
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCritical assessment of the exsolution process in Cu-doped SrTiO3 by a combined spectroscopic approach†
Pietro
Mariani
a,
Xiao
Sun
 bc,
Simone
Mascotto
*c,
Luisa
Raimondo
a,
Adele
Sassella
a,
Damiano
Monticelli
d,
Enrico
Berretti
e,
Alessandro
Lavacchi
e,
Matus
Stredansky
f,
Cinzia
Cepek
f,
Silvia
Mostoni
a,
Carlo
Santoro
a,
Barbara
Di Credico
a,
Roberto
Scotti
a and
Massimiliano
D'Arienzo
*a
bc,
Simone
Mascotto
*c,
Luisa
Raimondo
a,
Adele
Sassella
a,
Damiano
Monticelli
d,
Enrico
Berretti
e,
Alessandro
Lavacchi
e,
Matus
Stredansky
f,
Cinzia
Cepek
f,
Silvia
Mostoni
a,
Carlo
Santoro
a,
Barbara
Di Credico
a,
Roberto
Scotti
a and
Massimiliano
D'Arienzo
*a
aUniversity of Milano-Bicocca, Dept. of Materials Science, INSTM, Via R. Cozzi 55, 20125 Milano, Italy. E-mail: massimiliano.darienzo@unimib.it
bDeutsches Elektronen-Synchrotron DESY, Notkestr. 85, 22607 Hamburg, Germany
cInstitut für Integrierte Naturwissenschaften, Univertsität Koblenz, Germany. E-mail: mascotto@uni-koblenz.de
dUniversity of Insubria, Dept. of Science and High Technology, Via Valleggio 11, Como, Italy
eInstitute of Chemistry of the Organometallic Compounds – National Research Council (ICCOM-CNR), Via Madonna del Piano, 10, 50019 Sesto Fiorentino (FI), Italy
fIstituto Officina dei Materiali-CNR Laboratorio TASC, Strada Statale 14, km 163.4, I-34012, Trieste, Italy
First published on 22nd November 2024
Abstract
The surface transformation and defect evolution of Cu-doped SrTiO3 upon copper exsolution have been studied by exploiting a multi-technique approach which integrates, for the first time, common methods describing exsolution like XAS, XPS and STEM with unconventional strategies, namely electron paramagnetic resonance (EPR) and UV-Vis diffuse reflectance (UV-DRS). XAS and EPR indicated that copper is present in the matrix in a disordered coordination environment as amorphous Cu2O and CuO located at the surface and as substitutional Cu2+ lattice species with a distorted octahedral structure. Interestingly, EPR unveiled that, during exsolution, Cu2+ surface sites with disordered coordination primarily migrate undergoing selective reduction, while a delay is observed for the lattice defects. UV-DRS resulted in a valid alternative to HRTEM to determine the size of exsolved nanoparticles by tracking the plasmon resonance effect. Moreover, when XANES showed the complete regain of the pristine state of Cu after reoxidation, both UV-DRS and EPR highlighted that the original features are not entirely restored. These outcomes suggest that the chemical environment of exsolvable species is much more heterogeneous and the exsolution process much less straightforward than expected. Thus, alternative and original characterization techniques should be exploited to provide a solid methodological benchmark for an effective evaluation of this phenomenon.
Introduction
Supported metal nanoparticles play a fundamental role in catalysis due to their high surface area and reactivity that enable high reaction rates and process efficiency.1 The usual synthetic route for these catalysts follows suspension impregnation techniques. The latter intrinsically engender loosely bonded nanoparticles that easily migrate over the substrate surface leading to coalescence or sintering during high-temperature and prolonged operation. This process causes catalyst deactivation.2 Another ageing mechanism is surface poisoning caused by the deposition of carbon and sulphur compounds, with the consequent activity decrease of the catalyst surface.3 Due to the irreversible nature of these processes, traditional catalyst recycling and even substitution are still expensive procedures to pursue.4 These considerations become particularly relevant for perovskite oxides (ABO3), an important class of materials widely employed in several applications, including photovoltaics, heterogeneous (photo)catalysis and energy storage.5,6To overcome these issues, over the last decade a new approach design has been introduced. This procedure provides a relatively simple, single-step, synthetic pathway to support nanoparticles on perovskite oxide materials.7–9 This route entails the possibility of incorporating, under oxidizing conditions, catalyst cations in the B-site of the perovskite lattice and then, upon subsequent reduction, partly exsolve them forming nanoparticles. The process, commonly known as redox exsolution,10 was initially proposed only for Pd nanoparticles11 and successively extended to several noble and non-noble transition metals (Ni, Co, Fe, Cu, Mn, and Pt), leading to a rapid growth of interest from the scientific community.12
The crystallographic alignment between the exsolved nanoparticles and the host phases provides their partial embedding (“socketed”) in the support phase. The result is sintering-resistant nanoparticles with the ability to redissolve into the lattice of the oxide support under an oxidative atmosphere. Such reversibility allows for regeneration of the materials through redox treatment, also alleviating agglomeration and enhancing the overall lifetime. This process fosters the utilization of these materials in a circular economy perspective.13–15 Moreover, the specific features of socketing and alignment protect the nanoparticles from poisoning mechanisms, preventing also sintering and coalescence processes.16
SrTiO3-based parental structures have been widely adopted for exsolution because of their high chemical stability and well-known defect chemistry.17,18 In particular, the control of composition,19 morphological features,20 exposed crystal surfaces13 and, specifically, degree of nonstoichiometry have been demonstrated as key triggers of exsolution in these materials. In fact, as comprehensively described by Irvine et al., exsolution emerges preferentially on the surface when highly A-site-deficient perovskites (A/B < 1) are employed.19 In detail, when an A-site-deficient perovskite is reduced, oxygen vacancies 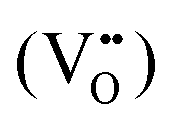 are introduced. For example, in A1−αBO3−δ (α symbolizes the A-site deficiency while δ the number of oxygens removed per formula unit of perovskite, respectively),
are introduced. For example, in A1−αBO3−δ (α symbolizes the A-site deficiency while δ the number of oxygens removed per formula unit of perovskite, respectively), 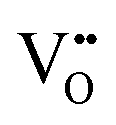 destabilizes the perovskite lattice and spontaneous exsolution of B-site species may locally be induced in an attempt to restore the stoichiometry. The deliberate doping with acceptor metal dopants goes in the same direction, generating additional
destabilizes the perovskite lattice and spontaneous exsolution of B-site species may locally be induced in an attempt to restore the stoichiometry. The deliberate doping with acceptor metal dopants goes in the same direction, generating additional 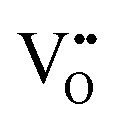 , which serve as preferential nucleation sites for exsolved nanoparticles.21
, which serve as preferential nucleation sites for exsolved nanoparticles.21
Therefore, engineering and monitoring the defect chemistry of the host oxide appear a challenging task to link the phenomena occurring at the nanoscale with the macroscopic nanoparticle exsolution.
To date, most of the efforts in the literature are devoted to the screening of the exsolution process in host oxides with controlled composition and, consequently, with different degrees of non-stoichiometry.19Ex situ imaging studies with conventional, high-resolution or scanning transmission electron microscopy (STEM) and scanning electron microscopy (SEM) techniques have been widely exploited.22,23 More complex in situ X-ray diffraction (XRD) or absorption spectroscopy (XAS) studies, often requiring synchrotron radiation, were also presented.24,25 However, since the process itself may cause structural evolution in the host oxide compositions, such as Sr segregation26 or further defect formation,27,28 a certain degree of uncertainty in determining point defect generation and evolution is still entailed in these methods.
In this context, electron paramagnetic resonance (EPR) may provide a sensitive spectroscopic method to elucidate the defect structure of transition-metal doped perovskite oxides, in terms of the coordination environment and aggregation state of the exsolving metal ions.29 This technique can also reveal the dynamic processes involving oxygen and strontium vacancies within these materials.30 Moreover, since the EPR detection limit even for a standard X-band setup is typically on the order of 1011 spins,31 far below the defect concentrations typically relevant as dopant levels for exsolution (0.5–10.0 mol%),32,33 it appears to be a useful strategy to evaluate the extent of the exsolution reversibility.
Furthermore, UV-Vis diffuse reflectance (UV-DRS) can effectively help to characterize the local titanium environment and the oxygen defect, as well as to gain more information about the coordination and aggregation state of dopant metal species in the perovskite lattice.34 In this frame, Kittilstved et al.35 exploited UV-DRS to track metal (Cr, Fe, Mn)-doped SrTiO3 native point defects, such as oxygen vacancies or Ti3+, which are known to give rise to visible light absorption in bulk SrTiO3. In particular, it was suggested that the broad absorption feature in the visible region that tails into the near-infrared region in the Cr-doped SrTiO3 may be attributed to electronic transitions involving V×O, 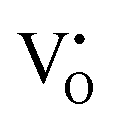 and
and 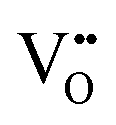 species introduced during synthesis. In fact, these peaks have also been observed in SrTiO3 after vacuum annealing.
species introduced during synthesis. In fact, these peaks have also been observed in SrTiO3 after vacuum annealing.
Despite the potentiality of EPR and UV-DRS in monitoring the changes in the electronic structures of both intrinsic defects and extrinsic dopants in perovskite materials, to the best of our knowledge, there are no studies which attempted to follow exsolution in metal-doped SrTiO3 taking advantage also of these spectroscopic techniques.
In this scenario, the present work aims at the investigation of surface transformation and defect evolution of Cu-doped SrTiO3 systems upon metallic copper exsolution, with special emphasis on the possibility of studying the process via UV-DRS and EPR measurements.36–41
In detail, A-site-deficient perovskite oxides of the type Sr0.95Ti1−αCuαO3−δ with α = 0, 0.005, 0.05, 0.15 were successfully synthesized by using a tailored sol–gel method, while surface decoration with small metallic nanoparticles was achieved through exsolution. The copper-based system was selected as a model low-cost alternative material for a plethora of catalytic reactions such as soot combustion,42 NOx storage and reduction,43 water gas shift reaction44 or CO2 hydrogenation.45,46 Besides, Cu(II) ions are paramagnetic species, thus giving rise to a peculiar fingerprint in the EPR spectra as a function of their coordination environment.
A multi-technique approach, combining temperature-programmed reduction (TPR), XAS, XPS and STEM, was exploited to follow the exsolution pathway of the Cu species. Interestingly, these consolidated techniques for the description of the exsolution were integrated with unconventional methods, namely EPR and UV-DRS, which proved to be original and effective strategies to critically understand and unravel hidden aspects of the phenomenon, thanks to the possibility of monitoring the modifications of the inner sphere copper coordination and the evolution of oxygen during the exsolution process.
The results, besides evoking a heterogeneous chemical environment for the exsolvable species and rather more complex phenomena than expected, may lay the background for a new methodological route for a more effective evaluation of exsolution in doped perovskites.
Materials and methods
Materials
Strontium nitrate (99%, Acros Organics), titanium(IV) isopropoxide (97%, Alfa Aesar), copper(II) acetate (98%, Alfa Aesar), anhydrous citric acid (99.6%, Acros Organics), glycerol (99%, Alfa Aesar), and glacial acetic acid (99.7%, Fisher Scientific) were used as received without further purification.Synthesis of perovskite materials
A modified Pechini synthesis route47 was utilized for the preparation of Sr0.9Ti1−αCuαO3±δ (Cu-STO) perovskite nanoparticles (α stands for the percentage of doping in the B-site). In preference to the conventionally used ethylene glycol, glycerol was selected as the polyol, due to its better chelating and cross-linking features.47,48 In a typical synthesis titanium(IV) isopropoxide (Ti(OCH(CH3)2)4) was added in 15 mL of glycerol and stirred at room temperature for 60 minutes; then 7.82 g of citric acid was added, and the temperature was increased to 60 °C for 60 minutes in an oil bath. Successively, Sr(NO3)2 and Cu(CO2CH3)2 were dissolved in 1–2 mL of water and added to the Ti solution, in a series, at 60 min intervals under stirring. The final molar ratio of the total amount of metal cations to glycerol and to citric acid was set to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:33.3.
:8:33.3.
After 2 more hours of stirring, the temperature of the solution was increased to 130 °C and left to stir again for 2 hours in order to foster the polyol (glycerol) and the polycarboxylic acid (citric acid) polycondensation reaction. This procedure resulted in the generation of a sticky polyester gel.
Finally, the precursor gel was calcined in a muffle furnace at 400 °C for 2 hours and then at 900 °C for 2 hours (ramp rate: 2 °C min−1). The obtained powders were then treated with 200 mL of aqueous acetic acid solution (5 vol%) and stirred for 1 h to remove surface carbonate impurities, and finally washed with deionized water until reaching a neutral pH. The prepared perovskites were named xCu-STO, where x refers to the nominal mol mol−1 percentage of the Cu-dopant in the B-site.
Exsolution procedure
Exsolution/reduction treatments were carried out using a U-shaped quartz tube filled with 200 mg of each sample. First, 5% H2/Ar was chosen as the reducing gas, and the flow rate was set at 20 mL min−1. A ramping rate of 10 °C min−1 was employed to reach the final temperatures of 200 °C, 400 °C, 600 °C, 800 °C or 900 °C with a dwell time of 1 h. Samples that underwent the reduction treatment at 900 °C were labelled as xCu-STO_r, where r stands for the reducing procedure.Re-oxidation process
The re-oxidation process was performed by putting the sample in a round bottom quartz tube inserted in a tubular oven, under an O2 flow of around 100 ml min−1. The process was performed at a temperature of 50 °C higher than the reduction temperature and maintained at this temperature for 2 hours to allow the complete reoxidation and redispersion of the metal in the lattice. Samples that underwent re-oxidation at 950 °C were labelled as xCu-STO_ox, where ox stands for the re-oxidation treatment.Materials characterization
Powder X-ray powder diffraction (PXRD) patterns were recorded using a Rigaku Miniflex 600. The acquisition was performed using a Cu source (40 kV, 15 mA), scanning in the 10–90° 2θ range, with a step size of 0.02 degrees and an angular velocity of 1.0 degree per minute. Instrumental PDXL-2 software was used for the sake of comparison with reference diffraction patterns from the ICDD database. The mean crystallite sizes were calculated from the full width at half-maximum (fwhm) of the most intense reflection using the Scherrer equation.The concentration of the metal ions (Sr, Ti, and Cu) was determined with an inductively coupled plasma optical emission spectrometer (ICP-OES, Optima 7000 DV, PerkinElmer, MA, United States).
The temperature-programmed reduction measurements under a H2 environment (H2-TPR) were performed with a Micromeritics AutoChem 2920 analyzer. The catalyst (∼100 mg) was pretreated under a 5% H2 stream balanced in Ar (20 mL min−1) at room temperature for 2 h. Then, the sample was heated up to 120 °C with a ramp rate of 10 °C min−1 and a dwell time of 1 h. Then the temperature was increased up to 950 °C at a heating rate of 10 °C min−1 and a second isotherm of 1 h was performed. The amount of consumed H2 was measured using a thermal conductivity detector (TCD).
UV-DRS spectra were recorded in the spectral range from 300 to 1400 nm using a PerkinElmer Lambda 1050+ UV/Vis/NIR spectrophotometer equipped with a 10 cm diameter integrating sphere on the as-prepared powdered samples, after the thermal reducing treatment at each temperature and after re-oxidation at 950 °C.
The surface chemical composition of the Cu-STO powders was investigated by X-ray photoelectron spectroscopy (XPS). The measurements were performed on the samples in powder form, fixed on the sample holder using UHV-compatible carbon tape. The reduced 5Cu-STO_r sample at 900 °C was transported and mounted in the XPS UHV apparatus under a nitrogen atmosphere, paying utmost attention to minimizing any possible modifications/re-oxidation due to atmospheric exposure. The XPS spectra were acquired under ultrahigh vacuum (base pressure: ∼5 × 10−10 mbar) at RT in a normal emission geometry using a conventional Mg X-ray source (hν = 1253.6 eV) and a hemispherical electron energy analyzer (120 mm by PSP: total energy resolution ∼0.8 eV). Due to charging effects, all binding energies (BE) were calibrated by fixing the BE of the Ti 2p3/2 core level relative to STO at 458.3 eV.49 Survey scans were acquired in the 0–1100 eV energy range, and detailed scans were recorded in the BE regions corresponding to O 1s, C 1s, Sr 3d, Ti 2p, and Cu 2p core levels. The XPS spectra were analyzed by fitting the experimental data using integral background and Voigt components, corresponding to different oxidation states and chemical environments. Details of the fitting procedure and parameters are provided in the ESI.† The atomic ratio was calculated from the peak area and the sensitivity factor of our instrument.
A Thermo Fisher Talos F200X G2 was used for high-resolution scanning transmission electron microscopy (STEM) imaging with an acceleration voltage of 200 kV and a resolution of 4096 × 4096 pixels without any objective apertures. Super X spectrometers equipped with silicon drift detectors of 30 mm2 were used to collect energy dispersive X-ray analysis (EDX) maps with a collection angle of 0.7 s rad.
X-ray absorption spectra at the Cu K-edge (ECu = 8979 eV) were acquired at the P64 beamline at the Deutsches Elektronen Synchrotron (DESY, Hamburg, Germany).50 A Si(111) double crystal monochromator was employed for the energy scans around the respective metal absorption edge. The EXAFS spectra were collected in a fluorescence geometry using gas ionization chambers to measure the photon intensities before and after the interaction with the samples. The materials were diluted in cellulose, pressed into pellets of around 28 μm, and then covered in Kapton tape before placing them in the beam path. Normalization of the obtained XANES data was carried out by performing pre-edge and post-edge background subtraction in the Athena software using the AUBACK algorithm.51 The EXAFS analysis was performed using a Python script ‘xaesa’ provided by the beamline.
EPR measurements were performed by using a Bruker EMX spectrometer operating at the X-band frequency and equipped with an Oxford cryostat working in the temperature range of 4–298 K. A tailored home-made system was utilized, where the sample in the EPR tube can be directly heated up and connected both to a high-vacuum pumping system and to a controlled gas feed (Ar/H2 95/5 wt% and O2) to provide a reducing or oxidation atmosphere. Spectra were recorded at a power of 5–63 mW under in vacuo conditions (p < 10−5 mbar) and at 130 K both on the as-prepared powders and after their thermal treatment at 200 °C, 400 °C, 600 °C, 800 °C or 900 °C under an Ar/H2 atmosphere. To check the reversibility of the process, EPR survey was also performed on the same perovskite materials after re-oxidation in O2 at 950 °C.
Results and discussion
Structural and morphological characterization of parental materials
The crystalline phase structures of the as-prepared materials were assessed using PXRD patterns and are summarized in Fig. 1.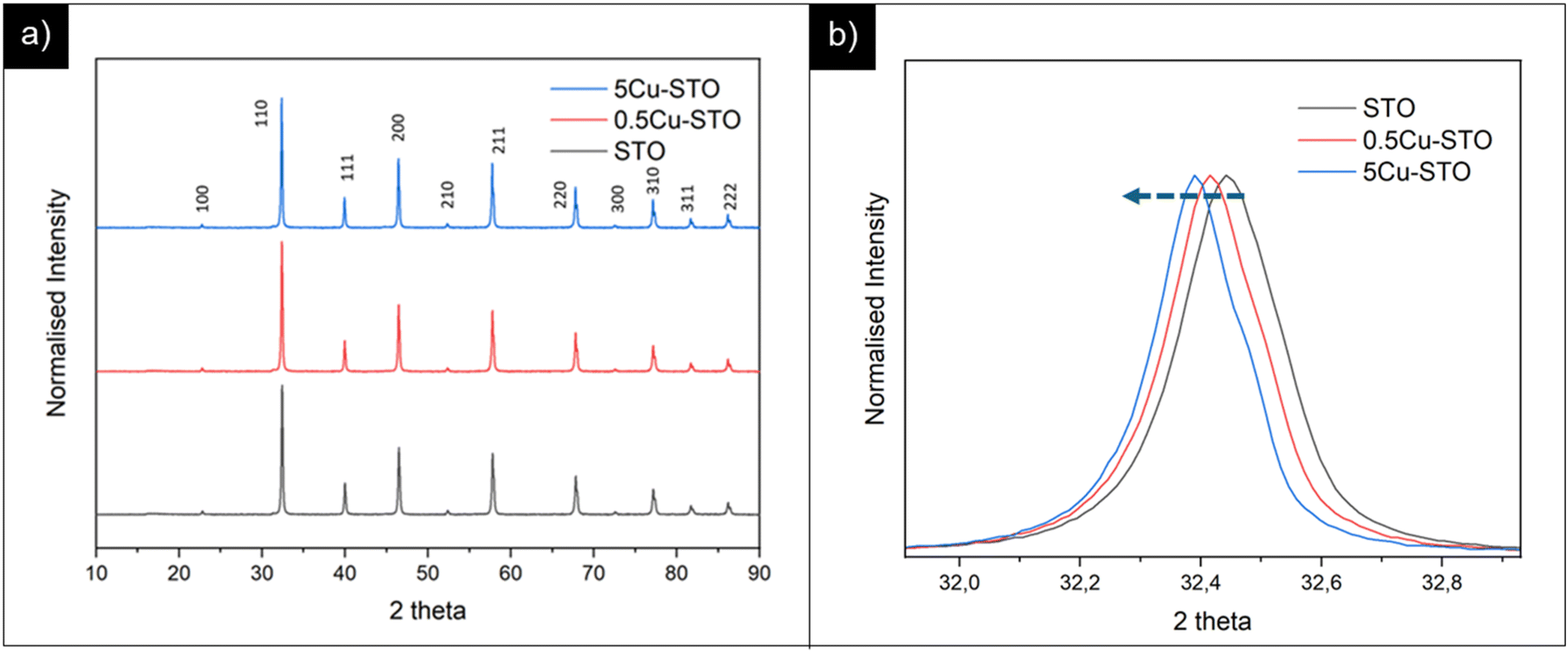 | ||
| Fig. 1 (a) PXRD patterns of pristine STO and of copper doped perovskite parental structures. (b) Highlight on the main reflection shift before and after copper doping. | ||
All the samples display similar structures corresponding to the cubic Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m structure of the SrTiO3 perovskite oxide without any indication of other phase segregation.
m structure of the SrTiO3 perovskite oxide without any indication of other phase segregation.
Interestingly, we found that the peak positions for the SrTiO3 phase in the doped samples slightly shifted to a lower angle compared with those of the pristine matrix. This observation indicates an increase of the cell volume upon incorporation of larger Cu2+ ions in the smaller Ti4+ sites (Cu2+ = 0.73 Å > Ti4+ = 0.61 Å).52 Fitting of PXRD data (Fig. S1†) gave more quantitative information on of these structural changes upon dopant incorporation, revealing a slight increase in the lattice parameter a for the cubic perovskite from 3.904 and 3.910 Å for pristine STO and 5Cu-STO. This demonstrates that a successful doping of Cu atoms into the STO lattice is generally attained.
To clarify the status of the Cu dopant, we performed X-ray absorption spectroscopy (XAS) experiments. The spectra of the as-prepared 0.5Cu-STO and 5Cu-STO were measured around the Cu K-edge (E = 8979 eV, Fig. 2). Compared to the absorption edge of the Cu foil (8979 eV), the peak positions of the first derivative of the XANES curves of 0.5Cu-STO and 5Cu-STO are obtained at 8981.5 and 8983.5 eV, respectively. These values are slightly higher than that of Cu0. Moreover, the energies at their half-maximum values are higher than those of Cu foil and Cu2O. Moreover, the absorption spectrum of 0.5Cu-STO overlaps with that of CuO, indicating that Cu in 0.5.Cu-STO is present as Cu(II), whereas in 5Cu-STO a blue shift is evident, thus indicating that the average copper valence state is slightly above +2. This shift is possibly explained by the different and more complex chemical environments of copper species in this sample. The shape of the XANES curves of both doped materials are very similar and agree with other literature works on Cu-doping in SrTiO3.53 A careful comparison with the spectral features of the reference materials shows that copper does not completely substitute Ti in the perovskite lattice and some impurities are present. In fact, the pre-edge feature at 8983 eV in 0.5Cu-STO reveals the presence of Cu2O contaminations, whereas the shoulder at 8987 eV clearly visible in 5Cu-STO is indicative of CuO traces. To further confirm our conclusions, we analyzed the Fourier transformation of extended X-ray absorption fine structure (EXAFS) spectra of these samples (see the ESI†). The coordination numbers obtained from the fittings are listed in Table S2.† For 0.5Cu-STO and 5Cu-STO the patterns can be fitted using two shells with Cu–O bond lengths of about 1.95 Å and 1.59/2.25 Å, respectively. This bond length matches those of the Cu–O bond in CuO and Cu2O. However, the coordination number is reduced from 4 to 3.14 for 5Cu-STO. This finding might be indicative of substitutional copper (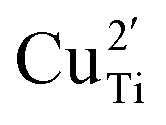 or
or 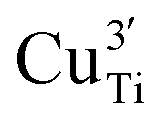 ) along with one or two oxygen vacancies
) along with one or two oxygen vacancies 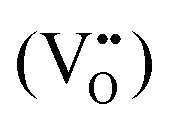 .
.
 | ||
| Fig. 2 (a) X-ray absorption spectra. (b and c) SEM images of the 0.5Cu-STO and 5Cu-STO parental structures, respectively. | ||
Representative SEM micrographs of 0.5Cu-STO and 5Cu-STO are reported in Fig. 2b and c. 0.5Cu-STO displays grains of size varying between ∼100 and ∼200 nm, while larger particles (lengths ranging from ∼200 to ∼400 nm) were detectable for 5Cu-STO powders.
Monitoring copper exsolution from SrTiO3
The exsolution process was firstly monitored with mass spectrometry to gain insights into the reduction of the materials during the high-temperature treatment with hydrogen. In particular, the evolution of the water signal was followed (Fig. 3a) since it is directly correlated with the release of lattice oxygen and thus with the progressive reduction of copper ions according to the reactions:| H2 + O2− → H2O + 2e− |
| Cu2+ + e− → Cu+ |
| Cu+ + e− → Cu0 |
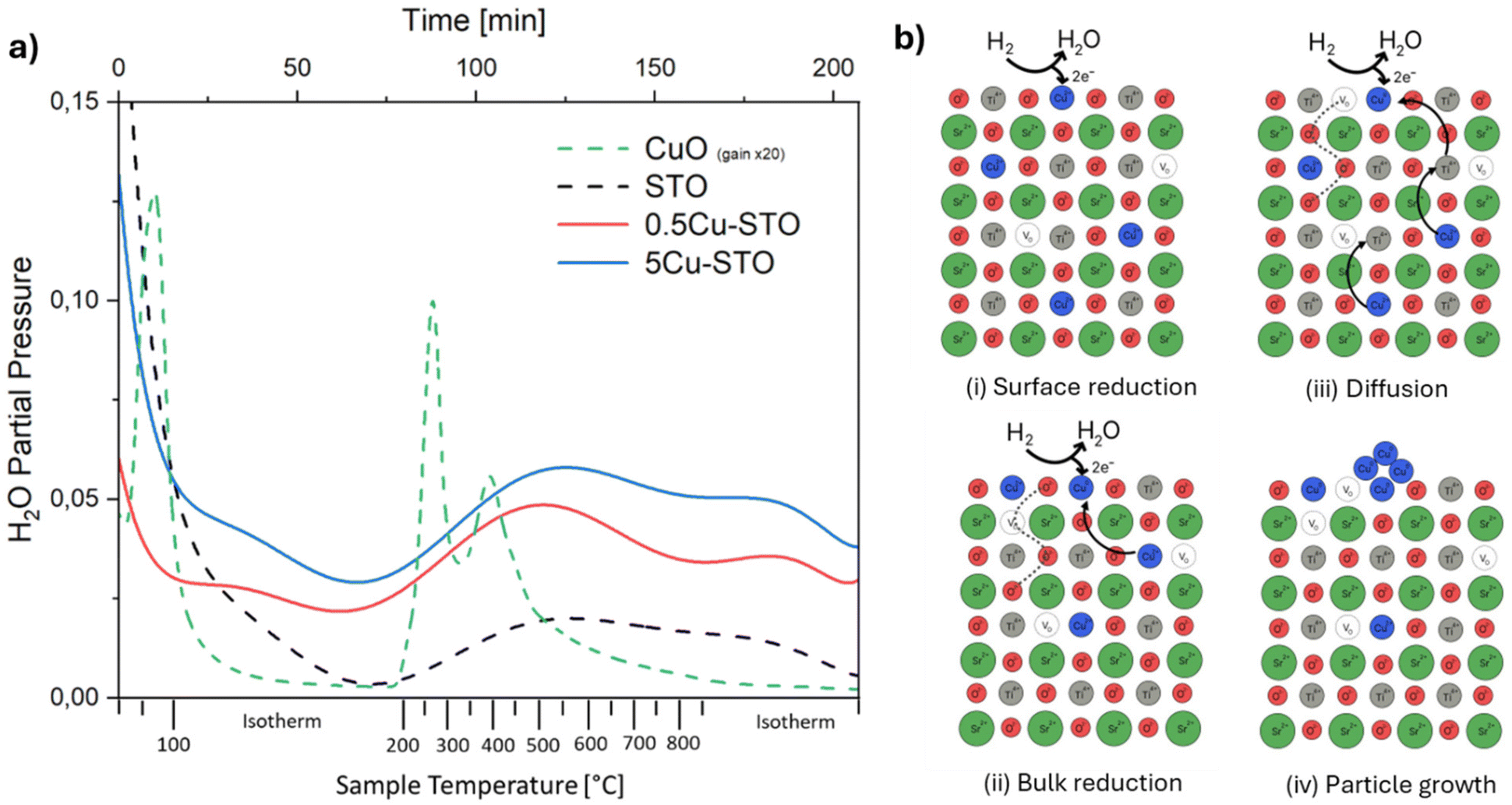 | ||
| Fig. 3 (a) Water evolution during exsolution from mass spectrometry for Cu-STO powders, CuO–alumina and bare STO reference samples. (b) Simplified representation of the expected Cu exsolution process from SrTiO3. | ||
To provide a benchmark for understanding the phenomena occurring in the doped STO samples, undoped SrTiO3 and a mixture of CuO (5% mol mol−1) in commercial alumina were initially analyzed as control samples. The samples display high water desorption during the isothermal treatment at 120 °C which is ascribed to adsorbed water from the environment.
The TPR profile of reference copper oxide shows two main peaks at T ≈ 270 °C and ≈420 °C, which can be attributed to the reduction of Cu2+ species in different chemical environments. It must be mentioned that literature studies generally indicate CuO reactions in H2 with the occurrence of a first reduction from Cu(II) to Cu(I) at temperatures between 210 and 270 °C, and a second reduction stage at higher temperatures from Cu(I) to metallic copper.54,55
For Cu-STO materials, two very broad features in the temperature range of 250–870 °C ascribable to two different reduction steps can be detected. By comparing these TPR profiles with that of the reference CuO sample, we may suggest that the first main signal involves surface copper species with different coordination environments i.e. different reduction abilities. The second shoulder occurring during the isothermal step at 870 °C is most likely ascribed to the lattice oxygen release from the perovskite matrix,56 which fosters the migration of the doping cation toward the surface and its consequent reduction to the elemental state. This passage is followed by spontaneous nucleation that drains further metal atoms from the bulk enlarging the nanoparticle and starting the growth process. The overall expected exsolution process of Cu from Cu-SrTiO3 is schematized in Fig. 3b.
Interestingly, the same reduction behavior is observed also for the bare A-site deficient undoped STO material. Here the two main signals are likely ascribed to the release of oxygen from the surface (T = 250–600 °C) and the lattice (T = 870 °C) in an attempt to restore the stoichiometric perovskite structure.
The reduction process of the copper species was assessed in detail with XAS (Fig. 4). After the reduction procedures, both 0.5Cu-STO and 5Cu-STO show shifts towards lower energies in the half-maximum positions of the spectra. These results indicate that the Cu dopant in both samples is partially reduced to Cu+ or even to metallic Cu. However, after the reduced samples were re-oxidized under an O2 atmosphere at 950 °C, 0.5Cu-STO (Fig. 4a) showed only a small shift compared to 5Cu-STO. This suggests that the exsolved Cu cannot easily get completely re-oxidized most likely because the reduction process occurs also in the bulk of the SrTiO3 matrix.25,57 However, for the re-oxidized 5Cu-STO the XANES spectrum (Fig. 4b) overlaps with that of the as-prepared sample showing that the exsolution procedure for 5Cu-STO is reversible and surface dominant.
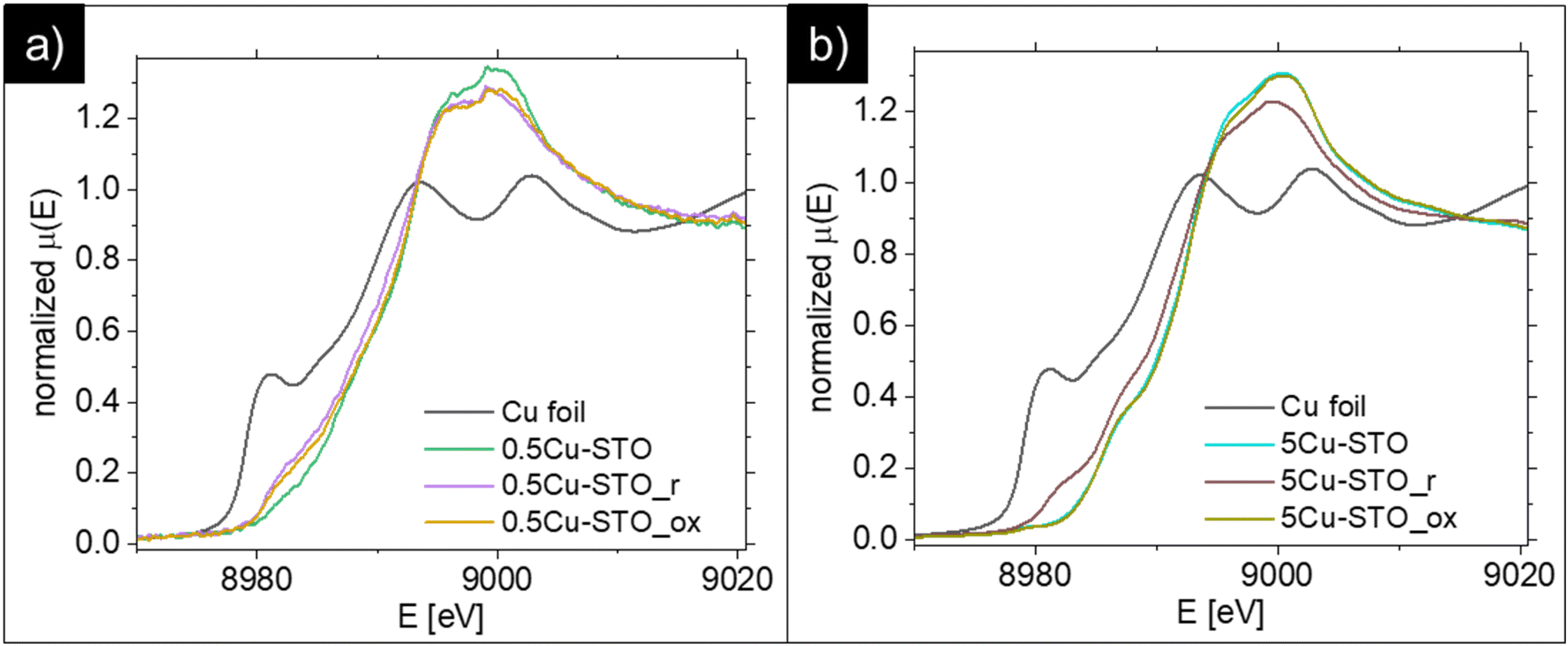 | ||
| Fig. 4 Comparison of the XANES spectra of metallic Cu foil and (a) 0.5Cu-STO and (b) 5Cu-STO before and after reduction or re-oxidation. | ||
During reduction, the Cu-dopant in the as-prepared samples can be reduced to Cu+ or Cu. By fitting the XANES spectra (see Fig. S2 in the ESI†) using a linear combination of the as-prepared sample, metallic Cu, Cu2O, and CuO references, one can obtain the quantitative change in the composition of the reduced and re-oxidized samples. In Table 1, the results attained from the best fit are listed. As can be seen about 71% of 0.5Cu-STO remains in its as-prepared structure during the reduction. 9% of the Cu dopant was reduced to metallic Cu and 13.4% to Cu2O. 5Cu-STO shows similar behavior after reduction with a larger ratio of metallic Cu compared to Cu2O. After re-oxidation the ratio of metallic Cu, Cu2O and CuO decreases slightly for 0.5Cu-STO. However, about 95% of Cu in the Cu-STO re-oxidized sample recovers to its as-prepared state with about 6% remaining in the CuO structure. These results further confirm that 5Cu-STO can be more easily reduced and re-oxidized than 0.5Cu-STO.
| Sample | 0.5Cu-STO | Metallic Cu | Cu2O | CuO |
|---|---|---|---|---|
| 0.5Cu-STO_ap | 1.000 | — | — | — |
| 0.5Cu-STO_r | 0.712 | 0.090 | 0.134 | 0.064 |
| 0.5Cu-STO_ox | 0.816 | 0.070 | 0.077 | 0.037 |
| 5Cu-STO_ap | 1.000 | — | — | — |
| 5Cu-STO_r | 0.686 | 0.131 | 0.090 | 0.093 |
| 5Cu-STO_ox | 0.938 | 0.000 | 0.000 | 0.062 |
The analysis of EXAFS spectra (Fig. S3, ESI†) supports these conclusions, assessing that Cu in the as-prepared 0.5Cu-STO, 5Cu-STO and the re-oxidized 5Cu-STO is in the Cu2+ state while, after reduction, it is partially reduced to Cu+. Moreover, 0.5Cu-STO was not re-oxidized as for 5Cu-STO. Information on the Cu–Cu distance cannot be determined from EXAFS because of insufficient data quality for the fitting.
Complementary to the above analysis, TEM-EDX investigation allowed monitoring the morphology evolution of Cu-STO powders in the exsolution procedure. In detail, as a representative example, the STEM images and the EDX elemental maps of the 5Cu-STO sample before and after thermal reduction under H2/Ar at 600 °C or 900 °C are reported in Fig. 5. Irregular aggregates of cubic-like nanoparticles of a few hundred nm in size can be easily observed. EDX maps of the pristine sample reveal a homogeneous distribution of the elements and, in particular, of the copper dopant in the whole matrix (Fig. 5a). Upon exsolution at 600 °C, the segregation process of Cu starts, as witnessed by the appearance of irregular shaped nanoparticles with a Cu-rich phase on the surface of SrTiO3 aggregates (Fig. 5b). The increase of the reduction temperature up to 900 °C fosters further copper migration to the surface. The higher ionic diffusion and the additional oxygen release from the parental matrix at that temperature explain the formation of larger nanoparticles (Fig. 5c).56,58
 | ||
| Fig. 5 STEM images and corresponding EDX elemental maps of 5Cu-STO powders: (a) as-prepared and after exsolution at (b) 600 and (c) 900 °C. | ||
A similar investigation was carried out for 0.5Cu-STO powders (see Fig. S5 ESI†). However, owing to the low copper concentration in the sample, only a few and small Cu-rich phase aggregates were detected.
The elemental compositions and oxidation states of the elements in bare and Cu-doped SrTiO3 perovskites were inspected by XPS, measuring the Sr 3d, Ti 2p, Cu 2p3/2, and O 1s (see Fig. S4 in ESI†) XPS core levels before and after reduction and upon re-oxidation in air, as shown in Fig. 6.
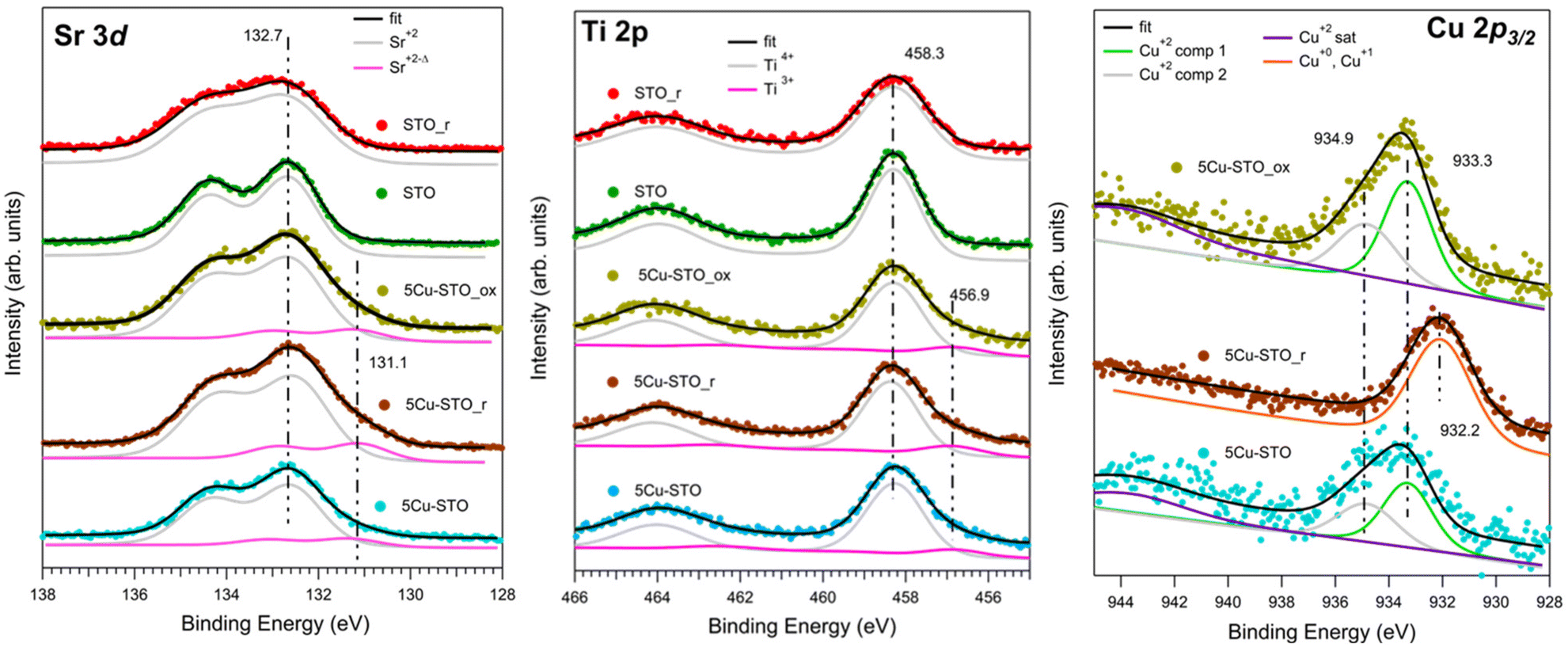 | ||
| Fig. 6 XPS core level spectra of: Sr 3d (left), Ti 2p (center) and Cu 2p3/2 (right). The intensities of all the spectra have been normalized to the total Ti 2p intensity. Superimposition of the experimental data (dots) is shown with the fit results (black line) and the fit components, all including the background signal. To better visualize the results, an offset has been summed to all components. See the text for more details. | ||
The reference STO sample shows the typical XPS features of this compound (Fig. 6, green spectra). In this case the Sr 3d and Ti 2p core levels consist of a single doublet, where the Sr 3d3/2 and Ti 2p3/2 BE lie at, respectively, 132.7 eV and 458.3 eV, corresponding to Sr2+ and Ti4+ in SrTiO3.49 The reduction treatment (STO_r sample, Fig. 6 red spectra) induces in both levels a relevant broadening (FWHM from 1.3 to 1.8 eV in Sr 3d, FWHM from 1.3 eV to 1.6 eV in Ti 2p), while the BEs remain unchanged. This is compatible with an enhancement of the disorder and the co-existence of many different chemical environments (e.g. segregation of Sr surface species).26,59
Cu doping induces the presence of a further component at lower BE in both Sr 3d and Ti 2p core levels in all investigated samples: as-prepared (5Cu-STO, light blue spectra), after reduction (5Cu-STO_r, brown spectra) and after air oxidation (5Cu-STO_ox, yellow spectra). In particular, the additional Sr 3d3/2 level at 131.1 eV, slightly increasing in intensity upon reduction, might be again attributed to the enrichment on the surface of Sr species, as testified by the relevant broadening of the spectral features. The component of Ti 2p3/2 at 456.9 eV is instead compatible with the presence of Ti3+ sites.60 It has to be mentioned that these reduced components may be also due to the presence of strong inhomogeneity of the sample, which could induce different charging effects.
The Cu 2p3/2 core level spectra give direct evidence of the Cu reduction. The spectra of the sample as prepared (5Cu-STO) and after oxidation (5Cu-STO_ox) are, as observed in XAS, very similar, with the presence of a broad peak at around 933.5 eV and its satellites at about 944 eV. Such spectra have been fitted using two components at 933.3 and 934.9 eV and their satellites at 944.0 (5Cu-STO) and 943.8 eV (5Cu-STO_ox) and are compatible with the presence of Cu2+.61 Upon reduction the spectrum changes significantly: the satellites are no more visible and the main peak shifts at 932.2 eV, pointing to the presence of Cu0 and Cu+ species,59 in full agreement with XAS results. The rather broad FWHM (2.3 eV) may be due to different chemical environments of the Cu atoms, in agreement with TPR data. We note that after re-oxidation the spectrum lineshape follows that of the as-prepared sample in agreement with XAS results. Such findings may be a first hint of the possibility of restoring the initial characteristics of the doped perovskite, indicating a partial reversibility of the exsolution process.
The atomic ratios of the metal elements were calculated and are summarized in Table S3.†
In the case of 5Cu-STO_r and 5Cu-STO_ox the relative intensities between Sr and Ti suggest a partial segregation of Sr at the perovskite surface leading to a slight enhancement of its intensity. The segregation of Sr (i.e. generation of strontium vacancies, 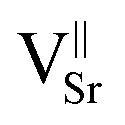 ) at the surface of STO is widely reported in the literature.62 Such an effect is observed only in the presence of Cu. The value obtained for bare 5Cu-STO is 1 ± 0.1%, in agreement with the quantities used during synthesis.
) at the surface of STO is widely reported in the literature.62 Such an effect is observed only in the presence of Cu. The value obtained for bare 5Cu-STO is 1 ± 0.1%, in agreement with the quantities used during synthesis.
UV-DRS can be a valid alternative technique to gain information about the coordination and aggregation state of copper metal species in the perovskite and lattice to characterize the local titanium environment and the oxygen defect. For this, diffuse-reflectance (DR) spectra of both undoped and 5Cu-STO powders as a function of reduction and re-oxidation treatments were collected and are reported in Fig. 7 in terms of −log(DR), which permits us to better observe the different spectral features.
 | ||
| Fig. 7 Diffuse reflectance (DR) spectra of (a) undoped STO and (b) 5Cu-STO samples as prepared (green lines), after reduction under H2/Ar at 900 °C (red lines) and after subsequent re-oxidation in O2 at 950 °C (blue lines). Inset in (b) shows the evolution of the spectra upon reduction as a function of the dopant level in Cu-STO powders. The noisy signal and the step around 860 nm are unavoidable instrumental artefacts. | ||
In Fig. 7a, the spectrum of the bare STO sample (green line) is observed to be dominated by the SrTiO3 band edge at 3.31 eV, as obtained from the Tauc plot (Fig. S6 and Table S4†). Upon reduction under H2/Ar at 900 °C, a new broad feature extending throughout the near-IR region and approximately centered at ∼1.2 eV (∼1000 nm) is noticeable (Fig. 7a, red line), which, on the basis of previous studies on chemically reduced and vacuum annealed undoped SrTiO3 and TiO2,27,34,63 can be assigned to Ti3+ lattice defects and oxygen vacancies. Reduction also makes the powder color change from white to grey (inset Fig. 7a), indeed in agreement with the lower diffuse reflectance in the whole visible range (Fig. S7†). Finally, upon re-oxidation under an O2 atmosphere, the complete annihilation of the signal is observed (blue line), with the spectrum becoming featureless, exactly as that of the as-prepared powders and the powder becomes white again.
Concerning the Cu-doped materials, while no relevant spectral features were observed for 0.5Cu-STO, the as-prepared 5Cu-STO sample displays, in addition to the expected SrTiO3 band edge, a slight shift to lower energy (3.27 eV from the Tauc plots, Fig. S6 and S7†) and a broad absorption band in the visible range centered at about 750 nm that tails into the near-infrared region (Fig. 7b, green line). This feature certainly depends on the copper addition to the perovskite matrix and can be mainly ascribed to the d–d transition (2Eg → 2T2g) of octahedral Cu2+ species.64,65
By forcing copper exsolution through thermal reduction at 900 °C, a new absorption band centered at around 610 nm appears (Fig. 7b, red line), accompanied by a color change of the powders from light to dark grey. This behavior might be assigned to an enhanced generation of Ti3+ lattice defects and oxygen vacancies upon reduction, and to the simultaneous formation of Cu0 nanoparticles at the perovskite surface, as testified by XAS and TEM images, which can give rise to a localized surface plasmon resonance (LSPR) effect.66,67
To prove this hypothesis, a Cu-STO sample with a nominal mol mol−1 percentage of dopant in the B-site of 15% was ad hoc synthesized and subjected to the same reducing and oxidation treatments undergone by the other perovskite powders. The spectrum collected on 15Cu-STO revealed a sharpening and shift at a slightly lower wavelength of the absorption band at ∼600 nm (see the inset in Fig. 7b), thus supporting the presence of metallic Cu nanoparticles at the material surface.68
Finally, after re-oxidation, the spectral profile is gradually recovered (Fig. 7b, blue line).
These results can improve the knowledge on the aggregation state of dopant metal species in the perovskite lattice, thus providing a valid protocol for monitoring the exsolution process. Indeed, they give the experimental assessment of the possibility of exploiting DR for detecting the changes of the electronic structure and the intrinsic defect of the samples.
To investigate the evolution during the exsolution process of the defect structure of both undoped and Cu-doped STO samples, a comprehensive X-band EPR investigation was performed.
In particular, the spectra were acquired under vacuum conditions (p < 10−5 mbar) at 130 K, after subjecting the samples to a reducing treatment under H2/Ar at different temperatures (from 200 up to 900 °C) and upon re-oxidation at 950 °C under an oxygen atmosphere. Useful hints can be retrieved in terms of the coordination environment and aggregation state of the copper ions as well as the dynamic processes involving the generation of oxygen and strontium vacancies.
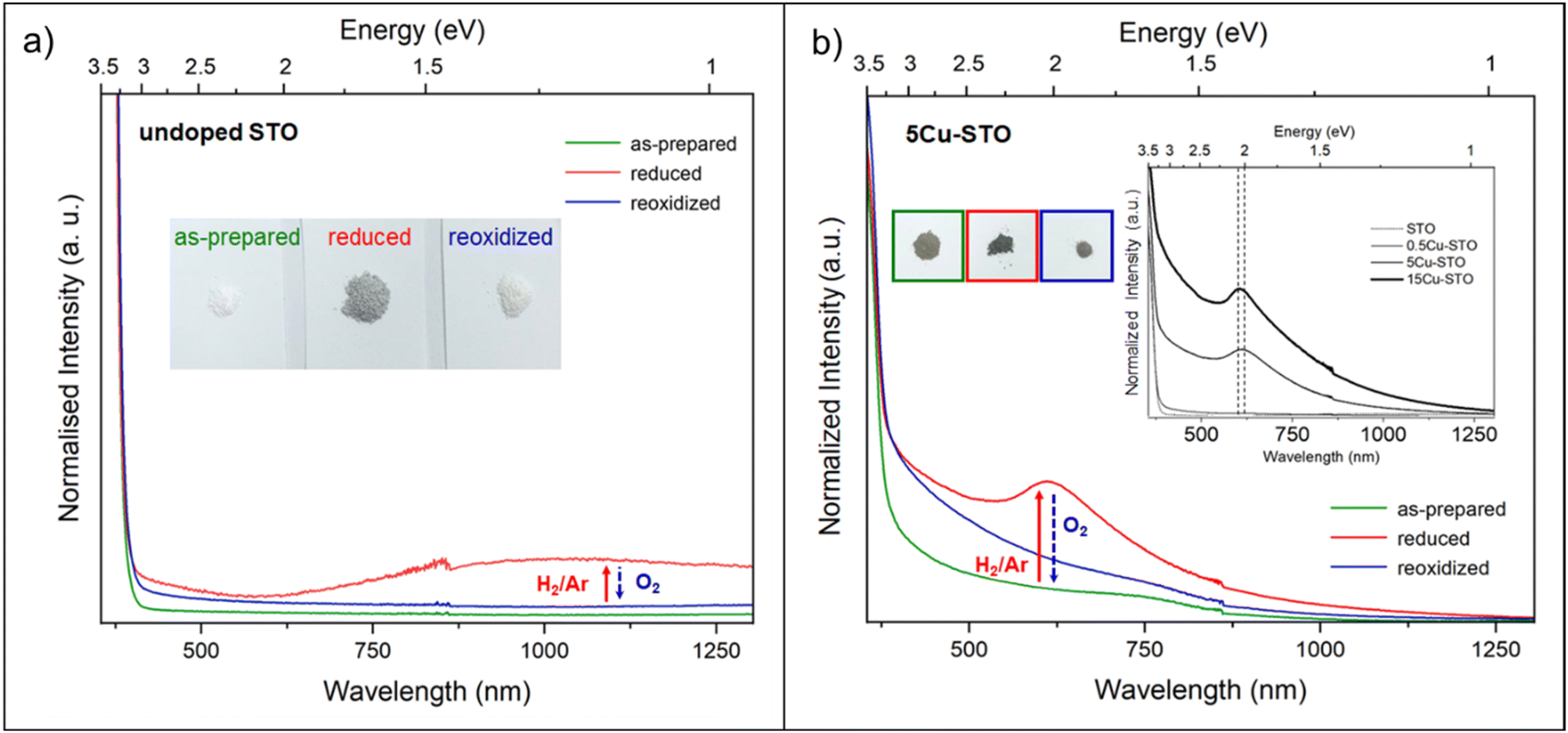
The spectra collected for the bare STO sample are reported in Fig. 8. The as-prepared powders, besides a very weak signal at a higher field with g ≈ 1.97 attributable to residual Ti3+ centers, display overlapped resonances attributable to different paramagnetic species at g = 1.99–2.01, whose assignment is still controversial in the frame of the limited EPR literature available for SrTiO3.69–74
 | ||
| Fig. 8 (a) EPR spectra at 130 K of the as-prepared (a.p.) STO nanopowders, after thermal reduction at different temperatures under H2/Ar and subsequent re-oxidation (re-ox) under an O2 atmosphere at 950 °C. (b) Experimental (exp.) and simulated (sim.) EPR spectra at 130 K of the as-prepared STO powders in the high field region and deconvolution of signals into centers I and II. | ||
Thermal treatment under a reducing atmosphere provides the chance to discriminate the nature and suggest a fair attribution of these superimposed species (Fig. 8a). Upon increasing the reduction temperature, all the spectral features increase in intensity and, in particular, a sharp and isotropic line at g ≈ 2.0 (labeled with *) appears. The g values and the relative contribution of the different species to the spectrum (calculated as % of the total intensity of the overlapping signals at g = 1.99–2.01) were calculated by signal simulation (Table 2 and Fig. 8b).
| Center | Contribution (%) | g value | |
|---|---|---|---|
| I | O− | 70.0% | g ⊥ = 2.0124 |
| g ‖ = 1.9865 | |||
| II |
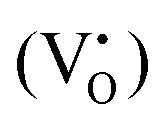
|
30.0% | g iso = 1.9988 |
Two different paramagnetic defects were identified: one axial signal (center I) and one isotropic line (center II). According to previous studies59,75 and in agreement with UV-DRS and XPS investigations, they have been attributed to paramagnetic defects located in proximity to the surface of the SrTiO3 particles. In particular, species I with axial symmetry can be associated with O− centers localized in proximity to Ti4+ species or to oxygen ions surrounding 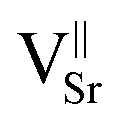 defects, in agreement with the A-site deficiency of the parental perovskite and with the Sr-segregation as assessed by XPS. The isotropic species II is assigned to singly ionized oxygen vacancies
defects, in agreement with the A-site deficiency of the parental perovskite and with the Sr-segregation as assessed by XPS. The isotropic species II is assigned to singly ionized oxygen vacancies 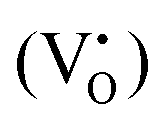 , probably generated from the removal of lattice oxygen upon thermal reduction, which is consistent also with a considerable enhancement of the signal of Ti3+ at g ≈ 1.97 upon reduction. Moreover, a very weak and broad bump at 344 mT (g ∼ 1.95) can be detected. This signal at has been widely reported in the literature for titania-based systems76 and is connected to the presence of Ti(III) centers. However, its fair attribution is still under debate. In general, it has been ascribed to a peculiar symmetry of the Ti centers or the presence of surface heterogeneity.
, probably generated from the removal of lattice oxygen upon thermal reduction, which is consistent also with a considerable enhancement of the signal of Ti3+ at g ≈ 1.97 upon reduction. Moreover, a very weak and broad bump at 344 mT (g ∼ 1.95) can be detected. This signal at has been widely reported in the literature for titania-based systems76 and is connected to the presence of Ti(III) centers. However, its fair attribution is still under debate. In general, it has been ascribed to a peculiar symmetry of the Ti centers or the presence of surface heterogeneity.
Interestingly, re-oxidation under an oxygen atmosphere leads to an almost complete recovery of the initial spectral features, except for Ti3+ centers which appear more stable (Fig. 8a). This again recalls the trend observed in the DR spectra.
By doping the perovskite even with a poor amount of copper (0.5Cu-STO), besides an increase of the intensity of the resonances of O− centers, singly ionized oxygen vacancies and Ti3+ defects, a very complex set of signals attributable to different Cu2+ species appear in the spectra (Fig. 9a).
 | ||
| Fig. 9 (a) EPR spectra at 130 K of the as-prepared (a.p.) 0.5Cu-STO nanopowders, after thermal reduction at different temperatures under H2/Ar and subsequent re-oxidation (re-ox) under an O2 atmosphere at 950 °C. (b) Highlight of the Cu2+ spectral region: i, ii, and iii indicate three types of isolated copper centers, whose corresponding structures are depicted in the inset. | ||
In detail, the magnification of the Cu2+ spectral region is reported in Fig. 9b. The spectrum of the as-prepared sample (a.p.) is characterized by the overlapping of at least four superimposed signals: (i) a broad, asymmetric, and unstructured line centered at g ≈ 2, typical of dipolar coupled Cu2+ species with a disordered coordination environment,77–80 reasonably connected to the CuO traces detected by XANES in Cu-STO; (ii) at least three superimposed signals due to distinct isolated copper centers (i, ii and iii) exhibiting a well resolved “parallel” (z) hyperfine structure, whose corresponding g-tensor and hyperfine tensor (A‖) values are reported in Table 3. The “perpendicular” (x, y features) hyperfine coupling consists of several superimposed and broadened lines, which makes a precise determination of the given contributing component signals by spectra simulation very hard.
| Species | g ‖ value | A ‖ value (MHz) |
|---|---|---|
| Center i | 2.234 | 118.2 |
| Center ii | 2.373 | 349.7 |
| Center iii | 2.326 | 408.9 |
Based on the g and A‖ parameters, these latter species can be associated with Cu2+ substitutional defects in the perovskite lattice with a different coordination with oxygen species.30,37,39,81 In detail, center i can be assigned to the copper center with a slightly distorted octahedral symmetry, while centers ii and iii were attributed, according to the model proposed by Eichel et al., to octahedrally coordinated Cu2+ centers with an associated oxygen vacancy (i.e.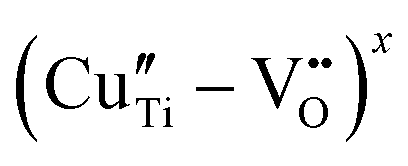 ) or, more in general, with a pronounced Jahn Teller tetragonal distortion (see the sketch of the possible structure in the inset of Fig. 9b).30,37,39,82,83
) or, more in general, with a pronounced Jahn Teller tetragonal distortion (see the sketch of the possible structure in the inset of Fig. 9b).30,37,39,82,83
Thermal annealing under a reducing atmosphere helped to get clearer information on the evolution of these copper centers during the exsolution process. By increasing the reduction temperature, the broad line free of a hyperfine structure attributable to coupled Cu2+ species promptly decreases in intensity and, at 900 °C, disappears leaving only weak signals related to the isolated copper sites constituting the final spectrum (see the red line in Fig. 9b corresponding to the sample treated under H2/Ar at 900 °C). This may suggest that, during exsolution, the Cu2+ sites with a disordered coordination environment are probably located at the surface of the perovskite and primarily migrate undergoing selective reduction to Cu(I) or zero valent diamagnetic species. Instead, a sort of “delay” is observed for the other lattice defects, since more thermal energy is required in order to impart them mobility and subsequent reduction. Re-oxidation in oxygen results in a partial recovery of the initial resonances (re-ox spectrum in Fig. 9a), thus supporting a feasible but limited reversibility of the procedure.
This fits well with the quantitative changes in the samples’ composition retrieved from XANES data, which reveal, besides the reduction of Cu(II) to Cu(I) and metallic copper, that a remarkable fraction of 0.5Cu-STO remains in its as-prepared structure during exsolution (see Fig. 4 and Table 1). Moreover, these outcomes are in line with the TPR outputs, where the signal of the lattice oxygen species, which could somehow activate the mobility/reduction of substitutional Cu-species, is only visible at high temperatures (see Fig. 3).
On increasing the amount of copper acceptor dopant (i.e. 5Cu-STO sample) a similar behavior can be observed (Fig. 10). In this case, the broad feature associated with dipolarly coupled Cu2+ species is much more intense and dominates the spectrum of the as-prepared material (Fig. 10a). Nevertheless, the distinct hyperfine structure of the isolated Cu(II) centers (i, ii and iii) is also clearly visible (Fig. 10b). Analogous to 0.5Cu-STO, a marked depletion of the copper resonances is observed upon reduction of 5Cu-STO, and only the features of isolated centers remain in the final spectrum, thus corroborating a preferential and faster reduction occurring for the disordered Cu species. Interestingly, in this case, a slight increase of the intensity of the signals assigned to oxygen defects and Ti3+ species can be observed upon reduction as a result of the removal of lattice oxygen and Sr segregation at the perovskite surface, as pointed out by UV-DRS and XPS. Finally, also in this case, the treatment under an oxidative atmosphere enables us to partially regain the initial spectral features (re-ox spectrum in Fig. 10a).
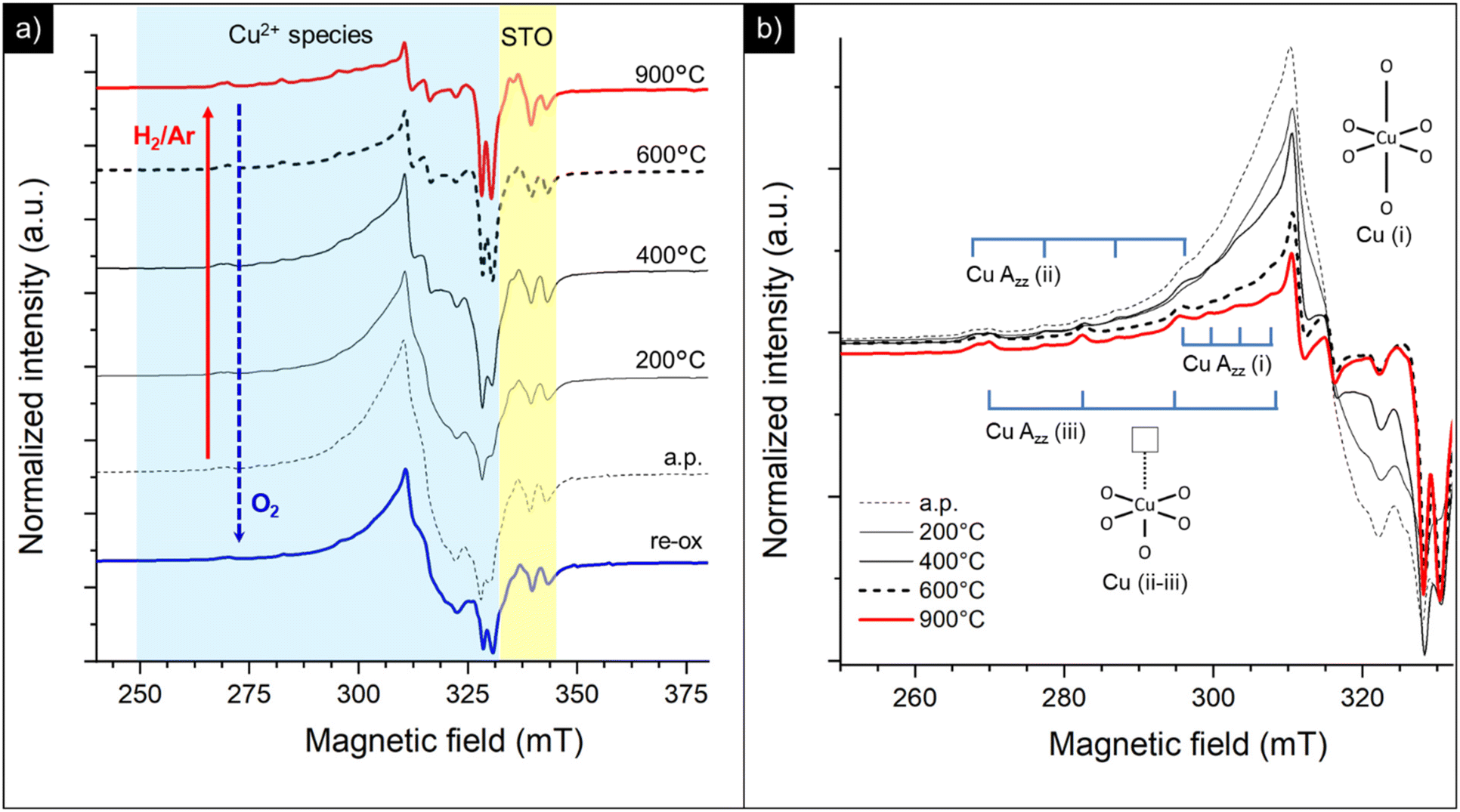 | ||
| Fig. 10 (a) EPR spectra at 130 K of the as-prepared (a.p.) 5Cu-STO nanopowders, after reduction under H2/Ar and subsequent re-oxidation (re-ox) at 950 °C. (b) Highlight of the Cu2+ spectral region: i, ii, and iii indicate three types of isolated Cu centers, whose structures are depicted in the inset. | ||
In summary, EPR investigation provides very useful hints on the evolution of Cu coordination and speciation during exsolution even at a very low doping level and with a high resolution, thus complementing the information gained utilizing more complex techniques like XAS.
Conclusions
In this work, the combination of different spectroscopic techniques (XAS, XPS, DR-UV and EPR) sheds new light on the exsolution and the reincorporation process of copper nanoparticles in SrTiO3. Although from XRD and TEM-EDX, which are the standardly employed methods in the analysis of exsolved materials, the copper dopant appears homogeneously distributed within the perovskite matrix, we showed that it exists at least in two different configurations. Independently from the dopant concentration in the matrix (0.5 and 5 wt%) the existence of copper in a distorted octahedral environment, ascribed to amorphous Cu2O and CuO likely located on the material surface, and of substitutional acceptor species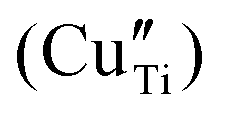 in the lattice was demonstrated by combining XANES and EPR. Surface oxide species reduced at much lower temperature (T = 200–400 °C) than lattice species (T > 800 °C). This inhomogeneity in the local environment and reduction temperature of Cu is expected to be reproduced in the nanoparticle properties (e.g. sintering resistance and support anchoring), thus possibly affecting the final catalytic application. EPR spectroscopy, in agreement with XPS suggestion, revealed also that both the doping and the high-temperature reduction were associated with the generation of oxygen defects and a change of the oxidation state not only of copper but also of the Ti-site. These results demonstrate that the free electrons originating from the lattice oxygen release also reduce the parental matrix and not exclusively the aliovalent defects, which might be relevant in catalytic applications of exsolved materials such as photocatalysis. UV-DRS proved to be a valid alternative method to TEM to unveil the segregation of exsolved nanoparticles by the plasmon resonance effect. Moreover, when XANES showed for 5Cu-STO the almost complete regain of the pristine state of Cu after reoxidation, EPR and UV-DRS analyses clearly pointed out that the original state of Cu after reoxidation is not entirely restored, leading to a highly disordered environment.
in the lattice was demonstrated by combining XANES and EPR. Surface oxide species reduced at much lower temperature (T = 200–400 °C) than lattice species (T > 800 °C). This inhomogeneity in the local environment and reduction temperature of Cu is expected to be reproduced in the nanoparticle properties (e.g. sintering resistance and support anchoring), thus possibly affecting the final catalytic application. EPR spectroscopy, in agreement with XPS suggestion, revealed also that both the doping and the high-temperature reduction were associated with the generation of oxygen defects and a change of the oxidation state not only of copper but also of the Ti-site. These results demonstrate that the free electrons originating from the lattice oxygen release also reduce the parental matrix and not exclusively the aliovalent defects, which might be relevant in catalytic applications of exsolved materials such as photocatalysis. UV-DRS proved to be a valid alternative method to TEM to unveil the segregation of exsolved nanoparticles by the plasmon resonance effect. Moreover, when XANES showed for 5Cu-STO the almost complete regain of the pristine state of Cu after reoxidation, EPR and UV-DRS analyses clearly pointed out that the original state of Cu after reoxidation is not entirely restored, leading to a highly disordered environment.
The use of uncommon spectroscopic techniques to describe the exsolution process evidenced that the chemical environment of exsolvable species is much more heterogeneous and the exsolution process much less straightforward than usually expected. Therefore, we believe it should be good praxis to not exclusively rely on standard techniques to describe the exsolution approach, but instead to widen the characterization of these systems with spectroscopic, laboratory-scale methods.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the European Union – NextGenerationEU, Italian National Recovery and Resilience Plan, Mission 4, Component 2, Investment 1.5 “Innovation Ecosystems”, project MUSA (grant No. ECS 000037). S. Mascotto and X. Sun gratefully acknowledge the funding support from BMBF (SITCOM project, grant No. 05K22GU3) and the DFG (grant No. MA-7146-7-1). The authors acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. The authors would like to thank Aleksandr Kalinko for his support at the P64 beamline and the fruitful discussions about the data analysis.References
- B. Yan, Q. Wu, J. Cen, J. Timoshenko, A. I. Frenkel, D. Su, X. Chen, J. B. Parise, E. Stach, A. Orlov and J. G. Chen, Highly active subnanometer Rh clusters derived from Rh-doped SrTiO3 for CO2 reduction, Appl. Catal., B, 2018, 237, 1003, DOI:10.1016/j.apcatb.2018.06.074.
- M. D. Argyle and C. H. Bartholomew, Heterogeneous Catalyst Deactivation and Regeneration: A Review, Catalysts, 2015, 5, 145, DOI:10.3390/catal5010145.
- D. K. Niakolas, Sulfur poisoning of Ni-based anodes for Solid Oxide Fuel Cells in H/C based fuels, Appl. Catal., A, 2014, 486, 123, DOI:10.1016/j.apcata.2014.08.015.
- M. Miceli, P. Frontera, A. Macario and A. Malara, Recovery/Reuse of Heterogeneous Supported Spent Catalysts, Catalysts, 2021, 11, 591, DOI:10.3390/catal11050591.
- D. Mateo, J. Albero and H. García, Titanium-Perovskite-Supported RuO2 Nanoparticles for Photocatalytic CO2 Methanation, Joule, 2019, 3, 1949, DOI:10.1016/j.joule.2019.06.001.
- C. Yang, J. Li, Y. Lin, J. Liu, F. Chen and M. Liu, In Situ Fabrication of CoFe Alloy Nanoparticles Structured (Pr0.4Sr0.6)3(Fe0.85Nb0.15)2O7 Ceramic Anode for Direct Hydrocarbon Solid Oxide Fuel Cells, Nano Energy, 2015, 11, 704, DOI:10.1016/j.nanoen.2014.12.001.
- Y. F. Sun, Y. L. Yang, J. Chen, M. Li, Y. Q. Zhang, J. H. Li, B. Hua and J. L. Luo, Toward a rational photocatalyst design: a new formation strategy of co-catalyst/semiconductor heterostructures via in situ exsolution, Chem. Commun., 2018, 54, 1505, 10.1039/C7CC08797G.
- A. J. Carrillo, K. J. Kim, Z. D. Hood, A. H. Bork and J. L. M. Rupp, La0.6Sr0.4Cr0.8Co0.2O3 Perovskite Decorated with Exsolved Co Nanoparticles for Stable CO2 Splitting and Syngas Production, ACS Appl. Energy Mater., 2020, 3, 4569, DOI:10.1021/acsaem.0c00249.
- D. Neagu, E. I. Papaioannou, W. K. W. Ramli, D. N. Miller, B. J. Murdoch, H. Ménard, A. Umar, A. J. Barlow, P. J. Cumpson, J. T. S. Irvine and I. S. Metcalfe, Demonstration of chemistry at a point through restructuring and catalytic activation at anchored nanoparticles, Nat. Commun., 2017, 8, 1855, DOI:10.1038/s41467-017-01880-y.
- K. Kousi, C. Tang, I. S. Metcalfe and D. Neagu, Emergence and Future of Exsolved Materials, Small, 2021, 17, 2006479, DOI:10.1002/smll.202006479.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Self-regeneration of a Pd-perovskite catalyst for automotive emissions control, Nature, 2002, 418, 6894, DOI:10.1038/nature00893.
- Y. Gao, J. Wang, Y. Q. Lyu, K. Lam and F. Ciucci, In situ growth of Pt3Ni nanoparticles on an A-site deficient perovskite with enhanced activity for the oxygen reduction reaction, J. Mater. Chem. A, 2017, 5, 14, 10.1039/C7TA00349H.
- D. Neagu, T. Oh, D. N. Miller, H. Ménard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution, Nat. Commun., 2015, 6, 8120, DOI:10.1038/ncomms9120.
- D. Burnat, R. Kontic, L. Holzer, P. Steiger, D. Ferri and A. Heel, Smart material concept: reversible microstructural self-regeneration for catalytic applications, J. Mater. Chem. A, 2016, 30, 11939, 10.1039/C6TA03417A.
- S. Li, Q. Qin, K. Xie, Y. Wang and Y. Wu, High-performance fuel electrodes based on NbTi0.5M0.5O4 (M = Ni, Cu) with reversible exsolution of the nano-catalyst for steam electrolysis, J. Mater. Chem. A, 2013, 1, 8984, 10.1039/C3TA10404D.
- Y. R. Jo, B. Koo, M. J. Seo, J. K. Kim, S. Lee, K. Kim, J. W. Han, W. Jung and B. J. Kim, Growth Kinetics of Individual Co Particles Ex-solved on SrTi0.75Co0.25O3-δ Polycrystalline Perovskite Thin Film, J. Am. Chem. Soc., 2019, 141, 6690, DOI:10.1021/jacs.9b01882.
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, From material design to mechanism study: Nanoscale Ni exsolution on a highly active A-site deficient anode material for solid oxide fuel cells, Nano Energy, 2016, 27, 499, DOI:10.1016/j.nanoen.2016.07.013.
- T. S. Oh, E. K. Rahani, D. Neagu, J. T. S. Irvine, V. B. Shenoy, R. J. Gorte and J. M. Vohs, Evidence and Model for Strain-Driven Release of Metal Nanocatalysts from Perovskites during Exsolution, J. Phys. Chem. Lett., 2015, 6, 24, DOI:10.1021/acs.jpclett.5b02292.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. S. Irvine, In situ growth of nanoparticles through control of non-stoichiometry, Nat. Chem., 2013, 5, 11, DOI:10.1038/nchem.1773.
- T. Ruh, D. Berkovec, F. Schrenk and C. Rameshan, Exsolution on perovskite oxides: morphology and anchorage of nanoparticles, Chem. Commun., 2023, 59, 3948, 10.1039/D3CC00456B.
- D. Neagu, J. T. S. Irvine, J. Wang, B. Yildiz, A. K. Opitz, J. Fleig, Y. Wang, J. Liu, L. Shen, F. Ciucci, B. A. Rosen, Y. Xiao, K. Xie, G. Yang, Z. Shao, Y. Zhang, J. Reinke, T. A. Schmauss, S. A. Barnett, R. Maring, V. Kyriakou, U. Mushtaq, M. N. Tsampas, Y. Kim, R. O'Hayre, A. J. Carrillo, T. Ruh, L. Lindenthal, F. Schrenk, C. Rameshan, E. I. Papaioannou, K. Kousi, I. S. Metcalfe, X. Xu and G. Liu, Roadmap on exsolution for energy applications, J. Phys.: Energy, 2023, 5, 031501, DOI:10.1088/2515-7655/acd146.
- D. Neagu, V. Kyriakou, I.-L. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur-Piet, I. S. Metcalfe, P. Vernoux, M. C. M. van de Sanden and M. N. Tsampas, In Situ Observation of Nanoparticle Exsolution from Perovskite Oxides: From Atomic Scale Mechanistic Insight to Nanostructure Tailoring, ACS Nano, 2019, 13(11), 12996, DOI:10.1021/acsnano.9b05652.
- E. Calì, M. P. Thomas, R. Vasudevan, J. Wu, O. G. Diaz, K. Marquardt, E. Saiz, D. Neagu, R. R. Unocic, S. C. Parker, B. S. Guiton and D. J. Payne, Real-time insight into the multistage mechanism of nanoparticle exsolution from a perovskite host surface, Nat. Commun., 2023, 14, 1754, DOI:10.1038/s41467-023-37212-6.
- S. Shah, J. Hong, L. Cruz, S. Wasantwisut, S. R. Bare and K. L. Gilliard-AbdulAziz, Dynamic Tracking of NiFe Smart Catalysts using In Situ X-Ray Absorption Spectroscopy for the Dry Methane Reforming Reaction, ACS Catal., 2023, 13, 6, DOI:10.1021/acscatal.2c05572.
- B. Rudolph, A. I. Tsiotsias, B. Ehrhardt, P. Dolcet, S. Gross, S. Haas, N. D. Charisou, M. A. Goula and S. Mascotto, Nanoparticle Exsolution from Nanoporous Perovskites for Highly Active and Stable Catalysts, Adv. Sci., 2023, 10, 2205890, DOI:10.1002/advs.202205890.
- B. Kayaalp, K. Klauke, M. Biesuz, A. Iannaci, V. M. Sglavo, M. D'Arienzo, H. Noei, S. Lee, W. Jung and S. Mascotto, Surface Reconstruction Under the Exposure of Electric Fields Enhances the Reactivity of Donor-Doped SrTiO3, J. Phys. Chem. C, 2019, 123, 16883, DOI:10.1021/acs.jpcc.9b04620.
- H. Tan, Z. Zhao, W. Zhu, E. N. Coker, B. Li, M. Zheng, W. Yu, H. Fan and Z. Sun, Oxygen Vacancy Enhanced Photocatalytic Activity of Pervoskite SrTiO3, ACS Appl. Mater. Interfaces, 2014, 6, 21, DOI:10.1021/am5051907.
- B. Liu, V. R. Cooper, H. Xu, H. Xiao, Y. Zhang and W. J. Weber, Composition Dependent Intrinsic Defect Structures in SrTiO3, Phys. Chem. Chem. Phys., 2014, 16(29), 15590, 10.1039/C4CP01510J.
- Y. Gao, Z. Lu, T. L. You, J. Wang, L. Xie, J. He and F. Ciucci, Energetics of Nanoparticle Exsolution from Perovskite Oxides, J. Phys. Chem. Lett., 2018, 9(13), 3772, DOI:10.1021/acs.jpclett.8b01380.
- R. A. Eichel, E. Erünal, M. D. Drahus, D. M. Smyth, J. van Tol, J. Acker, H. M. Kungl and J. Hoffmann, Local variations in defect polarization and covalent bonding in ferroelectric Cu2+-doped PZT and KNN functional ceramics at the morphotropic phase boundary, Phys. Chem. Chem. Phys., 2009, 11, 8698, 10.1039/B905642D.
- G. Feher, Sensitivity Considerations in Microwave Paramagnetic Resonance Absorption Techniques, Bell Syst. Tech. J., 1956, 36, 2 Search PubMed.
- S. A. Horlick, Y. L. Huang, I. A. Robinson and E. D. Wachsman, Controlling exsolution with a charge-balanced doping approach, Nano Energy, 2021, 87, 106193, DOI:10.1016/j.nanoen.2021.106193.
- E. Calì, G. Kerherve, F. Naufal, K. Kousi, D. Neagu, E. I. Papaioannou, M. P. Thomas, B. S. Guiton, I. S. Metcalfe, J. T. S. Irvine and D. J. Payne, Exsolution of Catalytically Active Iridium Nanoparticles from Strontium Titanate, ACS Appl. Mater. Interfaces, 2020, 12(33), 37444, DOI:10.1021/acsami.0c08928.
- W. L. Harrigan and K. R. Kittilstved, Reversible modulation of the Cr3+ spin dynamics in colloidal SrTiO3 nanocrystals, J. Phys. Chem. C, 2018, 122(46), 26652, 10.1039/d0na00106f.
- K. A. Lehuta and K. R. Kittilstved, Reversible control of the chromium valence in chemically reduced Cr-doped SrTiO3 bulk powders, Dalton Trans., 2016, 45, 10034, 10.1039/C6DT00706F.
- R. F. Howe, Electron paramagnetic resonance spectroscopy of catalytic surfaces, Colloids Surf., A, 1993, 72, 353, DOI:10.1016/0927-7757(93)80485-W.
- R. A. Eichel, Defect structure of oxide ferroelectrics—valence state, site of incorporation, mechanisms of charge compensation and internal bias fields, J. Electroceram., 2007, 19, 1, DOI:10.1007/s10832-007-9068-8.
- R. A. Eichel, Structural and dynamic properties of oxygen vacancies in perovskite oxides—analysis of defect chemistry by modern multi-frequency and pulsed EPR techniques, Phys. Chem. Chem. Phys., 2011, 13, 368, 10.1039/B918782K.
- R. A. Eichel, P. Erhart, P. Träskelin, K. Albe, H. Kungl and M. J. Hoffmann, Defect-Dipole Formation in Copper-Doped PbTiO3 Ferroelectrics, Phys. Rev. Lett., 2008, 100, 095504, DOI:10.1103/PhysRevLett.100.095504.
- K. A. Schönau, L. A. Schmitt, M. Knapp, H. Fuess, R. A. Eichel, H. Kungl and M. J. Hoffmann, Nanodomain structure of Pb[Zr1−xTix]O3 at its morphotropic phase boundary: Investigations from local to average structure, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 184117, DOI:10.1103/PhysRevB.75.184117.
- E. Garribba and G. Micera, The Determination of the Geometry of Cu(II) Complexes: An EPR Spectroscopy Experiment, J. Chem. Educ., 2006, 83, 8, DOI:10.1021/ed083p1229.
- S. Yuan, P. Mklaudeau and V. Pemchonb, Catalytic combustion of diesel soot particles on copper catalysts supported on TiO2. Effect of potassium promoter on the activity, Appl. Catal., B, 1994, 3, 319, DOI:10.1016/0926-3373(94)00005-0.
- S. Mohan, P. Dinesha and S. Kumar, NOx reduction behaviour in copper zeolite catalysts for ammonia SCR systems: A review, Chem. Eng. J., 2020, 384, 123253, DOI:10.1016/j.cej.2019.123253.
- Z. Zhang, X. Chen, J. Kang, Z. Yu, J. Tian, Z. Gong, A. Jia, R. You, K. Qian, S. He, B. Teng, Y. Cui, Y. Wang, W. Zhang and W. Huang, Nat. Commun., 2021, 12, 4331, DOI:10.1038/s41467-021-24621-8.
- N. Zhou, Y. M. Yin, Z. Chen, Y. Song, J. Yin, D. Zhou and Z. F. Ma, Suppressing the Voltage Fading of Li[Li0.2Ni0.13Co0.13Mn0.54]O2 Cathode Material via Al2O3 Coating for Li-Ion Batteries, J. Electrochem. Soc., 2018, 165, A1648, DOI:10.1149/2.0441809jes.
- W. Qi, C. Ruan, G. Wu, Y. Zhang, Y. Wang, K. Xie and Y. Wu, Int. J. Hydrogen Energy, 2014, 39, 5485, DOI:10.1016/j.ijhydene.2014.01.108.
- B. Kayaalp, S. Lee, K. Klauke, J. Seo, L. Nodari, A. Kornowski, W. Jung and S. Mascotto, Template-free mesoporous La0.3Sr0.7Ti1(-xFexO3±δ for CH4 and CO oxidation catalysis, Appl. Catal., B, 2019, 245, 536, DOI:10.1016/j.apcatb.2018.12.077.
- E. Poffe, H. Kaper, B. Ehrhardt, L. Gigli, D. Aubert, L. Nodari, S. Gross and S. Mascotto, Understanding Oxygen Release from Nanoporous Perovskite Oxides and Its Effect on the Catalytic Oxidation of CH4 and CO, ACS Appl. Mater. Interfaces, 2021, 13(21), 25483, DOI:10.1021/acsami.1c02281.
- R. P. Vasquez, Surf. Sci. Spectra, 1992, 1, 17, DOI:10.1116/1.1247685.
- W. A. Caliebe, V. Murzin, A. Kalinko and M. Görlitz, High-flux XAFS-beamline P64 at PETRA III, AIP Conf. Proc., 2019, 2054, 060031, DOI:10.1063/1.5084662.
- B. Ravel and M. Newville, Data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537, DOI:10.1107/S0909049505012719.
- L. Rizzato, J. Cavazzani, A. Osti, M. Scavini and A. Glisenti, Cu-Doped SrTiO3 Nanostructured Catalysts for CO2 Conversion into Solar Fuels Using Localised Surface Plasmon Resonance, Catalysts, 2023, 13, 1377, DOI:10.3390/catal13101377.
- Z. Bao, V. Fung, J. Moon, Z. D. Hood, M. Rochow, J. Kammert, F. Polo-Garzo and Z. Wu, Revealing the interplay between “intelligent behavior” and surface reconstruction of non-precious metal doped SrTiO3 catalysts during methane combustion, Catal. Today, 2023, 416, 113672, DOI:10.1016/j.cattod.2022.03.012.
- J. J. Rodriguez, J. Y. Kim, J. C. Hanson, M. Pérez and A. I. Frenkel, Reduction of CuO in H2: In Situ Time-Resolved XRD Studies, Catal. Lett., 2003, 85, 247, DOI:10.1023/A:1022110200942.
- P. Gwóźdź, A. Łącz, A. Mizera and E. Drożdż, Some Aspects of Cu Incorporation into SrTiO3 Structure, J. Therm. Anal. Calorim., 2022, 147, 9949, DOI:10.1007/s10973-022-11306-7.
- B. Rudolph, A. I. Tsiotsias, B. Ehrhardt, P. Dolcet, S. Gross, S. Haas, N. D. Charisou, M. A. Goula and S. Mascotto, Nanoparticle Exsolution from Nanoporous Perovskites for Highly Active and Stable Catalysts, Adv. Sci., 2023, 10, 2205890, DOI:10.1002/advs.202205890.
- K. Kousi, D. Neagu, L. Bekris, E. I. Papaioannou and I. S. Metcalfe, Endogenous Nanoparticles Strain Perovskite Host Lattice Providing Oxygen Capacity and Driving Oxygen Exchange and CH4 Conversion to Syngas, Angew. Chem., Int. Ed., 2020, 59, 2510, DOI:10.1002/anie.201915140.
- A. I. Tsiotsias, B. Ehrhardt, B. Rudolph, L. Nodari, S. Kim, W. C. Jung, N. D. Charisiou, M. A. Goula and S. Mascotto, Bimetallic Exsolved Heterostructures of Controlled Composition with Tunable Catalytic Properties, ACS Nano, 2022, 16, 8904, DOI:10.1021/acsnano.1c11111.
- J. Scholz, A. Garbujo, B. Kayaalp, K. Klauke, A. Glisenti and S. Mascotto, Functional Nanostructured Perovskite Oxides from Radical Polymer Precursors, Inorg. Chem., 2019, 58, 15942, DOI:10.1021/acs.inorgchem.9b02460.
- M. de Simone, E. Snidero, M. Coreno, G. Bongiorno, L. Giorgetti, M. Amati and C. Cepek, Oxidation of nanostructured Ti films produced by low energy cluster beam deposition: An X-ray Photoelectron Spectroscopy characterization, Thin Solid Films, 2012, 520, 4803, DOI:10.1016/j.tsf.2011.10.075.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. Smart, St. C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257, 2717, DOI:10.1016/j.apsusc.2010.10.051.
- A. G. Shard, Detection limits in XPS for more than 6000 binary systems using Al and Mg Kα X-rays, Surf. Interface Anal., 2014, 46, 175, DOI:10.1002/sia.5406.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, Self-Doped Ti3+ Enhanced Photocatalyst for Hydrogen Production under Visible Light, J. Am. Chem. Soc., 2010, 132(34), 11856, DOI:10.1021/ja103843d.
- R. Hierl, H. Knözinger and H. P. Urbach, Surface properties and reduction behavior of calcined. CuOAl2O3 and CuO-NiOAl2O3 catalysts, J. Catal., 1981, 69, 475, DOI:10.1016/0021-9517(81)90183-4.
- B. R. Strohmeier, D. E. Leyden, R. S. Field and D. M. Hercules, Surface spectroscopic characterization of CuAl2O3 catalysts, J. Catal., 1985, 94, 514, DOI:10.1016/0021-9517(85)90216-7.
- M. J. Kale, T. Avanesian and P. Christopher, Direct Photocatalysis by Plasmonic Nanostructures, ACS Catal., 2014, 4, 116, DOI:10.1021/cs400993w.
- Z. Li, G. Zhu, W. Zhang, L. Zhu, B. Cao, J. Gao, X. Shi, Y. Huang, P. Liu and M. Hojamberdiev, Dual-Functional Copper (Cu0/Cu2+)-Modified SrTiO3-δ Nanosheets with Enhanced Photothermal Catalytic Performance for CO2 Reduction and H2 Evolution, Chem. Eng. J., 2023, 452, 139378, DOI:10.1016/j.cej.2022.139378.
- G. Y. Yao, Z. Y. Zhao, Q. L. Liu, X. D. Dong and Q. M. Zhao, Theoretical calculations for localized surface plasmon resonance effects of Cu/TiO2 nanosphere: Generation, modulation, and application in photocatalysis, Sol. Energy Mater. Sol. Cells, 2020, 208, 110385, DOI:10.1016/j.solmat.2019.110385.
- K. Xie, N. Umezawa, N. Zhang, P. Reunchan, Y. Zhang and J. Ye, Self-doped SrTiO3−δ photocatalyst with enhanced activity for artificial photosynthesis under visible light, Energy Environ. Sci., 2011, 4, 4211, 10.1039/C1EE01594J.
- A. Troup, J. Mort, S. Grammatica and D. J. Sandman, Direct observation of superexchange in a disordered molecular solid, Solid State Commun., 1980, 33, 91, DOI:10.1016/0038-1098(80)90703-6.
- I. Bykov, M. Makarova, V. Trepakov, A. Dejneka, L. Yurchenko, L. Yurchenko, A. Jäger and L. Jastrabik, Intrinsic and Impurity Defects in Chromium-doped SrTiO3 Nanopowders: EPR and NMR Study, Phys. Status Solidi B, 2013, 250, 821, DOI:10.1002/pssb.201200871.
- C. Oliva, L. Bonoldi, S. Cappelli, L. Fabbrini, I. Rossetti and L. Forni, Effect of Preparation Parameters on SrTiO3±δ Catalyst for the Flameless Combustion of Methane, J. Mol. Catal. A: Chem., 2005, 226, 33, DOI:10.1016/j.molcata.2004.09.023.
- X. Zhou, N. Liu, T. Yokosawa, A. Osvet, M. E. Miehlich, K. Meyer, E. Spiecker and P. Schmuki, Intrinsically Activated SrTiO3: Photocatalytic H2 Evolution from Neutral Aqueous Methanol Solution in the Absence of Any Noble Metal Cocatalyst, ACS Appl. Mater. Interfaces, 2018, 10(35), 29532, DOI:10.1021/acsami.8b08564.
- Z. Wei, M. Zhao, Z. Yang, X. Duan, G. Jiang, G. Li, F. Zhang and Z. Hao, Oxygen vacancy-engineered titanium-based perovskite for boosting H2O activation and lower-temperature hydrolysis of organic sulfur, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2217148120, DOI:10.1073/pnas.2217148120.
- B. Kayaalp, K. Klauke, M. Biesuz, A. Iannaci, V. M. Sglavo, M. D'Arienzo, H. Noei, S. Lee, W. Jung and S. Mascotto, Surface Reconstruction Under the Exposure of Electric Fields Enhances the Reactivity of Donor-Doped SrTiO3, J. Phys. Chem. C, 2019, 123, 16883, DOI:10.1021/acs.jpcc.9b04620.
- S. Livraghi, M. Rolando, S. Maurelli, M. Chiesa, M. C. Paganini and E. Giamello, Nature of Reduced States in Titanium Dioxide as Monitored by Electron Paramagnetic Resonance. II: Rutile and Brookite Cases, J. Phys. Chem. C, 2014, 118, 22141, DOI:10.1021/jp209075m.
- S. Fujiwara, S. Katsumata and T. Seki, Electron exchange as evidenced by electron spin resonance of copper(II) confined in molecular space, Phys. Chem., 1967, 71, 115, DOI:10.1021/j100860a016.
- J. P. Donoso, C. J. Magon, J. F. Lima, O. R. Nascimento, E. Benavente, M. Moreno and G. Gonzalez, Electron Paramagnetic Resonance Study of Copper-Ethylenediamine Complex Ion Intercalated in Bentonite, J. Phys. Chem. C, 2013, 117(45), 24042, DOI:10.1021/jp408658d.
- S. Radhakrishnan, S. Smet, C. V. Chandran, S. P. Sree, K. Duerinckx, G. Vanbutsele, J. A. Martens and E. Breynaert, Prediction of Cu Zeolite NH3-SCR Activity from Variable Temperature 1H NMR Spectroscopy, Molecules, 2023, 28, 6456, DOI:10.3390/molecules28186456.
- J. F. Boas, R. H. Dunhill, J. R. Pilbrow, R. C. Srivastava and T. D. Smith, Electron spin resonance studies of copper(II) hydroxy-carboxylic acid chelates in aqueous and non-aqueous solutions, J. Chem. Soc. A, 1969, 94, 10.1039/J19690000094.
- P. Politzer, J. Martinez, J. S. Murray, M. C. Concha and A. Toro-Labbé, An electrostatic interaction correction for improved crystal density prediction, Mol. Phys., 2009, 107, 2095, DOI:10.1080/00268970903156306.
- S. Maurelli, M. Ruszak, S. Witkowski, P. Pietrzyk, M. Chiesa and Z. Sojka, Spectroscopic CW-EPR and HYSCORE investigations of Cu2+ and O2− species in copper doped nanoporous calcium aluminate (12CaO·7Al2O3), Phys. Chem. Chem. Phys., 2010, 12, 10933, 10.1039/C0CP00084A.
- A. Godiksen, F. N. Stappen, P. N. R. Vennestrøm, F. Giordanino, S. B. Rasmussen, L. F. Lundegaard and S. Mossin, Coordination environment of copper sites in Cu-CHA zeolite investigated by electron paramagnetic resonance, J. Phys. Chem. C, 2014, 118, 23126, DOI:10.1021/jp5065616.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi02391a |
| This journal is © the Partner Organisations 2025 |