
Interface engineering of highly stable CeO2/CoFe@C electrocatalysts for synergistically boosting overall alkaline water splitting performance†
Waleed
Yaseen‡
a,
Karim
Harrath‡
b,
Guangya
Li
a,
Bashir Adegbemiga
Yusuf
a,
Suci
Meng
*a,
Meng
Xie
 a,
Iltaf
Khan
c,
Jimin
Xie
ad,
Changkun
Xia
a and
Yuanguo
Xu
*a
a,
Iltaf
Khan
c,
Jimin
Xie
ad,
Changkun
Xia
a and
Yuanguo
Xu
*a
aSchool of Materials Science & Engineering, School of Chemistry and Chemical Engineering, School of Pharmacy, Jiangsu University, Zhenjiang 212013, China. E-mail: mengsc@ujs.edu.cn; xuyg@ujs.edu.cn
bFundamental Science Center of Rare Earths, Ganjiang Innovation Academy, Chinese Academy of Sciences, Ganzhou 341000, China
cSchool of Environmental & Chemical Engineering, Jiangsu University of Science and Technology, Zhenjiang 212003, PR China
dJiangsu Jiangke Graphene Research Institute Co., Ltd, Jiangsu Jiangke Composite Material Co., Ltd, China
First published on 16th November 2024
Abstract
Electrochemical water splitting produces “green hydrogen,” a clean, sustainable fuel that can eventually contribute to carbon neutrality. However, the big challenge to the widespread adoption of water-splitting technology is the complex synthesis routes that involve harmful or expensive chemicals and sluggish reaction kinetics. This work presents a scalable and environmentally friendly solvent-free strategy for in situ synthesis of highly dispersed CeO2/CoFe nanoparticles encapsulated within 3D hierarchically porous carbon heterostructures (CeO2/CoFe@C) via a simple pyrolysis process. The optimized Ce20/CoFe@C/750 catalyst shows low overpotentials of 114 and 191 mV at 10 mA cm−2 toward the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), respectively, in 1.0 M KOH. Two-electrode systems achieve a cell voltage of 1.508@10 mA cm−2 with robust stability over 500 h in 1.0 M KOH. This notable performance is attributed to the hierarchically porous nanosheet architecture with a superhydrophilic surface that facilitates mass transport, and rapid H2/O2 gas bubble escape, and the synergistically coupled CeO2/CoFe heterointerface and abundant oxygen vacancies boost overall activity, particularly for the OER. Additionally, experimental results indicate that the optimum performance depends critically on the effect of changing Ce concentration. Density functional theory (DFT) calculations suggest that optimizing the CeO2/CoFe interface triggered CeO2 reconstruction, where oxygen migration to CoFe created vacancies. Also, this reduction of the Ce site at the interface and the availability of d and f orbitals contribute to bonding and antibonding adsorbates, thereby moderating their adsorption energy and boosting OER activity. This study demonstrates the significance of rational design concepts in catalyst structure optimization, resulting in noticeably improved overall water-splitting performance.
Introduction
The development of low-cost, highly efficient electrocatalysts for the HER and OER remains a critical challenge for sustainable hydrogen generation by water splitting. A major hurdle is the sluggish OER kinetics at the anode following the four-electron oxidation pathway, requiring a high overpotential for water splitting.1,2 This higher overpotential significantly impacts the overall efficiency of H2 production. Although there has been significant progress in the development of effective materials such as platinum group materials (PGMs) for the HER and Ru/Ir-derived catalysts for the OER, their dependence on costly precious metals restricts their widespread applications. Therefore, attention is directed towards developing catalysts that are cost-effective, long-lasting, and highly efficient, preferably using sustainable manufacturing techniques.3,4Recently, researchers have explored several Earth-abundant electrocatalysts, such as transition metal (TM) oxides,5 phosphates,6 carbides,7 alloys,8 borides,9 sulfides,10 and others, to find long-lasting electrodes capable of efficiently performing alkaline water splitting.11 Among them, CoFe bimetallic alloy nanoparticles (NPs) encapsulated within porous carbon structures (CoFe@C) have attracted significant attention as a promising alternative to expensive catalysts for water splitting,12 because the addition of Fe to Co creates a strong synergistic effect, modifying the Co structure and generating active defects that enhance catalytic activity.13 Additionally, building a three-dimensional (3D) hierarchical carbon nanosheet structure allows the creation of a well-organized network of channels that improves conductivity, prevents metal NPs from leaching, exfoliating, and agglomerating by encapsulating them, and provides a pathway for efficient electron transport within the catalyst.14,15 However, several CoFe-based catalysts suffer from low OER activity due to inherent limitations (electronic structure and oxygen binding) or NPs aggregation hindering active site exposure.16 For example, Xiao et al. reported ultrafine FeCo NPs-embedded boron/nitrogen-co-doped 2D porous carbon nanosheets with self-assembled hierarchically porous structures and obtained good performance and stability for the HER and OER in alkaline media.12 Zeng et al. reported single-crystalline CoFe alloy NPs encapsulated in N-doped carbon nanotubes using the pyrolysis method and obtained notable HER and OER activity with low overpotentials of 110 and 292 mV at 10 mA cm−2 in 1.0 M KOH.17 Thus, designing an adequate heterostructure using CoFe alloy NPs to expose sufficient active sites and enable rapid electron transport is crucial for enhancing the overall efficiency of the catalysts.
Ceria (CeO2) is a very promising material for electrocatalysis due to its being readily available on Earth, making it a more ecologically friendly and economically viable option than traditional catalysts.18 Also, the introduction of CeO2 into CoFe alloy NPs leads to a wide range of oxidation states (Ce4+/Ce3+), generating high concentrations of oxophilic Ce3+ sites, which are often accompanied by oxygen vacancies.19 The existence of these oxygen vacancy defects, Fe/Co cation vacancies, and multivacancy defects can regulate the electronic structures of the catalyst, improve its conductivity, optimize the adsorption of reaction intermediates, and ultimately lower the reaction energy barrier.20,21 However, pristine CeO2 exhibits restricted electrical conductivity and limited active sites for both the HER and the OER, which constrains its capacity to serve as a bifunctional catalyst.22 Thus, combining CeO2 with CoFe alloy NPs via interfacial engineering creates a heterointerface, aiming to overcome limitations and enhance electrocatalytic performance. Xu et al. synthesized an urchin-like NiCo2O4@CeO2 electrocatalyst and obtained remarkable OER performance with a low overpotential of 228 mV@10 mA cm−2 in 1.0 M KOH. The abundant Ce3+ sites with potential oxygen vacancies in NiCo2O4@CeO2 are responsible for boosting the OER activity by promoting electron transport.23 Therefore, maximizing the active surface area of the CoFe–CeO2 heterostructure encapsulated within a hierarchical carbon nanosheet structure is critical for obtaining robust OER performance. Previous studies reveal that the introduction of oxygen vacancies and Ce3+ ions optimizes the electronic structure, weakens the O–O bond, and reduces the adsorption energy of reaction intermediates, thereby facilitating the adsorption and activation of molecular oxygen, thus accelerating the OER kinetics.24,25 Heterostructures, specifically, provide unique interfaces that enhance charge transfer and stability, which in conjunction with oxygen vacancies, significantly boost the catalytic efficiency for electrochemical water splitting.26 However, incorporating bimetallic CoFe alloy NPs with CeO2 for water-splitting applications remains less explored.
Herein, we report a scalable and environmentally friendly solvent-free strategy for the in situ synthesis of highly dispersed CeO2/CoFe NPs encapsulated within 3D hierarchically porous carbon heterostructures (CeO2/CoFe@C) via a simple pyrolysis process. To optimize the catalyst performance, the effects of varying Ce doping levels (10, 20, 30, and 40% by weight) and pyrolysis temperatures (650, 750, and 850 °C) in combination with CoFe@C were systematically investigated. The results indicate that the optimum performance of CeO2/CoFe@C electrocatalysts depends critically on the effect of changing Ce concentration. The optimized Ce20/CoFe@C/750 catalyst prepared at 750 °C shows low overpotentials of 114 and 191 mV at 10 mA cm−2 toward the HER and OER, respectively, in 1.0 M KOH. Two electrode systems achieve a cell voltage of 1.508@10 mA cm−2 with robust stability over 500 h in a 1.0 M KOH solution. This remarkable performance is attributed to the following key factors: (1) 3D hierarchically porous architecture, in conjunction with electronic modulation, allows for efficient mass transport and facilitates the escape of generated H2/O2 gas bubbles, (2) the CeO2–CoFe interface can promote synergistic effects, improving the overall catalytic activity, (3) the superhydrophilic surface allows for good water wetting, ensuring good contact between the water and the catalyst for efficient water splitting, and (4) optimizing the CeO2/CoFe interface triggered CeO2 reconstruction, where oxygen migration to CoFe created vacancies which boost the OER activity during water splitting.
2. Experimental section
2.1 Materials
Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O ≥ 99%), iron(III) chloride (Fe(NO3)3·9H2O ≥ 99%), cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O), citric acid monohydrate (C6H8O7·H2O ≥ 99%), tartaric acid (C4H6O6 ≥ 99%), absolute ethanol (C2H5OH), potassium hydroxide (KOH ≥ 86%), and Ni foam (NF) were used. M/s Shanghai Chemical Co., Ltd, China provided all the chemicals and laboratory equipment. The entire experiment used DI water.2.2 Preparation of the Ce/CoFe@C electrocatalyst
The Ce/CoFe@C electrocatalyst was prepared by a simple pyrolysis method without using any toxic organic solvents. The synthesis process involved mixing different precursor materials, including Co(NO3)2·6H2O, Fe(NO3)3·9H2O, citric acid (acting as a chelating agent), and tartaric acid (functioning as both a complexing and a pore-forming agent), which were combined in a mortar in a specific ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:10 (Co:Fe:C). To introduce varying cerium salt contents (wt%), different amounts relative to the total mixture (10, 20, 30, and 40%) were incorporated into the mortar mixture (Table S1†). After thorough mixing, the mixture was dried in an oven at 80 °C and finally ground into a powder form for further processing. The uniformly mixed composite was then pyrolyzed first at 350 °C for 30 min and subsequently at 750 °C for 2 h under argon (Ar) at a rate of 3.0 °C min−1 (Fig. 1a). The samples were denoted as Cex/CoFe@C/750 (x = 10, 20, 30, and 40). Additionally, to determine the optimal pyrolysis temperature, a sample with 20% Ce salt content was subjected to pyrolysis at three different temperatures, 650, 750, and 850 °C, and denoted as Ce20/CoFe@C/T (T = 650, 750, and 850). The as-prepared samples were repeatedly rinsed with DI water after pyrolysis. Furthermore, for comparison, pristine CoFe alloys and CeO2 were also synthesized without cerium salt and Co and Fe precursors and were denoted as CoFe@C/750 and CeO2@C, while keeping other parameters the same.
:1:10 (Co:Fe:C). To introduce varying cerium salt contents (wt%), different amounts relative to the total mixture (10, 20, 30, and 40%) were incorporated into the mortar mixture (Table S1†). After thorough mixing, the mixture was dried in an oven at 80 °C and finally ground into a powder form for further processing. The uniformly mixed composite was then pyrolyzed first at 350 °C for 30 min and subsequently at 750 °C for 2 h under argon (Ar) at a rate of 3.0 °C min−1 (Fig. 1a). The samples were denoted as Cex/CoFe@C/750 (x = 10, 20, 30, and 40). Additionally, to determine the optimal pyrolysis temperature, a sample with 20% Ce salt content was subjected to pyrolysis at three different temperatures, 650, 750, and 850 °C, and denoted as Ce20/CoFe@C/T (T = 650, 750, and 850). The as-prepared samples were repeatedly rinsed with DI water after pyrolysis. Furthermore, for comparison, pristine CoFe alloys and CeO2 were also synthesized without cerium salt and Co and Fe precursors and were denoted as CoFe@C/750 and CeO2@C, while keeping other parameters the same.
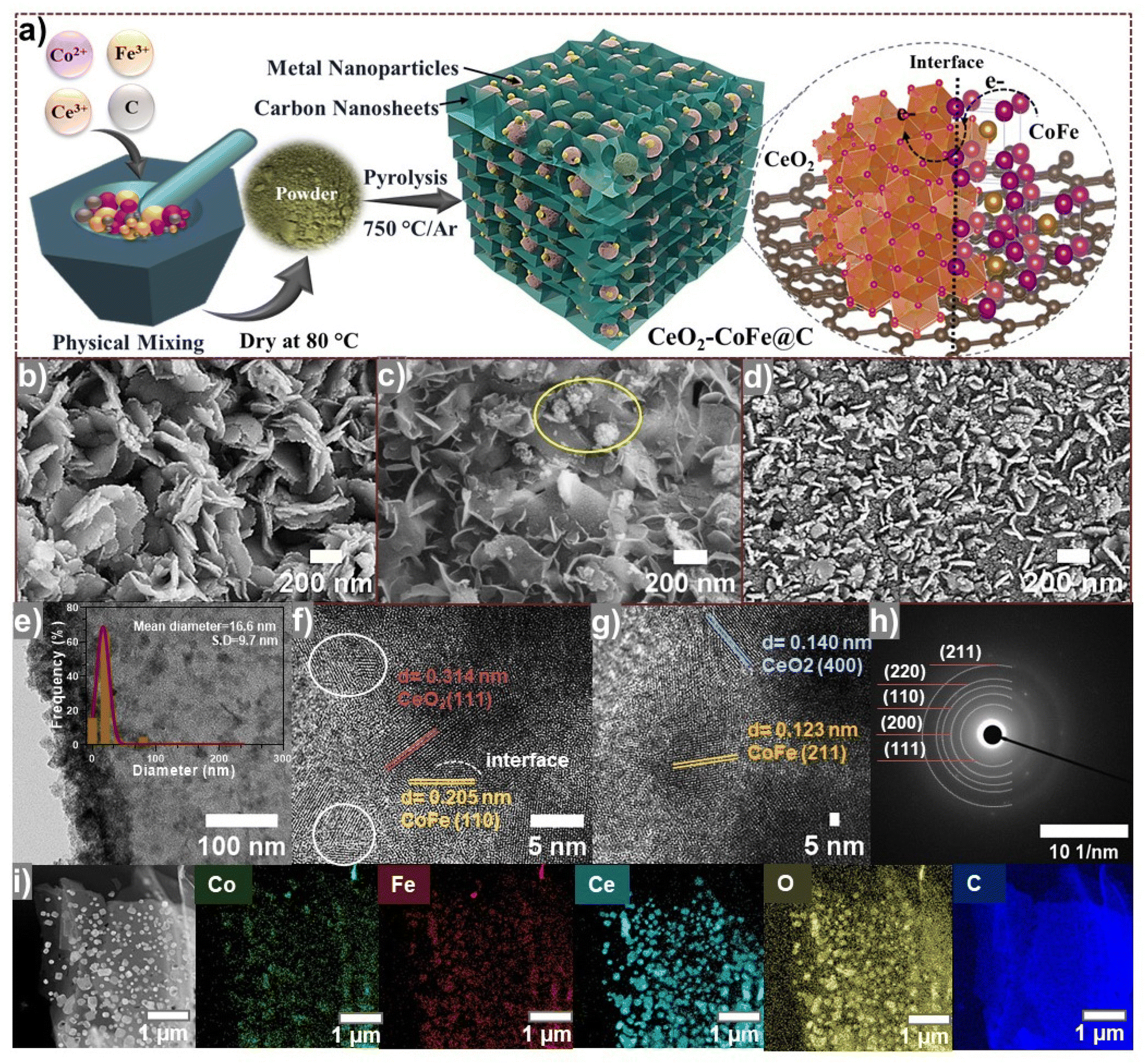 | ||
| Fig. 1 (a) Schematic illustration of the Ce/CoFe@C catalyst. SEM images of (b) CoFe@C/750, (c) Ce20/CoFe@C/750, and (d) Ce40/CoFe@C/750. (e–h) TEM and HR-TEM images with SAED patterns of the Ce20/CoFe@C/750 sample and (i) corresponding TEM images with EDS elemental mapping. | ||
2.3. Characterization of the fabricated electrocatalysts
Advanced analytical equipment was used to study the components and surface features of the electrocatalysts. The crystalline phases of the synthesized materials were determined using X-ray diffraction (XRD) with a LabX-XRD-6100 SHIMADZU instrument. The XRD analysis employed monochromatized Cu–K radiation with a scan rate of 5° per minute, an accelerator power of 40 kV, and a 2-theta range of 10° to 80°. Scanning electron microscopy (SEM) model Gemini 300 (ZEISS, Germany) was used to observe the surface properties of the catalyst. Energy dispersive spectroscopy (EDS) using an SU8100 (Hitachi, Japan) was performed to analyze the elemental composition. The structural characteristics and particle dispersion were determined using transmission electron microscopes (TEM), models JEOL 2100F and HT7700 (Hitachi, Japan). At temperatures ranging from 35 to 1000 °C, a TGA Q5000 differential thermal analyzer was used to conduct a thermogravimetric (T.G.) study in the air. The JESX320 from JEOL was used to accomplish electron paramagnetic resonance (EPR) analysis. The Quantachrome Instruments N2 Autosorb iQ was employed to measure the specific surface and porosity. The ESCALAB 250xi was employed to analyze the composition of elements and chemical states using the X-ray photoelectron spectroscopy (XPS) technique. The contact angle (CA) was measured using an KSV CM200 instrument. Raman analysis was performed using a 532 nm DXR Laser-Spectrometer (Thermo Fisher Corporation). A CHI-760e workstation was employed to evaluate the electrochemical data. The stability of the flow cell electrolyzer was evaluated using the DHElecChem (DH7000) workstation.2.4 Electrochemical measurements
The electrochemical test was carried out on a CHI 760e workstation using a three-electrode arrangement, where mercury/mercury oxide (Hg/HgO), a graphite rod, and the catalysts on NF served as the reference, counter, and working electrodes, respectively. The reference electrode was calibrated before testing in a three-electrode setup (Fig. S1†). The catalyst ink was prepared by mixing 10 mg of catalyst with 250 μL of ethanol, 725 μL of deionized water, and 25 μL of a 5% Nafion solution and ultrasonicated for 30 min. Then, 100 μL of the prepared ink was evenly applied onto the NF (surface area 1 cm2) electrode with a mass loading of ∼1 mg cm−2. To assess the electroactivity of the samples, linear sweep voltammetry (LSV) was employed at a slow scan rate of 5 mV s−1 in 1.0 M KOH. The working electrode was pre-activated using cyclic voltammetry (CV) at a rate of 50 mV s−1 for 100 cycles before the LSV analysis. All LSV curves were corrected with 95% iR-compensation, and the potentials were transformed to the reversible hydrogen electrode (RHE) scale using eqn (1):| ERHE(V) = EHg/HgO + 0.0592 × pH + 0.098 V | (1) |
The Tafel slope was calculated according to eqn (2):
| η = a + blog(j) | (2) |
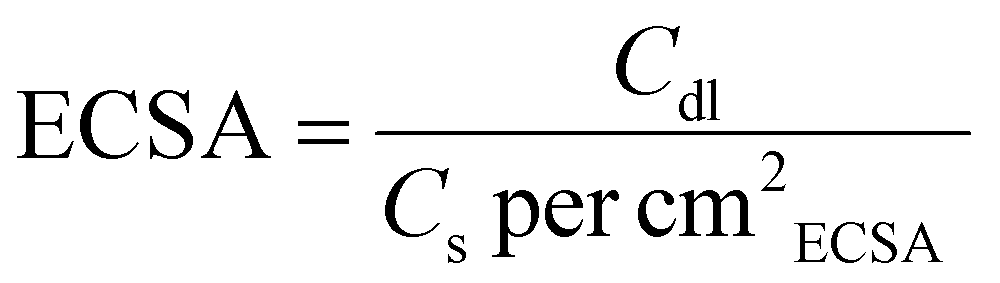 | (3) |
Here, Cs represents the specific capacitance for a standard working electrode, typically between 40 and 80 μF cm−2 in 1.0 M KOH for Ni-based electrode materials. This study employed a standard value of 40 μF cm−2 for calculating the ECSA.27,28 Multi-current stability tests for the HER/OER were carried out by chronopotentiometry (CP). Chronoamperometry (CA) i–t tests were performed to evaluate the long-term stability for the HER and the OER. For overall water splitting, benchmark electrodes, Pt/C for the HER and RuO2 for the OER, were prepared by drop-casting a suspension onto an NF electrode. The stability of the electrode system was evaluated for 500 h at 30 °C in 1.0 M KOH. The water drainage method was used to measure the volume of H2 and O2 generated. Faradaic efficiency was calculated using the method described in the ESI.† An anion exchange membrane (AEM)-based flow cell (1 × 1 cm2 surface area) was assembled using an AMI-7001 membrane (0.45 mm thickness).
2.5 Computational methods
Spin-polarized DFT + U calculations with the Perdew–Burke–Ernzerhof (RPBE) exchange–correlation functional29 were conducted using the Vienna ab initio simulation package (VASP). The projector augmented wave method (PAW)30,31 with a plane-wave kinetic energy cutoff of 500 eV was employed, with a Gaussian smearing of 0.05 eV. The Hubbard correction of DFT + U was applied for Co, Fe, and Ce atoms with a value of 3.0 eV for Co and Fe and 4.5 eV for Ce.32–34 The Brillouin zone was sampled with a 3 × 3 × 1 K-point mesh for geometry optimization and a 6 × 6 × 1 K-point mesh to calculate the PDOS. The CeO2 (100) surface was modeled with a (3 × 3) supercell, and the FeCo (110) surface was modeled with a (3 × 3) supercell. A vacuum layer of 15 Å was added to avoid periodic interactions. The CeO2/FeCo interface was modeled using the optimized structure of CeO2 (100) and FeCo (110). The atomic positions were optimized until the forces were less than 0.03 eV Å−1, and all atoms were allowed to relax during geometry optimization. The structure of isolated molecules (O2, H2, and H2O) was optimized within a unit cell measuring 15 Å × 15 Å × 15 Å, with only the Γ-point utilized.35 Grimme's method was employed to simulate the effects of van der Waals corrections, which were implemented with Becke–Jonson damping.36,37The surface energy Esurface was calculated according to eqn (4):
| Esurface = (Eslab − nECeO2)/2A | (4) |
| Step I: OH− + ^{*} → ^{*}\kern-3pt OH + e− | (5) |
| Step II: ^{*}\kern-3pt OH + OH− → ^{*}\kern-3pt O + H2O + e− | (6) |
| Step III: ^{*}\kern-3pt O + OH− → ^{*}\kern-3pt OOH + e− | (7) |
| Step IV: ^{*}\kern-3pt OOH + OH− → ^{*}\kern-2pt + O2 + H2O + e− | (8) |
3. Results and discussion
3.1. Structural analysis and synthesis strategy
The CeO2/CoFe@C electrocatalysts were successfully synthesized through a simple, scalable pyrolysis method without using any solvent, as shown in Fig. 1(a).During high-temperature pyrolysis under an Ar atmosphere, the carbon precursor transforms into a 3D hierarchically ordered porous nanosheet structure, while the metal ions undergo in situ transformation to form CeO2 and binary CoFe alloy NPs encapsulated within the carbon matrices containing abundant CeO2–CoFe interfaces. The structural order of the CeO2/CoFe@C heterostructure was modulated by controlling the concentration of Ce ions from Ce(NO3)3·6H2O precursors.40
An in-depth investigation of the evolution of the morphology of the CeO2/CoFe@C electrocatalyst was performed through SEM and TEM analysis, as illustrated in Fig. 1(b–g). The CoFe@C/750 catalyst displayed a self-assembled 3D hierarchical architecture comprising ultrathin carbon nanosheets decorated with metal NPs across its surface (Fig. 1b and Fig. S2a–c†). The incorporation of Ce (Fig. 2b) significantly altered the hierarchical nanosheet shape, resulting in a disordered and shattered morphology (Fig. 1c and d). The presence of metal NPs, both encapsulated within and decorating the surface of the carbon nanosheets, could potentially influence their morphology.41 The SEM images indicate that the 3D hierarchical nanosheet structure of the Ce10/CoFe@C/750 catalyst remains well-maintained with a 10 wt% Ce loading (Fig. S3a–c†). However, with higher Ce loadings (20 and 30 wt%), there is a gradual encapsulation of the nanosheets within the agglomerates of metal NPs (Fig. 1c and Fig. S3d–f†). The Ce30/CoFe@C/750 sample displays an obvious presence of the nanosheets within the densest agglomerates of metal particles (Fig. 1d and Fig. S3g–i†). The larger metal agglomerates hinder the accessibility of the electroactive sites on the nanosheet (Fig. 1d). The SEM images acquired at various resolutions are shown in Fig. S3(a–i).† These results suggest a critical role in optimizing the Ce concentration to achieve the desired control over the sample morphology.42 TEM analysis further confirms the presence of a porous structure within the nanosheet assembled in the Ce20/CoFe@C/750 catalyst (Fig. 1e and Fig. S4a, b†). Small NPs with an average diameter of 16.6 nm were observed along the surfaces of the Ce20/CoFe@C/750 catalyst, appearing encapsulated in the carbon matrices (inset Fig. 1e and Fig. S4a, b†). The lattice structure of the Ce20/CoFe@C/750 catalyst was further examined using HR-TEM at a scale of 5 nm (Fig. 1f and g). HR-TEM analysis identified abundant, well-defined heterointerfaces within the electrocatalyst. These interfaces could play a crucial role in boosting electrocatalytic activity.43 Notably, clear lattice d-spacings of 0.314 and 0.140 nm were detected, corresponding to the (1 1 1) and (4 0 0) crystal planes of CeO2, respectively, while lattice fringes with an interplanar spacing of 0.205 and 0.123 nm correspond to the lattice planes of (1 1 0) and (2 1 1), confirming the formation of the CoFe alloy, respectively. The selected-area electron diffraction (SAED) pattern reveals the presence of multiple polycrystalline rings (Fig. 1h), consistent with the expected diffraction pattern for CeO2 and CoFe alloys. STEM imaging (Fig. 1i) and the corresponding EDS mapping confirmed the presence of Fe, Co, Ce, O, and C elements in the Ce20/CoFe@C/750 sample. These results support the encapsulation of NPs within the carbon matrix and indicate a homogeneous distribution of elements throughout the sample, as evidenced by the EDS mapping patterns (Fig. 1i and S5†).
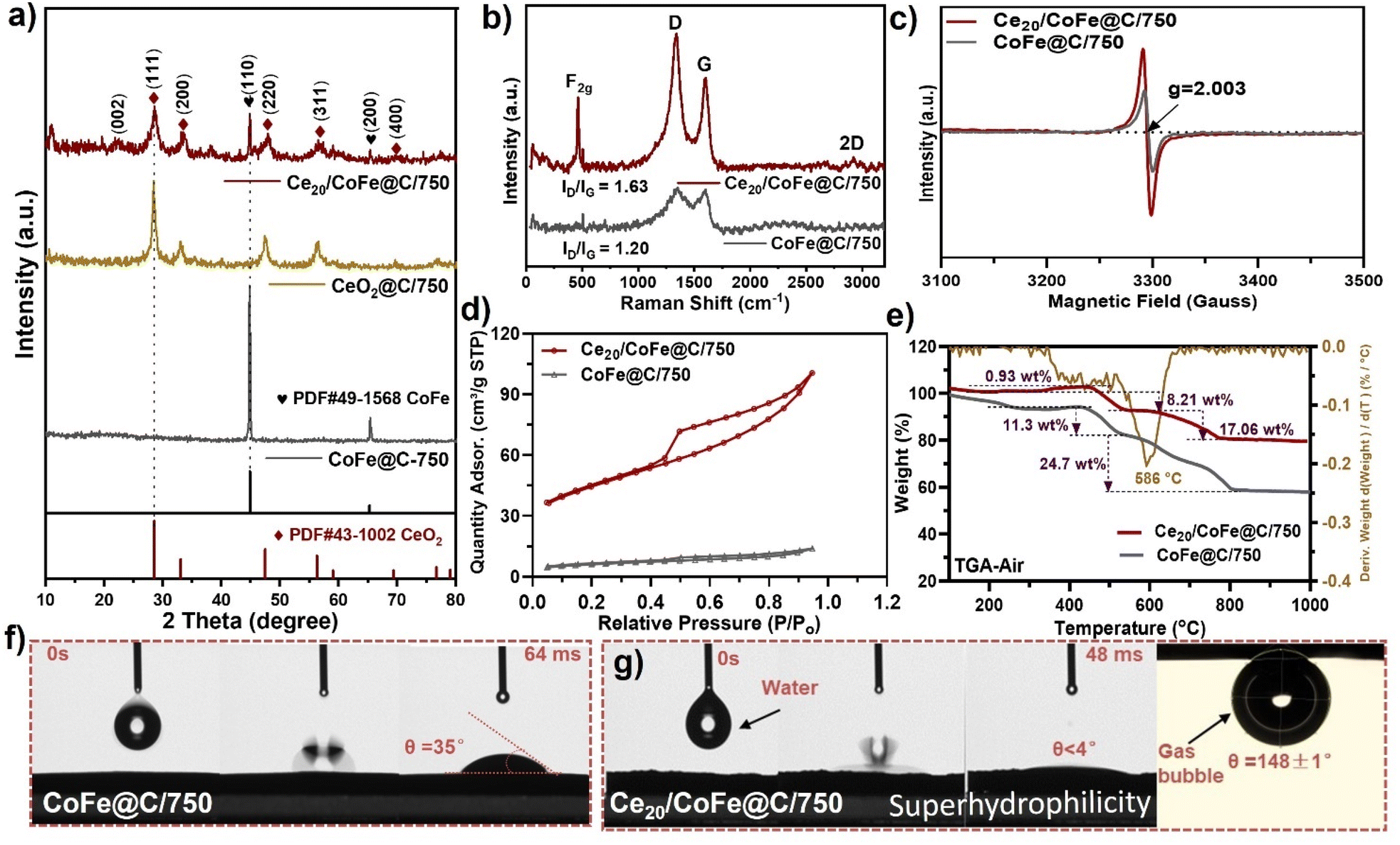 | ||
| Fig. 2 (a) XRD pattern of the as-prepared samples. (b) Raman spectra of the as-prepared samples, (c) EPR analysis of CoFe@C/750 and Ce20/CoFe@C/750, (d) N2 adsorption–desorption isotherms, (e) TGA analysis of Ce20/CoFe@C/750 and (f and g) contact angle measurement of CoFe@C/750 and Ce20/CoFe@C/750. | ||
XRD analysis was performed to assess the crystal structure properties of the as-prepared samples, as shown in Fig. 2(a) and Fig. S6(a and b).† The XRD pattern of the catalyst without adding Ce-salt confirms the formation of a binary CoFe alloy (Fig. 2a). The peaks located at 2θ = 44.8° and 65.3° corresponded to the (1 1 0) and (2 0 0) crystal planes of binary CoFe (JCPDF no. 49-1568).44 Upon incorporating varying Ce wt%, namely Ce10/CoFe@C/750, Ce20/CoFe@C/750, and Ce30/CoFe@C/750, the samples exhibit sharp diffraction peaks at 2θ = 28.5°, 33.0°, 47.4°, 56.3°, 59.0°, 69.4°, 76.7°, and 79.0° corresponding to the (1 1 1), (2 0 0), (2 2 0), (3 1 1), (2 2 2), (4 0 0), (3 3 1), and (4 2 0) crystal planes of CeO2 (JCPDF no. 43-1002), as shown in Fig. 2(a) and Fig. S6(a).† The strongest diffraction peak at 2θ = 26° corresponds to the crystal plane (0 0 2) interlayer spacing of graphite (JCPDS no. 41-1487)45 (Fig. 2a). These results verify the existence of the CoFe alloy, CeO2, and carbon. Besides that, XRD results of the Ce20/CoFe@C/750 sample at different temperatures show a phase transition influenced by temperature (Fig. S6b†). At a temperature of 500 °C, the patterns display diffraction peaks that correspond to the phases of CeFeO3 (JCPDS no. 22-0166) and CoO (JCPDS no. 42-1300)46,47 (Fig. S6b†). As the temperature increases (650, 750, and 850 °C), the XRD patterns show a shift toward a predominant combination of binary CoFe alloy and CeO248 (Fig. S6b†). The observed phase transition in the catalyst with increasing calcination temperature is due to enhanced thermal stability, particle sintering, and structural restructuring.49 Significantly, the positions of the diffraction peaks for the CoFe alloy and CeO2 phases remain relatively unchanged at these higher temperatures, indicating excellent phase stability. Nevertheless, the fluctuations in peak intensity are most likely due to changes in the proportional presence of these phases and/or the effect of crystallite size.50
In the Raman spectra, as shown in Fig. 2(b), a sharp peak at ∼461 cm−1 corresponds to the F2g Raman active mode of the fluorite-type lattice in CeO2, and a broad peak at 560 cm−1 is indexed to the oxygen vacancies present in Ce20/CoFe@C/750.23,51,52 In contrast, F2g bands were absent in the Raman spectrum of the CoFe@C/750 sample (Fig. 2b). Meanwhile, the D-band (A1g mode of finite-sized graphite) and G-band (E2g vibration mode of sp2 carbon) are also detected at ∼1335 and ∼1590 cm−1, respectively (Fig. 2b). The peak ratio value of ID/IG is calculated to be about 1.63 and 1.20 for Ce20/CoFe@C/750 and CoFe@C/750, respectively, indicating a high degree of graphitization of Ce20/CoFe@C/750, which is favourable for enhancing electronic conductivity.53
Furthermore, EPR analysis was used to determine the presence of oxygen vacancies by analyzing the electron spins of atoms that possess vacancies inside the molecule and monitoring changes in their electron spin state.54 The Ce20/CoFe@C/750 sample exhibited the highest peak intensity at ∼g = 2.003, suggesting the largest concentration of oxygen vacancies compared to the CoFe@C/750 sample, revealing that numerous oxygen vacancies might be created during the growth of CeO2–CoFe heterostructures. It has been widely suggested that the formation of oxygen vacancies may significantly control the surface electronic state of the active sites and regulate the adsorption energies of the reaction intermediates, resulting in accelerated charge transfer rates and increased intrinsic activity.52,55 These vacancies, with their unsaturated coordination structure, provide numerous active sites, potentially enhancing catalytic activity.56 Furthermore, the pore-size distribution and specific surface area of the as-prepared catalysts were assessed through N2 adsorption–desorption measurements (Fig. 2d and Fig. S6c†). The specific SBET surface area of Ce20/CoFe@C/750 was 154.2 m2 g−1, larger than that of CoFe@C/750 (21.8 m2 g−1), confirming the presence of mesopores and macropores formed by the lamellar structure stacking, which exhibited an H3-hysteresis loop (Fig. 2d).57 In contrast, the pore-size distribution of Ce20/CoFe@C/750 and CoFe@C/750 is quite similar (Fig. S6c†), further confirming the co-existence of mesopores and macropores on the surface. The porous structure is likely formed as a result of the release of gaseous products from the thermal degradation of tartaric acid during the production process.58,59
The thermal properties of the as-prepared CoFe@C/750 and Ce20/CoFe@C/750 samples were evaluated by TGA in the temperature range of 50–1000 °C under air. The weight loss occurring in both samples in the range of 150–400 °C in the TGA curve (Fig. 2e) undergoes progressive oxidation, leading to the evolution of CO2 under the air atmosphere.60,61 At high temperatures, beyond 400 °C, the weight loss is probably due to the oxidation of the CoFe alloy NPs or continued carbon decomposition. The estimated weight loss of Ce20/CoFe@C/750 and CoFe@C/750 is assessed to be ∼17.6 and ∼24.7 wt%, respectively, indicating the existence of metal content in the final product (Fig. 2e and Fig. S6d†).62 Meanwhile, the wettability of the electrode surface is a crucial factor in assessing the mass transfer behaviour at the interface between the electrode and electrolyte.63 The exceptional efficiency in transferring mass played a crucial role in boosting the catalytic reaction rate. The Ce20/CoFe@C/750 sample displayed superhydrophilicity with a contact angle of almost θ ∼ 4°, further confirming its remarkable electrolyte wettability, which is valuable for good bubble release and fast mass transfer (Fig. 2g).64 Besides, the CoFe@C/750 samples exhibit a larger contact angle of θ ∼ 35° (Fig. 2f), demonstrating the advantageous role of 3D diffusion of heterogeneous nanosheets in reducing the bubble adhesion force. In addition, Ce20/CoFe@C/750 showed a superaerophobic surface with a bubble contact angle of θ = 148 ± 1°, as shown in Fig. 2(g). The results demonstrate that the incorporation of CeO2 can result in the good adsorption of H2O molecules. Also, a hierarchical carbon nanosheet structure further improves wettability by increasing the number of channels through which H2O may penetrate, bringing the electrolyte into closer contact with the catalyst.65
To further evaluate the change in the chemical valence state and electronic structure, XPS spectra of Ce20/CoFe@C/750 and CoFe@C/750 were examined. The survey XPS spectrum of Ce20/CoFe@C/750 confirms the existence of a predominant peak of Co 2p, Fe 2p, Ce 3d, O 1s, and C 1s (Fig. 3a). The core-level Co-2p XPS spectra of Ce20/CoFe@C/750 can be deconvoluted into Co3+ spin–orbit doublet peaks (781.1 and 796.5 eV) and Co2+ doublet peaks (783.5 and 797.8 eV), along with two shakeup satellite peaks (787.8 eV and 803.9 eV), as shown in (Fig. 3b).16,66 The Co 2p spectrum of Ce20/CoFe@C/750 exhibits a slight positive shift for both Co3+ and Co2+ species, indicating an increase in the oxidation state of Co due to charge transfer from Ce. This modification of the electronic structure can enhance catalytic activity by influencing adsorption and desorption properties. Similarly, the Fe-2p spectrum (Fig. 3c) displays Fe2+ doublet peaks (710.9 and 724.7 eV) and Fe3+ doublet peaks (713.2 and 727.7 eV) along with the shakeup satellite peaks at B.E of 718.3 and 732.9 eV.67,68 Metals with high valence states are often a result of the partial transfer of electrons between metal atoms and oxygen atoms on the surface.17 Interestingly, the peaks of Fe2+ and Fe3+ in Ce20/CoFe@C/750 exhibit a positive shift (∼0.2 eV) compared with that in CoFe@C/750. This shift might be due to a partial positive shift within the FeII state or potentially a minor electron transfer from Fe to Co, influenced by their slightly different electronegativities.69 This decrease in electron density at the Fe sites could lead to a weaker bond with the adsorbed *OOH intermediate, potentially enhancing the OER activity.70
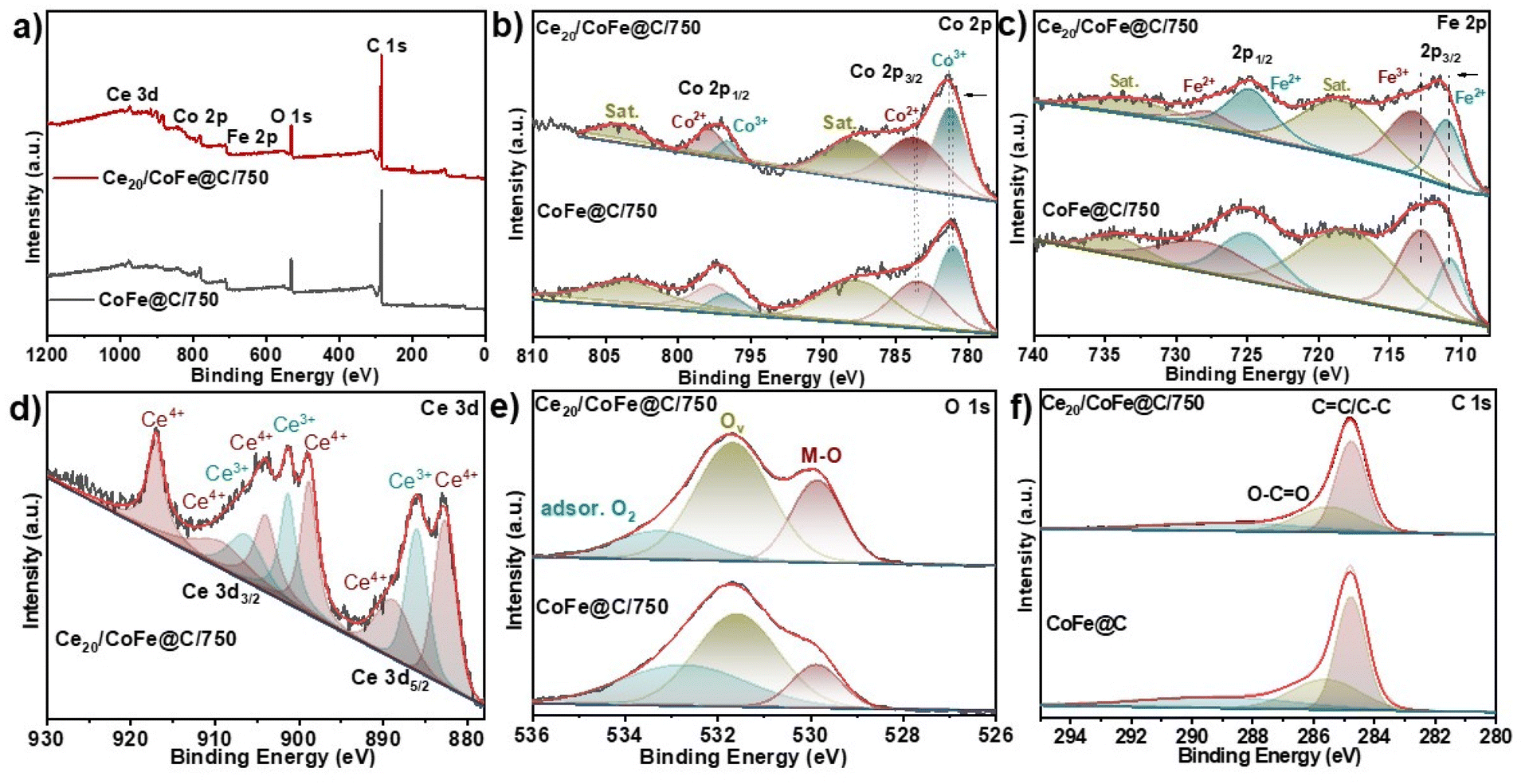 | ||
| Fig. 3 XPS spectra of Ce20/CoFe@C/750 and CoFe@C/750 samples: (a) survey spectrum, (b) Co 2p, (c) Fe 2p, (d) Ce 3d, (e) O 1s, and (f) C 1s. | ||
Fig. 3d describes the Ce 3d spectrum of Ce20/CoFe@C/750; the peaks at 882.6, 888.7, 898.8, 903.9, and 906.2 and 917.0 eV are attributed to Ce4+, whereas the peaks at 885.9, 901.3, and 909.1 eV are assigned to Ce3+, which indicates the coexistence of Ce3+ and Ce4+ valence states.71 Notably, Ce3+ plays a fascinating role in CeO2 by interacting with nearby atoms like Co using its lone electron in the 4f1 orbital. Additionally, the Ce3+ ion's positive charge needs to be balanced (charge compensation). This process creates oxygen vacancies on the ceria surface, which can then capture more oxygen molecules, potentially enhancing the catalyst's performance.72 As shown in Fig. 3(e), O 1s can be deconvoluted into three peaks. The peaks positioned at B.E of 529.8, 531.6, and 533.3 eV correspond to the metal–oxygen bond, oxygen vacancies (Ov), and adsorbed oxygen, respectively.24,72 It is noteworthy that the oxygen vacancy content in Ce20/CoFe@C/750 is 69.5%, higher than that of 51.3% in CoFe@C/750 (Fig. S7†). This indicates that the addition of CeO2 facilitates the generation of a larger number of oxygen vacancies, which is advantageous for the adsorption and conversion of oxygen-containing intermediates favourable for electrocatalytic activity.43,73Fig. 3(f) displays the C 1s XPS spectrum of Ce20/CoFe@C/750, which contains four fitted peaks. The peak at 284.8 eV belongs to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C bonds, and the peak located at 285.4 eV is associated with the O–CO bond, and the other two weak peaks correspond to the CO and C–O bonds.74–76 The atomic percentages of Co, Fe, Ce, O, and C of Ce20/CoFe@C/750 are determined to be 2.05, 2.44, 1.44, 14.03, and 80.03 at%, respectively (Table S2†).
C/C–C bonds, and the peak located at 285.4 eV is associated with the O–CO bond, and the other two weak peaks correspond to the CO and C–O bonds.74–76 The atomic percentages of Co, Fe, Ce, O, and C of Ce20/CoFe@C/750 are determined to be 2.05, 2.44, 1.44, 14.03, and 80.03 at%, respectively (Table S2†).
3.2. OER electrocatalytic performance in alkaline media
Generally, the OER is the most significant barrier to water electrolysis efficacy. The OER performance of the as-prepared samples was evaluated in N2-saturated 1.0 M KOH, as shown in Fig. 4(a–h). Fig. 4(a) displays the LSV curves of CeO2/CoFe@C catalysts. To optimize the OER activity, the effect of different Ce concentrations on the surface properties of CeO2/CoFe@C electrocatalysts was evaluated. The results revealed that the introduction of 20 wt% Ce into electrocatalysts containing Co:Fe:C in a 1:1:10 atomic ratio exhibited optimal performance for the OER. Impressively, the Ce20/CoFe@C/750 electrode exhibits superior OER performance with an overpotential of only 191 mV to reach a current density of 10 mA cm−2, greatly surpassing Ce10/CoFe@C/750 (η10 = 244 mV), Ce30/CoFe@C/750 (η10 = 256 mV), Ce40/CoFe@C/750 (η10 = 283 mV) and CoFe@C/750 (η10 = 298 mV), and commercial RuO2 (η10 = 324 mV) as well as recently reported TM-based catalysts (Fig. 4a, b and Table S6†). Fig. S8† shows the LSV curves before and after iR-correction. To explore the optimal conditions, the OER activity of the as-prepared catalysts obtained at different annealing temperatures was also investigated (Fig. S9a†). The Ce20/CoFe@C/750 catalyst pyrolyzed at 750 °C showed a significantly lower overpotential compared to Ce20/CoFe@C/650 (η10 = 270 mV) and Ce20/CoFe@C/850 (η10 = 261 mV). The synergistic combination of CeO2–CoFe NPs dispersed on high-surface-area carbon nanosheets and superhydrophilic surface features facilitates efficient mass transport that enhances intrinsic OER activity, leading to remarkable OER performance. Additionally, optimizing the CeO2/CoFe interface triggered CeO2 reconstruction, where oxygen migration to CoFe created vacancies further promoting electron transport essential for the OER process.
 | ||
| Fig. 4 (a) LSV polarization curves for the OER in 1.0 M KOH; (b) histogram of η10, η50, η100, and η200 of various catalysts; (c) consistent Tafel slopes; (d) Nyquist-plot (inset: the suitable circuit model); (e) Cdl value of the as-synthesized samples; (f) multi-current process for Ce20/CoFe@C/750CP stability at different current densities; (g) OER stability test for 200 h at a high current density of 135 mA cm−2 (inset: LSV before and after the test); and (h) comparison of our catalyst with previously reported TM-based OER catalysts. | ||
Furthermore, Tafel slopes were extracted from LSV polarization curves to understand the OER kinetics. As shown in Fig. 4(c) and S9(b),† the corresponding Tafel slope of Ce20/CoFe@C/750 is 38.8 mV dec−1, which is smaller than those of Ce10/CoFe@C/750 (51.8 mV dec−1), Ce30/CoFe@C/750 (62.6 mV dec−1), Ce40/CoFe@C/750 (50.4 mV dec−1), CoFe@C/750 (54.4 mV dec−1), Ce20/CoFe@C/650 (67.3 mV dec−1) and Ce20/CoFe@C/850 (45.3 mV dec−1). A smaller Tafel slope suggests rapid OER kinetics, potentially offering significant insights for developing OER electrocatalysts. Tafel-plot measurements were subsequently employed to investigate the kinetics and mechanism of the OER. Notably, the OER pathway for the catalysts in alkaline electrolytes is thoroughly understood. Typically, OH− species undergo initial adsorption on the active catalyst sites of the CeO2–CoFe interface (M* + OH− → *M–OH + e−), where M* represents an active site. The charge rearrangement at the interface is involved in promoting the generation of *M–O species (*M–OH + OH− → *M–O + H2O + e−), which subsequently combines with OH− to form *OOH (*M–O + OH− → *M–OOH + e−). Finally, O2 is generated by incorporating M*–OOH and OH− (*M–OOH + OH− → O2 + *M + H2O + e−).77,78 The rate-determining step significantly relies on the corresponding Tafel slope.79 The observed Tafel slope of 38.8 mV dec−1 suggests that forming adsorbed oxygen intermediates (*O) is the rate-determining step in the OER (Fig. 4c). This finding highlights the beneficial effect of the electronic interaction between the CeO2 and CoFe heterointerface, which facilitates the adsorption of OH− ions and weakens the O–H bond, ultimately enhancing the OER kinetics.
Nyquist plots of the EIS spectra were fitted using an equivalent circuit (inset Fig. 4d). In the high-frequency range, the intersection of the curve with the real axis signifies the solution resistance (Rs), while the magnitude of the semicircular arc reflects the charge transfer resistance (Rct) at the contact interfaces.80,81 As shown in Fig. 4(d), Ce20/CoFe@C/750 exhibits the lowest Rct of 6.82 Ω, which is smaller than that of Ce10/CoFe@C/750 (47.2 Ω), Ce30/CoFe@C/750 (21.9 Ω), Ce40/CoFe@C/750 (90.7 Ω), and CoFe@C/750 (157.1 Ω), indicating that the addition of CeO2 significantly improves the charge transfer mobility, making it highly favorable for alkaline OER activity. Meanwhile, the Rct values of other samples prepared under different synthesis temperatures were 72.5 Ω (Ce20/CoFe@C/650 ) and 46.7 Ω (Ce20/CoFe@C/850), as shown in Fig. S9(c) and Table S4.† Fig. S10† presents the Bode plot, exhibiting a typical single time-constant response at high frequencies, characteristic of the OER kinetics. The lower phase angle observed for the Ce20CoFe@C/750 catalyst suggests improved OER reaction kinetics. The result implies that the Ce20/CoFe@C/750 catalyst prepared with a Ce-salt concentration of 20 wt% relative to the total mixture and 750 °C temperature shows an optimum value, demonstrating swift dynamics at the electrode–electrolyte interface and a robust charge-transfer capacity. Moreover, the ECSA was determined by Cdl measurement with CV in the non-faradaic area at various scan rates (Fig. S11a–g†). Ce20/CoFe@C/750 exhibits a higher Cdl value of 24.7 mF cm−2 compared to Ce10/CoFe@C/750 (22.7 mF cm−2), Ce30/CoFe@C/750 (14.5 mF cm−2), Ce40/CoFe@C/750 (11.7 mF cm−2), CoFe@C/750 (10.7 mF cm−2), Ce20/CoFe@C/650 (13.2 mF cm−2) and Ce20/CoFe@C/850 (17.6 mF cm−2), signifying a larger active surface area than the other catalysts (Fig. 4e and Fig. S9d†). The estimated ECSA are 617, 567, 362, 292, 267, 330 and 440 cm2ECSA for Ce10/CoFe@C/750, Ce30/CoFe@C/750, Ce40/CoFe@C/750, CoFe@C/750, Ce20/CoFe@C/650, and Ce20/CoFe@C/850, respectively (Table S3†). Multi-step CP testing was used to evaluate the stability of the Ce20/CoFe@C/750 catalyst at current densities ranging from 50 mA cm−2 to 250 mA cm−2 for 10 h. As shown in Fig. 4(f), the electrode demonstrated consistent potential under a constant current density, signifying effective mass transfer performance and mechanical strength in promoting the OER even at a high current. Furthermore, the i–t amperometric test was used to confirm the long-term stability of the Ce20/CoFe@C/750 catalyst (Fig. 4g). At a current density of about 135 mA cm−2, the i–t curve exhibited continuously stable performance for 200 h with a slight current loss and maintained a high retention of about 98% with only a 2% performance decay. Almost the same LSV can be obtained before and after 200 h of testing, indicating its good durability (inset Fig. 4g). Both stability test results confirmed the excellent durability of the Ce20/CoFe@C/750 catalyst. The OER performance of Ce20/CoFe@C/750 is comparable to that of established TM-based electrocatalysts, as illustrated in Fig. 4(h). XRD, SEM, and XPS analyses were carried out for the Ce20/CoFe@C/750 electrode after OER to investigate its structural changes following extended stability testing (Fig. S12a–f†). XRD analysis in Fig. S12(a)† verifies the identical peak patterns after the OER stability test, which further confirms the outstanding phase stability of the Ce20/CoFe@C/750 catalyst. Some additional peaks are due to the oxidation of surface species to oxides and hydroxides during the OER.40,82 Furthermore, the SEM analysis conducted for the post-OER stability test revealed the sustained stability of the electrode surface. As illustrated in Fig. S12(b and c),† the hierarchical nanosheet structure was covered with a white surface deposit, likely attributed to the catalyst's oxidation during the OER process. Nevertheless, the catalyst's inherent nanosheet structure remained intact, indicating robust structural stability. Furthermore, the XPS data of Ce20/CoFe@C/750 after the OER were recorded. As shown in Fig. S12(d),† Co3+ and Co2+ peaks after the OER indicate a reduction in the oxidation state, likely due to surface restructuring or interaction with adsorbates. Previous studies have shown that Ce doping increased the presence of the Co3+ oxidation state (Fig. S12f†). This enhancement occurred due to electron transfer from Co2+ to Ce4+, resulting in the formation of stable cerium in Ce3+; the Co3+ centers functioned as the active sites for the OER83 (Fig. S12f†). In contrast, the stable Fe 2p peak oxidation state almost remains unchanged after the OER stability test (Fig. S12e†). These findings indicate that the Ce20/CoFe@C/750 electrode demonstrates favorable catalytic properties and robust long-term stability.
3.3. HER electrocatalytic performance in alkaline media
The as-prepared CeO2/CoFe@C catalyst was further evaluated in 1.0 M KOH for the HER at a 5 mV s−1 scan rate (Fig. 5a–f and Fig. S14a–d†). Fig. 5(a) displays the obtained LSV curves of the various as-prepared catalysts. As expected, Pt/C exhibited the best HER catalytic activity with a low overpotential of 56 mV at a current density of 10 mA cm−2. Among the as-prepared samples, Ce20/CoFe@C/750 exhibited good HER catalytic activity, achieving a current density of 10 mA cm−2 at a low overpotential of 114 mV as compared to Ce10/CoFe@C/750 (η10 = 147 mV), Ce30/CoFe@C/750 (η10 = 225 mV), Ce40/CoFe@C/750 (η10 = 215 mV) and CoFe@C/750 (η10 = 201 mV), demonstrating the important role of CeO2 integration with the CoFe alloy in HER performance improvement, as shown in Fig. 5(a) and Table S5.† Fig. S13† shows the LSV curves before and after iR-correction. In addition, the catalyst prepared at different temperatures revealed activity of 196 and 189 mV at 10 mA cm−2 for Ce20/CoFe@C/650 and Ce20/CoFe@C/850, which confirms that 750 °C is the optimal temperature for obtaining the best HER performance (Fig. S14a†). The improved HER performance is attributed to the CeO2/CoFe NPs embedded within hierarchically structured carbon nanosheets. Table S6† lists the HER performances of recently reported non-noble metal-based electrocatalysts.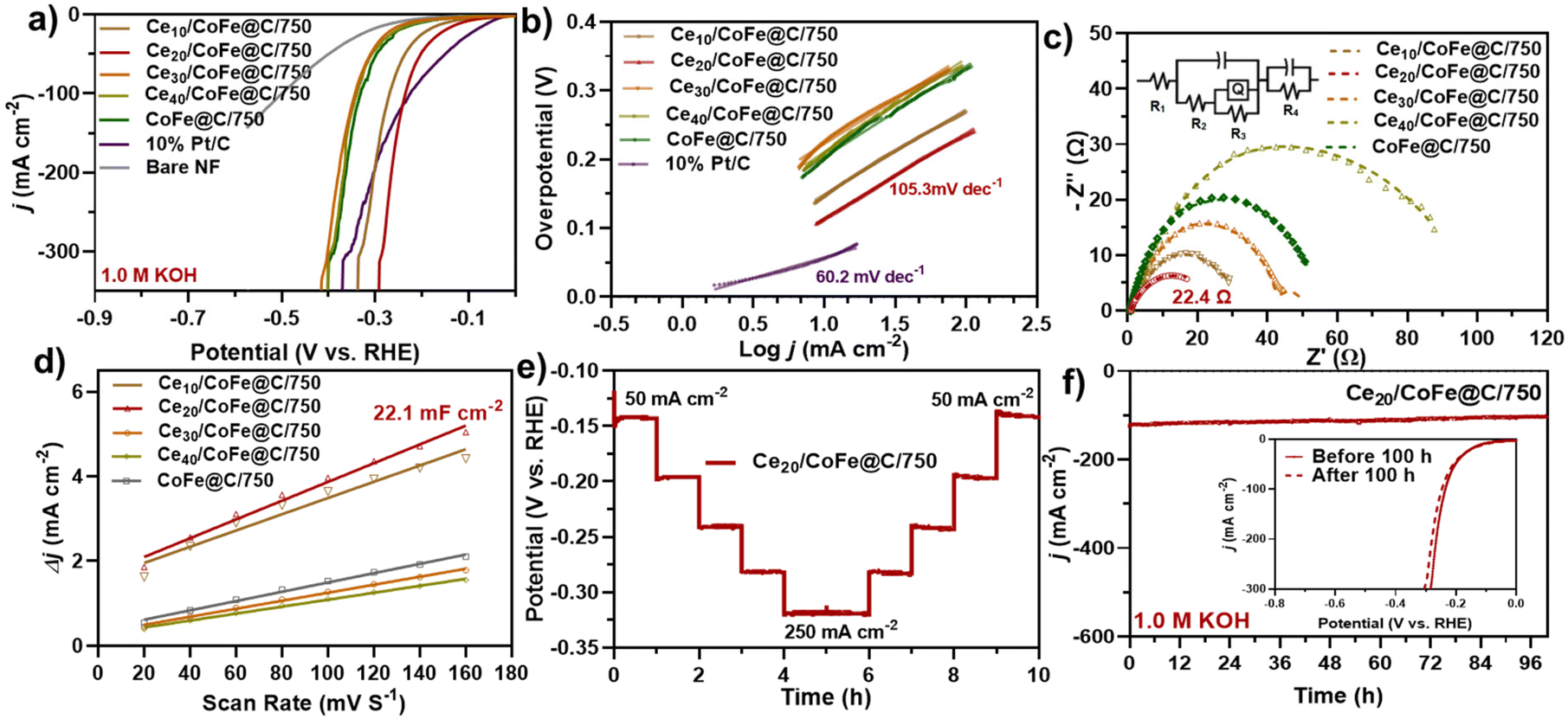 | ||
| Fig. 5 (a) LSV polarization curves for the HER in 1.0 M KOH; (b) consistent Tafel slopes, (c) Nyquist-plot (inset: the suitable circuit model), (d) Cdl value of the as-synthesized samples, (e) Multi-current CP stability process of Ce20/CoFe@C/750 catalyst for 10 h, and (f) stability test for 100 h at a high current density of 100 mA cm−2 (inset: LSV before and after the test). | ||
Furthermore, the Tafel slope was obtained from the linear fitting of LSV curves to gain insight into reaction kinetics, as shown in Fig. 5(b). A lower Tafel value signifies an accelerated rate of reaction kinetics and is beneficial to the reaction. As shown in Fig. 5(b), Ce20/CoFe@C/750 displays a lower Tafel slope of 105 mV dec−1, compared to Ce10/CoFe@C/750 (123.9 mV dec−1), Ce30/CoFe@C/750 (129.1 mV dec−1), Ce40/CoFe@C/750 (136.3 mV dec−1) and CoFe@C/750 (132.2 mV dec−1), indicating the fastest reaction kinetics. As shown in Fig. S14(b),† the other prepared samples obtained at different synthesis temperatures display Tafel slopes of 121 mV dec−1 (Ce20/CoFe@C/650) and 115.0 mV dec−1 (Ce20/CoFe@C/850). These results suggested that the surface of Ce20/CoFe@C/750 followed the Volmer–Heyrovsky mechanism during the HER, in which the Heyrovsky step (H3O+ + e− + H* → H2 + H2O) is typically regarded as the rate-limiting step.84,85 EIS measurements were carried out to demonstrate faster kinetics during the HER process using an equivalent circuit model (inset Fig. 5c). The Ce20/CoFe@C/750 catalysts exhibited the lowest Rct value of 22.4 Ω compared to the Ce10/CoFe@C/750 (25.8 Ω), Ce30/CoFe@C/750 (41.4 Ω), Ce40/CoFe@C/750 (82.8 Ω), CoFe@C/750 (39.4 Ω), Ce20/CoFe@C/650 (84.4 Ω), and Ce20/CoFe@C/850 (37.6 Ω) catalysts, implying that the abundant unsaturated coordination active sites on edges, CeO2–CoFe heterointerface, and the porous structure can enhance the charge transfer and dynamic processes (Fig. 5c and Fig. S14c†). Table S4† lists the values of Rs and Rct of all as-prepared catalysts. Fig. S15† presents a corresponding EIS Bode plot, showing a lower phase angle for the Ce20CoFe@C/750 catalyst that suggests improved HER reaction kinetics.86 Furthermore, the Cdl value was obtained in the non-faradaic region using CV and was employed to determine the ECSA of the electrode (Fig. 5d and Fig. S14d†). Ce20/CoFe@C/750 exhibits a higher Cdl value of 22.1 mF cm−2 than Ce10/CoFe@C/750 (19.1 mF cm−2), Ce30/CoFe@C/750 (9.45 mF cm−2), Ce40/CoFe@C/750 (8.19 mF cm−2), CoFe@C/750 (10.9 mF cm−2), Ce20/CoFe@C/650 (18.1 mF cm−2) and Ce20/CoFe@C/850 (19.8 mF cm−2), as shown in Fig. 5(d) and S16(a–g).† The estimated ECSA can be further calculated using Cdl/Cs, where Cs is the specific capacitance generally in the range of 20 to 60 μF cm−2 for a flat electrode (real surface area = 1 cm2).87,88 In this context, the mean value of 40 μF cm−2 was considered for the ECSA measurements. Accordingly, the ECSA was calculated to be 477, 552, 236, 204, 272, 452, and 495 cm2ECSA for Ce10/CoFe@C/750, Ce30/CoFe@C/750, Ce40/CoFe@C/750, CoFe@C/750, Ce20/CoFe@C/650, and Ce20/CoFe@C/850, respectively (Table S5†). The long-term stability of electrocatalysts is an essential attribute for a wide range of industrial applications. The CP technique was utilized to examine the stability of the best-optimized sample. As shown in the inset of Fig. 5(e), the HER overpotential increased with increasing current density and functioned well during a multi-current process (20 to 250 mA cm−2), specifically under high current conditions. Likewise, the long-term HER stability of Ce20/CoFe@C/750 was further evaluated via continuous amperometric i–t testing for 100 h at −100 mA cm−2, as shown in Fig. 5(f). Ce20/CoFe@C/750 showed excellent HER stability even after 100 h of continuous operation (Fig. 5f), with a negligible difference in the LSV curve before and after testing (inset Fig. 5f). Due to exceptional catalytic stability, highly active metal sites, and the synergistic effect at the CeO2/CoFe interface, and a porous nanosheet structure, the as-prepared catalysts exhibit enhanced electrical conductivity and efficient charge transfer, contributing to their overall effectiveness.
3.4. Overall water-splitting performance in alkaline media
Inspired by its excellent performance in both the OER and the HER, the Ce20/CoFe@C/750 heterostructure was directly employed as both the anode and the cathode to evaluate its efficiency under real water-splitting conditions. The LSV curve depicted in Fig. 6(a) illustrates the excellent performance of an as-prepared bifunctional Ce20/CoFe@C/750(+)∥Ce20/CoFe@C/750(−) catalyst, with a cell potential of 1.508 V to reach a large current density of 10 mA cm−2. For comparison, the benchmark catalyst Pt/C(−)∥RuO2(+) obtained a current density of 10 mA cm−2 at an applied voltage of 1.562 V under the same conditions (Fig. 6a). Compared to benchmark catalysts, the Ce20/CoFe@C/750 electrode exhibits superior performance. An H-type electrolytic cell (Fig. S17†) was utilized to measure the volumes of H2 and O2 produced during water electrolysis through the water drainage method. At a given constant current of 100 mA cm−2, the Ce20/CoFe@C/750∥Ce20/CoFe@C/750 electrolytic cell produces about 69 mL of H2 and 34 mL of O2 after 100 min, exhibiting a linear relationship with a volume ratio of approximately 2:1 identical to the theoretical value, demonstrating the efficient energy utilization of the cell (Fig. 6b). In contrast to the theoretically calculated gas production volume, the Ce20/CoFe@C/750 electrode exhibits consistent faradaic efficiency of 97–100% (Fig. 6b) for water electrolysis.
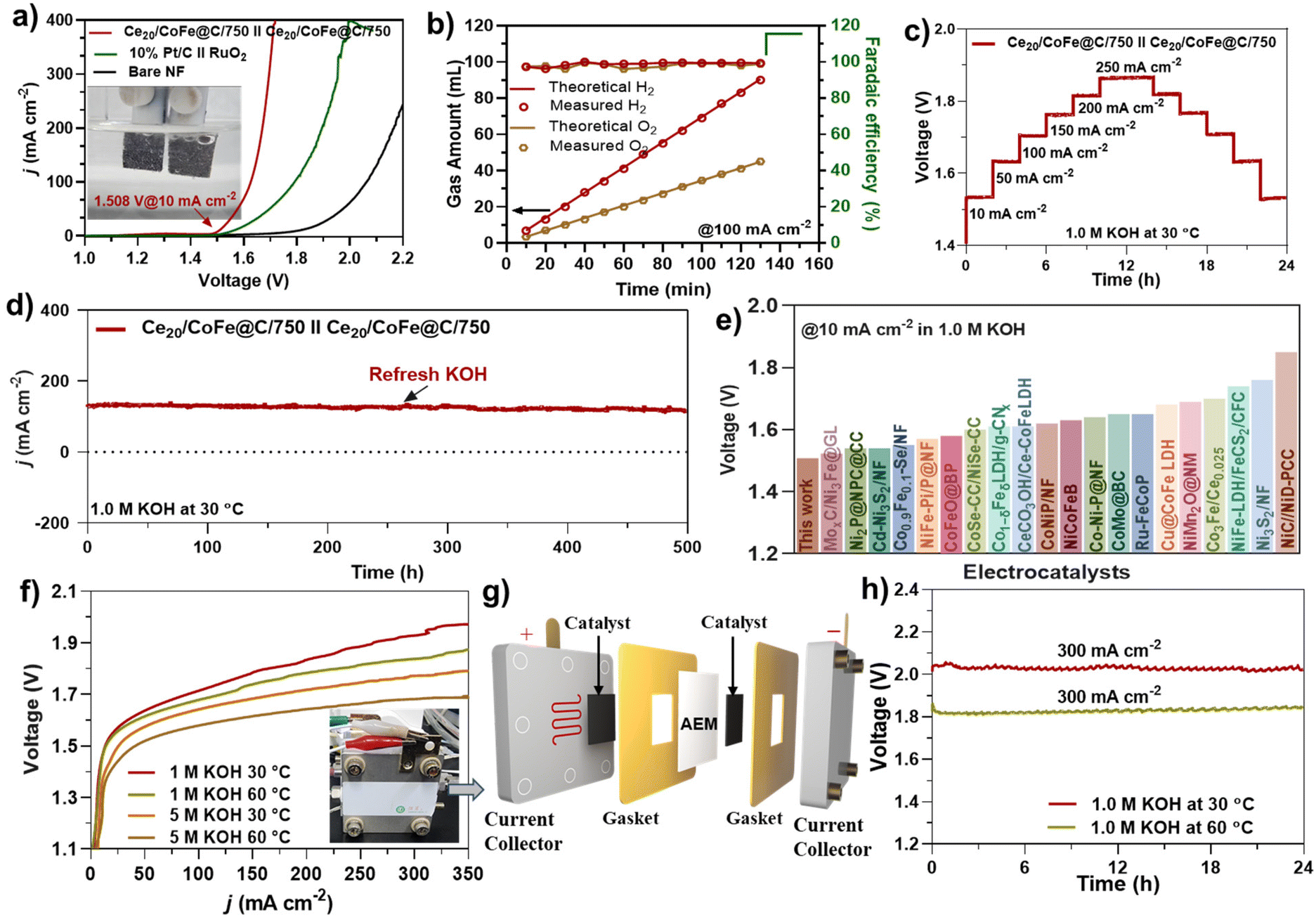 | ||
| Fig. 6 Overall water splitting testing using a glass cell in 1.0 M KOH: (a) LSV curves of Ce20/CoFe@C/750∥Ce20/CoFe@C/750 and 10%-Pt/C∥RuO2, (b) H2 and O2 amounts collected at 100 mA cm−2 by the water drainage method and faradaic efficiency calculations, (c) multi-current stability test, (d) long-term stability testing at 125 mA cm−2 for 500 h, and (e) comparison of the Ce20/CoFe@C/750 catalyst with data in the literature (Table S6†). AEM-based flow cell electrolyzer: (f) LSV curves of the Ce20/CoFe@C/750 catalyst under different electrolyte and temperature conditions, (g) arrangement of various components in the assembly of an AEM electrolyzer, and (h) durability of the AEM electrolyzer using the Ce20/CoFe@C/750 catalyst at 300 mA cm−2 under 30 and 60 °C in 1.0 M KOH. | ||
A multi-current CP stability test was performed to evaluate the durability of the Ce20/CoFe@C/750 electrode in 1.0 M KOH. After 24 h of testing at different current densities, the Ce20/CoFe@C/750 electrode revealed robust stability even at high current densities (Fig. 6c). To assess the practical viability of the synthesized electrocatalyst, extended stability tests were also conducted under real water splitting conditions using amperometric i–t testing in 1.0 M KOH at 30 °C. As evident in Fig. 6(d), the Ce20/CoFe@C/750 electrode exhibits exceptional stability over 500 h of testing with a negligible decay rate of about 8%, signifying its long-lasting performance for water-splitting applications. Fig. 6(e) highlights the performance comparison of the synthesized and previously reported TM-based electrode materials for overall alkaline water splitting (Table S6†). To meet industrial standards, an AEM electrolyzer was assembled using the Ce20/CoFe@C/750 catalyst as both the cathode and the anode, as shown in Fig. 6(f–h). The LSV curves of the AEM Ce20/CoFe@C/750∥Ce20/CoFe@C/750 electrolyzer were analyzed under various electrolyte and temperature conditions, as shown in Fig. 6(f). At an electrolysis temperature of 60 °C in 5 M KOH, the electrolyzer achieved a current density of 350 mA cm−2 at a cell voltage of 1.688 V. Additionally, the AEM electrolyzer demonstrated excellent durability, with negligible voltage variation even after 24 h of operation at 60 °C and a current density of 0.3 A cm−2 (Fig. 6h). These results confirm the potential of the Ce20/CoFe@C/750 catalyst for efficient and stable industrial electrolyzer applications.
3.5. Origin of enhanced OER activity at the CoFe/CoO2 interface
To gain insight into the remarkable OER electrocatalytic activity and identify structure–activity interactions of the CoFe/CeO2 heterointerface compared to the isolated CeO2 (100) and FeCo (110) models, DFT simulations were conducted (Fig. 7a–i and 8a–h). Given that the CeO2 (100) surface can expose either Ce or O atoms as surface terminals, surface energy for both possibilities was calculated. In line with previous reports, the surface with O terminals exhibits superior stability compared to the Ce-terminal surface.89 Based on the observed superior stability, the O-terminal surface was chosen for subsequent enhanced OER investigations (Fig. 7a–d). Fig. 7(e) illustrates the OER energy profile on the CeO2 (100) model. It is evident that the CeO2 (100) catalyst exhibits a high barrier energy of 2.65 eV, associated with a potential-limiting step of 1.42 V (Fig. 7e). This indicates the challenge of OER processing on the CeO2 (100) surface at low electrode potential, where the determining step is the transformation of *OH to *O. In contrast, introducing an oxygen vacancy into the CeO2 (100) model (Fig. 7f) significantly reduced the barrier energy to 2.12 eV and the potential determining step to 0.89 V (Fig. 7g). Notably, the *OH to *O transformation remained the rate-limiting step.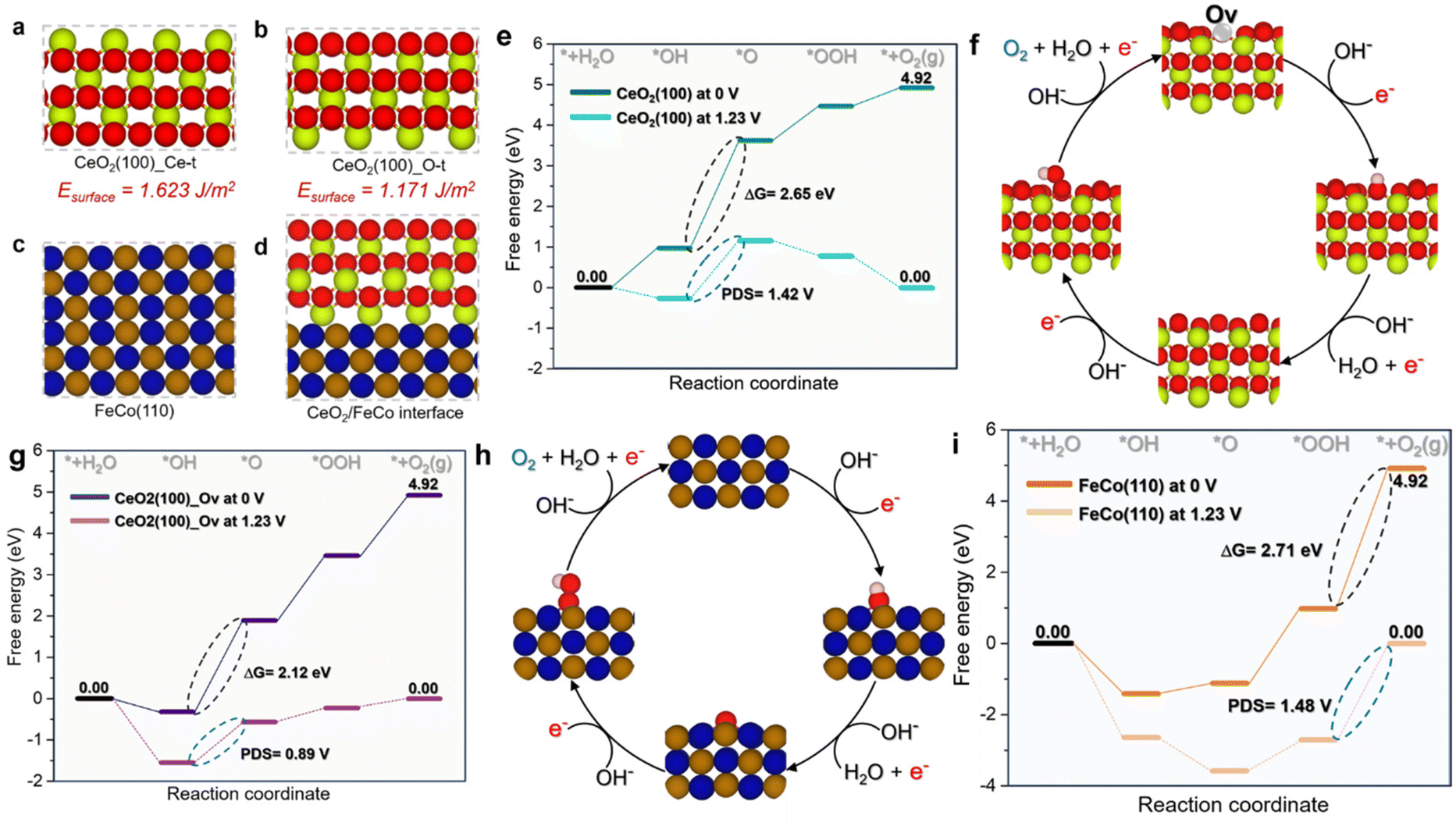 | ||
| Fig. 7 Catalyst surface models investigated in this work. (a) CeO2_Ce-t, (b) CeO2_O-t, (c) FeCo (110), (d) CeO2/FeCo interface, (e) OER energy profile of CeO2 (100) calculated at 0 V and 1.23 V, (f) OER mechanism pathway of CeO2(100)_Ov, (g) OER energy profile of CeO2 (100)_Ov calculated at 0 V and 1.23 V, (h) OER mechanism pathway of FeCo(110)_Ov and (i) OER energy profile of FeCo(110) calculated at 0 V and 1.23 V. | ||
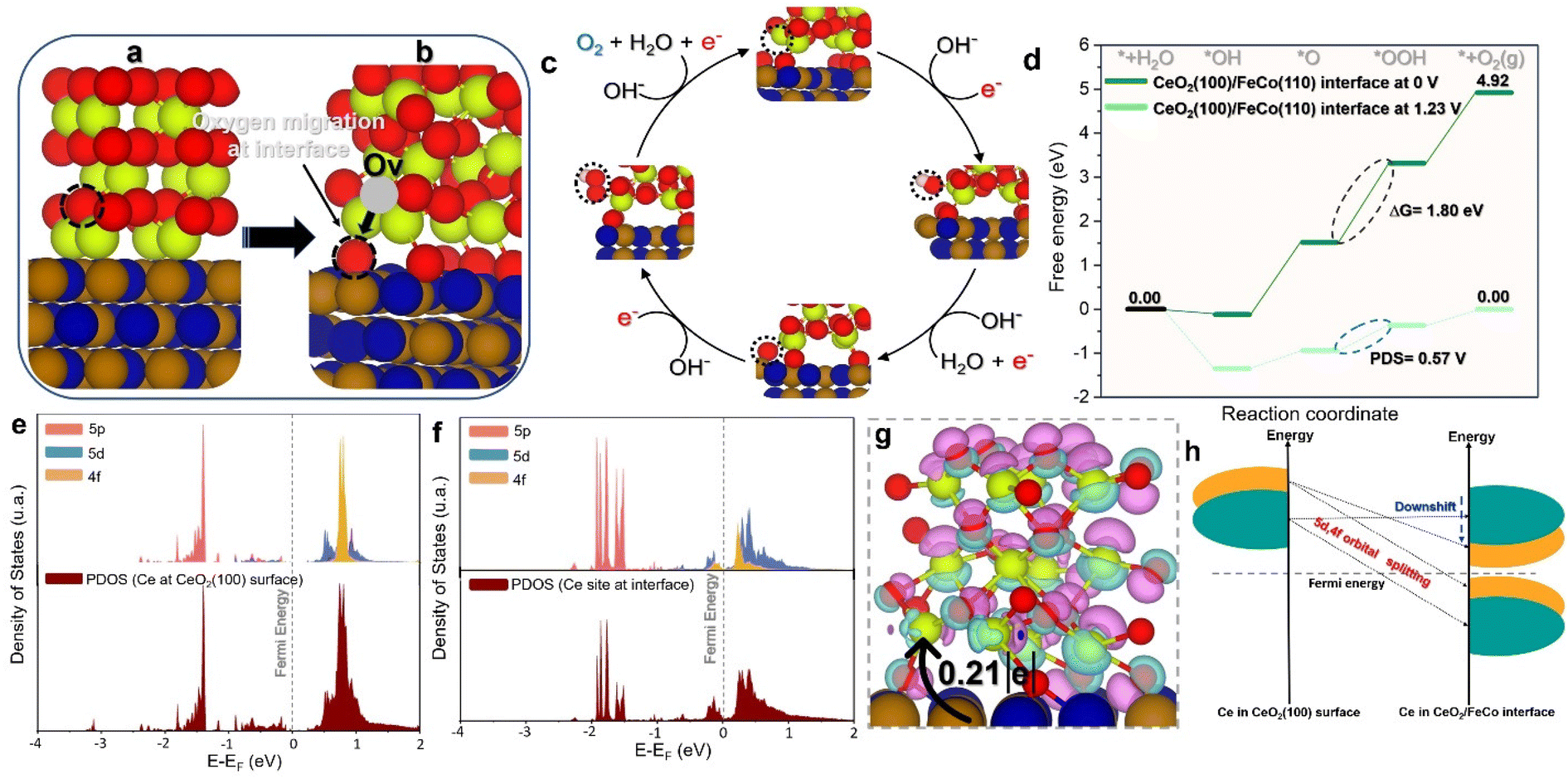 | ||
| Fig. 8 (a and b) CeO2 (100) reconstruction at the CeO2/CoFe interface, (c) OER mechanism pathway at the CeO2/CoFe interface, (d) OER energy profile of the CeO2/FeCo interface calculated at 0 V and 1.23 V, (e) partial density of states (PDOS) of the d and f-projected DOS of Ce active sites at the CeO2/CoFe interface, (f) charge density differences at the CeO2/CoFe interface (light blue represents the electron gain and purple color presents the electron loss (isosurface = 0.1)) and (g) schematic representation of the d and f orbital states at the CeO2/CoFe interface after reconstruction and charge transfer. | ||
Moving to the CoFe (110) surface, as depicted in Fig. 7(h and i), the OER energy profile shows a high barrier energy of 2.71 eV, associated with a potential-limiting step of 1.48 V. In contrast to the CeO2 (100) models, the limiting step switched to the transformation of *OOH to the O2 product. This is not surprising given the high affinity of Fe and Co sites for oxygen and their propensity to be oxidized, leading to a high adsorption energy of oxygenate intermediates, which hinders the formation of the O2 product. Investigation of the CeO2/CoFe interface revealed an intriguing phenomenon during the optimization process, as shown in Fig. 8(a and b). Interestingly, as depicted in Fig. 8(a and b), the optimization of the interface revealed a reconstruction phenomenon where oxygen atoms from CeO2 migrated to CoFe, creating oxygen vacancies at the CeO2 interface. This could be beneficial for the adsorption of OER intermediates. Therefore, in Fig. 8(c and d), the OER mechanism was investigated at the interface and it was found that the limiting step is associated with the transformation of the *O intermediate to *OOH intermediate, with an energy of 1.80 eV and a potential-determining step of 0.57 V. This is favorable at low electrode potential and suggests that the Ce site at the interface can moderate the adsorption energy of OER intermediates to minimize the barrier energies.
To fundamentally understand the effect of CeO2 reconstruction at the interface on the OER energy profile, the electronic properties of the Ce site at the interface were thoroughly investigated. Fig. 8(e and f) show the partial density of states (PDOS) for the Ce site in the CeO2 (100) model and the CeO2/CoFe interface model, respectively. It is evident that in the CeO2 (100) model the LUMO is composed of 5d and 4f orbitals, which do not contribute to the HOMO orbitals, indicating the high oxidation state of the Ce atom. In contrast, at the Ce interface site, the 5d and 4f orbitals contribute to both LUMO and HOMO orbitals and are much closer to the Fermi energy. This indicates the reduction of the Ce site at the interface and the availability of d and f orbitals to contribute to bonding and antibonding adsorbates, thereby moderating their adsorption energy and boosting the OER activity.
The CeO2 reconstruction at the interface serves to moderate the occupation of the 5d and 4f Ce orbitals, splitting them to contribute to both LUMO and HOMO orbitals as shown in Fig. 8(g), thus controlling the adsorption–desorption process during the OER. Furthermore, the charge difference density of the CeO2/CoFe interface was calculated and is shown in Fig. 8(h) for a deeper understanding of the interfacial interaction. The results confirm the reduction of Ce sites at the interface, with a charge transfer of 0.21e occurring from FeCo to CeO2. These results highlight the role of the interface in tuning the electronic properties of Ce atoms for better OER activity by moderating the occupation of 5d and 4f orbitals. Overall, this phenomenon can be generalized to other chemical reactions by controlling the reducibility of metal oxides through engineering interfaces with highly reductive materials such as FeCo, especially with low-reducible metal oxides.
In brief, the Ce20/CoFe@C/750 electrode shows excellent performance and durability for overall water splitting. This is due to both its favorable electronic structure and its geometric design, which increase the number of active sites and improve mass transport. Here are some key considerations for improving the effectiveness and longevity of the material. (1) The CoFe–CeO2 heterostructure, with its hierarchically porous carbon nanostructure, offers distinct geometric advantages. (2) CoFe/CeO2 NPs encapsulated in the carbon structure provide a large number of active sites, significantly increasing the surface area and stability of the reaction. (3) The near-zero contact angle of the catalyst, signifying its superhydrophilicity surface features, facilitates efficient mass transport of ions and water molecules to and from the active sites. (4) Optimization of the interface reveals a reconstruction phenomenon where oxygen atoms from CeO2 migrated to CoFe, creating oxygen vacancies at the CeO2 interface, which could be beneficial for the adsorption of OER intermediates. (5) Reduction of the Ce site at the interface and the availability of d and f orbitals contribute to bonding and antibonding adsorbates, thereby moderating their adsorption energy and boosting OER activity. These geometric benefits complement the electronic structural advantages of the CoFe–CeO2 heterostructure, leading to outstanding activity for the OER and potentially for the HER as well.
4. Conclusions
In summary, a bifunctional CeO2/CoFe@C heterostructure with abundant interfaces was successfully fabricated via a scalable one-step pyrolysis method without using any solvent for the HER and OER. Experimental results revealed that incorporating CeO2 into the CoFe@C architecture noticeably enhances the electrocatalyst's performance, predominantly for the OER, by increasing the electrochemically active surface area and introducing abundant oxygen vacancies, thereby facilitating electron transport. Also, the superhydrophilic surface promotes improved electrolyte penetration and gas diffusion within the catalyst. With the above advantages, the optimized Ce20/CoFe@C/750 achieved 10 mA cm−2 at low overpotentials of 114 and 191 for the HER and the OER, respectively, in 1.0 M KOH. Additionally, the Ce20/CoFe@C/750 electrode, as both the anode and the cathode, operates at a low voltage of 1.508 V to achieve 10 mA cm−2 with stable operation over 500 h in 1.0 M KOH. DFT results demonstrate that CeO2–CoFe interfaces improve electron distribution at the heterointerface, optimizing the adsorption/desorption of intermediate species and leading to enhanced OER activity. The results also revealed that systematic variation in Ce content was found to exert a distinct influence on the electronic structure and surface properties of the catalyst, leading to a direct correlation with the observed changes in electrocatalytic activity. This research will help contribute to the rational development of durable and high-performance Earth-abundant metal-based electrocatalysts for sustainable energy-related applications.Safety statement
No uncommon hazards are noted.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (W2433031 and 22350410392) and Fundamental Research and Development Plan of Zhenjiang City in 2024 (Industry Foresight and Common Key Technologies GY2024027 and GJ2024012).References
- S. M. Pawar, B. S. Pawar, B. Hou, J. Kim, A. T. Aqueel Ahmed, H. S. Chavan, Y. Jo, S. Cho, A. I. Inamdar, J. L. Gunjakar, H. Kim, S. Cha and H. Im, Self-Assembled Two-Dimensional Copper Oxide Nanosheet Bundles as an Efficient Oxygen Evolution Reaction (OER) Electrocatalyst for Water Splitting Applications, J. Mater. Chem. A, 2017, 5, 12747–12751 RSC.
- C.-Z. Yuan, S. Huang, H. Zhao, J. Li, L. Zhang, Y. Weng, T.-Y. Cheang, H. Yin, X. Zhang and S. Ye, Vacancy Defect Tuning of Electronic Structures of Transition Metal (Hydr)Oxide-Based Electrocatalysts for Enhanced Oxygen Evolution, Energy Adv., 2023, 2, 73–85 RSC.
- B. He, Y. Cao, K. Lin, M. Wu, Y. Zhu, X. Cui, L. Hu, Y. Yang and X. Liu, Enhanced Bulk and Interfacial Charge Transfer in Fe:VOPO4 Modified Mo:BiVO4 Photoanodes for Photoelectrochemical Water Splitting, eScience, 2024, 100242 CrossRef.
- Z. Song, Q. Wang, J. Li, K. Adair, R. Li, L. Zhang, M. Gu and X. Sun, Single–atom Surface Anchoring Strategy via Atomic Layer Deposition to Achieve Dual Catalysts with Remarkable Electrochemical Performance, EcoMat, 2023, 5, e12351 CrossRef CAS.
- J. Saravanan, M. Pannipara, A. G. Al-Sehemi, S. Talebi, V. Periasamy, S. S. Shah, M. A. Aziz and G. Gnana Kumar, Flower-like CuO/NiO Nanostructures Decorated Activated Carbon Nanofiber Membranes for Flexible, Sensitive, and Selective Enzyme-Free Glucose Detection, J. Mater. Sci.: Mater. Electron., 2021, 32, 24775–24789 CrossRef CAS.
- Y. Zhang, J. Wu, B. Guo, H. Huo, S. Niu, S. Li and P. Xu, Recent Advances of Transition–metal Metaphosphates for Efficient Electrocatalytic Water Splitting, Carbon Energy, 2023, 5(12), e375 CrossRef CAS.
- Y. Yu, J. Zhou and Z. Sun, Novel 2D Transition–Metal Carbides: Ultrahigh Performance Electrocatalysts for Overall Water Splitting and Oxygen Reduction, Adv. Funct. Mater., 2020, 30(47), 2000570 CrossRef CAS.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Self-Supported Transition-Metal-Based Electrocatalysts for Hydrogen and Oxygen Evolution, Adv. Mater., 2020, 32, e1806326 CrossRef PubMed.
- Y. Jiang and Y. Lu, Designing Transition-Metal-Boride-Based Electrocatalysts for Applications in Electrochemical Water Splitting, Nanoscale, 2020, 12, 9327–9351 RSC.
- M. Li, S. Zhou, R. Sun, S. Han and J. Jiang, Hierarchical Electronic Coupling Engineering of Bimetallic Sulfide Driven by CoFe Bimetal MOFs Anchored on MXene Promotes Efficient Overall Water Splitting, Fuel, 2024, 358, 130256 CrossRef CAS.
- Y. S. Cho, D. Rhee, J. Lee, S. Y. Jung, J. Eom, V. Mazanek, B. Wu, T. Kang, S. Baek and H. Choi, Electronic and Electrocatalytic Applications Based on Solution–processed Two–dimensional Platinum Diselenide with Thickness–dependent Electronic Properties, EcoMat, 2023, 5(8), e12358 CrossRef CAS.
- L. Xiao, Y. Wang, T. Fu, Q. Liu, F. Guo, Y. Zhang, M. Li, X. Bo and T. Liu, Facile Synthesis of Ultrafine Iron-Cobalt (FeCo) Nanocrystallite-Embedded Boron/Nitrogen-Codoped Porous Carbon Nanosheets: Accelerated Water Splitting Catalysts, J. Colloid Interface Sci., 2024, 654, 150–163 CrossRef CAS PubMed.
- M. Li, T. Liu, X. Bo, M. Zhou and L. Guo, J.A Novel Flower-like Architecture of FeCo@NC-Functionalized Ultra-Thin Carbon Nanosheets as a Highly Efficient 3D Bifunctional Electrocatalyst for Full Water Splitting, Mater. Chem. A, 2017, 5, 5413–5425 RSC.
- I. S. Amiinu, Z. Pu, X. Liu, K. A. Owusu, H. G. R. Monestel, F. O. Boakye, H. Zhang, S. Mu, H. G. R. Monestel, F. O. Boakye, H. Zhang and S. Mu, Multifunctional Mo–N/C@MoS2 Electrocatalysts for HER, OER, ORR, and Zn–Air Batteries, Adv. Funct. Mater., 2017, 27(44), 1702300 CrossRef.
- Y. Ren, K. Chen, Y. Zhang, D. Shi, Q. Wu, D. Liang, C. Hu and H. Li, N-Doped Carbon Confined CoFe@Pt Nanoparticles with Robust Catalytic Performance for the Methanol Oxidation Reaction, J. Mater. Chem. A, 2022, 10, 13345–13354 RSC.
- S. Ali Shah, L. Xu, R. Sayyar, T. Bian, Z. Liu, A. Yuan, X. Shen, I. Khan, A. Ali Tahir and H. Ullah, Growth of MoS2 Nanosheets on M@N-Doped Carbon Particles (M = Co, Fe or CoFe Alloy) as an Efficient Electrocatalyst toward Hydrogen Evolution Reaction, Chem. Eng. J., 2022, 428, 132126 CrossRef CAS.
- X. Zeng, M. J. Jang, S. M. Choi, H.-S. Cho, C.-H. Kim, N. V. Myung and Y. Yin, ingle-Crystalline CoFe Nanoparticles Encapsulated in N-Doped Carbon Nanotubes as a Bifunctional Catalyst for Water Splitting, Mater. Chem. Front., 2020, 4, 2307–2313 RSC.
- G. Lu, H. Zheng, J. Lv, G. Wang and X. Huang, Review of Recent Research Work on CeO2-Based Electrocatalysts in Liquid-Phase Electrolytes, J. Power Sources, 2020, 480, 229091 CrossRef CAS.
- E. L. Lawrence, B. D. A. Levin, T. Boland, S. L. Y. Chang and P. A. Crozier, Atomic Scale Characterization of Fluxional Cation Behavior on Nanoparticle Surfaces: Probing Oxygen Vacancy Creation/Annihilation at Surface Sites, ACS Nano, 2021, 15, 2624–2634 CrossRef CAS PubMed.
- K. Bhattacharyya and A. A. Auer, Oxygen Evolution Reaction Electrocatalysis on Cobalt(Oxy)Hydroxide: Role of Fe Impurities., J. Phys. Chem. C, 2022, 126, 18623–18635 CrossRef CAS.
- M. Chen, N. Kitiphatpiboon, C. Feng, A. Abudula, Y. Ma and G. Guan, Recent Progress in Transition-Metal-Oxide-Based Electrocatalysts for the Oxygen Evolution Reaction in Natural Seawater Splitting: A Critical Review, eScience, 2023, 3, 100111 CrossRef.
- Y. Huang, Y. Zhang, J. Hao, Y. Wang, J. Yu, Y. Liu, Z. Tian, T.-S. Chan, M. Liu, W. Li and J. Li, Tuning the Coordination Environment of Fe Atoms Enables 3D Porous Fe/N-Doped Carbons as Bifunctional Electrocatalyst for Rechargeable Zinc-Air Battery, J. Colloid Interface Sci., 2022, 628, 1067–1076 CrossRef CAS PubMed.
- S. Xu, P. Zhang, R. Zhao, J. Wook Bae, H. Li, J. Yong Lee and P. J. Yoo, Engineered Oxidation States in NiCo2O4@CeO2 Nanourchin Architectures with Abundant Oxygen Vacancies for Enhanced Oxygen Evolution Reaction Performance, Chem. Eng. J., 2024, 482, 148787 CrossRef CAS.
- Y. Hu, W. Liu, K. Jiang, L. Xu, M. Guan, J. Bao, H. Ji and H. Li, Constructing a CeO2−x @CoFe-Layered Double Hydroxide Heterostructure as an Improved Electrocatalyst for Highly Efficient Water Oxidation, Inorg. Chem. Front., 2020, 7, 4461–4468 RSC.
- Z. Huang, W. Li, J. Jiang, W. Zhou, M. Zhang, R. Mao, Z. Wang, J. Xie and Z. Hu, Cerium Oxide Boosted CoFe-N Codoped Carbon Nanotubes with Abundant Oxygen-Vacancies toward Efficient Oxygen Reduction and Methanol Oxidation Reaction, Colloid Interface Sci., 2024, 654, 164–173 CrossRef CAS PubMed.
- Y. Hu, W. Liu, K. Jiang, L. Xu, M. Guan, J. Bao, H. Ji and H. Li, Constructing a CeO2−x@CoFe-Layered Double Hydroxide Heterostructure as an Improved Electrocatalyst for Highly Efficient Water Oxidation, Inorg. Chem. Front., 2020, 7, 4461–4468 RSC.
- W. Zhu, W. Chen, H. Yu, Y. Zeng, F. Ming, H. Liang and Z. Wang, NiCo/NiCo–OH and NiFe/NiFe–OH Core Shell Nanostructures for Water Splitting Electrocatalysis at Large Currents, Appl. Catal., B, 2020, 278, 119326 CrossRef CAS.
- M. Wang, W. Zhang, F. Zhang, Z. Zhang, B. Tang, J. Li and X. Wang, Theoretical Expectation and Experimental Implementation of In Situ Al-Doped CoS2 Nanowires on Dealloying-Derived Nanoporous Intermetallic Substrate as an Efficient Electrocatalyst for Boosting Hydrogen Production, ACS Catal., 2019, 9, 1489–1502 CrossRef CAS.
- A. J. R. Hensley, K. Ghale, C. Rieg, T. Dang, E. Anderst, F. Studt, C. T. Campbell, J.-S. McEwen and Y. Xu, DFT-Based Method for More Accurate Adsorption Energies: An Adaptive Sum of Energies from RPBE and VdW Density Functionals, J. Phys. Chem. C, 2017, 121, 4937–4945 CrossRef CAS.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- K. Harrath, Z. Yao, Y.-F. Jiang, Y.-G. Wang and J. Li, Tailoring the Active-Site Spacing of a Single-Atom Catalyst for CH 4 -to-CH 3 OH Conversion: The Co1/UiO-66 MOF as an Exemplary Model, J. Phys. Chem. C, 2024, 128, 5579–5589 CrossRef CAS.
- T. Wu, Q. Deng, H. A. Hansen and T. Vegge, Mechanism of Water Splitting on Gadolinium-Doped CeO2(111): A DFT + U Study, J. Phys. Chem. C, 2019, 123(9), 5507–5517 CrossRef CAS.
- F. O. Boakye, K. Harrath, M. Tabish, G. Yasin, K. A. Owusu, S. Ajmal, W. Zhang, H. Zhang, Y.-G. Wang and W. Zhao, Phosphorus Coordinated Co/Se2 Heterointerface Nanowires: In situ Catalyst Reconstruction toward Efficient Overall Water Splitting in Alkaline and Seawater Media, J. Alloys Compd., 2023, 969, 172240 CrossRef CAS.
- L. Bahri, F. Mbarki and K. Harrath, Understanding the Direct Methane Conversion to Oxygenates on Graphene-Supported Single 3d Metal Atom Catalysts, Chem. Pap., 2023, 77, 3759–3767 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, Density Functional Theory with London Dispersion Corrections, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2011 Search PubMed.
- X.-Q. Gong, A. Selloni, M. Batzill and U. Diebold, Steps on Anatase TiO2 (101), Nat. Mater., 2006, 5, 665–670 CrossRef CAS PubMed.
- I. C. Man, H. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- W. Shan, H. Guo, C. Liu and X. Wang, Controllable Preparation of CeO2 Nanostructure Materials and Their Catalytic Activity, J. Rare Earths, 2012, 30, 665–669 CrossRef CAS.
- C. Mao, Z. Shi, J. Peng, L. Ou, Y. Chen and J. Huang, Hierarchically Porous Carbonized Wood Decorated with MoNi 4 –Embedded MoO2 Nanosheets: An Efficient Electrocatalyst for Water Splitting, Adv. Funct. Mater., 2024, 34(8), 2308337 CrossRef CAS.
- Y. Pan, X. Zhang, T. Wu, B. Shao, T. Li, Q. He, Z. Chen, L. Zhou, S. Liu, X. Huang and Z. Liu, Application of 3D Hierarchical Porous NiCo-Spinel Nanosheet Array for Enhancement of Synergistic Activation of Peroxymonosulfate: Degradation, Intermediates, Mechanism and Degradation Pathway of Tetracycline, Chem. Eng. J., 2024, 481, 148506 CrossRef CAS.
- X. Ding, J. Yu, W. Huang, D. Chen, W. Lin and Z. Xie, Modulation of the Interfacial Charge Density on Fe2P–CoP by Coupling CeO2 for Accelerating Alkaline Electrocatalytic Hydrogen Evolution Reaction and Overall Water Splitting, Chem. Eng. J., 2023, 451, 138550 CrossRef CAS.
- F. Pan, Z. Shen, X. Cao, Y. Zhang, C. Gong, J. Wu, J. Zhang, H. Liu, X. Li and Y. Zhao, Ordered Mesoporous Carbon with Binary CoFe Atomic Species for Highly Efficient Oxygen Reduction Electrocatalysis, Nanoscale, 2024, 16, 8960–8967 RSC.
- Z. Zhang, Z. Wang, J. Lu, J. Lyu, X. Zhuge, K. Luo, Y. Ren, A. Shahzad, W. Lei and D. Liu, Enhancing Electrochemical Performance of Aluminum–Oxygen Batteries with Graphene Aerogel Cathode, Small Methods, 2024, 8, 2301225 CrossRef CAS PubMed.
- Y. Feng, M. Wang, Q. Meng, Z. Wang, Z. Bu, X. Chen and Y. Zhang, Constructing a CeFeO3/LaFeO3/ZnIn2S4 Double Z-Scheme Heterojunction for Photocatalytic Atrazine Degradation under Visible Light Irradiation, Colloids Surf., A, 2024, 134229 CrossRef CAS.
- X. Meng, Y. Dong, Q. Hu and Y. Ding, Co Nanoparticles Decorated with Nitrogen Doped Carbon Nanotubes for Boosting Photocatalytic Water Splitting, ACS Sustainable Chem. Eng., 2019, 7, 1753–1759 CrossRef CAS.
- S. Jiang, R. Zhang, H. Liu, Y. Rao, Y. Yu, S. Chen, Q. Yue, Y. Zhang and Y. Kang, Promoting Formation of Oxygen Vacancies in Two-Dimensional Cobalt-Doped Ceria Nanosheets for Efficient Hydrogen Evolution, J. Am. Chem. Soc., 2020, 142, 6461–6466 CrossRef CAS PubMed.
- S. Kogularasu, Y. Lee, B. Sriram, S. Wang, M. George, G. Chang-Chien and J. Sheu, Unlocking Catalytic Potential: Exploring the Impact of Thermal Treatment on Enhanced Electrocatalysis of Nanomaterials, Angew. Chem., 2024, 136, e202311806 CrossRef.
- K. Liu, Y. Liao, P. Wang, X. Fang, J. Zhu, G. Liao and X. Xu, Lattice Capacity-Dependent Activity for CO 2 Methanation: Crafting Ni/CeO2 Catalysts with Outstanding Performance at Low Temperatures, Nanoscale, 2024, 16, 11096–11108 RSC.
- Z. Wu, M. Li, J. Howe, H. M. Meyer and S. H. Overbury, Probing Defect Sites on CeO2 Nanocrystals with Well-Defined Surface Planes by Raman Spectroscopy and O2 Adsorption, Langmuir, 2010, 26(21), 16595–16606 CrossRef CAS PubMed.
- T. Li, J. Yin, D. Sun, M. Zhang, H. Pang, L. Xu, Y. Zhang, J. Yang, Y. Tang and J. Xue, Manipulation of Mott−Schottky Ni/CeO2 Heterojunctions into N–Doped Carbon Nanofibers for High–Efficiency Electrochemical Water Splitting, Small, 2022, 18(13), 2106592 CrossRef CAS PubMed.
- S. Guo, J. Wang, Y. Sun, L. Peng and C. Li, Interface Engineering of Co3O4/CeO2 Heterostructure in situ Embedded in Co/N-doped Carbon Nanofibers Integrating Oxygen Vacancies as Effective Oxygen Cathode Catalyst for Li-O2 Battery, Chem. Eng. J., 2023, 452, 139317 CrossRef CAS.
- K. Yan, C. Wen, R. Li, B. Zhang, T. Liu, Q. Liu and Z. Zhou, Morphological Optimized CeO2 and Cu-Doped CeO2 Nanocrystals for Hydrogen Production by Solar Photo-Thermochemical Water Splitting Based on Surface Photoinduced Oxygen Vacancies, Appl. Surf. Sci., 2023, 636, 157779 CrossRef CAS.
- L. Zhang, Y. Lei, W. Xu, D. Wang, Y. Zhao, W. Chen, X. Xiang, X. Pang, B. Zhang and H. Shang, Highly Active and Durable Nitrogen-Doped CoP/CeO2 Nanowire Heterostructures for Overall Water Splitting, Chem. Eng. J., 2023, 460, 141119 CrossRef CAS.
- S. S. Shah, M. A. Aziz, M. Ali, A. S. Hakeem and Z. H. Yamani, Advanced High-Energy All-Solid-State Hybrid Supercapacitor with Nickel-Cobalt-Layered Double Hydroxide Nanoflowers Supported on Jute Stick-Derived Activated Carbon Nanosheets, Small, 2024, 20, 2306665 CrossRef CAS PubMed.
- J. Yang, J. Shi, Y. Wu, H. Liu, Z. Liu, Q. You, X. Li, L. Cong, D. Liu, F. Liu, Y. Jiang, N. Lin, W. Zhang and H. Lin, Heterostructure CoFe@(Co0.5Fe0.5)S@NCNT Anchored on Rice Husk-Based Hierarchical Porous Carbon as a Bifunctional Cathode Catalyst for Zn–Air Batteries, J. Mater. Chem. A, 2024, 12(20), 11907–11919 RSC.
- X. Zheng, Y. Li, L. Zhang, L. Shen, Y. Xiao, Y. Zhang, C. Au and L. Jiang, Insight into the Effect of Morphology on Catalytic Performance of Porous CeO2 Nanocrystals for H2S Selective Oxidation, Appl. Catal., B, 2019, 252, 98–110 CrossRef CAS.
- W. Yaseen, N. Ullah, M. Xie, W. Wei, Y. Xu, M. Zahid, C. J. Oluigbo, B. A. Yusuf and J. Xie, Cobalt–Iron Nanoparticles Encapsulated in Mesoporous Carbon Nanosheets: A One-Pot Synthesis of Highly Stable Electrocatalysts for Overall Water Splitting, Int. J. Hydrogen Energy, 2021, 46, 5234–5249 CrossRef CAS.
- T. Li, S. Li, Q. Liu, Y. Tian, Y. Zhang, G. Fu and Y. Tang, Hollow Co3O4/CeO2 Heterostructures in Situ Embedded in N-Doped Carbon Nanofibers Enable Outstanding Oxygen Evolution, ACS Sustainable Chem. Eng., 2019, 7, 17950–17957 CrossRef CAS.
- I. A. Buliyaminu, M. A. Aziz, S. S. Shah, A. K. Mohamedkhair and Z. H. Yamani, Preparation of Nano-Co3O4-Coated Albizia Procera-Derived Carbon by Direct Thermal Decomposition Method for Electrochemical Water Oxidation, Arabian J. Chem., 2020, 13, 4785–4796 CrossRef CAS.
- N. Gao, B. Li, Y. Zhang, W. Li, X. Li, J. Zhao, W. Yue, Z. Xing and B. Wang, CoFe Alloy-Decorated Interlayer with a Synergistic Catalytic Effect Improves the Electrochemical Kinetics of Polysulfide Conversion, ACS Appl. Mater. Interfaces, 2021, 13(48), 57193–57203 CrossRef CAS PubMed.
- T. Li, F. Gu, X. H. Chen, Q. Zhang, H. C. Fu, H. Q. Luo and N. B. Li, Engineered Superhydrophilic/SuperaerophobicCatalyst: Two-Dimensional Co(OH)2−CeO2 Nanosheets Supported on Three-Dimensional Co Dendrites for Overall Water Splitting, Inorg. Chem., 2023, 62, 2784–2792 CrossRef CAS PubMed.
- X. Guo, M. Li, L. Qiu, F. Tian, L. He, S. Geng, Y. Liu, Y. Song, W. Yang and Y. Yu, Engineering Electron Redistribution of Bimetallic Phosphates with CeO2 Enables High-Performance Overall Water Splitting, Chem. Eng. J., 2023, 453, 139796 CrossRef CAS.
- S. M. Abu Nayem, S. Shaheen Shah, N. Sultana, M. Abdul Aziz and A. J. Saleh Ahammad, Electrochemical Sensing Platforms of Dihydroxybenzene: Part 2− Nanomaterials Excluding Carbon Nanotubes and Graphene, Chem. Rec., 2021, 21, 1073–1097 CrossRef CAS PubMed.
- Z. Cui, X. Liang, P. Wang, P. Zhou, Q. Zhang, Z. Wang, Z. Zheng, Y. Liu, Y. Dai and B. Huang, In Situ Integration of Fe3N@Co4N@CoFe Alloy Nanoparticles as Efficient and Stable Electrocatalyst for Overall Water Splitting, Electrochim. Acta, 2021, 395, 139218 CrossRef CAS.
- Y. Fan, Y. Sun, X. Zhang and J. Guo, Synergistic Effect between Sulfur and CoFe Alloys Embedded in N-Doped Carbon Nanosheets for Efficient Hydrogen Evolution under Neutral Condition, Chem. Eng. J., 2021, 426, 131922 CrossRef CAS.
- D. Guo, S. Han, R. Ma, Y. Zhou, Q. Liu, J. Wang and Y. Zhu, In Situ Formation of Iron-Cobalt Sulfides Embedded in N,S-Doped Mesoporous Carbon as Efficient Electrocatalysts for Oxygen Reduction Reaction, Microporous Mesoporous Mater., 2018, 270, 1–9 CrossRef CAS.
- Y. Hu, P. Wu, Y. Yin, H. Zhang and C. Cai, Effects of Structure, Composition, and Carbon Support Properties on the Electrocatalytic Activity of Pt-Ni-Graphene Nanocatalysts for the Methanol Oxidation, Appl. Catal., B, 2012, 111, 208–217 CrossRef.
- F. Chen, H. Liu, F. Yang, S. Che, N. Chen, C. Xu, N. Wu, Y. Sun, C. Yu and Y. Li, Multifunctional Electrocatalyst Based on MoCoFe LDH Nanoarrays for the Coupling of High Efficiency Electro-Fenton and Water Splitting Process, Chem. Eng. J., 2023, 467, 143274 CrossRef CAS.
- W. Li, L. Zhao, C. Wang, X. Lu and W. Chen, Interface Engineering of Heterogeneous CeO2−CoO Nanofibers with Rich Oxygen Vacancies for Enhanced Electrocatalytic Oxygen Evolution Performance, ACS Appl. Mater. Interfaces, 2021, 13, 46998–47009 CrossRef CAS PubMed.
- A. Sivanantham, P. Ganesan and S. Shanmugam, A Synergistic Effect of Co and CeO2 in Nitrogen-Doped Carbon Nanostructure for the Enhanced Oxygen Electrode Activity and Stability, Appl. Catal., B, 2018, 237, 1148–1159 CrossRef CAS.
- D. Ji, L. Fan, L. Tao, Y. Sun, M. Li, G. Yang, T. Q. Tran, S. Ramakrishna and S. Guo, The Kirkendall Effect for Engineering Oxygen Vacancy of Hollow Co3O4 Nanoparticles toward High-Performance Portable Zinc–Air Batteries., Angew. Chem., Int. Ed., 2019, 131(39), 13978–13982 CrossRef.
- W. Ma, W. Li, H. Zhang and Y. Wang, N-Doped Carbon Wrapped CoFe Alloy Nanoparticles with MoS2 Nanosheets as Electrocatalyst for Hydrogen and Oxygen Evolution Reactions, Int. J. Hydrogen Energy, 2023, 48(58), 22032–22043 CrossRef CAS.
- W. Yaseen, M. Xie, B. A. Yusuf, Y. Xu, N. Ullah, M. Rafiq, A. Ali and J. Xie, Synergistically Coupling of Co/Mo2C/Co6Mo6C2@C Electrocatalyst for Overall Water Splitting: The Role of Carbon Precursors in Structural Engineering and Catalytic Activity, Appl. Surf. Sci., 2022, 579, 152148 CrossRef CAS.
- J. Bao, Z. Wang, J. Xie, L. Xu, F. Lei, M. Guan, Y. Huang, Y. Zhao, J. Xia and H. Li, The CoMo-LDH Ultrathin Nanosheet as a Highly Active and Bifunctional Electrocatalyst for Overall Water Splitting, Inorg. Chem. Front., 2018, 5, 2964–2970 RSC.
- X. Xie, L. Du, L. Yan, S. Park, Y. Qiu, J. Sokolowski, W. Wang and Y. Shao, Oxygen Evolution Reaction in Alkaline Environment: Material Challenges and Solutions, Adv. Funct. Mater., 2022, 32(21), 2110036 CrossRef CAS.
- S. Liu, X. Zheng, T. Jiang and W. Chen, Non-Noble Metal Nanocatalysts for Oxygen Evolution Reaction, in Encyclopedia of Nanomaterials, Elsevier, 2023, pp. 590–609 Search PubMed.
- W. Yaseen, S. Meng, W. Li, M. Xie, M. Rafiq, B. Adegbemiga Yusuf, S. A. Shah, I. Khan, J. Xie and Y. Xu, Facile Synthesis of CoMoO4/CoMoB/Boron-Doped Carbon Nanocomposite as a Highly Durable Bifunctional Electrocatalyst for Overall Water Splitting, Int. J. Hydrogen Energy, 2024, 51, 565–577 CrossRef CAS.
- S. Li, T. Chen, J. Wen, P. Gui and G. Fang, In Situ Grown Ni9S8 Nanorod/O-MoS2 Nanosheet Nanocomposite on Carbon Cloth as a Free Binder Supercapacitor Electrode and Hydrogen Evolution Catalyst, Nanotechnology, 2017, 28, 445407 CrossRef PubMed.
- Y. He, J. Shen, Q. Li, X. Zheng, Z. Wang, L. Cui, J. Xu and J. Liu, In situ Growth of VS4 Nanorods on Ni-Fe Sulfides Nanoplate Array towards Achieving a Highly Efficient and Bifunctional Electrocatalyst for Total Water Splitting, Chem. Eng. J., 2023, 474, 145461 CrossRef CAS.
- A. Zou, Y. Tang, C. Wu, J. Li, H. Meng, Z. Wang, Y. Ma, H. An, H. Zhong, Q. Zhang, X. Zhang, J. Xue, X. Wang and J. Wu, Understanding the Origin of Reconstruction in Transition Metal Oxide Oxygen Evolution Reaction Electrocatalysts, ChemSusChem, 2024, 17, e202301195 CrossRef CAS.
- P. Aggarwal, B. Singh and A. Paul, Pore Size and Electronic Tuning in Cerium-Doped CoFe-LDH for the Oxygen Evolution Reaction, Mater. Adv., 2023, 4(19), 4377–4389 RSC.
- Y. Wang, Q. Ye, L. Lin, Y. Zhao and Y. Cheng, NiFeRu/C and Ru, Fe-Ni5P4/C as Complementary Electrocatalyst for Highly Efficient Overall Water Splitting, J. Colloid Interface Sci., 2023, 651, 1008–1019 CrossRef CAS.
- H.-M. Yang, H.-Y. Wang, M.-L. Sun and Z.-Y. Yuan, Interface Engineering of Bifunctional Nickel Hydroxide/Nickel Phosphide Heterostructure for Efficient Intermittent Hydrazine-Assisted Water Splitting, Chem. Eng. J., 2023, 475, 146134 CrossRef CAS.
- K. Ojha, S. Saha, H. Kolev, B. Kumar and A. K. Ganguli, Composites of Graphene-Mo2C Rods: Highly Active and Stable Electrocatalyst for Hydrogen Evolution Reaction, Electrochim. Acta, 2016, 193, 268–274 CrossRef CAS.
- L. Zhuang, Y. Jia, H. Liu, Z. Li, M. Li, L. Zhang, X. Wang, D. Yang, Z. Zhu and X. Yao, Sulfur–Modified Oxygen Vacancies in Iron–Cobalt Oxide Nanosheets: Enabling Extremely High Activity of the Oxygen Evolution Reaction to Achieve the Industrial Water Splitting Benchmark, Angew. Chem., Int. Ed., 2020, 59, 14664–14670 CrossRef CAS.
- Z. Wei, M. Guo and Q. Zhang, Scalable Electrodeposition of NiFe-Based Electrocatalysts with Self-Evolving Multi-Vacancies for High-Performance Industrial Water Electrolysis, Appl. Catal., B, 2023, 322, 122101 CrossRef CAS.
- C. Y. Zhou, D. Wang and X. Q. Gong, A DFT+: U Revisit of Reconstructed CeO2(100) Surfaces: Structures, Thermostabilities and Reactivities, Phys. Chem. Chem. Phys., 2019, 21(36), 19987–19994 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi02487g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |