
Li3V2(PO4)3 particles embedded in a N and S co-doped porous carbon cathode for high performance lithium storage: an experimental and DFT study†
Jinggao
Wu‡
 *ab,
Canyu
Zhong‡
c,
Xiaofan
Chen
a and
Jing
Huang
*d
*ab,
Canyu
Zhong‡
c,
Xiaofan
Chen
a and
Jing
Huang
*d
aHunan Engineering Laboratory for Preparation Technology of Polyvinyl Alcohol Fiber Materials, Huaihua Key Laboratory for Preparation of Ceramic Materials and Devices, College of Chemistry and Materials Engineering, Huaihua University, Huaihua 418000, P. R. China. E-mail: jinggaowu@foxmail.com
bChongqing Key Laboratory for Advanced Materials and Technologies of Clean Energies, School of Materials and Energies, Southwest University, Chongqing 400715, P.R. China
cPanzhihua Engineering Technology Research Center for Graphene, Panzhihua University, Panzhihua 617000, P.R. China
dState Key Laboratory of Silkworm Genome Biology, Key Laboratory of Sericultural Biology and Genetic Breeding, Ministry of Agriculture and Rural Affairs, College of Biotechnology, P.R. China. E-mail: hj41012@163.com
First published on 11th November 2024
Abstract
Li3V2(PO4)3 (LVP) coated with N and S co-doped carbon (NSC) was investigated by DFT calculation, suggesting that NSC significantly enhances electronic conductivity and lowers the energy barrier to Li+ migration in comparison to LVP-embedded in pristine carbon. To experimentally confirm the theoretical prediction, three types of LVP particle embedded in N and S co-doped porous carbon (LVP@NSC) materials with various nitrogen and sulfur molar ratios (N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S = 1:1, 1:2 and 2:1) were prepared by a facile freeze-drying-assisted wet chemical route associated with a post-annealing process. When used as a cathode for a lithium-ion battery (LIB), the designed LVP@NSC with N:S = 1:2 exhibits outstanding high rate capacities of 124.4 and 107.85 mA h g−1, respectively, at 2 and 20 C in a voltage window of 3.0–4.3 V, and an ultralong cycling stability of 500 times at 20 C while retaining a reversible capacity of 100.22 mA h g−1, possibly due to its smallest charge transfer resistance and highest Li+ migration coefficient, which is in good agreement with the theoretical prediction. This work not only reveals the critical role of an interaction mechanism between NSC and LVP, but also offers great potential for high-energy density LIB applications.
:S = 1:1, 1:2 and 2:1) were prepared by a facile freeze-drying-assisted wet chemical route associated with a post-annealing process. When used as a cathode for a lithium-ion battery (LIB), the designed LVP@NSC with N:S = 1:2 exhibits outstanding high rate capacities of 124.4 and 107.85 mA h g−1, respectively, at 2 and 20 C in a voltage window of 3.0–4.3 V, and an ultralong cycling stability of 500 times at 20 C while retaining a reversible capacity of 100.22 mA h g−1, possibly due to its smallest charge transfer resistance and highest Li+ migration coefficient, which is in good agreement with the theoretical prediction. This work not only reveals the critical role of an interaction mechanism between NSC and LVP, but also offers great potential for high-energy density LIB applications.
1. Introduction
With increasing demand for higher energy density, environmental pollution and the unsustainability of fossil fuels, a new type of alternative energy source is urgently required.1 Lithium-ion batteries (LIBs), as a new form of energy storage and conversion, have achieved great success in applications in all aspects of people's lives due to their environmental friendliness, high energy density and capacity retention.2–5 However, electrode materials, especially the cathode, are a key factor limiting the large-scale application of LIBs. Researchers have been working on the search for novel cathode materials with higher specific capacity, safety, high rate capabilities and long cycle life. Among the available cathode materials, lithium metal phosphate compounds, especially LVP, have received a great deal of research interest as advanced next-generation cathode materials for LIBs, ascribed to their low environmental impact, improved safety characteristics, excellent thermal stability, high operating voltage and outstanding theoretical capacity (197 mA h g−1, when all three Li+ deintercalated) which means much higher power density.6 However, their poor cyclability and lower intrinsic electronic conductivity (2 × 10−8 S cm−1) restrict their practical application.7Generally, there are three strategies to overcome these drawbacks: (a) designing nanostructures to increase the active sites for Li+ storage and shortening the Li+ migration distance, which could improve the rate properties,8,9 according to the formula t = L2/D (where t is the Li+ migration time, L is the Li+ migration length and D is the Li+ migration coefficient);10 (b) doping at the cation or anion site to improve the intrinsic electronic conductivity and Li+ migration;11,12 (c) surface modifications, such as carbon coating,13 graphene modification,14 or metal15 and nonelectroactive metal oxides.16 Among all the strategies mentioned above, carbon coating is the most common method to be employed because of its low-cost, facile synthesis, and the coated carbon layer could also play a key role in enhancing electronic conductivity, alleviating the growth of LVP particles during a long-term, high-temperature calcination process.17 However, a pristine carbon coating usually cannot achieve the optimum electrochemical performance, and further modification is often required before coating. Recently, it has been reported that a carbon matrix substitutionally doped by adventitious nitrogen and sulfur heteroatoms can enhance its electrochemical catalytic activity. For example, the synergic effects of N and S co-doping in a carbon framework facilitates the properties of K+ storage,18 supercapacitance,19 oxygen reduction20 and CO2 capture.21 Surprisingly, to the best of our knowledge, the effects of N and S co-doped carbon, further coated on LVP, on the electrochemical performance for lithium storage have not yet been investigated. In addition, motivated by our DFT calculation results, the LVP@NSC has significantly enhanced electronic conductivity and a much lower Li+ migration energy barrier than pristine carbon-coated LVP, we thought it would be valuable to be explored.
Herein, a facile freeze-drying-assisted wet chemical route followed by an annealing method was employed to synthesize three types of LVP particle embedded in N,S-co-doped porous carbon materials with various nitrogen and sulfur molar ratios. Pristine carbon-coated and without carbon layer coated LVP smaples were also synthesized for comparison. The designed N:S = 1:2 sample displays outstanding electrochemical properties. Its structure, morphology and the enhancement mechanism were accordingly investigated.
2. Experimental and calculation method
The chemical reagents and solvents were purchased from Aladdin Reagent Co., Ltd (Shanghai, P.R.China), and used as received without any further processing.2.1. Electrode material preparation
LVP@NSC with various atomic ratios of nitrogen and sulfur (N:S = 1:1, 2:1 and 1:2) were prepared via a facile freeze-drying-assisted wet chemical route, as schematically illustrated in Fig. 2a. In a typical procedure, 0.245 g of citric acid anhydrous (CAA) was initially dissolved in deionized water (DI-H2O) and ethyl alcohol absolute (EAA) mixed solvent [V(H2O):(C2H5OH) = 5:3] under vigorous stirring at room temperature to obtain a clear solution. Then, 0.032 g of N-phenyl-p-phenylenediamine (C12H12N2) and 0.086 g of dibenzyl disulfide (C14H14S2), which were used as nitrogen and sulfur sources, were added into the above clear solution in sequence. After continuous stirring for a certain number of minutes, ammonium vanadate (NH4VO3), lithium hydroxide monohydrate (LiOH·H2O) and ammonium dihydrogen phosphate (NH4H2PO4) with molar ratio 3.02:2:3 were finally added. The mixture was then heated at 80–85 °C in a water bath for several hours with continuous stirring to evaporate the mixed solvent until a wet gel was obtained. Subsequently, the obtained gel precursors were vacuum-dried in a laboratory freeze-dryer for 24 h, since freeze-drying has been considered a good technique to improve the long-term stability of colloidal nanoparticles.22 After being ground and pelletized, the precursors were initially heated to 350 °C in a tubular furnace with flowing argon for 4 h and at 800 °C for 8 h to yield the N:S = 1:2 product (LVP@C-NS12). The calcination temperature was selected by thermogravimetric-derivative thermal analysis (TGA/DTA, Fig. S1†). For comparison, N:S = 1:1, N:S = 2:1 and pure C-coated LVP samples (denoted LVP@C-NS11, LVP@C-NS21 and LVP@C-PURE, respectively) were prepared by the same route with the corresponding stoichiometry. A pristine LVP sample (without carbon layer coating, denoted LVP-PURE) was also prepared using the same route in the absence of CAA, C12H12N2 or C14H14S2, except for calcination under a 10% H2/Ar atmosphere.
2.2. Material characterization
Powder X-ray diffraction (XRD) was performed using an X-ray diffractometer (MAXima-XRD-7000) with Cu Kα radiation (λ = 1.5406 nm). The data were recorded using continuous scanning mode with a step size of 0.02 over the 2θ range of 10–50° at room temperature. The surface morphology was observed via a scanning electron microscope (SEM, JSM-6700F) and a transmission electron microscope (TEM, JEM-2100). X-ray photoelectron spectroscopy (XPS) was conducted on a spectrometer (Escalab 250xi) with monochromatic Al Kα radiation to investigate the surface chemical state. Thermogravimetric measurements (Mettler-Toledo TGA) were carried out with a flow of Ar at a heating rate of 10 K min−1 from room temperature to 800 °C. Nitrogen adsorption–desorption isotherms were conducted at 77 K (ASAP 2020).2.3. Electrochemical measurements
The working electrode was prepared by mixing 75 wt% of the active material, 15 wt% of SP and 10 wt% of polyvinylidene fluoride (PVDF) binder. A slurry of carbon layer modified LVP and commercial biomass hard carbon cast with N-methyl-2-pyrrolidone (NMP) solvent was pasted onto carbon-coated aluminum foil and copper foil current collectors, respectively. These electrodes were then dried in a vacuum at 120 °C for 12 h. Electrochemical tests were operated using CR 2032 coin cells assembled in an Ar-filled glove box with 1 M LiPF6 in ethylene carbonate (EC), diethyl-carbonate (DEC) and ethyl methyl carbonate (EMC) (1:1:1 by volume) electrolyte with 5% fluoroethylene carbonate (FEC). Lithium metal foil was used as the anode and Celgard 2400 as the separator. The cells were aged for 8 h before they were subjected to electrochemical cycling. Galvanostatic charge/discharge tests were carried out using a LAND battery test system (CT2001A, Wuhan, China) in the voltage windows 3.0–4.3 and 3.0–4.8 V (vs. Li+/Li) under various current densities. The mass of active materials was calculated based on the mass of the LVP. Cyclic voltammograms (CV) were studied using an electrochemical workstation (CHI 760E, Shanghai, China) at a scan rate of 0.2 mV s−1. Electrochemical impedance spectroscopy (EIS) data was collected on the CHI 760E electrochemical workstation, over a frequency range from 1 MHz to 100 mHz with an applied amplitude of 5 mV. For the full battery test, commercial biomass hard carbon was used as the counter electrode and reference electrode, which initially underwent lithium insertion activation via several cycles of charge and discharge with current densities of 20, 50, 100 and 20 mA g−1 in sequence for the successive full battery assembly. The full cell test was charged by a constant current followed by a constant voltage model (1 min at 4.3 V) at the initial cycle for cell activation, and discharged by a constant current model, over a voltage range of 3.0–4.3 V at various current densities. All the electrochemical measurements were performed at room temperature.
2.4. DFT calculation details
The first principles calculations in this work were performed using density functional theory (DFT) within the projector augmented wave (PAW) method,23 with an energy cutoff of 400 eV, as implemented in the VASP package.24,25 The Perdew–Burke–Ernzerholf (PBE) form26 based on the general gradient approximation (GGA) was adopted for the exchange–correlation functional. Porous carbon, either undoped or doped with nitrogen and sulfur and adsorbed on the surface of LVP electrodes, was modeled by single-layer graphene, which has also been employed in previous literature.27 The most energetically favorable configurations of the nitrogen and sulfur co-doped carbon layer were tested using the spin-polarized GGA method without any symmetry constraints (Fig. S2†). Surface energy calculations were also carried out for the (010) and (001) surfaces of the 2 × 1 × 1 monoclinic LVP slab configuration, since the migration of lithium ions is primarily along the a-axis direction, according to previous classic molecular dynamics calculation results.28 The outer six atom 1st–6th layers were allowed to relax, while the remaining internal 7th–12th layers were frozen in bulk positions to simulate the bulk properties of the material, with a vacuum layer of at least 12 Å along the c-axis to minimize undesirable interactions between the adjacent images (Fig. S3†). The most energetically favorable nitrogen and sulfur co-doped carbon (NSC) layer and LVP (001) surface were used to construct the interface atomic model between lithium vanadium phosphate and the carbon layer. Two isolated carbon atoms were introduced at the LVP interface to simulate amorphous carbon for subsequent DFT calculations. A small lattice mismatch between the two surfaces indicates a relatively reasonable interface configuration. Brillouin-zone integrations were approximated by using the special k-point sampling of the Γ-centered Monkhorst–Pack scheme29 with a k-point mesh resolution of 2π × 0.04 Å, but a denser 2π × 0.02 Å was used for the density of states (DOS) calculation. The self-consistent calculations were performed using the tetrahedron method with Blöchl corrections until the total energy converged to 10−4 eV and forces acting on every atom became less than 0.03 eV Å−1. Dipole corrections30 were employed in all the slab models and Grimme DFT-D2 van der Waals corrections31 were also taken into account in the interface atomic model. Based on the single vacancy hopping mechanism, the climbing image nudged elastic band (CiNEB) method32 was used to determine the minimum energy path and saddle points of Li+ migration with energy and force convergence criteria of 10−4 eV and 0.05 eV Å−1, respectively. The Brillouin-zone sampling used here was restricted to a single Γ point to keep the computational costs to a reasonable level.3. Results and discussion
The electrochemical performance of LVP@NSC was initially investigated by DFT calculation. As shown in Fig. 1a and b, compared to pristine carbon, the N,S-co-doped carbon shows a denser charge distribution, indicating that higher electron conduction activity carbon can be obtained via N,S-co-doping. Therefore, the differential charge density of both the carbon-coated LVPs was further calculated for a qualitative analysis of the charge transfer between interfaces. The differential charge density was calculated from: Δρ = ρAB − ρA − ρB, where ρAB, ρA and ρB are the charge density of carbon-coated LVP, LVP and carbon with and without N,S-co-doping adsorbed on the surface, respectively.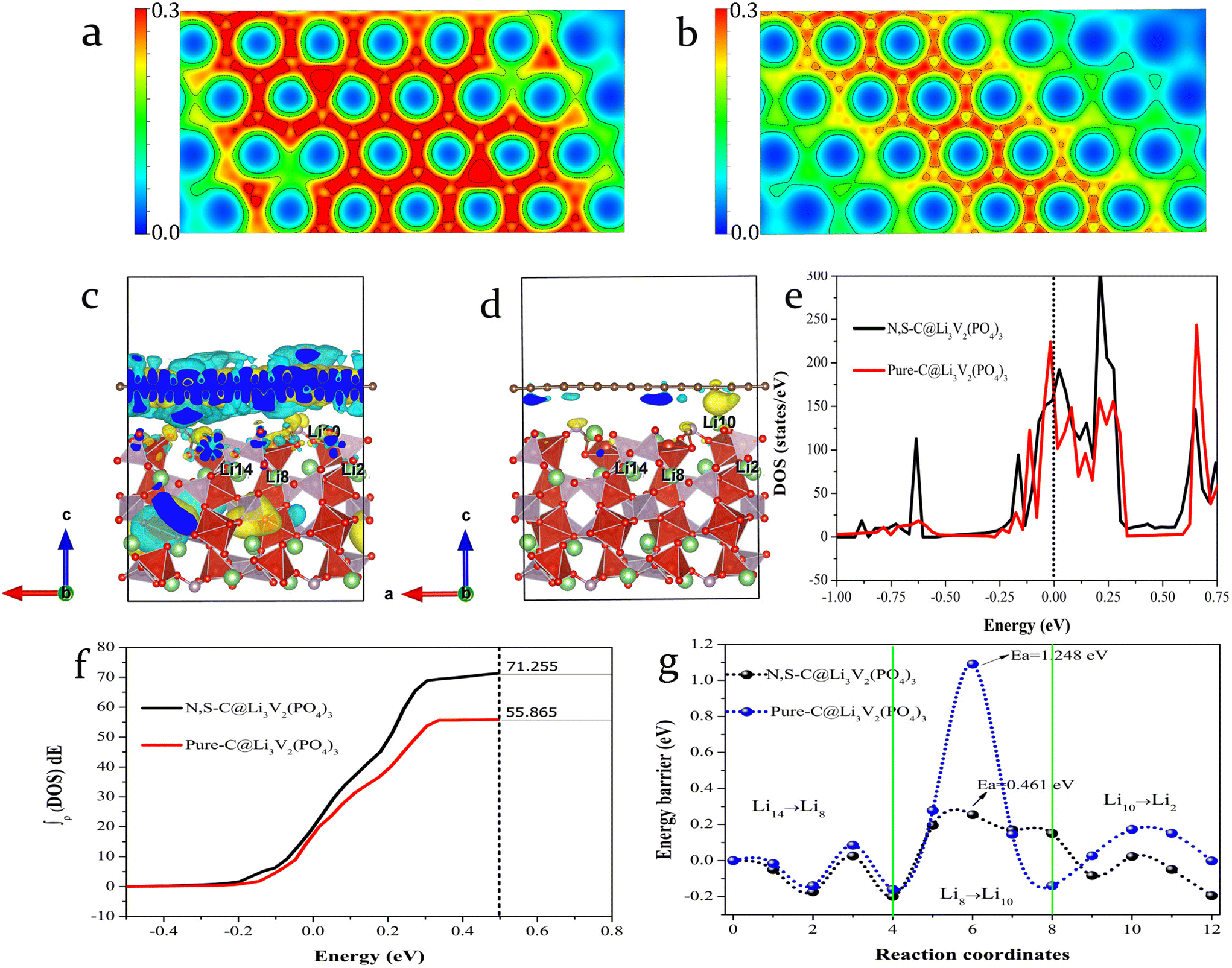 | ||
| Fig. 1 Charge density distribution of (a) N,S-co-doped carbon and (b) pristine carbon. The differential charge density of the carbon-coated LVP structures: (c) N,S-co-doped carbon-coated and (d) pristine carbon-coated structures. Yellow and grey-blue represent density excess and depletion, respectively. The value of the isosurface was set to 0.00066 e Å−3. (e) TDOS of the two coated samples predicted by non-spin polarization calculation. The vertical dashed line indicates the Fermi level, which is set to zero. The corresponding ITDOS from Ef −0.5 to Ef +0.5 eV are given in (f). (g) Potential energy curves for Li+ migration along the a-axis. | ||
Significant charge transfer occurs at the interface between the N,S-co-doped carbon and LVP (Fig. 1c), while the undoped sample shows only minimal charge transfer (Fig. 1d). The integral total densities of states (ITDOS) obtained by integrating the total density of states (TDOS) from Ef −0.5 to Ef +0.5 eV, reveal that the electron count near the Fermi level is 71.255 for the N,S-co-doped sample and 55.865 for the pristine carbon-coated LVP sample (Fig. 1e and f). This significant increase in electron count near the Fermi level suggests enhanced electrical conductivity at the LVP and coated carbon layer interface after N,S-co-doping.33 It is well known that Li+ migration is an important factor affecting the rate capabilities of LIBs. Thus, we further investigated the energy barrier to Li+ migration along the a-axis, since it is the main migration direction in LVP, according to previous literature reports.28 The energy barriers to Li+ migration in the N,S-co-doped carbon layer and pristine carbon-coated LVP were calculated to be 0.461 and 1.248 eV, respectively (Fig. 1g), indicating that Li+ migrate more readily, due to the N,S-co-doping carbon layer coating effect.
Fig. 2b and Fig. S4† display the XRD patterns of the synthesized samples. The coated LVP samples exhibit the same diffraction patterns, indicating minimal structural changes after nitrogen and sulfur doping. The diffraction peaks of all the LVP samples can be well indexed to standard monoclinic LVP (PDF# 01-072-7074, space group: P21/n)34 without any other impurities. Besides, no diffraction peaks of carbon were detected in Fig. 2b, which could be due to its amorphous structure or very small nanocrystals. The major diffraction peaks are sharp and clear, whereas the diffraction peaks of the LVP@C-NS12 sample are weaker than those of the other samples, suggesting its relatively low crystallinity that may show a relatively small particle size distribution with better lithium ion kinetics. Further structural information about the coated carbon layer could be obtained from the Raman spectra (Fig. 2c). A small band appearing at 1000 cm−1 is attributed to the symmetric stretching mode of the PO43− anion.35 A typical D band resulting from the sp3-hybridized disordered carbon atoms and the G band from sp2-hybridized graphitic carbon atoms could be recorded at roughly 1350 cm−1 and 1580 cm−1, respectively.36 The relative intensity ratio between the D and G bands (ID/IG) can be used to assess the contents of the defect and disorder level in carbon materials.37 The values are 1.032, 1.041, 1.022 and 0.995 for LVP@C-NS11, LVP@C-NS12 LVP@C-NS21 and LVP@C-PURE, respectively. Generally, an increased ID/IG ratio indicates a higher defect density in the surface carbon-coated layer.38 Notably, all the LVP@NSC samples exhibit higher ID/IG values than LVP@C-PURE, suggesting that the co-doped carbon layers may introduce more electrochemically active sites for Li+ migration and storage, resulting in better electrochemical properties for the LVP cathode.39,40
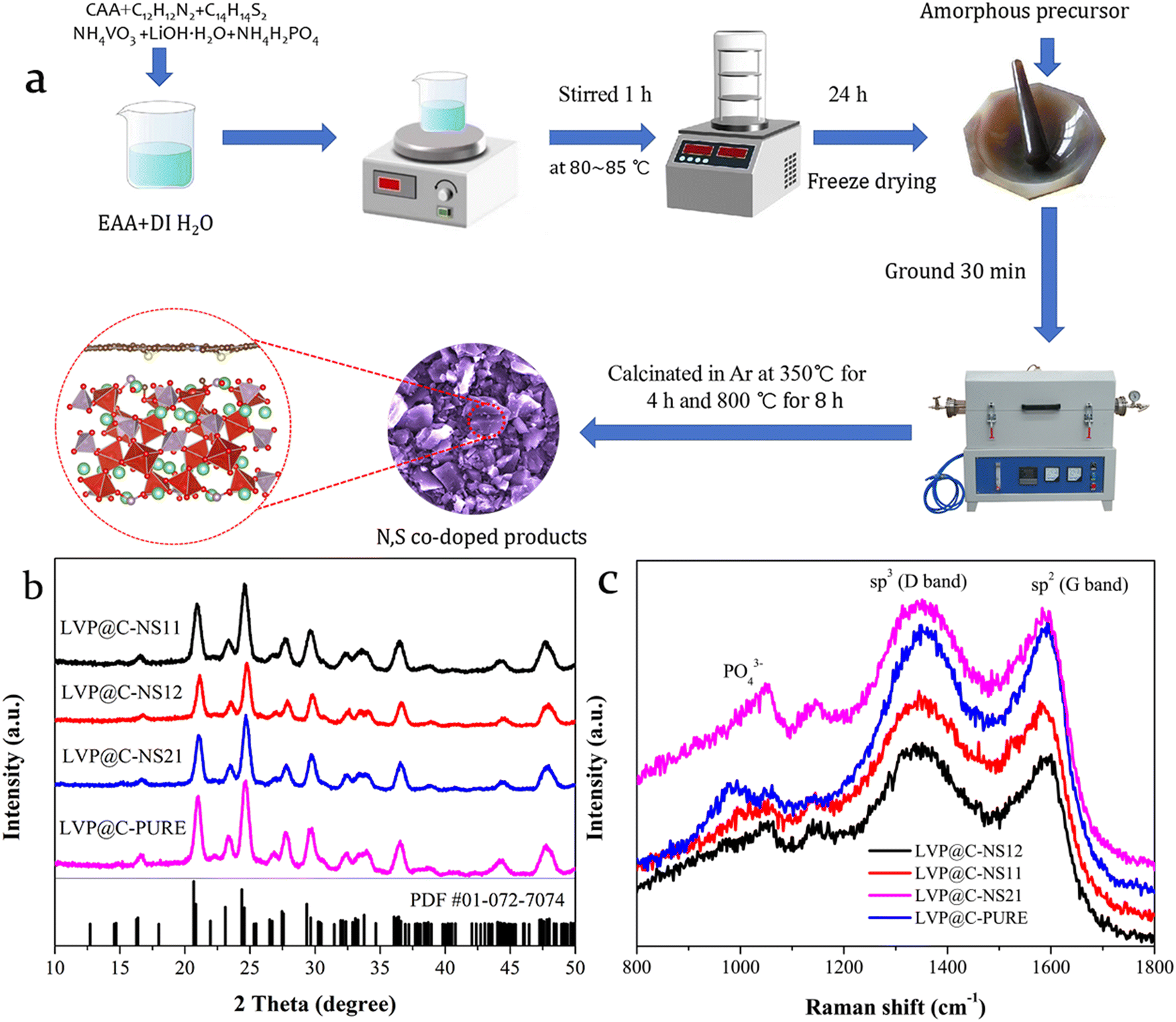 | ||
| Fig. 2 (a) Graphical illustration of the fabrication process of LVP@NSC. (b) XRD patterns and (c) Raman spectra of LVP@C-PURE and various N:S-doped LVP@C samples. | ||
In order to confirm the effects of N and S doped carbon on the morphology of LVP, LVP@C-NS12 and LVP@C composites were investigated using SEM. As shown in Fig. 3a and b, some aggregation with irregular shaped morphology can be observed for both samples, since the relatively long high-temperature sintering process coarsens the grains.41 In Fig. 3c, the TEM image reveals that LVP@C-NS12 consists of many irregularly shaped LVP nanograins with sizes in the range from 7 to 200 nm embedded in the nitrogen and sulfur co-doped porous carbon matrix. This speculation can be confirmed by high-resolution TEM (HRTEM), where the observed interplanar distance is about 2.058 Å, which agrees well with the (320) crystalline plane of monoclinic LVP (Fig. 3c, inset). To further reveal the nature of the N,S-co-doped carbon layer and the elemental distribution of LVP particles, we took typical LVP@C-NS12 as an example to conduct elemental analysis by energy-dispersive spectroscopy (EDS), as shown in Fig. 3d. The N, S and C elements share similar and homogenous distributions, manifesting a uniform doping of nitrogen and sulfur in carbon layers, with efficient hybridization on the LVP particle surfaces. Moreover, as the bright and dark spots correspond to the LVP crystal phase and the grain boundary, respectively, the homologous distribution of elements of V, P and O implies that the LVP particles are not single crystalline but consist of many nanoparticles. Additionally, the precise carbon percentages of LVP@C-NS12 were evaluated by organic elemental analysis (OEA). The carbon content was 6.22% and the presence of nitrogen and sulfur was also detected, and their contents were 0.744% and 0.561%, respectively.
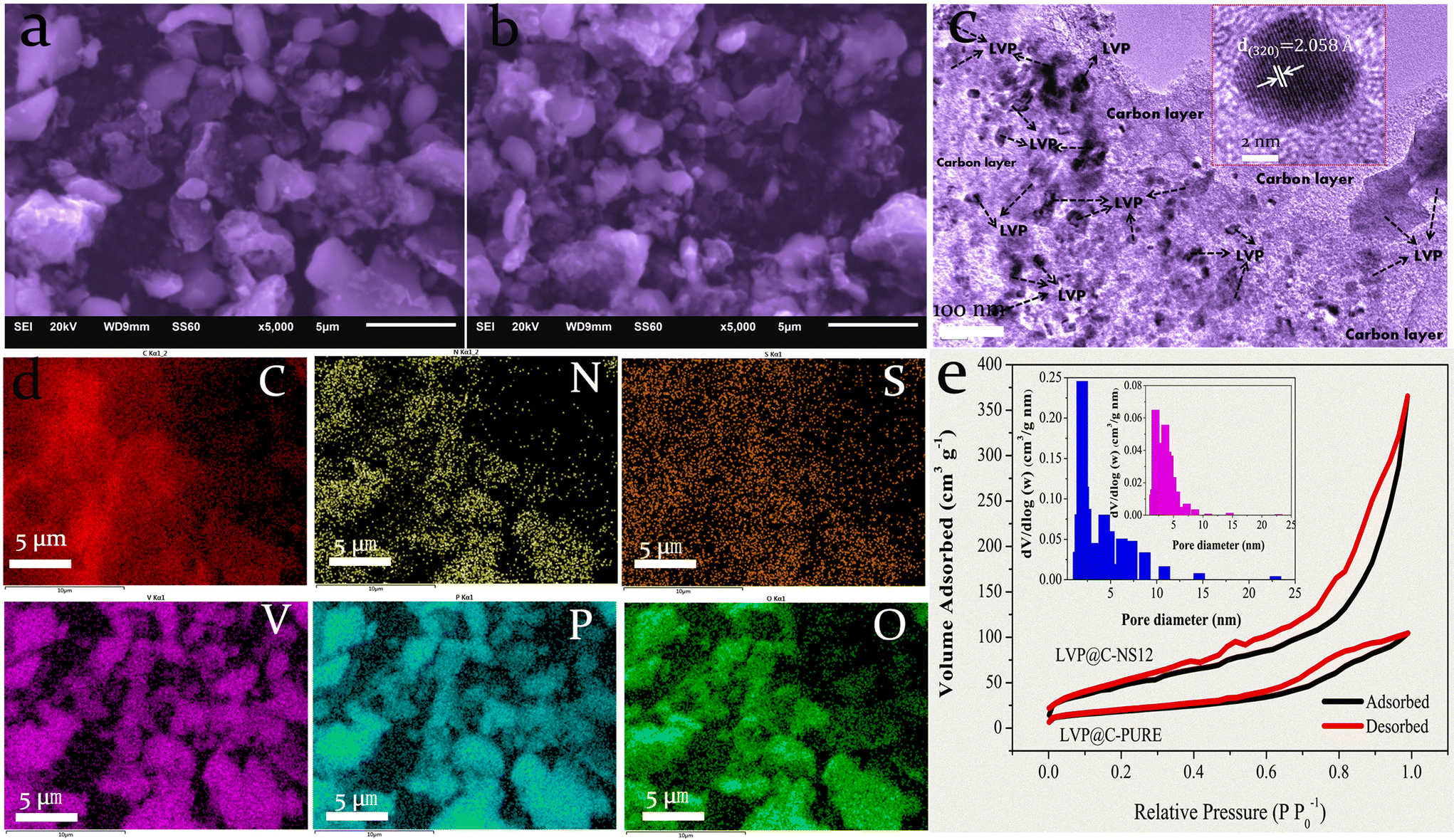 | ||
| Fig. 3 SEM characterization of (a) typical LVP@C-NS12 and (b) LVP@C-PURE samples. (c) TEM image of LVP@C-NS12; the inset shows the corresponding HRTEM image. (d) Elemental mappings of LVP@C-NS12, which were recorded from (a). (e) Nitrogen adsorption–desorption isotherms and the corresponding BJH pore size distribution (inset). | ||
The pore size distribution of the LVP@C-NS12 and LVP@C-PURE samples was characterized using the Brunauer–Emmett–Teller (BET) method. Both samples exhibit nitrogen adsorption–desorption isotherms resembling type IV, with noticeable hysteresis loops at medium and high pressures (P/P0 are 0.3–1 and 0.6–1, respectively), indicating the presence of mesopores. According to the Barrett–Joyner–Halenda (BJH) method, the pore size distribution of the LVP@C-NS12 sample mesopores is narrow, and the size range is mainly concentrated at 2–3 nm, while the pore size distribution of the LVP@C-PURE sample is relatively broad, with a relatively concentrated distribution in the range of 2–6 nm, but the number of mesopores in the sample is much lower than that of the LVP@C-NS12 sample. This may be due to the co-doping of the foreign atoms N and S in the LVP@C-NS12 sample, which introduces more defects into the carbon layer, which is consistent with previous Raman spectroscopy results.
The surface chemical composition and electronic state of the LVP@C-NS12 sample were further explored by XPS characterization. The existence of the C, N, S, V, Li, P and O elements is found in Fig. 4a, revealing that the N and S atoms have been successfully doped into the carbon matrix, in accordance with the EDS results. Fig. 4b shows the N 1s XPS spectra, which reveal the formation of graphitic, pyrrolic and pyridinic nitrogen structures doped in the carbon matrix, with binding energies located at 401.40, 400.35 and 398.44 eV, respectively.42 Additionally, as shown in Fig. 4c, two peaks appeared at 164.02 and 165.24 eV, which are assigned to S 2p3/2 and S 2p1/2 of –C–S–C– covalent bonds, respectively, indicating the effective integration of S into the carbon framework, while the last fitted peak at 168.70 eV could be attributed to oxidized sulfur species (–C–SO3–C– or –C–SO4–C–).18,43–45
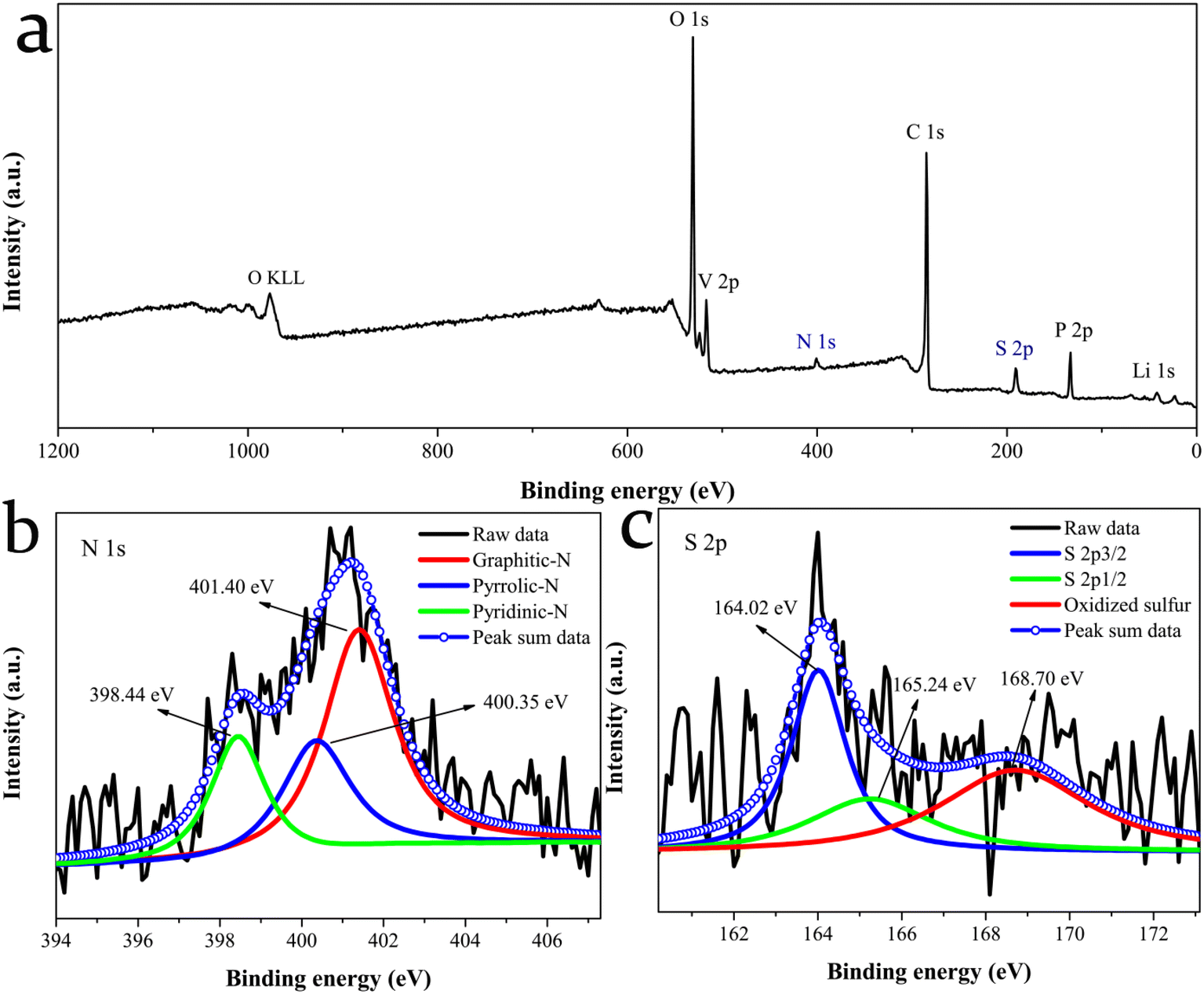 | ||
| Fig. 4 (a) XPS survey spectrum. (b) N 1s and (c) S 2p spectra of the LVP@C-NS12 sample. | ||
The electrochemical performance of the prepared samples for LIBs was evaluated using coin-type half-cells, with the as-prepared samples as the working electrode and Li foil as both the counter and reference electrodes. Galvanostatic charge–discharge (GCD) measurements were conducted using a LAND battery test system. Fig. 5a–d present the representative GCD voltage curves of the as-prepared electrodes at various current rates ranging from 0.5 to 20C (1C = 133 mA h g−1) within the voltage window 3.0–4.3 V (vs. Li+/Li), each sustained for 5 cycles. All the electrodes exhibit three charge and discharge plateaus, which are three typical characteristics of LVP-based materials, corresponding to the two phase transitions during the GCD process (as detailed in the CV analysis below). With the increasing current densities, the LVP@C-PURE electrode not only shows an obviously larger potential drop with the plateaus gradually becoming indistinct, but also delivers much lower Li+ storage capabilities, attributed to the stronger electrode polarization compared with that of LVP@NSC electrodes. Fig. 5e summarizes the rate capabilities of the electrodes, highlighting that the LVP@C-NS12 electrode delivers the highest reversible capacities at all tested current densities. For example, the electrode can deliver specific capacities of 122.61, 124.40 and 107.85 mA h g−1 at current rates of 0.5, 2 and 20C, respectively. The initial relatively low capacity and coulombic efficiency could be ascribed to the incomplete activation process.46 In contrast, the LVP@C-PURE electrode can only achieve 110.81, 109.89 and 84.25 mA h g−1 at the corresponding current rates. Although its initial capacity is higher than that of the LVP@C-NS21 electrode, greater fading behavior can be observed with increased current density. Fig. 5f and g demonstrate that the LVP@C-NS12 electrode exhibits excellent long-term cycling stability at both 1 C and 20 C compared to all the other electrodes. After 100 cycles at 1 C and 500 cycles at 20 C, stable reversible capacities of 117.42 and 100.22 mA h g−1 can be maintained, corresponding to capacity retentions of 94.83% and 86.1%, respectively. Therefore, it is believed that the LVP@NSC electrode with a moderate molar ratio of N and S element modification is capable of decreasing the polarization and then delivering stable charge and discharge under even a high current rate. As a comparison, the setup without a carbon-coated LVP-PURE electrode shows poor rate performance and extremely low cycle discharge specific capacity (Fig. S5†). This indicates that carbon coating plays an extremely key role in improving the electrochemical lithium storage performance of LVP.
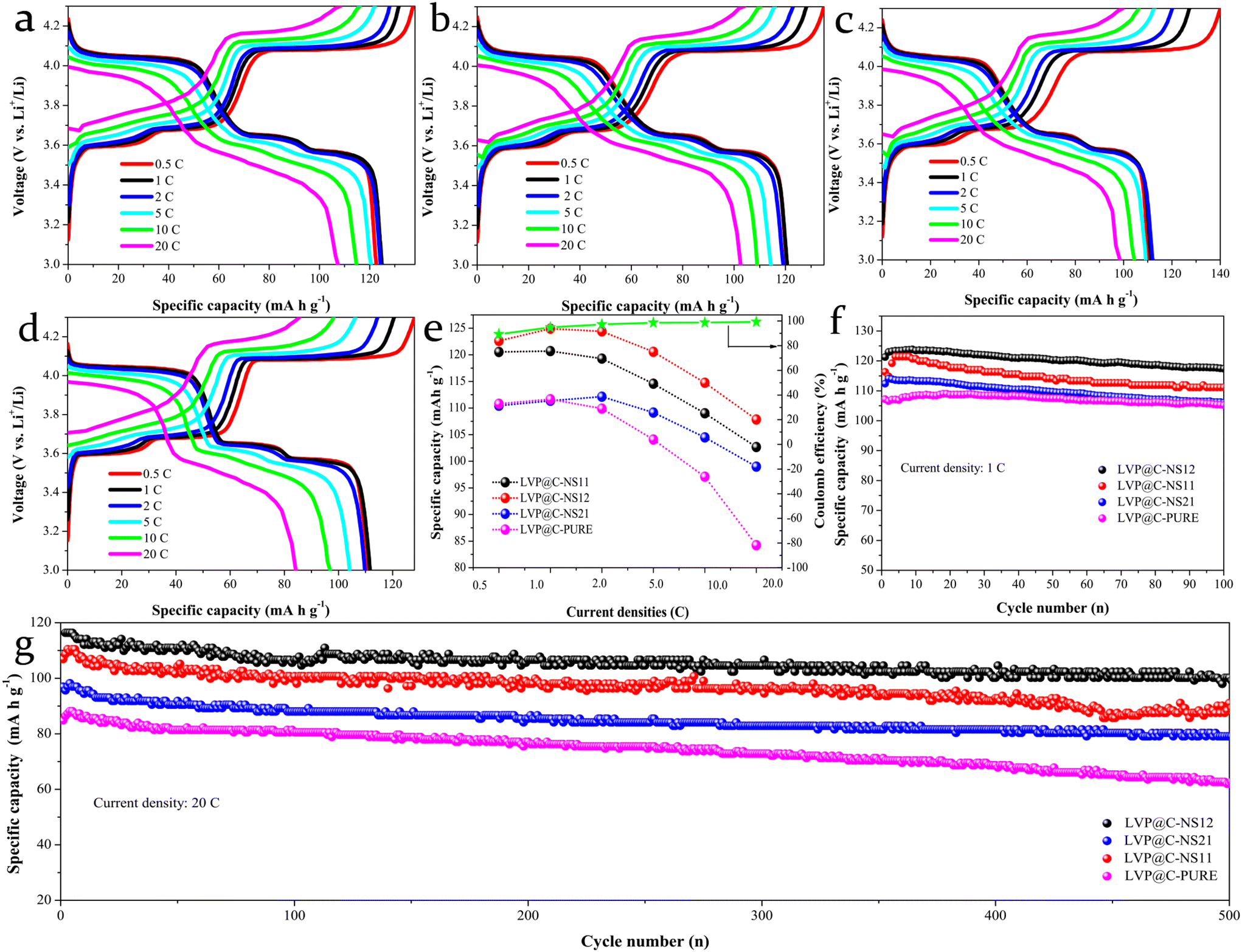 | ||
| Fig. 5 GCD curves of the as-prepared electrodes at various current densities: (a) LVP@C-NS12, (b) LVP@C-NS11, (c) LVP@C-NS21 and (d) LVP@C-PURE. (e) In a comparison of various rate capabilities, the coulombic efficiency of LVP@C-NS12 was also demonstrated. Capacity retentions of the as-obtained electrodes at: (f) 1 C and (g) 20 C with ultralong cycle life and ultrahigh capacity retention. The GCD curves were tested in the voltage range 3.0–4.3 V (1 C = 133 mA h g−1). | ||
To further investigate the fundamental electrochemical behavior of the as-prepared samples for LIBs, and reveal the reason for the disparity in electrochemical performances of each electrode, CV and EIS measurements were conducted. Fig. 6a shows the 3rd cycled CV curves of LVP@C-PURE and various N:S doped LVP@C electrodes at a scan rate of 0.2 mV s−1 in the voltage window between 3.0 and 4.3 V (vs. Li+/Li). Three obvious redox couple peaks relating to a series of reversible phase transitions appear in all the curves. The corresponding electrochemical reactions can be expressed by the following equations:47,48
| Li3V2(PO4)3 ↔ Li2.5V2(PO4)3 + 0.5Li+ + 0.5e− | (1) |
| Li2.5V2(PO4)3 ↔ Li2V2(PO4)3 + 0.5Li+ + 0.5e− | (2) |
| Li2V2(PO4)3 ↔ LiV2(PO4)3 + Li+ + e−. | (3) |
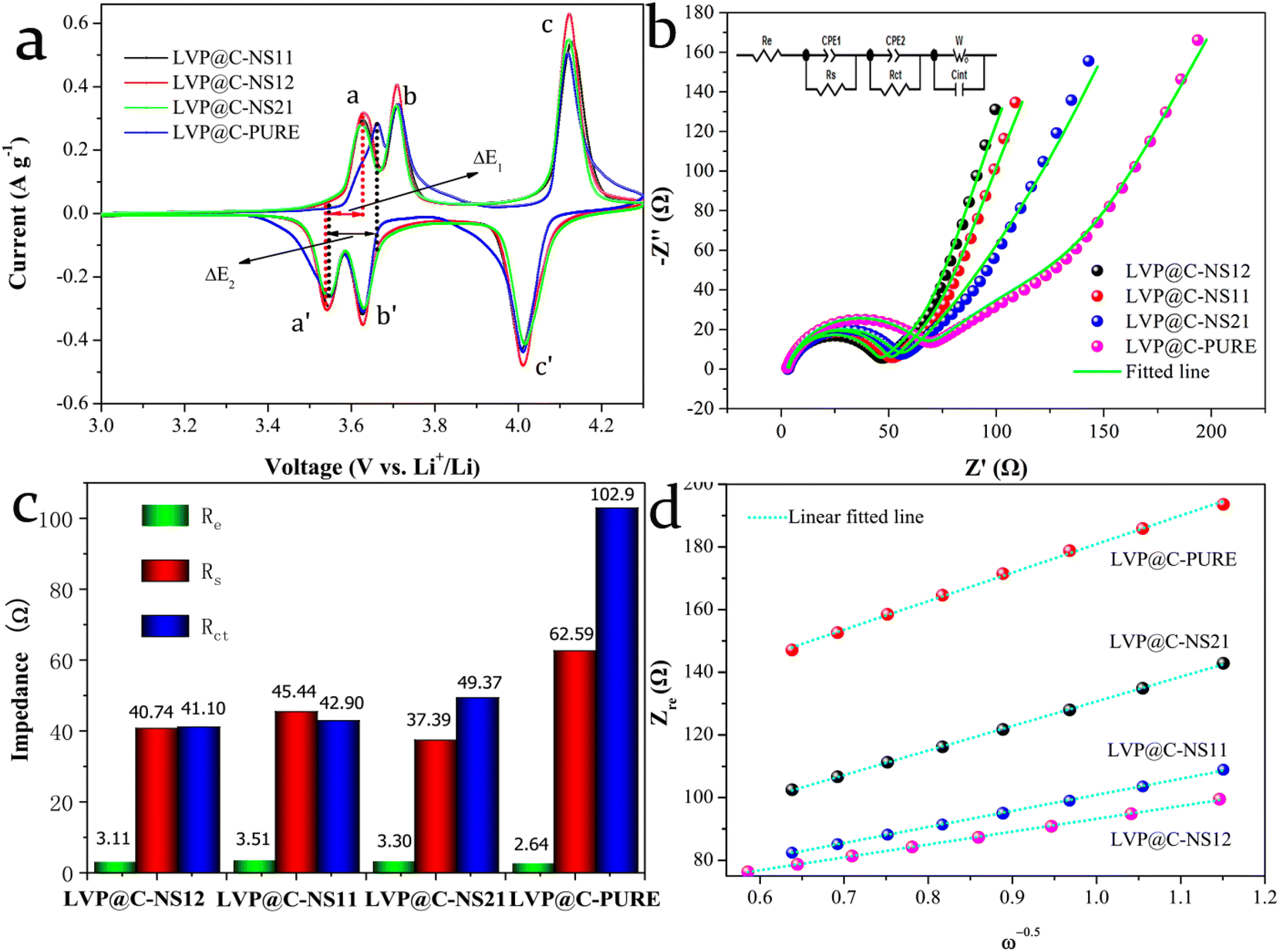 | ||
| Fig. 6 (a) CV curves of LVP@C-PURE and various N:S doped LVP@C electrodes at a scan rate of 0.2 mV s−1. (b) Nyquist plots of LVP@C and various N:S doped LVP@C samples after 3 CV cycles (3.0–4.3 V); inset is the equivalent circuit corresponding to the impedance spectra. (c) The fitted EIS parameters of LVP@C-PURE and various N:S doped LVP@C electrodes. (d) Relationship between Zre and ω−1/2 in the low-frequency region. | ||
The two oxidation peaks, labeled a and b, observed during the charge process, correspond to the delithiation of the first Li+, with the vanadium ion from V3+ oxidized to V3+/V4+ (eqn (1) and (2)). Another sharp oxidation peak, which is labeled c, results from the removal of the second Li+ with the vanadium ion from V3+/V4+ completely oxidized to V4+ (eqn (3)). In the following discharge process, the first sharp reduction peak, which is labeled c′, originates from the initial reintercalation of Li+ into LiV2(PO4)3, while the other reduction peaks b′ and a′ correspond to reinsertion of the second lithium ion. It is noticeable that, compared to the LVP@C-PURE electrode, all the N,S-co-doped carbon-coated electrodes exhibit significantly lower polarization. For instance, in the first pair of redox peaks a and a′, ΔE2 = 118 mV compared to ΔE1 = 86 mV. This reduced polarization in N,S-co-doped electrodes is attributed to enhanced reaction kinetics,49 which indicates improved electrical conductivity at the electrode/electrolyte interface after nitrogen and sulfur co-modification. Additionally, the sharpest redox peaks, along with the largest integrated area, means that the most favorable electrochemical specific capacity could be obtained with the LVP@C-NS12 electrode. These results are very consistent with our GCD measurements above.
Fig. 6b shows Nyquist plots at the discharge state, with a potential of 3.0 V after 3 CV cycles at a scan rate of 0.2 mV s−1. The inset is the equivalent circuit model to fit the Nyquist plots. The Nyquist plots can be divided into three parts: (a) one semicircle over high to medium frequencies is related to the resistance of the electrolyte (Re) and the contact resistance (Rs); (b) one depressed semicircle over medium to low frequencies is associated with the charge transfer resistance (Rct) at the electrode/electrolyte interface; (c) one linear region in the low-frequency range is related to the Warburg impedance (Zw), which could reveal the Li+ migration coefficient within the electrode. Note that Rs and Rct are typically connected in parallel with the constant phase element (CPE), which accounts for the non-ideal behavior of the double-layer capacitance in the equivalent circuit diagram. The Warburg impedance (Zw), however, can be connected either in parallel or in series with the Li+ insertion/extraction capacitance (Cint), which results from the consumption and accumulation of Li+ in the active material, depending on the specific environment of the corresponding system. The Li+ migration coefficients (DLi+, cm2 s−1) for the LVP electrodes can be estimated from the slope of the inclined line in the low-frequency region using the following equation:50
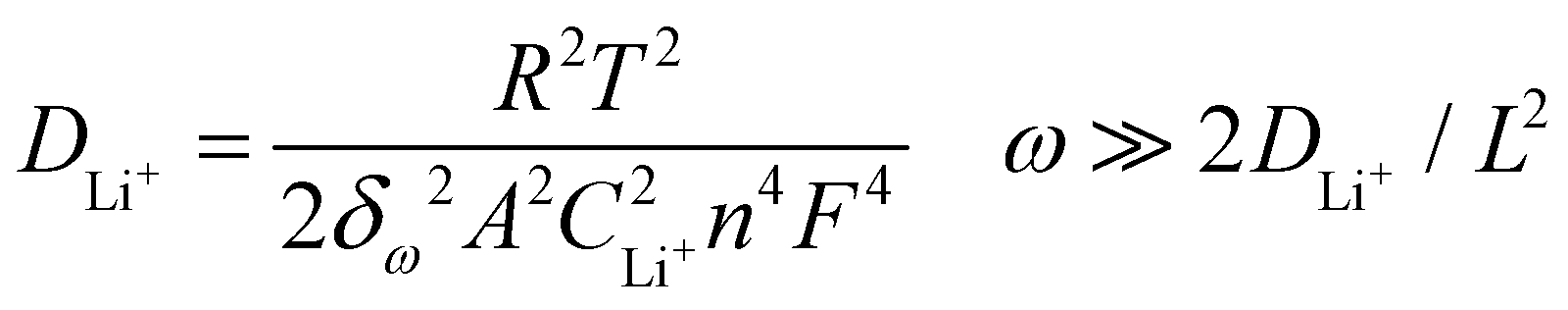 | (4) |
485.337 C mol−1) and CLi+ is the molar concentration of Li+ in the corresponding electrode material (1.49 × 10−2 mol cm−3), which is calculated from the volume of LVP (889.926 Å3).34n is the number of electrons per formula unit during the redox reaction (here, equal to 2), ω represents the angular frequency in the low-frequency region (ω = 2πf), L represents the thickness of the electrode and δω is the Warburg coefficient, which can be obtained from the correlation between impedance and frequency, according to the formula:| Zre = Re + Rct + δωω−0.5. | (5) |
The fitted EIS parameters are given in Fig. 6c. All the electrodes show almost the same Re value, however, as for Rs and Rct, all the doped carbon-coated electrodes show smaller values. Among these, the LVP@C-NSC12 electrode exhibits the most favorable impedance characteristics. Although the Rs of the LVP@C-NSC21 electrode is only 37.39 Ω, its larger Rct (49.37 Ω) inhibits charge transfer, resulting in a lower exchange current density with poor electrode reaction reversibility.41 The relationship between Zre and ω−1/2 in the low-frequency region is shown in Fig. 6d. The slopes of the fitted lines give the values of δω, which were estimated to be 41.01, 46.87, 71.708 and 83.225 Ω cm2 s−0.5 for LVP@C-NSC12, LVP@C-NSC11, LVP@C-NSC21 and LVP@C-PURE, respectively. Based on these δω values, their Li+ migration coefficients (DLi+) can be further calculated to be 3.355 × 10−15, 2.568 × 10−15, 1.097 × 10−15 and 8.145 × 10−15 cm2 s−1, respectively. From the above results, the LVP@C-NS12 electrode demonstrates the best Li+ migration capability followed by the LVP@C-NS11, LVP@C-NS21 and LVP@C-PURE electrodes in sequence, well in line with our DFT theoretical calculation analysis in Fig. 1 and electrochemical measurement results in Fig. 5. The superior Li+ migration capability of the LVP@C-NS12 electrode could be attributed to its excellent electrode reaction reversibility, stemming from its lowest Rct value, which promotes faster charge compensation and enhances Li+ migration rates. Moreover, due to its low crystallinity, the smaller particle size of LVP@C-NS12 can substantially shorten Li+ transport distance in the crystal structure. Therefore, these combined factors contribute to its outstanding electrochemical performance as a cathode material for LIBs.
The electrochemical properties of the LVP@C-NS12 electrode were also investigated in the potential range from 3.0 to 4.8 V with the LVP@C-PURE electrode for comparison. As presented in Fig. 7a, the charging curves of the LVP@C-NS12 electrode in the voltage range 3–4.8 V show an additional voltage platform around 4.60 V, but no corresponding voltage platform could be observed in the subsequent discharging plot, which can be seen clearly from the corresponding differential capacity plot of the LVP@C-NS12 electrode at 0.5 C (Fig. 7c, inset). This phenomenon can be attributed to the solid-solution reaction for the third Li+ deintercalation, since for the third Li+ deintercalation phase only average vanadium valence (+4.5) is observed, indicating that the mixed V4+/V5+ state exists and there is no charge ordering. Such charge disorder will result in Li+ reinsertion disorder during discharging. Therefore, solid-solution behavior characterized by an S-shaped curve is observed during the discharge process.51 The corresponding electrochemical reactions can be further expressed by the equation:47,52
| LiV2(PO4)3 ↔ V2(PO4)3 + Li+ + e−. | (6) |
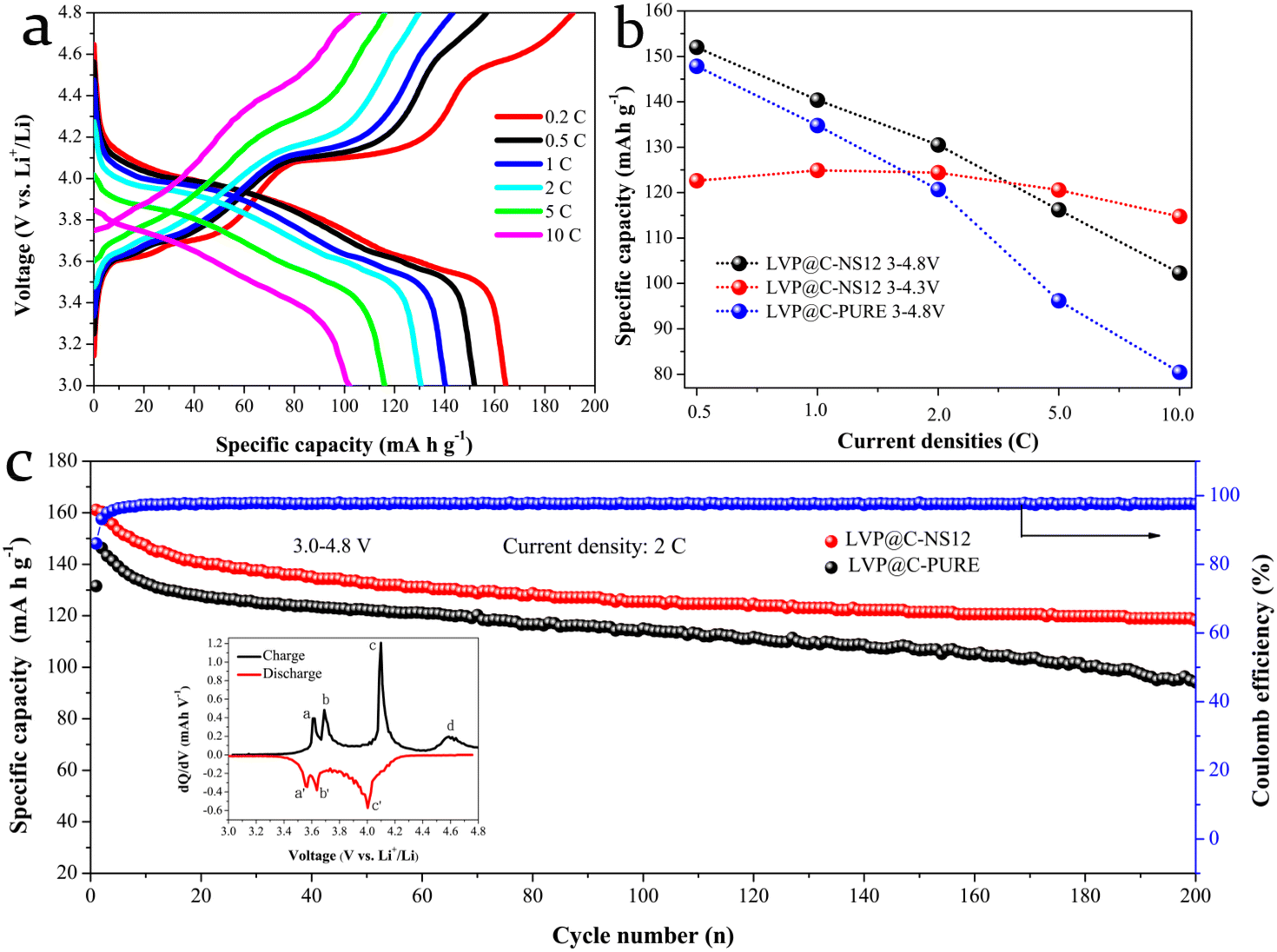 | ||
| Fig. 7 (a) DCG curves of the LVP@C-NS12 electrode at various current densities within the voltage range from 3 to 4.8 V. (b) Comparison of the rate capabilities of the LVP@C-PURE and LVP@C-NS12 electrodes, and the LVP@C-NS12 electrode in different potential windows. (c) Capacity retentions of the LVP@C-PURE and LVP@C-NS12 electrode at 2 C in the voltage range from 3 to 4.8 V (1 C = 197 mA h g−1). The inset is the differential capacity plot of LVP@C-NS12 at 0.2 C current density. | ||
This equation is associated with the valence state of the vanadium ion changing between V4+ and V5+, but the Li+ reinsertion process in this stage has been recognized as kinetically difficult due to the limited ionic and electronic conductivity of the V2(PO4)3 framework.17,53 The rate capabilities are compared in Fig. 7b. The LVP@C-NS12 electrode shows better rate performance than the LVP@C-PURE electrode. Specific capacities of 164.38, 151.98, 140.37, 130.52, 116.18 and 102.29 mA h g−1 can be obtained at current densities of 0.2, 0.5, 1, 2, 5 and 10 C, respectively. However, its high capacity performance is gradually lost after 5 C, compared to that of charging and discharging at the upper cutoff voltage of 4.3 V vs. Li+/Li. This can be attributed to the decomposition of the electrolyte at high voltage and severe volumetric changes after the removal of all three Li+ to form the V2(PO4)3 configuration.34 As illustrated in Fig. 7c, the LVP@C-NS12 electrode also demonstrates superior cycling behavior at 2 C, with nearly 100% coulombic efficiency after several activation processes in the initial cycles. The specific capacity of 118.22 mA h g−1 can be maintained after 200 cycles (with capacity retention of 73.37%).
A lithium-ion full battery with commercially available biomass hard carbon directly coated on Cu foil as an anode and LVP@C-NS12 coated on carbon-coated Al foil as a cathode was assembled to evaluate the possibility of its practical application. Surprisingly, it could achieve ignorable reversible discharge capacity. This may be attributed to the limited lithium source in the full cell being substantially consumed. On the one hand, the formation of the SEI film needs to consume Li+. On the other hand, due to the special disordered structure of biomass hard carbon, the Li+ cannot be reversibly released after insertion into it. Therefore, we initially used lithium foil as the external lithium source for electrochemical pre-lithiation for the biomass hard carbon at low current density. This activation process enabled the biomass hard carbon to adopt a more ordered structure, while the formation of a relatively stable SEI film reduced subsequent Li+ loss in the fully assembled battery.
Fig. 8a shows an optical photograph of an LED bulb powered by the coin-type full battery. A schematic diagram of the full battery structure is given in Fig. 8b. The prepared LVP@C-NS12 was used as the cathode, pre-lithiated biomass hard carbon as the anode. The electrolyte and separator adopted here are the same as those of the half-cell. The assembled full battery has an open circuit voltage of around 2.6–2.8 V. Thus, we employed a galvanostatic mode to charge to 4.3 V at a current density of 0.2 C, followed by an additional 1-minute charge at constant voltage (4.3 V). Fig. 8c and d show the CV curves for the full cell in the potential window of 3–4.3 V and the GCD curves for the 1st, 2nd and 10th cycles at 0.2 C, respectively. Three distinct pairs of redox peaks, labeled a and a′, b and b′, c and c′, are observed in the charge and discharge process, corresponding to three pairs of voltage platforms, which are located at around 3.2 and 3.1 V, 3.4 and 3.3 V, 3.9 and 3.8 V in the GCD curves, respectively. These are the typical characteristic peaks of the LVP electrodes. The lower platforms are due to the matching of the hard carbon anode. The discharge curves of the initial three cycles are basically overlapped, indicating that it has good electrochemical reversibility. Fig. 8e presents the GCD curves at 0.2, 0.5, 1, 2, 5 and 10 C. At the corresponding current densities, the full battery can achieve discharge capacities of 107.17, 90.57, 65.85, 53.34 and 43.43 mA h g−1. When the current density returns to 0.2 C, a discharge capacity of 105.72 mA h g−1 can still be obtained (Fig. 8f). The good capacity reproducibility indicates that it has a good and large rate capability. Fig. 8g depicts the cycling stability test conducted at a current density of 2 C. The lower coulombic efficiency in the first few cycles is similar to that in the hard carbon half-cell test (Fig. S6†), which could be ascribed to the electrode activation process taking place. The initial specific discharge capacities of the first and second cycles were 63.87 and 53.04 mA h g−1, respectively, and remained 43.61 mA h g−1 after 80 cycles. The capacity retention is 82.2%, compared with the reversible capacity of the second cycle, suggesting its better cycle stability. However, it is worth noting that, although the discharge specific capacity (63.87 mA h g−1) obtained in the first cycle of the cycling performance test of the full battery at a current density of 2 C is basically equivalent to the discharge specific capacity (65.85 mA h g−1) obtained at the same current density in the rate test, the discharge specific capacity in the second cycle significantly decreases. Additionally, the overall electrochemical lithium storage performance of the full battery is not as good as that of the half battery. The reason for this may be that, as mentioned above, the commercial biomass hard carbon used here makes it relatively difficult for insertion of Li+ in full battery mode: that is, the reversibility of Li+ deintercalation in the electrode is poor. In the stability cycle test, it has not been activated by a small current as in the rate test, resulting in relatively low discharge specific capacity in the subsequent cycle test at 2 C current density. The unstable coulombic efficiency also leads to a certain degree of fluctuation in specific capacity, which has also been also observed in the literature.54,55
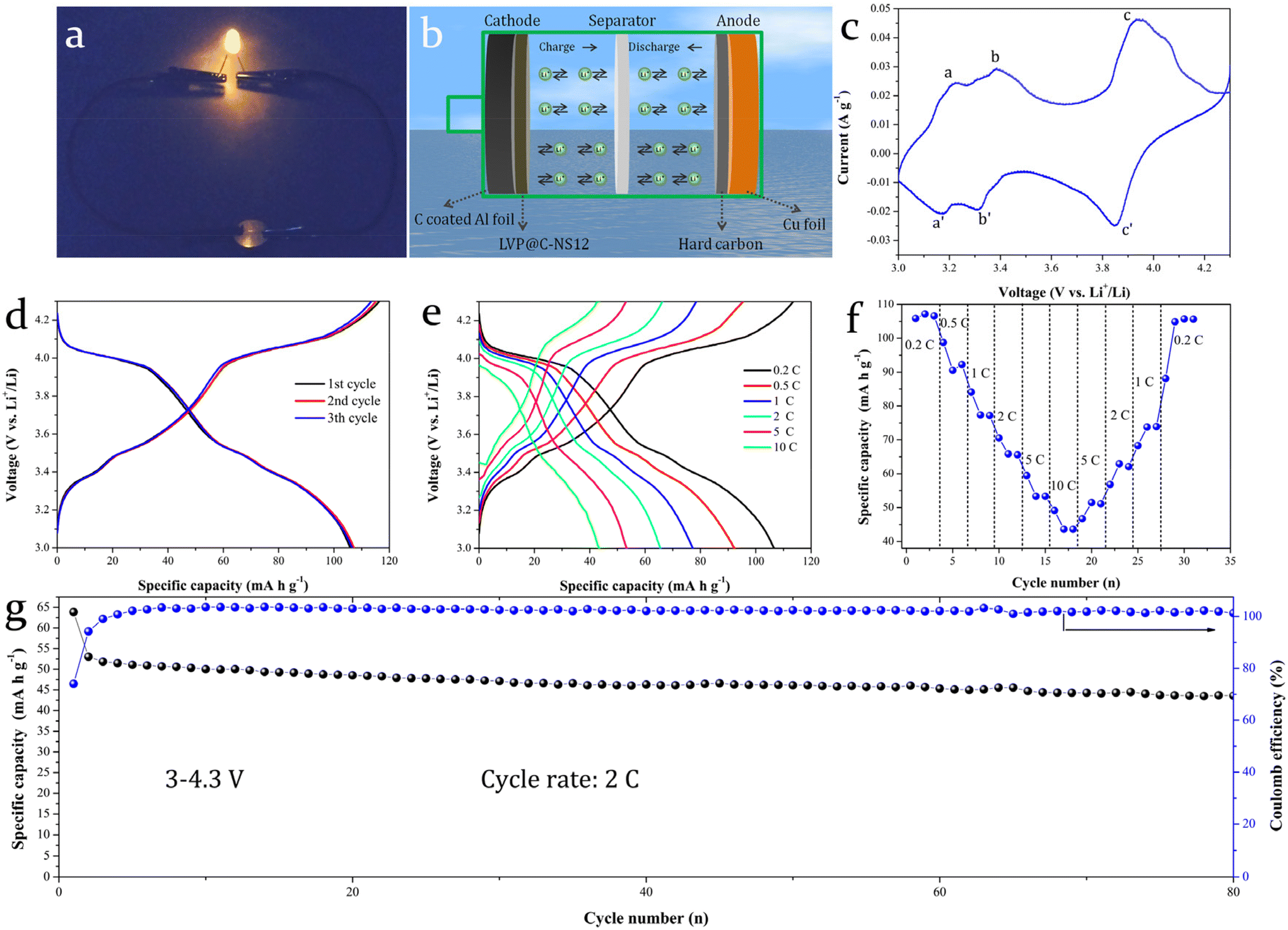 | ||
| Fig. 8 (a) Optical photograph of LED bulb powered by a full battery. (b) Schematic diagram of the full battery structure. Electrochemical performance test of the full battery: (c) CV curve at a scan rate of 0.05 mV s−1 where the voltage window is 3–4.3 V (vs. Li+/Li). (d) GCD curves at 1st, 2nd and 10th cycles at a current density of 0.2 C (1 C = 133 mA h g−1). (e) GCD curves under various current densities. (f) Rate properties from 0.2 C to 10 C and then returning to 0.2 C. (g) Cycle performance at a current density of 2 C with the corresponding coulombic efficiency. | ||
As a result, for the first time, the DFT calculation method was employed in our work to predict the feasibility and explain the possible intrinsic mechanism of N,S-co-doped C and further encapsulated LVP at the atomic level. Remarkably, the specific capacity and cycling capabilities of the designed LVP@C-NS12 electrode are comparable to or even higher than those of many state-of-the-art reports in recent years, as listed in Table S2 (ESI).† By combining our experimental and calculation results, the enhanced electrochemical behavior of LVP@C-NS12 could be ascribed to the following aspects: (i) LVP particles with decreased size effectively shorten the Li+ transport length and lower its energy barriers. (ii) The increased electron near the Fermi level improves the electrical conductivity of the whole composites. Meanwhile, the increased interface charge transfer between the doped carbon layer and LVP could further reduce the resistance to Li+ passing through the interface and entering into the active sites. (iii) The defective N,S-doped carbon network could increase the probability of contact between the electrode and electrolyte, and accommodate the strain of the volume variation of the LVP crystal during the repeated Li+ uptake and release processes.
4. Conclusions
In summary, the effect of N,S-co-doped carbon and further coated LVP materials as cathodes for LIBs was investigated by joint theoretical calculation and experimental validation. The results reveal that the LVP embedded in an N,S-co-doped carbon matrix shows significantly enhanced electronic conductivity and much lower Li+ migration energy barrier than in the pristine carbon matrix. The designed LVP@-NS12 sample shows the best electrochemical performance. Its smallest charge transfer resistance and highest Li+ migration coefficient were confirmed by electrochemical analysis. This work not only reveals the critical role of the interaction mechanism between N,S-co-doped carbon and LVP for the first time, but also sheds light on the design and modification of electrode materials for more advanced potential for high energy density LIB applications.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the financial support from the Natural Science Foundation of Hunan Province of China (2021JJ30538); Scientific Research Foundation of Hunan Provincial Education Department (23C0301) and Key Projects of Applied Characteristic Disciplines of “Double First-Class” in Materials Science & Engineering of Huaihua University (19CKA006).References
- L. Chen, Z. Liu, W. Yang, S. Wu, Y. Li, Y. Zhang, L. Zeng and H. Fan, Micro-mesoporous cobalt phosphosulfide (Co3S4/CoP/NC) nanowires for ultrahigh rate capacity and ultrastable sodium ion battery, J. Colloid Interface Sci., 2024, 666, 416–423 CrossRef CAS PubMed
.
- J. Wang and W. Azam, Natural resource scarcity, fossil fuel energy consumption, and total greenhouse gas emissions in top emitting countries, Geosci. Front., 2024, 15, 101757 CrossRef CAS
- M. H. Hossain, M. A. Chowdhury, N. Hossain, M. A. Islam and M. H. Mobarak, Advances of lithium-ion batteries anode materials-A review, Chem. Eng. J. Adv., 2023, 16, 100569 CrossRef
- C. Zhao, P. Nie, Z. Sun, H. Geng, H. Yu, M. Yu, M. Yang, Z. Luo and X. Wu, SbPS4: A novel anode for high-performance sodium-ion batteries, Chin. Chem. Lett., 2022, 33, 470–474 CrossRef
- S. Wu, Y. Li, L. Chen, Y. Zhang, L. Zeng and H. Fan, Hexapod cobalt phosphosulfide nanorods encapsulating into multiple hetero-atom doped carbon frameworks for advanced sodium/potassium ion battery anodes, Chin. Chem. Lett., 2024, 24, 109796 CrossRef
- X. Li, X. Du and M. D. Xiao, Three–Dimensional Holey Graphene Enwrapped Li3V2(PO4)3/N-Doped Carbon Cathode for High-Rate and Long-Life Li-Ion Batteries, ChemSusChem, 2022, 15, 202201459 CrossRef PubMed
- M. Ding, C. Cheng, Q. Wei, Y. Hu, Y. Yan, K. Dai, J. Mao, J. Guo, L. Zhang and L. Mai, Carbon decorated Li3V2(PO4)3 for high-rate lithium-ion batteries: Electrochemical performance and charge compensation mechanism, J. Energy Chem., 2021, 53, 124–131 CrossRef CAS
- Y. Xia, L. Yu, C. Lu, J. Zhu and W. Zhang, Passion fruit-like structure endows Li3V2(PO4)3@C/CNT composite with superior cyclic stability and rate performance, J. Alloys Compd., 2020, 859, 157806 CrossRef
- B. Dunn, H. Kamath and J. M. Tarascon, Electrical Energy Storage for the Grid: A Battery of Choices, Science, 2011, 334, 928–935 CrossRef CAS PubMed
- H. Liu, Y. Liang, C. Wang, D. Li, X. Yan, C. W. Nan and L. Z. Fan, Priority and prospect of sulfide-based solid-electrolyte membrane, Adv. Mater., 2023, 35, 2206013 CrossRef CAS PubMed
- Z. Huang, P. Luo and H. Zheng, Design of Ti4+doped Li3V2(PO4)3/C fibers for lithium energy storage, Ceram. Int., 2022, 48, 8325–8330 CrossRef CAS
- M. Liang, L. Li, X. Cui, S. Qi, L. Wang, H. Dong, X. Chen, Y. Wang, S. Chen and G. Wang, Ru-and Cl-Codoped Li3V2(PO4)3 with Enhanced Performance for Lithium-Ion Batteries in a Wide Temperature Range, Small, 2022, 18, 2202151 CrossRef CAS PubMed
- C. Sun, S. Rajasekhara, Y. Dong and J. B. Goodenough, Hydrothermal synthesis and electrochemical properties of Li3V2(PO4)3/C-based composites for lithium-ion batteries, ACS Appl. Mater. Interfaces, 2011, 3, 3772–3776 CrossRef CAS PubMed
- Y. S. Hu, X. Ma, P. Guo, F. Jaeger and Z. H. Wang, 3D graphene-encapsulated Li3V2(PO4)3 microspheres as a high-performance cathode material for energy storage, J. Alloys Compd., 2017, 723, 873–879 CrossRef CAS
- J. Yan, H. Fang, X. D. Jia and L. Z. Wang, Copper incorporated in Li3V2(PO4)3/C cathode materials and its effects on high-rate Li-ion batteries, J. Alloys Compd., 2018, 730, 103–109 CrossRef CAS
- M. Choi, I. H. Yoon, I. H. Jo, H. S. Kim, B. S. Jin, Y. M. Lee and W. K. Choi, The effect of SiO2 nanoparticles in Li3V2(PO4)3/graphene as a cathode material for Li-ion Batteries, Mater. Lett., 2015, 160, 206–209 CrossRef CAS
- X. Rui, Q. Yan, M. Skyllas-Kazacos and T. M. Lim, Li3V2(PO4)3 cathode materials for lithium-ion batteries: A review, J. Power Sources, 2014, 258, 19–38 CrossRef CAS
- M. Qin, C. Chen, B. Zhang, J. Yan and J. Qiu, Ultrahigh Pyridinic/Pyrrolic N Enabling N/S Co-Doped Holey Graphene with Accelerated Kinetics for Alkali-Ion Batteries, Adv. Mater., 2024, 2407570 CrossRef CAS PubMed
- Q. Zhou, Q. Chen, W. Xu, F. Wang, X. Du, Y. Zhou, Y. Zhan and M. Jiang, Nitrogen and sulfur co-doped carbonized lignin nanotubes for supercapacitor applications, Chem. Eng. J., 2024, 496, 154126 CrossRef CAS
- X. Zhou, M. Wu, K. Chen, H. Wang, Z. Wen and S. Ci, γ-Fe2O3 decorating N, S co-doped carbon nanosheets as a cathode electrocatalyst for different-scenario fuel cells, Inorg. Chem. Front., 2024, 11, 4625–4637 RSC
- M. Cao, Y. Shu, Q. Bai, C. Li, B. Chen, Y. Shen and H. Uyama, Design of biomass-based N, S co-doped porous carbon via a straightforward post-treatment strategy for enhanced CO2 capture performance, Sci. Total Environ., 2023, 884, 163750 CrossRef CAS PubMed
- H. Schoof, J. Apel, I. Heschel and G. Rau, Control of pore structure and size in freeze-dried collagen sponges, J. Biomed. Mater. Res., 2001, 58, 352–357 CrossRef CAS PubMed
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- Z. J. Ding, L. Zhao, L. M. Suo, Y. Jiao, S. Meng, Y. S. Hu, Z. X. Wang and L. Q. Chen, Towards understanding the effects of carbon and nitrogen-doped carbon coating on the electrochemical performance of Li4Ti5O12 in lithium ion batteries: a combined experimental and theoretical study, Phys. Chem. Chem. Phys., 2011, 13, 15127–15133 RSC
- S. Lee and S. S. Park, Atomistic Simulation Study of Monoclinic Li3V2(PO4)3 as a Cathode Material for Lithium Ion Battery: Structure, Defect Chemistry, Lithium Ion Transport Pathway, and Dynamics, J. Phys. Chem. C, 2012, 116, 25190–25197 CrossRef CAS
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef
- L. Bengtsson, Dipole correction for surface supercell calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12301–12304 CrossRef CAS
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS
- Y. Gao, J. Ma, X. Wang, X. Lu, Y. Bai, Z. Wang and L. Chen, Improved electron/Li-ion transport and oxygen stability of Mo-doped Li2MnO3, J. Mater. Chem. A, 2014, 2, 4811–4818 RSC
- S.-C. Yin, H. Grondey, P. Strobel, M. Anne and L. F. Nazar, Electrochemical property: structure relationships in monoclinic Li3-yV2(PO4)3, J. Am. Chem. Soc., 2003, 125, 10402–10411 CrossRef CAS PubMed
- C. Y. Nan, J. Lu, C. Chen, Q. Peng and Y. D. Li, Solvothermal synthesis of lithium iron phosphate nanoplates, J. Mater. Chem., 2011, 21, 9994–9996 RSC
- M. J. Im, J. I. Kim, S. K. Hyeong, B. J. Moon and S. Bae, From Pristine to Heteroatom–Doped Graphene Quantum Dots: An Essential Review and Prospects for Future Research, Small, 2023, 19, 2304497 CrossRef CAS PubMed
- R. Chang, P. Hao, H. Qu, J. Xu and J. Ma, A fire resistant MXene-based flexible film with excellent Joule heating and electromagnetic interference shielding performance, J. Colloid Interface Sci., 2024, 654, 437–445 CrossRef CAS PubMed
- Z. S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. L. Feng and K. Mullen, Three-Dimensional Nitrogen and Boron Co-doped Graphene for High-Performance All-Solid-State Supercapacitors, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS PubMed
- J. Wu, M. Xu, C. Tang, G. Li and H. He, F-Doping effects on carbon-coated Li3V2(PO4)3 as a cathode for high performance lithium rechargeable batteries: combined experimental and DFT studies, Phys. Chem. Chem. Phys., 2018, 20, 15192–15202 RSC
- W. Cong, G. Ziyang, S. Wei, Xu Qunjie and L. Haimei, B-doped Carbon Coating Improves the Electrochemical Performance of Electrode Materials for Li-ion Batteries, Adv. Funct. Mater., 2014, 24, 5511–5521 CrossRef
- J. G. Wu, M. W. Xu, C. Tang, G. N. Li, H. He and C. M. Li, F-Doping effects on carbon-coated Li3V2(PO4)3 as a cathode for high performance lithium rechargeable batteries: combined experimental and DFT studies, Phys. Chem. Chem. Phys., 2018, 20, 15192–15202 RSC
- Y. Liu, Y. Qiao, G. Wei, S. Li, Z. Lu, X. Wang and X. Lou, Sodium storage mechanism of N, S co-doped nanoporous carbon: Experimental design and theoretical evaluation, Energy Storage Mater., 2018, 11, 274–281 CrossRef
- Y. Liu, Y. Qiao, G. Y. Wei, S. Li, Z. S. Lu, X. B. Wang and X. D. Lou, Sodium storage mechanism of N, S co-doped nanoporous carbon: Experimental design and theoretical evaluation, Energy Storage Mater., 2018, 11, 274–281 CrossRef
- C. Wang, Z. Guo, W. Shen, A. Zhang, Q. Xu, H. Liu and Y. Wang, Application of sulfur-doped carbon coating on the surface of Li3V2(PO4)3 composites to facilitate Li-ion storage as cathode materials, J. Mater. Chem. A, 2015, 3, 6064–6072 RSC
- D. Zhang, G. Huang, H. Zhang, Z. Zhang, Y. Liu, F. Gao, Z. Shang, C. Gao, Y. Zhou and S. Fu, Soft template-induced self-assembly strategy for sustainable production of porous carbon spheres as anode towards advanced sodium-ion batteries, Chem. Eng. J., 2024, 495, 153646 CrossRef CAS
- P. Ge, H. S. Hou, S. J. Li, L. Yang and X. B. Ji, Tailoring Rod-Like FeSe2 Coated with Nitrogen-Doped Carbon for High-Performance Sodium Storage, Adv. Funct. Mater., 2018, 28, 1801765 CrossRef
- Q. Wei, Q. An, D. Chen, L. Mai, S. Chen, Y. Zhao, K. M. Hercule, L. Xu, A. Minhas-Khan and Q. Zhang, One-pot synthesized bicontinuous hierarchical Li3V2(PO4)3/C mesoporous nanowires for high-rate and ultralong-life lithium-ion batteries, Nano Lett., 2014, 14, 1042–1048 CrossRef CAS PubMed
- T. Ruan, S. Lu, J. Lu, J. Niu and R. Li, Unraveling the intercalation chemistry of multi-electron
reaction for polyanionic cathode Li3V2(PO4)3, Energy Storage Mater., 2023, 55, 546–555 CrossRef
- X. Li, H.-C. Lin, W.-J. Cui, Q. Xiao and J.-B. Zhao, Fast Solution-Combustion Synthesis of Nitrogen-Modified Li4Ti5O12 Nanomaterials with Improved Electrochemical Performance, ACS Appl. Mater. Interfaces, 2014, 6, 7895–7901 CrossRef CAS PubMed
- S. Wang, Y. Yu, S. Fu, H. Li and J. Huang, Regulating the non-effective carriers transport for high-performance lithium metal batteries, J. Energy Chem., 2024, 92, 132–141 CrossRef CAS
- Y. Lokeswararao, A. C. Dakshinamurthy, A. K. Budumuru and C. Sudakar, Influence of Nano-fibrous and Nano-particulate Morphology on the Rate Capability of Li3V2(PO4)3/C Li-ion Battery Cathode, Mater. Res. Bull., 2023, 166, 112331 CrossRef CAS
- S.-C. Yin, H. Grondey, P. Strobel, H. Huang and L. F. Nazar, Charge ordering in lithium vanadium phosphates: electrode materials for lithium-ion batteries, J. Am. Chem. Soc., 2003, 125, 326–327 CrossRef CAS PubMed
- J. Shin, J. Yang, C. Sergey, M. S. Song and Y. M. Kang, Carbon Nanofibers Heavy Laden with Li3V2(PO4)3 Particles Featuring Superb Kinetics for High-Power Lithium Ion Battery, Adv. Sci., 2017, 4, 1700128 CrossRef PubMed
- Z. Y. Gu, J. M. Cao, K. Li, J. Z. Guo, X. T. Wang, S. H. Zheng, X. X. Zhao, B. Li, S. Y. Li and W. L. Li, 2D Exfoliation Chemistry Towards Covalent Pseudo-Layered Phosphate Framework Derived by Radical/Strain-Synergistical Process, Angew. Chem., 2024, 136, 202402371 CrossRef
- Z.-Y. Gu, X.-T. Wang, X.-X. Zhao, J.-M. Cao, Y.-L. Heng, S.-H. Zheng, Y. Liu, J.-Z. Guo, S.-Z. Wang and X.-L. Wu, Advanced K3V2 (PO4)2O2F cathode for rechargeable potassium-ion batteries with high energy density, Appl. Phys. Lett., 2024, 124, 183904 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi01916d |
| ‡ These authors equally contributed to this work. |
| This journal is © the Partner Organisations 2025 |