
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilizing Rh nanoparticles using a TiO2 array with oxygen vacancies for high-performance pH-wide hydrogen evolution†
Guangxia
Wang
 a,
Wenhui
Wang
a,
Yaozhong
Yu
a,
Tao
Hu
*a,
Juan
Ding
*b and
Yongming
Sui
*c
a,
Wenhui
Wang
a,
Yaozhong
Yu
a,
Tao
Hu
*a,
Juan
Ding
*b and
Yongming
Sui
*c
aSchool of Applied Physics and Materials, Wuyi University, Jiangmen 529020, P. R. China. E-mail: hutaowyu@sina.com
bJiaxing Nanhu University, Jiaxing City 314001, Zhejiang Province, P. R. China. E-mail: juanding@jxnhu.edu.cn
cState Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, P. R. China. E-mail: suiym@jlu.edu.cn
First published on 4th February 2025
Abstract
Active and durable catalysts are crucial for the hydrogen evolution reaction (HER). In the volcano plot, rhodium (Rh) and platinum (Pt) are positioned closely, while the activity and stability of Rh are much inferior to those of Pt. Here, we developed a Rh/TiO2/Ti electrocatalyst to mitigate Pt dependency. The in situ formed TiO2 with oxygen vacancies was prepared utilizing Ti foam as the Ti source. The TiO2 support can confine Rh nanoparticles and ensure charge accumulation at the Rh site, while Rh can enhance the conductivity of TiO2. DFT calculations demonstrate that the strong interaction between Rh and TiO2 could regulate the electronic structure of Rh, thereby accelerating water dissociation and optimizing the adsorption strength of *H. Consequently, compared to Rh/Ti, Rh/TiO2/Ti exhibits significantly enhanced HER activity, requiring overpotentials of only 37.3 and 34.3 mV to reach 10 mA cm−2 in the alkaline and acidic HER, respectively. Furthermore, the vertical nanoarray structure, surface roughness, and metal–support interaction endow Rh/TiO2/Ti with remarkable durability. It works in both alkaline and acidic media for 120 hours at 10 mA cm−2 with unnoticeable activity decline, outperforming commercial Pt/C. This work provides a strategy to overcome the activity and stability limitations of Rh-based catalysts, providing references for promising catalysts for water electrolysis.
1. Introduction
As a clean and sustainable energy carrier, hydrogen holds great promise due to its high-energy density. Electrocatalytic water splitting offers a promising avenue for the production of eco-friendly hydrogen.1,2 The industrial application of water electrolysis technology primarily depends on the excellent electrocatalysts, which can exhibit superior catalytic performance toward the acidic and alkaline HER.3,4 In an acidic electrolyte, the kinetics of the HER is rapid, while the harsh acidic environment lowers the stabilities of the catalysts, significantly limiting the widespread application of acidic water splitting.5 Compared to acidic water electrolysis, alkaline water electrolysis stands as a more favorable method for achieving large-scale hydrogen production.3,6 Nevertheless, the sluggish kinetics of the Volmer step for the alkaline HER will result in 2–3 orders of inferior catalytic performance than that in acidic media.7–9 Pt-based materials have served as benchmark electrocatalysts toward the HER, but their scarcity and the inferior catalytic stability in alkaline media severely hindered widespread applications.2 Therefore, there is an urgent need to develop a variety of catalysts to reduce the risk of dependency on a single catalyst.According to the volcano plot, rhodium (Rh) exhibits physicochemical properties resembling those of Pt. Notably, the inherent high corrosion resistance in diverse electrolytes make Rh-based catalysts promising alternatives for the HER.10 Given the scarcity and high cost of Rh, several approaches have been investigated to further boost the activity and stability of Rh to elevate the atom utilization efficiency under wide pH conditions.11–14 Among them, an effective approach is to create heterostructures by depositing catalyst nanoparticles onto a support, which features an interaction between the support and catalyst nanoparticles. Heterogeneous catalysts provide stable anchoring sites for catalyst nanoparticles to ensure their durability through strong catalyst–support interactions. Additionally, the electron transfer at the catalyst–support interface can finely regulate the electronic properties of the catalyst and optimize the adsorption/desorption energy of the reaction intermediates.15 Consequently, supported catalysts offer advantages in both stability and cost-effectiveness.16–18 The support materials hold a crucial position in determining the electrocatalytic performance.3 Metal oxides, particularly Ti-based supports, have emerged as attractive support materials replacing carbon-based supports due to their strong metal–support interactions.16 However, the further improvement of HER activity is hindered by the inadequate conductivity of metal oxides. In view of the merits of the support and metal, it is anticipated to address the electrical conductivity issue by the fine distribution of tiny metal nanoparticles on the support.17,19–21 Most of the reported catalysts are in powder form and require organic binders to be fastened onto working electrodes. Consequently, they encounter challenges such as mass transport limitations, significant performance decline, and susceptibility to detachment.22–24 A promising approach is direct synthesis of the active components onto highly conductive substrates, facilitating rapid electron transport and subsequently accelerating the reaction kinetics.25,26 For instance, Liberato Manna et al. electrodeposited Ru nanoparticles on Cu nanoplatelet arrays vertically grown on Ti meshes. The resulting Ru@Cu-TM electrode demonstrated high performance towards the alkaline HER.1 Furthermore, Liberato Manna engineered an electrode composed of TiO2/Cu nanorods decorated with Ru–Cu, achieving stable HER activity in 1 M NaOH.27 Nevertheless, a deeper investigation is still required to understand how the relationship between metals and the supports impacts their catalytic activity and stability.
In this work, we constructed a robust Rh/TiO2/Ti electrocatalyst consisting of small-sized Rh anchored on vertical TiO2 nanoarrays through metal–support interactions. Notably, TiO2 nanoarrays have abundant oxygen vacancies (VO) and are grown in situ on Ti foam without an additional Ti source. Additionally, the vertical orientation and mechanical robustness of the TiO2 nanoarray facilitate effective reactant diffusion and gas transport. Consequently, the unique structure and catalyst–support interaction endow Rh/TiO2/Ti with excellent HER activity and stability (120 hours). Rh/TiO2/Ti exhibits low overpotentials of 37.3 and 34.3 mV to reach 10 mA cm−2 in alkaline and acidic electrolytes, respectively. These performances are much superior to that of Rh/Ti, as well as the commercial Pt/C catalyst and reported Rh-based catalysts. The experimental results and density functional theory (DFT) calculations indicate that the charge transfer between Rh and TiO2 could modulate the d band center of Rh and regulate the relative strength of reactants and intermediates, resulting in exceptional performance of Rh/TiO2/Ti. Therefore, we proposed a strategy to overcome the activity and stability limitations of Rh-based catalysts, which provides references for the development of potential catalysts for practical water electrolysis.
2. Experimental section
2.1 Materials
Hydrochloric acid (HCl, 36.0–38.0%) was obtained from Guangzhou Chemical Reagent Factory. Rhodium(III) chloride trihydrate (Cl3H6O3Rh, 98%) was purchased from Bide pharm. Ethylene glycol (EG, 99%) and carbamide (urea, 99%) were from Macklin and Aladdin, respectively. Commercial Pt/C (20 wt%) was obtained from Hesen. All chemicals were used directly as obtained, without additional purification steps. Deionized water, with a resistivity of 18 MΩ cm, was sourced from an ultra-pure water purification system.2.2 Sample preparation
By altering the solvent, such as mixing ethylene glycol and water in volume ratios of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1, and 1:2, and using pure water as the solvent, TiO2 with different morphologies were obtained.
:1, 1:1, and 1:2, and using pure water as the solvent, TiO2 with different morphologies were obtained.
In this process, replacing TiO2/Ti with Ti foam allows for the synthesis of a Rh/Ti catalyst.
2.3 Electrochemical measurements
The hydrogen evolution performance was evaluated using a CHI 660E electrochemical workstation (Chenhua, China). A carbon rod acted as the counter electrode, and a 1 cm × 2 cm Rh/TiO2/Ti plate (with the lower 1 cm × 1 cm portion submerged in the electrolyte) was employed as the working electrode. A calomel electrode (SCE) was used as the reference electrode for acidic conditions, while a mercury/mercury oxide electrode (Hg/HgO) was used for alkaline conditions. Linear scanning voltammetry (LSV) curves were recorded at a scan rate of 5 mV s−1 with 90% iR-compensation in both acidic and alkaline electrolytes. EIS measurements were conducted in the frequency range of 10000 Hz to 1 Hz with an amplitude of 5 mV at −0.29 V (vs. SCE) in an acidic electrolyte and −0.98 V (vs. Hg/HgO) in an alkaline electrolyte. To evaluate the durability of the catalyst, the chronopotentiometry (CP) test was conducted at a steady current density of −10 mA cm−2. The electrochemical double-layer capacitance (Cdl) was assessed from cyclic voltammetry (CV) scans at various rates ranging from 25 to 150 mV s−1. All potentials in this study were referenced to the reversible hydrogen electrode (RHE) as follows:| E (vs. RHE) = E (vs. SCE) + 0.0591 × pH + 0.2415 |
| E (vs. RHE) = E (vs. Hg/HgO) + 0.0591 × pH + 0.0977. |
3. Results and discussion
3.1 Material characterization
The self-supported catalyst of Rh/TiO2/Ti was prepared by a two-step process as illustrated in Fig. 1a. Initially, TiO2 nanoarrays were vertically grown in situ on titanium (Ti) foam using a hydrothermal technique. In this process, Ti foam was used as the Ti source, and ethylene glycol (EG) with a small amount of added HCl served as the solvent. In the second step, Rh nanoparticles were deposited onto TiO2 nanoarrays with EG and urea as reducing agents to obtain the final catalyst Rh/TiO2/Ti.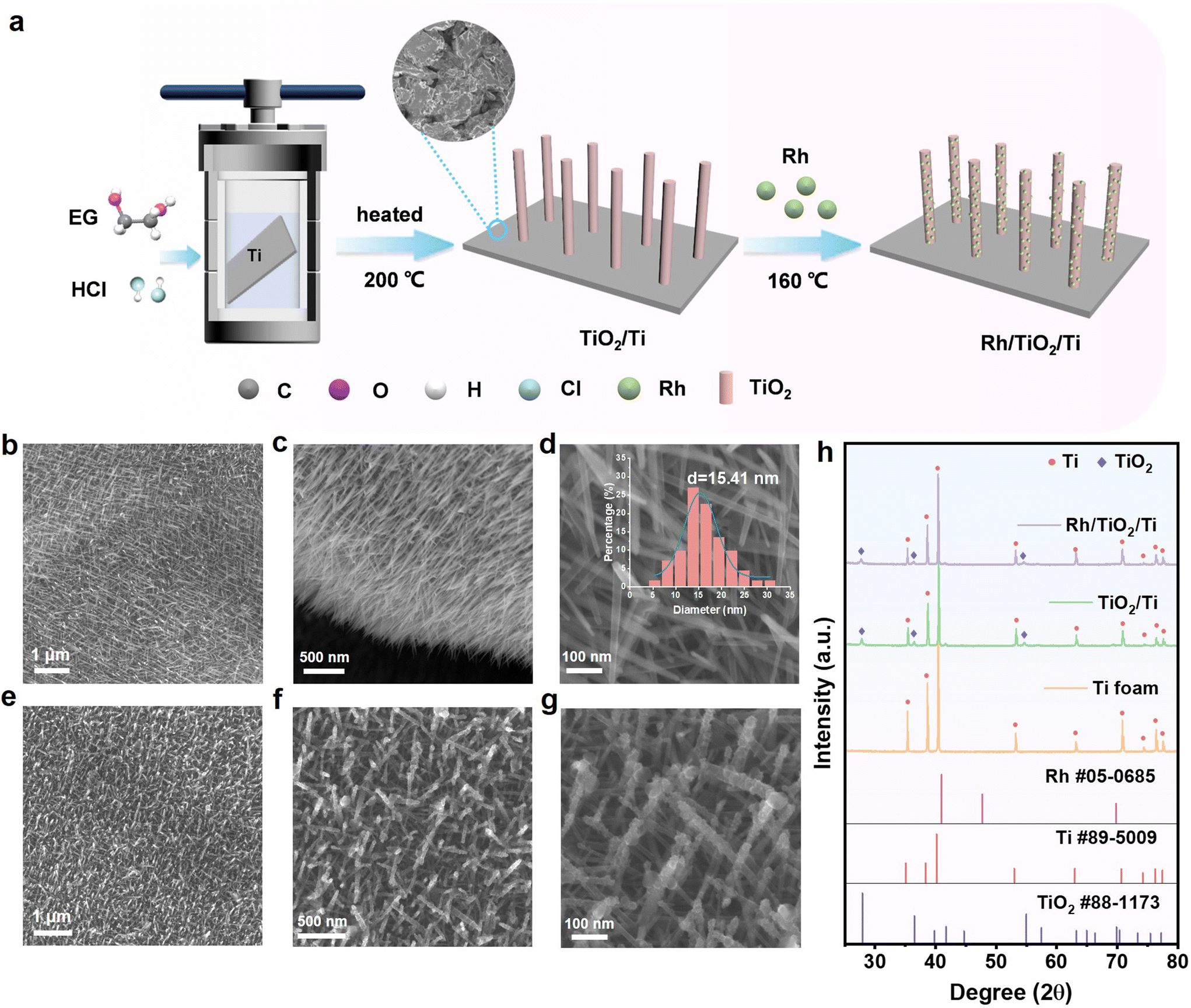 | ||
| Fig. 1 (a) Schematic diagram of the preparation process of Rh/TiO2/Ti. (b–d) SEM images of TiO2/Ti. Inset shows the diameter size distribution diagram. (e–g) SEM images of Rh/TiO2/Ti. (h) XRD patterns of Rh/TiO2/Ti, TiO2/Ti and Ti foam. | ||
The scanning electron microscopy (SEM) technique was employed to investigate the morphological features of the prepared samples. As shown in Fig. 1b–d, representative SEM images illustrate the one-dimensional (1D) morphology of TiO2/Ti, featuring a uniform diameter of 15.41 nm and a smooth surface. Images from different angles confirm the complete encapsulation of Ti foam by the TiO2 nanoarray layer, which not only prevents the Ti foam from hydrogenation-induced degradation but also offers favorable anchoring sites for metals.28 The surface of TiO2 nanoarrays become rougher after the deposition of Rh nanoparticles, as depicted in Fig. 1e–g. To confirm the successful deposition of Rh nanoparticles, elemental mapping and EDX spectroscopy were conducted and the images are provided in Fig. S1.† The elements of Ti, O, and Rh are distributed uniformly and the mass ratio of Rh is 0.64% as tested by EDX, far higher than the value of 0.03% measured by ICP-MS testing. It might be attributed to the fact that the Ti substrate cannot be detected totally during the EDX test because of the complete coverage by TiO2 arrays. According to the ICP-MS result, the mass loading of Rh is 35 μg cm−2. These results demonstrate the successful anchoring of Rh nanoparticles on the surface of the vertical TiO2 array.
To investigate the influencing factors in the preparation of TiO2 nanoarrays, we conducted explorations focusing on solvents and their proportions. As shown in Fig. S2a,† when pure water is used as the solvent, the TiO2 rods are relatively coarse without a vertical growth morphology. The photograph reveals severe fragmentation of the Ti substrate. By adding a small amount of ethylene glycol to the aqueous solution, the fragmentation of the Ti sheet is significantly alleviated. When the volume ratio of water to EG is 1:1, robust and thinner nanorods begin to appear. As the volume of ethylene glycol in the solvent increases to 80%, uniformly thick and dense TiO2 nanoarrays are obtained, accompanied by more black spots visible on the electrode photograph. When the solvent is entirely EG, as previously described, vertically grown nanorrays with a diameter of 15.41 nm can be observed. The photograph of the electrode is shown in Fig. S1f;† it exhibited a pure black color. The difference in morphology caused by different solvents might be attributed to differences in polarity and reaction rates.
We also studied the effect of the substrate (TiO2/Ti sheet, TiO2/Ti foam, and Ti foam) on the distribution of Rh nanoparticles. The SEM images of the as-obtained TiO2/Ti sheet, Rh/TiO2/Ti sheet, and Rh/Ti foam are illustrated in Fig. S3.† When Ti foam is replaced with a Ti sheet, uniform and vertically grown TiO2 nanoarrays could still be obtained using the same method (Fig. S3a–c†). Upon the loading of Rh, the structure of TiO2 nanoarrays remains unaffected, while an augmentation in surface roughness is observed (Fig. S3d–f†). When the Rh-loaded substrate is Ti foam, as depicted in Fig. S3g–i,† it is evident that Rh particles grown directly on Ti foam exhibit noticeable agglomeration and possess significantly larger dimensions in comparison with those particles grown on TiO2/Ti foam. Therefore, TiO2 nanoarrays serve as an effective anchoring medium for Rh nanoparticles, facilitating a more refined and homogeneous distribution.
X-ray diffraction (XRD) patterns of Rh/TiO2/Ti and other samples are presented in Fig. 1h. It can be observed that the peaks of Ti foam match well with the standard peaks of Ti (Ti PDF #89-5009). Comparing Rh/TiO2/Ti, TiO2/Ti, and Ti foam, it was observed that the TiO2 nanoarrays exhibit characteristic diffraction peaks corresponding to TiO2 (PDF #88-1173), which are marked with diamonds in the diagram. Specifically, no additional diffraction peaks are observable after the deposition of Rh nanoparticles onto TiO2/Ti, possibly due to the low loading quantity of Rh or its excellent dispersion.
Further confirmation of the Rh/TiO2/Ti structure was achieved through Transmission Electron Microscopy (TEM) images. In Fig. 2a, the distinct lattice fringes of TiO2 validate its crystallinity. Fig. 2b presents the corresponding Selected Area Electron Diffraction (SAED) pattern, suggesting the single-crystal structure of TiO2. The lattice spacing is measured along α and β directions in Fig. 2c. As depicted in Fig. 2d and e, the lattice distance of 0.325 nm and 0.224 nm correspond to the (110) and (200) planes of rutile TiO2, respectively. Fig. 2f–i demonstrates the successful loading of Rh nanoparticles onto TiO2. The particle size distribution was analyzed as shown in Fig. S4.† It shows that the average particle size of Rh is around 3.33 nm. Moreover, the High-Resolution TEM (HRTEM) image reveals a close interfacial contact between the Rh nanoparticles and TiO2. This finding also suggests that TiO2 possesses the capability to hinder the agglomeration of Rh, thereby ensuring the abundance of catalytically active sites. The structural characterization of the Rh nanoparticles within the marked area in Fig. 2f, as shown in Fig. 2h, reveals the existence of the Rh (111) plane. The elemental mapping shown in Fig. 2i demonstrates the homogeneous distribution of Ti, O, and Rh within Rh/TiO2/Ti. These morphologies and microstructural characterization indicate the successful preparation of TiO2/Ti and Rh/TiO2/Ti nanoarrays.
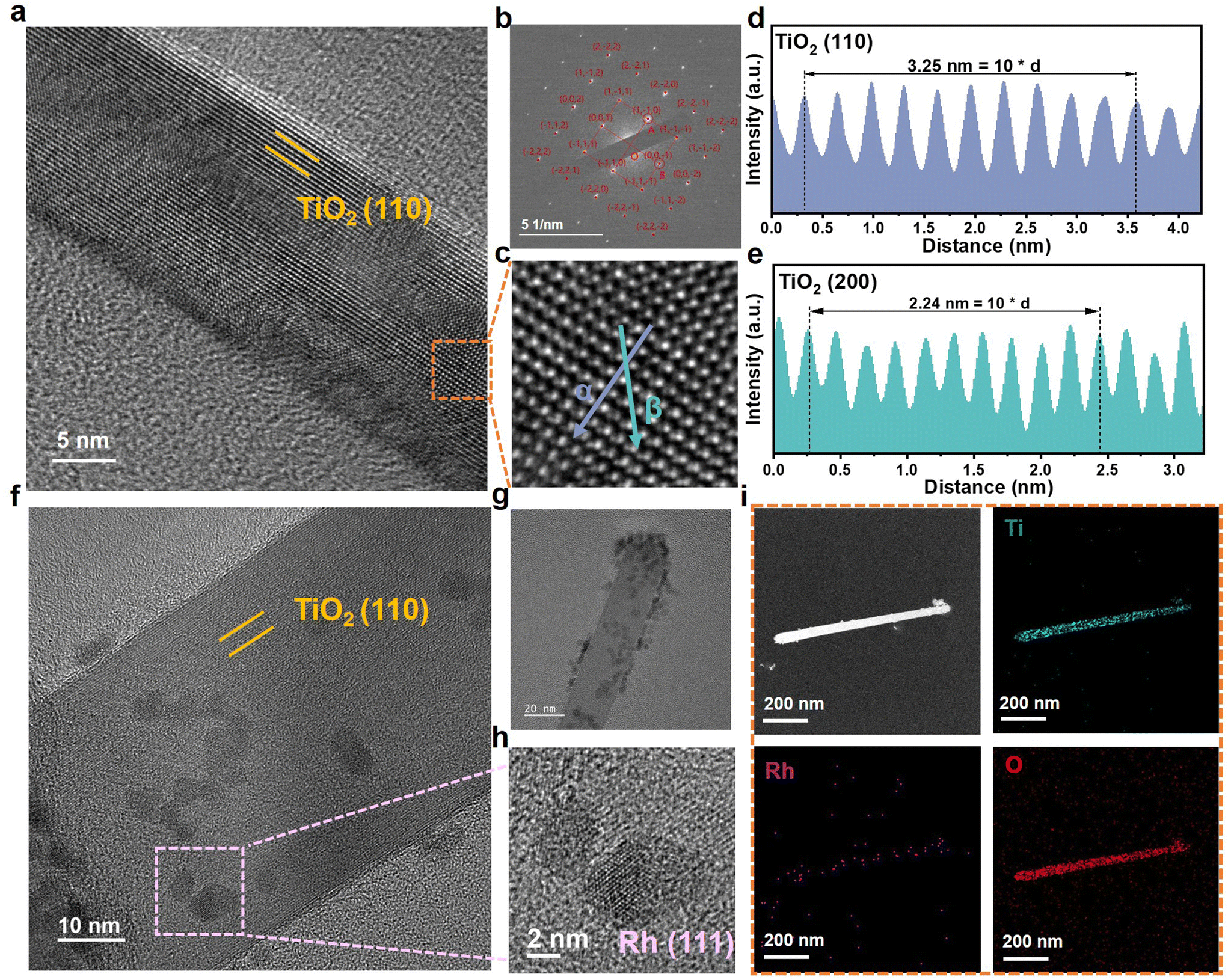 | ||
| Fig. 2 Morphological and structural characterization of TiO2/Ti and Rh/TiO2/Ti nanoarrays grown in situ on Ti foam. (a) The HRTEM image, (b) the corresponding SAED, (c) the enlarged HRTEM image labeled with α and β arrows, and (d and e) the corresponding intensity profiles and the lattice fringe spacing of the TiO2 support; (f) the HRTEM image and (g) the TEM image of Rh/TiO2/Ti nanoarrays. (h) HRTEM of the Rh site, and (i) EDX elemental mapping profiles of Rh/TiO2/Ti. | ||
X-ray photoelectron spectroscopy (XPS) was used to reveal the electronic configurations and the surface compositions of Rh/TiO2/Ti. As shown in Fig. 3, the XPS spectrum of Rh/TiO2/Ti verifies the coexistence of Rh, Ti, and O. The high-resolution Rh 3d spectra of Rh/TiO2/Ti and Rh/Ti are depicted in Fig. 3a and both Rh/TiO2/Ti and Rh/Ti exhibit four peaks. The peaks at about 306.89 eV (Rh 3d5/2) and 311.55 eV (Rh 3d3/2) are indicative of metallic Rh of Rh/TiO2/Ti; while those at 307.03 eV (Rh 3d5/2) and 311.69 eV (Rh 3d3/2) correspond to metallic Rh in Rh/Ti. The remaining peaks are attributed to Rh3+. Notably, the Rh 3d peaks in Rh/TiO2/Ti undergo a negative shift of about 0.14 eV compared to Rh/Ti, indicative of more electron transfer between TiO2 and Rh, demonstrating the strong metal–support interaction between Rh sites and the TiO2 support.25,28 The Ti 2p XPS spectrum of Rh/TiO2/Ti (Fig. 3b) distinctly shows two peaks at around 458.83 and 464.49 eV, which correspond to Ti 2p3/2 and Ti 2p1/2 of Ti4+, respectively. Compared to TiO2/Ti, the binding energy of Ti 2p in Rh/TiO2/Ti exhibits a shift towards higher values by approximately 0.3 eV, which is attributed to the electronic transfer from TiO2 to Rh. As presented in Fig. 3c, the O 1s XPS spectra of Rh/TiO2/Ti and TiO2/Ti can be fitted with two peaks of lattice oxygen (OL) and oxygen deficiency (VO).5,9,25,29 The lattice oxygen is associated with the Ti–O–Ti bond in the TiO2 layer. By comparing the integrated areas of the fitted peaks, it is evident that the incorporation of Rh increases the VO/OL ratio from 36.3% in TiO2/Ti to 55.5% in Rh/TiO2/Ti. This observation indicates a higher concentration of oxygen vacancies in Rh/TiO2/Ti. As reported, the presence of oxygen vacancies might enhance the electronic conductivity and modulate the electronic environment, thereby boosting the HER activity of the catalyst.17 Specifically, a notable shift of the OL peak towards a higher binding energy by 0.22 eV is observed in Rh/TiO2/Ti (529.99 eV) compared to TiO2/Ti (529.77 eV). The positive shift of OL and Ti 2p further reveals the transferring of electrons from TiO2 to Rh, providing more evidence for the metal–support interaction between Rh and TiO2.28 The higher electron density around Rh atoms in Rh/TiO2/Ti might result in a weaker surface binding with H atoms, and thus enhance the HER activity.30,31 The formation energies of VO and VTi in Rh/TiO2/Ti were calculated. As shown in Fig. 3d, the formation energies of VTi is significantly higher than VO, indicating the easier formation of VO in Rh/TiO2/Ti.
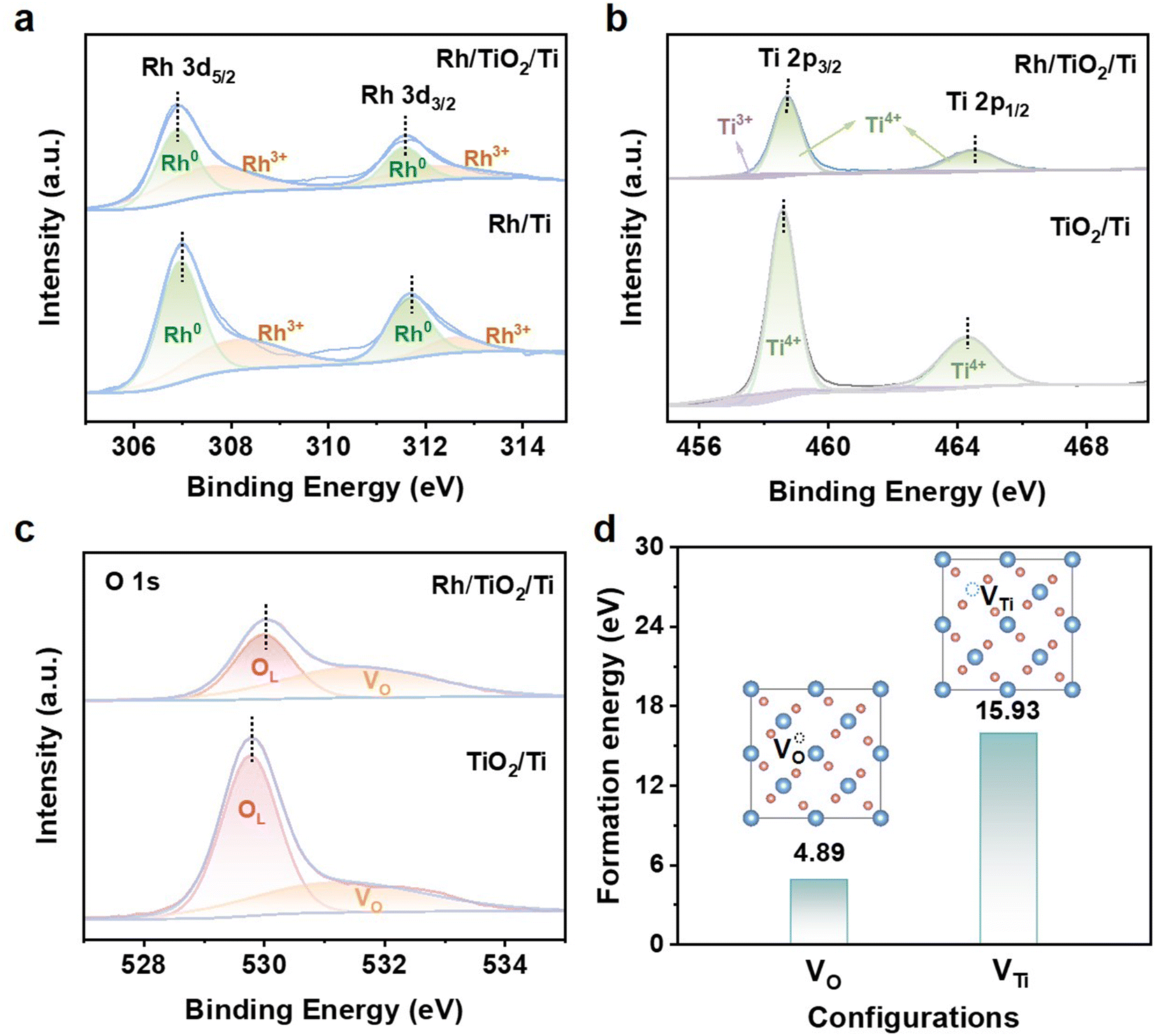 | ||
| Fig. 3 XPS spectra of Rh/TiO2/Ti, TiO2/Ti and Rh/Ti. (a) Rh 3d, (b) Ti 2p, and (c) O 1s. (d) Formation energy of VO and VTi in Rh/TiO2/Ti. | ||
3.2 Electrocatalytic performance
In 1.0 M KOH electrolyte, the electrochemical HER performance of the binder-free Rh/TiO2/Ti electrode was evaluated using a three-electrode system. Firstly, the hydrogen evolution performances of Rh/TiO2/Ti with various Rh loadings were tested. As shown in Fig. S5,† the hydrogen evolution activity and mass activity achieve the highest values when the Rh loading is 35 μg cm−2. Then, TiO2/Ti, Rh/Ti, and commercial Pt/C catalysts were evaluated under identical conditions for comparison. Fig. 3a–c present polarization curves, comparison of overpotentials, and Tafel slopes of different catalysts toward the HER. The lower overpotential and Tafel slope suggest that Rh/TiO2/Ti possesses superior activity and favorable HER kinetics, potentially attributed to the strong interaction between Rh and the TiO2 support. Additionally, polarization curve tests were conducted on the Rh/TiO2/Ti sheet catalyst and the Rh/Ti sheet catalyst, both grown in situ on Ti sheet substrates. As illustrated in Fig. S6,† the activity of the Rh/TiO2/Ti sheet surpasses that of the Rh/Ti sheet, suggesting that the incorporation of TiO2 may provide a stronger metal–support interaction, thereby enhancing the performance of Rh. Compared to the Rh/TiO2/Ti sheet, the enhanced activity of Rh/TiO2/Ti foam can be ascribed to the fact that Ti foam can enhance both the number of active sites and the permeation of electrolytes.To further explore the charge transfer kinetics, electrochemical impedance spectroscopy (EIS) was conducted. The EIS spectra presented in Fig. 4d and Fig. S7† reveal that TiO2/Ti exhibits the highest resistance, while Rh/TiO2/Ti demonstrates the lowest charge transfer resistance. The result is reasonable due to the inherently low conductivity of the semiconductor. Furthermore, this observation implies that the loading of Rh metal onto TiO2 can improve the conductivity of the semiconductor, thereby accelerating the charge transfer.
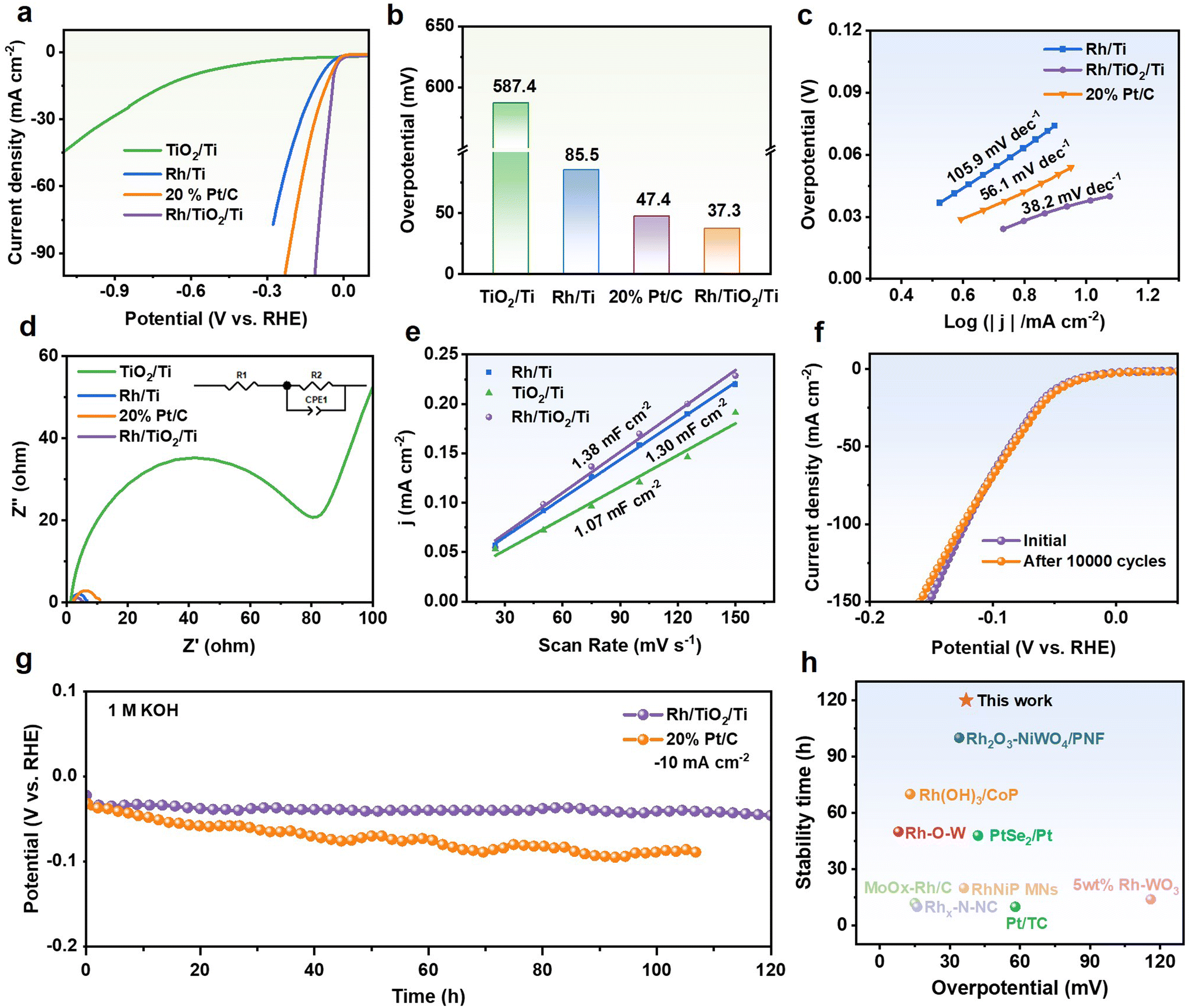 | ||
| Fig. 4 HER performances in 1.0 M KOH solution. (a) Polarization curves, (b) comparison of overpotentials at a current density of −10 mA cm−2, and (c) the corresponding Tafel plots of Rh/TiO2/Ti, TiO2/Ti, Rh/Ti and commercial Pt/C. (d) Nyquist plots of Rh/TiO2/Ti, TiO2/Ti, Rh/Ti and commercial Pt/C. The enlarged plots are shown in Fig. S7.† (e) Electrochemical double-layer capacitance of Rh/TiO2/Ti, TiO2/Ti, Rh/Ti. The stability tests for Rh/TiO2/Ti. (f) Polarization curves before and after 10000 CV cycles. (g) The chronopotentiometry curves of Rh/TiO2/Ti and commercial Pt/C at −10 mA cm−2. (h) The overpotentials and stability time comparison between Rh/TiO2/Ti and recently published HER electrocatalysts at a current density of −10 mA cm−2, with respect to Table S1.† | ||
Electrochemical surface area (ECSA) is a crucial parameter for assessing the catalytically active sites, which determines the specific activity to a large extent.25 The electrochemical double-layer capacitance (Cdl), which is commonly considered to be proportional to the ECSA, can be derived from cyclic voltammetry (CV) curves recorded at various scan rates within the non-faradaic region (Fig. S8†). The results imply that Rh/TiO2/Ti possesses a larger electrochemically active area and more catalytically active sites, which are closely related to its excellent catalytic activity (Fig. 4e).
Apart from catalytic activity, Rh/TiO2/Ti also demonstrates remarkable long-term catalytic durability during the alkaline HER process. In Fig. 4f, the polarization curve of Rh/TiO2/Ti exhibits almost no shift after 10000 CV cycles. The chronopotentiometry (CP) test at −10 mA cm−2 also indicates that Rh/TiO2/Ti possesses excellent electrochemical durability. As depicted in Fig. 4g, during a long-term test of 120 hours, the overpotential of the Rh/TiO2/Ti catalyst hardly increases. Furthermore, SEM and XRD characterization studies after cycling and CP test reveal that the Rh/TiO2/Ti catalyst retains its original crystal structure and morphology (Fig. S9–11†), which confirms its structure stability. Impressively, compared to previously reported HER electrocatalysts, the Rh/TiO2/Ti catalyst exhibits a lower overpotential and superior catalytic stability (Fig. 4h and Table S1†), thereby highlighting its efficient utilization of precious metal resources.
To investigate the hydrogen evolution performance of Rh/TiO2/Ti in a wide range of pH values, we evaluated its activity and stability in 0.5 M H2SO4. As depicted in Fig. 5a–c, Rh/TiO2/Ti exhibits an overpotential comparable to that of Pt and possesses the lowest Tafel slope of 25.5 mV dec−1. The Nyquist plot (Fig. 5d and Fig. S12†) reveals that Rh/TiO2/Ti exhibits a reduced charge transfer resistance, attributed to the incorporation of Rh metal nanoparticles. As depicted in Fig. S13† and Fig. 5e, Rh/TiO2/Ti possesses the highest Cdl value, indicating that it has more active surface sites in acidic electrolytes when compared with Rh/Ti and TiO2/Ti. Furthermore, Rh/TiO2/Ti also demonstrates remarkable stability in a severe acidic environment. As illustrated in Fig. 5f, the polarization curve of Rh/TiO2/Ti exhibits no significant change after 7500 CV cycles, suggesting its durability. Additionally, Rh/TiO2/Ti demonstrates a surprising stability in the HER for nearly 120 hours, with a negligible activity decay at −10 mA cm−2. This performance exceeds the operating life of commercial Pt/C. Compared to recently reported noble metal-based catalysts, Rh/TiO2/Ti exhibits superior performance in terms of both overpotential and stability (Fig. 5h; Table S2†). As a binder-free electrode, the synthesized Rh/TiO2/Ti catalyst demonstrates excellent activity and durability in both alkaline and acidic environments as concluded above. This highlights its notable competitiveness when compared to recently reported high-efficiency Rh-based electrocatalysts.
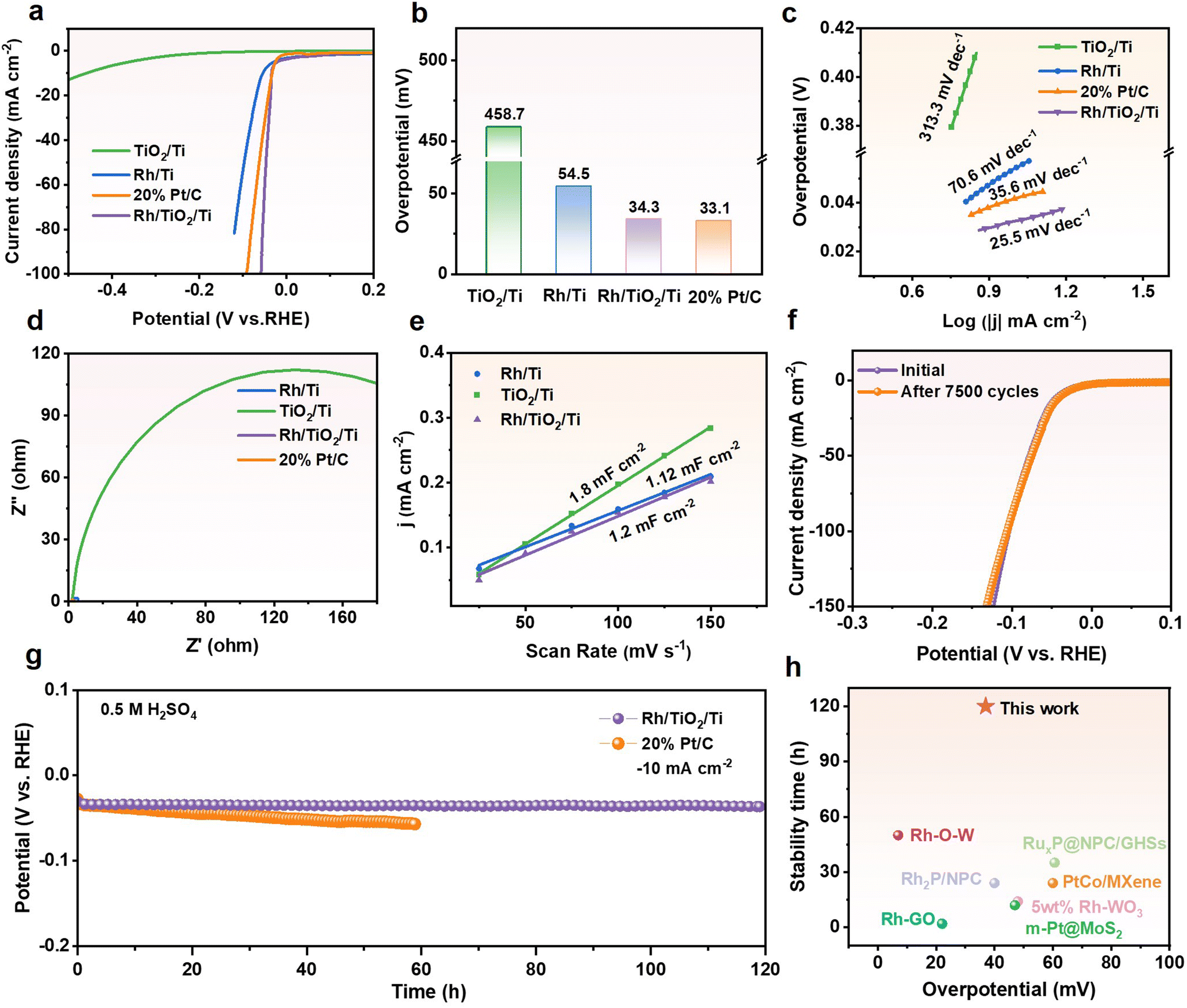 | ||
| Fig. 5 Electrochemical HER performances in 0.5 M H2SO4 solution. (a) Polarization curves, (b) comparison of overpotentials at −10 mA cm−2, and (c) the corresponding Tafel plots of Rh/TiO2/Ti, TiO2/Ti, Rh/Ti and commercial Pt/C. (d) Nyquist plots of Rh/TiO2/Ti, TiO2/Ti, Rh/Ti and commercial Pt/C. The enlarged plots are presented in Fig. S12.† (e) Electrochemical double-layer capacitance of Rh/TiO2/Ti, TiO2/Ti, Rh/Ti. The stability tests for Rh/TiO2/Ti. (f) Polarization curves before and after 7500 CV cycles. (g) Comparison of chronopotentiometry curves of Rh/TiO2/Ti and commercial Pt/C at −10 mA cm−2. (h) Comparison of the overpotential and duration of Rh/TiO2/Ti and recently published HER electrocatalysts at −10 mA cm−2, with respect to Table S2.† | ||
The aforementioned electrochemical results reveal that the vertical Rh/TiO2/Ti nanoarrays play a pivotal role in the catalyst-liquid/gas interface interactions, subsequently influencing the hydrogen evolution performance.25 To explore the mechanism contributing to the exceptional activity and stability of catalyst, the liquid contact angle measurements were conducted to compare the wettability of Rh/TiO2/Ti and TiO2/Ti surfaces. As depicted in Fig. S14,† Rh/TiO2/Ti exhibits a smaller contact angle at the same time point, indicating its enhanced hydrophilicity and superior wettability towards electrolytes. Consequently, this enhancement facilitates improved interfacial interaction between the electrolyte and the electrode surface. According to the solid–liquid–gas interface theory, micro- and nanoscale roughness reduces the contact area between bubbles and the electrode. This reduction subsequently leads to a decrease in the adhesion force between the catalyst and the bubbles, thereby imparting robust mechanical stability to the electrocatalyst.32,33 The rapid bubble detachment from the electrode surface is crucial for maintaining the catalyst activity under prolonged operating conditions, as bubble accumulation can obstruct active sites and diminish reaction efficiency. Compared to TiO2/Ti, the loading of Rh nanoparticles markedly increases the surface roughness of Rh/TiO2/Ti. Therefore, the synergistic effect of the superior wettability and the surface roughness of the Rh/TiO2/Ti electrocatalyst contributes to its efficient and stable operation during hydrogen evolution reactions.34
DFT calculations were carried out to elucidate the electron transfer process, d-band center of Rh, and the mechanism of the enhanced HER catalytic performance. As depicted in Fig. 6a, three theoretical models of Rh/Ti, Rh/TiO2 without VO, and Rh/TiO2 were constructed. To further confirm the charge redistribution on catalysts, we conducted charge density difference analyses. As shown in Fig. 6b, in Rh/TiO2, approximately each Rh atom acquires 0.273 electrons, while in Rh/Ti, each Rh atom gains 0.267 electrons, indicative of more electron transfer between TiO2 and Rh. These findings are in alignment with the XPS findings presented in Fig. 3. Prior studies have shown that electron transfer from the support to Rh can modulate the d-band center of Rh. Therefore, we analyzed the projected density of states (PDOS) of the d orbital for Rh. As displayed in Fig. 6c, the d-band center of Rh in Rh/TiO2/Ti is closer to the Fermi level compared to Rh/Ti. It enables Rh to be more electron-rich, thereby facilitating enhanced adsorption of reactants and contributing to a reduction in the activation energy barrier.29,35–37 In comparison with Rh/TiO2/Ti without VO, the d-band center of Rh in Rh/TiO2/Ti is downshifted by 0.25 eV. According to the d-band center theory, this negative shift generally results in easy release of the H intermediate and thus improve the HER kinetics.
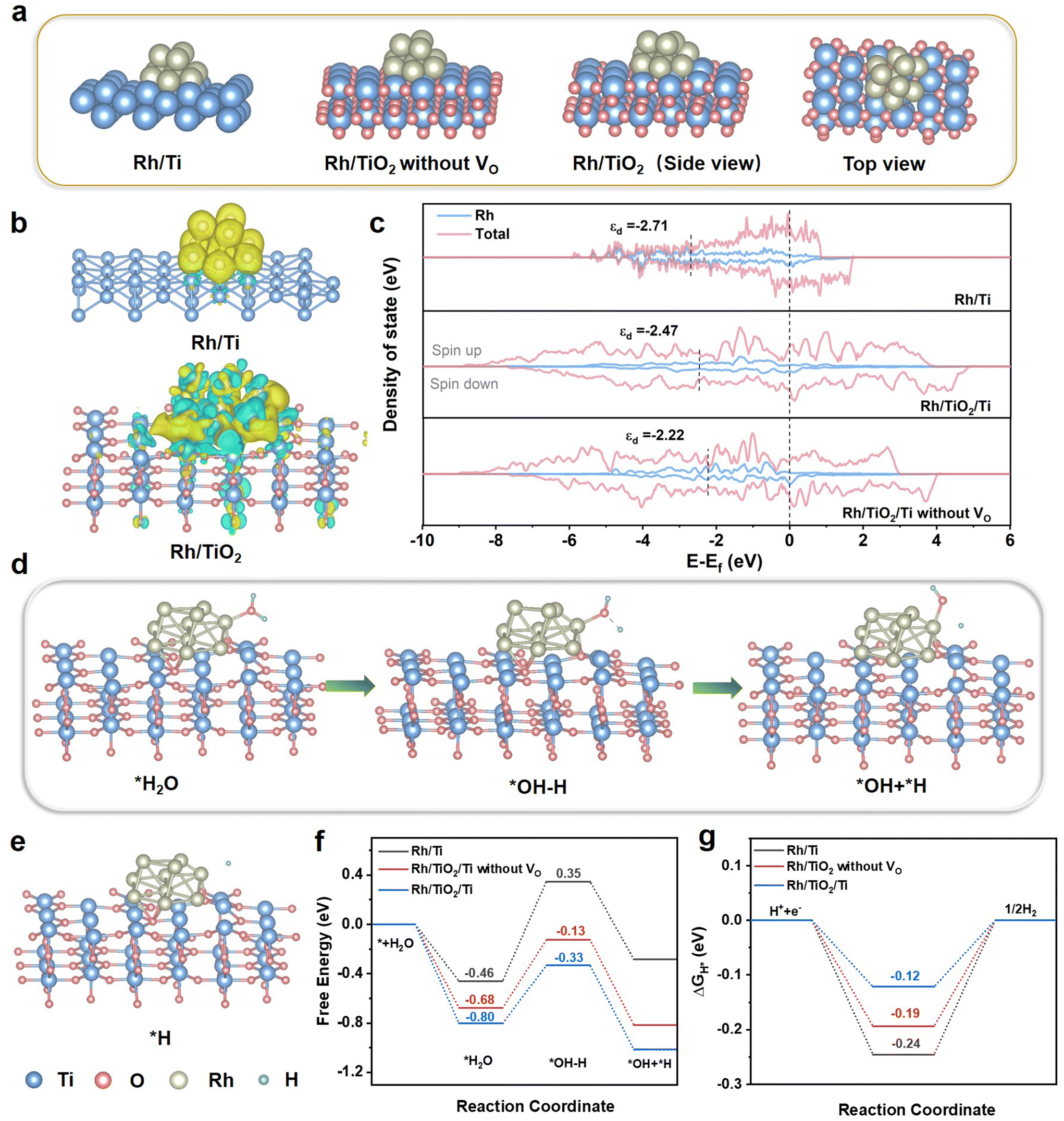 | ||
| Fig. 6 Theoretical models and calculations. (a) The theoretical model of Rh/Ti, Rh/TiO2 without VO, side view of Rh/TiO2, and top view of Rh/TiO2. (b) Difference charge density analyses of Rh/Ti and Rh/TiO2. (c) PDOS plots for Rh/Ti, Rh/TiO2/Ti without VO, and Rh/TiO2/Ti. Schematic diagram of the HER processes on Rh/TiO2 in (d) alkaline electrolyte and (e) acidic electrolyte. (f) Water absorption and dissociation energy, and (g) the ΔGH* value of Rh/Ti, Rh/TiO2/Ti and Rh/TiO2/Ti without VO. | ||
To unravel the fundamental mechanism for the improved catalytic activity, the reaction procedure models and the calculated free energy profiles for the optimized structures during the HER are shown in Fig. 6d–g. Generally, the efficiency of the alkaline HER process relies on both the water dissociation step and the H* adsorption/desorption behaviors.14,36,38,39 Thus, the water adsorption and dissociation on the catalyst surfaces were calculated.40 The water adsorption energy for Rh/TiO2/Ti is −0.80 eV, smaller than those of the Rh/Ti (−0.46 eV) and the Rh/TiO2 without VO (−0.68 eV) (Fig. 6f). The stronger water adsorption capability of Rh/TiO2/Ti is beneficial for the proceeding of the HER, since it can facilitate the Volmer reaction.14,41 For the following water dissociation, it does not occur readily on catalyst surfaces due to the uphill energy barrier.14 The Rh/TiO2/Ti shows a more favorable water dissociation energy of −0.33 eV as compared to Rh/Ti (0.35 eV) and Rh/TiO2 without VO (−0.13 eV). The Gibbs free energy of H* intermediates (ΔGH*) is widely acknowledged as a key descriptor for evaluating the catalytic performance of HER.41 Theoretically, a near-zero ΔGH* is crucial for maintaining a viable balance between H* adsorption and desorption.2,14 As shown in Fig. 6g, the ΔGH* value of Rh/TiO2/Ti is closer to the optimal value of 0 (−0.12 eV), higher than those of Rh/Ti (−0.24 eV) and Rh/TiO2/Ti without VO (−0.19 eV). These findings suggest that Rh/TiO2/Ti is beneficial for the water dissociation, H* adsorption and H2 desorption due to the interaction between TiO2 and Rh, which can effectively facilitate HER pathways.
4. Conclusion
In summary, we have reported highly efficient HER catalysts by anchoring Rh nanoparticles onto self-supported TiO2 nanoarrays with abundant VO. The design of combining Rh and the TiO2 support can ensure the electrical conductivity as well as modulate the electronic environment of Rh, which strengthens water adsorption and optimizes the adsorption energy of intermediates. Optimized Rh/TiO2/Ti demonstrates overpotentials of only 37.3 and 34.3 mV in alkaline and acidic media, respectively. Additionally, the vertical nanoarray structure, surface roughness, and the strong metal–support interaction endow the Rh/TiO2/Ti catalyst with robust stability for over 120 hours, surpassing the performance of the benchmark Pt/C catalyst and other Rh-based catalysts. The strategy presented in this study holds potential for the development of various hybrid supports tailored for a multitude of electrocatalytic applications.Data availability
All data that support the findings of this study are provided in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Guangdong Basic and Applied Basic Research Foundation (2020A1515110120), the Science Foundation of High-Level Talents of Wuyi University (2019AL029), the Open Project of State Key Laboratory of Superhard Materials, Jilin University (No. 202010), the “Lingyan” R&D Program of Zhejiang Province (No. 2023C01238, No. 2023C01133), and the Key R&D Program of Jiaxing City (No.2023BZ10002).References
- Y. Zuo, S. Bellani, M. Ferri, G. Saleh, D. V. Shinde, M. I. Zappia, R. Brescia, M. Prato, L. De Trizio, I. Infante, F. Bonaccorso and L. Manna, High-performance alkaline water electrolyzers based on Ru-perturbed Cu nanoplatelets cathode, Nat. Commun., 2023, 14, 2625 CrossRef PubMed.
- D. Yue, T. Feng, Z. Zhu, S. Lu and B. Yang, Ir Single Atom-Doped Ni2P Anchored by Carbonized Polymer Dots for Robust Overall Water Splitting, ACS Catal., 2024, 14, 3006–3017 CrossRef CAS.
- T. Chao, W. Xie, Y. Hu, G. Yu, T. Zhao, C. Chen, Z. Zhang, X. Hong, H. Jin, D. Wang, W. Chen, X. Li, P. Hu and Y. Li, Reversible hydrogen spillover at the atomic interface for efficient alkaline hydrogen evolution, Energy Environ. Sci., 2024, 17, 1397–1406 RSC.
- Y. Wang, Y. Liu, Z. Zhao, Z. Zheng, A. M. Balu, R. Luque and K. Yan, Topological iron silicide with H* intermediate modulated surface for efficient electrocatalytic hydrogenation of nitrobenzene in neutral medium, Mater. Today, 2023, 66, 84–91 CrossRef CAS.
- H. Zhu, Y. Wang, Z. Jiang, B. Deng, Y. Xin and Z. J. Jiang, Defect Engineering Promoted Ultrafine Ir Nanoparticle Growth and Sr Single–Atom Adsorption on TiO2 Nanowires to Achieve High–Performance Overall Water Splitting in Acidic Media, Adv. Energy Mater., 2024, 14, 2303987 CrossRef CAS.
- Y. Zhu, M. Klingenhof, C. Gao, T. Koketsu, G. Weiser, Y. Pi, S. Liu, L. Sui, J. Hou, J. Li, H. Jiang, L. Xu, W.-H. Huang, C.-W. Pao, M. Yang, Z. Hu, P. Strasser and J. Ma, Facilitating alkaline hydrogen evolution reaction on the hetero-interfaced Ru/RuO2 through Pt single atoms doping, Nat. Commun., 2024, 15, 1447 CrossRef CAS PubMed.
- Y. Jiang, J. Leng, S. Zhang, T. Zhou, M. Liu, S. Liu, Y. Gao, J. Zhao, L. Yang, L. Li and W. Zhao, Modulating Water Splitting Kinetics via Charge Transfer and Interfacial Hydrogen Spillover Effect for Robust Hydrogen Evolution Catalysis in Alkaline Media, Adv. Sci., 2023, 10, 2302358 CrossRef CAS PubMed.
- S. Pan, C. Li, T. Xiong, Y. Xie, F. Luo and Z. Yang, Hydrogen spillover in MoOxRh hierarchical nanosheets boosts alkaline HER catalytic activity, Appl. Catal., B, 2024, 341, 123275 CrossRef CAS.
- J. Chen, C. Chen, M. Qin, B. Li, B. Lin, Q. Mao, H. Yang, B. Liu and Y. Wang, Reversible hydrogen spillover in Ru-WO3−x enhances hydrogen evolution activity in neutral pH water splitting, Nat. Commun., 2022, 13, 5382 CrossRef CAS PubMed.
- Q. Mao, K. Deng, H. Yu, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, In Situ Reconstruction of Partially Hydroxylated Porous Rh Metallene for Ethylene Glycol–Assisted Seawater Splitting, Adv. Funct. Mater., 2022, 32, 2201081 CrossRef CAS.
- Y. Zheng, B. Zhang, T. Ma, R. Yan, W. Geng, Z. Zeng, Y. Zhang and S. Li, Nitrided Rhodium Nanoclusters with Optimized Water Bonding and Splitting Effects for pH-Universal H2-Production, Small, 2024, 20, 2307405 CrossRef CAS PubMed.
- K. Deng, Q. Mao, W. Wang, P. Wang, Z. Wang, Y. Xu, X. Li, H. Wang and L. Wang, Defect-rich low-crystalline Rh metallene for efficient chlorine-free H2 production by hydrazine-assisted seawater splitting, Appl. Catal., B, 2022, 310, 121338 CrossRef CAS.
- X. Yang, B. Ouyang, L. Zhao, Q. Shen, G. Chen, Y. Sun, C. Li and K. Xu, Ultrathin Rh Nanosheets with Rich Grain Boundaries for Efficient Hydrogen Oxidation Electrocatalysis, J. Am. Chem. Soc., 2023, 145, 27010–27021 CrossRef CAS PubMed.
- J. Wu, J. Fan, X. Zhao, Y. Wang, D. Wang, H. Liu, L. Gu, Q. Zhang, L. Zheng, D. J. Singh, X. Cui and W. Zheng, Atomically Dispersed MoOx on Rhodium Metallene Boosts Electrocatalyzed Alkaline Hydrogen Evolution, Angew. Chem., Int. Ed., 2022, 61, e202207512 CrossRef CAS PubMed.
- X. Tang, A. Yu, Q. Yang, H. Yuan, Z. Wang, J. Xie, L. Zhou, Y. Guo, D. Ma and S. Dai, Significance of Epitaxial Growth of PtO2 on Rutile TiO2 for Pt/TiO2 Catalysts, J. Am. Chem. Soc., 2024, 146, 3764–3772 CrossRef CAS PubMed.
- S. Zaman, M. Khalid and S. Shahgaldi, Advanced Electrocatalyst Supports for Proton Exchange Membrane Water Electrolyzers, ACS Energy Lett., 2024, 9, 2922–2935 CrossRef CAS.
- C. Zhang, H. Song, Z. Wang, Q. Ye, D. Zhang, Y. Zhao, J. Ma and Y. Cheng, Titanium Dioxide and N–Doped Carbon Hybrid Nanofiber Modulated Ru Nanoclusters for High–Efficient Hydrogen Evolution Reaction Electrocatalyst, Small, 2024, 2311667 CrossRef CAS PubMed.
- H.-S. Oh, H. N. Nong, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araújo, E. Willinger, R. Schlögl, D. Teschner and P. Strasser, Electrochemical Catalyst–Support Effects and Their Stabilizing Role for IrOx Nanoparticle Catalysts during the Oxygen Evolution Reaction, J. Am. Chem. Soc., 2016, 138, 12552–12563 CrossRef CAS PubMed.
- S. M. Alia, S. Stariha and R. L. Borup, Electrolyzer Durability at Low Catalyst Loading and with Dynamic Operation, J. Electrochem. Soc., 2019, 166, F1164–F1172 CrossRef CAS.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, In situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- Y. Qin, T. Yu, S. Deng, X.-Y. Zhou, D. Lin, Q. Zhang, Z. Jin, D. Zhang, Y.-B. He, H.-J. Qiu, L. He, F. Kang, K. Li and T.-Y. Zhang, RuO2 electronic structure and lattice strain dual engineering for enhanced acidic oxygen evolution reaction performance, Nat. Commun., 2022, 13, 3784 CrossRef CAS PubMed.
- K. Du, L. Zhang, J. Shan, J. Guo, J. Mao, C.-C. Yang, C.-H. Wang, Z. Hu and T. Ling, Interface engineering breaks both stability and activity limits of RuO2 for sustainable water oxidation, Nat. Commun., 2022, 13, 5448 CrossRef CAS PubMed.
- L. Yang, X. Zhang, L. Yu, J. Hou, Z. Zhou and R. Lv, Atomic Fe–N4/C in Flexible Carbon Fiber Membrane as Binder-Free Air Cathode for Zn–Air Batteries with Stable Cycling over 1000 h, Adv. Mater., 2022, 34, 2105410 CrossRef CAS PubMed.
- S. Chandrasekaran, M. Khandelwal, F. Dayong, L. Sui, J. S. Chung, R. D. K. Misra, P. Yin, E. J. Kim, W. Kim, A. Vanchiappan, Y. Liu, S. H. Hur, H. Zhang and C. Bowen, Developments and Perspectives on Robust Nano- and Microstructured Binder-Free Electrodes for Bifunctional Water Electrolysis and Beyond, Adv. Energy Mater., 2022, 12, 2200409 CrossRef CAS.
- L. Zhou, Y. Shao, F. Yin, J. Li, F. Kang and R. Lv, Stabilizing non-iridium active sites by non-stoichiometric oxide for acidic water oxidation at high current density, Nat. Commun., 2023, 14, 2514 CrossRef PubMed.
- W. Li, H. Zhang, M. Hong, L. Zhang, X. Feng, M. Shi, W. Hu and S. Mu, Defective RuO2/TiO2 nano-heterostructure advances hydrogen production by electrochemical water splitting, Chem. Eng. J., 2022, 431, 134072 CrossRef CAS.
- Y. Zuo, S. Bellani, G. Saleh, M. Ferri, D. V. Shinde, M. I. Zappia, J. Buha, R. Brescia, M. Prato, R. Pascazio, A. Annamalai, D. O. de Souza, L. De Trizio, I. Infante, F. Bonaccorso and L. Manna, Ru–Cu Nanoheterostructures for Efficient Hydrogen Evolution Reaction in Alkaline Water Electrolyzers, J. Am. Chem. Soc., 2023, 145, 21419–21431 CrossRef CAS PubMed.
- H. Yuan, J. Li, Z. Tang, Y. Wang, T. Wu, M. Huang, L. Zhao, Z. Zhao, H. Liu, C. Xu, X. Liu and W. Zhou, Enhanced interfacial stability of Pt/TiO2/Ti via Pt-O bonding for efficient acidic electrolyzer, Chem. Eng. J., 2024, 492, 152339 CrossRef CAS.
- L. Wang, Z. Mao, X. Mao, H. Sun, P. Guo, R. Huang, C. Han, X. Hu, A. Du and X. Wang, Engineering Interfacial Pt—O—Ti Site at Atomic Step Defect for Efficient Hydrogen Evolution Catalysis, Small, 2023, 20, 2309791 CrossRef PubMed.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Metal–organic frameworks as selectivity regulators for hydrogenation reactions, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- N. Yang, H. Cheng, X. Liu, Q. Yun, Y. Chen, B. Li, B. Chen, Z. Zhang, X. Chen, Q. Lu, J. Huang, Y. Huang, Y. Zong, Y. Yang, L. Gu and H. Zhang, Amorphous/Crystalline Hetero-Phase Pd Nanosheets: One-Pot Synthesis and Highly Selective Hydrogenation Reaction, Adv. Mater., 2018, 30, e1803234 CrossRef PubMed.
- N. Han, K. R. Yang, Z. Lu, Y. Li, W. Xu, T. Gao, Z. Cai, Y. Zhang, V. S. Batista, W. Liu and X. Sun, Nitrogen-doped tungsten carbide nanoarray as an efficient bifunctional electrocatalyst for water splitting in acid, Nat. Commun., 2018, 9, 924 CrossRef PubMed.
- L. Wu, F. Zhang, S. Song, M. Ning, Q. Zhu, J. Zhou, G. Gao, Z. Chen, Q. Zhou, X. Xing, T. Tong, Y. Yao, J. Bao, L. Yu, S. Chen and Z. Ren, Efficient Alkaline Water/Seawater Hydrogen Evolution by a Nanorod-Nanoparticle-Structured Ni-MoN Catalyst with Fast Water-Dissociation Kinetics, Adv. Mater., 2022, 34, 2201774 CrossRef CAS PubMed.
- X. Kang, F. Yang, Z. Zhang, H. Liu, S. Ge, S. Hu, S. Li, Y. Luo, Q. Yu, Z. Liu, Q. Wang, W. Ren, C. Sun, H.-M. Cheng and B. Liu, A corrosion-resistant RuMoNi catalyst for efficient and long-lasting seawater oxidation and anion exchange membrane electrolyzer, Nat. Commun., 2023, 14, 3607 CrossRef CAS PubMed.
- L. M. Cao, L. H. Yu, H. B. Huang, C. J. Gao, X. Huang, X. F. Zhang, X. H. Zhang, Z. Y. Du and C. T. He, A Molecular Engineered Strategy to Remolding Architecture of RuP2 Nanoclusters for Sustainable Hydrogen Evolution, Adv. Funct. Mater., 2024, 2411111 CrossRef CAS.
- S. Zhu, X. Qin, F. Xiao, S. Yang, Y. Xu, Z. Tan, J. Li, J. Yan, Q. Chen, M. Chen and M. Shao, The role of ruthenium in improving the kinetics of hydrogen oxidation and evolution reactions of platinum, Nat. Catal., 2021, 4, 711–718 CrossRef CAS.
- H. Hu, Z. Zhang, Y. Zhang, T. Thomas, H. Du, K. Huang, J. P. Attfield and M. Yang, An ultra-low Pt metal nitride electrocatalyst for sustainable seawater hydrogen production, Energy Environ. Sci., 2023, 16, 4584–4592 Search PubMed.
- Y. Liu, X. Liu, A. R. Jadhav, T. Yang, Y. Hwang, H. Wang, L. Wang, Y. Luo, A. Kumar, J. Lee, H. T. D. Bui, M. Gyu Kim and H. Lee, Unraveling the Function of Metal-Amorphous Support Interactions in Single-Atom Electrocatalytic Hydrogen Evolution, Angew. Chem., Int. Ed., 2022, 61, e202114160 Search PubMed.
- H. Luo, P. Yu, G. Li and K. Yan, Topological quantum materials for energy conversion and storage, Nat. Rev. Phys., 2022, 4, 611–624 Search PubMed.
- G. Lin, Z. Zhang, Q. Ju, T. Wu, C. U. Segre, W. Chen, H. Peng, H. Zhang, Q. Liu, Z. Liu, Y. Zhang, S. Kong, Y. Mao, W. Zhao, K. Suenaga, F. Huang and J. Wang, Bottom-up evolution of perovskite clusters into high-activity rhodium nanoparticles toward alkaline hydrogen evolution, Nat. Commun., 2023, 14, 280 Search PubMed.
- H. Hu, Z. Zhang, L. Liu, X. Che, J. Wang, Y. Zhu, J. Paul Attfield and M. Yang, Efficient and durable seawater electrolysis with a V2O3- protected catalyst, Sci. Adv., 2024, 10, eadn7012 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi03077j |
| This journal is © the Partner Organisations 2025 |