particularly stable complexes with predictable geometries due to their strong σ-bond donating and π-accepting nature. Additionally, pyridine-derived ligands inherently provide a flat, rigid, and aromatic scaffold for π-stacking interactions. To further enhance interactions between monomers, hydrogen bonding groups (e.g., amides) or extended π-systems are often synthetically attached. However, other ligand motifs, such as acetylides, were also employed in the past.30Within the class of metallosupramolecular polymers, the group of Fernández has contributed many inspiring contributions, starting with a report on the cooperative supramolecular polymerization of a charge-neutral, square-planar, oligophenyleneethynylene (OPE)-based bispyridyldichlorido Pd(II) complex 1A in methylcyclohexane, as shown in Fig. 2a.31 In addition to the pronounced π–π stacking enabled by the rigid aromatic ligands, density functional theory (DFT) calculations revealed strong Pd–Cl interactions stabilizing the pseudo-parallel aggregation while some intermetallic σ-bonding interaction between the dz2 orbitals of the central Pd(II) atoms of neighbouring molecules was observed as well (left inlay of Fig. 2a). A fully parallel-stacked conformation with close metallophilic Pd–Pd contacts is ruled out by the energy gain from two Pd–Cl bonds formed per monomer addition and is further hindered by the steric demand of the out-of-plane chlorine co-ligands. Follow-up works introduced the heavier analogue Pt(II) with similar ligand motifs 1B, revealing a shift in the aggregation mode from pseudo-parallel stacks to a slipped conformation (left inlay of Fig. 2a).32 This shift was driven by cooperative π–π, C–H⋯Cl, and π(alkyne)–Pt interactions and involve σ- and π-electron donation from the alkyne moiety to Pt(II), as well as δ- and π-back donations (right inlay of Fig. 2a). However, secondary Van der Waals (VdW) interactions of the aliphatic C12 chains turned out to be the determining factor as the same Pt complex equipped with only methyl groups formed parallel stacks again. Furthermore, exchanging the aliphatic C12 chains by tetraethylene glycol chains reversed the preferential packing modes for Pd (1C)33 and Pt (1D)34 as well. As there are numerous competing non-covalent interactions in these systems, the formation of both the thermodynamically favored pseudo-parallel and the kinetically favored slipped aggregation modes was furthermore found to be, among other factors, dependent on the solvent,35 and the halogen co-ligands36 for Pd and Pt. Therefore, the final stacking geometry of a particular system is defined by the interplay of the individual energetic contributions of various interaction types, with metallophilic interactions being a factor but not the determining one.
 | ||
Fig. 2 (a) Pt(II) (1A,C) and Pd(II) (1B,D) complexes comprising different solubilizing chains as factors leading to the competition of a slipped and a parallel molecular packing. Left inlay: DFT-optimized geometry of a dimer of 1A with electron density of the HOMO−2 (B3LYP/6-31G*/LANL2DZ level). Reproduced from ref. 31 with permission from American Chemical Society, copyright 2013. Right inlay: PM6 geometry-optimized structure of a dimer of 1B and 1C. Copyright 2016 Wiley. (b) Molecular structures of the trans and cis isomers of 2, and a schematic representation of the supramolecular assembly of 2 triggered by coordination isomerism. Inlay of dispersion-corrected PM6 optimized hexameric stack of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of trans and cis isomers of 2. Reproduced from ref. 37 with permission from Wiley, copyright 2019. :1 mixture of trans and cis isomers of 2. Reproduced from ref. 37 with permission from Wiley, copyright 2019. | ||
In a related study, the group of Fernández has successfully utilized the UV-triggered geometric isomerism in square-planar Pt(II) complexes 2, as shown in Fig. 2b.37 For this purpose, they employed azobenzene moieties, as they promote conformational freedom through minimal steric repulsion in the cis-conformation, achieving an optimal balance between isomerization and aggregation ability. The 1-dimensional assembly of the trans conformers showed a slipped aggregation stabilized by aromatic and N–H⋯Cl interactions without direct participation of the Pt centers. Hence, the coordination isomerism that develops over time could act orthogonally, without affecting the stacking interactions. In a 2:1 mixture of trans- and cis-complexes, the latter would function as end-capping units. While partially interacting with the preceding monomer, the cis conformers prevent propagation by interrupting the alkyl chain shell around the aromatic units. Upon UV light irradiation, all cis conformers could be converted back into trans conformers, allowing elongation to resume and enabling the formation of long fibers once again. Therefore, the inherent coordination isomerism could be utilized to regulate supramolecular polymerization.
Studies by Li and coworkers, employing comparable monodentate pyridine-based ligands equipped with solubilizing units and amide linkers, took a closer look at the differences in the bonding behavior of Pd(II) and Pt(II) (Fig. 3).38 While, once again, VdW interactions, π–π stacking, and hydrogen bonding assist the stacking, the difference in assembly could be directly attributed to the metal employed, as it was the only variable that was changed. Absorption spectroscopy, wide-angle X-ray diffraction (WAXD), and extended X-ray absorption fine structure (EXAFS) experiments revealed a higher propensity for aggregation of Pt(II) (3A) than Pd(II) (3B), as indicated by shorter intermetallic bonds. A follow-up study showed how the metal centers could even be brought closer together through a shift from H- to J-type aggregates by changing the amide linker in the ligands to ester bonds.39
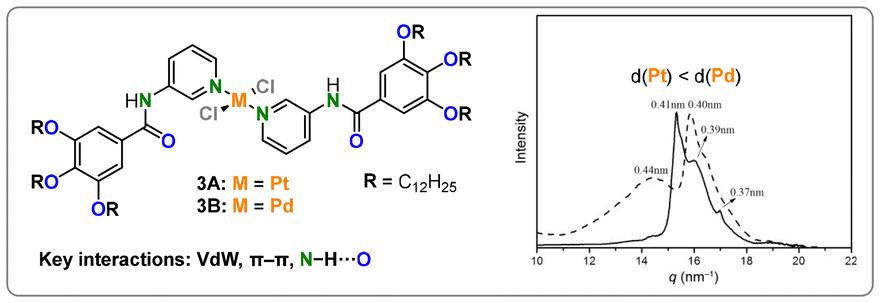 | ||
| Fig. 3 Structure and WAXD spectra of 3A (—) and 3B (- - -), normalized intensity was used. Reproduced from ref. 38 with permission from Wiley, copyright 2014. | ||
The utilization of pyridyl-based ligands for supramolecular polymers, however, is not limited to late d8 metals, as nicely demonstrated in a recent study by the group of Fu.40 They reported the dynamic chirality inversion of different types of MSPs formed from Ag+ (AgNO3) (4A) and Al3+ (AlCl3) (4B) complexes of pyridyl-conjugated cholesterol (PVPCC) as presented in Fig. 4. Additionally, a whole range of metal ions (Co2+, Cu2+, Zn2+, Mn2+, Ni2+, Bi3+; chlorine salts) could be employed to form gels under ultrasonication as well as thermal treatment in p-xylene/n-butanol while only Bi3+ and Zn2+ exhibited chirality inversion, similar to Ag+ and Al3+, respectively. They found the MSP of the Ag(I)/PVPCC complex 4A to exhibit a negative cotton effect in the circular dichroism (CD) spectra after ultrasonication and a positive CD absorption after thermal treatment, indicating a chirality inversion. For the kinetically controlled aggregation mode of 4A (Ag-SP I) after sonication, a single crystal structure was obtained, revealing a slipped stack featuring Ag–π coordination, as well as π–π interactions, VdW forces, and hydrogen bonding (upper inlay Fig. 4). After thermal treatment, the Ag-SP II, driven by metallophilic Ag–Ag interactions and a strongly positive CD signal, was obtained. In fact, they observed that after cooling to room temperature, Ag-SP I was first formed and subsequently transformed into Ag-SP II. This transformation was found to be dependent on the cooling rate; slow cooling (0.5–1 K min−1) led to higher-order assemblies of Ag-SP I, slowing down the conversion to the thermodynamically favored Ag-SP II (Fig. 4).
 | ||
| Fig. 4 Structure of PVPCC ligand, schematic representation of the supramolecular assembly of Ag(I)/PVPCC complex 4A with inlay (top) of solid-state structure of 4A and schematic representation of the assembly of Al(III)/PVPCC complex 4B with inlay (bottom) of the morphologic evolution of 4B monitored by SEM. Reproduced from ref. 40 with permission from Springer Nature, copyright 2024. | ||
In the second part of the study, the consecutive chirality inversion pathway of the differently behaving 4B (Al(III)/PVPCC) assembly in p-xylene/EtOH was investigated, as summarized in the bottom inlay of Fig. 4. Here again, two different aggregation modes were found, with Al-SP I being formed after mixing the starting materials and solvents. Al-SP I was found to have a micron-sheet structure by scanning electron microscopy (SEM) (bottom inlay Fig. 4). After heating to 100 °C, the first aggregated state underwent a chirality inversion to form Al-SP II, which exhibits a nanotube structure; this inversion was found to reverse over a period of 9 hours. It turned out that in Al-SP II, EtOH molecules are coordinated to the Al(III) metal, which detach upon heating, enabling the formation of Al-SP I, and re-coordinate after cooling to room temperature. Lastly, the circularly polarized luminescence (CPL) of the complexes was investigated and exemplarily employed for dynamic information encryption in a co-assembled system of Ag(I)/PVPCC and the fluorescent dye thioflavin T (ThT). The latter was needed to improve the CPL performance. Although this was only a proof of concept, it showcases the potential of dynamic chirality inversion promoted by coordinative interactions in assembled systems for applications in information encryption.
The group of Zang has developed supramolecular polymers based on metallophilic interactions in a heterobimetallic Au(I)–Cu(I) system (Fig. 5).41 They employed cationic N-heterocyclic carbene–Au(I) complexes bearing amino acid residues. The alanine-derived complex 5A (Au(Ala)2) exhibited chiral nanofibrous structures with P- and M-helicity for the D- and L-isomers, respectively, as observed by SEM and CD spectroscopy (Fig. 5a). Upon the addition of [CuI2]− anions (using K+@[18-Crown-6] as the counter cation), however, co-assemblies were formed via Au–Cu interactions, leading to a chirality inversion and a change in the nanoarchitectures from lamellar to columnar chiral packing as determined by X-ray diffraction experiments. The valine-derived complex 5B (Au(Val)2), bearing isopropyl groups, on the other hand, formed achiral stacked nanosheets that transformed into twisted chiral nanoribbons upon the addition of [CuI2]− anions (Fig. 5c and d). Lastly, the phenylalanine-derived complex 5C (Au(Phe)2) showed achiral fibers that only transformed into achiral rods upon the addition of [CuI2]− anions (Fig. 5e and f). Thus, the large sterically hindered amino acid residue failed to generate morphological supramolecular chirality. Overall, the geometry and steric demand of the ligands drastically influence the ability of these systems to assemble into helical superstructures. Computational studies on the involved interactions revealed a combination of metallophilic Au–Cu contacts, typical anion–cation d10–d10 binding energies, and N–H⋯I hydrogen bonds between the amino acid moieties and iodine ligands. Furthermore, the Au–Cu interactions enhanced the CPL of the complexes, displaying emission lifetimes ranging from hundreds to thousands of nanoseconds. This study, therefore, demonstrates the effective use of metallophilic interactions in heterobimetallic systems to modulate morphology and induce intriguing photophysical properties.
 | ||
| Fig. 5 Structure of 5A (Au(Ala)2), 5B (Au(Val)2) and 5C (Au(Phe)2) and respective SEM images before (a, c and e) and after (b, d and f) [CuI2]− addition. Inlay of schematic representation of the helicity inversion of 5C from Individual self-assembly to co-assembly with columnar chiral packing induced by [CuI2]−. Reproduced from ref. 41 with permission from American Chemical Society, copyright 2023. | ||
Bidentate ligands (LMX2)
Transitioning from monodentate to bidentate ligands, the group of Fernández has contributed several interesting reports regarding Pt(II) bipyridine complexes in recent years.42–45 Building on their previously introduced monodentate OPE ligand system in bispyridyldichlorido Pt(II) complexes, a structurally similar but covalently connected bipyridine ligand was introduced (6A) to investigate the structure–property relationships in terms of coordination geometry changes (Fig. 6a).42 While the monodentate ligands (6B) were oriented in a “linear” trans-conformation, the “V-shaped” bipyridine motif enforced a cis-conformation. Both monomers were assembled in methylcyclohexane. While the linear monomer was known to form slipped aggregates without direct metallophilic Pt–Pt interactions due to the out-of-plane arrangement of the chlorine co-ligands at the Pt(II) center, the V-shaped bipyridine-based system was found to assemble into nearly parallel H-type aggregates with short intermetallic Pt–Pt contacts, as determined by UV-Vis and fluorescence spectroscopy studies, where characteristic metal–metal-to-ligand charge-transfer (MMLCT) bands were observed. The predetermined planarity of the system ultimately led to an altered aggregation energy landscape, as it reduced the energy penalty for nucleation, with dimers or small oligomers resembling an on-pathway intermediate for subsequent polymerization. Lastly, the different packing modes caused distinct morphologies: the V-shaped monomer formed flexible fibers, while the linear one formed poorly solvated bundles of stiff fibers (see atomic force microscopy (AFM) images Fig. 6a).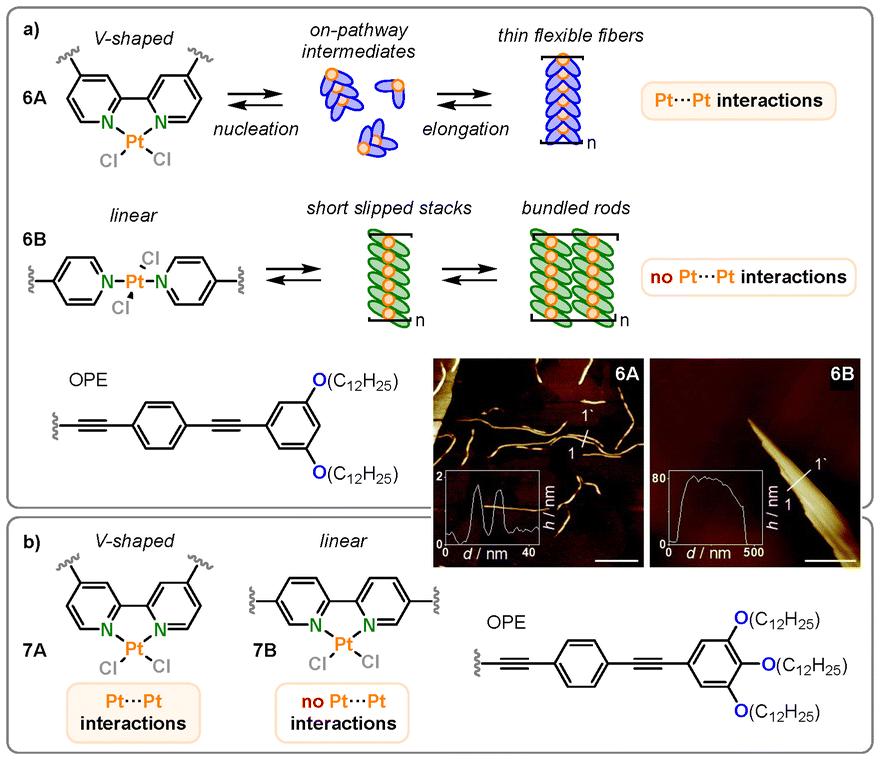 | ||
| Fig. 6 (a) Structures and AFM height images on HOPG of the V-shaped (6A) and linear (6B) Pt(II) OPE complexes and schematic representation of their supramolecular polymerization in MCH. Reproduced from ref. 42 with permission from Royal Society of Chemistry, copyright 2021. (b) Structures of the bipyridine-based V-shaped (7A) and linear (7B) Pt(II) OPE complexes. Reproduced from ref. 45 with permission from American Chemical Society, copyright 2023. | ||
In follow-up works, related ligands were employed for assembly in organic solvents as well as in aqueous media.43–45 They compared bipyridine-based ligands where the position of the π-extended OPE units differed, but the Pt(II) remained in a cis-conformation at all times (7A, 7B; Fig. 6b). Again, they found that in all cases, only the V-shaped complexes formed assemblies through metallophilic interactions, while the linear monomers did not show any Pt–Pt contacts. Combined, these studies revealed that, ultimately, the shape of the ligand (V-shaped vs. linear) determines the stacking mode rather than the coordination isomerism of the metal center.
A rather unusual structural motif was recently reported by the group of Freire, where methoxy-substituted phenylacetylene units were assembled into 1-dimensional helical fibers by the addition of Ag(I) ions 8 (Fig. 7).46 The Ag(I) ions are chelated by the terminal alkyne and methoxy units, and are located between the planes of the rigid aromatic phenylacetylene, thus adding coordinative Ag–OMe and Ag–π interactions, as well as argentophilic Ag–Ag interactions, to the ligand-centered π–π stacking (inlay of Fig. 7). Combined, these interactions led to stable aggregation in chloroform. As the monomers lack any solubilizing chains, these aggregates would be considered crystalline materials rather than supramolecular polymers. However, by using (R)-sulfoxide units with stereogenic sulfur centers as an unconventional source of chirality in the helical assembly, fibrous aggregates with a uniform M-helicity were successfully induced (Fig. 7). Interestingly, when the Ag(I)/ligand ratio exceeded 1:1, a second binding event was observed, where a second Ag(I) ion was chelated by the sulfoxide group. This enabled cross-linking, resulting in interconnected chiral fibers leading to a 3-dimensional network structure (see SEM image Fig. 7) and macroscopic gelation. Accordingly, Ag(I) can be regarded as a functional element capable of both connecting individual ligands and cross-linking the assemblies through various coordinative interactions.
 | ||
| Fig. 7 Structure of chiral phenylacetylene monomer 8 with AFM height image of 8 (1.0/0.6 ligand/Ag+ molar ratio), SEM image of 8 (1.0/4.0 ligand/Ag+ molar ratio) and inlay of Ag(I) complex 8 as well as the aggregation model for 8 showing the involved interactions and M chiral helix. Reproduced from ref. 46 with permission from Wiley, copyright 2024. | ||
Tridentate ligands (LMX)
When considering tridentate ligands applied in supramolecular assembly, one predominantly finds pyridine-based pincer ligands in combination with d8 transition metals (Pd(II), Pt(II)), as these form highly stable complexes and give rise to interesting photophysical properties through extended metallophilic interactions. Supramolecular polymers based on metallophilic interactions of Rh(II) complexes with N-donor pincer ligands are also known.47 An additional co-ligand provides the opportunity to easily attach solubilizing or tethering units. As nitrogen-based tridentate pincer ligands have been long known and widely investigated, it is not surprising that a variety of different ligands have been employed, ranging from (cyclometallated) terpyridine48 to bzimpy (2,6-bis(benzimidazol-2′-yl)pyridine),49 2,6-di(triazol)pyridine,50 and 2,6-di(tetrazol)pyridine.51Impressive work by the group of Che serves as a good example of the versatility that such heavy metal-containing monomers can offer.23 They synthesized a series of cationic pincer-type Pd(II) and Pt(II)-isocyanide complexes with different weakly coordinating counterions ([PF6]−, [ClO4]− and [OTf]−) and found that these complexes underwent supramolecular polymerization upon the addition of water to acetonitrile solutions, resulting in the formation of two distinct aggregation modes (Fig. 8a). The kinetic product was found to be a stack of J-aggregated dimers (Agg1) with close contacts to the counterions (9A,B; inlay Fig. 8a). With time or when seeds—pre-formed nucleating templates—were added, the kinetic barrier of loosening the contact with the respective counterion led to the transition into the thermodynamic aggregation mode (Agg2), where direct metallophilic interactions formed a linear (M–M)n chain (9C,D; inlay Fig. 8a). Interestingly, while the formation of Agg1 followed an isodesmic polymerization model, Agg2 formed via a cooperative mechanism. Since both Pt(II) and Pd(II) are metals with a d8 electron configuration that can form intermetallic Pt–Pd bonds, the authors could readily co-assemble both monomers into (multi-)block polymers, as shown in Fig. 8b. This was done by adding, for instance, seeds of Pt(II) in the Agg2 state to Pd(II) monomers, which led to the living elongation of the seeds at both ends. Using this technique, polymers with up to seven blocks were obtained, as visualized by confocal microscopy. They also found that at increased temperatures, multidimensional seeds were formed, leading to multidimensional block polymers.
 | ||
| Fig. 8 (a) Structure of M-1-X and M-2-X pincer-type Pd(II) and Pt(II)-isocyanide complexes with inlay of solid state structures of Pd-1-OTf 9A, Pd-1-PF69B, Pd-2-PF69C and Pt-2-PF69D. Hydrogen atoms are omitted for clarity. (b) Schematic representation of the preparation of (multi-)block PtII and PdII supramolecular copolymers and confocal images of one- and multidimensional block polymers in the merged channel. (c) Emission color of [Pt·Pd] co-assemblies (center), Pd-2-ClO4 (right) and Pt-1-PF6 assemblies (left) in the Agg2. Reproduced from ref. 23 with permission from Elsevier, copyright 2020. | ||
This class of complexes is known to exhibit delocalized metal–metal-to-ligand charge transfer (3MMLCT) transitions upon aggregation.23 However, while the pure aggregates of Pd(II) and Pt(II) in Agg2 showed only weak phosphorescence, doping a solution of Pd(II) monomers with 0.5–3% Pt(II) monomers led to a strong increase in emission efficiency (from ∼3% to >70%) upon aggregation (Fig. 8c). This phenomenon can be explained by external heavy-atom spin–orbit coupling from the Pt centres. The Pt 5dz2 and Pd 4dz2 orbitals can mix due to their identical symmetry, which allows for a significant overlap that results in substantial involvement of Pt(II) orbitals in the frontier orbitals. Consequently, it was proposed that the external Pt-atom perturbation facilitates the radiative decay pathway of phosphorescence in the Pd assemblies, considerably enhancing the emission efficiency. With this, the work nicely demonstrates the targeted combination and utilization of structurally similar ligand motifs, coordinatively similar metals, and energetically controlled aggregation modes to form (multi-)block and multidimensional copolymers and coassemblies with enhanced emission performance.
A very recent report by the group of Jung demonstrates how the incorporation of secondary metals and ligands can drastically determine the assembly pathway and morphology.52 They prepared a chiral monoalkynylplatinum(II) diterpyridine complex 10 with one terpyridine site remaining unoccupied, leaving room for the coordination of secondary metals, as shown in Fig. 9. The monometallic compound assembled into nanoparticles (SP-I, step (a) Fig. 9), driven by its amphiphilic structure, based solely on π–π interactions and H-bonding as the kinetic product. However, when Ag+ ions were introduced, metallophilic Pt–Pt and Ag–Ag interactions were found, leading to the formation of one-dimensional left-handed fibers (SP-II, step (b) Fig. 9). The removal of Ag+ with NaCl did not lead back to SP-I but rather to a third aggregation mode, SP-III, where intermetallic Pt–Pt interactions were even more pronounced. Thus, introducing metallophilic interactions through reversible Ag+ additions overcomes a kinetic barrier, enabling the formation of the thermodynamic product of the monometallic 10.
 | ||
| Fig. 9 Structure of 10 and the secondary ligands (bpy and DA18C6) as well as the proposed formation mechanism of SPs from 10 in the (a) absence and (b and c) presence of secondary metal ions (Ag+ and Fe2+) and (d and e) partial chemical structures of the 10 complexes with (d) Ag+ and (e) Fe2+ ions obtained by DFT calculation. Reproduced from ref. 52 with permission from Royal Society of Chemistry, copyright 2024. | ||
Having proven the concept, Fe3+ ions were employed, leading to the formation of a third type of left-handed helical fibers, SP-IV (step (c) Fig. 9). These could also be obtained from an on-pathway mechanism by directly exchanging Ag+ ions in SP-II with Fe3+ ions. Mechanistic studies using the photoluminescence intensities of the respective characteristic bands revealed an isodesmic polymerization mechanism for the π–π-dominated monometallic SP-I, while SP-II and SP-IV showed cooperative behavior correlated to the dominating metallophilic Pt–Pt/Ag–Ag interactions. Lastly, competing ligands bpy (4,4′-dimethyl-2,2′-bipyridine) or DA18C6 (1,14-diaza-18-crown-6) were introduced, which did not result in morphological changes but rather caused a lag time in the polymerization kinetics of SP-II and SP-IV due to the competitive complexation of the secondary metal ions. Altogether, the structural incorporation of a second binding pocket enabled the formation of four distinct aggregation modes, guided by the coordination of different secondary metals, which could be leveraged to navigate through the energy landscape of self-assembly (see SP-I to SP-III).
Tetradentate ligands (LM)
Undoubtedly, the most widely studied tetradentate ligands in metal-containing monomers for supramolecular polymers are metalloporphyrins, as they provide a rigid planar ligand scaffold with an expanded π-system. In addition to π–π interactions facilitated by the porphyrin scaffold, the central forces connecting supramolecular stacks of (metallo)porphyrins often involve additional hydrogen bonding units, such as amides, or coordinative interactions with the central metal ion. A recent review by the group of Lee on porphyrin-based supramolecular polymers has summarized the various classes of polymers reported.53 It becomes evident that whenever the porphyrin units are connected through coordinative interactions, this is achieved by linking coordinating ligands, mostly pyridine derivatives, to the porphyrin scaffold, which then axially bind to the metal center of another monomer, as shown in Fig. 10.54 Therefore, the monomers do not stack vertically but are laterally connected. The number of free binding sites depends on the metal ion employed, as cobalt(II), for example, prefers a hexagonal coordination and can polymerize with two additional axial ligands, while Zn(II) forms a pentacoordinate geometry and polymerizes with one axial ligand.54–57 In contrast, d9 Cu(II) complexes exhibit only weak coordination to axial ligands. | ||
| Fig. 10 Schematic representation of exemplary supramolecular polymer formation through hexagonal coordination (e.g. Co(II)) or square pyramidal coordination (e.g. Zn(II)). | ||
When the bridging ligand is directly attached to the porphyrin ligand, the π-systems can still come into contact, and a slipped J-aggregated stacking mode is observed. This naturally occurs in metallochlorin dyes in bacteriochlorophylls, where hydroxy groups form coordinative bridges between the magnesium(II) porphyrin units.58
An inspiring example of the functional use of the metal center in metalloporphyrin-based supramolecular polymers has been reported by the group of Sugiyasu.59 First, zinc(II) porphyrins 11A were polymerized into fibers and subsequently depolymerized upon the addition of 4-dimethylaminopyridine (DMAP) as an external base axially coordinating to the zinc(II) center and blocking aggregation (Fig. 11a).60 When structurally identical copper(II) porphyrin monomers 11B were added to seeds of the zinc(II) polymers, however, the depolymerization pathway was blocked via the formation of end-caps. Tetragonally planar coordinated copper(II) does not show binding affinity to axial ligands, as its d9 electronic configuration leads to strong ligand binding in the equatorial plane due to a single vacancy in the 3dx2−y2 orbitals, limiting the availability of electrons for axial coordination.61 Thereby, they were able to synthesize stabilized, end-capped triblock polymers.
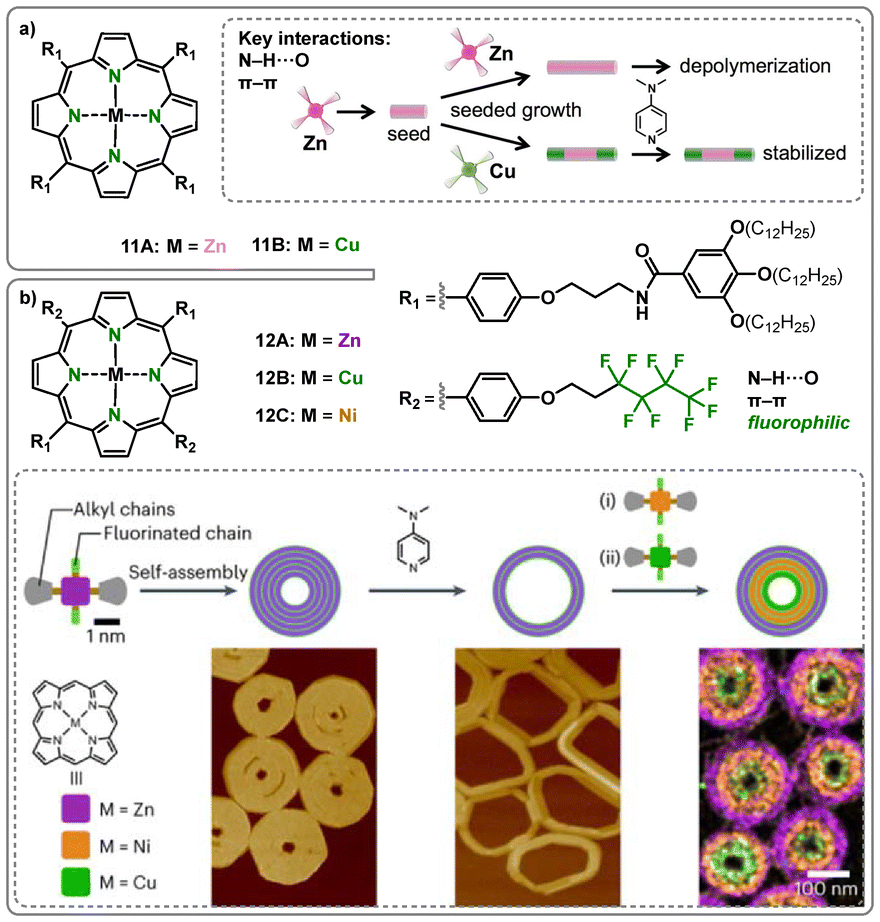 | ||
| Fig. 11 (a) Structure of Zn(II) 11A and Cu(II) 11B porphyrin-based monomers and inlay with a schematic representation of the (de-)polymerization of 11A and copolymer formation with 11B blocking depolymerization. Reproduced from ref. 60 with permission from American Chemical Society, copyright 2018. (b) Structure of Zn(II) 12A, Cu(II) 12B and Ni(II) 12C porphyrin-based monomers and inlay with a synthetic scheme towards the triblock SCTs together with the respective AFM images and energy dispersive X-ray spectroscopy in scanning transmission electron microscopy (STEM-EDS) image. Reproduced from ref. 63 with permission from Springer Nature, copyright 2023. | ||
In a more recent study, this phenomenon was extended to another class of metalloporphyrins bearing two aliphatic and two shorter fluorinated side chains.62 These zinc(II) porphyrins 12A were found to assemble into double-stranded Archimedean spirals and concentric toroids (Fig. 11b). In addition to the one-dimensional H-aggregations through hydrogen bonds and π–π stacking, the two-dimensional assembly was enabled by fluorine-specific interactions of the fluorinated chains, which represented nucleation sites for the next ring-layer. Furthermore, structurally similar monomers comprising copper(II) (12B) and nickel(II) (12C) could be added to form di- and triblock supramolecular concentric toroids (SCT).63 It was found that the addition of a block occurred preferentially on the inside of the structure due to a higher density of fluorinated chains on the concave site. Leveraging this phenomenon, the susceptibility of the zinc(II) porphyrins to disaggregation through DMAP was used for controlled partial depolymerization of the concentric toroids and subsequent block polymer formation on the inside of the structures. Thus, the metal-specific depolymerizability of the Zn(II) porphyrins could be utilized in an orthogonal fashion to the interactions building the SCTs enabling up to triblock SCTs as an exceptional 2-dimensional morphological motif.
Another well-established tetradentate ligand in coordination chemistry is the Salphen Schiff base ligand, which, in contrast to porphyrins, remains less explored in the field of supramolecular polymerisation. The group of Yam recently reported supramolecular polymers formed from amphiphilic platinum(II) Salphen complexes in DMSO/water mixtures.64,65 In this system, the square-planar coordination geometry of Pt(II), combined with the planar chelating bisphenoxy-imine ligand, promotes directed cooperative self-assembly through metallophilic Pt–Pt and π–π stacking interactions.
Building on this concept, the group of Vives developed a transient dissipative system in which assembly is controlled by the presence or absence of zinc(II) ions (Fig. 12).21 They synthesized terpyridine-linked Pt(II) Salphen complexes 13 (terpy(Pt-Salphen)2) equipped with solubilizing aliphatic chains that function as molecular tweezers (Fig. 12). In these tweezers, the terpyridine unit adopts a ‘W’-shaped open conformation due to repulsion between nitrogen lone pairs. This fully planar form was found to self-assemble into a fibrillar network through one-dimensional stacking, driven by metallophilic and π–π interactions in aromatic solvents (i.e. benzene and toluene). Upon coordination of zinc(II) (ZnCl2) 13 transitions into a closed ‘U’-shaped conformation. The resulting loss of planarity and reduced π–π interaction surface disrupts its aggregation ability. Remarkably, this transformation is reversible upon the addition of tris(2-aminoethyl)amine (tren), which preferentially coordinates with zinc(II). This enabled the development of a multicomponent dissipative system in which 2-cyano-2-phenylpropanoic acid (Fig. 12, inlay)—an acid that decomposes over time via a decarboxylation reaction in the presence of a base—temporarily modulates the pH. The resulting protonation of the acid-sensitive [Zn(Tren)]Cl2 complex releases zinc(II) ions, triggering disassembly within approximately 30 minutes, followed by reassembly over the course of 10 hours. This study highlights the potential of molecular tweezers as stimuli-responsive gelators via metal based supramolecular interactions in a dissipative system.
 | ||
| Fig. 12 Structure of terpy(Pt-Salphen)2-based tweezers 13 and schematic representation of sol–gel transitions through the mechanical switching of the molecular tweezers. Inlay of transient disassembly and reassembly controlled by the chemical fuel 2-cyano-2-phenylpropanoic acid in a multicomponent system composed of tweezers 13 and [Zn(Tren)]Cl2 in toluene. Reproduced from ref. 21 with permission from American Chemical Society, copyright 2025. | ||
Earlier studies reporting supramolecular assemblies of Salphen complexes, however, employ zinc(II), as it tends to form pentacoordinate structures when bound to square-planar ligands, thereby enabling coordinative supramolecular assemblies.66,67 Furthermore the Zn(II) Salphen system offers an exceptionally straightforward synthesis, and the absence of ligand-field stabilization in the d10 electron configuration of zinc(II) allows for highly flexible coordination geometries, accommodating coordination numbers of four—preferentially five—and even a weak sixth coordination, known as spodium bonding.68–70 Within the pseudo-planar geometry of the Salphen bisphenoxyimine ligand, however, the Zn(II) ion remains in a coordinatively unsaturated state.66 This results in pronounced Lewis acidity, which is typically satisfied by either the association of an external Lewis base or dimerization through intermolecular Zn–O interactions between the Zn(II) centre of one and a phenoxy group of another complex.71,72 However, the formation of linear polymeric structures through (ZnO)n motifs has also been reported.73–76 The group of MacLachlan has first described fibrous aggregated of Zn(II) Salphen complexes, in which aliphatic chains had been attached to the backbone of the Salphen ligand to ensure solubility during supramolecular polymerization (14, Fig. 13a).73,74 Their study demonstrated that the primary interaction driving the assembly was the Zn–O bond rather than π–π interactions, as the respective Ni(II) complex did not form aggregates, and bulky tert-butyl groups, as well as replacement of phenoxy groups with thiolates, disrupted the assembly. A few subsequent studies of related Zn(II) Salphen complexes described similar structural motifs forming fibrous aggregates.75–78
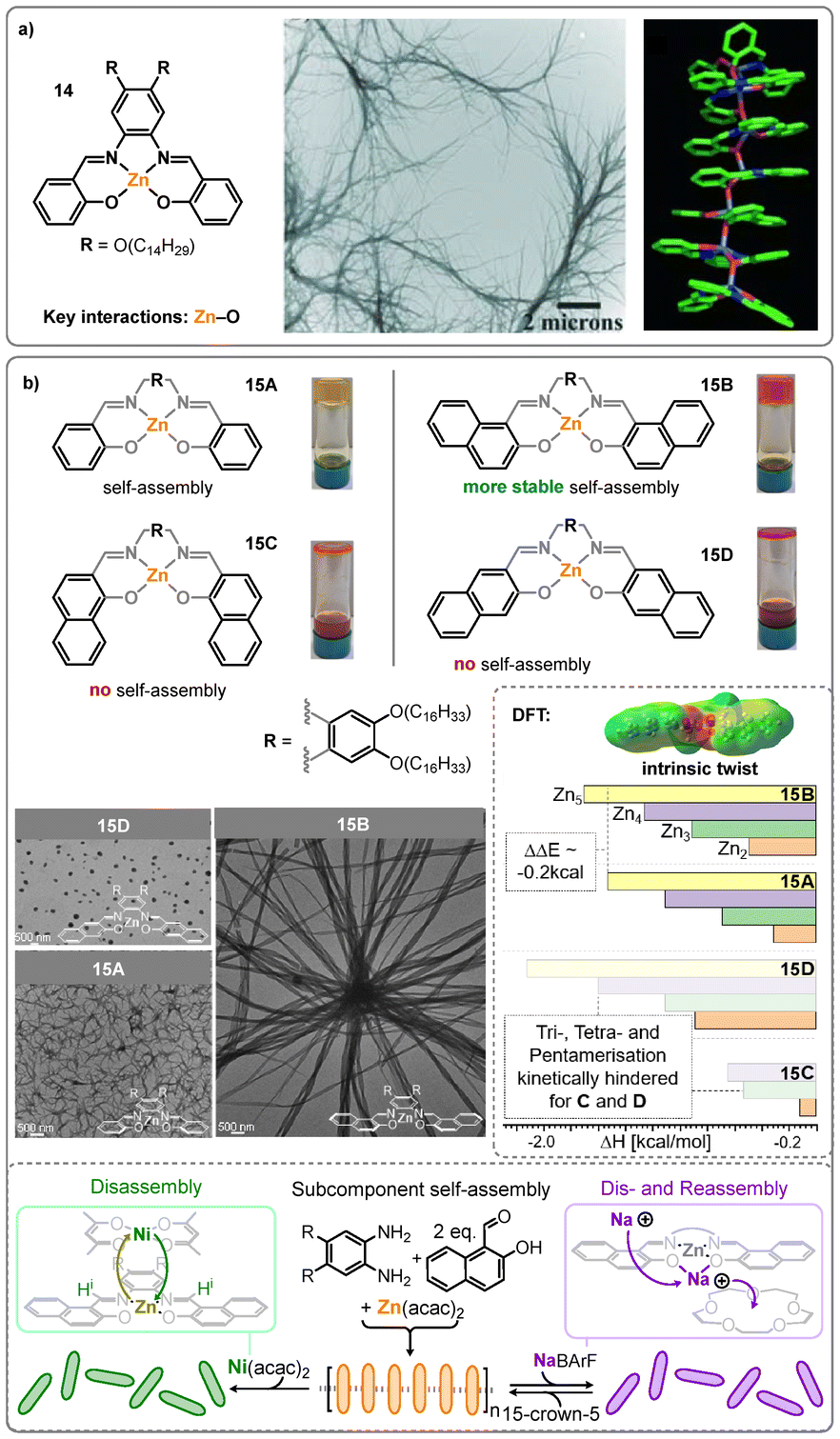 | ||
| Fig. 13 (a) Structure of Zn(II) Salphen complex 14, TEM image and PM3 optimized heptameric structure of 14 (alkyl chains excluded). Reproduced from ref. 73 with permission from Wiley, copyright 2007. (b) Structures and corresponding photographs of gels/solutions of 15A–D in toluene as well as TEM images of 15A, 15B, and 15D deposited from THF solution. Upper inlay of DFT results showing intrinsically twisted structure of 15B and dimerization and oligomerization enthalpies; faded bars correspond to optimizations that required manual adjustment as outlined in the publication. Bottom inlay with schematic representation of subcomponent self-assembly in toluene as well as coordination-induced dis- and reassembly. Reproduced from ref. 79 with permission from Wiley, copyright 2025. | ||
In a recent study by our group, the central Zn(II) Salphen motif was functionalized with π-extended naphthyl units in different orientations (15B–D; see Fig. 13b) and compared to the parent Zn(II) Salphen complex 15A.79 While the aggregation of complexes 15C and 15D was fully suppressed, complex 15B exhibited pronounced self-assembly into large fibrous aggregates, as determined by atomic force microscopy (AFM), transmission electron microscopy (TEM), and electron tomography (ET). A comprehensive analysis of the polymerization process—employing techniques such as variable-temperature NMR and static wide-angle X-ray scattering (WAXS)—revealed simultaneous aggregation of individual monomers into one-dimensional stacks and three-dimensional packing of these fibers into bundles with diameters of several hundred nanometers. This process commenced at a nucleation temperature of approximately 71–75 °C, significantly higher than that of the parent complex 15A (52–57 °C). Computational DFT studies indicated that the enhanced stability of the aggregates formed by 15B is due to an intrinsic twist in the monomeric complex (see upper inlay of Fig. 13b). This twisted conformation was found to be essential for providing the spatial arrangement required for supramolecular polymer formation via Zn–O bonds. As a result, the energy penalty associated with deforming from a planar monomer geometry (as seen in 15A) is offset, increasing the overall energy gain upon assembly. In contrast, although 15D can theoretically form even more thermodynamically stable polymers, its polymerization is kinetically hindered by a high energy barrier associated with torsional deformation. The same behavior was observed for complex 15C. As these complexes can be synthesized through a simple one-pot imine condensation reaction in the presence of a Zn(II) ion, it was possible to achieve assembly directly from the subcomponents in toluene (see bottom inlay of Fig. 13b). Additionally, the flexible coordination environment of the Zn(II) ion allowed for transmetallation with Ni(II), resulting in disassembly, as evidenced by liquefaction and complete loss of fluorescence intensity. Disassembly was also induced by the addition of sodium ions, which exhibit labile coordination and could be sequestered by 15-crown-5, enabling fully reversible disassembly.
Notable contributions came from the group of Kleij, who enhanced the strength of the assembly by introducing two coordinative Zn–O bonds in bis-Zn(II) Salphen complexes (16A, Fig. 14a).20 Their work revealed highly stable 1-dimensional stacks in both solution and the solid phase, which exhibited complete inertness towards dissociation via addition of external coordinating bases such pyridine. This stability arose from monomer association constants being orders of magnitude higher than those for pyridine coordination. Scanning tunneling microscopy (STM) enabled the study of a single layer of the assembled structure on highly ordered pyrolytic graphite (HOPG) and 1,2,4-trichlorobenzene (TCB)/THF 95:5 (v/v), revealing extended domains of long lamellar stacks of 16A. Additionally, the respective Ni(II) complex 16B was investigated, which did not form intermolecular assemblies but was found adsorbed with its extended conjugated planes parallel to the graphite surface.
A distinct class of laterally connected bis-Zn(II) Salphen complexes (17) was found to form unique morphologies, including nanosized rings interconnected by large nanorods (Fig. 14b).80 These structures mimic a biological neural network and hold potential for use in nanoelectronic circuits for transmitting electrical signals. Unlike the previously discussed assemblies, these structures were mainly driven by vdW and π–π interactions as the respective Ni(II) derivatives showed a similar aggregation behavior. Together, the studies reveal the potential of the Salphen ligand system to form versatile morphologies, primarily through zinc-centered interactions, offering a platform for responsiveness (i.e., switchable aggregation) via coordinative flexibility.
 | ||
| Fig. 14 (a) Structure of bis-Zn(II) and Ni(II) Salphen complexes 16A–B and STM topography of 16A on HOPG–TCB interface. Reproduced from ref. 20 with permission from American Chemical Society, copyright 2012. (b) Structure of bis-Zn(II) Salphen complex 17 and TEM image (scale bar: 5 μm). Reproduced from ref. 80 with permission from Springer Nature, copyright 2013. | ||
A recent study from the group of Adhikari reports a class of supramolecular polymers built from Cu(I) guanosine (G)-quadruplexes 18 (G4·Cu+), as shown in Fig. 15.22 Hoogsteen-type hydrogen-bonded cyclic tetramers of guanosine (G-quartets), stabilized by a central metal ion and π–π stacking, represent a non-canonical secondary structure commonly found in telomeric DNA.81,82 However, their assemblies and gels are typically labile due to weak tetramer–tetramer interactions. By introducing in situ generated Cu(I) from Cu(OAc)2via the addition of ascorbic acid (Asc) (with an overall ratio of G:Cu2+:Asc = 1:0.75:0.56), robust self-assemblies in water were achieved, exhibiting tolerance to dilution (viscous up to [18] = 10−4 M), temperature (≥95 °C), and time (≥36 h). The stability arises from cuprophilic interactions between the Cu(I) cores, assisted by π–π stacking of the guanosine units. The Cu(I) ions are coordinated through C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Cu+ interactions with the carbonyl oxygens of the four guanosine units connected through hydrogen bonding thereby defining a tetradentate ligand plane. A combination of TEM, UV-Vis, and CD spectroscopy revealed that polymer formation occurred within the first 15 minutes of mixing the subcomponents, independent of temperature, while macroscopic gelation required 24 hours at room temperature due to cross-linking of the one-dimensional fibers caused by partial dehydration. This gelation process could be accelerated to 8 hours when conducted at 50 °C. At temperatures above 50 °C, the supramolecular polymers underwent an irreversible phase transition from a transparent gel to a cloudy solution, with a lower critical solution temperature (LCST) of 56 °C. This thermoresponsive behavior is attributed to the spontaneous dehydration of the polymer chains, resulting from the disruption of hydrogen bonds between water molecules and the peripheral hydroxyl groups of the guanosine units. Furthermore, the redox responsiveness of the system was demonstrated by oxidizing Cu(I) to Cu(II) via the addition of H2O2, leading to the formation of a green solution and complete disassembly as the driving cuprophilic interactions are specific to Cu(I). This process could subsequently be reversed by adding ascorbic acid, which restored gelation. Altogether, the incorporation of Cu(I) and the resulting cuprophilic interactions led to enhanced stability compared to other, e.g., K+ (G)-quadruplexes, along with unusual LCST behavior and redox responsiveness.
O⋯Cu+ interactions with the carbonyl oxygens of the four guanosine units connected through hydrogen bonding thereby defining a tetradentate ligand plane. A combination of TEM, UV-Vis, and CD spectroscopy revealed that polymer formation occurred within the first 15 minutes of mixing the subcomponents, independent of temperature, while macroscopic gelation required 24 hours at room temperature due to cross-linking of the one-dimensional fibers caused by partial dehydration. This gelation process could be accelerated to 8 hours when conducted at 50 °C. At temperatures above 50 °C, the supramolecular polymers underwent an irreversible phase transition from a transparent gel to a cloudy solution, with a lower critical solution temperature (LCST) of 56 °C. This thermoresponsive behavior is attributed to the spontaneous dehydration of the polymer chains, resulting from the disruption of hydrogen bonds between water molecules and the peripheral hydroxyl groups of the guanosine units. Furthermore, the redox responsiveness of the system was demonstrated by oxidizing Cu(I) to Cu(II) via the addition of H2O2, leading to the formation of a green solution and complete disassembly as the driving cuprophilic interactions are specific to Cu(I). This process could subsequently be reversed by adding ascorbic acid, which restored gelation. Altogether, the incorporation of Cu(I) and the resulting cuprophilic interactions led to enhanced stability compared to other, e.g., K+ (G)-quadruplexes, along with unusual LCST behavior and redox responsiveness.
 | ||
| Fig. 15 Structure of Cu(I) guanosine (G)-quadruplex 18 and inlay of its supramolecular polymer showing a thermoresponsive LCST transition and redox responsive reversible depolymerization. Reproduced from ref. 22 with permission from Wiley, copyright 2025. | ||
Conclusions and outlook
The field of metallosupramolecular polymers (MSPs) has seen significant advancements, driven by the interplay between metal based and ligand based intermolecular interactions. Unlike purely organic supramolecular polymers, MSPs exhibit unique properties such as redox activity, optoelectronic functionality, and stimuli-responsiveness, which arise from the incorporation of metal centers. These characteristics make MSPs highly versatile, enabling dynamic control over self-assembly, morphology, and emission behavior. The reviewed studies illustrate the diverse range of ligands (i.e., monodentate, bidentate, tridentate, and tetradentate) each contributing to distinct assembly behaviors, from one-dimensional fibers to multidimensional block copolymers and gel networks. The inherent dynamic nature of the metal–ligand bonds, in conjunction with secondary interactions like π–π stacking, hydrogen bonding, and metallophilic contacts, was not only found to enhance stability and stimuli-responsiveness (light, temperature) but also enables sophisticated behaviors such as chirality inversion, living polymerization, and reversible (de)polymerization.These unique attributes position metallosupramolecular assemblies as versatile platforms for tuning optical, electronic, and mechanical properties at the nanoscale. Accordingly, potential applications of MSPs span multiple disciplines. In optoelectronics, MSPs can serve as tunable light-emitting materials (i.e., CP-OLEDs),40,83 sensors,66,84 and conductive nanowires85 due to their inherent charge transport properties and metal-centered luminescence. In the biomedical field, MSPs hold promise for drug delivery,86 bioimaging,87 and responsive hydrogels,88 provided that biocompatible metals and aqueous-stable assemblies are employed. Catalysis is another promising avenue, where MSPs could act as self-assembling catalytic scaffolds, enhancing reaction selectivity and efficiency through spatial control of active sites.89,90 Additionally, their ability to undergo reversible transitions upon external stimuli (light, pH, redox conditions) makes them ideal candidates for smart materials, including molecular switches,91 adaptive coatings and films,92,93 and data encryption systems.94,95 To fully harness these applications, future research must address key challenges such as enhancing the robustness of self-assembly, improving processability, and developing scalable synthetic routes. Ultimately, continued innovation in MSP design will lead to next-generation materials that bridge fundamental supramolecular and coordination chemistry with advanced functional applications.
Data availability
This contribution does not contain any new data.Conflicts of interest
There are no conflicts to declare.References
- T. Aida, E. Meijer and S. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed
.
- J. S. Moore, Curr. Opin. Colloid Interface Sci., 1999, 4, 108–116 Search PubMed
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef
-
J.-M. Lehn, in Supramol. Chem, VCH, Weinheim New York, 1995 Search PubMed
- C. Fouquey, J. M. Lehn and A. M. Levelut, Adv. Mater., 1990, 2, 254–257 CrossRef CAS
- T. Aida, A. Takemura, M. Fuse and S. Inoue, J. Chem. Soc., Chem. Commun., 1988, 391–393 RSC
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. K. Hirschberg, R. F. Lange, J. K. Lowe and E. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed
- D. van der Zwaag, T. F. de Greef and E. Meijer, Angew. Chem., Int. Ed, 2015, 54, 8334–8336 CrossRef CAS PubMed
- R. D. Mukhopadhyay and A. Ajayaghosh, Science, 2015, 349, 241–242 CrossRef CAS PubMed
- L. Brunsveld, B. J. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed
- H.-Q. Peng, W. Zhu, W.-J. Guo, Q. Li, S. Ma, C. Bucher, B. Liu, X. Ji, F. Huang and J. L. Sessler, Prog. Polym. Sci., 2023, 137, 101635 CrossRef CAS
- T. F. de Greef and E. Meijer, Nature, 2008, 453, 171–173 CrossRef CAS PubMed
- D. Mandal, A. N. Shirazi and K. Parang, Org. Biomol. Chem., 2014, 12, 3544–3561 RSC
- N. J. Sinha, M. G. Langenstein, D. J. Pochan, C. J. Kloxin and J. G. Saven, Chem. Rev., 2021, 121, 13915–13935 CrossRef CAS PubMed
- C. Kulkarni, E. Meijer and A. R. Palmans, Acc. Chem. Res., 2017, 50, 1928–1936 CrossRef CAS PubMed
- F. V. Gruschwitz, T. Klein, M. T. Kuchenbrod, N. Moriyama, S. Fujii, I. Nischang, S. Hoeppener, K. Sakurai, U. S. Schubert and J. C. Brendel, ACS Macro Lett., 2021, 10, 837–843 CrossRef CAS PubMed
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed
- G. Ghosh, P. Dey and S. Ghosh, Chem. Commun., 2020, 56, 6757–6769 RSC
- G. Salassa, M. J. Coenen, S. J. Wezenberg, B. L. Hendriksen, S. Speller, J. A. Elemans and A. W. Kleij, J. Am. Chem. Soc., 2012, 134, 7186–7192 CrossRef CAS PubMed
- P. Msellem, G. Groslambert, L. Miton, M. Pomes-Hadda, N. J. Van Zee, C. Guibert and G. Vives, J. Am. Chem. Soc., 2025, 147, 5360–5367 CrossRef CAS PubMed
- N. Sahu, C. Guchhait, I. Mohanta, V. Suriyaa and B. Adhikari, Angew. Chem., 2025, 137, e202417508 CrossRef
- Q. Wan, W.-P. To, X. Chang and C.-M. Che, Chem, 2020, 6, 945–967 CAS
- N. Bäumer, J. Matern and G. Fernández, Chem. Sci., 2021, 12, 12248–12265 RSC
- R. D. Mukhopadhyay and A. Ajayaghosh, Chem. Soc. Rev., 2023, 52, 8635–8650 RSC
- S. Ma, J. Xu, S. Sohrabi and J. Zhang, J. Mater. Chem. A, 2023, 11, 11572–11606 RSC
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed
- G. Ghosh, T. Ghosh and G. Fernández, ChemPlusChem, 2020, 85, 1022–1033 Search PubMed
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS
- Y.-J. Tian, E. Meijer and F. Wang, Chem. Commun., 2013, 49, 9197–9199 RSC
- M. J. Mayoral, C. Rest, V. Stepanenko, J. Schellheimer, R. Q. Albuquerque and G. Fernández, J. Am. Chem. Soc., 2013, 135, 2148–2151 CrossRef CAS PubMed
- N. K. Allampally, M. J. Mayoral, S. Chansai, M. C. Lagunas, C. Hardacre, V. Stepanenko, R. Q. Albuquerque and G. Fernández, Chem. – Eur. J., 2016, 22, 7810–7816 CrossRef CAS PubMed
- C. Rest, A. Martin, V. Stepanenko, N. K. Allampally, D. Schmidt and G. Fernández, Chem. Commun., 2014, 50, 13366–13369 RSC
- C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700–705 Search PubMed
- J. P. Coelho, J. Matern, R. Q. Albuquerque and G. Fernández, Chem. – Eur. J., 2019, 25, 8960–8964 CrossRef CAS PubMed
- J. Matern, N. Bäumer and G. Fernández, J. Am. Chem. Soc., 2021, 143, 7164–7175 CrossRef CAS PubMed
- N. Bäumer, K. K. Kartha, N. K. Allampally, S. Yagai, R. Q. Albuquerque and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 15626–15630 CrossRef PubMed
- M. Chen, C. Wei, J. Tao, X. Wu, N. Huang, G. Zhang and L. Li, Chem. – Eur. J., 2014, 20, 2812–2818 CrossRef CAS PubMed
- M. Chen, C. Wei, X. Wu, M. Khan, N. Huang, G. Zhang and L. Li, Chem. – Eur. J., 2015, 21, 4213–4217 CrossRef CAS PubMed
- K. Fu, Y. Zhao and G. Liu, Nat. Commun., 2024, 15, 9571 CrossRef CAS PubMed
- Y.-J. Wang, X.-Y. Shi, P. Xing and S.-Q. Zang, JACS Au, 2023, 3, 565–574 CrossRef CAS PubMed
- N. Bäumer, K. K. Kartha, S. Buss, I. Maisuls, J. P. Palakkal, C. A. Strassert and G. Fernández, Chem. Sci., 2021, 12, 5236–5245 RSC
- P. Sutar, I. Maisuls, Z. Fernández, C. A. Strassert and G. Fernández, Chem. – Eur. J., 2024, 30, e202403287 CrossRef CAS PubMed
- N. Bäumer, K. K. Kartha, S. Buss, J. P. Palakkal, C. A. Strassert and G. Fernández, Org. Chem. Front., 2021, 8, 4138–4143 RSC
- P. Sutar, T. Dünnebacke, Z. Fernández, T. Krüger, C. Rest and G. Fernández, Precis. Chem., 2023, 1, 332–340 CrossRef CAS
- F. Rey-Tarrío, S. Simón-Fuente, J. M. Cuerva, D. Miguel, M. Ribagorda, E. Quiñoá and F. Freire, Angew. Chem., 2024, 136, e202318454 CrossRef
- A. K.-W. Chan, D. Wu, K. M.-C. Wong and V. W.-W. Yam, Inorg. Chem., 2016, 55, 3685–3691 CrossRef CAS PubMed
- Z. Gao, P. A. Korevaar, R. Zhong, Z. Wu and F. Wang, Chem. Commun., 2018, 54, 9857–9860 RSC
- C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136–12143 CrossRef CAS PubMed
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10 CrossRef CAS PubMed
- M. E. Robinson, D. J. Lunn, A. Nazemi, G. R. Whittell, L. De Cola and I. Manners, Chem. Commun., 2015, 51, 15921–15924 RSC
- M. Kim, H. Choi, M. Kim, S. Kim, S. Yun, E. Lee, J. Cho, S. H. Jung and J. H. Jung, Chem. Sci., 2024, 15, 19729–19738 RSC
- H. Lee, H. Park, D. Y. Ryu and W.-D. Jang, Chem. Soc. Rev., 2023, 52, 1947–1974 RSC
- U. Michelsen and C. A. Hunter, Angew. Chem., 2000, 112, 780–783 CrossRef
- A. Satake, O. Shoji and Y. Kobuke, J. Organomet. Chem., 2007, 692, 635–644 CrossRef CAS
- A. Satake, Y. Suzuki, M. Sugimoto and Y. Kuramochi, Chem. – Eur. J., 2020, 26, 669–684 CrossRef CAS PubMed
- F. Gutzeit, M. Dommaschk, N. Levin, A. Buchholz, E. Schaub, W. Plass, C. Näther and R. Herges, Inorg. Chem., 2019, 58, 12542–12546 CrossRef CAS PubMed
- S. Ogi, C. Grzeszkiewicz and F. Würthner, Chem. Sci., 2018, 9, 2768–2773 RSC
- S. H. Jung, D. Bochicchio, G. M. Pavan, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2018, 140, 10570–10577 CrossRef CAS PubMed
- F. Helmich, C. C. Lee, M. M. Nieuwenhuizen, J. C. Gielen, P. C. Christianen, A. Larsen, G. Fytas, P. Leclère, A. Schenning and E. Meijer, Angew. Chem., Int. Ed., 2010, 49, 3939–3942 CrossRef CAS PubMed
- J. Cremers, S. Richert, D. V. Kondratuk, T. D. Claridge, C. R. Timmel and H. L. Anderson, Chem. Sci., 2016, 7, 6961–6968 RSC
- N. Sasaki, M. F. Mabesoone, J. Kikkawa, T. Fukui, N. Shioya, T. Shimoaka, T. Hasegawa, H. Takagi, R. Haruki and N. Shimizu, Nat. Commun., 2020, 11, 3578 CrossRef PubMed
- N. Sasaki, J. Kikkawa, Y. Ishii, T. Uchihashi, H. Imamura, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2023, 15, 922–929 CrossRef CAS PubMed
- H. Zhang, M. H.-Y. Chan, J. Lam, Z. Chen, M.-Y. Leung, E. K.-H. Wong, L. Wu and V. W.-W. Yam, Chem. Sci., 2024, 15, 8545–8556 RSC
- H. Zhang, M. H.-Y. Chan, J. Lam, M.-Y. Leung, L. Wu and V. W.-W. Yam, Org. Chem. Front., 2025, 12, 1733–1747 RSC
- I. P. Oliveri and S. Di Bella, Chemistry, 2023, 5, 119–137 CrossRef CAS
- A. W. Kleij, Dalton Trans., 2009, 4635–4639 RSC
- A. Singer and D. Atwood, Inorg. Chim. Acta, 1998, 277, 157–162 CrossRef CAS
- A. Erxleben, Coord. Chem. Rev., 2003, 246, 203–228 CrossRef CAS
- R. M. Gomila, A. Bauzá, T. J. Mooibroek and A. Frontera, CrystEngComm, 2021, 23, 3084–3093 Search PubMed
- X. Song, H. Yu, X. Yan, Y. Zhang, Y. Miao, K. Ye and Y. Wang, Dalton Trans., 2018, 47, 6146–6155 Search PubMed
- X. Yan, X. Song, X. Mu and Y. Wang, New J. Chem., 2019, 43, 15886–15891 Search PubMed
- J. K. H. Hui, Z. Yu and M. J. MacLachlan, Angew. Chem., Int. Ed., 2007, 46, 7980–7983 CrossRef CAS PubMed
- J. K.-H. Hui and M. J. MacLachlan, Dalton Trans., 2010, 39, 7310–7319 RSC
- I. P. Oliveri and G. Malandrino, Dalton Trans., 2014, 43, 10208–10214 Search PubMed
- M. Piccinno, C. A. Angulo-Pachón, P. Ballester, B. Escuder and A. Dalla Cort, RSC Adv., 2016, 6, 57306–57309 RSC
- I. P. Oliveri, S. Failla, G. Malandrino and S. Di Bella, J. Phys. Chem. C, 2013, 117, 15335–15341 CrossRef CAS
- J. K. H. Hui, Z. Yu, T. Mirfakhrai and M. J. MacLachlan, Chem. – Eur. J., 2009, 15, 13456–13465 CrossRef CAS PubMed
- M. R. Stühler, H. Makki, D. Silbernagl, M. Dimde, K. Ludwig, B. E. Tegner, C. Greve, K. Rausch, E. M. Herzig, A. Köhler and A. J. Plajer, Adv. Funct. Mater., 2025 Search PubMed
- M. V. Escárcega-Bobadilla, G. A. Zelada-Guillén, S. V. Pyrlin, M. Wegrzyn, M. M. Ramos, E. Giménez, A. Stewart, G. Maier and A. W. Kleij, Nat. Commun., 2013, 4, 2648 CrossRef PubMed
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed
- L. Stefan and D. Monchaud, Nat. Rev. Chem., 2019, 3, 650–668 CrossRef
- X. S. Zhong, L. Yuan, X. J. Liao, J. J. Hu, S. Xing, S. Q. Song, J. Q. Xi and Y. X. Zheng, Adv. Mater., 2024, 36, 2311857 Search PubMed
- M. E. Germain and M. J. Knapp, J. Am. Chem. Soc., 2008, 130, 5422–5423 Search PubMed
- X. Zhang, F. Gärisch, Z. Chen, Y. Hu, Z. Wang, Y. Wang, L. Xie, J. Chen, J. Li and J. V. Barth, Nat. Commun., 2022, 13, 442 Search PubMed
- X. Zhang, J. Zhuo, D. Wang and X. Zhu, Chem. – Eur. J., 2025, e202404617 CrossRef CAS PubMed
- Y. Hai, J.-J. Chen, P. Zhao, H. Lv, Y. Yu, P. Xu and J.-L. Zhang, Chem. Commun., 2011, 47, 2435–2437 RSC
- J. A. Sánchez-Fernández, Polymers, 2023, 15, 1365 CrossRef PubMed
- L. N. Neumann, M. B. Baker, C. M. Leenders, I. K. Voets, R. P. Lafleur, A. R. Palmans and E. Meijer, Org. Biomol. Chem., 2015, 13, 7711–7719 RSC
- Z.-J. Gong, Y. S. Narayana, Y.-C. Lin, W.-H. Huang, W.-N. Su, Y.-P. Li, M. Higuchi and W.-Y. Yu, Appl. Catal., B, 2022, 312, 121383 Search PubMed
- F. Xu and B. L. Feringa, Adv. Mater., 2023, 35, 2204413 CrossRef CAS PubMed
- S. Mondal, D. C. Santra, S. Roy, Y. S. Narayana, T. Yoshida, Y. Ninomiya and M. Higuchi, ACS Appl. Mater. Interfaces, 2023, 15, 42912–42919 CrossRef CAS PubMed
- M. Higuchi, J. Mater. Chem. C, 2014, 2, 9331–9341 RSC
- H. Yang, S. Li, J. Zheng, G. Chen, W. Wang, Y. Miao, N. Zhu, Y. Cong and J. Fu, Adv. Mater., 2023, 35, 2301300 CrossRef CAS PubMed
- K. Fu, D.-H. Qu and G. Liu, J. Am. Chem. Soc., 2024, 146, 33832–33844 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |