
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotothermally-driven cobalt catalysed atom transfer radical polymerisation enables isocyanide copolymer synthesis†
Cristina
Preston-Herrera
and
Erin E.
Stache
 *
*
Department of Chemistry, Princeton University, Princeton, New Jersey 08544, USA. E-mail: estache@princeton.edu
First published on 30th May 2025
Abstract
Design and synthesis of degradable polymers is a burgeoning area of research to combat plastic waste buildup. Here, a cobalt-catalysed atom transfer radical polymerisation (ATRP) turned on by photothermal conversion is used to synthesize photodegradable non-alternating acrylate-isocyanide copolymers. A simple one-step protocol enables control over copolymerisation with good chain-end fidelity under mild conditions. Although fundamentally a ground state reaction, we demonstrate temporal control via a unique localized heating strategy. Lastly, we demonstrate the advantage of this polymerisation compared to more traditional ATRP protocols.
Introduction
As the rate of plastic production continues to rise, it is necessary to develop synthetic methods to lessen the future accumulation of these materials.1–4 With end-of-life considerations in mind, current methods have focused on synthesizing photodegradable polymers via copolymerisation with carbon monoxide (CO) to enable environmental photodegradation via Norrish-type cleavages.5–11 However, this method is limited to copolymerisation with ethylene due to the rate of decarbonylation with more polar monomers.6,12–15 Commercially available isocyanides have shown to be a worthy C1 alternative for synthesizing photodegradable copolymers.13,16–18Our group first reported using isocyanides as comonomers to synthesize photodegradable copolymers via cobalt-mediated radical polymerisation (CMRP).12 The use of cobalt hydride (CoIII-H) enabled rapid initiation of polymerisation, bypassing the standard induction period in conventional CMRP.19–21 Elimination of the induction period was essential to maximize copolymerisation efficiency while minimizing isocyanide ligation to CoII-Salen.12 However, a carefully timed addition of isocyanide to the polymerisation mixture was necessary to achieve control over the reaction and prevent unwanted side reactivity. This multi-step procedure is further limited by stoichiometric amounts of cobalt and low monomer conversions (Scheme 1 top). While this method was a key step toward photodegradable polymers, its limitations in reaction efficiency hinder the potential for adoption in polymer synthesis. Thus, highlighting the need for improved strategies to access photodegradable isocyanide copolymers.
 | ||
| Scheme 1 Comparison of previous work in cobalt catalysed isocyanide copolymerisation (top) and this work (bottom). | ||
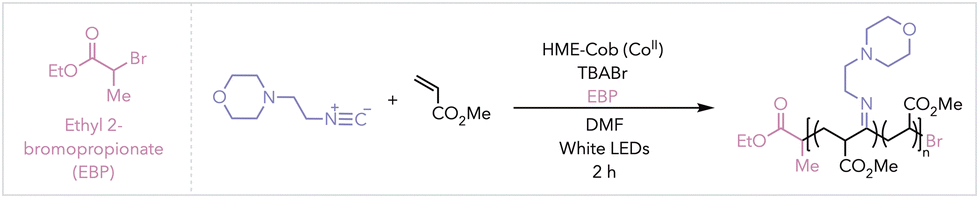
Atom transfer radical polymerisation (ATRP) is an alternative method as it is extensively used in controlled radical polymerisations (CRP).22,23 Recently, we reported using heptamethyl ester cobyrinate (HME-Cob) as a competent cobalt catalyst for ATRP.24 This vitamin B12 derivative was thermally activated to facilitate the reversible halogen exchange of growing polymer chains using 5 mol% of catalyst. Lower concentrations of HME-Cob (down to 10 ppm) were achieved under photothermal conditions.25
Photothermal conditions also gave access to temporal control, where HME-Cob converted light into heat while maintaining an overall ambient solution temperature. Once the light is removed, the localized heating at the catalyst surface dissipates, eliminating the temperature gradient and halting polymerization—thereby affording temporal control.25–28 Given these conditions, we hypothesized that using HME-Cob would enable a simplified method for isocyanide copolymer synthesis. Low catalyst concentrations and one less open coordination site than Co-salen, used in CMRP, would prevent isocyanide ligation, therefore allowing for higher conversion of monomer all in a single step addition. Under light activation, HME-Cob can also be leveraged to enable temporal control over the copolymerisation (Scheme 1 bottom).
Results and discussion
To determine if HME-Cob could be an effective catalyst in the presence of isocyanides, we applied our previously optimised photothermal conditions to a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 feed ratio of methyl acrylate (MA) to p-toluenesulfonylmethyl isocyanide (TOSMIC). Excitingly, MA conversion reached 67%, with TOSMIC conversion at 19%, resulting in a copolymer containing 3.1% isocyanide incorporation (Fig. 1A, entry 1). Under an identical feed ratio, CMRP protocols resulted in only 37% MA conversion due to termination or catalyst deactivation by TOSMIC with similar incorporation.12 In the absence of photoinitiator phenyl-bis(2,4,6-trimethylbenzoyl)phosphine oxide (BAPO), low monomer conversions, broader dispersity, and lower initiator efficiencies are seen (Fig. 1A entry 3). In line with our earlier findings, catalyst regeneration plays a critical role in maintaining copolymerization control and achieving high molecular weights in ATRP.24,25,27,29,30 We proceeded to investigate the copolymerisation of MA with TOSMIC by targeting various degrees of polymerisation (DP). Using a 9 to 1 MA to isocyanide feed ratio, incorporations of TOSMIC ranged from 2.4 to 3.1%, slightly decreasing at higher DPs (Fig. 1B). Gratifyingly, higher molecular weights were achieved by increasing the DPs up to 23.4 kg mol−1 (Fig. 1C). However, the dispersity (Đ) with TOSMIC remained high, with Đ > 1.50 (Fig. 1D).
:1 feed ratio of methyl acrylate (MA) to p-toluenesulfonylmethyl isocyanide (TOSMIC). Excitingly, MA conversion reached 67%, with TOSMIC conversion at 19%, resulting in a copolymer containing 3.1% isocyanide incorporation (Fig. 1A, entry 1). Under an identical feed ratio, CMRP protocols resulted in only 37% MA conversion due to termination or catalyst deactivation by TOSMIC with similar incorporation.12 In the absence of photoinitiator phenyl-bis(2,4,6-trimethylbenzoyl)phosphine oxide (BAPO), low monomer conversions, broader dispersity, and lower initiator efficiencies are seen (Fig. 1A entry 3). In line with our earlier findings, catalyst regeneration plays a critical role in maintaining copolymerization control and achieving high molecular weights in ATRP.24,25,27,29,30 We proceeded to investigate the copolymerisation of MA with TOSMIC by targeting various degrees of polymerisation (DP). Using a 9 to 1 MA to isocyanide feed ratio, incorporations of TOSMIC ranged from 2.4 to 3.1%, slightly decreasing at higher DPs (Fig. 1B). Gratifyingly, higher molecular weights were achieved by increasing the DPs up to 23.4 kg mol−1 (Fig. 1C). However, the dispersity (Đ) with TOSMIC remained high, with Đ > 1.50 (Fig. 1D).
 | ||
| Fig. 1 (A) Polymerisation conditions and results for photothermal copolymerisation of methyl acrylate (MA) with p-toluenesulfonylmethyl isocyanide or 2-morpholinoethyl isocyanide (MEI) with and without phenyl-bis(2,4,6-trimethylbenzoyl)phosphine oxide (BAPO). (B) Chart showing isocyanide incorporation for a 9 to 1 ratio of methyl acrylate to TOSMIC (blue data) or MEI (lavender data) at different degrees of polymerisation (DP). (C) Chart showing molecular weight for copolymers made with TOSMIC (blue data) and MEI (lavender data). (D) Dispersity results over different DPs of TOSMIC (blue data) and MEI (lavender data). Reaction conditions using 0.028 mmol of EBP: [MA]/[Isocyanide]/[HME]/[EBP]/[TBABr]/[BAPO] = 180/20/0.05/1/0.1/0.1 in 50 vol% DMF. Reactions were prepared in a glovebox and brought out of the glovebox to be irradiated with white LEDs (relative intensity ∼0.2 W) for 2 h with air-cooling. | ||
We hypothesized that the loss of control with TOSMIC may result from the highly reactive electrophilic imidoyl radical, leading to side reactions such as hydrogen atom transfer (HAT), β-scission, or deactivation of the nucleophilic HME-Cob catalyst (Fig. S5†).12,31–33 Consequently, control over the copolymerisation becomes harder to maintain – especially at higher concentrations of monomer, with respect to the concentration of catalyst and initiator (Fig. S6†). We envisioned that exchanging the isocyanide for commercially available 2-morpholinoethyl isocyanide (MEI) may improve efficiency by proceeding through a more stable imidoyl radical.16,18,34,35 The ethyl group on MEI may help stabilize the imidoyl radical, reducing side reactions, improving control and achieving higher MA conversion compared to TOSMIC (Fig. S7†). Copolymerisation with MEI improved MA conversion with decreased dispersity, albeit lower incorporation (Fig. 1A entry 2). In the absence of BAPO, the same relative improvements in polymerization were observed. In targeting a range of DPs, the dispersity remained lower across all ranges of molecular weights at incorporations ranging from 1.1 to 1.5% (Fig. 1B–D).
Using the more efficient polymerisation with MEI, we examined control reactions in our system (Table 1). Without photoinitiator, no polymerisation was observed without adding HME-Cob or alkyl halide initiator, ethyl 2-bromopropionate (EBP) (Table 1 entries 2 and 3). In the absence of light, <10% monomer conversion is observed, consistent with our previous results (Table 1 entry 4). These controls show the need for light, catalyst, and alkyl halide initiator to facilitate the copolymerisation. Excluding additional bromide anion, tetrabutylammonium bromide (TBABr) resulted in higher molecular weights and poor efficiency (Table 1 entry 5). Increasing the concentration of TBABr improved the control over the polymerisation, by favouring the deactivation of growing polymer chains (Fig. S8†).24,29,36
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Control | % MA conv. | % MEI conv. | M n,exp | Đ | % Ieff | % MEIth | % MEIexp | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Standard reaction conditions: MA/ISO/EBP/HME-Cob (CoII)/TBABr = 180/20/1/0.05/0.1 in 50 vol% DMF. Conditions for 0.028 M stock of pre-reduced HME-Cob (CoII) in DMF: [HME-Cob (CoIII)]/[Zn]/[NH4Cl] = 1/0.5/1.5. Polymerisation conditions: [MA]/[MEI]/[HME-Cob (CoII)]/[EBP]/[TBABr] = 180/20/0.05/1/0.1 in 50 vol% DMF. Monomer conversion from 1H NMR in CDCl3. Conversions used to calculate theoretical molecular weight (Mn,th) and theoretical MEI incorporation (MEIth). Mn,th = (DPMA × MWMA × Conv.MA) + (DPMEI × MWMEI × Conv.MEI) + MWEBP. MEIth = (DPMEI × Conv.MEI)/((DPMEI × Conv.MEI) + (DPMA × Conv.MA)). Experimental molecular weights (Mn,exp.) obtained via gel permeation chromatography (GPC). Initiator efficiency (Ieff) = Mn,th/Mn,exp. 1H NMR of precipitated samples used to for MEI % incorporation (MEIexp). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Standard | 41 | 5 | 11000 |
1.54 | 61 | 1.2 | 1.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | No HME-Cob | 0 | 0 | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | No EBP | 0 | 0 | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | No light | 7 | 2 | 14200 |
1.44 | 10 | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | No TBABr | 33 | 6 | 20300 |
1.83 | 27 | 1.9 | 1.0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
We next sought to increase the incorporation of MEI by varying the feed ratio of MA to MEI. Increasing the MA:MEI ratio from 9:1 to 1:4 resulted in increasing MEI incorporations from 1% to 20%, respectively (Fig. 2A). The higher incorporations led to a decrease in initiator efficiencies to 41%, likely due to catalyst deactivation as the concentration of MEI increases (Fig. S10†). Higher incorporations also led to lower molecular weights, also seen with polymerisations via photo iniferter reversible addition-fragmentation chain transfer (PI-RAFT) and CMRP.12,16 Kinetics of the copolymerisation using a 9:1 MA:MEI feed ratio showed a plateau in monomer conversion after 90 minutes, with efficiency increasing throughout the polymerisation (Fig. 2B). Finally, to demonstrate photothermal temporal control, on/off cycles of light irradiation were performed (Fig. 2C). Satisfyingly, no monomer conversion occurred in the dark, indicating that MEI does not contribute to monomer conversion in the absence of light and that reactivity resumes when light is restored. Retention of chains was further supported via chain extension with tert-butyl acrylate (tBA) using a PMA-r-MEI macroinitiator. Here, a significant change in the molecular weight from 4.3 kg mol−1 to 26.2 kg mol−1 and a narrowing of dispersity is seen after tBA polymerisation. Minimal amounts of macroinitiator (7.5%) were inactive, as indicated by the GPC trace, indicating good chain end retention for copolymers made with MEI.
 | ||
| Fig. 2 (A) Graph showing the percent of MEI incorporation at different ratios of MA to MEI. (B) Kinetics of 180:20 (MA:MEI) copolymerisation. (C) Temporal control for a 180:20 (MA:MEI) copolymerisation, with three 15-minute off cycles and 15-minute on cycles. (D) Use of copolymer made with MA and MEI as a macroinitiator for chain extension with tert-butyl acrylate. Polymerisation conditions: reaction conditions using 0.028 mmol of EBP: [MA]/[Isocyanide]/[HME]/[EBP]/[TBABr]/[BAPO] = 180/20/0.05/1/0.1/0.1 in 50 vol% DMF. Reactions were prepared in the glovebox and brought out of the glovebox to react under white LEDs (relative intensity ∼0.2 W) with air-cooling for 2 h. Aliquots for each time point and on/off cycle were taken in an N2 purge syringe for crude NMR and GPC analysis. Polymerisation conditions using 0.029 mmol macroinitiator: [tBA]/[pMA-r-MEI]/[HME]/[TBABr]/[BAPO] = 150/1/0.05/1/0.1/0.1 in 50 vol% DMF. Reactions were prepared in the glovebox and brought out of the glovebox to react under white LEDs (relative intensity ∼0.2 W) with air-cooling for 2 h. | ||
Having successfully used HME-Cob in photothermal ATRP for isocyanide copolymers, we next sought to compare our method to a more traditional ATRP protocol. Copolymerisation of MEI and MA catalysed by cuprous bromide (CuBr) and an amine ligand, PMDETA (N,N,N′,N′′,N′′-pentamethyldiethylenetriamine), resulted in an uncontrolled polymerisation with molecular weight three times higher than theoretical (Fig. 3A, entry 1). The lack of control in this polymerisation can be attributed to MEI ligation to the copper complex, causing the deactivation of the catalyst.37,38 This is seen in the UV-Vis spectrum of Cu/PMDETA that shows absorbance features at 300–400 nm; however, upon the addition of MEI, this feature substantially decreased (Fig. 3B). In contrast, when looking at the UV-Vis spectrum for HME-Cob, only a slight decrease in absorbance is noticed at 470 nm, the peak that is characteristic of a CoII species for HME-Cob, in the presence of MEI (Fig. 3C). This minor change in intensity indicates that MEI does not ligate to the cobalt catalyst as easily as it does to Cu/PMDETA, leading to improved polymerisation outcomes.
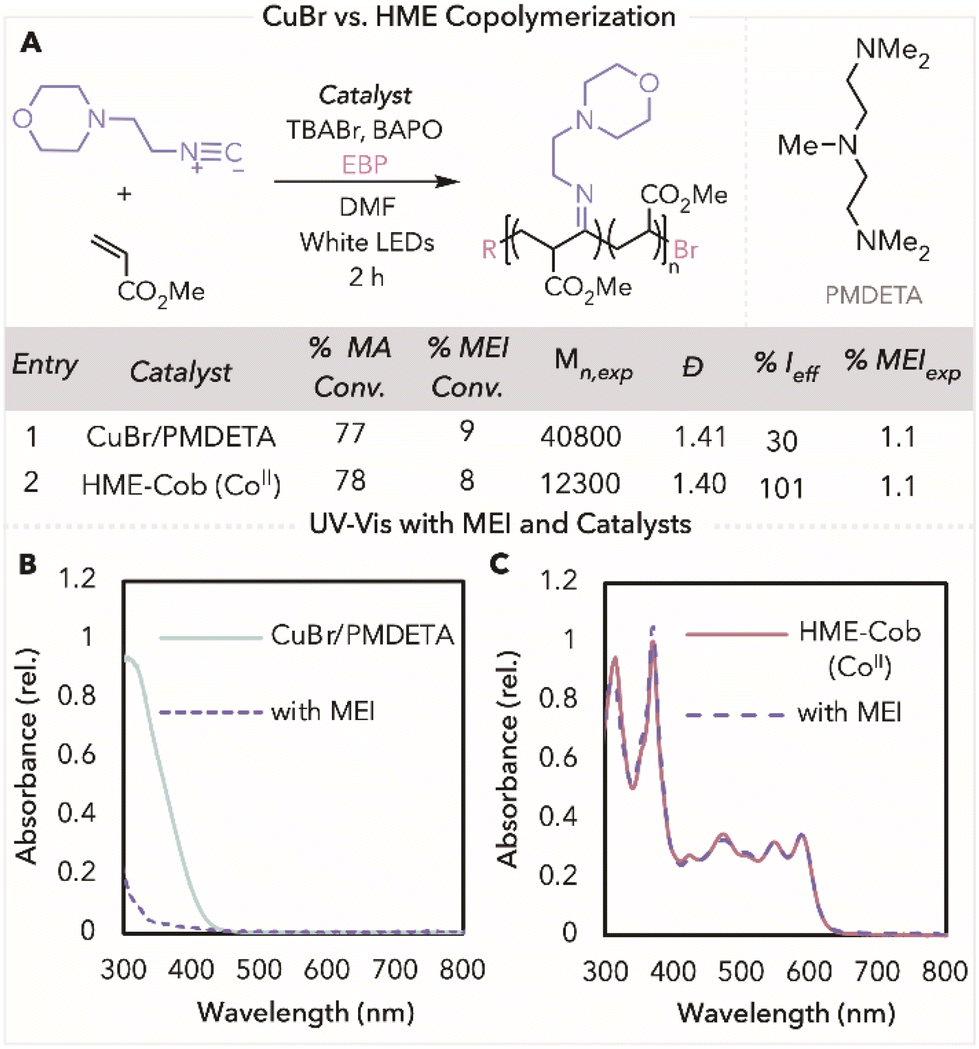 | ||
| Fig. 3 (A) General scheme of polymerisation using CuBr/PMDETA or HME with copolymerisation results. UV-Vis of (B) 597 μM CuBr/PMDETA with and without MEI and (C) 50 μM HME with and without MEI. Polymerisation conditions: reaction conditions with CuBr using 0.028 mmol of EBP: [MA]/[Isocyanide]/[CuBr]/[PMDETA]/[EBP]/[TBABr]/[BAPO] = 180/20/0.05/0.06/1/0.1/0.1 in 50 vol% DMF. Reaction conditions using with HME using 0.028 mmol of EBP: [MA]/[Isocyanide]/[HME]/[EBP]/[TBABr]/[BAPO] = 180/20/0.05/1/0.1/0.1 in 50 vol% DMF. Reactions were prepped in the glovebox and brought out of the glovebox to react under white LEDs (relative intensity ∼0.2 W) with air-cooling for 2 h. UV-Vis conditions for 597 μM of CuBr in 50/50 DMF/MA: [CuBr]/[PEMDETA]/[MEI] = 1/1.1/400. UV-Vis conditions for 50 μM of HME in 50/50 DMF/MA: [HME]/[MEI] = 1/400. | ||
Conclusions
Here, we show the first example of an ATRP system amenable to isocyanide copolymerisation under mild, white light irradiation. This method gave copolymers with isocyanide incorporations ranging from 1–20%. Molecular weights ranged from 3.0–30.0 kg mol−1 for copolymers with 1% isocyanide incorporation. This method also appears favourable over copper-mediated ATRP with the addition of isocyanides showing minimal impact on the catalytic reactivity of HME-Cob. This work further demonstrates how degradable isocyanide copolymers can be synthesized using a highly versatile ATRP method.Author contributions
CP wrote the manuscript and conducted experiments, EES supervised the research. Both CP and EES edited and approved the final manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under Award CHE-2337228. C. P. thanks the Ted Taylor family for an Edward C. Taylor Fellowship graduate fellowship. Special thanks to Dr Sajjad Dadashi-Silab for guidance on Cu-catalysed ATRP and Lejla Čamdžić for help in NMR analysis.References
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. D. Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515–1518 CrossRef CAS PubMed.
- A. S. Pottinger, R. Geyer, N. Biyani, C. C. Martinez, N. Nathan, M. R. Morse, C. Liu, S. Hu, M. de Bruyn, C. Boettiger, E. Baker and D. J. McCauley, Science, 2024, 386, 1168–1173 CrossRef CAS PubMed.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. D. Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- S. T. Schwab, M. Baur, T. F. Nelson and S. Mecking, Chem. Rev., 2024, 124, 2327–2351 CrossRef CAS PubMed.
- A. M. Ibrahim, in Plastics: Photodegradations and Mechanisms, ed. V. Sivasankar and T. G. Sunitha, Springer, Cham, 2024, pp. 25–49 Search PubMed.
- F. Y. Xu and J. C. W. Chien, Macromolecules, 1993, 26, 3485–3489 CrossRef CAS.
- T. O. Morgen, M. Baur, I. Göttker-Schnetmann and S. Mecking, Nat. Commun., 2020, 11, 3693 CrossRef CAS PubMed.
- S. G. Bond and J. R. Ebdon, Polymer, 1994, 35, 4079–4082 CrossRef CAS.
- E. Drent, R. van Dijk, R. van Ginkel, B. van Oort and R. I. Pugh, Chem. Commun., 2002, 964–965 RSC.
- L. Čamdžić and E. E. Stache, J. Am. Chem. Soc., 2023, 145, 20311–20318 CrossRef PubMed.
- L. Čamdžić, C. A. Haynes and E. E. Stache, Chem, 2024, 10, 1357–1370 Search PubMed.
- A. Nakamura, K. Munakata, T. Kochi and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 8128–8129 CrossRef CAS PubMed.
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215–5244 CrossRef CAS PubMed.
- L. Čamdžić and E. E. Stache, Macromolecules, 2024, 57, 9250–9256 CrossRef.
- A. Ogawa and Y. Yamamoto, Beilstein J. Org. Chem., 2024, 20, 2114–2128 CrossRef CAS PubMed.
- M. Minozzi, D. Nanni and P. Spagnolo, Curr. Org. Chem., 2007, 11, 1366–1384 CrossRef CAS.
- S. Dadashi-Silab and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 13311–13318 CrossRef CAS PubMed.
- C.-H. Peng, T.-Y. Yang, Y. Zhao and X. Fu, Org. Biomol. Chem., 2014, 12, 8580–8587 RSC.
- L. E. N. Allan, M. R. Perry and M. P. Shaver, Prog. Polym. Sci., 2012, 37, 127–156 CrossRef CAS.
- M. Destarac, Polym. Chem., 2018, 9, 4947–4967 RSC.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS PubMed.
- S. Dadashi-Silab, C. Preston-Herrera and E. E. Stache, J. Am. Chem. Soc., 2023, 145, 19387–19395 CrossRef CAS PubMed.
- C. Preston-Herrera, S. Dadashi-Silab, D. G. Oblinsky, G. D. Scholes and E. E. Stache, J. Am. Chem. Soc., 2024, 146, 8852–8857 CrossRef CAS PubMed.
- M. E. Matter, C. Tagnon and E. E. Stache, ACS Cent. Sci., 2024, 10, 1460–1472 CrossRef CAS PubMed.
- S. Dadashi-Silab, C. Preston-Herrera, D. G. Oblinsky, G. D. Scholes and E. E. Stache, J. Am. Chem. Soc., 2024, 146, 34583–34590 CrossRef CAS PubMed.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem. Catal., 2022, 2, 52–83 CrossRef CAS.
- D. Konkolewicz, A. J. D. Magenau, S. E. Averick, A. Simakova, H. He and K. Matyjaszewski, Macromolecules, 2012, 45, 4461–4468 CrossRef CAS.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- P. M. Blum and B. P. Roberts, J. Chem. Soc., Chem. Commun., 1976, 535–536 RSC.
- Y. Nakamura, R. Lee, M. L. Coote and S. Yamago, Macromol. Rapid Commun., 2016, 37, 506–513 CrossRef CAS PubMed.
- F. P. Pruchnik and S. A. Duraj, Isocyanide Complexes, in Organometallic Chemistry of the Transition Elements, Springer, Boston, MA, 1990, pp. 617–645 Search PubMed.
- Z. Szablan, T. Junkers, S. P. S. Koo, T. M. Lovestead, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2007, 40, 6820–6833 CrossRef CAS.
- S. Sharma, A. P. Pandey and A. Sharma, Adv. Synth. Catal., 2020, 362, 5196–5218 CrossRef CAS.
- K. Grudzień, W. Nogaś, G. Szczepaniak and K. Grela, Polyhedron, 2021, 199, 115090 CrossRef.
- V. Vlasova, Yu. N. Toikka, V. V. Suslonov, A. S. Smirnov and N. A. Bokach, Russ. J. Gen. Chem., 2024, 94, 1682–1688 CrossRef CAS.
- G. Szczepaniak, J. Piątkowski, W. Nogaś, F. Lorandi, S. S. Yerneni, M. Fantin, A. Ruszczyńska, A. E. Enciso, E. Bulska, K. Grela and K. Matyjaszewski, Chem. Sci., 2020, 11, 4251–4262 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00422e |
| This journal is © The Royal Society of Chemistry 2025 |