
Electron diffraction unveils the 2D metal-radical framework of two molecule-based magnets†
Emre
Yörük‡
 b,
Constance
Lecourt§
a,
Dominique
Housset
c,
Yuuta
Izumi
d,
Wai Li
Ling
c,
Stéphanie
Kodjikian
b,
Evgeny
Tretyakov
e,
Katsuya
Inoue
d,
Kseniya
Maryunina¶
d,
Cédric
Desroches
a,
Holger
Klein
b and
Dominique
Luneau
*a
b,
Constance
Lecourt§
a,
Dominique
Housset
c,
Yuuta
Izumi
d,
Wai Li
Ling
c,
Stéphanie
Kodjikian
b,
Evgeny
Tretyakov
e,
Katsuya
Inoue
d,
Kseniya
Maryunina¶
d,
Cédric
Desroches
a,
Holger
Klein
b and
Dominique
Luneau
*a
aLaboratoire des Multimatériaux et Interfaces (UMR 5615), Université Claude Bernard Lyon 1, 69100 Villeurbanne, France. E-mail: dominique.luneau@univ-lyon1.fr
bUniversité Grenoble Alpes and CNRS, Institut Néel, Grenoble, France
cUniversité Grenoble Alpes, CEA, CNRS, IBS, 38000 Grenoble, France
dWPI-SKCM2, CResCent and Department of Chemistry, Hiroshima University, Kagamiyama-1-3-1, Higashihiroshima, Hiroshima, 739-8526, Japan
eN. D. Zelinsky Institute of Organic, Chemistry, Leninsky Avenue 47, Moscow 119991, Russian Federation
First published on 26th November 2024
Abstract
Low-dose electron diffraction has been instrumental in determining the crystal structures of two compounds with metal-radical coordination frameworks {[MnII2(NITIm)3]CF3SO3·CH3OH}n (1) and {[MnII2(NITImMe2)3]ClO4}n (2) that could never be grown to a crystal size large enough for single-crystal X-ray diffraction characterization. The compounds crystallize as nanocrystals upon addition of triflate (1) and perchlorate (2) anions and coordination of manganese(II) with bis-chelate nitronyl nitroxide radicals NITImH (1) and NITImHMe2 (2) which are respectively 2-(2-imidazolyl)- and 2-(4,5-dimethylimidazol-2-yl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol-3-oxide-1-oxyl. The two compounds have layered crystal structures in which cationic 2D metal-radical coordination polymers {[MnII2(NITIm)3]+}n (1) and {[MnII2(NITImMe2)3]+}n (2) are separated by layers of triflate (1) or perchlorate (2) anions. Magnetic measurements evidence a ferrimagnetic behavior within the 2D metal-radical sheets due to alternating antiferromagnetically coupled spins (SMn2+ = 5/2 and Sradical = 1/2). Both compounds exhibit a long-range 3D ordering of weak-ferromagnetic type due to spin canting with Curie temperatures Tc = 45 K (1) and 40 K (2). This is associated with a field-induced metamagnetic transition from antiferromagnetic to ferromagnetic coupling of 2D metal-radical sheets. Studies of the crystal structures allows to rationalize how the molecular structure of nitronyl nitroxide radicals and of the counter-anions along with crystal packing affect the magnetic behavior related to interlayer distance and framework flexibility. These results are striking evidence that electron crystallography is a unique tool to solve structures of metal–organic compounds crystallizing as nanocrystals even with nitronyl nitroxide radical components too sensitive to typical electron doses. Overcoming the crystal size barrier, it allows the validation of chemical synthesis and the establishment of magneto-structural relationships fostering new advances in the design of molecule-based magnets.
Introduction
Molecule-based magnet exploration was initiated nearly forty years ago fostering the design of supramolecular assemblies of open-shell molecules that hold an overall magnetization in the solid state.1,2–11 They may be purely organic, organometallic charge-transfer or coordination compounds.12 So far, most fruitful development has relied on coordination chemistry, thanks to a wide variety of paramagnetic centers comprising metal ions and radical ligands when the directionality of the metal–ligand bonds facilitates a better control of the atom arrangement and magnetic interactions among spin carriers.1,13 Along with this approach, the building of coordination polymers favors the emergence of a network of magnetic exchange interactions among the spin carriers while a gain in the long-range magnetic ordering temperature is expected with increasing the coordination polymer dimensionality i.e. formation of 1D (chain), 2D (layered) and 3D metal–organic frameworks. Clearly, crystal structure is a prerequisite to check the supramolecular assemblies as well as for the systematic study of magnetic-structural relationships, and a key factor on controlling functional properties. The difficulty and often impossibility for most compounds with 2D and 3D metal–organic frameworks to grow single crystals large enough for X-ray crystallography (>103 μm3) has pushed scientists in the field to essentially focus on magnetic single-molecules (0D). This has resulted in significant successes in the design of single-molecule and single-ion magnets14 and the control of their ground spin state, but in the meantime investigations of metal–organic materials with 3D magnetic ordering has stagnated.The crystal-size barrier has also hampered our investigations of molecular magnets based on the metal-radical approach.2 We have previously designed imidazole-substituted derivatives of nitronyl nitroxide radicals (Fig. 1) that can coordinate with almost any 3d and 4f metal ion.15 Furthermore, following deprotonation of the imidazole moiety they become bis-chelating bridging ligands that allow the formation of metal-radical coordination polymers. Over the years and repeated attempts, we have succeeded in obtaining single crystals for several compounds that have 1D (chain) and 2D (layered) metal-radical frameworks, enabling us to determine their crystal structures by X-ray diffraction and to study their magneto-structural relationships.16–18 However, this has only been achieved for a small number of compounds, specifically with Mn2+ ions, while the crystal structure of many others could not be elucidated because the crystal sizes were too small for X-ray diffraction experiments, even with synchrotron radiation sources.
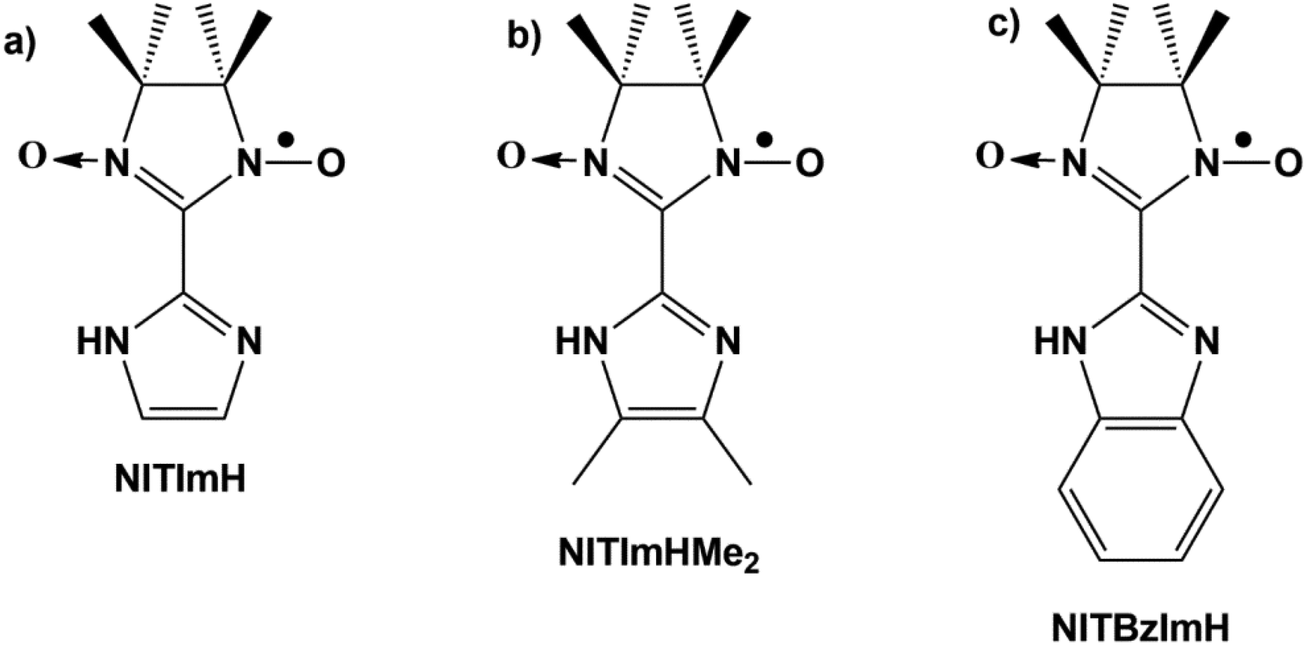 | ||
| Fig. 1 Schematic representation of nitronyl nitroxide radicals substituted with (a) imidazole (NITImH), (b) 4,5-dimethylimidazole (NITImHMe2), and (c) benzimidazole (NITBzImH ). | ||
Among the few compounds whose crystal structures could be elucidated, most are composed of 2D layered Mn-radical frameworks in which the Mn2+ and nitroxide spins are antiferromagnetically coupled. Remarkably and despite these similarities, different types of magnetic behavior are observed which depend on the substituents present on the nitronyl nitroxide radical or on the counter-anions that intercalate in-between the 2D Mn-radical sheets. Thus, {[MnII2(NITBzIm)3]X}n coordination compounds based on benzimidazole-substituted radical NITBzImH (2-(2-benzimidazolyl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol-3-oxide-1-oxyl, Fig. 1c) with the counter-anions X being either ClO4− (3) or alkylsulfonate,18 exhibit long-range magnetic ordering. The highest Curie temperature (Tc = 55 K) has been observed for {[MnII2(NITBzIm)3]ClO4}n (3) with perchlorate anions. In contrast, changing the nitronyl nitroxide radical substituent from BzImH to imidazole (ImH) (Fig. 1a), led to a series of compounds with structurally similar 2D-coordination frameworks {[MnII2(NITIm)3]X}n (X = ClO4− (4), BF4− (5) and PF6− (6)) but who undergo thermally-induced valence tautomeric (VT) conversions instead.17,19,20 For the each of the three compounds, temperature-dependent oxidation states are observed. There is a high temperature region where all manganese ions are in the +II oxidation state and are coordinated by three NITIm− anion-radicals. Upon cooling, they oxidize to the +III state, while electrons are transferred to the radicals to reach a low temperature region where two third of radicals are reduced (NITImred2−) for charge equilibrium, and only one third are still persistent (NITIm−). Therefore, the compounds are better formulated as {[MnII2−yMnIIIy(NITIm)3−y(NITImred)y]X}n where y can take any value between 0 and 2 when going from the high to the low temperature regions. The extent of the high and low temperature regions, the smoothness of conversion and the number of steps in between the two regions depend on the counter-anions interlayering the 2D manganese(II)-nitroxide sheets.
These previous studies have led us to consider the distance between the mean planes of the 2D metal-radical frameworks {[MnII2(radical)3]}n sheets as a key parameter in determining whether the compounds undergo long-range magnetic ordering or exhibit VT conversion. To test these assumption, we investigated here two different structural modifications of {[MnII2(NITIm)3]ClO4}n (4). In compound 1 perchlorate was substituted by the bulkier triflate anion while keeping the same radical derivative NITImH. In compound 2 the size of nitroxide radical was increased by adding two methyl substituents on the imidazole ring to give NITImHMe2 (Fig. 1b),21 while keeping the perchlorate anions. Despite numerous attempts, only powders of nano-sized crystals were obtained for compounds 1 and 2 preventing structure determination by single crystal X-ray diffraction. Moreover, our attempts to use powder X-ray diffraction failed to provide a reliable crystal structure, even when the structural motif [MnII2(NITIm)3] from the crystal structure of 4 was used as a reference building block.
Because compounds 1 and 2 could never be grown to a crystal size large enough for single-crystal X-ray diffraction characterization we overcome the crystal size barrier by using electron diffraction (ED) to determine their crystal structures. In the last two decades there have been tremendous methodological advances in the ED technique, based on possibilities of recording quasi-kinematical diffraction intensities via 3D electron diffraction (3D ED)22,23 together with the development of fast hybrid detectors enabling low dose continuous rotation ED data collection.24 These developments have significantly facilitated the structure determination of nanocrystalline samples in material science and chemistry.25–27 This has particularly been useful for metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) that often crystallize in the form of nanocrystalline powders.28–33 Moreover, use of low-dose electrons and cryogenic conditions has allowed to minimize electron beam damage on MOFs.34–42 These advances in 3D ED should benefit to coordination chemists, particularly in the field of molecule-based magnets for which obtaining crystals suitable for X-ray crystallography may be difficult.
Here, we show how ED was successful in determining the crystal structure of two novel magnetic compounds with 2D metal-radical coordination frameworks {[MnII2(NITIm)3]CF3SO3·CH3OH}n (1) and {[MnII2(NITImMe2)3]ClO4}n (2) where NITImH and NITImHMe2 are respectively 2-(2-imidazolyl)- (Fig. 1a) and 2-(4,5-dimethylimidazol-2-yl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol-3-oxide-1-oxyl (Fig. 1b). The molecular replacement technique was used with the [MnII2(NITIm)3] moiety as a search model to phase the crystallographic data and provided a structure with nearly atomic resolution. This major result allows us to first validate the chemical synthesis even from nanocrystals then to establish magneto-structural relationships for these two compounds exhibiting both a long-range 3D ordering of weak-ferromagnetic associated with spin canting and a field-induced metamagnetic transition.
Results and discussion
Synthesis and crystal growth
Compounds {[MnII2(NITIm)3]CF3SO3·CH3OH}n (1) and {[MnII2(NITImMe2)3]ClO4}n (2) were synthesized following the procedure described previously to yield either VT compounds {[MnII2(NITIm)3]X}n with X = ClO4− (4); BF4− (5), PF6− (6) or compounds exhibiting long range magnetic ordering {[MnII2(NITBzIm)3]ClO4}n (3) with NITBzImH (Fig. 1c).16–18 To this end, nitronyl nitroxide radicals (NITImH or NITImHMe2) were first mixed with manganese(II) acetate in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio in methanol, where manganese(II) acetate acts both to provide metal ions and to deprotonate the imidazole moiety. Then compounds were slowly precipitated as polycrystalline powders upon addition of sodium triflate (NaOTf) for 1 or sodium perchlorate (NaClO4) for 2. Laboring efforts to grow single crystals of {[MnII2(NITIm)3]OTf}n (1) and {[MnII2(NITImMe2)3]ClO4}n (2) large enough for X-ray diffraction studies have been unsuccessful. Methanol diffusion into a water solution of the reactants, which allowed crystal growth for previously studied compounds 4–6, was not effective for complexes 1 and 2. Furthermore, although nitronyl nitroxide radicals are quite stable under standard conditions, they should be handle with care, which limits the scope of crystallization techniques to be used. For example, solvothermal synthesis which are common for MOFs cannot be used.
:2 ratio in methanol, where manganese(II) acetate acts both to provide metal ions and to deprotonate the imidazole moiety. Then compounds were slowly precipitated as polycrystalline powders upon addition of sodium triflate (NaOTf) for 1 or sodium perchlorate (NaClO4) for 2. Laboring efforts to grow single crystals of {[MnII2(NITIm)3]OTf}n (1) and {[MnII2(NITImMe2)3]ClO4}n (2) large enough for X-ray diffraction studies have been unsuccessful. Methanol diffusion into a water solution of the reactants, which allowed crystal growth for previously studied compounds 4–6, was not effective for complexes 1 and 2. Furthermore, although nitronyl nitroxide radicals are quite stable under standard conditions, they should be handle with care, which limits the scope of crystallization techniques to be used. For example, solvothermal synthesis which are common for MOFs cannot be used.
3D ED data collection
We used 3D electron diffraction for the structure determination from nano-sized crystalline samples. Diffraction data were collected under low-dose conditions on a Tecnai F20 TEM (transmission electron microscope) operated at 200 kV equipped with a Medipix3RX hybrid pixel detector (Amsterdam scientific instruments) as described in the ESI Section 3.† Preliminary beam damage assessment showed that crystallinity loss became visible already at 1.0 e− Å−2. Thus, pre-exposure was significantly reduced by searching the crystalline particles digitally on low magnification TEM images, and by centering the crystals (eucentric height tuning) at very low dose with the help of the Medipix3RX detector. The total accumulated dose during data acquisition on the crystals of compound 1 was 4.0 e Å−2 (crystal 1), 3.4 e Å−2 (crystal 2), and 3.8 e Å−2 (crystal 3), while the compound 2 crystal was irradiated with 3.1 e Å−2.Crystal structures
Compound 1 crystallizes in the monoclinic space group P21 and compound 2 in the orthorhombic space group P212121 (Tables S1 and S2†). Interestingly, crystal structures of previously described 2D manganese-radical frameworks were also found to crystallize in these non-centrosymmetric space groups (Table S3†).16,17,19,20 The resolution, less than 1.2 Å for most data, was not high enough for ab initio structure solution.43,44 The crystal structure was then solved by the molecular replacement method using the structural motif [MnII2(NITIm)3] from the crystal structure of {[MnII2(NITIm)3]ClO4}n (4) at 300 K (CSD 100351) as a search model. This allowed the structure determination of both compounds (Fig. 2) and their refinement against 3D ED data, using the kinematical approximation. The detailed description of the structure solution and refinement is provided in the ESI.† The X-ray powder diffraction patterns and the simulated one from the refined structures are in good agreement for compound 2 (Fig. S2†) while for compound 1 there is agreement for the positions of the Bragg peaks but discrepancy for their intensities (Fig. S1†). This is attributed to the preferential orientation of the nanocrystals, which are platelet-shaped. For compound 2 the structure of the perchlorate anion was also taken from the crystal structure of 4 at 300 K, while for compound 1 the structure of the triflate anion was taken from a published crystal structure elsewhere.45 Both anions could be placed unambiguously in the residual {Fobs − Fcalc} Coulomb potential map.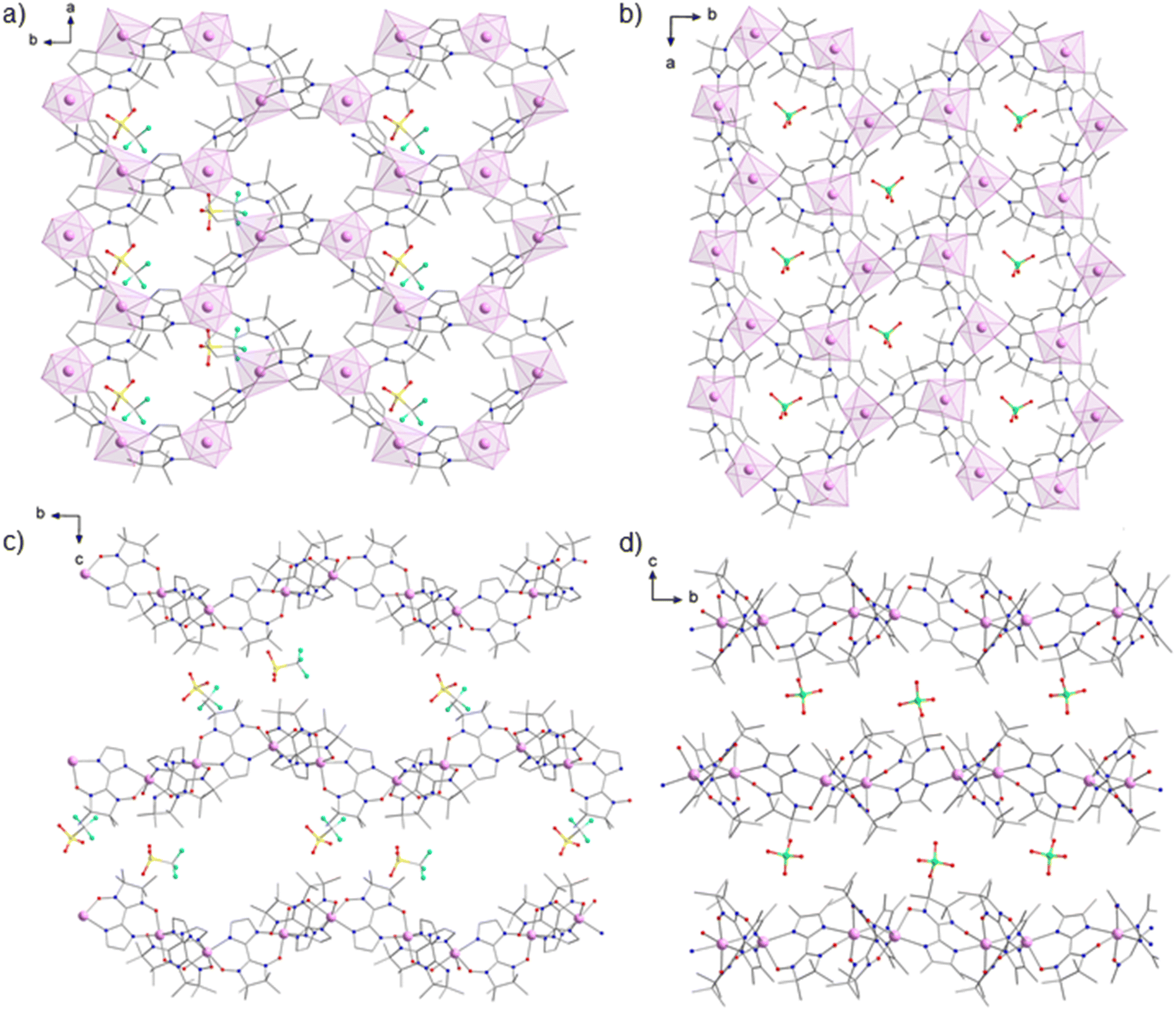 | ||
| Fig. 2 View of the 2D metal-radical frameworks: (a) {[MnII2(NITIm)3]OTf}n (1) and (b) {[MnII2(NITImMe2)]ClO4}n (2) along the c axis direction layered with (c) OTf− (1) and (d) ClO4− (2) anions viewed along the a axis direction. Mn2+ ions are depicted in pink with coordination polyhedra (a and b). Atoms are depicted as follows: Mn, pink; O, red; N, dark blue, S, yellow, F, light blue and Cl, green; H atoms and CH3OH solvent molecules are omitted for clarity. | ||
Compounds 1 and 2 crystallize as layered structures. The sheets are made of 2D positively charged coordination polymers {[MnII2(NITIm)3]+}n (1) and {[MnII2(NITImMe2)3]+}n (2) with honeycomb-like structures growing along the ab plane direction (Fig. 2a and b). These sheets are separated along the c axis by layers of OTf− (1) or ClO4− (2) counter-anions layers (Fig. 2c and d). CH3OH solvent molecules were found together with triflate anions in inter-sheets space in compound 1 while compound 2 crystallizes as a solvent-free layered structure.
In structure of 1 there are two crystallographically independent {[MnII2(NITIm)3]OTf·CH3OH} moieties in the asymmetric unit, leading to four Mn(II) centers denoted Mn1, Mn2, Mn3 and Mn4 (Fig. 3a and Fig. S3†). For compound 2, only one {[MnII2(NITImMe2)3]ClO4}n moiety is found in the asymmetric unit with two crystallographically independent Mn(II) centers denoted Mn1 and Mn2 (Fig. 3b). As for any coordination polymers built on metal centers with three coordinated bis-chelating ligands, the 2D honeycomb-like structure is made of the alternation of Λ and Δ coordination stereoisomers. Here the Λ conformation for Mn1 (1 and 2) and Mn4 (1), and Δ conformation for Mn2 (1 and 2) and Mn3 (1) (Fig. 3 and Fig. S3†). For compound 2, the radicals coordinated to the Mn1 and Mn2 metallic centers are in the mer conformation, as observed in other analogous compounds 3–6.17–19 Interestingly for compound 1, the radicals coordinated to the Mn1 and Mn3 centers are in the mer arrangement while the ones bound to Mn2 and Mn4 are in the fac conformation. Among compounds with 2D Mn-radical frameworks, compound 1 is the first to show the fac arrangement of the radical. The fac arrangement was also found in the isolated complex [MnII(NITIm)3](ClO4)2.46
 | ||
| Fig. 3 (a) View of one cation moiety [MnII2(NITIm)3]+ in compound 1 showing manganese ions Mn1 and Mn2 with respectively fac-Δ and mer-Λ alternating conformations. Symmetry related atoms: (i) : (−1 + x, y, z); (ii) : (−x, −0.5 + y, −z); (iii) : (−x, 0.5 + y, −z); (iv) : (1 + x, y, z). (b) View of the cation moiety [MnII2(NITImMe2)3]+ in compound 2 showing manganese ions Mn1 and Mn2 with Δ and Λ alternating conformation but both with mer arrangement of the ligands. Symmetry-related atoms: (ix) : (−1 − x, −0.5 + y, −0.5 − z); (x) : (−x, −0.5 + y, −0.5 − z); (xi) : (−1 − x, 0.5 + y, −0.5 − z); (xii) : (−x, 0.5 + y, −0.5 − z). Atoms are depicted as follows: Mn, pink; O, red; N, blue; H atoms are omitted for clarity. | ||
In compound 1, the {[MnII2(NITIm)3]}n sheets are highly corrugated along the b direction (Fig. 2c and Fig. S4†). The Mn2 and Mn4 are ±1.43 Å apart from the mean plane containing the Mn1 or Mn3 (Fig. S5†). Moreover, and quite remarkably, the successive corrugated sheets are not stacked in the classical manner of crest on crest (trough on trough), but are staggered in an almost crest on trough stacking. This appears to be due to intermolecular bonding involving the –CF3 and –SO3− moieties of the triflate anions, which brings the crests of the lower 2D-sheets closer to the troughs of the upper ones (Fig. 2c). This yields empty channels running along the a axis direction (Fig. S6†). This is in contrast to the nearly flat {[MnII2(NITImMe2)3]}n 2D sheets in compound 2 (Fig. 2d and Fig. S7†), where Mn1 and Mn2 just slightly deviate (±0.36 Å) from their mean plane (Fig. S8†), reminiscent of what was observed in the structure of analogous compounds 3–6.17–20
In both compounds, each Mn is hexacoordinated by three O atoms and three N atoms from three chelated nitronyl nitroxide radicals with the Mn–O and Mn–N bond lengths falling in the 1.98–2.3 Å range for compound 1 (Tables S4 and S5†) and in the 2.00–2.32 Å range for compound 2 (Tables S6 and S7†). These bond lengths are typical for Mn(II) coordination with N- and O-based ligands, particularly with Mn-nitroxide compounds.46
The structural features within the metal-radical frameworks are constrained by the chelation of the Mn2+ ions to the radicals with slight variations within the family of different coordination polymers depending on the type of nitronyl nitroxide radicals. Thus, in 1D systems {[MnII(NITIm)]}n built from NITImH radical, the Mn–Mn distances between the first neighbors and the second neighbors were found to be 6.33–6.40 Å and 9.59–9.94 Å respectively within the infinite chain.46 Similar Mn–Mn distances were also found for 2D compound 4 above 300 K (6.21–6.28 Å and 9.88–11.30 Å respectively).17 These structural parameters are also observed within the plane in compounds 1 and 2. The Mn–Mn first and second neighbor distances are respectively in the ranges 6.31–6.40 Å and 9.83–11.81 Å for 1 and 6.46–6.48 Å and 10.32–11.95 Å for 2 (Tables S3 and S5†). In compounds 1 and 2 the Mn centers build a hexagonal network where the hexagons are either aligned but slightly elongated along the b axis (1) (Fig. S5†) or tilted and forming zigzag along the b direction (2) (Fig. S8†). Although the unit cell of compound 1 and 2 contains the same number of [MnII2(NITIm)3] or {[MnII2(NITImMe2)3]}, the a parameter and cell volume of compound 1 are larger than those of compound 2, as shown in Table S2.† This suggests that OTf− anion requires more space than ClO4− anion and the six Me-substitutions per formula unit for complex 2. This may be also the reason why compound 1 show alternation of the mer and fac arrangements of the radicals.
Relevant structural features to consider for magneto-structural relationships is the dihedral angle α between the plane (P2) going through atoms of the nitronyl nitroxide moiety (O–N–C–N–O) and the plane (P1) going through the coordination atoms (O–Mn–N) as shown on Fig. S9.† Indeed, the unpaired electron of nitronyl nitroxide is located in the π* orbital which is perpendicular to the P2 plane going through atoms O–N–C–N–O. Therefore, as the dihedral angle α decreases, this increases the overlap of the π* and d magnetic orbitals and, as a result, the magnetic interaction between the radical and Mn(II) is expected to become more antiferromagnetic (Fig. S9†).47 In compound 1 (Table S9†) the α dihedral angles are in the range 12.37–46.12° for Mn1–Mn2 and in the range 16.80–35.75° for Mn3 and Mn4. In compound 2 they are in the range (4.54–38.56°). It is also important to examine the dihedral angle or torsion angle φ between the P2 plane and the (P3) plane passing through the N–C–N atoms of the imidazole substituent to assess the deviation from coplanarity of the nitronyl nitroxide radical. In compound 1 the dihedral angles φ (Fig. S9†) are in the range 10.23–24.15° for Mn1–Mn2 (Table S8a†) and in the range 10.45–19.06° for Mn3 and Mn4 (Table S8b†) while for compound 2 it is in the range 7.18–14.27° (Table S10†). These values are relatively small compared with 48.4° found for the nitronyl nitroxyde alone and may be ascribe to the constraint effect of coordination.
Magnetic studies
Magnetic measurements were carried out on bulk crystalline powders, whose X-ray diffraction patterns were checked for agreement with those calculated from electron diffraction crystal structures, as described in the section on crystal structures (Fig. S1 and S2†). {[Mn2(NITIm)3]OTf·CH3OH}n (1) and {[Mn2(NITImMe2)3]ClO4}n (2) exhibit similar magnetic behavior. At 300 K under a magnetic field of 1 kOe, the χT product is 7.45 cm3 K mol−1 (1) (Fig. 4) and 7.65 cm3 K mol−1 (2) (Fig. S10†). These values are well below what would be expected (9.875 cm3 K mol−1) for the uncorrelated spins comprising two Mn2+ (S = 5/2) and three radicals (S = 1/2). For both compounds, upon cooling, χT increases continuously to reach a maximum value around 40 K, a value that is magnetic field-dependent. The inverse of the magnetic susceptibility (1/χ) versus temperature are linear in the temperature range 150–300 K (Fig. 4 (1) and Fig. S10† (2)), following a Curie–Weiss behavior with θ values of 83 K (1) and 87 K (2) and Curie constant C = 5.37 cm3 K mol−1 (1) and C = 5.40 cm3 K mol−1 (2). These behaviors are similar to those shown by {[Mn2(NITBzIm)3]ClO4}n (3).18 This is due to alternating spins S = 5/2 and S = 1/2 which are antiferromagnetically coupled within the 2D metal-radical frameworks. In the temperature range studied, the 2D networks behave like ferromagnetically coupled Stot = 7/2 effective units resulting from two Mn2+ ions antiferromagnetically coupled with three radicals. The magnetic susceptibility was fitted using the mixed quantum-classical Heisenberg model reported for a 2D honeycomb structure with alternating Mn2+ and Cu2+ ions.48 A good agreement with experimental data was obtained for both compounds 1 and 2 in the temperature range 300–150 K, (Fig. 4 and Fig. S10†), which is well within the limit of the given reliability of the model. The best fit gives an antiferromagnetic interaction J = −44.2(4) cm−1 (H = −2JΣSiSj) (with grad = 2.00(2) and gMn = 2.08(2)) between the Mn2+ ion and the radical of compound 1 (Fig. 4) and J = −44.9(5) cm−1 (H = −2JSiSj) with grad = 2.00(2) and gMn = 2.00(2) between the Mn2+ ion and the radical of compound 2 (Fig. S10†). Below 150 K the model is unable to fit the experimental data (Fig. 4) as it does not take into account the three-dimensional magnetic ordering. The goodness of fit value is further supported by these J values, which are of the same order of magnitude as those found in the mononuclear complex [Mn(NITImH)3](ClO4)2 (J = −53(2) cm−1)46 and in 1D compounds with bridging NITIm− nitroxide radicals (−43(3) < J < −86(5) cm−1).49 For such a ferrimagnetic behavior, a minimum of χT is expected at 365 K, but this temperature is above the thermal stability limit for both compounds.48 | ||
| Fig. 4 χT (left, plain square) and 1/χ (right, open squares) vs. T for {[Mn2(NITIm)3]OTf·CH3OH}n (1) at magnetic field H = 1 kOe. The blue (χT) and red (1/χ) solid lines represent the fit of the data with a mixed quantum-classical Heisenberg model as described in the text.48 | ||
For both compounds, the first magnetization curves are sigmoidal. They show two magnetic regions, which are reminiscent of a metamagnetic transition.50 At low magnetic fields (region I), the magnetization increases almost linearly (insert Fig. 5 and Fig. S11†) up to a bend at 225 Oe for 1 and at 100 Oe for 2, as determined by a zero of the second derivative. Beyond these respective values (region II), the magnetization increases asymptotically to reach 6.58μB and 6.23μB at 15 kOe for compounds 1 and 2, respectively (Fig. 5). Further then, they continue to increase slowly, reaching 6.72μB (1) and 6.41μB (2) at 50 kOe, but do not reach the expected M = 7μB saturation value for an effective Stot = 7/2. Similar magnetic behavior was also found for other 2D and 1D coordination polymer-based compounds based on Mn2+ ions and nitronyl nitroxide radicals and it was ascribed to spin-canting within the 2D layers.18,49,51–53
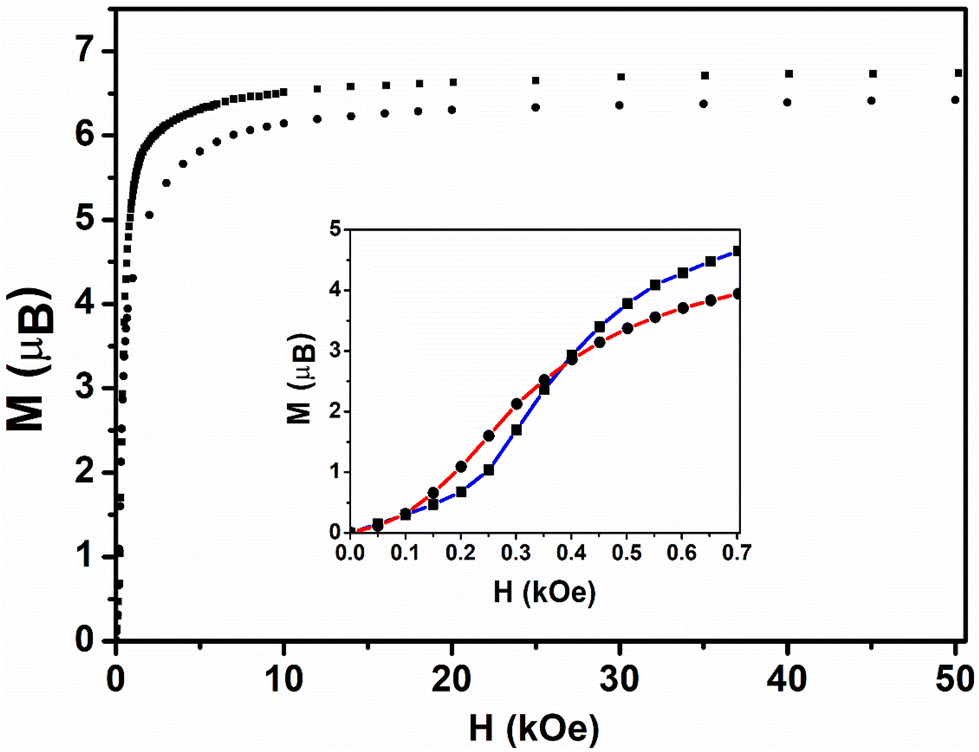 | ||
| Fig. 5 Field dependence of the magnetization for 1 (square) and 2 (circle) at 2 K (1st magnetization). The insert zooms in on the behavior in the low magnetic field range (0–700 Oe). | ||
Zero field cool (ZFC) and field cool (FC) measurements and AC measurements confirm the existence of the two magnetic regions evidenced by first magnetization curves (insert Fig. 5 and Fig. S11†). For compound 1 the ZFC/FC curves shows the onset of bifurcation only for magnetic field above 500 Oe, that is beyond the linear region I of the first magnetization, with a divergence of 3.5μB at 2 K (Fig. 6). For compound 2 bifurcation between the ZFC/FC curves is already observed from 100 Oe with a divergence of 2μB at 2 K (Fig. S16†). The first derivative on FC provides Curie temperatures Tc = 45 K for 1 (Fig. 6) and Tc = 40 K for 2 (Fig. S16†), which are lower than those found for compound 3 (Tc = 55 K).18 It is comparable to the Curie temperature (Tc = 46 K) found for assemblies of Mn2+ ions and trinitroxide radicals with a quartet ground state.54
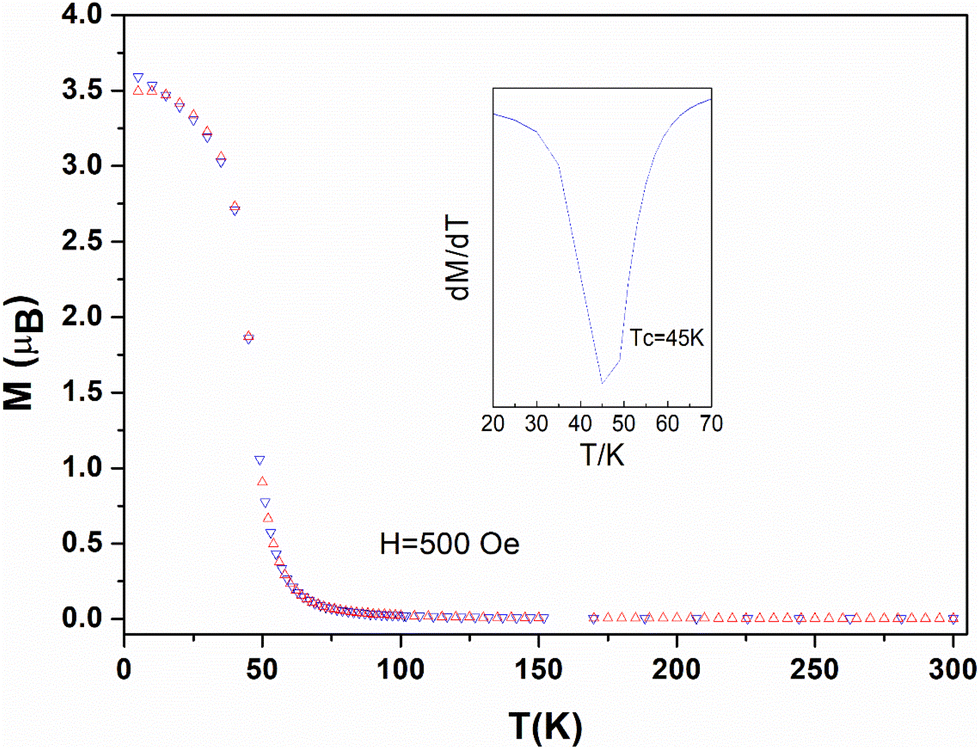 | ||
| Fig. 6 Zero field cool (red triangles) and field cool (blue triangles) magnetization for 1 in a magnetic field of H = 500 Oe. The insert shows the derivative (dT/dM) in the Tc region. | ||
The AC susceptibility data are shown in Fig. 7 (1) and Fig. S17† (2). There are frequency-independent peaks at 43 K for 1 and 39 K for 2 both in the real part (χ′) and imaginary part (χ′′) which are the signature of the onset of three-dimensional long-range ferromagnetic ordering and are in good agreement with Curie temperature found from ZFC/FC measurements. The bulges in the real part (χ′) and peaks in imaginary part (χ′′) visible for 1 at lower temperatures which are little frequency-dependent, are ascribed to slow relaxation of magnetization within the 2D layers.
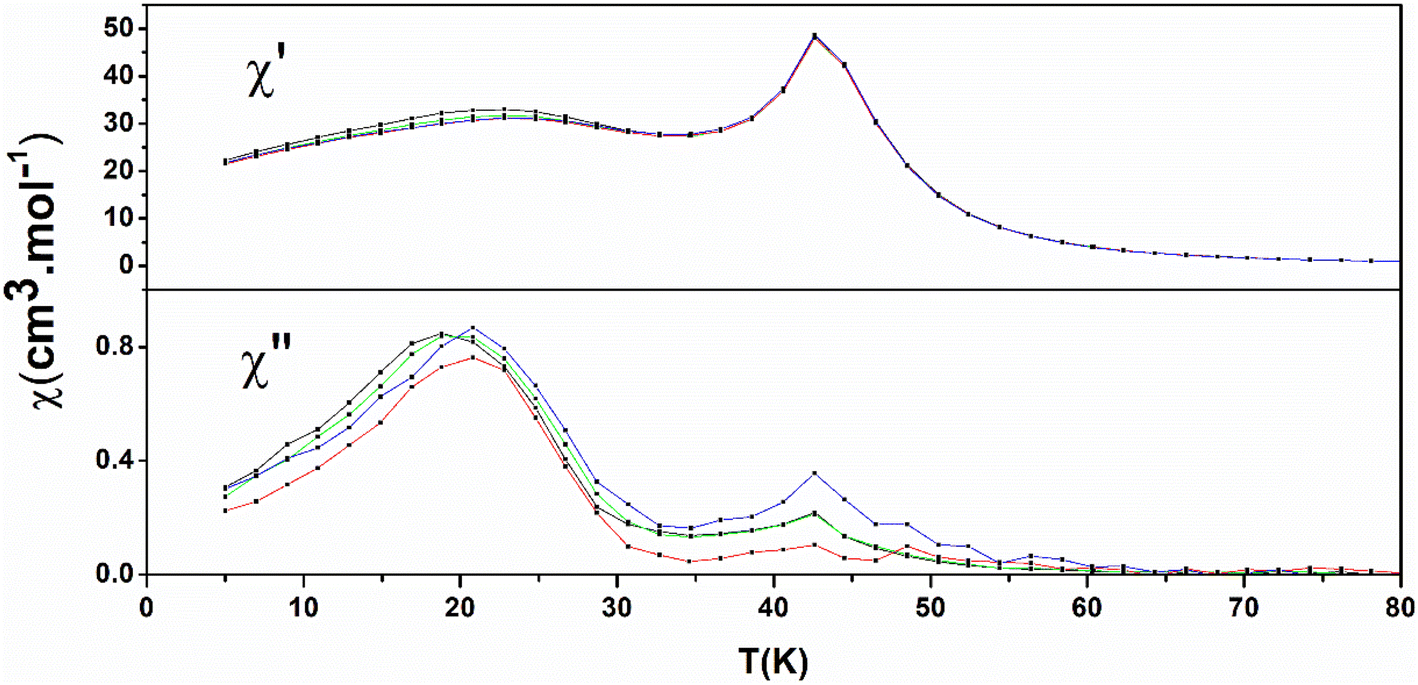 | ||
| Fig. 7 Temperature dependence of the real part χ′ and imaginary part χ′′ of the susceptibility at AC field frequencies 10 (black), 100 (green), 500 (red), and 1400 (blue) Hz and 0 Oe applied dc for compound 1. | ||
Three-dimensional ferromagnetic ordering is confirmed by cycling the field-dependence of the magnetization for both compounds 1 and 2 which exhibit a butterfly-shaped hysteresis that tightens but does not vanishes at zero magnetic field and gives a small but coercive field of 50 Oe at 2 K (Fig. 8 and 9). Reports of metamagnets exhibiting coercive field is not unusual55–60 despite it is not expected as the hysteresis should be confined above the field induce transition.61
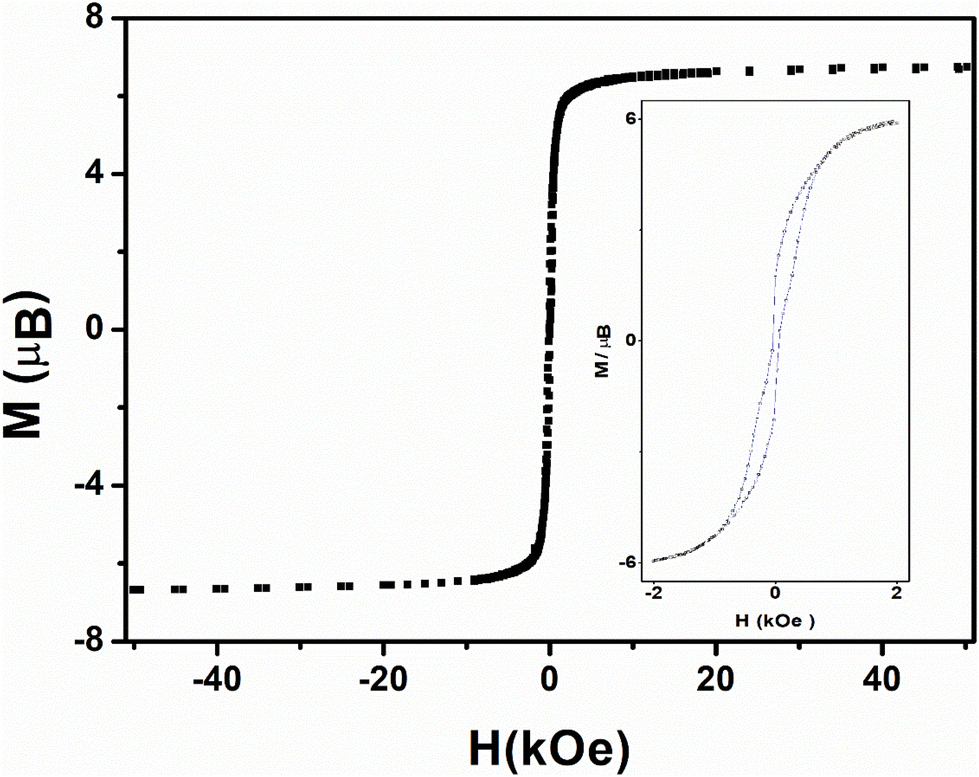 | ||
| Fig. 8 Field dependence of the magnetization upon cycling for 1 (square) at 2 K. The inset zooms in on the hysteresis loop. | ||
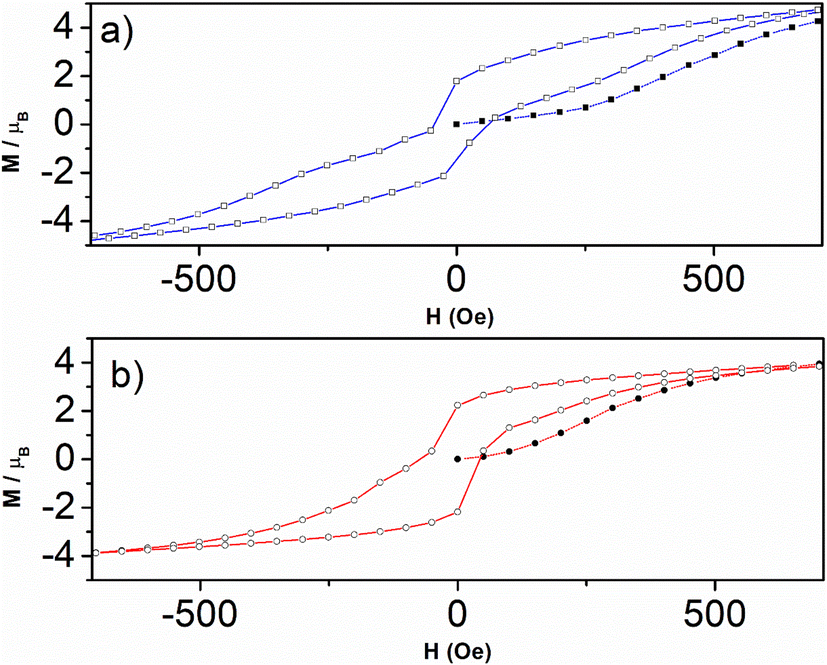 | ||
| Fig. 9 Hysteresis loop zoom (−600 to 600 Oe) for 1 (a, square) and for 2 (b, circle) with first magnetization for 1 (a, filled square) and for 2 (b, filled circle) at scan rate 0.5 Oe s−1. | ||
The explanation for such a coercive but weak field in our case is that once the spins have flipped into the ferromagnetic phase, this induces an increase in magnetic anisotropy, so that their reversal to the antiferromagnetic phase requires more energy. In other words, the sweep rate used in the measurements (0.5 Oe s−1) may be faster than the relaxation time, so the spins may not follow. This results in the constriction around the zero field without canceling out.55,62–64 This may depend on chemical factors, but also on the size and shape of the grains in the compounds.
The hysteresis loops close up at 4μB for both compounds 1 and 2 (Fig. 8), far from the magnetization maximum values of 6.72μB (1) and 6.41μB (2) reached at 50 kOe (Fig. 5), and this argues for spin-canting within the 2D metal-radical framework sheets. In addition, the hysteresis loops for both compounds deviate from the initial magnetization curves. This behavior, together with the temperature dependence of the ZFC magnetic susceptibilities (Fig. S12–S16†) at different magnetic fields, which supports two different magnetic phases, also argues in favor of a metamagnetic transition. The 2D spin-canted metal-radical framework sheets {[MnII2(NITIm)3]+}n (1) and {[MnII2(NITImMe2)3]+}n (2), which are antiferromagnetically coupled below 500 Oe for 1 and 100 Oe for 2, undergo a magnetic field-induced transition to ferromagnetic coupling at higher fields, with the onset of long-range weak ferromagnetic ordering. Metamagnetic transitions in molecule-based magnets have been reported elsewhere,58,63,65–70,71–73 including several cases with spin-canting coexistence.58,71–73 Similar behavior was supported by neutron diffraction studies of previously reported enantiopure 1D-chain molecular ferromagnets.51–53 From the neutron study, the magnetic structure of this compound can be described as stacked canted ferromagnetic sheets that are antiferromagnetically coupled in zero field, but ferromagnetically coupled when a magnetic field is applied.52,53 This was ascribed as due to long-ranged but weak antiferromagnetic dipolar coupling prevailing between the layers with canted spin at low magnetic field while a ferromagnetic coupling between the layers is prevails at high fields. The same magnetic field-induced reversal between antiferromagnetic and ferromagnetic long-range ordering near Tc should occur in compounds 1 and 2. The origin of magnetic anisotropy associated with spin canting is still questionable owing Mn2+ ions and nitronyl nitroxide radicals are not expected to have anisotropy. As in reported Mn-radical chain compounds the dipolar interaction is hold as the main source of the magnetic anisotropy in these layered compounds.47,49,53,74,75 Moreover, the Dzyaloshinskii–Moriya (DMI) antisymmetric exchange interaction may be at the origin of the canted spin as this compound crystallizes in non-centrosymmetric space groups (P21 and P212121).76,77
The fact that compounds 1 and 2, as well as 3, do not exhibit VT behavior like compounds 4–6 supports our hypothesis that the distance between the 2D metal-radical framework layers is a key parameter for inducing the intra-sheet electron-charge transfer. In the previously reported compounds with the NITIm− radical (4–6), the largest interlayer distance was 10.85 Å at room temperature for compound 6 with the PF6− anion, which was the largest anion in the series. For compounds 1–3, this interlayer distance at room temperature (Table S3†) is larger by 0.5 Å for 2 and by more than 1.5 Å for 1 and 3. Moreover, compound 6 was stabilized in the MnIII redox tautomeric form at room temperature {[MnIII2(NITIm)(NITRed)2]PF6}n.19 This suggests that the PF6− anion may induce a higher chemical pressure than the ClO4− and BF4− anions, favoring the stabilization of the redox tautomeric phase characterized by a smaller volume.78 In contrast, compounds 1–3 are stabilized in the MnII tautomeric form {[MnII2(radical)3]X}n (radical: NITIm− (1), NITImMe2− (2), NITBzIm− (3); X = ClO4− (2, 3) or OTf− (1)) and over the whole temperature range (2–300 K). We can see that the densities of compounds 1–3 are lower than those of 4–6 (Table S3†), indicating a decrease in solid state compactness, which may induce less chemical pressure favoring the high-spin tautomer for compounds 1–3. This agrees with the stabilization of the MnIII redox tautomer at higher temperature when a mild external pressure is applied on compounds 4–6.20
As with spin crossover and other phenomena involving electron transfer, features of valence tautomeric conversion such as abruptness and hysteresis depend on cooperativity and flexibility.79–81 Cooperativity is attributed to weak intermolecular interactions and electrostatic contributions and is expected to decrease with increasing interlayer distance in our 2D frameworks.82,83 The flexibility of the molecular system can be seen as its ability to “breathe”. That is being capable of structure transformation upon electron relocation as this involves many small changes in bond lengths and bond angles.79–81
The fact that compounds 1 and 2, as well as 3, do not exhibit VT may be due to a lower flexibility of their 2D metal-radical layers. For 2 and 3, because of the higher steric hindrance of the dimethyl (NITImMe2−) and benzimidazole (NITBzIm−) substituents compared to the NITIm− radical. For compound 1, the triflate anions bind the layers more strongly through intermolecular bonds involving the –CF3 and –SO3− moieties. Thus, it appears that in compounds 1–3, greater separation in-between the 2D metal-radical layers, lower densities and enhanced rigidity combine to stabilize the compounds as {[MnII2(radical)3]X}n.
Conclusion
We have succeeded in synthetizing two new complexes, {[MnII2(NITIm)3]OTf·CH3OH}n (1) and {[MnII2(NITImMe2)3]ClO4}n (2) crystallizing as layered compounds made of 2D metal-radical frameworks {[MnII2(radical)3]+}n with interleaved layers of anions (X) for electro-neutrality and succeed to unveil the crystal structure by using 3D ED. The result is all the more remarkable given that these two compounds form only nano-sized crystalline powders, which could never be grown to a crystal size allowing X-ray diffraction characterization of a single crystal, and are furthermore electron-sensitive due to their organic radical nitronyl nitroxide components.The magnetic characterization shows that the two compounds exhibit a ferrimagnetic behavior within the 2D metal-radical frameworks due to an alternation of antiferromagnetically coupled spins (SMn2+ = 5/2 and Sradical = 1/2). Furthermore, they highlight a field-induced metamagnetic transition from antiferromagnetic coupling (region I) to predominantly ferromagnetic coupling (region II) associated with change of spin-canting angle between the 2D metal-radical layers. Both compounds undergo a long-range 3D weak ferromagnetic ordering in region II with the Curie temperature Tc = 45 K (1) and 40 K (2). This behavior differs from that of the analogous compounds 4–6, which have a similar layered structure, but different anions or substituents on the radical, and exhibit valence tautomerism. Comparative crystal structure analysis highlights the complex interplay between factors such as interlayer distance, anion size, chemical pressure, and framework flexibility to stabilize compounds 1 and 2 as the {[MnII2(radical)3]X}n tautomers.
Moreover, this work illustrates, how crucial it is to obtain crystal structures, and how electron crystallography can be used to cross the crystal size barrier and to elucidate complex structures even with beam-sensitive nanoscale crystals. Even when the resolution does not match the criteria for solving the structure by ab initio methods, other phasing methods such as molecular replacement can be successfully used. Herein we show that by providing a nearly atomic resolution on nanocrystals of compounds 1 and 2, 3D ED was the key to ultimately allowing us to establish magneto-structural correlations for these two compounds. Recent methodological progress in electron diffraction should provide further breakthroughs in the field of molecule-based magnets and other area related to coordination polymers in which conventional X-ray diffraction techniques cannot be used to provide accurate 3D structural information. For our part, we can now envisage systematic studies with metals other than manganese, and tackling the design of 3D compounds based on the same metal-radical approach where crystal structure characterization is essential to ensure we get the right architecture and develop advanced materials.
Experimental
All experimental details on synthesis and characterization including powder XRD patterns and additional Tables and Figures for crystal structures and magnetism are in the ESI† supported by additional references.16–20,84–88Author contributions
E. Yörük., D. Housset., W. L. Ling, S. Kodjikian and H. Klein: ED studies; C. Lecourt, Y. Izumi, C. Desroches, K. Inoue, E. Tretyakov, K. Maryunina: synthesis and magnetic studies. D. Luneau: project conception, discussion and supervision.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors dedicate this paper to the memory of Victor Ovcharenko for his seminal work in the field of molecular magnetism and the metal-radical approach. We thank Dr Guy Schoehn for setting up and maintaining the IBS/ISBG EM platform and for discussion and Emmanuelle Neumann for training on the F20 cryo-electron microscope. IBS acknowledges integration into the Interdisciplinary Research Institute of Grenoble (IRIG, CEA). We acknowledge the support of JSPS Summer Program 2017 for an internship fellowship in Hiroshima University to C. L. We thank the Faculté des Sciences at Université Claude Bernard Lyon 1 for internship support to Y. I. This work was supported by CNRS collaborative research program (PRC 2017-2019 No. 1536), by the Russian Science Foundation (grant No. 21-73-20079) and Magdesign Project (ANR-22-CE29-0018-03). Platforms of the Grenoble Instruct-ERIC center (ISBG; UAR 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology (PSB), supported by FRISBI (ANR-10-INBS-0005-02) and GRAL, financed within the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). The IBS-ISBG electron microscope facility is supported by the Région Auvergne-Rhône-Alpes, Fondation pour la Recherche Médicale (FRM), Fonds FEDER and the GIS-Infrastructures en Biologie Santé et Agronomie (IBiSA). This work was also supported by Grant-in-Aid for Scientific Research (No. 25220803 and 22H02053), CResCent (Chirality Research Center) in Hiroshima University (the MEXT program for promoting the enhancement of research universities, Japan) and Core-to-Core Program, A. Advanced Research Networks.References
- O. Kahn, Molecular Engineering of Coupled Polynuclear Systems - Orbital Mechanism of the Interaction between Metallic Centers., Inorg. Chim. Acta, 1982, 62, 3–14 CrossRef CAS.
- A. Caneschi, D. Gatteschi, R. Sessoli and P. Rey, Toward Molecular Magnets - The Metal-Radical Approch, Acc. Chem. Res., 1989, 22, 392–398 CrossRef CAS.
- E. Coronado, F. Palacio and J. Veciana, Molecule-Based Magnetic Materials, Angew. Chem., Int. Ed., 2003, 42, 2570–2572 CrossRef CAS PubMed.
- K. Inoue and H. Iwamura, Ferromagnetic and Ferrimagnetic Ordering in a 2-Dimensional Network Formed by Manganese(II) and 1,3,5-Tris P(N-tert-Butyl-N-oxyamino)Phenyl Benzene, J. Am. Chem. Soc., 1994, 116, 3173–3174 CrossRef CAS.
- O. Kahn, Chemistry and physics of supramolecular magnetic materials, Acc. Chem. Res., 2000, 33, 647–657 CrossRef CAS PubMed.
- M. Kinoshita, Ferromagnetism of Organic Radical Crystals, Jpn. J. Appl. Phys., 1994, 33, 5718–5733 CrossRef CAS.
- J. S. Miller, Organometallic- and organic-based magnets: New chemistry and new materials for the new millennium, Inorg. Chem., 2000, 39, 4392–4408 CrossRef CAS.
- P. Perlepe, I. Oyarzabal, A. Mailman, M. Yquel, M. Platunov, I. Dovgaliuk, M. Rouzieres, P. Negrier, D. Mondieig, E. A. Suturina, M. A. Dourges, S. Bonhommeau, R. A. Musgrave, K. S. Pedersen, D. Chernyshov, F. Wilhelm, A. Rogalev, C. Mathoniere and R. Clerac, Metal-organic magnets with large coercivity and ordering temperatures up to 242 degrees C, Science, 2020, 370, 587–591 CrossRef CAS.
- H. Tamaki, Z. J. Zhong, N. Matsumoto, S. Kida, M. Koikawa, N. Achiwa, Y. Hashimoto and H. Okawa, Design of Metal-Complex Magnets - Syntheses and Magnetic-Properties of Mixed-Metal Assemblies (Nbu4 MCr(Ox)3)X (Nbu4+=Tetra(Normal-Butyl)Ammonium Ion Ox(2−)= Oxalate Ion M=Mn2+,Fe2+,Co2+,Ni2+,Cu2+,Zn2+), J. Am. Chem. Soc., 1992, 114, 6974–6979 CrossRef CAS.
- E. V. Tretyakov and V. I. Ovcharenko, The chemistry of nitroxide radicals in the molecular design of magnets, Russ. Chem. Rev., 2009, 78, 971–1012 CrossRef CAS.
- E. V. Tretyakov, V. I. Ovcharenko, A. O. Terent’ev, I. B. Krylov, T. V. Magdesieva, D. G. Mazhukin and N. P. Gritsan, Conjugated nitroxides, Russ. Chem. Rev., 2022, 91, RCR5025 CrossRef CAS.
- D. Luneau, Molecular magnets, Curr. Opin. Solid State Mater. Sci., 2001, 5, 123–129 CrossRef CAS.
- J. Ferrando-Soria, J. Vallejo, M. Castellano, J. Martinez-Lillo, E. Pardo, J. Cano, I. Castro, F. Lloret, R. Ruiz-Garcia and M. Julve, Molecular magnetism, quo vadis? A historical perspective from a coordination chemist viewpoint, Coord. Chem. Rev., 2017, 339, 17–103 CrossRef CAS.
- R. Sessoli, H. L. Tsai, A. R. Schake, S. Y. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, High-Spin Molecules - [Mn12O12(O2CR)16(H2O)4], J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS.
- D. Luneau and P. Rey, Magnetism of metal-nitroxide compounds involving bis-chelating imidazole and benzimidazole substituted nitronyl nitroxide free radicals, Coord. Chem. Rev., 2005, 249, 2591–2611 CrossRef CAS.
- K. Fegy, D. Luneau, T. Ohm, C. Paulsen and P. Rey, Two-dimensional nitroxide-based molecular magnetic materials, Angew. Chem., Int. Ed., 1998, 37, 1270–1273 CrossRef CAS PubMed.
- A. Lannes, Y. Suffren, J. B. Tommasino, R. Chiriac, F. Toche, L. Khrouz, F. Molton, C. Duboc, I. Kieffer, J. L. Hazemann, C. Reber, A. Hauser and D. Luneau, Room Temperature Magnetic Switchability Assisted by Hysteretic Valence Tautomerism in a Layered Two-Dimensional Manganese Radical Coordination Framework, J. Am. Chem. Soc., 2016, 138, 16493–16501 CrossRef CAS PubMed.
- D. Luneau, A. Borta, Y. Chumakov, J.-F. Jacquot, E. Jeanneau, C. Lescop and P. Rey, Molecular magnets based on two-dimensional Mn(II)–nitronyl nitroxide frameworks in layered structures, Inorg. Chim. Acta, 2008, 361, 3669–3676 CrossRef CAS.
- C. Lecourt, Y. Izumi, L. Khrouz, F. Toche, R. Chiriac, N. Bélanger-Desmarais, C. Reber, O. Fabelo, K. Inoue, C. Desroches and D. Luneau, Thermally-induced Hysteretic Valence Tautomeric Conversions in Solid State via Two-step Labile Electron Transfers in Manganese-Nitronyl Nitroxide 2D-Frameworks, Dalton Trans., 2020, 49, 15646–15662 RSC.
- C. Lecourt, Y. Izumi, K. Maryunina, K. Inoue, N. Belanger-Desmarais, C. Reber, C. Desroches and D. Luneau, Hypersensitive pressure-dependence of the conversion temperature of hysteretic valence tautomeric manganese-nitronyl nitroxide radical 2D-frameworks, Chem. Commun., 2021, 57, 2376–2379 RSC.
- V. Romanov, I. Bagryanskaya, N. Gritsan, D. Gorbunov, Y. Vlasenko, M. Yusubov, E. Zaytseva, D. Luneau and E. Tretyakov, Assembly of Imidazolyl-Substituted Nitronyl Nitroxides into Ferromagnetically Coupled Chains, Crystals, 2019, 9, 219 CrossRef CAS.
- U. Kolb, T. Gorelik, C. Kübel, M. T. Otten and D. Hubert, Towards automated diffraction tomography:: Part I -: Data acquisition, Ultramicroscopy, 2007, 107, 507–513 CrossRef CAS.
- E. Mugnaioli, T. Gorelik and U. Kolb, “Ab initio” structure solution from electron diffraction data obtained by a combination of automated diffraction tomography and precession technique, Ultramicroscopy, 2009, 109, 758–765 CrossRef CAS.
- I. Nederlof, E. van Genderen, Y. W. Li and J. P. Abrahams, A Medipix quantum area detector allows rotation electron diffraction data collection from submicrometre three-dimensional protein crystals, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2013, 69, 1223–1230 CrossRef CAS.
- M. Gemmi, E. Mugnaioli, T. E. Gorelik, U. Kolb, L. Palatinus, P. Boullay, S. Hovmöller and J. P. Abrahams, 3D Electron Diffraction: The Nanocrystallography Revolution, ACS Cent. Sci., 2019, 5, 1315–1329 CrossRef CAS.
- T. Gruene, J. J. Holstein, G. H. Clever and B. Keppler, Establishing electron diffraction in chemical crystallography, Nat. Rev. Chem., 2021, 5, 660–668 CrossRef CAS PubMed.
- B. L. Nannenga and T. Gonen, MicroED opens a new era for biological structure determination, Curr. Opin. Struct. Biol., 2016, 40, 128–135 CrossRef CAS.
- M. Feyand, E. Mugnaioli, F. Vermoortele, B. Bueken, J. M. Dieterich, T. Reimer, U. Kolb, D. de Vos and N. Stock, Automated Diffraction Tomography for the Structure Elucidation of Twinned, Sub-micrometer Crystals of a Highly Porous, Catalytically Active Bismuth Metal-Organic Framework, Angew. Chem., Int. Ed., 2012, 51, 10373–10376 CrossRef CAS PubMed.
- Y. B. Zhang, J. Su, H. Furukawa, Y. F. Yun, F. Gándara, A. Duong, X. D. Zou and O. M. Yaghi, Single-Crystal Structure of a Covalent Organic Framework, J. Am. Chem. Soc., 2013, 135, 16336–16339 CrossRef CAS.
- T. Rhauderwiek, H. S. Zhao, P. Hirschle, M. Döblinger, B. Bueken, H. Reinsch, D. De Vos, S. Wuttke, U. Kolb and N. Stock, Highly stable and porous porphyrin-based zirconium and hafnium phosphonates - electron crystallography as an important tool for structure elucidation, Chem. Sci., 2018, 9, 5467–5478 RSC.
- H. L. Nguyen, Reticular design and crystal structure determination of covalent organic frameworks, Chem. Sci., 2021, 12, 8632–8647 RSC.
- Z. H. Huang, T. Willhammar and X. D. Zou, Three-dimensional electron diffraction for porous crystalline materials: structural determination and beyond, Chem. Sci., 2021, 12, 1206–1219 RSC.
- B. Q. Yu, W. L. Li, X. Wang, J. H. Li, R. B. Lin, H. L. Wang, X. Ding, Y. C. Jin, X. Y. Yang, H. Wu, W. Zhou, J. P. Zhang and J. Z. Jiang, Observation of Interpenetrated Topology Isomerism for Covalent Organic Frameworks with Atom-Resolution Single Crystal Structures, J. Am. Chem. Soc., 2023, 145, 25332–25340 CrossRef CAS.
- F. Banihashemi, G. H. Bu, A. Thaker, D. Williams, J. Y. S. Lin and B. L. Nannenga, Beam-sensitive metal-organic framework structure determination by microcrystal electron diffraction, Ultramicroscopy, 2020, 216, 113048 CrossRef CAS.
- M. O. Cichocka, Z. Z. Liang, D. W. Feng, S. Back, S. Siahrostami, X. Wang, L. Samperisi, Y. J. Sun, H. Y. Xu, N. Hedin, H. Q. Zheng, X. D. Zou, H. C. Zhou and Z. H. Huang, A Porphyrinic Zirconium Metal-Organic Framework for Oxygen Reduction Reaction: Tailoring the Spacing between Active-Sites through Chain-Based Inorganic Building Units, J. Am. Chem. Soc., 2020, 142, 15386–15395 CrossRef CAS PubMed.
- J. H. Dou, M. Q. Arguilla, Y. Luo, J. Li, W. Z. Zhang, L. Sun, J. L. Mancuso, L. M. Yang, T. Y. Chen, L. R. Parent, G. Skorupskii, N. J. Libretto, C. Y. Sun, M. C. Yang, P. V. Dip, E. J. Brignole, J. T. Miller, J. Kong, C. H. Hendon, J. L. Sun and M. Dinca, Atomically precise single-crystal structures of electrically conducting 2D metal-organic frameworks, Nat. Mater., 2021, 20, 222–228 CrossRef CAS.
- M. Ge, Y. Z. Wang, F. Carraro, W. B. Liang, M. Roostaeinia, S. Siahrostami, D. M. Proserpio, C. Doonan, P. Falcaro, H. Q. Zheng, X. D. Zou and Z. H. Huang, High-Throughput Electron Diffraction Reveals a Hidden Novel Metal-Organic Framework for Electrocatalysis, Angew. Chem., Int. Ed., 2021, 60, 11391–11397 CrossRef CAS.
- J. Hynek, P. Brázda, J. Rohlícek, M. G. S. Londesborough and J. Demel, Phosphinic Acid Based Linkers: Building Blocks in Metal-Organic Framework Chemistry, Angew. Chem., Int. Ed., 2018, 57, 5016–5019 CrossRef CAS.
- S. Leubner, H. S. Zhao, N. Van Velthoven, M. Henrion, H. Reinsch, D. E. De Vos, U. Kolb and N. Stock, Expanding the Variety of Zirconium-based Inorganic Building Units for Metal-Organic Frameworks, Angew. Chem., Int. Ed., 2019, 58, 10995–11000 CrossRef CAS PubMed.
- N. Portolés-Gil, A. Lanza, N. Aliaga-Alcalde, J. A. Ayllón, M. Gemmi, E. Mugnaioli, A. M. López-Periago and C. Domingo, Crystalline Curcumin bioMOF Obtained by Precipitation in Supercritical CO2 and Structural Determination by Electron Diffraction Tomography, ACS Sustainable Chem. Eng., 2018, 6, 12309–12319 CrossRef.
- Y. C. Wang, S. Takki, O. Cheung, H. Y. Xu, W. Wan, L. Öhrström and A. K. Inge, Elucidation of the elusive structure and formula of the active pharmaceutical ingredient bismuth subgallate by continuous rotation electron diffraction, Chem. Commun., 2017, 53, 7018–7021 RSC.
- T. M. Yang, T. Willhammar, H. Y. Xu, X. D. Zou and Z. H. Huang, Single-crystal structure determination of nanosized metal-organic frameworks by three-dimensional electron diffraction, Nat. Protoc., 2022, 17, 2389–2413 CrossRef CAS.
- G. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 2015, 27, 3–8 CrossRef.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, Crystal structure determination and refinement via SIR2014, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS.
- C. Yang, F. Mehmood, T. L. Lam, S. L. F. Chan, Y. Wu, C. S. Yeung, X. G. Guan, K. Li, C. Y. S. Chung, C. Y. Zhou, T. T. Zou and C. M. Che, Stable luminescent iridium(III) complexes with bis(N-heterocyclic carbene) ligands: photo-stability, excited state properties, visible-light-driven radical cyclization and CO2 reduction, and cellular imaging, Chem. Sci., 2016, 7, 3123–3136 RSC.
- K. Fegy, N. Sanz, D. Luneau, E. Belorizky and P. Rey, Proximate Nitroxide Ligands in the Coordination Spheres of Manganese(II) and Nickel(II) Ions. Precursors for High-Dimensional Molecular Magnetic Materials, Inorg. Chem., 1998, 37, 4518–4523 CrossRef CAS.
- A. Caneschi, D. Gatteschi and P. Rey, The Chemistry and Magnetic-Properties of Metal Nitronyl Nitroxide Complexes, Prog. Inorg. Chem., 1991, 39, 331–429 Search PubMed.
- J. Leandri, Y. Leroyer, S. V. Meshkov, Y. Meurdesoif, O. Kahn, B. Mombelli and D. Price, Thermodynamics of a mixed quantum-classical Heisenberg model in two dimensions, J. Phys.: Condens. Matter, 1996, 8, L271–L278 CrossRef CAS.
- K. Fegy, D. Luneau, E. Belorizky, M. Novac, J.-L. Tholence, C. Paulsen, T. Ohm and P. Rey, 1D Manganese(II) Derivatives of an Imidazole-Substituted Nitronyl Nitroxide. An Approach toward Molecular Magnetic Materials of High Dimensionality, Inorg. Chem., 1998, 37, 4524–4532 CrossRef CAS.
- E. Stryjewski and N. Giordano, Metamagnetism, Adv. Phys., 1977, 26, 487–650 CrossRef CAS.
- M. Minguet, D. Luneau, E. Lhotel, V. Villar, C. Paulsen, D. B. Amabilino and J. Veciana, An enantiopure molecular ferromagnet, Angew. Chem., Int. Ed., 2002, 41, 586–589 CrossRef CAS.
- E. Lhotel, D. B. Amabilino, C. Sporer, D. Luneau, J. Veciana and C. Paulsen, Intrinsic avalanches and collective phenomena in a Mn(II)-free radical ferrimagnetic chain, Phys. Rev. B:Condens. Matter Mater. Phys., 2008, 77 CrossRef CAS.
- E. Lhotel, V. R. Simonet, E. B. Canals, D. B. Amabilino, C. Sporer, D. Luneau, J. Veciana and C. Paulsen, Subtle competition bewteen ferromagnetic and antiferromagnetic order in Mn(II)-free radical ferrimagnetic chain, Phys. Rev. B:Condens. Matter Mater. Phys., 2007, 75, 104429 CrossRef.
- K. Inoue, T. Hayamizu, H. Iwamura, D. Hashizume and Y. Ohashi, Assemblage and alignment of the spins of the organic trinitroxide radical with a quartet ground state by means of complexation with magnetic metal ions. A molecule-based magnet with three-dimensional structure and high T-c of 46 K, J. Am. Chem. Soc., 1996, 118, 1803–1804 CrossRef CAS.
- B. W. Lovett, S. J. Blundell, H. Kumagai and M. Kurmoo, Cobalt(II)-hydroxide terephthalate - a metamagnet with a large coercive field, Synth. Met., 2001, 121, 1814–1815 CrossRef CAS.
- C. Dendrinou-Samara, J. P. S. Walsh, C. A. Muryn, D. Collison, R. E. P. Winpenny and F. Tuna, Evidence of Spin Canting, Metamagnetism, Negative Coercivity and Slow Relaxation in a Two-Dimensional Network of {Mn6} Cages, Eur. J. Inorg. Chem., 2018, 485–492 CrossRef CAS.
- K. C. Mondal, G. E. Kostakis, Y. H. Lan, C. E. Anson and A. K. Powell, Spin-Canting and Metamagnetic Behavior in a New Species from the Hydrothermal Co(II)-trans-3-Pyridylacrylate System, Inorg. Chem., 2009, 48, 9205–9213 CrossRef CAS.
- M. H. Zeng, W. X. Zhang, X. Z. Sun and X. M. Chen, Spin canting and metamagnetism in a 3D homometallic molecular material constructed by interpenetration of two kinds of cobalt(II)-coordination-polymer sheets, Angew. Chem., Int. Ed., 2005, 44, 3079–3082 CrossRef CAS.
- J. Boonmak, M. Nakano, N. Chaichit, C. Pakawatchai and S. Youngme, Spin Canting and Metamagnetism in 2D and 3D Cobalt(II) Coordination Networks with Alternating Double End-On and Double End-to-End Azido Bridges, Inorg. Chem., 2011, 50, 7324–7333 CrossRef CAS PubMed.
- D. Shao, Y. Zhou, Q. Pi, F. X. Shen, S. R. Yang, S. L. Zhang and X. Y. Wang, Two-dimensional frameworks formed by pentagonal bipyramidal cobalt(II) ions and hexacyanometallates: antiferromagnetic ordering, metamagnetism and slow magnetic relaxation, Dalton Trans., 2017, 46, 9088–9096 RSC.
- M. L. Taliaferro, F. Palacio and J. S. Miller, The magnetic behaviors of the metamagnetic and ferromagnetic phases of [Fe(C5Me5)2][TCNQ] (TCNQ = 7,7,8,8-tetracyano-p-quinodimethane). Determination of the phase diagram for the metamagnetic phase, J. Mater. Chem., 2006, 16, 2677–2684 RSC.
- O. Cador, D. Price, J. Larionova, C. Mathoniere, O. Kahn and J. V. Yakhmi, Dc and ac magnetic properties of the two-dimensional molecular-based ferrimagnetic materials A(2)M(2) Cu(opba) (3)center dot nsolv A(+)=cation, M-II=Mn-II or Co-II, opba equals ortho-phenylenebis(oxamato) and solv equals solvent molecule, J. Mater. Chem., 1997, 7, 1263–1270 RSC.
- N. Motokawa, S. Matsunaga, S. Takaishi, H. Miyasaka, M. Yamashita and K. R. Dunbar, Reversible Magnetism between an Antiferromagnet and a Ferromagnet Related to Solvation/Desolvation in a Robust Layered [Ru2]2TCNQ Charge-Transfer System, J. Am. Chem. Soc., 2010, 132, 11943–11951 CrossRef CAS.
- A. Paduanfilho, C. C. Becerra and F. Palacio, Hysteresis at the spin-flop transition in the antiferromagnets K2Fe(Cl1-xBrx)5·H2O, Phys. Rev. B:Condens. Matter Mater. Phys., 1991, 43, 11107–11111 CrossRef CAS.
- R. D. Willett, C. P. Landee, R. M. Gaura, D. D. Swank, H. A. Groenendijk and A. J. Vanduyneveldt, Magnetic-properties of one-dimensional spin 1/2 ferromagnets-metagnetic behavior of (C6H11NH3)CuCl3, J. Magn. Magn. Mater., 1980, 15–18, 1055–1056 CrossRef CAS.
- A. Paduanfilho and R. L. Carlin, Phase-boundary of metamagnetic N(CH3)4 NiBr3 (TMNB), Physica B+C, 1987, 145, 16–20 CrossRef CAS.
- H. O. Stumpf, Y. Pei, L. Ouahab, F. Leberre, E. Codjovi and O. Kahn, Crystal-Structure and Metamagnetic Behavior of the Ferrimagnetic Chain Compound MnCu(opba)(H2O)2-DMSO (opba=O-Phenylenebis(oxamato) and DMSO=Dimethyl Sulfoxide), Inorg. Chem., 1993, 32, 5687–5691 CrossRef CAS.
- P. Zhou, B. G. Morin, A. J. Epstein, R. S. McLean and J. S. Miller, Spin Frustration and Metamagnetic Behavior in a Molecular-Based Quasi-1D Ferrimagnetic Chain - (MnTPP)(TCNE), J. Appl. Phys., 1993, 73, 6569–6571 CrossRef CAS.
- O. Kahn, E. Bakalbassis, C. Mathoniere, M. Hagiwara, K. Katsumata and L. Ouahab, Metamagnetic behavior of the novel bimetallic ferromagnetic chain compound MnNi(NO2)(4)(en)(2) (en equals ethylenediamine), Inorg. Chem., 1997, 36, 1530–1531 CrossRef CAS.
- A. S. Ovchinnikov, I. G. Bostrem, V. E. Sinitsyn, A. S. Boyarchenkov, N. V. Baranov and K. Inoue, Quantum dissipation theory of slow magnetic relaxation mediated by domain-wall motion in the one-dimensional chain compound [Mn(hfac)2BNOH], Phys. Rev. B:Condens. Matter Mater. Phys., 2006, 74, 174427 CrossRef.
- M. Kurmoo, H. Kumagai, M. A. Green, B. W. Lovett, S. J. Blundell, A. Ardavan and J. Singleton, Two modifications of layered cobaltous terephthalate: Crystal structures and magnetic properties, J. Solid State Chem., 2001, 159, 343–351 CrossRef CAS.
- E. Q. Gao, Z. M. Wang and C. H. Yan, From manganese(II)-azido layers to a novel three-dimensional molecular magnet: spin canting and metamagnetism, Chem. Commun., 2003, 1748–1749 RSC.
- X. Y. Wang, L. Wang, Z. M. Wang, G. Su and S. Gao, Coexistence of spin-canting, metamagnetism, and spin-flop in a (4,4) layered manganese azide polymer, Chem. Mater., 2005, 17, 6369–6380 CrossRef CAS.
- A. Caneschi, D. Gatteschi, P. Rey and R. Sessoli, Structure and Magnetic Properties of Ferrimagnetic Chains Formed by Manganese(II) and Nitronyl Nitroxide, Inorg. Chem., 1988, 27, 1756–1761 CrossRef CAS.
- A. Caneschi, D. Gatteschi, P. Rey and R. Sessoli, Structure and magnetic-ordering of a ferrimagnetic helix formed by manganese(II) and a nitronyl nitroxide radical, Inorg. Chem., 1991, 30, 3936–3941 CrossRef CAS.
- I. Dzyaloshinsky, A thermodynamic theory of weak ferromagnetism of antiferromagnetics, J. Phys. Chem. Solids, 1958, 4, 241–255 CrossRef CAS.
- T. Moriya, Anisotropic superexchange interaction and weak ferromagnetism, Phys. Rev., 1960, 120, 91–98 CrossRef CAS.
- G. G. Levchenko, V. Ksenofontov, A. V. Stupakov, H. Spiering, Y. Garcia and P. Gutlich, Pressure effect on temperature induced high-spin-low-spin phase transitions, Chem. Phys., 2002, 277, 125–129 CrossRef CAS.
- P. Gutlich, A. Hauser and H. Spiering, Thermal and optical switching of iron(II) complexes, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef.
- J. R. Jiménez, M. Tricoire, D. Garnier, L. M. Chamoreau, J. von Bardeleben, Y. Journaux, Y. L. Li and R. Lescouëzec, A new {Fe4Co4} soluble switchable nanomagnet encapsulating Cs+: enhancing the stability and redox flexibility and tuning the photomagnetic effect, Dalton Trans., 2017, 46, 15549–15557 RSC.
- O. Sato, J. Tao and Y. Z. Zhang, Control of magnetic properties through external stimuli, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS.
- M. Kepenekian, B. Le Guennic and V. Robert, Primary Role of the Electrostatic Contributions in a Rational Growth of Hysteresis Loop in Spin-Crossover Fe(II) Complexes, J. Am. Chem. Soc., 2009, 131, 11498–11502 CrossRef CAS.
- H. Spiering, in Spin Crossover in Transition Metal Compounds III, ed. P. Gutlich and H. A. Goodwin, 2004, vol. 235, pp. 171–195 Search PubMed.
- P. Emsley and K. Cowtan, Coot: model-building tools for molecular graphics, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- C. Hirel, K. E. Vostrikova, J. Pecaut, V. I. Ovcharenko and P. Rey, Nitronyl and imino nitroxides: Improvement of Ullman's procedure and report on a new efficient synthetic route, Chem. – Eur. J., 2001, 7, 2007–2014 CrossRef CAS.
- W. Kabsch, XDS, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS.
- O. Kahn, Molecular Magnetism, VCH, New York, 1993 Search PubMed.
- O. Kovalevskiy, R. A. Nicholls, F. Long, A. Carlon and G. N. Murshudov, Overview of refinement procedures within REFMAC5: utilizing data from different sources, Acta Crystallogr., Sect. D:Struct. Biol., 2018, 74, 215–227 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2330280 (1) and 2330279 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi02257b |
| ‡ Present address: Smart Materials Lab, New York University Abu Dhabi, 129188 Abu Dhabi United Arab Emirates. |
| § Present address: Institut Parisien de Chimie Moléculaire (UMR 8232), Sorbonne Université, 75252 Paris cedex 05, France. |
| ¶ Present address: International Tomography Center SB RAS, 3A Institutskaya Street, Novosibirsk 630090, Russian Federation and Novosibirsk State University, Pirogova Str., 2, Novosibirsk 630090, Russian Federation. |
| This journal is © the Partner Organisations 2025 |