
Pentagonal-bipyramidal dysprosium(III) complexes with two apical phosphine oxide ligands and equatorial pentadentate N3O2 Schiff-base ligands: breakdown of the apical magnetic axiality by a strong equatorial crystal field†
Tamara A.
Bazhenova
 a,
Vyacheslav A.
Kopotkov
a,
Denis V.
Korchagin
a,
Elena A.
Yureva
a,
Mikhail V.
Zhidkov
a,
Alexei I.
Dmitriev
a,
Ilya A.
Yakushev
b,
Nikolay N.
Efimov
b,
Konstantin A.
Babeshkin
b,
Vladimir S.
Mironov
*c and
Eduard B.
Yagubskii
*a
a,
Vyacheslav A.
Kopotkov
a,
Denis V.
Korchagin
a,
Elena A.
Yureva
a,
Mikhail V.
Zhidkov
a,
Alexei I.
Dmitriev
a,
Ilya A.
Yakushev
b,
Nikolay N.
Efimov
b,
Konstantin A.
Babeshkin
b,
Vladimir S.
Mironov
*c and
Eduard B.
Yagubskii
*a
aFederal Research Centre of Problems of Chemical Physics and Medicinal Chemistry, FRC PCP MC RAS, 1 Academician Semenov av., Chernogolovka, 142432, Russian Federation. E-mail: yagubski@gmail.com
bKurnakov Institute of General and Inorganic Chemistry, IGIC RAS, 31 Leninskii av., Moscow, 119071, Russian Federation
cNational Research Center “Kurchatov Institute”, 1 Academician Kurchatov sq., Moscow, 123182, Russian Federation. E-mail: mirsa@list.ru
First published on 12th November 2024
Abstract
A series of three new seven-coordinate pentagonal-bipyramidal (PBPY-7) Dy(III) complexes, [Dy(LCH3)(Cy3PO)2]ClO4·CH3CN (1), [Dy(L2(t-Bu))(Ph3PO)2]ClO4·0.63C2H5OH (2), and [Dy(LOCH3)(Ph3PO)2]ClO4·2H2O (3), including various chelating pentadentate ligands with [N3O2]2− binding node in the equatorial plane, LCH3 = [2,6-diacetylpyridine bis(acetylhydrazone)]2−, L2(t-Bu) = [2,6-diacetylpyridine bis(3,5di-tert-butylbenzoylhydrazone)]2−, and LCH3 = [2,6-diacetylpyridine bis(4-methoxybenzoylhydrazone)]2−, and two apical ligands Cy3PO and Ph3PO were synthesized and characterized structurally and magnetically. The ac magnetic measurements indicated the single-molecule-magnet (SMM) behavior of 1–3 with energy barriers of Ueff ≈ 318–350 K. Ab initio calculations and crystal-field (CF) analysis showed that the ground states of 1–3 were a nearly pure Ising type Kramers doublet (KD0) |±15/2〉eq with the long magnetic axis lying in the equatorial plane of N3O2, which was the opposite of high-performance PBPY-7 Dy(III) SMMs (Ueff > 1000 K), where the long magnetic axis of KD0 |±15/2〉 invariably pointed toward apical ligands. This difference is due to competition between the apical and equatorial CFs, which have been quantitatively examined with CF calculations. We show that the turning of the long magnetic axis (gz ∼ 19.6) from apical ligands (z) to the equatorial plane (xy) is due to crossover between the oblate |±15/2〉 and prolate |±1/2〉 ground states of the Dy(III) ion, which occurs at the negative ratio of B20/B40 < −0.07 of the two axial CF parameters B20 and B40. Complexes 1–3 correspond to this case due to the strong equatorial CF of the negatively charged chelate node of [N3O2]2− producing a large positive CF parameter B40 and negative B20. In this case, the SMM properties of 1–3 arise from distortions of the PBPY-7 complex (namely, from a large O1–Dy–O2 bond angle of ∼100° in the N3O2 pentagon of 1–3) that mix the lowest |±1/2〉 state and low-lying low-mJ states to produce the equatorial KD0 |±15/2〉eq. This highlights a breakdown of the apical magnetic axiality, since the SMM performances of 1–3 are governed by a strong equatorial CF and distortions rather than by high D5h symmetry and strong apical ligands. Some ways to improve the SMM efficiency of 1–3 and related PBPY-7 Dy(III) complexes are discussed.
1. Introduction
Mononuclear lanthanide complexes have been extensively explored as high-performance single-molecule magnets (SMMs) in the past few decades,1–10 since the discovery of slow magnetic relaxation with a high spin-reversal barrier in double decker lanthanide complexes [LnPc2]− (Ln3+ = Tb, Dy, and Ho; Pc = phthalocyanine derivatives) in 2003.1 Because of the unique electronic structure of lanthanide ions with an open 4fN electronic shell possessing a large unquenched orbital moment and high magnetic anisotropy, Ln-SMMs have great prospects in emerging advanced technologies such as high-density data storage, quantum computing, and molecular spintronics.11–16 Presently, myriads of lanthanide SMMs with a wide variety of structures and magnetic properties have been synthesized and characterized.1–10 In this research field, major efforts have been focused on monometallic lanthanide-based SMMs (termed single-ion magnets, Ln-SIMs), with which incredible progress has been made to increase the effective spin-reversal barrier (Ueff) and blocking temperature (TB), particularly in Dy(III) complexes, up to Ueff ∼ 1500 cm−1 and TB = 60–80 K.17–19 In monometallic Ln-SIMs comprising single magnetic Ln3+ ions, the barrier Ueff is controlled solely by the crystal-field (CF) splitting of the ground (2J + 1) multiplet of the open 4fN shell, without the engagement of spin coupling with other magnetic centers. The characteristic CF splitting pattern is specified by the type and symmetry of the coordination environment and by the nature of the lanthanide ion.7In general, two conditions are essential to obtain the highest SMM performance of mononuclear Ln-based complexes, namely, maximal total CF splitting energy (ΔECF) of the ground J-multiplet and perfect axiality of the CF potential of 4f-electrons.7,20–22 In terms of CF theory, this implies that the axial CF parameters Bk0 (k = 2, 4, 6) should be as large as possible with vanishing non-axial parameters Bkq = 0, |q| > 0. The axial CF suppresses the quantum tunneling of magnetization (QTM) and spin–phonon relaxation processes, and thus leads to higher Ueff and TB values.7 These conditions are best met for an axially compressed coordination geometry of the Dy3+ ion having an oblate-shaped 4f-electron density.5–10,23 In fact, there are only three groups of Dy(III) complexes with the highest Ueff barriers, the quasi-linear sandwich metallocene complexes17–19,24 (and some low-coordinate complexes25), the pentagonal-bipyramidal (PBPY-7) complexes with D5h symmetry26–29 and hexagonal-bipyramidal complexes.30–32 Formally, despite the exceptionally high SMM performance of some hexagonal-bipyramidal Dy(III) complexes,30–32 they do not ensure perfect axiality of the crystal field (even at the regular D6h symmetry) due to the presence of the non-axial CF terms B6±6, in contrast to linear (D∞h) and PBPY-7 (D5h) complexes providing perfect axiality of the CF potential. However, this interesting topic is beyond the scope of our paper, which focuses on PBPY-7 Dy(III) complexes.
The first group of complexes holds the current record for the barrier Ueff and blocking temperature TB.17–19 However, even if the maximum SMM performance of PBPY-7 Dy complexes is lower than that in record-holding bis-cyclopentadienyl compounds, they show more diverse structures and magnetic properties with the barrier Ueff up to 1469 cm−1.26–28 An advantage of the PBPY-7 lanthanide complexes is that they are much more readily available synthetically using a variety of monodentate or polydentate ligands in the apical and equatorial positions.29 This enables efficient control and fine tuning of the apical and equatorial components of the CF potential. Particularly, adopting ligands with short Dy–O bonds in the apical positions (such as Cy3PO, Ph3PO, MeO−, Me3SiO−) produces a strong apical CF, resulting in an exceptionally high CF splitting energy (ΔECF > 1000 cm−1) and nearly pure Ising KDs with minimal mixing of ±mJ states.26–29
These remarkable properties have attracted great interest in lanthanide PBPY-7 complexes, which have become a popular research field. To date, numerous PBPY-7 Dy(III) complexes have been reported, including those with high energy barriers (Ueff > 1000 cm−1) and blocking temperatures (TB ∼ 10–20 K).29 However, as noted by many researchers, the SMM performance of these complexes is highly sensitive to the ratio between the CF components originating from the apical and equatorial ligands.26–29 The apical and equatorial CF components act in opposite ways for the oblate dysprosium ion. More specifically, the electron density distribution of Dy3+ has an oblate shape for the mJ = ±15/2 state, but as mJ decreases, the shape gradually transforms from oblate to prolate for the mJ = ±1/2 state.23 As a result, apical ligands minimize the energy of the highest-mJ state of |±15/2〉 and maximize the energy of the lowest-mJ state of |±1/2〉, resulting in a parabolic profile of the E vs. mJ diagram with the |±15/2〉 ground state and |±1/2〉 top state (Fig. S1a, ESI†). This corresponds to the easy axis magnetic anisotropy of the PBPY-7 Dy complex. In contrast, the CF of equatorial ligands stabilizes the prolate mJ = ±1/2 state and maximizes the energy of the oblate mJ = ±15/2 state (Fig. S1b, ESI†); this corresponds to easy plane magnetic anisotropy. In line with this, competition between the apical (CFap) and equatorial (CFeq) crystal field components leads to considerable changes in the pattern of the spin energy diagram and in the effective energy barrier, Ueff, as the equatorial CF increases due to shorter metal–ligand distances in the equatorial plane of the PBPY-7 complex. Accordingly, there are three cases of E vs. mJ diagrams, resulting in different magnetic behavior and SMM characteristics of the PBPY-7 Dy complexes.
1.1 Case I: dominant apical CF (CFap ≫ CFeq)
This case concerns a family of PBPY-7 complexes with short apical bonds (∼2.1 Å) and long equatorial bonds (>2.5 Å), where the apical crystal field, CFap, is much stronger than the equatorial crystal field, CFeq, CFap ≫ CFeq. The strongest apical field, CFap, is generated by negatively charged apical ligands with a short Dy–O bond (2.05–2.15 Å), such as BuO−, PhO−, Me3SiO− and MeO−, while a weak equatorial CF is formed by five neutral pyridine molecules with long Dy–N distances in the equatorial plane, 2.5–2.6 Å. These complexes exhibit the highest barrier (Ueff > 1000 K) and blocking temperature, [Dy(t-BuO)2(py)5]BPh4 (Ueff = 1815 K, TB = 14 K),26 [Dy(Me3SiO)2(py)5]BPh4 (Ueff = 1596 K, TB = 15 K),33 [Dy(CH3CH(C6F5)O)2(THF)5]BPh4·2THF (Ueff = 2113 K, TB = 22 K),27 and [Dy(PhO)2(py)5]BPh4 (Ueff = 1302 K, TB = 13 K).33 The E vs. mJ diagram has a parabolic profile with the lowest |±15/2〉 and top |±1/2〉 states (Fig. 1a). The ground-state Kramers doublet KD0 |±15/2〉 has nearly perfect axiality, as quantified by the Ising g-tensor with gz ∼ 19.8 and vanishingly small transverse components, gx, gy (<10−2).26,28,33 A strong apical CF also retains the magnetic axiality of several excited KDs (up to KD4–5), which have nearly pure |±mJ〉 character. In this case, spin relaxation occurs through highly excited |±mJ〉 states close to the upper KD7 |±1/2〉 (Fig. 1a), thereby pushing up the barrier Ueff toward to the top KD7 with mJ = ±1/2.7,26–28,33 In concert with the large total CF splitting energy of the 6H15/2 multiplet of the Dy3+ ion, this results in a very high barrier Ueff. Indeed, experimental data and theoretical calculations show that Ueff approximately corresponds to the energy position of the top |±1/2〉 state. The long magnetic axis of the |±15/2〉 ground state is parallel to the apical C5 axis, and the overall magnetic anisotropy has a distinct easy-axis characteristic.26–29,33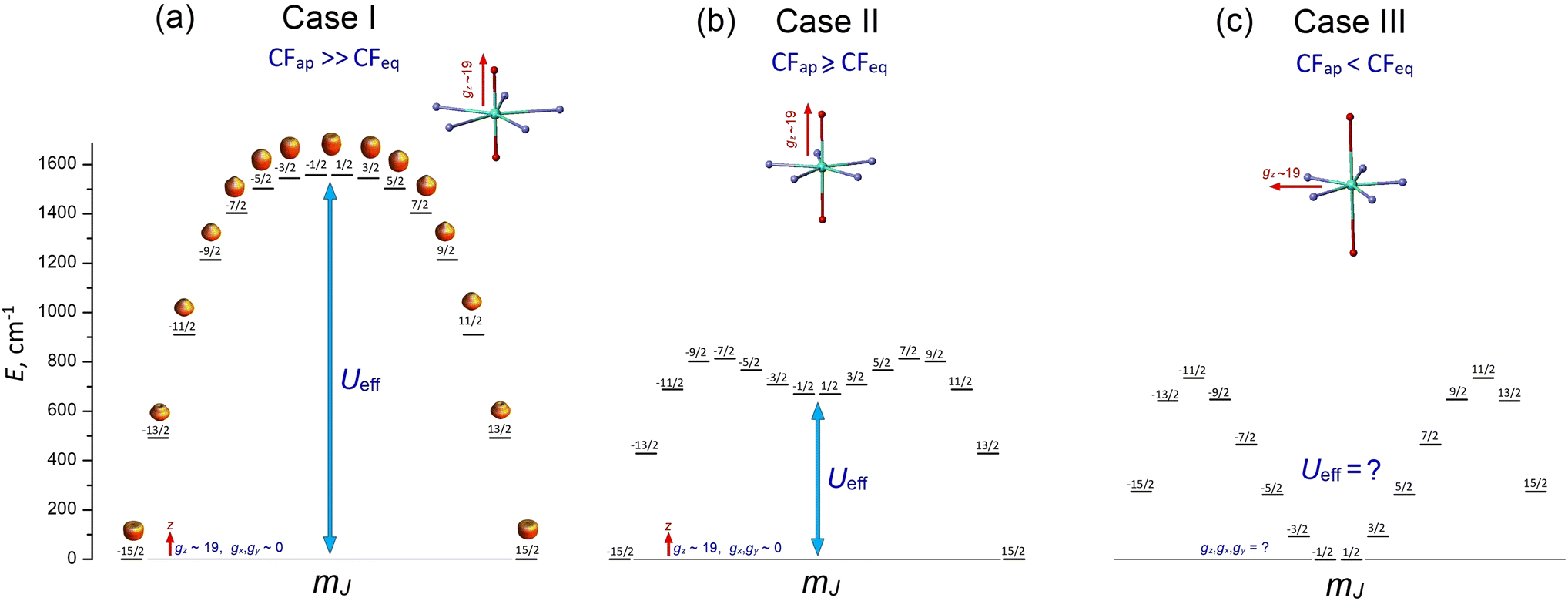 | ||
| Fig. 1 Evolution of the E vs. MJ diagram of PBPY-7 Dy(III) complexes with decreasing ratio between the apical crystal field component, CFap, and the equatorial crystal field component, CFeq, (a) CFap ≫ CFeq, (b) CFap > CFeq, (c) CFap < CFeq. | ||
1.2 Case II: moderately strong apical CF (CFap > CFeq)
This group includes numerous PBPY-7 Dy(III) complexes in which the apical CF component still prevails, but the balance between the apical CFap and equatorial CFeq components of the CF is less marked than in Case I. The decrease in the CFap/CFeq ratio may be caused by the enhancement of the equatorial crystal field, CFeq, due to shorter equatorial metal–ligand distances (2.3–2.4 Å), which occurs in PBPY-7 complexes [DyL2X5] with X = THF, H2O ([Dy(Cy3PO)2(H2O)5]3+,34,35 [Dy(CyPh2PO)2(H2O)5]3+,36 and [Dy(pz)2(THF)5]+),37 or with mixed axial ligands Y1, Y2 (one strong, one weak) in [Dy(Y1Y2)X5] complexes with Y1/Y2 = –OCMe3, –OSiMe3, –OPh, Cl− or Br−; X = THF, Py.33,38 A number of PBPY-7 complexes [Dy(Y1Y2)L] with polydentate equatorial ligands L can also be attributed to this group.39–41 In this group of PBPY-7 complexes, the ground state remains KD0 |±15/2〉, but the non-axial |±1/2〉 state (with gz = 1.32, gx = gy = 10.59) is no longer the highest in energy among the |±mJ〉 states due to the sagging of the top of the E vs. mJ diagram caused by a stronger equatorial field (Fig. 1b). As a result, the total CF splitting energy, ΔECF, of the 6H15/2 multiplet decreases significantly (ΔECF < 800 cm−1) and the effective barrier, Ueff, is further reduced, in accordance with a lower energy of the non-axial |±1/2〉 state (Fig. 1b). In fact, Ueff typically ranges between 400 and 1000 K.34–41 The ground state KD0 |±15/2〉 and a few excited states (|±13/2〉, |±11/2〉) retain high axiality, although their transverse components, gx and gy, may be more pronounced compared to Case I due to the reduced overall axiality of the CF. Case II complexes exhibit easy axis magnetic anisotropy parallel to the polar C5 axis of the bipyramid. As in Case I, the energy barrier, Ueff, is close to the energy of the non-axial |±1/2〉 state.33–351.3. Case III: the apical field is weaker than the equatorial field (CFap < CFeq)
This group comprises PBPY-7 complexes with weak apical ligands (such as Cl, Br)33,42,43 and rather numerous Dy(III) complexes with polydentate ligands.43,44 Of special note are PBPY-7 complexes with the strongest equatorial CF comprising pentadentate Schiff-base ligands based on 2,6-diacetylpyridine45–48 (see below). Complexes of this type reveal low barriers (Ueff < 300 K, in most cases <100 K) as well as overall low CF splitting energies (600–700 cm−1 or lower).33,42–48 In this group, the magnetic properties change drastically due to crossover between the |±1/2〉 and |±15/2〉 ground states in the E vs. mJ diagram (Fig. 1c). More specifically, under the strong equatorial field, the prolate |±1/2〉 state drops in energy and goes to the ground state, while the |±15/2〉 state with the oblate electron cloud becomes an excited state due to enhanced repulsion from the strong equatorial ligands. As a result, the overall magnetic anisotropy of the PBPY-7 Dy(III) complex transforms from negative (easy-axis) to positive (easy-plane). Accordingly, in Case III PBPY-7 Dy complexes, the driving force behind barrier formation and SMM behavior differs considerably from that in Cases I and II.As in the high-performance PBPY-7 Dy-SIMs with a strong axial CF, distinct SMM behavior in Case III complexes occurs only when the ground-state KD0 has a dominant |±15/2〉 character and nearly perfect Ising g-tensor with gz ∼ 19 and very small gx, gy components. However, now the orientation of the long magnetic z axis (which, according to ab initio software, is taken to be the common spin quantization axis)22 turns from the apical C5 axis to the equatorial plane. In other words, the nature of the equatorial KD0 |±15/2〉eq in Case III complexes differs drastically from that for the axial KD0 |±15/2〉 in Case I and II complexes. This is evident from the fact that with the old spin quantization z axis parallel to the polar C5 axis, the ground equatorial KD0 |±15/2〉eq is mainly composed of the |±1/2〉, |±3/2〉 and |±5/2〉 states, which are all low-lying in the E vs. mJ diagram in Case III (Fig. 1c), which is opposite to Cases I and II, where |±15/2〉 is the lowest state (Fig. 1a and b). Importantly, the formation of the highly anisotropic equatorial Ising-type ground state, |±15/2〉eq, is now governed not by the strength of the axial ligands (measured by the axial CF parameters Bk0), but by the non-axial CF parameters Bkq, q ≠ 0, which mix the low-lying states, |±1/2〉, |±3/2〉, |±5/2〉, …, to produce the equatorial KD0 |±15/2〉eq. Below, we show that this mixing is mainly due to the asymmetry of the equatorial pentagon formed by monodentate or polydentate equatorial ligands (see section 3.5 for details).
In this paper, we report the syntheses, characterization, and magnetic properties of three new seven-coordinate pentagonal-bipyramidal Dy(III) complexes, [Dy(LCH3)(Cy3PO)2]ClO4·CH3CN (1), [Dy(L2(t-Bu))(Ph3PO)2]ClO4 0.63C2H5OH (2), and [Dy(LOCH3)(Ph3PO)2]ClO4·2H2O (3), including various chelating pentadentate ligands with [N3O2]2− binding node in the equatorial plane, LCH3 = [2,6-diacetylpyridine bis(acetylhydrazone)]2−, L2(t-Bu) = [2,6-diacetylpyridine bis(3,5di-tert-butylbenzoylhydrazone)]2−, and LCH3 = [2,6-diacetylpyridine bis(4-methoxybenzoylhydrazone)]2−, and OPCy3 or OPPh3 in the apical positions. As in our previous works,49,50 as well as in ref. 45–48, we took advantage of pentadentate chelating LR ligands based on 2,6-diacetylpyridine, which provided a rigid pentagonal equatorial plane around the Ln(III) ions in the heptacoordinated complexes and left the axial positions free, thereby enabling us to use various axial ligands (Fig. 2).
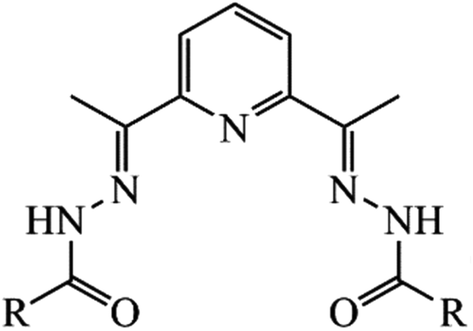 | ||
| Fig. 2 2,6-Diacetylpyridine-based acyclic ligands used in this work: R = CH3 (2,6-diacetylpyridine bis(acetylhydrazone), H2LCH3), R = 4-OCH3C6H4 (2,6-diacetylpyridine bis(4-methoxybenzoylhydrazone), H2LOCH3), R = 3,5-di-tBuC6H3 (2,6-diacetylpyridine bis(3,5 di-tert-butylbenzoylhydrazone), H2L2(t-Bu)). | ||
Originally, the objective of this work was to investigate the impact of the apical ligands Ph3PO and Cy3PO on the SMM characteristics, as well as to compare our results with those of previously reported related complexes with similar equatorial ligands and different combinations of axial ligands.46–49 In the present study, we show that, like in the PBPY-7 Dy complexes reported in ref. 45–48, the ground state of complexes 1–3 is KD0 |±15/2〉eq (gx ∼ 19.6, gy, gz ∼ 0) with the long magnetic axis lying in the equatorial xy plane; therefore, all of these complexes fall under Case III (Fig. 1c). Then, after closer inspection of the magneto-structural correlations in numerous PBPY-7 Dy complexes from Cases I, II and III reported in the literature,29 we have concluded that a more in-depth analysis of the origin of SMM behavior in Case III complexes is highly advisable.
Following the above considerations, herein we report the magnetic properties of complexes 1–3 and provide a detailed theoretical study of the origin of the spin-reversal barrier, Ueff, in Case III complexes in terms of CF theory for lanthanide ions. We show that in Case III complexes, a strong equatorial CF results in a completely different scenario for the origin of SMM behavior, in which distortions of the PBPY-7 geometry (especially the asymmetry of the coordination pentagon in the equatorial plane) are more important than the CF strength of the apical ligands, which is opposite to Cases I and II, in which distorted PBPY-7 geometry is highly undesirable since it tends to decrease Ueff and TB due to the departure from perfect magnetic axiality. Therefore, these results indicate that Case III complexes, including our complexes 1–3, experience a breakdown of the strong apical magnetic axiality, which is crucial for reaching the highest Ueff and TB values, as occurs in the record PBPY-7 Dy(III) complexes, [Dy(t-BuO)2(py)5]BPh4 (Ueff = 1815 K, TB = 14 K),26 and [Dy(CH3CH(C6F5)O)2(THF)5]BPh4·2THF (Ueff = 2113 K, TB = 22 K).28 From a wider point of view, this refers to the fact that the oblate Dy3+ ion prefers a strong apical CF and a weak equatorial CF to have maximal Ueff and TB values, while in Case III dysprosium complexes the situation is just the opposite. As a result, the PBPY-7 Dy(III) complexes with the long magnetic axis lying equatorially have little chance of being high-performance SMMs. Even though the negative impact of a strong equatorial CF on the SMM performance of PBPY-7 Dy complexes is currently well-recognized,5–10 the specific mechanism behind the origin of the pure Ising-type ground state KD0 |±15/2〉eq in the equatorial plane of Case III complexes (i.e., with CFeq > CFap) is explored for the first time in this paper. To the best of our knowledge, this issue has not been previously addressed in the literature, despite much research activity in the field of PBPY-7 Ln-SMMs.
2. Experimental section
2.1 Materials and methods
2,6-Diacetylpyridine, Et3N, Dy(ClO4)3·6H2O, 3,5-di-tert-butylbenzoic acid, 4-methoxybenzoic acid, thionyl chloride, hydrazine hydrate solution (50–60% N2H4), acetic hydrazide, tricyclohexylphosphine oxide (Cy3PO), and triphenylphosphine oxide (Ph3PO) were purchased from commercial sources and used without further purification. Methanol was dried upon refluxing with magnesium methoxide, followed by distillation. Acetonitrile was distilled over calcium hydride. Diethyl ether was distilled over LiAlH4. All the solvents were purged with argon and stored over 3 Å molecular sieves prior to use.IR spectra of solid samples were recorded with a PerkinElmer SPECTRUM TWO FT-IR spectrometer in the range of 4000–600 cm−1. Elemental analyses were performed by the Analytical Department service at the Federal Research Center of Problems of Chemical Physics and Medicinal Chemistry RAS using a Vario MICRO Cube analyzer.
2.2 Synthetic procedures
Methyl-3,5-di-tert-butylbenzoate was prepared from 3,5-di-tert-butylbenzoic acid, methanol and thionyl chloride according to a previously published procedure.51 3,5-Di-tert-butylbenzoic acid hydrazide was synthesized from methyl-3,5-di-tert-butylbenzoate and hydrazine hydrate in refluxing methanol.52 The 4-methoxybenzoic acid hydrazide was in a similar manner.The ligands 2,6-diacetylpyridine bis(acetylhydrazone), H2LCH3, 2,6-diacetylpyridine bis(3,5 di-tert-butylbenzoylhydrazone), H2L2(t-Bu), and 2,6-diacetylpyridine bis(4-methoxy-benzoylhydrazone), H2LOCH\3, were prepared through a ketone–hydrazine condensation reaction between 1 equiv. of 2,6-diacetylpyridine and 2 equiv. of acetic hydrazide, 3,5-di-tert-butylbenzoic acid hydrazide or 4-methoxybenzoic acid hydrazide, respectively, in 96% ethanol, according to procedures in the literature with minor modification.53 The yield was over 90% for all three ligands.
H 2 L 2(t-Bu) found: C, 75.57; H, 8.55; N, 11.04%. Calc. for C39H53N5O2: C, 75.12; H, 8.5; N, 11.23%. FT-IR νmax/cm−1: 3170m, 2959s, 2929m, 1658vs, 1593m, 1529s, 1449s, 1362s, 1312m, 1244vs, 1156s, 1121m, 1077m, 860m, 814m, 738m, 707vs.
H 2 L OCH 3 found: C, 65.62; H, 5.71; N, 15.35%. Calc. for C25H25N5O4: C, 65.35; H, 5.48; N, 15.24%. FT-IR νmax/cm−1: 3411m, 3218m, 2296m, 1646s, 1606s, 1580m, 1547s, 1502vs, 1455m, 1365m, 1285s, 1250vs, 1176vs, 1145m, 1120m, 1025s, 919m, 834vs, 758m.
Anal. found: C, 52.35; H, 7.32; N, 6.87%. Calc. for DyC51H86N6O8P2Cl (Mm 1171.18 g mol−1): C, 52.25; H, 7.34; N, 7.17%.
FT-IR νmax/cm−1: 2928s, 2853m, 1579m, 1515s, 1446m, 1388vs, 1333m, 1321m, 1083vs, 892m, 853m, 675m, 622vs (Fig. S2, ESI†).
FT-IR νmax/cm−1: 2962m, 1557m, 1504s, 1438s, 1407m, 1368vs, 1275s, 1140vs, 1119s, 1075vs, 997m, 890m, 820m, 762m, 726vs, 706s, 692vs, 622vs, 538vs (Fig. S3, ESI†).
Anal. found: C, 56.31; H, 4.17; N, 5.41%. Calc. for C61H57ClDyN5O12P2, (Mm 1312 g mol−1): C, 55.79; H, 4.34; N, 5.34%.
FT-IR νmax/cm−1: 3032w, 1586m, 1494s, 1438s, 1362vs, 1352m, 1249s, 1164s, 1140vs, 1083vs, 1040vs, 998s, 765s, 724vs, 684vs, 618vs, 538vs (Fig. S4, ESI†).
2.3 X-ray data collection, structure solution, and refinement
Single-crystal X-ray data for complexes 1 and 2 were collected using a Bruker D8 Venture Photon single-crystal diffractometer equipped with an Incoatec IμS 3.0 microfocus sealed tube (Mo Kα radiation, λ = 0.71073 Å), and for compound 3 using a Bruker SMART APEX II instrument (Mo Kα radiation, λ = 0.71073 Å) in φ- and ω-scan mode at the Center for Collective Use of the Kurnakov Institute RAS (Moscow, Russia). The raw data for 1–3 were treated with APEX3 software;54 experimental intensities were corrected for absorption effects using SADABS.55 The crystal structures of 1–3 were solved by direct methods56 and refined by full-matrix least-squares on F2 using the OLEX2 structural data visualization57 and analysis program suite.58 Non-hydrogen atoms were refined with anisotropic thermal parameters (except disordered methyl and tert-butyl groups in structures 2 and 3 with low occupancies of disordered atom positions, which were refined using isotropical approximations). Geometrical restraints (SADI, DFIX) and restraints on atomic displacement parameters (SIMU, RIGU, ISOR, DELU, EADP) were also applied on disordered atoms of structures 2 and 3. All hydrogen atoms were placed in the calculated positions and refined using the riding model with dependent isotropic thermal parameters with Uiso(H) = 1.5Ueq(C) for the methyl or hydroxyl groups and with Uiso(H) = 1.2Ueq(C) for other hydrogen atoms. Table S1 in the ESI† summarizes crystal data and structure refinement details for 1–3. Full crystallographic data for the crystal structures of 1–3 are given in CCDC deposition numbers 2239615–2239617, respectively.†The powder XRD patterns for the complexes were recorded at room temperature on an Aeris diffractometer (Malvern PANalytical B.V., Netherlands). The powder XRD measurements showed that polycrystalline samples of the complexes were monophase crystalline materials corresponding to single crystal data (Figs. S5–S7, ESI†).
2.4 Magnetic measurements
The dc and ac magnetic properties were measured by using the Quantum Design PPMS-9 physical property measuring system (complexes 1, 2) and the vibrating-sample magnetometer of a Cryogen Free Measurement System (CFMS, Cryogenic Ltd, UK) in the case of 3. The temperature dependence of the magnetic moment, M(T), was measured at T = 2–300 K in a dc magnetic field of B = 0.5 T. The field dependences of the magnetic moment, M(H), were obtained at temperatures of 2, 3 and 5 or 6 K in the dc magnetic field range of 0–5 T. The ac magnetic behavior of the complexes was studied within the frequency range of 10–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz in the absence and under the application of a dc magnetic field. The measurements were carried out on polycrystalline samples moistened with mineral oil (Fomblin YR 1800) to prevent orientation of the crystals in a dc magnetic field. The prepared samples were sealed in plastic bags. The experimental data were corrected for the sample holder and mineral oil. The diamagnetic contribution from the substance was calculated using Pascal's constants.
000 Hz in the absence and under the application of a dc magnetic field. The measurements were carried out on polycrystalline samples moistened with mineral oil (Fomblin YR 1800) to prevent orientation of the crystals in a dc magnetic field. The prepared samples were sealed in plastic bags. The experimental data were corrected for the sample holder and mineral oil. The diamagnetic contribution from the substance was calculated using Pascal's constants.
2.5 Computational details
Ab initio calculations were performed using the OpenMolcas program.59 The [.ANO-RCC…8s7p5d3f2g1h.] basis set for the Dy atom, [.ANO-RCC…5s4p2d1f.] for the P atom, [.ANO-RCC…4s3p2d1f] for the N and O atoms, [.ANO-RCC…2s1p.] for C atoms and [.ANO-RCC…1s.] for H atoms were employed. The ground state f-electron configuration for Dy(III) is 4f9 with the 6H15/2 multiplet as the ground state. Initially, we generated the guess orbitals and from there the seven Dy(III) based starting orbitals were selected to perform the CASSCF calculations, where 9 electrons were in the 7 active orbitals with an active space of CAS (9,7). Using this active space, 21 sextets were computed using the configuration interaction (CI) procedure. After that, all 21 sextets were mixed using the RASSI-SO module to compute the spin–orbit states. After computing these spin–orbit states, using the SINGLE_ANISO code60a the corresponding g-tensors and CF parameters for the eight low-lying Kramers doublets (KDs) were extracted. The Cholesky decomposition for two electron integrals was employed throughout the calculations to reduce the disk space. Crystal-field calculations were performed in terms of conventional CF theory using the Wybourne notation for the CF parameters Bkq, as described elsewhere.60b3 Results and discussion
3.1 Synthesis
Most of the seven-coordinate high-barrier Dy(III) SMMs described to date contain five neutral weak-field ligands as equatorial ligands and two anionic or neutral strong-field ligands at the axial positions. There are not many 4f element complexes with D5h symmetry containing chelating pentadentate ligands in the equatorial plane.45–48,61 In contrast to 3d transition-metal ions, PBPY-7 coordination geometry is rare among 4f complexes with pentadentate acyclic ligands because rare Earth ions prefer large coordination numbers (8–10) and variable coordination geometries. The composition and structures of compounds formed by the interaction of dysprosium salts with planar pentadentate ligands strongly depend on the reaction conditions, and it is possible to obtain 10-, 9-, 8- and 7-coordinate complexes.45–50,62 To obtain the dysprosium seven-coordinate complex in a targeted manner, it is necessary to strictly control the synthesis conditions, in particular the ratio of reagents, the order of base addition, and the type of anion of the initial dysprosium salt.In this work, Dy(ClO4)3 6H2O was used as the starting dysprosium salt. During the synthesis, the ratio of the reaction components, Dy(ClO4)3:H2L:OPPh3:Et3N = 1:1:2:2, was strictly enforced, and the deprotonating Et3N base was added to the reaction medium approximately 1 h after the addition of the dysprosium salt to the ligand mixture. The point is that we have previously shown that preliminary deprotonation of the H2L ligand by adding Et3N to the ligand before the dysprosium salt leads to the formation of a stable and well crystallized charge-neutral mononuclear complex, [Dy(HL)(L)], in the reaction medium as the main product, in which the Ln3+ ion binds two charged pentadentate ligands.62 To avoid the formation of such complexes, it is highly desirable not to exceed the ratio of H2L/Dy(ClO4)3 = 1 during synthesis, and also to introduce Et3N after adding the metal salt to the mixture of H2L and OPPh3 ligands.
Following these general rules, we have synthesized a series of three new heptacoordinated mononuclear Dy PBPY-7 complexes, including various pentadentate ligands with [N3O2]2− binding node in the equatorial plane, H2LCH3, H2LOCH3, and H2L2(t-Bu), and OPPh3 or OPCy3 in the apical positions. All the complexes are characterized by elemental analysis, infrared spectroscopy, magnetic measurements and X-ray crystallography. Single crystals of compounds 1–3 are obtained by slow evaporation of the filtered mother liquors. The yields are quite high for the lanthanide complexes (65–75%). Elemental analyses of the complexes were consistent with the composition of [Dy(L)(OPPh3)2(ClO4)]·solv, where L denotes a dideprotonated ligand molecule. All the complexes are air-stable in the crystalline state. The phase purity of the samples of complexes used for magnetic measurements was confirmed by the X-ray powder diffraction data (Figs. S5–S7, ESI†).
Pentadentate ligands with the binding units N3O2, H2LCH3, H2LOCH3, and H2L2(t-Bu) used in this work differ in that different substituents are introduced into the phenyl groups of the H2LOCH3 and H2L2(t-Bu) ligands, which affect the electronic and steric properties of the resulting ligand. The use of slightly different pentadentate ligands with the same binding site allows us to determine whether the peculiarities of the equatorial ligand structure affect the properties of this series of complexes as monomolecular magnets.
All pentadentate ligands used in this work were obtained by the standard condensation reaction of 2,6-diacetylpyridine with two equivalents of the corresponding hydrazide. Hydrazides for the synthesis of H2LOCH3 and H2L2(t-Bu) ligands were synthesized beforehand from commercially available substituted phenylcarboxylic acids. The phenylcarboxylic acids were converted into the corresponding methyl esters, which were then converted into hydrazides by treatment with hydrazine hydrate in alcohol medium under heating. The ketone–hydrazine condensation with 2,6-diacetylpyridine yielded the final ligands. The synthesized hydrazides and ligands were characterized by elemental analysis, NMR, and IR spectra (Figs. S8–S10, ESI†).
3.2 Crystal structure descriptions
The crystal structures of compounds 1–3 were established by the single crystal X-ray diffraction technique. According to X-ray diffraction data, compounds 1 [Dy(LCH3)(Cy3PO)2](ClO4)·CH3CN (Fig. 3), 2 [Dy(L2(t-Bu))(Ph3PO)2](ClO4)·0.63C2H5OH (Fig. 4), and 3 [Dy(LOCH3)(Ph3PO)2]ClO4·2H2O (Fig. 5) crystallize in three different space groups, P21/n, P21/c, and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , respectively, with two independent molecular units in 1 (1a, 1b) and 2 (2a, 2b) and one in 3, see Table 1 and Fig. 6.
, respectively, with two independent molecular units in 1 (1a, 1b) and 2 (2a, 2b) and one in 3, see Table 1 and Fig. 6.
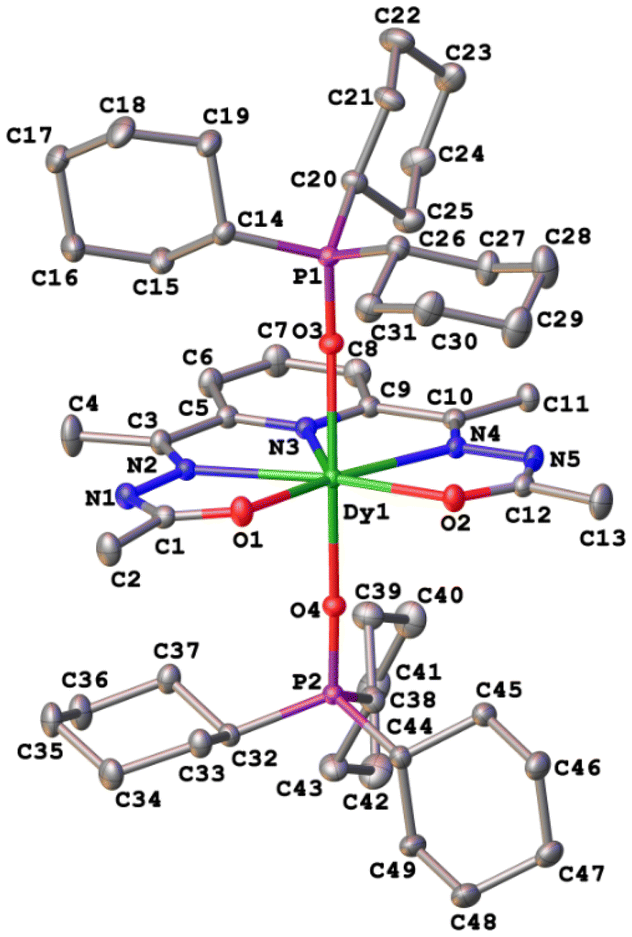 | ||
| Fig. 3 Crystal structure and atom numbering scheme of the cationic part of complex 1. ClO4− anions and hydrogen atoms are omitted for clarity. Atomic displacement parameters are shown at the 50% probability level. | ||
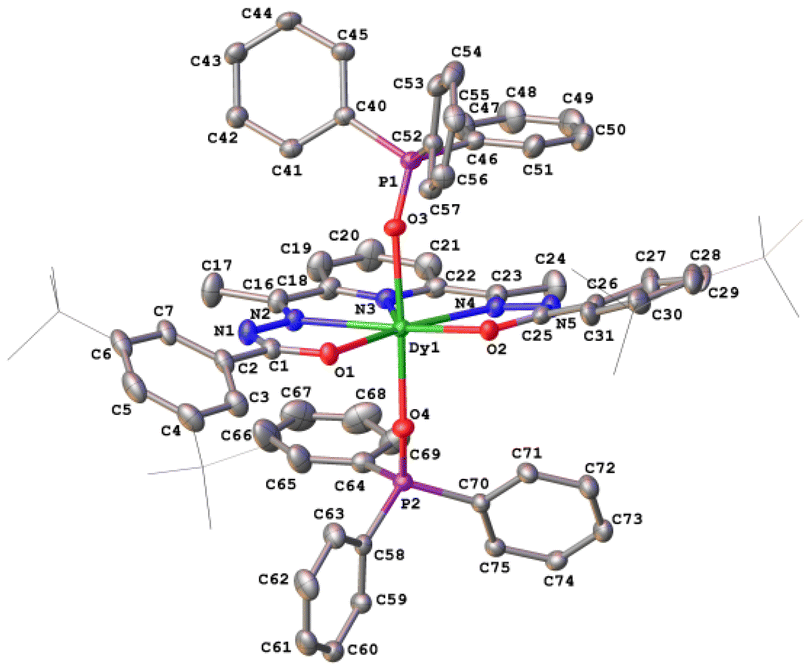 | ||
| Fig. 4 Crystal structure and atom numbering scheme of the cationic part of complex 2. ClO4− anions, ethanol molecules, disordering and hydrogen atoms are omitted for clarity. Bulky tert-butyl groups are shown schematically. Atomic displacement parameters are given at the 35% probability level. | ||
 | ||
| Fig. 5 Crystal structure and atom numbering scheme of the cationic part of complex 3. ClO4− anions, disordering of one methoxy group and hydrogen atoms are omitted for clarity. Atomic displacement parameters are given at the 35% probability level. | ||
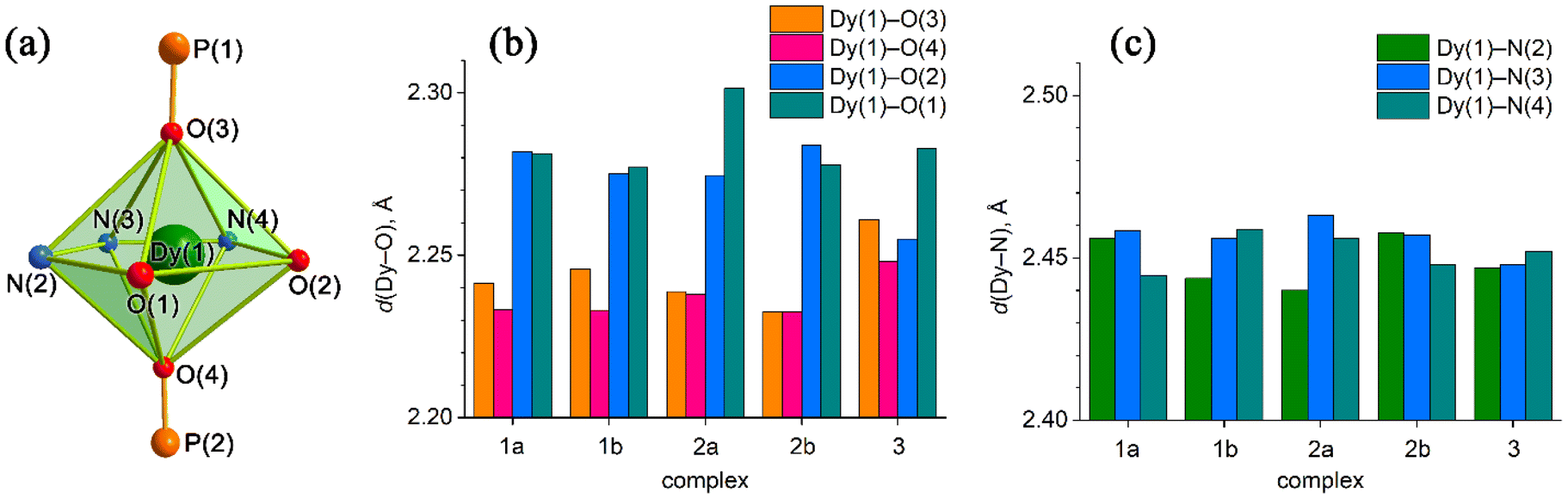 | ||
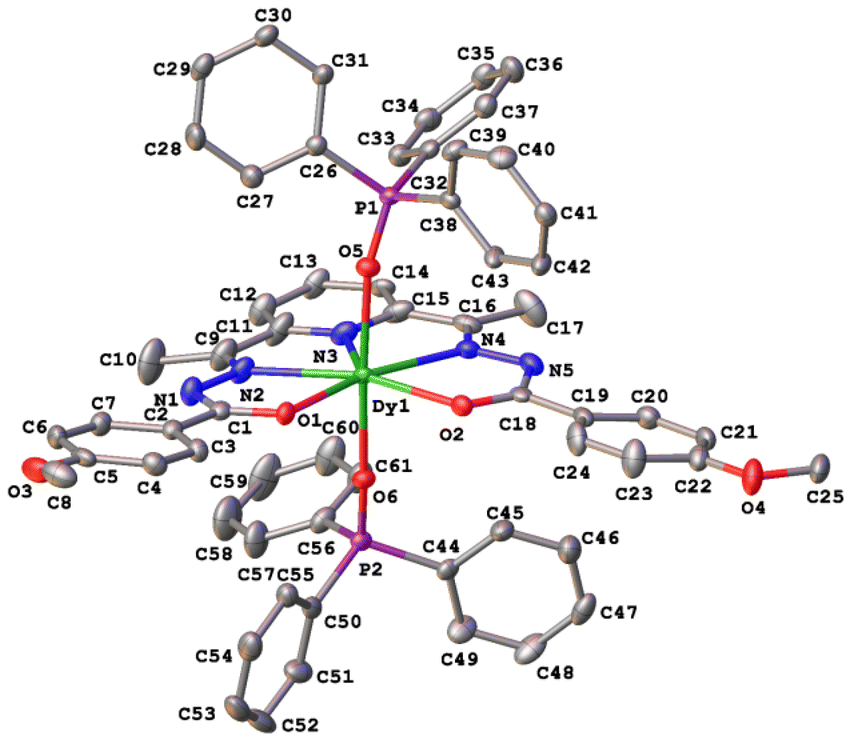
| Fig. 6 (a) Coordination polyhedron of Dy(III) with atom numbering. (b) Axial Dy(1)–O(3)/O(4) for 1a, 1b, 2a, and 2b and Dy(1)-O(5)/O(6) for 3 (Fig. 5), as well as equatorial Dy(1)–O(1)/O(2) distances. (c) Equatorial Dy–N distances. | ||
| Bond length | 1 (1a, 1b) | 2 (2a, 2b) | 3, Fig. 6 |
|---|---|---|---|
| Dy(1)–O(1) | 1a, 2.2813(11) | 2a, 2.3013(17) | 2.283(3) |
| Dy(2)–O(1A) | 1b, 2.2770(11) | 2b, 2.2779(15) | |
| Dy(1)–O(2) | 1a, 2.2819(11) | 2a, 2.2746(17) | 2.255(3) |
| Dy(2)–O(2A) | 1b, 2.2750(11) | 2b, 2.2839(15) | |
| Dy(1)–O(3) | 1a, 2.2415(12) | 2a, 2.2379(17) | 2.261(3) [Dy(1)−O(5)] |
| Dy(2)–O(3A) | 1b, 2.2458(11) | 2b, 2.2325(15) | |
| Dy(1)–O(4) | 1a, 2.2334(11) | 2a, 2.2388(17) | 2.248(3) [Dy(1)−O(6)] |
| Dy(2)–O(4A) | 1b, 2.2331(11) | 2b, 2.2326(15) | |
| Dy(1)–N(2) | 1a, 2.4561(13) | 2a, 2.440(2) | 2.447(3) |
| Dy(2)–N(2A) | 1b, 2.4437(14) | 2b, 2.4578(18) | |
| Dy(1)–N(3) | 1a, 2.4584(13) | 2a, 2.463(2) | 2.448(3) |
| Dy(2)–N(3A) | 1b, 2.4560(13) | 2b, 2.4571(18) | |
| Dy(1)–N(4) | 1a, 2.4446(14) | 2a, 2.456(2) | 2.452(3) |
| Dy(2)–N(4A) | 1b, 2.4588(14) | 2b, 2.4478(18) |
Each structure could be described by the general formula [Dy(L)(OPR3)2](ClO4), thus being a cation–anion complex with different solvent molecules but the same coordination environment around the central dysprosium atoms. Dysprosium atoms are surrounded by two oxygen atoms and three nitrogen atoms of the pentadentate ligand in the equatorial plane and by two oxygen atoms of the R3PO ligand, which complete coordination number seven (Figs. 3–5).
Continuous shape measure analysis (CShM) was performed for complexes 1–3 to elucidate the real geometry of the hepta-coordinate environment around the Dy(III) ion.63 The analysis showed that the local geometry of the Dy(III) ions in the complexes was pentagonal bipyramidal with pseudo-D5h symmetry for the [DyN3O4] core (Table S2, ESI† and Fig. 6). Deviations from pentagonal-bipyramidal geometry are between 1.154 and 1.569 (Table S2, ESI†).
The two non-equivalent types of oxygen atoms, which are bonded to Dy, form two different types of Dy–O bonds, as illustrated in Table 1 and Fig. 6: Dy–O(ap) distances are somewhat shorter than those of the Dy–O(eq).
The interatomic bond lengths of Dy with the polydentate equatorial ligand (Dy–N (2.440(2)–2.4588(14) Å) and Dy–O (2.255(3)–2.3013(17) Å)) for complexes 1–3 are close to the values found in previously described Dy complexes with the dianionic form of pentadentate N3O2-type Schiff base ligands.45,46,50 However, it should be noted that these Dy–O(eq) distances for complexes 1–3 are noticeably shorter than those in pentagonal-bipyramidal Dy complexes with magnetization barriers extending to 1000 K and even higher, which contain monodentate weak-field ligands (water, tetrahydrofuran (Dy–O 2.327(5)–2.453(3)), pyridine, pentaazomacrocycles) in the equatorial plane and strong-field ligands in the axial positions (phosphine/silano oxides, acyl/aryl alkoxides, aryl/acyl phosphonamides).26,28,33,34,64 At the same time, the Dy–O(ap) bond lengths in 1–3 (Table 1) are comparable to the Dy–O(ap) distances in the previously described seven-coordinate dysprosium complexes with O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PR3 ligands in axial positions.34,46
PR3 ligands in axial positions.34,46
Crystal structure 1 contains two crystallographically independent molecules, but they are almost identical. In complex 2, the independent molecules differ. In particular, the axial angles are different. For complex 1, the Oap–Dy–Oap bond angle between oxygen atoms of the axial ligands is 170.91(4)–171.49(4)°; for 2 and 3 it is 169.96(6)–176.71(6)° and 174.43(10)°, respectively (Tables S3–S5, ESI†).
The crystal packing of compounds 1 and 2 does not contain any obvious intermolecular interactions or π-stacked moieties due to high steric hindrance of the axial ligands. In contrast, a single π–π stacking interaction between the phenyl groups of neighboring cations of [Dy(LOCH3)(Ph3PO)2]+ (symmetry code: 2 − x, 1 − y, 1 − z) with a centroid-to-centroid distance of 3.742 Å was detected in 3 (Fig. S11, ESI†). It should be noted that the minimum distances between the dysprosium ions in the crystal structures of 1–3 are 10.949, 10.328 and 10.429 Å, respectively, i.e., the dysprosium ions are well separated in the crystal structures.
3.3 Static magnetic properties
The dc magnetic susceptibility data for complexes 1–3 were measured in the temperature range of 2.0–300 K under an applied field of 5000 Oe (Fig. 7). The χMT values at 300 K are 14.11, 13.92 and 13.64 cm3 K mol−1 for 1, 2 and 3, respectively, which are in agreement with the expected value of 14.17 cm3 K mol−1 for an isolated Dy(III) ion (6H15/2, g = 4/3). The χMT values decrease gradually upon cooling from room temperature to ∼10 K, and then decrease abruptly, reaching 7.13, 7.22 and 7.96 cm3 K mol−1 at 2 K for 1, 2 and 3, respectively (Fig. 7). Magnetization (M) versus field curves for 1, 2 and 3 at different temperatures are shown in the insets of Fig. 7. All complexes have the same behavior of M(H) dependences with similar values of 5.16NAμB, 5.05NAμB and 5.04NAμB for 1, 2 and 3, respectively, at 2 K and 5 T.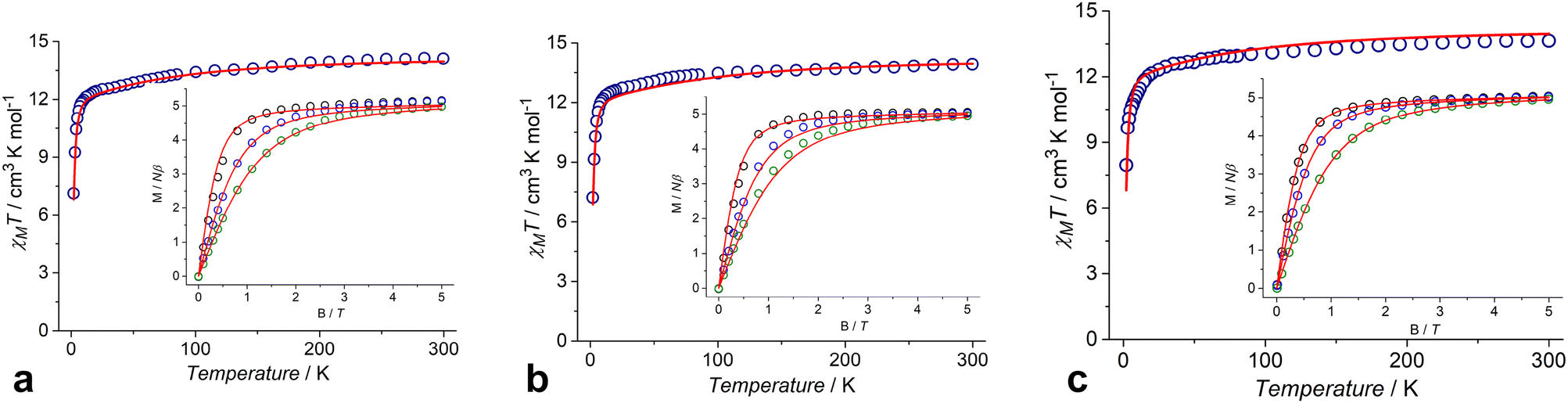 | ||
| Fig. 7 Temperature dependences of χMT for 1 (a), 2 (b) and 3 (c). Insets: Magnetization vs. field plots at different temperatures (2, 4, 6 K for 1, 2 and 2, 3, 5 K for 3). The red solid lines show the corresponding results of the SA-CASSCF/SO-RASSI/SINGLE_ANISO calculations. | ||
Magnetic hysteresis cycles at different temperatures and different magnetic field sweep rates have been studied using complex 3 as an example. At 2 K and a sweep rate of 50 Oe s−1, complex 3 exhibits a tight-waist hysteresis loop (S12, ESI†) with small coercivity and remanence values of 40 Oe and 0.05μB, respectively, indicating the occurrence of an effective QTM. As expected for SMM behavior, the hysteresis and remanent magnetization become larger (260 Oe and 0.25μB, respectively) when the sweep rate is increased to 150 Oe s−1 at the same 2 K temperature (S12, ESI†). In addition, the loop width (coercive field) and remanence values decrease significantly with increasing temperature from 2 K to 5 K for both 50 Oe s−1 and 150 Oe s−1 sweep rates (S13, ESI†), which is also characteristic of SMMs.
3.4 Dynamic magnetic properties
Complexes 1–3 reveal dynamic magnetic behavior at zero dc field (Fig. 8 and S14, ESI†). For complexes 1 and 2, the maximum frequency dependences of the out-of-phase ac susceptibility are characterized by an asymmetric broadening with a “tail” in the high-frequency region. This kind of broadening cannot be described by the α parameter in the generalized Debye model,65 so the two-component model has to be used to fit the experimental data with good quality. For complex 3, the generalized Debye model is sufficient. Best fit parameters are shown in Tables S6–S8, ESI.†
are characterized by an asymmetric broadening with a “tail” in the high-frequency region. This kind of broadening cannot be described by the α parameter in the generalized Debye model,65 so the two-component model has to be used to fit the experimental data with good quality. For complex 3, the generalized Debye model is sufficient. Best fit parameters are shown in Tables S6–S8, ESI.†
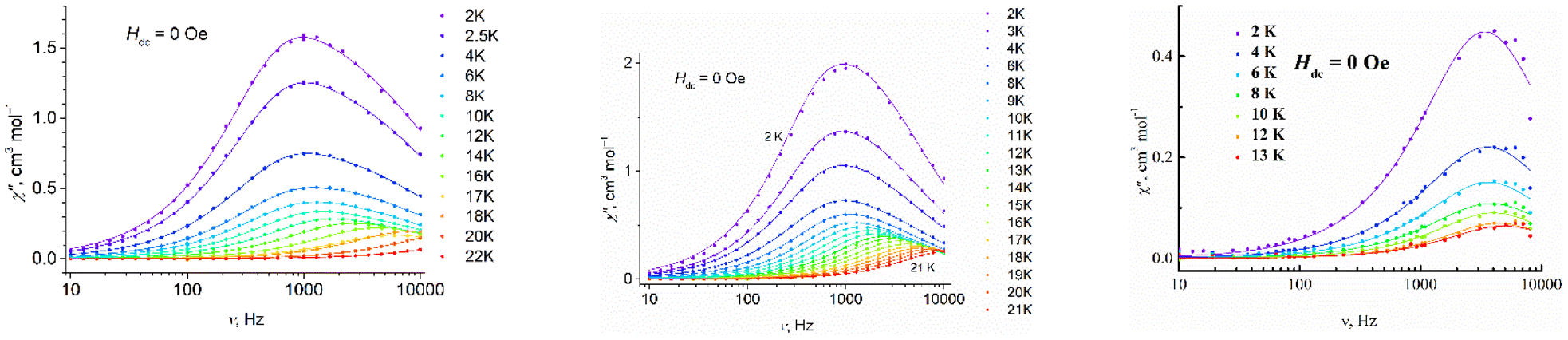 | ||
Fig. 8 Frequency dependence of the out-of-phase magnetic susceptibility 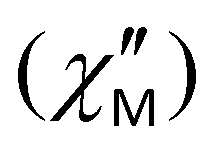 for 1–3 at different temperatures at zero dc field. The solid lines are the best fits (see text). for 1–3 at different temperatures at zero dc field. The solid lines are the best fits (see text). | ||
The difference in the magnetic behavior of the complexes could be attributed to differences in their crystal structures. The asymmetric unit contains two crystallographically independent molecules for complexes 1 and 2, and only one independent molecule for complex 3. However, some of the pentagonal- and hexagonal-bipyramidal complexes described containing two independent molecules per asymmetric unit cell showed no asymmetric broadening of the maximum frequency dependences.66,67
It was already noted earlier that asymmetric broadening arose only when QTM dominated over other relaxation processes.66,68–71 According to ref. 72, even in the case where only one peak was observable, the second underlying relaxation time could be extracted with the two-component Debye model. A single peak is observed for a small separation between τ1 and τ2 and high values of distribution parameter α. The separation is 0.2 ms for both 1 and 2 at 2 K (Fig. S15, ESI†); as the temperature rises, it sharply decreases. The distribution for the first relaxation time, τ1, is narrow (parameter α1 < 0.09), while α2 for the second relaxation time, τ2, is about twice as high (Tables S6 and S7, ESI†). We consider τ2 as a satellite of τ1, therefore, only the first relaxation time, τ1, is analyzed further in the text.
For complexes 1–3, the position of the maximum remains constant over the temperature range of 2–10 K (Fig. 8), which is characteristic for QTM. Above 10 K, temperature-dependent processes dominate, while the width of the maximum frequency dependence of 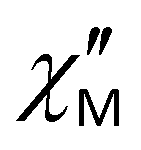 decreases.
decreases.
The contribution of QTM can be reduced by applying a static field; the frequency dependences of the in-phase 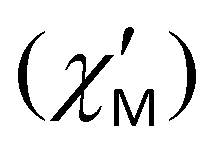 and out-of-phase ac susceptibility
and out-of-phase ac susceptibility  over the field range of 0–5000 Oe at a temperature of 10 K for 1 and 3, and 8 K for 2 are shown in Figs. S16–S18 and Tables S9–S11, ESI.† Two relaxation times for 1 and 2 are observed only at low fields. In fields of 1000 Oe for 1 and 500 Oe for 2, the broadening is almost absent and the experimental data are fitted by using the generalized Debye model.
over the field range of 0–5000 Oe at a temperature of 10 K for 1 and 3, and 8 K for 2 are shown in Figs. S16–S18 and Tables S9–S11, ESI.† Two relaxation times for 1 and 2 are observed only at low fields. In fields of 1000 Oe for 1 and 500 Oe for 2, the broadening is almost absent and the experimental data are fitted by using the generalized Debye model.
For complexes 1–3, the optimum applied field was estimated at Hdc = 1000 Oe. In the dc field, the QTM process is almost suppressed; the relaxation time is increased at low temperatures (Figs. 9 and S19 and Tables S12–S14, ESI†).
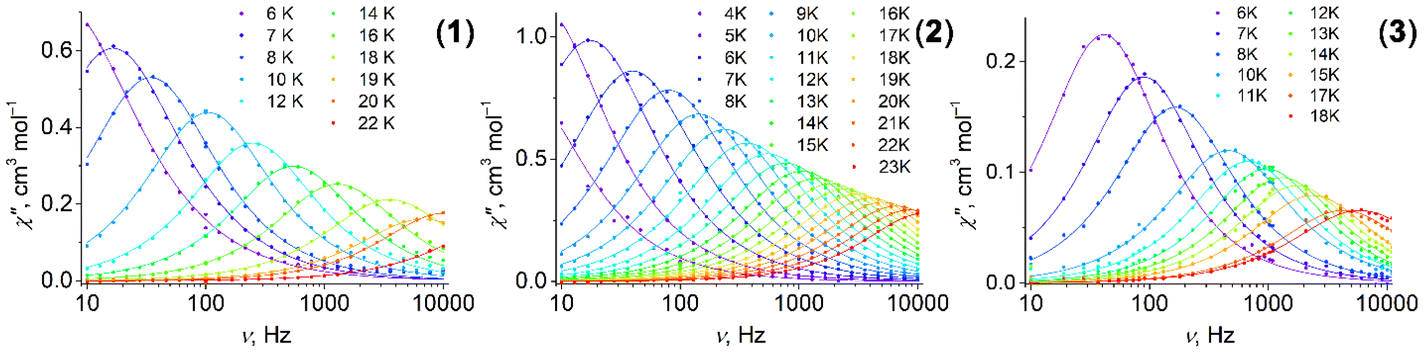 | ||
Fig. 9 Frequency dependences of the out-of-phase magnetic susceptibility  for 1–3 at different temperatures at a dc field of 1000 Oe. The solid lines are the best fits (see text). for 1–3 at different temperatures at a dc field of 1000 Oe. The solid lines are the best fits (see text). | ||
The dependences of relaxation time τ on the inverse temperature and the field for complex 1 are shown in Fig. 10a (Fig. S20, ESI† for 2 and 3) and Fig. 10b (Figs. S17d and S18b, ESI† for 2 and 3), respectively. Fitting of the data at zero field (Fig. 10a, black line) was performed according to eqn (1),73 which included one temperature-independent QTM process, and two temperature-dependent Raman and Orbach processes. Best-fit parameters are shown in Table 2.
| τ−1 = τ0−1exp(−Ueff/kBT) + CTn + τQTM−1 | (1) |
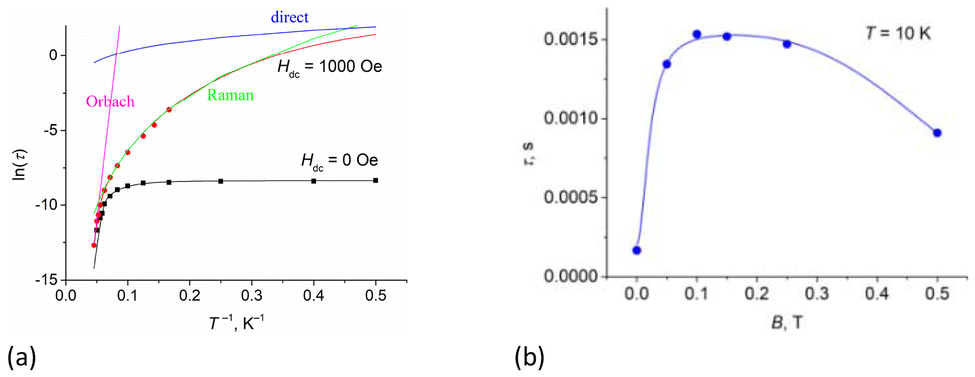 | ||
| Fig. 10 The inverse temperature (a) and field (b) dependences of the relaxation time, τ, for complex 1. | ||
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| a The Adirect parameters were fixed to the values defined in Table 3. | ||||||
| H dc, Oe | 0 | 1000 | 0 | 1000 | 0 | 1000 |
| U eff, K | 318 | 351 | 323 | 334 | 319 | 325 |
| τ 0, s | 3.6 × 10−13 | 3.7 × 10−13 | 2.9 × 10−11 | 2.9 × 10−11 | 4.7 × 10−12 | 6.9 × 10−12 |
| C Raman, s−1 K−n | 0.66 | 0.0022 | 1.0 | 0.08 | 0.58 | 0.11 |
| n Raman | 3.5 | 5.4 | 3.57 | 4.27 | 3.73 | 4.37 |
| τ QTM, s | 2.4 × 10−4 | — | 2.73 × 10−4 | — | 4 × 10−5 | — |
| A direct, K−1 T−4 s−1 | — | 919a | — | 536a | — | 780a |
The field dependence of τ (Fig. 10b) was fitted by eqn (2), which included two field-dependent processes, QTM and a direct one-phonon process. Best-fit parameters are shown in Table 3. Constant τRaman+Orbach implies the sum of Orbach and Raman contributions, because according to eqn (1) they are both field-independent. As follows from the values in Table 2, the main contribution to the τRaman+Orbach parameter is the Raman process.
| τ−1 = ATH4 + B1/(1 + B2H2) + τRaman+Orbach−1 | (2) |
| 1 | 2 | 3 | |
|---|---|---|---|
| T, K | 10 | 8 | 10 |
| A direct, K−1 T−4 s−1 | 919 | 536 | 780 |
| B 1, s−1 | 5358 | 3585 | 3655 |
| B 2, T−2 | 20.6 × 103 | 44.0 × 103 | 18 × 103 |
| τ Raman+Orbach −1, s−1 | 639 | 497 | 415 |
At a dc field of 1000 Oe, the relaxation time is the longest (Fig. 10b). The temperature dependence of the relaxation time at 1000 Oe (Fig. 10a, red line) was fitted with eqn (3), which included Orbach, Raman, and direct one-phonon processes. Best-fit parameters are shown in Table 2.
| τ−1 = τ0−1exp(−Ueff/kBT) + CTn + ATH4 | (3) |
In eqn (3), the parameter Adirect was fixed at the value determined from eqn (2) (Table 3). The blue line in Fig. 10a shows that the direct process makes a contribution only at low temperatures, so its inclusion in eqn (3) does not affect the fitting parameters of the Orbach and Raman processes.
The Raman process follows the τ−1 = CTn law, so the Raman process is considered to be field independent. However, as can be seen in Table 2, both CRaman and nRaman parameters differ in the zero and applied fields. Parameter CRaman is noticeably lower in the applied field (0.0022–0.11 s−1 K−n) compared to the zero field values (0.58–1.0 s−1 K−n). The degree, nRaman, varies over the range of 3.5–3.73 for the zero field and 4.27–5.4 for the dc field. The values of CRaman and nRaman are within the range commonly observed for the Raman process for DyIII SMMs.
Spin reversal energy barriers, Ueff = 318–351 K, are rather high for complexes 1–3. Unlike the Raman process, the fitting parameters for the Orbach process show field independence (Table 2). In Fig. 10a, the green line shows that the Raman process operates over almost the entire temperature range; Ueff observed is only relevant for magnetic relaxation at high temperatures (Fig. 10a, magenta line). This can potentially lead to misdetermination of the Orbach relaxation parameters if only zero-field measurements are used; therefore, dc-field measurements help to verify the fits. Correctly performed fits should give values of Ueff and τ0 parameters that are almost unaffected by whether a dc field is applied.34 For complexes 1–3, high-frequency ac magnetic measurements (up to 10000 Hz) made it possible to measure relaxation times up to 20 K and thus obtain accurate data for the Orbach relaxation process. Notably, the effective energy barriers (Ueff) in zero dc field for complexes 1–3 (Table 1), obtained from fitting with eqn (1), are close to the first excited Kramers doublets, as estimated from ab initio calculations (see section 3.5.1). For all three complexes, the magnetization barrier values in zero field and in the presence of a constant magnetic field are similar and over 320–350 K, indicating that the electronic effects associated with the nature of the substituents in the equatorial pentadentate ligands have almost no effect on the magnetic behavior of these mononuclear SMMs. Their magnetic properties are determined primarily by the geometry of the coordination environment of the Dy ions and the nature of the axial and equatorial ligands. The presence of two strong Cy3PO or Ph3PO donor ligands in the apical positions of complexes 1–3 leads to a slow relaxation of magnetization in the zero dc field and an increase in the magnetization barriers compared to similar PBPY-7 complexes containing two Cl− or Cl(Cy3PO/Ph3PO) in the axial positions.45,46 However, it should be noted that at the time this work was being prepared for publication, Sutter's group had published a PBPY-7 complex, [Dy(dapbh)(Cy3PO)2]BPh447 (where dapbh = 2,6-diacetylpyridine bis-benzoylhydrazone, R = C6H5, Fig. 1), similar to our complexes 1–3. Unlike 1–3, this complex showed a low value of magnetization barrier in a field of 750 Oe (Ueff = 51 K). Although the authors observed the 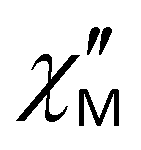 signal at 1000 Hz in the absence of a constant magnetic field, no data on the frequency or temperature dependence of
signal at 1000 Hz in the absence of a constant magnetic field, no data on the frequency or temperature dependence of 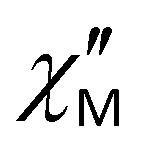 at zero field are given in the article; it is noted only that at zero field there is rapid relaxation due to QTM. It can be assumed that the difference in ac magnetic properties between our data for complexes 1–3 and this compound is due to the fact that the ac properties of [Dy(dapbh)(Cy3PO)2]BPh4 are studied over a narrow frequency range to 1500 Hz, whereas our study is conducted up to 10000 Hz. It should also be noted that the complex obtained by Sutter's group and our compounds 1–3 contain different counterions (BPh4vs. ClO4). The influence of the counterion and second coordination sphere on Ueff in the PBPY-7 complexes of Dy was observed in ref. 75.
at zero field are given in the article; it is noted only that at zero field there is rapid relaxation due to QTM. It can be assumed that the difference in ac magnetic properties between our data for complexes 1–3 and this compound is due to the fact that the ac properties of [Dy(dapbh)(Cy3PO)2]BPh4 are studied over a narrow frequency range to 1500 Hz, whereas our study is conducted up to 10000 Hz. It should also be noted that the complex obtained by Sutter's group and our compounds 1–3 contain different counterions (BPh4vs. ClO4). The influence of the counterion and second coordination sphere on Ueff in the PBPY-7 complexes of Dy was observed in ref. 75.
Although complexes 1–3 have rather high magnetization barriers, the height of these barriers is much lower than those for PBPY-7 Dy complexes containing monodentate weakly coordinating ligands (H2O, THF, pyridine) or macrocyclic neutral N5 ligands in the equatorial positions, and strong donor ligands (acyl/aryl alkoxides, aryl/acyl/phosphonamides, and silano/phosphine oxides) in the axial positions. Such a coordination environment of Dy ions, which have an oblate 4f electron density, provides strong apical and weak equatorial CF components, resulting in high values of Ueff extending to 1000 K and beyond.26–28,39,68,69,74 The lower values of magnetization barriers for complexes 1–3 compared to the PBPY-7 Dy complexes considered above are explained by the fact that in 1–3 the equatorial plane is formed by negatively charged pentadentate chelating ligands [N3O2]2−, which create a strong equatorial field competing with the apical crystal field (see the next sections for more detail). The difference in equatorial and apical Dy–O bond lengths in complexes 1–3 is approximately an order of magnitude smaller (0.02–04 Å, Fig. 6 and Table 1) compared to the same difference in the PBPY-7 Dy complexes with a homogeneous fully oxygen coordinated environment in the equatorial plane, [Dy(H2O)5(Cy3PO)2](Hal)3 (0.1 Å).34 The strong interaction in the equatorial plane between Dy and two O carrying negative charges leads to significant transverse anisotropy in complexes 1–3; the main magnetic axis in 1–3 is located in the equatorial plane, despite the presence of two strong axial ligands, whereas in all known PBPY-7 Dy complexes with weak monodentate equatorial ligands and strong apical ligands it is directed along the apical axis. A detailed theoretical analysis of the influence of different strengths of equatorial ligands and strong apical ligands on the electronic structure and SMM properties of PBPY-7 Dy complexes is presented below.
3.5 Computational study
The eight lowest Kramers doublets (KDs) and the g-tensors for 1–3 calculated using the SA-CASSCF/SO-RASSI/SINGLE_ANISO approach have been summarized in Table 4. The main magnetic anisotropy axes (gz) of ground KDs also lie on the equatorial plane due to the strong crystal field generated by the pentadentate ligand (Fig. S21, ESI†) as in recently studied Dy(III) complexes with the N3O2 equatorial ligand.46–48
| 1 | 2 | 3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KD | E | g x | g y | g z | E | g x | g y | g z | E | g x | g y | g z |
| 0 | 0.0 | 0.026 | 0.073 | 19.610 | 0.0 | 0.030 | 0.064 | 19.691 | 0.0 | 0.046 | 0.098 | 19.599 |
| 1 | 162.9 | 1.834 | 2.844 | 12.024 | 187.8 | 1.125 | 2.514 | 14.534 | 164.2 | 1.375 | 3.591 | 13.334 |
| 2 | 191.5 | 1.882 | 2.618 | 15.032 | 241.8 | 0.824 | 1.087 | 12.646 | 218.5 | 0.265 | 2.702 | 11.527 |
| 3 | 227.8 | 0.115 | 1.794 | 6.850 | 282.3 | 0.067 | 1.262 | 13.344 | 274.7 | 0.943 | 1.315 | 16.246 |
| 4 | 387.5 | 1.949 | 4.848 | 11.858 | 395.9 | 2.181 | 3.458 | 14.455 | 395.2 | 1.635 | 4.203 | 12.866 |
| 5 | 420.7 | 1.022 | 3.420 | 15.062 | 488.3 | 9.407 | 5.338 | 0.727 | 466.8 | 0.058 | 5.054 | 11.892 |
| 6 | 510.9 | 2.289 | 2.796 | 12.286 | 586.8 | 2.384 | 5.431 | 10.339 | 541.8 | 2.015 | 4.344 | 11.288 |
| 7 | 650.1 | 0.314 | 0.816 | 17.496 | 721.1 | 0.331 | 1.257 | 17.175 | 675.9 | 0.240 | 1.018 | 17.436 |
The calculated effective gzz components of g-tensors are 19.610, 19.691 and 19.599 for the ground KDs of 1, 2 and 3, respectively, which are close to the Ising-limit value of 20 for a pure MJ = 15/2 ground state, thus, all complexes have significant axial anisotropy for Dy(III) centers. The ground state wave functions of complexes 1 (95% |±15/2〉), 2 (96% |±15/2〉) and 3 (95% |±15/2〉) show the dominant contributions of the uniaxial magnetic anisotropy of the Dy(III) ions (Table 5).
| 1 | 2 | 3 | |
|---|---|---|---|
| KD0 | 0.948 |±15/2〉 | 0.962 |±15/2〉 | 0.948 |±15/2〉 |
| KD1 | 0.409 |±13/2〉 + 0.327 |±9/2〉 + 0.195 |±5/2〉 | 0.499 |±13/2〉 + 0.346 |±9/2〉 | 0.460 |±13/2〉 + 0.382 |±9/2〉 |
| KD2 | 0.301 |±7/2〉 + 0.245 |±1/2〉 + 0.149 |±3/2〉 | 0.425 |±7/2〉 + 0.292 |±11/2〉 + 0.112 |±5/2〉 | 0.463 |±7/2〉 + 0.412 |±11/2〉 |
| KD3 | 0.383 |±11/2〉 + 0.212 |±7/2〉 + 0.149 |±3/2〉 | 0.284 |±3/2〉 + 0.201 |±1/2〉 + 0.199 |±11/2〉 + 0.175 |±5/2〉 | 0.334 |±3/2〉 + 0.261 |±5/2〉 + 0.258 |±1/2〉 |
| KD4 | 0.454 |±13/2〉 + 0.313 |±9/2〉 + 0.123 |±5/2〉 | 0.355 |±13/2〉 + 0.321 |±9/2〉 + 0.138 |±7/2〉 | 0.413 |±13/2〉 + 0.309 |±9/2〉 |
| KD5 | 0.290 |±11/2〉 + 0.273 |±7/2〉 + 0.189 |±5/2〉+ 0.163 |±9/2〉 | 0.254 |±5/2〉 + 0.202 |±11/2〉 + 0.180 |±7/2〉+ 0.123 |±9/2〉 | 0.277 |±11/2〉 + 0.263 |±7/2〉 + 0.196 |±5/2〉+ 0.129 |±9/2〉 |
| KD6 | 0.347 |±3/2〉 + 0.287 |±5/2〉 + 0.155 |±11/2〉 + 0.143 |±7/2〉 | 0.316 |±3/2〉 + 0.251 |±5/2〉 + 0.157 |±7/2〉 + 0.147 |±11/2〉 | 0.338 |±3/2〉 + 0.265 |±5/2〉 + 0.135 |±11/2〉 + 0.112 |±7/2〉 |
| KD7 | 0.636 |±1/2〉 + 0.262 |±3/2〉 | 0.611 |±1/2〉 + 0.259 |±3/2〉 | 0.601 |±1/2〉 + 0.251 |±3/2〉 |
Strong mixing of the |±13/2〉 and |±9/2〉 (and |±5/2〉 for 1) wave functions (Table 5 and Fig. S22, ESI†) in the first excited KD1 leads to very large thermally assisted quantum tunneling (TA-QTM) values of 0.78, 0.63 and 0.84μB; as a result, all complexes have to relax via this state. Moreover, the first excited states, KD1, of all complexes show significant transverse anisotropy (gx,y are in the range of 1.1–3.6), suggesting that a significant QTM is operational at the first excited state.
It should be noted that the calculated energies of the first excited Kramers doublets, 234.6, 270.4 and 236.4 K, which in the cases under consideration correspond to the ab initio computed energy barriers for magnetization reversal, are slightly higher than those for similar systems with nonequivalent axial ligands.46 These values agree satisfactorily with the Ueff values obtained from the analysis of the temperature dependences of relaxation. Some discrepancies may be related to the influence of other relaxation mechanisms (Raman, QTM) on the accuracy of the approximation, since we observe a non-pure Orbach process for these systems.
Thus, replacing the second Cl− ion in the axial position by the neutral R3PO (R = Cy or Ph) ligand does not significantly affect the values of the spin-reversal barrier (Ueff). Since the main magnetic axis lies in the equatorial plane, the ligand field generated from the equatorial ligands mainly determines the properties of the complexes under consideration; the small differences in the energy spectrum, apparently, depend on the structure of the N3O2 ligands (see the next section for more details).
In order to analyze in more detail the reason for the strong influence of the ligand field of equatorial ligands, a LoProp charge analysis was carried out. The total charge on donor atoms in the equatorial plane is higher and it is unevenly distributed (Table 6). Previously,26 for D5h symmetric Dy(III) complexes, it was shown that, with a decrease in the negative charge on donor atoms in the equatorial plane and their equalization in absolute value upon transition to a homogeneous completely oxygen or nitrogen coordination environment, the Ueff values increased and the properties of such SIMs improved.
| Atom | 1 | 2 | 3 |
|---|---|---|---|
| Dy | 2.5288 | 2.5084 | 2.5307 |
| Oax | −1.1225/−1.1229 | −1.1019/−1.1067 | −1.1073/−1.1086 |
| Oeq | −0.8358/−0.8443 | −0.8532/−0.8339 | −0.8334/−0.8429 |
| NPy | −0.4141 | −0.3979 | −0.4190 |
| N | −0.2771/−0.2814 | −0.2722/−0.2749 | −0.2759/−0.2788 |
The CF parameters of all complexes was estimated using the Stevens formalism, 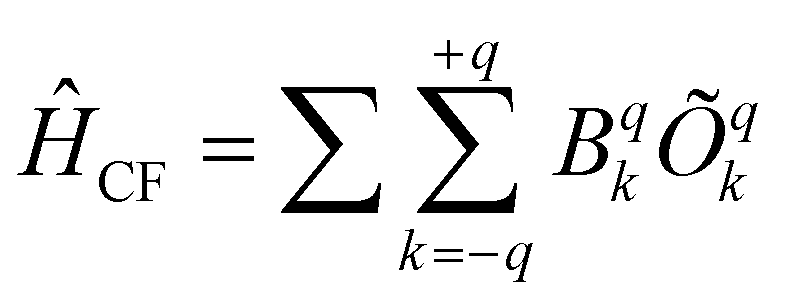 , where Bqk and Õqk are the calculated CF parameter and extended Stevens operator, as defined in ref. 76, respectively, as implemented in the SINGLE_ANISO code.60a The axial CF parameter B02 is found to be the largest in absolute value for all complexes (Table S17, ESI†). However, some Bq2 values are found to be comparable; as a result, it can lead to a loss of expected axiality.
, where Bqk and Õqk are the calculated CF parameter and extended Stevens operator, as defined in ref. 76, respectively, as implemented in the SINGLE_ANISO code.60a The axial CF parameter B02 is found to be the largest in absolute value for all complexes (Table S17, ESI†). However, some Bq2 values are found to be comparable; as a result, it can lead to a loss of expected axiality.
3.5.2.a General remarks. As pointed out above, Dy(III) PBPY-7 complexes with a strong equatorial CF (Case III, Fig. 1c, which involves our complexes 1–3 and similar Dy(III) complexes reported in ref. 45–48) can exhibit SMM behavior when the ground state has a dominant mJ = |±15/2〉 character with the long magnetic axis (gx ∼ 19.6) located in the equatorial xy plane; this state is referred to as |±15/2〉eq. From the point of view of the electron density, the equatorial |±15/2〉eq state (with gx ∼ 19.6, gy, gz ∼ 0) in Case III Dy(III) PBPY-7 complexes has the same oblate shaped electronic cloud as the axial |±15/2〉ap ground state (with gx, gy ∼ 0, gz ∼ 19.6) in Case I and II complexes differing only in the orientation of the cloud (Fig. 11a and b). However, there is a seminal difference between the equatorial |±15/2〉eq and apical |±15/2〉ap states concerning their mJ composition: namely, with a common spin quantization z axis directed along the pentagonal C5 axis of the bipyramid, KD0 |±15/2〉ap is a pure |±15/2〉 state (Fig. 11a), while KD0 |±15/2〉eq is a mixture of low-mJ states, |±15/2〉eq = 39.27% |±1/2〉 + 30.55% |±3/2〉 + 18.33% |±5/2〉 + 8.33% |±7/2〉 + … (Fig. 11b). At this point, it is important to note that this fact in itself highlights a drastic change in the origin of the |±15/2〉 ground state upon passing from Cases I and II to Case III Dy(III) PBPY-7 complexes. The key point is that the pure apical KD0 |±15/2〉ap arises with the perfect D5h symmetry of the PBPY-7 complex (Fig. 11a), whereas equatorial KD0 |±15/2〉eq results from some mJ mixing caused by non-axial CF terms Bkq, q ≠ 0, which can appear only in a distorted PBPY-7 geometry (Fig. 11b). Indeed, in the absence of non-axial CF terms Bkq (i.e., at perfect D5h symmetry), the ground state KD0 of the Case III complex has pure |±1/2〉 character with gz = 1.32, gx = gy = 10.59, which features positive (in-plane) magnetic anisotropy with no magnetic axiality (Fig. 1c). It is therefore an objective to establish the specific distortions of the PBPY-7 geometry that produce the equatorial KD0 |±15/2〉eq ground state in Case III complexes. Below in this section we examine this issue using several model approaches based on CF theory.
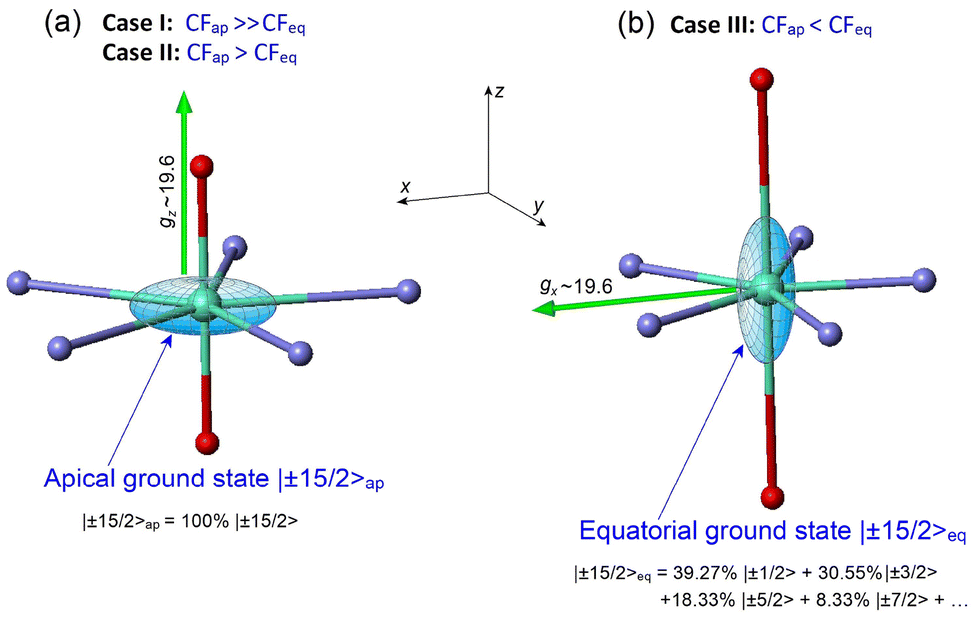 | ||
| Fig. 11 Difference between the nature of the |±15/2〉 ground state in Dy(III) PBPY-7 complexes for Cases I–III. (a) Cases I and II: KD0 |±15/2〉ap has pure |±15/2〉 character with the long magnetic axis (gz ∼ 19.6) pointing to the apical ligands, (b) Case III: KD0 |±15/2〉eq is a mixture of several low-mJ states with the long magnetic axis (gz ∼ 19.6) lying in the equatorial plane. In both cases, the electron density of KD0 |±15/2〉 has the same oblate shaped electronic cloud that differs only in orientation. | ||
3.5.2.b Simple CF model based on Bleaney's theory of magnetic anisotropy. A primary insight into the specific mechanism of the origin of the equatorial KD0 |±15/2〉eq due to the mixing of low-mJ states by the non-axial CF terms can be obtained with a simple CF model considering only the rank-2 CF parameters, B2q. In fact, this approach is closely related to Bleaney's theory,77 according to which the magnetic anisotropy of the lanthanide ion is determined by only two rank-2 CF parameters, the axial parameter B20 and the rhombic parameter B22, while higher-order CF parameters Bkq (k = 4 and 6) can be neglected.77
In the frame of this model, we consider the Dy(III) PBPY-7 complex with overall positive (in-plane) magnetic anisotropy, which is described by the negative axial CF parameter B20 < 0 (in the Wybourne notation) and we explore the evolution of the ground and excited CF electronic states with increasing rhombic CF parameter B22, the magnitude of which formally corresponds to the degree of distortion of the PBPY-7 complex in the equatorial plane (see the next section for more detail). For this, we perform comparative CF calculations at a fixed axial CF parameter of B20 = −1000 cm−1 and rhombic parameter B22 increasing from 0 to 600 cm−1 (Table 7 and Fig. 12).
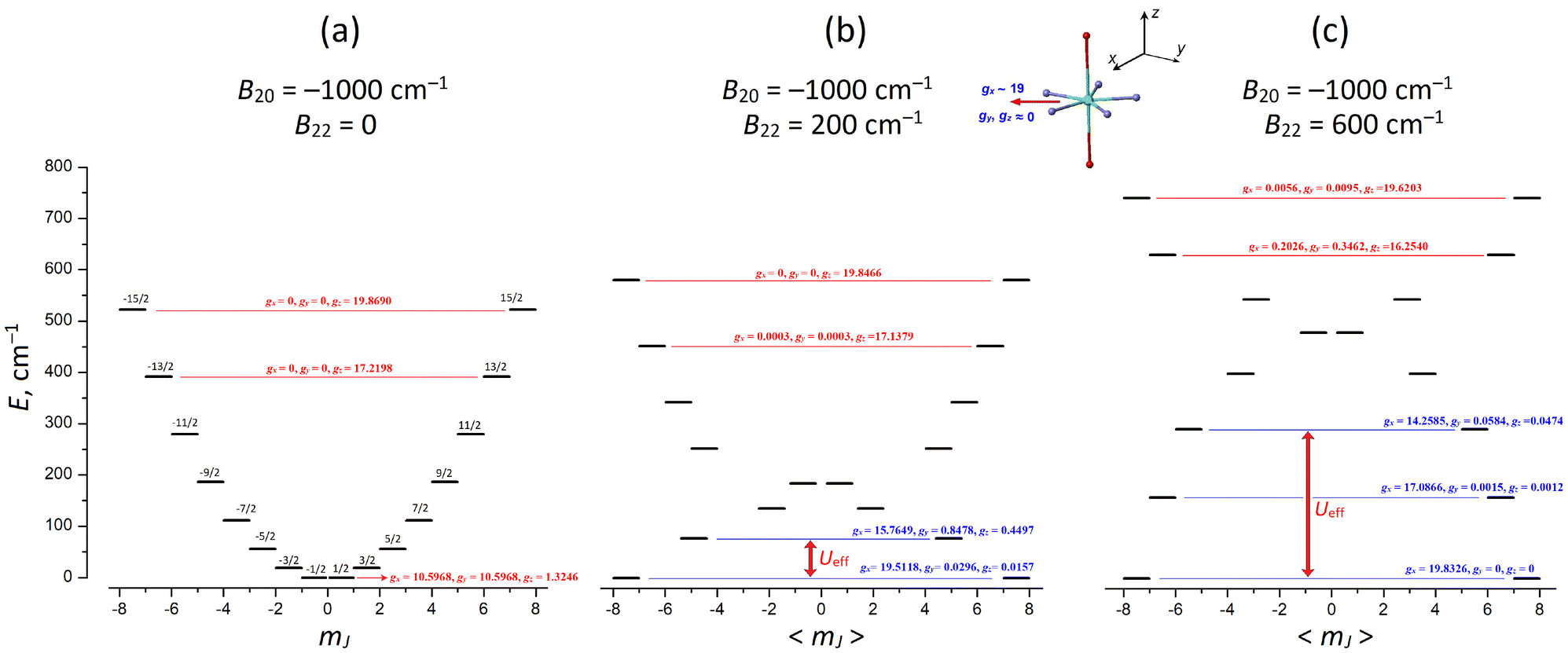 | ||
| Fig. 12 Formation of the equatorial KD0 |±15/2〉eq and the effective energy barrier, Ueff, for Case III PBPY-7 Dy(III) complexes upon increasing the rhombic CF parameter, B22 (which originates from distortions in the equatorial plane of the PBPY-7 complex). (a) Regular D5h PBPY-7 complex with the mJ = ±1/2 ground state; (b) moderately distorted complex, B22 = 200 cm−1; (c) strongly distorted PBPY-7 complex, B22 = 600 cm−1. At B22 > 200 cm−1, the low-mJ states are mixed by B22 more efficiently to produce the lowest purely Ising equatorial KD0 |±15/2〉eq with gx ≈ 19.8, gy, gz ≈ 0 (marked in blue). In all cases, the upper KDs (their g-tensors are marked in red) are rather insensitive to distortions, retaining their Ising-type nature with the long magnetic z axis parallel to the pentagonal C5 axis of the PBPY-7 complex. Effective energy barrier, Ueff, is indicated by the red arrow. CF calculations are performed with fixed axial CF parameter, B20 = −1000 cm−1, and varied B22 parameter (from 0 to 600 cm−1). | ||
| B 22, cm−1 | |||||||
|---|---|---|---|---|---|---|---|
| 0 | 20 | 50 | 100 | 200 | 400 | 600 | |
| State | E, cm−1, gx,y,z | ||||||
| KD0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| g x = 10.5968 | g x = 15.1092 | g x = 17.8625 | g x = 18.9831 | g x = 19.5118 | g x = 19.7635 | g x = 19.8326 | |
| g y = 10.5968 | g y = 5.5499 | g y = 1.8156 | g y = 0.3641 | g y = 0.0296 | g y = 0.0006 | g y = 0 | |
| g z = 1.3246 | g z = 1.0670 | g z = 0.5110 | g z = 0.1415 | g z = 0.0157 | g z = 0.0004 | g z = 0 | |
| KD1 | 18.6 | 21.1 | 30.6 | 48.5 | 76.9 | 120.0 | 157.0 |
| g x = 0 | g x = 4.7982 | g x = 8.8665 | g x = 12.8251 | g x = 15.7649 | g x = 16.8278 | g x = 17.0866 | |
| g y = 0 | g y = 4.7457 | g y = 6.9517 | g y = 4.4883 | g y = 0.8478 | g y = 0.0332 | g y = 0.0015 | |
| g z = 3.9738 | g z = 3.6894 | g z = 2.9572 | g z = 1.8423 | g z = 0.4497 | g z = 0.0233 | g z = 0.0012 | |
| KD2 | 55.9 | 57.8 | 65.6 | 85.9 | 135.2 | 219.9 | 290.4 |
| g x = 0 | g x = 0.2910 | g x = 1.6794 | g x = 5.0088 | g x = 9.8966 | g x = 13.5787 | g x = 14.2585 | |
| g y = 0 | g y = 0.2958 | g y = 1.7443 | g y = 5.0717 | g y = 5.4227 | g y = 0 055 | g y = 0.0584 | |
| g z = 6.6230 | g z = 6.5893 | g z = 6.3757 | g z = 5.4758 | g z = 3.2450 | g z = 0.4954 | g z = 0.0474 | |
| KD3 | 111.9 | 113.6 | 120.6 | 138.1 | 184.0 | 296.1 | 398.8 |
| g x = 0 | g x = 0.0053 | g x = 0.0802 | g x = 0.5945 | g x = 3.3895 | g x = 8.8745 | g x = 11.1474 | |
| g y = 0 | g y = 0.0054 | g y = 0.0836 | g y = 0.6450 | g y = 3.8569 | g y = 4.8268 | g y = 1.0299 | |
| g z = 9.2722 | g z = 9.2626 | g z = 9.2107 | g z = 9.0035 | g z = 7.7977 | g z = 3.6740 | g z = 0.8364 | |
| KD4 | 186.5 | 188.1 | 194.9 | 211.3 | 252.3 | 355.8 | 478.4 |
| g x = 0 | g x = 0 | g x = 0.0016 | g x = 0.0250 | g x = 0.3576 | g x = 3.3510 | g x = 6.7213 | |
| g y = 0 | g y = 0 | g y = 0.0017 | g y = 0.0272 | g y = 0.4216 | g y = 4.4267 | g y = 5.5485 | |
| g z = 11.9214 | g z = 11.9175 | g z = 11.8965 | g z = 11.8201 | g z = 11.4793 | g z = 9.1921 | g z = 5.1388 | |
| KD5 | 279.7 | 281.3 | 287.9 | 303.7 | 342.2 | 434.1 | 543.4 |
| g x = 0 | g x = 0 | g x = 0 | g x = 0.0005 | g x = 0.0152 | g x = 0.4121 | g x = 2.1786 | |
| g y = 0 | g y = 0 | g y = 0 | g y = 0.0005 | g y = 0.0179 | g y = 0.5782 | g y = 3.6028 | |
| g z = 14.5706 | g z = 14.5688 | g z = 14.5592 | g z = 14.5246 | g z = 14.3808 | g z = 13.6771 | g z = 11.5791 | |
| KD6 | 391.6 | 393.1 | 399.6 | 415.0 | 451.7 | 535.9 | 630.6 |
| g x = 0 | g x = 0 | g x = 0 | g x = 0 | g x = 0.0003 | g x = 0.0183 | g x = 0.2026 | |
| g y = 0 | g y = 0 | g y = 0 | g y = 0 | g y = 0.0003 | g y = 0.0256 | g y = 0.3462 | |
| g z = 17.2198 | g z = 17.2190 | g z = 17.2148 | g z = 17.1998 | g z = 17.1379 | g z = 16.8622 | g z = 16.2540 | |
| KD7 | 522.1 | 523.6 | 530.0 | 545.0 | 580.1 | 657.9 | 740.9 |
| g x = 0 | g x = 0 | g x = 0 | g x = 0 | g x = 0 | g x = 0.0003 | g x = 0.0056 | |
| g y = 0 | g y = 0 | g y = 0 | g y = 0 | g y = 0 | g y = 0.0004 | g y = 0.0095 | |
| g z = 19.8690 | g z = 19.8688 | g z = 19.8677 | g z = 19.8635 | g z = 19.8466 | g z = 19.7726 | g z = 19.6203 | |
In the regular D5h PBPY-7 complex (B22 = 0), the E vs. mJ energy diagram has a perfect parabolic profile with the lowest mJ = ±1/2 state and the upper mJ = ±15/2 state (Fig. 12a). As the rhombic CF parameter B22 increases, the easy-plane character of the ground-state g-tensor (KD0, gz = 1.32, gx = gy = 10.59 at B22 = 0) is transformed into a nearly perfect Ising one (gx = 19.80, gy, gy ∼ 0 at B22 = 500 cm−1) with the long magnetic x axis lying in the equatorial plane (Fig. 12b and c). Also note that with increasing rhombic parameter B22, the most drastic changes in g-tensors occur in the ground state KD0 and low-lying excited CF states (KD1–4), while the upper CF states (KD6 and KD7) change insignificantly (Table 7; in Fig. 12 their g-tensors are marked in red).
It is also important that the energy gap, ΔE01, between the ground KD0 and first excited KD1 increases with increasing B22, thereby enhancing the effective energy barrier, Ueff, and SMM performance. At strong distortions (B22 > 400 cm−1), not only the ground KD0 but also low-lying KDs acquire Ising character (Fig. 12c and Table 7); this implies that Ueff may increase beyond the ΔE01 energy gap due to suppression of thermally induced QTM processes in the excited KDs (Fig. 12c).
These results clearly show that in Case III PBPY-7 Dy(III) complexes with overall positive magnetic anisotropy, distortions in the equatorial plane lead to a larger effective barrier, Ueff, and improved SMM characteristics. However, this only occurs when the in-plane distortions are strong enough to efficiently mix the low-mJ states near the bottom of the parabolic E vs. mJ profile to form the equatorial KD0 |±15/2〉eq = 39.27% |±1/2〉 + 30.55% |±3/2〉 + 18.33% |±5/2〉 + 8.33% |±7/2〉 + … (Fig. 12 and Table 7).
3.5.2.c CF analysis of in-plane distortions resulting in the equatorial KD0 |±15/2〉eq ground state. In this section, we examine specific distortions of the PBPY-7 coordination sphere of the Dy(III) ion leading to a large rhombic parameter B22, which causes formation of the magnetically axial equatorial KD0 |±15/2〉eq ground state (Fig. 12 and Table 7). The impact of distortions on the CF energy spectra and SMM characteristics is analyzed with the superposition CF model,78,79 which relates the ‘global’ Bkq CF parameters to the actual coordination geometry of the metal ion in terms of ‘intrinsic’ CF parameters bk describing local metal–ligand interactions,
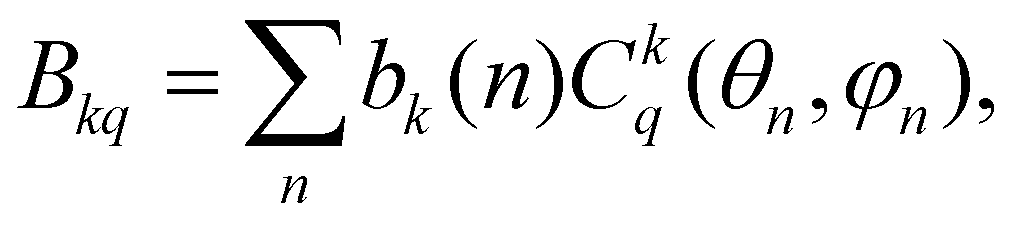 | (4) |
In these CF calculations, we used an idealized structure of the [DyN3O4] core, in which the apical O3–Dy–O4 group is strictly linear and the N3O2 pentagon (involving the equatorial atoms O1, O2, N2, N3 and N4) is planar. All atomic distances are set to the averaged experimental values for each group of atoms, namely 2.25 Å for apical O atoms, 2.45 Å for N atoms and 2.28 Å for the equatorial O atoms; the N–Dy–N bond angles are fixed at 72°, while the O1–Dy–O2 bond angle (φ) is variable (Fig. 13a). The intrinsic CF parameters bk for the equatorial N atoms are set to b2(N) = 1000, b4(N) = 300 and b6(N) = 50 cm−1. For the oxygen atoms, bk are set to b2(O) = 1200, b4(O) = 500 and b6(O) = 100 cm−1 at a reference distance of R0 = 2.26 Å; the radial dependence of the bk(O) parameters for the apical (O3, O4, R = 2.25 Å) and equatorial (O1, O2, R = 2.28 Å) oxygen atoms is described by bk(R) = bk(R0)(R0/R)t(k), where t(2) = 6, t(4) = 8 and t(6) = 11.78,79
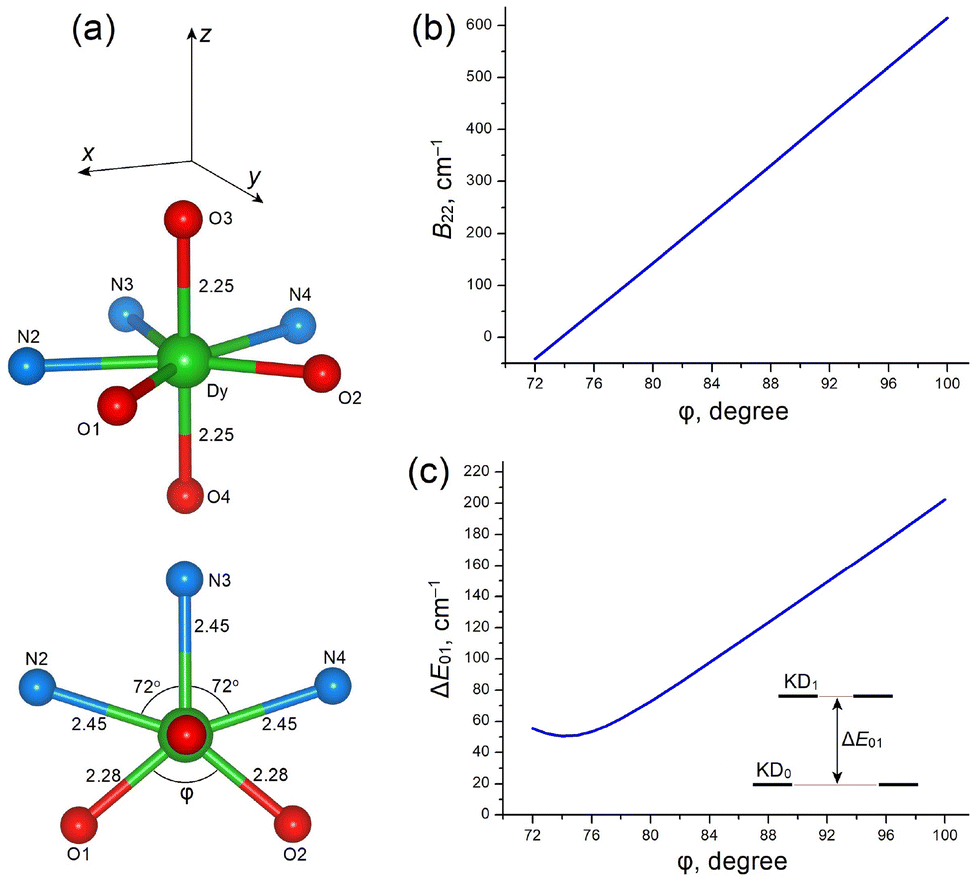 | ||
| Fig. 13 (a) Idealized structure of the DyN3O4 core of complexes 1–3 employed in CF calculations; all atomic distances are given in Å, (b) dependence of the rhombic CF parameter B22 on φ, the O1–Dy–O2 bond angle, (c) variation of the energy separation, ΔE01, of the first excited KD1 with increasing angle φ. | ||
Comparative CF calculations for various types of distortions showed that the strongest impact on the formation of the Ising equatorial KD0 |±15/2〉eq ground state was due to in-plane (xy) distortions of the equatorial N3O2 pentagon, which led to a large rhombic CF term B22. In this respect, it has also been found that angular in-plane distortions are more important than radial distortions. On the other hand, other types of distortion, including bending distortions of the apical O3–Dy–O4 group and out-of-plane (z) displacements of the equatorial O and N atoms are much less important. In particular, out-of-plane distortions in the N3O2 pentagon result mainly in the non-axial rank-2 CF parameters B2±1, which have a minor effect on the magnetic anisotropy compared to that of B22.
CF analysis indicates that the O1–Dy–O2 bond angle (φ) in the equatorial plane is a key structural parameter (Fig. 13a). We studied the influence of angle φ on the rhombic CF parameter B22, g-tensors of the ground and excited KDs, and the CF splitting energy patterns (Table 8 and Fig. 13). Increasing angle φ from 72° to 100° results in rapid linear growth of the rhombic parameter B22 from almost zero to about 600 cm−1 (Fig. 13b), which is followed by the formation of the equatorial KD0 |±15/2〉eq ground state with gx ≈ 19.6 (lying in the equatorial xy plane, Fig. 13a and Table 8). An increase of φ is also accompanied by nearly linear growth of the energy gap, ΔE01, between the ground KD0 and the first excited KD1, from ∼50 to ∼200 cm−1 (Fig. 13c and Table 8). The total CF splitting energy, ΔECF, also increases considerably, from 368 to 645 cm−1 (Table 8).
| KD0 | KD1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| φ | B 22, cm−1 | g x | g y | g z | ΔE01, cm−1 | g x | g y | g z | ΔECF, cm−1 |
| 72° | −41 | 6.388 | 14.367 | 1.129 | 55.5 | 1.003 | 6.759 | 3.592 | 368.4 |
| 80° | 143 | 17.309 | 2.356 | 0.601 | 72.7 | 11.067 | 2.413 | 3.474 | 391.7 |
| 88° | 331 | 18.903 | 0.346 | 0.133 | 123.3 | 14.512 | 1.546 | 1.613 | 470.6 |
| 92° | 426 | 19.204 | 0.153 | 0.067 | 149.3 | 15.511 | 0.974 | 0.801 | 523.1 |
| 96° | 520 | 19.404 | 0.073 | 0.035 | 175.6 | 16.067 | 0.599 | 0.429 | 582.2 |
| 100° | 614 | 19.542 | 0.037 | 0.019 | 202.2 | 16.407 | 0.381 | 0.268 | 645.3 |
At a large angle φ, close to the experimental value of ≈100° in complexes 1–3 (Tables S3–S5 ESI†), the ground-state g-tensor has almost pure Ising-type character (gx ≈ 19.6, gy, gz ≈ 0.02–0.03), which is inherent to the |±15/2〉 state. Note also that at φ ≈ 100° the energy gap is ΔE01 = 202 cm−1 and the overall CF energy is ΔECF = 645 cm−1, which are reasonably consistent with the results of ab initio calculations for 1–3 (Table 4). In contrast, at a small O1–Dy–O2 bond angle (φ < 80°), CF calculations reveal the poor axiality of KD0 (due to a large transverse component, gy, gz) and small energy separation, ΔE01, of the first excited KD1 (∼100 cm−1 or less, Table 8), which together lead to a degradation or even complete absence of SMM properties in PBPY-7 complexes with a more symmetric DyN3O4 core.
Similar CF calculations for analogous complexes with weaker apical ligands (Cl2, Cl/Ph3PO or Cl/Cy3PO)45,46 show fairly similar results. This indicates that in Case III PBPY-7 complexes, the SMM characteristics are rather insensitive to the nature of the apical ligands, as they are mainly governed by angular distortions in the equatorial plane. However, a detailed discussion of these issues is quite lengthy and thus it is beyond the scope of this article.
3.5.2.d Relationship between the axial CF parameters B20, B40 and B60 in Case I–III Dy(III) PBPY-7 complexes. Although the above model provides basic insights into the origin of the equatorial KD0 |±15/2〉eq due to mixing of low-mJ states by non-axial CF components, it does not describe the actual energy spectrum of KDs of PBPY-7 Dy(III) complexes due to the absence of higher-order CF terms, B4q and B6q. This is especially true for Case III PBPY-7 complexes, where the profile of the spin energy diagram is far from being parabolic due to the presence of large rank-4 CF parameters (Fig. 1c).49,80
In this section, we consider a more realistic CF model for Case III complexes involving the full set of Bkq CF parameters (k = 2, 4, 6). The key point is that in the regular D5h PBPY-7 complex, the CF Hamiltonian comprises only the axial CF terms, B20, B40 and B60; all non-axial terms, Bkq, are strictly zero under the symmetry conditions. Therefore, it is important to establish general relationships between B20, B40 and B60 when passing sequentially from Case I to Case II and III PBPY-7 Dy(III) complexes. To this end, we take advantage of the superposition CF model78,79 described in the previous section.
In a regular D5h PBPY-7 complex, there are two groups of intrinsic CF parameters related to the apical and equatorial ligands, bk(ap) and bk(eq) (k = 2, 4, 6). According to eqn (4), in the PBPY-7 Dy(III) complex, their contributions to the global CF parameters Bkq are given by
| B20 = 2b2(ap) − 2.5 b2(eq), | (5a) |
| B40 = 2b4(ap) + 1.875 b4(eq), | (5b) |
| B60 = 2b6(ap) − 1.5625 b6(eq). | (5c) |
It is important to note here that the contributions to the axial CF parameter B40 from the apical and equatorial ligands are summed, while those in parameters B20 and B60 are subtracted. This implies that B40 increases as the strength of the equatorial ligands increases, while B20 and B60 decrease, and at a certain point they change sign from positive to negative. In particular, in a perfectly symmetric PBPY-7 complex with equal metal–ligand distances (i.e., where bk(ap) = bk(eq) = bk for all metal–ligand pairs), the axial CF parameters Bk0 are expressed by B20 = −0.5b2, B40 = +3.875b4, and B60 = +0.4375b6. CF calculations show that this complex exhibits positive magnetic anisotropy due to the mJ = ±1/2 ground state and thus it falls under Case III (Fig. 1c). Therefore, in Case III, the axial CF parameter B40 > 0 is always positive and it dominates due to its far larger factor, eqn (5b). This is supported by experimental data for PBPY-7 lanthanide complexes with an equatorial ligand of DAPSC type, indicating B40 ∼ +1500 cm−1.80 In addition, these results show that the B60 parameter is of minor importance in Case III PBPY-7 complexes, both due to the small factor (0.4375) and because of the typical relationship of b6 < b2, b4 for the intrinsic CF parameters.79 Therefore, below in our model CF analysis of PBPY-7 Dy(III) complexes, the rank k = 6 B60 parameter can safely be neglected.
Thus, since only two CF parameters (B20 and B40) are active in symmetric D5h PBPY-7 complexes, the nature of the energy spectrum is determined only by their ratio, B20/B40. It is therefore important to find specific values for the B20/B40 ratio that define the boundaries between Cases I, II and III.
The Case I PBPY-7 complex has a parabolic profile of spin energy levels with the lowest KD0 |±15/2〉 and KD7 |±1/2〉 at the top of the parabola (Fig. 14a). The lower boundary of Case I is the point below which KD |±1/2〉 ceases to be the upper level. Calculations show that this occurs at B20/B40 = 1.35. On the other hand, the lower boundary of Case II corresponds to equal energies of KD |±15/2〉 and KD |±1/2〉. This point is reached at B20/B40 ≈ −0.07. Thus, these results quantify the limiting points of the realization of Cases I, II and III for PBPY-7 Dy(III) complexes in terms of CF parameters (Fig. 14):
| Case I: B20/B40 > 1.35, | (6a) |
| Case II: 1.35 ≥ B20/B40 ≥ −0.07, | (6b) |
| Case III: B20/B40 < −0.07. | (6c) |
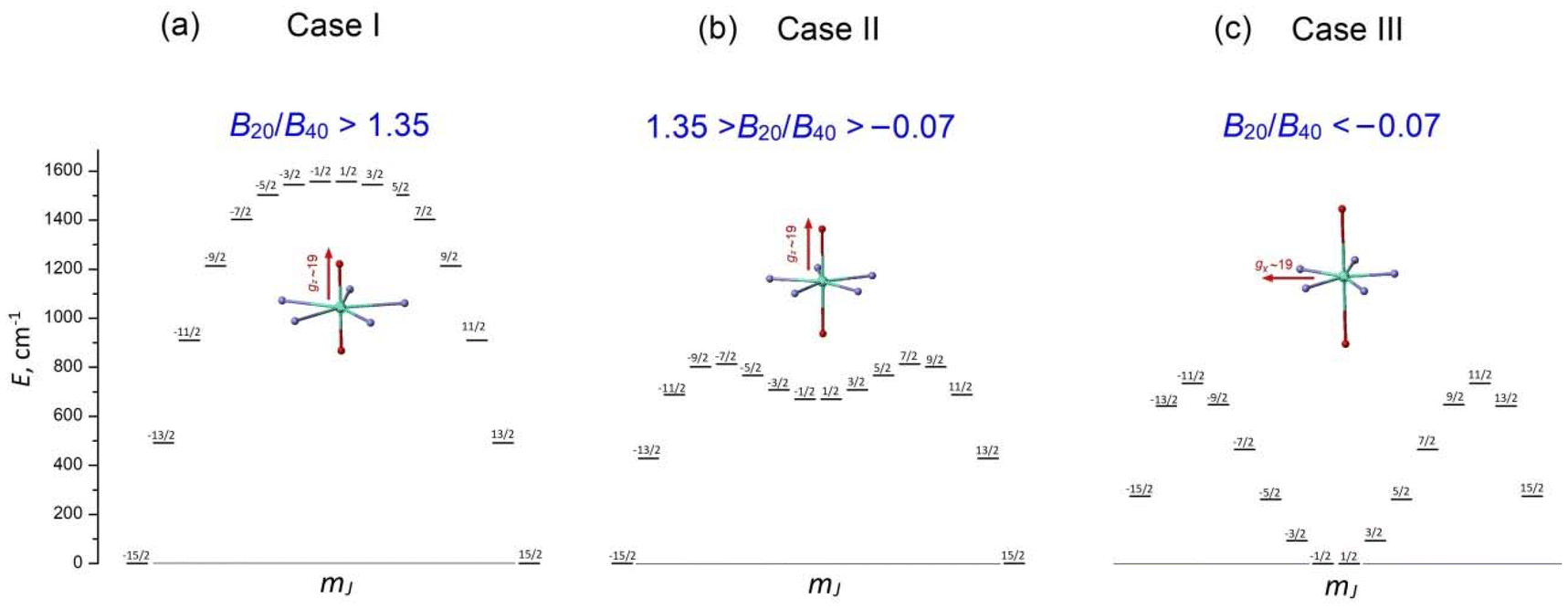 | ||
| Fig. 14 Criterion for the realization of Cases I, II and III of regular D5h PBPY-7 Dy(III) complexes in terms of CF theory. (a) Case I, B20/B40 > 1.35, (b) Case II, 1.35 > B20/B40 > −0.07, (c) Case III, B20/B40 < −0.07. In Case III, distortions in the equatorial plane (which operate through the rhombic CF parameter B22) mix the low-mJ states near the parabolic bottom of the E vs. mJ profile to produce the lowest equatorial KD0 |±15/2〉eq = 39.27% |±1/2〉 + 30.55% |±3/2〉 + 18.33% |±5/2〉 + 8.33% |±7/2〉 + … with gx ≈ 19.8, gy ≈ 0, gz ≈ 0 (see Table 7 and Fig. 12). | ||
In terms of intrinsic CF parameters bk, the PBPY-7 complex falls under Case III at 2b2(ap) − 2.5 b2(eq) < 0 (eqn (5a)), which corresponds to the ratio of b2(eq)/b2(ap) > 0.8. Hence, already with equal strengths of apical and equatorial ligands (b2(eq) = b2(ap)), the PBPY-7 complex definitely belongs to Case III.
According to eqn (6a–c), in the complexes of Cases I and II, the CF strength of the apical ligands should be significantly larger than that of the equatorial ligands. In particular, for this, the PBPY-7 complex must have short metal–ligand distances in the apical positions and long distances in the equatorial plane.
PBPY-7 complexes where the strength of the apical ligands is comparable to or weaker than that of equatorial ligands fall under Case III (Fig. 14c). Our complexes 1–3 and similar PBPY-7 Dy(III) complexes reported in ref. 45–48 certainly fall under Case III. Note that for Case III (Fig. 14c), the specific mechanism of formation of the equatorial KD0 |±15/2〉eq (Fig. 11b) remains essentially the same as in the case of the simplified model discussed above (Table 7 and Fig. 12). The reason is that the local energy structure of low-mJ states near the bottom of the diagram (Fig. 14c) is very close to that of the parabolic profile shown in Fig. 12a. Our CF calculations support this conclusion.
4. Conclusions
In summary, we report the syntheses, structures, and magnetic properties of three new Dy complexes obtained by the reactions of dysprosium perchlorate with mixed ligands, rigid pentadentate 2,6-diacetylpyridine bis(R-hydrazones) and monodentate tricyclohexylphosphine oxide (Cy3PO) or triphenylphosphine oxide (Ph3PO), in the presence of Et3N as a deprotonating base: [Dy(LCH3)(Cy3PO)2]ClO4·CH3CN (1), [Dy(L2(t-Bu))(Ph3PO)2]ClO4·0.63C2H5OH (2), and [Dy(LOCH3)(Ph3PO)2]ClO4·2H2O (3), where LCH3 = [2,6-diacetylpyridine bis(acetylhydrazone)]2−, L2(t-Bu) = [2,6-diacetylpyridine bis(3,5di-tert-butylbenzoylhydrazone)]2−, and LCH3 = [2,6-diacetylpyridine bis(4-methoxybenzoylhydrazone)]2−. The central Dy3+ ion is seven-coordinated by three N and two O atoms in the equatorial plane of the chelating pentadentate ligands and two O atoms of the apical ligands in all the structures. The CShM analysis showed that the local geometry of the Dy(III) ions in the complexes was PBPY-7 with pseudo-D5h symmetry for the [DyN3O4] core. For all three complexes, the magnetization barrier values in zero dc field and in the presence of a constant magnetic field are similar and are around 320–350 K, indicating that the nature of the substituents in the equatorial pentadentate ligands have almost no effect on the magnetic behavior of these mononuclear SMMs.The presence of two strong apical donor ligands, Cy3PO or Ph3PO, improves the SMM characteristics of 1–3 compared to similar PBPY-7 complexes containing two Cl− or Cl(Cy3PO/Ph3PO) in the apical positions,45,46 as it leads to a slow relaxation of magnetization in zero dc field and larger magnetization barriers. The ground states of 1–3 are almost pure Ising-type KD0 |±15/2〉 with very small transverse components (gx, gy ∼ 0, gz ∼ 19.6), which suppress the ground-state QTM processes. However, the first excited KD1 loses magnetic axiality (Table 4), which causes fast thermally assisted quantum tunneling of magnetization. As a result, the effective energy barriers (Ueff) for complexes 1–3 are close to the energy of the first excited Kramers doublets, KD1, as estimated from ab initio calculations.
Although complexes 1–3 have rather high magnetization barriers, Ueff, their heights are much lower than those of the record PBPY-7 Dy SMM complexes26–29,33 as well as other PBPY-7 Dy complexes containing monodentate weakly coordinating ligands with long Dy–O(N) distances (H2O, THF, pyridine) at the equatorial positions and strong donor ligands (acyl/aryl alkoxides, aryl/acyl/phosphonamides, silane/phosphine oxides) at the apical positions.34–41 It is also crucial that in all high-performance PBPY-7 Dy SMMs the long magnetic z axis invariably points to the apical ligands, whereas in PBPY-7 complexes with an equatorial ligand based on 2,6-diacetylpyridine (our complexes 1–3 and similar complexes reported in ref. 45–48), the long magnetic axis lies in the equatorial xy plane of the pentadentate LR ligand (Fig. 11b and Fig. S21, ESI†).
To elucidate the origin of these differences, we have examined in detail the influence of the competition between the CF strength of the apical and equatorial ligands in PBPY-7 Dy complexes on the CF splitting pattern and energy barrier, Ueff, in terms of CF theory. Depending on the ratio between the apical and equatorial CF components, PBPY-7 Dy complexes fall into three groups (shown as Cases I, II, and III in Fig. 1). In terms of the CF parameters Bkq, the ranges of Cases I–III are largely quantified by the ratio of the two axial CF parameters B20 and B40: B20/B40 > 1.35 (Case I, Fig. 14a), 1.35 > B20/B40 > −0.07 (Case II, Fig. 14b) and B20/B40 < −0.07 (Case III, Fig. 14c).
In Cases I and II, where the apical field prevails over the equatorial field (CFap ≫ CFeq, and CFap > CFeq, Fig. 1a and b, respectively), the ground state is the ‘apical’ Kramers doublet KD0 |±15/2〉ap with a long magnetic z axis directed toward the apical ligands. The maximum SMM performance in Cases I and II is reached at the ideal D5h symmetry of the PBPY-7 complex, which ensures perfect magnetic axiality of the ground KD0 |±15/2〉 and excited KDs |±mJ〉. However, in Case III (CFap < CFeq), a fundamentally different situation arises because the oblate |±15/2〉 ground state turns into the prolate |±1/2〉 ground state (Fig. 1c). As a result, the overall magnetic anisotropy of the complex switches from the easy-axis (z) to the easy-plane (xy) regime and the double-well potential turns into a single-well potential (Fig. 1c). This implies that the regular (D5h) PBPY-7 Dy complex from Case III is no longer a SMM (Fig. 1c). Therefore, in Case III, the necessary prerequisites for SMM behavior (i.e., double-well potential and barrier Ueff) can only arise due to distortions of the PBPY-7 structure of the complex; our complexes 1–3 definitely refer to Case III.
We have explored the specific mechanism of the origin of this ‘equatorial’ KD0 |±15/2〉eq (Fig. 11b) in the frame of CF theory. With the spin quantization z axis parallel to the apical axis, the ‘equatorial’ KD0 |±15/2〉eq (with gx ∼ 19.6 and gy, gz ∼ 0) is a mixture of low-mJ states (Fig. 11b). This mixing is mainly caused by the rhombic CF parameter B22 arising from distortions in the N3O2 equatorial plane, the most important of which is the deviation of the O1–Dy–O2 bond angle from 72° (Fig. 13). As B22 increases, the ground KD0 |±1/2〉 with easy-plane anisotropy (gx = gy = 10.59, gz = 1.32) progressively transforms into an ‘equatorial’ KD0 |±15/2〉eq with easy-axis anisotropy (with gx ∼ 19.6, gy, gz ∼ 0), in which the long magnetic x axis lies in the equatorial xy plane (Table 7 and Fig. 12). This occurs at large values of the rhombic parameter, B22 > 500 cm−1 (Table 7). In complexes 1–3 such values of B22 are reached due to the large O1–Dy–O2 bond angle, about 100° (Table 8 and Fig. 13b). Therefore, apart from the strong equatorial CF of the negatively charged (N3O2)2− chelate node of LR ligands, the large O1–Dy–O2 bond angle in the equatorial plane is the direct cause of the pronounced SMM behavior of 1–3.
Thus, the attribution of PBPY-7 complexes 1–3 and similar complexes45–48 to Case III highlights a breakdown of apical magnetic axiality, which plays a key role in the high SMM performance in Case I and II PBPY-7 Dy complexes. In other words, the main advantage of PBPY-7 complexes providing a strictly axial CF potential (i.e., with zero non-axial CF parameters Bkq, q ≠ 0) in the regular D5h geometry is completely lost in Case III, since here the SMM performance is governed by distortions in the N3O2 equatorial plane rather than by high D5h symmetry and strong apical ligands. As a result, Case III complexes can hardly be competitive with Case I and II complexes in terms of SMM performance due to the lower total CF splitting energy of the 6H15/2 multiplet and also because of the absence of magnetic axiality in the excited KDs.
It is also important to point out that the apical KD0 |±15/2〉ap and equatorial KD0 |±15/2〉eq have the same oblate shaped electron clouds, which differ only in orientation (Fig. 11), but have quite different origins. Turning the apical KD0 |±15/2〉ap into the equatorial KD0 |±15/2〉eq is a key trigger that switches between high SMM performance (Cases I and II) and low SMM performance (Case III) for PBPY-7 Dy(III) complexes.
In light of the above, when assessing the prospects of complexes 1–3 and other related complexes45–48 as performable SMMs, it should be emphasized that there is a general crucial problem concerning the rigid [N3O2]2−-type negatively charged ligands in the equatorial plane, which produce a strong equatorial CF exceeding the CF of apical ligands. Obviously, such a situation is extremely unfavorable because the oblate Dy3+ ion prefers a strong apical CF and weak equatorial CF to attain a maximal energy barrier, Ueff, and blocking temperature, TB.
In this work, we aimed to improve the SMM characteristics of the PBPY-7 Dy complexes with rigid N3O2-type Schiff-base ligands by enhancing the apical CF using strong neutral Cy3PO and Ph3PO donor ligands in the apical positions. However, despite some improvement in the SMM performance of 1–3, this was insufficient to change the overall positive (easy plane) magnetic anisotropy into a negative one (easy axis), since the long magnetic axis with gx ∼ 19.6 still lay in the equatorial xy plane of complexes 1–3 (Fig. S21, ESI†). Evidence for the dominance of the equatorial CF over the apical CF is the similarity of the Dy–O bond lengths in the equatorial plane of N3O2, 2.25–2.30 Å, to the distances to the apical ligands (Dy–O, 2.23–2.26 Å).
The unfavorable balance between the apical CF and equatorial CF can be improved by further enhancing the apical CF through the use of the strongest negatively charged donor ligands with the shortest Dy–X apical bonds (∼2.05–2.10 Å) occupying two apical positions, such as BuO−, PhO−, Me3SiO−, MeO− and AdO−. The only known example is the recently reported PBPY-7 anion complex, [Dy(LN3O2)(LADTP)2]− (H2LN3O2 = 2,6-diformylpyridine bis(4-phenylsemicarbazone), HLADTP = 4-(anthracen-9-yl)-2-(1,3-dithiolan-2-yl)phenol), which reveals a negative (easy-axis) magnetic anisotropy and ‘true’ mJ = ±15/2 ground state with the long gz axis pointing to the apical ligands.81
Alternatively, improved balance between the equatorial and apical CFs may be obtained by weakening the equatorial CF through the use of substituents with longer Dy–X bonds in the N3X2 chelate ring of the 2,6-diacetylpyridine-based ligand, such as LN5 in [Dy(LN5)(Ph3SiO)2]27 or LN3S2 in [Dy(LN3S2)(LADTP)2]− (H2LN3S2 = 2,6-diformylpyridine bis(4-phenylthiosemicarbazone)).81 The SMM characteristics of these complexes are much higher than those of 1–3 (Ueff > 1000 K vs. ∼350 K) and their long magnetic axes are oriented toward the apical ligands. The CF splitting patterns of these complexes suggest that they belong to Case II.
One more way to take advantage of the rigid structure of equatorial pentadentate ligands like N3O2 is to reduce the negative electric charge in the equatorial plane through the use of weaker neutral protonated ligands, H2N3O2. So far, no PBPY-7 Dy complexes with rigid neutral pentadentate H2N3O2 equatorial ligands have been described in the literature. Our further studies are aimed at realizing this approach.
Author contributions
Conceptualization, E. B. Y., V. S. M.; synthesis and characterization of the materials, T. A. B., V. A. K.; X-ray crystallography, formal analysis, I. A. Y.; measurement of dc and ac magnetic properties, formal analysis, M. V. Z., A. I. D., N. N. E., K. A. B.; ab initio calculations, analysis of dc properties, D. V. K.; analysis of the dynamic magnetic properties, E. A. Y.; crystal-field calculations, analysis single-ion magnet behavior of Dy PBPY-7 complexes, V. S. M.; writing–original draft preparation, T. A. B., I. A. Y., A. I. D., D. V. K., E. A. Y., V. S. M., E. B. Y.; writing–review and editing, V. S. M., E. B. Y.; supervision, project administration, E. B. Y. All authors have read and agreed to the published version of the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the main text of paper and its ESI.† Crystal-field calculations were performed using V. S. M.'s 4f-shell program suite described in V. S. Mironov and L. E. Li, J. Alloys Compd., 1998, 279, 83–92. More detailed data are available on request from the corresponding author, upon reasonable request. Crystallographic details are available in the ESI in CIF format. CCDC numbers 2239615, 2239616, and 2239617.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Ministry of Science and Higher Education within the state assignment for FRC PCP MC RAS, state registration numbers: 124013100858-3 (FFSG-2024-0009) and 124020200104-8 (FFSG-2024-0004). The work was partially performed using equipment of the Computational and Analytical Center of Collective Use of the FRC PCP MC RAS. The work was carried out within the state assignment of NRC ‘Kurchatov institute’.References
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-y. Koshihara and Y. Kaizu, Lanthanide Double-Decker Complexes Functioning as Magnets at the Single-Molecular Level, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- L. Sorace, C. Benelli and D. Gatteschi, Lanthanides in molecular magnetism: old tools in a new field, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Lanthanide single-molecule magnets, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- R. A. Layfield and M. Murugesu, Lanthanides and Actinides in Molecular Magnetism, Wiley, Hoboken, 2015 Search PubMed.
- S. T. Liddle and J. Van Slageren, Improving f-Element Single Molecule Magnets, Chem. Soc. Rev., 2015, 44, 6655–6669 RSC.
- K. L. M. Harriman and M. Murugesu, An Organolanthanide Building Block Approach to Single-Molecule Magnets, Acc. Chem. Res., 2016, 49, 1158–1167 CrossRef CAS PubMed.
- J.-L. Liu, Y.-C. Chen and M.-L. Tong, Symmetry Strategies for High Performance Lanthanide-Based Single-Molecule Magnets, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC.
- M. Feng and M.-L. Tong, Single Ion Magnets from 3d to 5f: Developments and Strategies, Chem. – Eur. J., 2018, 24, 7574–7594 CrossRef CAS PubMed.
- Z. Zhu, M. Guo, X.-L. Li and J. Tang, Molecular Magnetism of Lanthanide: Advances and Perspectives, Coord. Chem. Rev., 2019, 378, 350–364 CrossRef CAS.
- A. Zabala-Lekuona, J. M. Seco and E. Colacio, Single-Molecule Magnets: From Mn12-Ac to Dysprosium Metallocenes, a Travel in Time, Coord. Chem. Rev., 2021, 441, 213984 CrossRef CAS.
- (a) F. D. Natterer, K. Yang, W. Paul, P. Willke, T. Choi, T. Greber, A. J. Heinrich and C. P. Lutz, Reading and writing single-atom magnets, Nature, 2017, 543, 226–228 CrossRef CAS PubMed; (b) R. Sessoli, Single-atom data storage, Nature, 2017, 543, 189–190 CrossRef CAS PubMed.
- (a) L. Bogani and W. Wernsdorfer, Molecular spintronics using single-molecule magnets, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed; (b) S. Sanvito, Molecular spintronics, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- (a) M. N. Leuenberger and D. Loss, Quantum computing in molecular magnets, Nature, 2001, 410, 789–793 CrossRef CAS PubMed; (b) A. Gaita-Ariño, F. Luis, S. Hill and E. Coronado, Molecular spins for quantum computation, Nat. Chem., 2019, 11, 301–309 CrossRef PubMed.
- M. Atzori and R. Sessoli, The Second Quantum Revolution: Role and Challenges of Molecular Chemistry, J. Am. Chem. Soc., 2019, 141, 11339–11352 CrossRef CAS PubMed.
- E. Coronado, Molecular Magnetism: From Chemical Design to Spin Control in Molecules, Materials and Devices, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- E. Moreno-Pineda and W. Wernsdorfer, Measuring Molecular Magnets for Quantum Technologies, Nat. Rev. Phys., 2021, 3, 645–659 CrossRef CAS.
- T. Pugh, N. F. Chilton and R. A. Layfield, A Low-Symmetry Dysprosium Metallocene Single-Molecule Magnet with a High Anisotropy Barrier, Angew. Chem., Int. Ed., 2016, 55, 11082–11085 CrossRef CAS PubMed.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Molecular magnetic hysteresis at 60 kelvin in dysprosocenium, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamäki and R. A. Layfield, Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- L. Ungur and L. F. Chibotaru, Strategies toward High-Temperature Lanthanide-Based Single-Molecule Magnets, Inorg. Chem., 2016, 55, 10043–10056 CrossRef CAS PubMed.
- A. K. Bar, P. Kalita, M. K. Singh, G. Rajaraman and V. Chandrasekhar, Low-coordinate mononuclear lanthanide complexes as molecular nanomagnets, Coord. Chem. Rev., 2018, 367, 163–216 CrossRef CAS.
- L. Ungur and L. F. Chibotaru, Magnetic anisotropy in the excited states of low symmetry lanthanide complexes, Phys. Chem. Chem. Phys., 2011, 13, 20086–20090 RSC.
- S.-D. Jiang and S.-X. Qinc, Prediction of the Quantized Axis of Rare-Earth Ions: the Electrostatic Model with Displaced Point Charges, Inorg. Chem. Front., 2015, 2, 613–619 RSC.
- B. M. Day, F.-S. Guo and R. A. Layfield, Cyclopentadienyl Ligands in Lanthanide Single-Molecule Magnets: One Ring To Rule Them, All?, Acc. Chem. Res., 2018, 51, 1880–1889 CrossRef CAS PubMed.
- R. Sun, C. Wang, B. W. Wang, Z. M. Wang, Y. F. Chen and S. Gao, Low-coordinate bis(imidazolin-2-iminato) dysprosium(III) single-molecule magnets, Inorg. Chem. Front., 2023, 10, 485–492 RSC.
- Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, On Approaching the Limit of Molecular Magnetic Anisotropy: A Near-Perfect Pentagonal Bipyramidal Dysprosium(III) Single-Molecule Magnet, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef CAS PubMed.
- A. B. Canaj, S. Dey, C. Wilson, O. Céspedes, G. Rajaraman and M. Murrie, Engineering macrocyclic high performance pentagonal bipyramidal Dy(III) single-ion magnets, Chem. Commun., 2020, 56, 12037–12040 RSC.
- J. Long, A. O. Tolpygin, D. M. Lyubov, N. Y. Rad'kova, A. V. Cherkasov, Y. V. Nelyubina, Y. Guari, J. Larionova and A. A. Trifonov, High magnetization reversal barriers in luminescent dysprosium octahedral and pentagonal bipyramidal single-molecule magnets based on fluorinated alkoxide ligands, Dalton Trans., 2021, 50, 8487–8496 RSC.
- J.-P. Sutter, V. Béreau, V. Jubault, K. Bretosh, C. Pichon and C. Duhayon, Magnetic anisotropy of transition metal and lanthanide ions in pentagonal bipyramidal geometry, Chem. Soc. Rev., 2022, 51, 3280–3313 RSC.
- Z. Zhu, C. Zhao, T. Feng, X. Liu, X. Ying, X. L. Li, Y. Q. Zhang and J. Tang, Air-Stable Chiral Single-Molecule Magnets with Record Anisotropy Barrier Exceeding 1800 K, J. Am. Chem. Soc., 2021, 143, 10077–10082 CrossRef CAS PubMed.
- W. J. Xu, Q. C. Luo, Z. H. Li, Y. Q. Zhai and Y. Z. Zheng, Bis-Alkoxide Dysprosium(III) Crown Ether Complexes Exhibit Tunable Air Stability and Record Energy Barrier, Adv. Sci., 2024, 11, 2308548 CrossRef CAS PubMed.
- J. Corredoira-Vazquez, C. Gonzalez-Barreira, P. Oreiro-Martínez, A. M. García-Deibe, J. Sanmartín-Matalobos and M. Fondo, Synthesis and applications of lanthanoid complexes of pentadentate and hexadentate N5 and N6 macrocycles: A review, J. Rare Earths, 2024, 42, 1–15 CrossRef CAS.
- Y.-S. Ding, T. Han, Y.-Q. Zhai, D. Reta, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, A Study of Magnetic Relaxation in Dysprosium(III) Single-Molecule Magnets, Chem. – Eur. J., 2020, 26, 5893–5902 CrossRef CAS PubMed.
- Y.-C. Chen, J.-L. Liu, L. Ungur, J. Liu, Q.-W. Li, L.-F. Wang, Z.-P. Ni, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, Symmetry-Supported Magnetic Blocking at 20 K in Pentagonal Bipyramidal Dy(III) Single-Ion Magnets, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed.
- I. F. Dıáz-Ortega, J. M. Herrera, S. Dey, H. Nojiri, G. Rajaraman and E. Colacio, The effect of the electronic structure and flexibility of the counteranions on magnetization relaxation in [Dy(L)2(H2O)5]3+ (L = phosphine oxide derivative) pentagonal bipyramidal SIMs, Inorg. Chem. Front., 2020, 7, 689–699 RSC.
- Y.-C. Chen, J.-L. Liu, Y. Lan, Z.-Q. Zhong, A. Mansikkamäki, L. Ungur, Q.-W. Li, J.-H. Jia, L. F. Chibotaru, J.-B. Han, W. Wernsdorfer, X.-M. Chen and M.-L. Tong, Dynamic Magnetic and Optical Insight into a High Performance Pentagonal Bipyramidal DyIII Single-Ion Magnet, Chem. – Eur. J., 2017, 23, 5708–5715 CrossRef CAS PubMed.
- Z.-H. Li, Y.-Q. Zhai, W.-P. Chen, Q.-C. Luo, T. Han and Y.-Z. Zheng, Breaking the axiality of pentagonal-bipyramidal dysprosium(III) single-molecule magnets with pyrazolate ligands, Inorg. Chem. Front., 2020, 7, 4367–4376 RSC.
- Y.-S. Ding, K.-X. Yu, D. Reta, F. Ortu, R. E. P. Winpenny, Y.-Z. Zheng and N. F. Chilton, Field- and temperature-dependent quantum tunnelling of the magnetisation in a large barrier single-molecule magnet, Nat. Commun., 2018, 9, 3134–3144 CrossRef PubMed.
- J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, A Stable Pentagonal Bipyramidal Dy(III) Single-Ion Magnet with a Record Magnetization Reversal Barrier over 1000 K, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- Z. Jiang, L. Sun, Q. Yang, B. Yin, H. Ke, J. Han, Q. Wei, G. Xie and S. Chen, Excess axial electrostatic repulsion as a criterion for pentagonal bipyramidal DyIII single-ion magnets with high Ueff and TB, J. Mater. Chem. C, 2018, 6, 4273–4280 RSC.
- Y. Dong, L. Zhu, B. Yin, X. Zhu and D. Li, Regulating the magnetic properties of seven-coordinated Dy(III) single-ion magnets through the effect of positional isomers on axial crystal-field, Dalton Trans., 2021, 50, 17328–17337 RSC.
- J. Long, A. N. Selikhov, E. Mamontova, K. A. Lyssenko, Y. Guari, J. Larionova and A. A. Trifonov, Single-molecule magnet behaviour in a Dy(III) pentagonal bipyramidal complex with a quasi-linear Cl-Dy-Cl sequence, Dalton Trans., 2019, 48, 35–39 RSC.
- M. Li, H. Wu, Q. Yang, H. Ke, B. Yin, Q. Shi, W. Wang, Q. Wei, G. Xie and S. Chen, Experimental and Theoretical Interpretation on the Magnetic Behavior in a Series of Pentagonal-Bipyramidal DyIII Single-Ion Magnets, Chem. – Eur. J., 2017, 23, 17775–17787 CrossRef CAS PubMed.
- H. Wu, M. Li, B. Yin, Z. Xia, H. Ke, Q. Wei, G. Xie, S. Chen and S. Gao, Fine-tuning the type of equatorial donor atom in pentagonal bipyramidal Dy(III) complexes to enhance single-molecule magnet properties, Dalton Trans., 2019, 48, 16384–16394 RSC.
- A. K. Bar, P. Kalita, J.-P. Sutter and V. Chandrasekhar, Pentagonal-bipyramid Ln(III) complexes exhibiting single-ion-magnet behavior: A rational synthetic approach for a rigid equatorial plane, Inorg. Chem., 2018, 57, 2398–2401 CrossRef CAS PubMed.
- P. Kalita, N. Ahmed, A. K. Bar, S. Dey, A. Jana, G. Rajaraman, J.-P. Sutter and V. Chandrasekhar, Pentagonal bipyramidal Ln(III) complexes containing an axial phosphine oxide ligand: Field-induced single-ion magnetism behavior of the Dy(III) analogues, Inorg. Chem., 2020, 59, 6603–6612 CrossRef CAS PubMed.
- P. Kalita, N. Ahmed, S. Moorthy, V. Béreau, A. Kumar Bar, P. Kumar, P. Nayak, J.-P. Sutter, S. K. Singh and V. Chandrasekhar, Slow magnetic relaxation in a homoaxially phosphine oxide coordinated pentagonal bipyramidal Dy(III) complex, Dalton Trans., 2023, 52, 2804–2815 RSC.
- V. Singh, D. Das, S. Anga, J.-P. Sutter, V. Chandrasekhar and A. K. Bar, Rigid N3O2-Pentadentate Ligand-Assisted Octacoordinate Mononuclear Ln(III) Complexes: Syntheses, Characterization, and Slow Magnetization Relaxation, ACS Omega, 2022, 7, 25881–25890 CrossRef CAS PubMed.
- V. D. Sasnovskaya, V. A. Kopotkov, A. V. Kazakova, A. D. Talantsev, R. B. Morgunov, S. V. Simonov, L. V. Zorina, V. S. Mironov and E. B. Yagubskii, Slow magnetic relaxation in mononuclear complexes of Tb, Dy, Ho and Er with the pentadentate (N3O2) Schiff-base dapsc ligand, New J. Chem., 2018, 42, 14883–14893 RSC.
- T. A. Bazhenova, V. S. Mironov, I. A. Yakushev, R. D. Svetogorov, O. V. Maximova, Y. V. Manakin, A. B. Kornev, A. N. Vasiliev and E. B. Yagubskii, End-to-End Azido-Bridged Lanthanide Chain Complexes (Dy, Er, Gd, and Y) with a Pentadentate Schiff-Base [N3O2] Ligand: Synthesis, Structure, and Magnetism, Inorg. Chem., 2020, 59, 563–578 CrossRef CAS PubMed.
- G. Zhang, Y. Yu, Y. Zhao, X. Xie and C. Ding, Iron(III)/TEMPO-Catalyzed Synthesis of 2,5-Disubstituted 1,3,4-Oxadiazoles by Oxidative Cyclization under Mild Conditions, Synlett, 2017, 1373–1377 CAS.
- M. S. R. Murty, R. Penthala, S. Polepalli and N. Jain, Synthesis and biological evaluation of novel resveratrol-oxadiazole hybrid heterocycles as potential antiproliferative agents, Med. Chem. Res., 2016, 25, 627–643 CrossRef CAS.
- T. J. Giordano, G. J. Palenik, R. C. Palenik and D. A. Sullivan, Pentagonal-bipyramidal complexes. Synthesis and characterization of aqua(nitrato)[2,6-diacetylpyridine bis(benzoyl hydrazone)]cobalt(II) nitrate and diaqua[2,6-diacetylpyridine bis(benzoyl hydrazone)]nickel(II) nitrate dehydrate, Inorg. Chem., 1979, 18, 2445–2450 CrossRef CAS.
- APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, WI, USA, 2016 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXT-Integrated Space-Group and Crystal Structure Determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- (a) I. F. Galván, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov, R. K. Carlson, L. F. Chibotaru, J. Creutzberg, N. Dattani, M. G. Delcey, S. S. Dong, A. Dreuw, L. Freitag, L. M. Frutos, L. Gagliardi, F. Gendron, A. Giussani, L. González, G. Grell, M. Guo, C. E. Hoyer, M. Johansson, S. Keller, S. Knecht, G. Kovačević, E. Källman, G. L. Manni, M. Lundberg, Y. Ma, S. Mai, J. P. Malhado, P. A. Malmqvist, P. Marquetand, S. A. Mewes, J. Norell, M. Olivucci, M. Oppel, Q. M. Phung, K. Pierloot, F. Plasser, M. Reiher, A. M. Sand, I. Schapiro, P. Sharma, C. J. Stein, L. K. Sørensen, D. G. Truhlar, M. Ugandi, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, O. Weser, T. A. Wesołowski, P.-O. Widmark, S. Wouters, A. Zech, J. P. Zobel and R. Lindh, OpenMolcas: From Source Code to Insight, J. Chem. Theory Comput., 2019, 15, 5925–5964 CrossRef PubMed; (b) F. Aquilante, J. Autschbach, A. Baiardi, S. Battaglia, V. A. Borin, L. F. Chibotaru, I. Conti, L. De Vico, M. Delcey, I. F. Galván, N. Ferré, L. Freitag, M. Garavelli, X. Gong, S. Knecht, E. D. Larsson, R. Lindh, M. Lundberg, P. A. Malmqvist, A. Nenov, J. Norell, M. Odelius, M. Olivucci, T. B. Pedersen, L. Pedraza-González, Q. M. Phung, K. Pierloot, M. Reiher, I. Schapiro, J. Segarra-Martí, F. Segatta, L. Seijo, S. Sen, D.-C. Sergentu, C. J. Stein, L. Ungur, M. Vacher, A. Valentini and V. Veryazov, Modern quantum chemistry with [Open]Molcas, J. Chem. Phys., 2020, 152, 214117 CrossRef CAS PubMed.
- (a) L. F. Chibotaru and L. Ungur, Ab initio calculation of anisotropic magnetic properties of complexes. I. Unique definition of pseudospin Hamiltonians and their derivation, J. Chem. Phys., 2012, 137, 064112 CrossRef CAS PubMed; (b) B. G. Wybourne, Spectroscopic Properties of Rare Earths, John Wiley, New York, 1965 Search PubMed.
- T. A. Bazhenova, V. A. Kopotkov, D. V. Korchagin, Y. N. Manakin, L. V. Zorina, S. V. Simonov, I. A. Yakushev, V. S. Mironov, A. N. Vasiliev, O. V. Maximova and E. B. Yagubskii, A Series of Novel Pentagonal-Bipyramidal Erbium(III) Complexes with Acyclic Chelating N3O2 Schiff-Base Ligands: Synthesis, Structure, and Magnetism, Molecules, 2021, 26, 6908 CrossRef CAS PubMed.
- T. A. Bazhenova, I. A. Yakushev, K. A. Lyssenko, O. V. Maximova, V. S. Mironov, Y. N. Manakin, A. B. Kornev, A. N. Vasiliev and E. B. Yagubskii, Ten-Coordinate Lanthanide [Ln(HL)(L)] Complexes (Ln = Dy, Ho, Er, Tb) with Pentadentate N3O2-Type Schiff-Base Ligands: Synthesis, Structure and Magnetism, Magnetochemistry, 2020, 6, 60 CrossRef CAS.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE Program, version 2.1, Universitat de Barcelona, 2013 Search PubMed.
- P. Kalita, J. Acharyab and V. Chandrasekh, Mononuclear pentagonal bipyramidal Ln(III) complexes: Syntheses and magnetic properties, J. Magn. Magn. Mater., 2020, 498, 166098 CrossRef CAS.
- K. S. Cole and R. H. Cole, Dispersion and Absorption in Dielectrics I. Alternating Current Characteristics, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS.
- Y. Ma, Y.-Q. Zhai, Y.-S. Ding, T. Han and Y.-Z. Zheng, Understanding a pentagonal-bipyramidal holmium(III) complex with a record energy barrier for magnetisation reversal, Chem. Commun., 2020, 56, 3979–3982 RSC.
- Y.-S. Ding, W. J. A. Blackmore, Y.-Q. Zhai, M. J. Giansiracusa, D. Reta, I. Vitorica-Yrezabal, R. E. P. Winpenny, N. F. Chilton and Y.-Z. Zheng, Studies of the Temperature Dependence of the Structure and Magnetism of a Hexagonal-Bipyramidal Dysprosium(III) Single-Molecule Magnet, Inorg. Chem., 2022, 61, 227–235 CrossRef CAS PubMed.
- B. Zhang, X. Guo, P. Tan, W. Lv, X. Bai, Y. Zhou, A. Yuan, L. Chen, D. Liu, H.-H. Cui, R. Wang and X.-T. Chen, Axial Ligand as a Critical Factor for High-Performance Pentagonal Bipyramidal Dy(III) Single-Ion Magnets, Inorg. Chem., 2022, 61, 19726–19734 CrossRef CAS PubMed.
- S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, An air-stable Dy(III) single-ion magnet with high anisotropy barrier and blocking temperature, Chem. Sci., 2016, 7, 5181–5191 RSC.
- E. V. Gorshkov, D. V. Korchagin, E. A. Yureva, G. V. Shilov, M. V. Zhidkov, A. I. Dmitriev, N. N. Efimov, A. V. Palii and S. M. Aldoshin, Effect of Ligand Substitution on Zero-Field Slow Magnetic Relaxation in Mononuclear Dy(III) β-Diketonate Complexes with Phenanthroline-Based Ligands, Magnetochemistry, 2022, 8, 151 CrossRef CAS.
- A. V. Gavrikov, P. S. Koroteev, N. N. Efimov, Z. V. Dobrokhotov, A. B. Ilyukhin, A. K. Kostopoulos, A.-M. Ariciu and V. M. Novotortsev, Novel mononuclear and 1D-polymeric derivatives of lanthanides and (η6-benzoic acid)tricarbonylchromium: synthesis, structure and magnetism, Dalton Trans., 2017, 46, 3369–3380 RSC.
- D. Reta and N. F. Chilton, Extraction of “hidden” relaxation times from AC susceptibility data, Chem. Sq., 2020, 4, 3 Search PubMed.
- K. N. Shrivastava, Theory of Spin-Lattice Relaxation, Phys. Status Solidi B, 1983, 117, 437–458 CrossRef CAS.
- V. S. Parmar, D. P. Mills and R. E. P. Winpenny, Mononuclear Dysprosium Alkoxide and Aryloxide Single-Molecule Magnets, Chem. – Eur. J., 2021, 27, 7625–7645 CrossRef CAS PubMed.
- S. K. Gupta, S. Dey, Th. Rajeshkumar, G. Rajaraman and R. Murugavel, Deciphering the Role of Anions and Secondary Coordination Sphere in Tuning Anisotropy in Dy(III) Air-Stable D5h SIMs, Chem. – Eur. J., 2022, 28, e202103585 CrossRef CAS PubMed.
- C. Rudowicz, Transformation relations for the conventional Okq and normalised O’kq Stevens operator equivalents with k = 1 to 6 and −k ≤ q ≤ k, J. Phys. C: Solid State Phys., 1985, 18, 1415–1430 CrossRef.
- B. Bleaney, Nuclear magnetic resonance shifts in solution due to lanthanide ions, J. Magn. Reson., 1972, 8, 91–100 CAS.
- D. J. Newman, The orbit-lattice interaction for lanthanide ions. I. determination of empirical parameters, Aust. J. Phys., 1978, 31, 79–94 CrossRef CAS.
- D. J. Newman and B. Ng, The superposition model of crystal fields, Rep. Prog. Phys., 1989, 52, 699–762 CrossRef CAS.
- L. Spillecke, C. Koo, O. Maximova, V. S. Mironov, V. A. Kopotkov, D. V. Korchagin, A. N. Vasiliev, E. B. Yagubskii and R. Klingeler, Magnetic behavior of the novel pentagonal-bipyramidal erbium(III) complex (Et3NH)[Er(H2DAPS)Cl2]: high-frequency EPR study and crystal-field analysis, Dalton Trans., 2021, 50, 18143–18154 RSC.
- W. Deng, Y.-Q. Zhou, S.-N. Du, Z.-Y. Ruan, S.-G. Wu, J.-L. Liu and M.-L. Tong, From geometric to charge-distribution symmetry: deeper insights into lifting the performance of dysprosium single-ion magnets, Sci. China: Chem., 2023, 66, 1989–1996 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural, magnetic and ab initio calculation data. CCDC 2239615 (1), 2239616 (2) and 2239617 (3). For ESI and crystallographic data in CIF or other electronic formats, see DOI: https://doi.org/10.1039/d4qi02262a |
| This journal is © the Partner Organisations 2025 |