
PIFA-mediated cyclization of methyl(2-(1-phenylvinyl)phenyl)sulfane for the concise, flexible, and scalable de novo synthesis of C3-arylated benzo[b]thiophenes†
Xinya
Liu
a,
Olivier
Provot
*a,
Christine
Tran
*a,
Jean François
Soulé
 b and
Abdallah
Hamze
*a
b and
Abdallah
Hamze
*a
aCNRS, BioCIS, Université Paris-Saclay, 91400 Orsay, France. E-mail: abdallah.hamze@universite-paris-saclay.fr
bChimie ParisTech, CNRS, Institute of Chemistry for Life and Health Sciences, PSL University, 75005 Paris, France
First published on 12th November 2024
Abstract
The well-decorated arylated benzo[b]thiophene scaffold is a pivotal structural motif with diverse applications in medicinal chemistry. This paper outlines a de novo synthesis of C3-arylated benzo[b]thiophenes, showcasing its flexibility and efficiency. The methodology involves Pd-catalyzed coupling of N-tosylhydrazones with (2-bromophenyl)(methyl)sulfane derivatives, followed by cyclization under mild conditions using [bis-(trifluoroacetoxy)iodo]benzene (PIFA) to yield the desired C3-arylated benzo[b]thiophenes. Substrate scope investigations and a gram-scale reaction underscore this protocol's synthetic potential and scalability. The developed approach offers a concise and versatile strategy for accessing this important class of compounds, providing a valuable tool for medicinal chemistry and pharmaceutical research.
Introduction
The arylated benzo[b]thiophene scaffold is a pivotal structural motif with widespread applications in medicinal chemistry and pharmaceutical research.1 Among these, C3-arylated benzo[b]thiophenes are particularly noteworthy for their potent therapeutic potential (Fig. 1).2–6 However, in contrast to C2-arylated benzo[b]thiophenes, which are well represented in marketed drugs such as raloxifene—used for osteoporosis prevention and breast cancer risk reduction—the C3-arylated analogs are currently absent from the market. This gap in the chemical space can be attributed to the significant challenges associated with their synthesis.7–9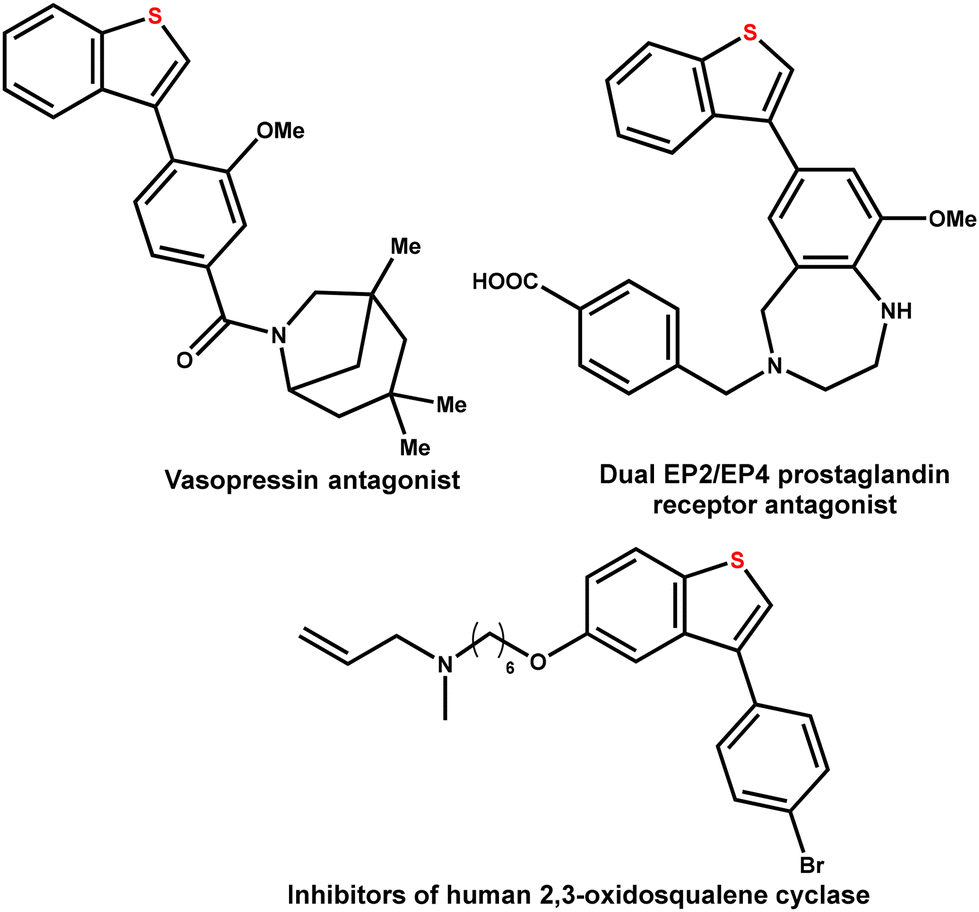 | ||
| Fig. 1 Importance of C3 functionalized benzo[b]thiophene scaffolds in biologically active molecules. | ||
The first approach involves employing starting materials that contain the benzothiophene core and performing cross-coupling reactions to diversify them. This can be achieved through classical cross-coupling arylation using 3-bromobenzo[b]thiophene and an organometallic reagent, typically catalyzed by palladium (Pd)10,11 or nickel (Ni)12 (Scheme 1A). However, only three examples of this method have been reported so far. Alternatively, recent advancements in C–H bond functionalization have been developed to bypass the pre-functionalization of the benzo[b]thiophene core.13,14 Nonetheless, achieving control over regioselectivity remains challenging, often resulting in C2-arylated products.10,15–17
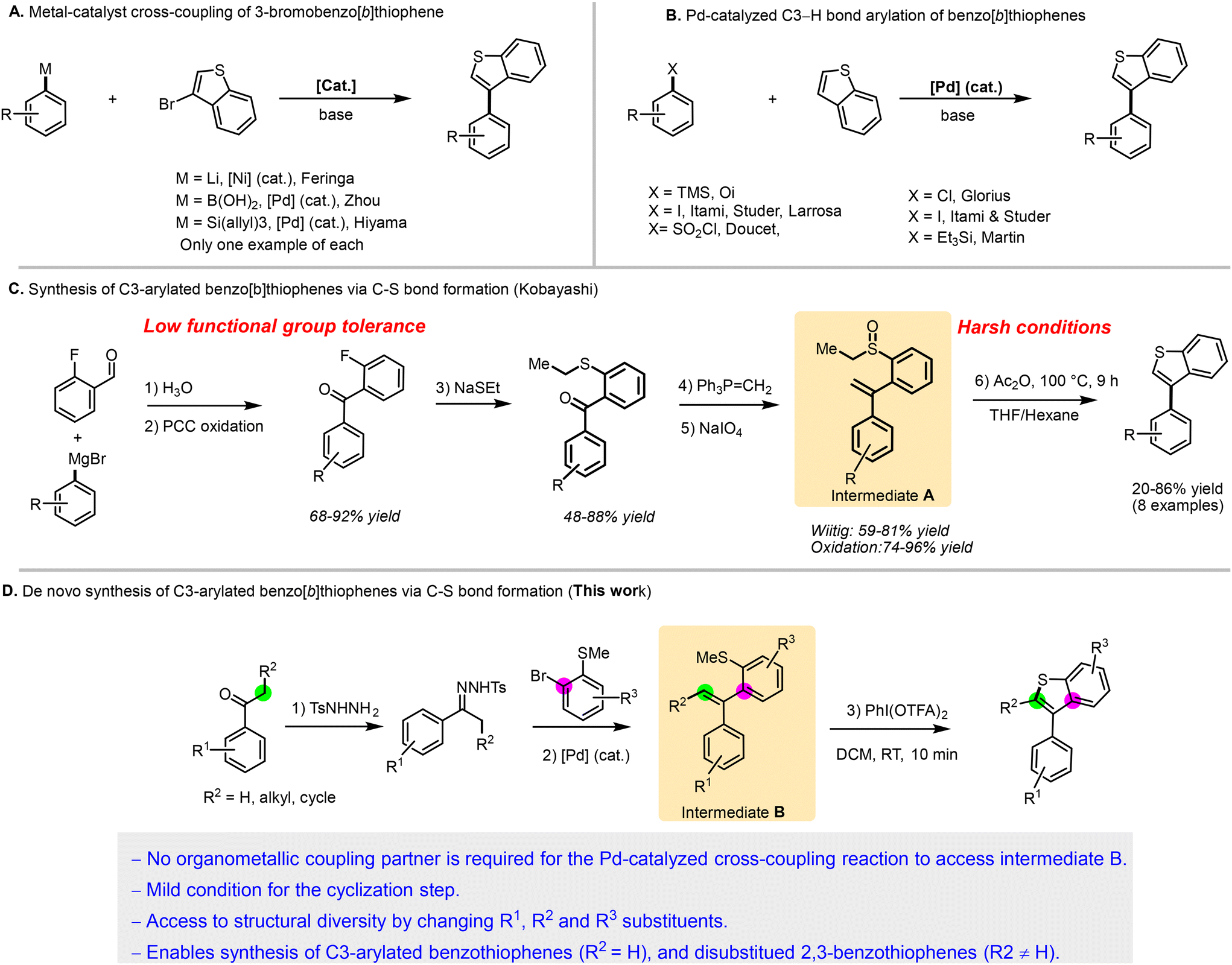 | ||
| Scheme 1 Different approaches leading to C3-arylated benzo[b]thiophenes. | ||
To achieve the C–H bond arylation of benzo[b]thiophenes at the C3 position, specialized catalysts, coupling partners, or conditions are required, which limit functional group tolerance (Scheme 1B).12,18,19 These methodologies are further constrained by the limited availability of commercial benzo[b]thiophenes, which have often low functional group diversity at positions 4 to 7. This limitation restricts the structural diversity exploration critical for comprehensive medicinal chemistry structure–activity relationship studies.
Consequently, synthesizing multi-substituted benzo[b]thiophenes, especially C3-arylated variants, remains a challenging task due to functional group tolerance and substitution pattern constraints.
A second approach for the preparation of C3-arylated benzo[b]thiophenes involves constructing the heteroarene ring via C–S bond formation. This method remains synthetically challenging and is not widely applicable, with relatively few reported examples. For instance, Kobayashi et al. provided an elegant method to access C3-arylated benzo[b]thiophenes through an interrupted Pummerer reaction of intermediate A (Scheme 1C).20 However, their synthetic scheme to prepare intermediate A is tedious, requiring five steps and involving reactions with low functional group compatibility, such as the Grignard reaction and oxidation. Moreover, the Pummerer reaction conditions are not compatible with sensitive functional groups due to the high temperatures required. These factors limit the synthesis of more complex, polyfunctionalized benzothiophenes. It should be noted that 3-substituted benzo[b]thiophenes can also be obtained starting from α-substituted 2-(1-phenylethylthio)styrenes, which are synthesized through a reaction between a free thiol and (1-bromoethyl)benzene.21
We hypothesized that intermediate B, which is closely related to intermediate A, could be synthesized more directly through Pd-catalyzed cross-coupling between N-tosylhydrazones and (2-bromophenyl)(methyl)sulfane derivatives (Scheme 1D). This Pd-catalysis approach offers remarkable tolerance to a diverse range of functional groups, enhancing the overall synthetic diversity. By employing a suitable oxidant, an interrupted Pummerer-like reaction can be triggered under milder conditions, facilitating the production of well-decorated 3-arylbenzo[b]thiophenes through a concise two-step protocol. The careful selection of the N-tosylhydrazones partner allows access to 2,3-disubstituted thiophenes, further expanding the synthetic possibilities and demonstrating the versatility of Pd-catalysis in complex molecule synthesis.
Results and discussion
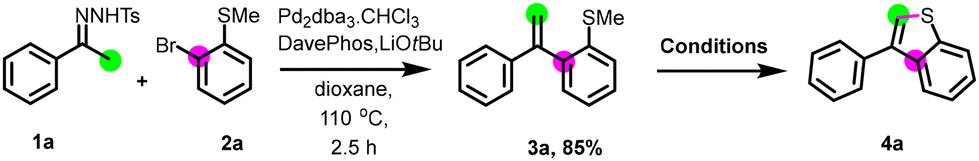
First, the preparation of intermediate 3a was achieved through the coupling between N-tosylhydrazone 1a and 2-bromothioanisole 2a using our established conditions.22 Next, we studied the cyclization step employing different additives, solvents, temperature, and time (Table 1). The utilization of Lewis acids such as BF3·Et2O, or strong acid like triflic acid in acetonitrile at room temperature for 24 h, did not lead to the formation of the desired product (entries 1 and 2). Intriguingly, the use of iron(III) chloride for a long time (60 h) gave 4a in 26% yield (entry 3). Elevating the reaction temperature to 80 °C for 3 h increased the yield up to 43%. However, extending the reaction time to 12 hours (entry 5) or substituting iron(III) chloride additive with iron(III) bromide (entry 6) failed to improve the yield of 4a. Attempts to use oxidants, such as tert-butyl hydroperoxide (TBHP, entry 7) or potassium persulfate (K2S2O8, entry 8) at room temperature, remained unsuccessful. Conducting the reaction with (diacetoxyiodo)benzene led to only 3% of 4a (entry 9). Switching to phenyliodine bis(trifluoroacetate) (PIFA) as oxidant increased the yield to 15% (entry 10). In the pursuit of optimization, we explored other solvents, utilizing PhI(OTFA)2. No reaction was observed in pentane or methyl tert-butyl ether, and a slight increase in the yield was observed when diethyl ether was used as the solvent (entries 11–13). Interestingly, under the same conditions, a significant increase in the yield was observed when CH2Cl2 was used as solvent, and 4a was obtained in a 75% yield (entry 14). Fine-tuning of the reaction conditions showed that the highest yield of 82% for the desired product (entry 15) was obtained by using 3 equivalents of PhI(OTFA)2 as an additive in CH2Cl2 (0.1 M) as the solvent, at room temperature and for a short time (10 minutes). Finally, we confirmed that the reaction does not work by switching PhI(OTFA)2 to FeCl3.|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Additive | Solvent | Temp °C | Time | Yield of 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 3a (0.2 mmol, 1.0 equiv.), additive (0.6 mmol, 3.0 equiv.), solvent (2.0 mL), under Ar. b Isolated yield. c Performing the reaction with 1 mL and 3 mL of CH2Cl2 led to a 71% and 60% yield respectively. d Accomplishment of the reaction with 2 equiv. of PhI(OTFA)2 led to a 56% yield. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | BF3·OEt2 | MeCN | r.t. | 24 h | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | TfOH | MeCN | r.t. | 24 h | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | FeCl3 | MeCN | r.t. | 60 h | 26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | FeCl3 | MeCN | 80 | 3 h | 43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | FeCl3 | MeCN | 80 | 12 h | 44 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | FeBr3 | MeCN | 80 | 12 h | 38 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | TBHP | MeCN | r.t. | 6 h | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | K2S2O8 | MeCN | r.t. | 6 h | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | PhI(OAc)2 | MeCN | r.t. | 6 h | 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | PhI(OTFA)2 | MeCN | r.t. | 6 h | 15 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | PhI(OTFA)2 | Pentane | r.t. | 6 h | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | PhI(OTFA)2 | MTBE | r.t. | 6 h | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | PhI(OTFA)2 | Et2O | r.t. | 6 h | 23 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | PhI(OTFA)2 | CH2Cl2 | r.t. | 6 h | 75 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | PhI(OTFA)2 | CH2Cl2 | r.t. | 10 min | 82c,d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | FeCl3 | CH2Cl2 | r.t. | 6 h | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Having optimized the process, we then investigated the scope of the reaction (Scheme 2) by preparing a library of C3-arylated benzo[b]thiophene using different N-tosylhydrazones derivatives. N-tosylhydrazones bearing para, meta, ortho electron-donating groups (1a–1g) reacted efficiently, and derivatives 3a–3g underwent cyclization, providing the corresponding C3-arylated benzo[b]thiophene 4a–4g in good yields. Apart from the nitro substituent on N-tosylhydrazone (1l), hydrazones having electron-withdrawing groups as para-substituents also provided intermediates 3h–3k in good to excellent yields. All (methyl)sulfane derivatives, including 3l, were cyclized to afford the products 4h–4l in a good yield, demonstrating no electronic dependency in the cyclization step. Remarkably, this method tolerates the presence of functional groups such as halogen, nitrile, and nitro paving the way for further post-functionalization. Additionally, this methodology proved suitable for the preparation of benzo[b]thiophene having 1,1′ biphenyl or naphthalene substituents (4m–4n). Moreover, N-tosylhydrazones derived from heterocycles such as 1-thiophen-2-yl-ethanone 1o, 3-acetylpyridine 1p, 3-acetyl-9-methylcarbazole 1q, and 2-acetylbenzofuran 1r were all effective for this transformation, giving the corresponding bis-heterocyclic compounds 4o–4r in good yield (Scheme 3).
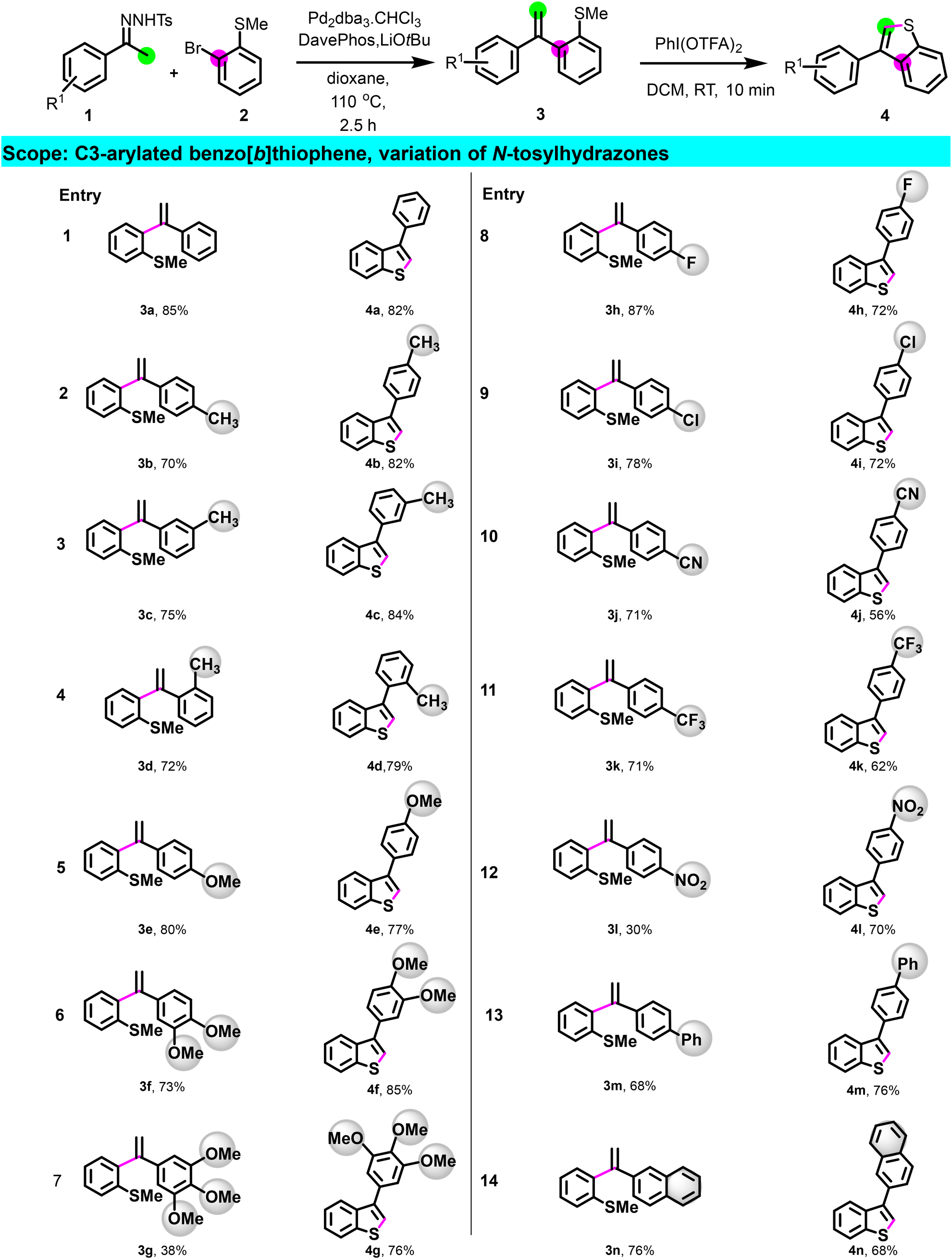 | ||
| Scheme 2 Synthesis of C3 arylated benzothiophenes, scope of N-tosylhydrazones. Standard conditions: first step: reactions were performed with 1 (2.75 mmol), 2 (2.5 mmol), Pd2dba3·CHCl3 (5 mol%), DavePhos (10 mol%), and LiOtBu (5.5 mmol), in 10 mL of dioxane at 110 °C in a sealed tube. Second step: 3a (0.2 mmol), PhI(OTFA)2 (0.6 mmol), CH2Cl2 (2 mL), rt, 10 min. | ||
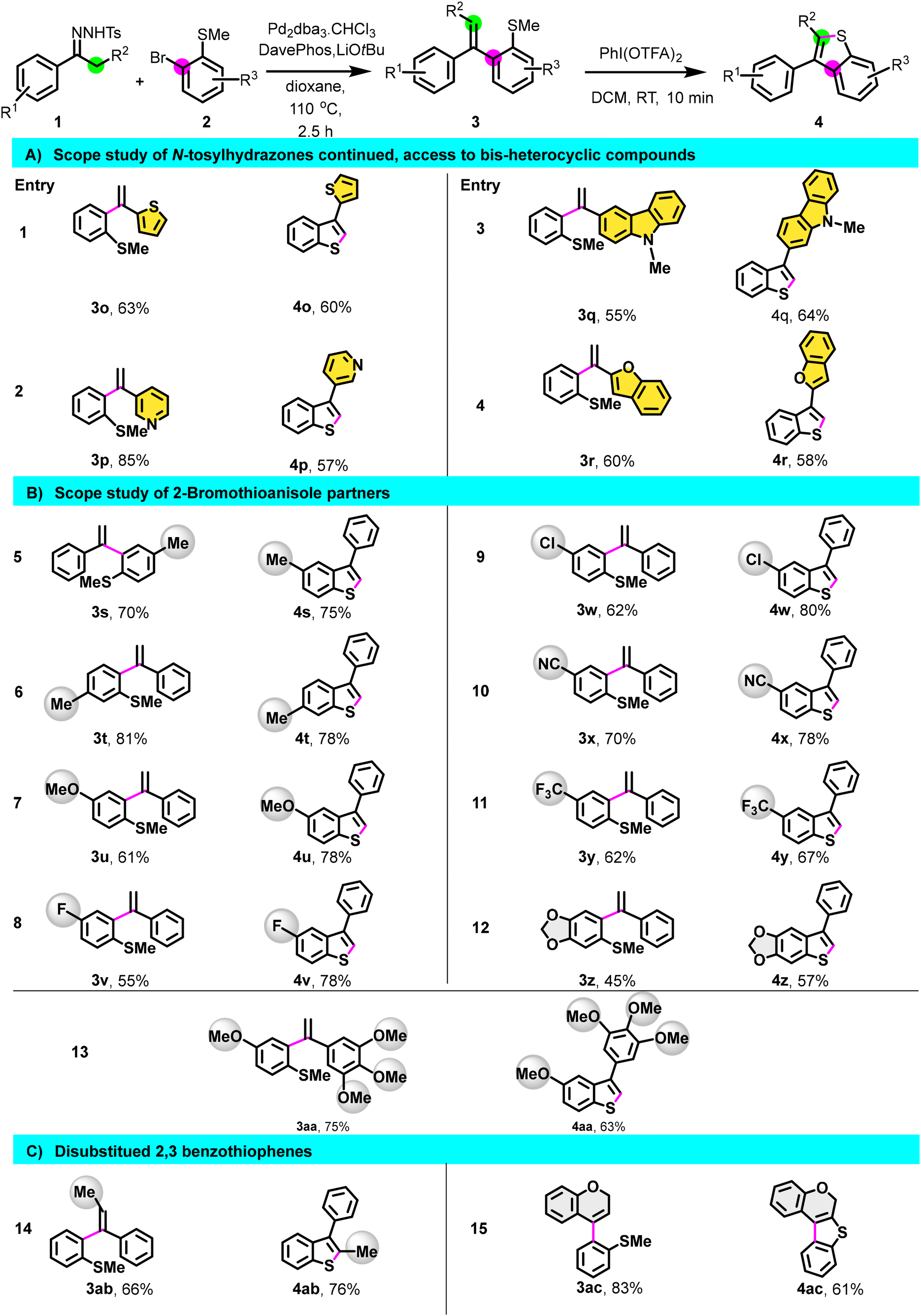 | ||
| Scheme 3 Substrate scope. Same conditions as in Scheme 2. | ||
We then shifted our focus to the scope study of 2-bromothioanisole partner (Scheme 3). Regardless of the nature of the R3 substituent, whether the electron-withdrawing group (EWG) or electron-donating group (EDG), the transformation proceeded successfully, resulting in the formation of derivatives 3s–3z.
Subsequently, the cyclization step was executed, resulting in the synthesis of 4s–4z in good yield. Interestingly, under our standard conditions, 7-phenylthieno[2′,3′:4,5]benzo[1,2-d][1,3]dioxole 4z and 4aa featuring a trimethoxyphenyl substituent were also obtained in a good yield (Scheme 3).
To broaden the scope of this reaction, we initiated the reaction using N-tosylhydrazones derived from propiophenone 1ab and 4-chromanone 1ac. Gratifyingly, following coupling and cyclization, disubstituted 2,3-benzothiophene derivatives 4ab and 4ac were obtained in a satisfactory yield (Scheme 3). All these challenging examples highlight the flexibility of this method compared to other approaches in generating molecular diversity.
To further explore the synthetic potential of our protocol, we conducted a gram-scale reaction using 2-bromothioanisole 2a and N-tosylhydrazone 1m as the substrates (Scheme 4, part A). Under our standard conditions, we isolated the desired product 3m at a yield of 78%, and obtained 1.49 g of 3-phenylbenzo[b]thiophene derivative 4m with an 80% yield, confirming its suitability for large-scale reactions. Furthermore, we showed that this transformation can proceed without the purification step of 3a (Scheme 4, part B). However, it is necessary to evaporate the dioxane before performing the cyclization step in CH2Cl2. In this case, compound 4a was obtained in a 65% isolated yield over the two steps.
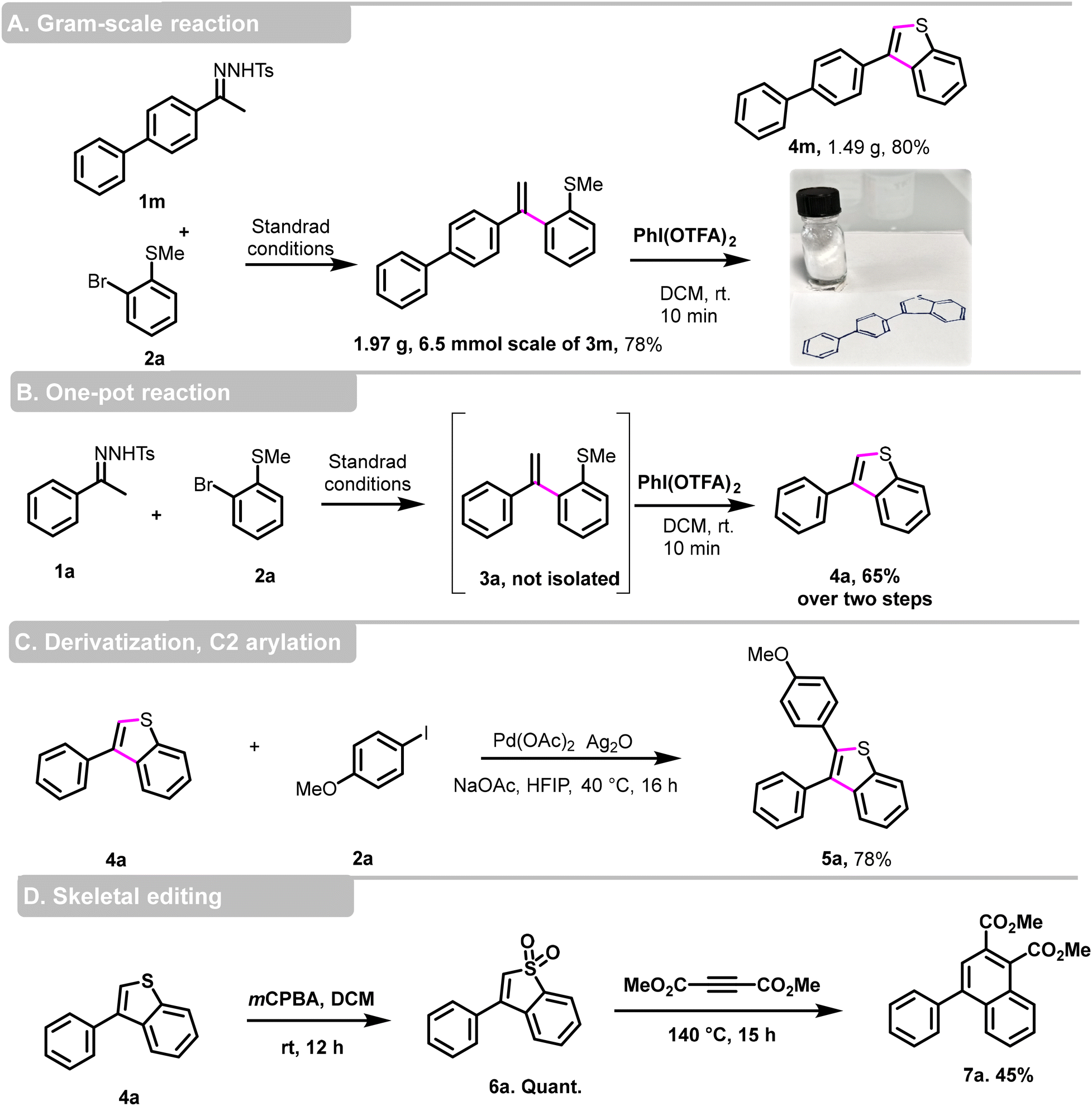 | ||
| Scheme 4 Scale-up, one-pot reaction, and synthetic transformations. | ||
To enhance the molecular diversity and high structural complexity, we achieved a C2-arylation of 3-phenylbenzo[b]thiophene 4a using Larrosa conditions23 and the 2,3 disubstituted benzo[b]thiophene 5a was obtained in a good 78% yield (Scheme 4, part C).
The field of ‘skeletal editing’ in synthetic chemistry is rapidly evolving, with researchers developing techniques to modify complex molecules by adding, deleting, or swapping individual atoms in a compound's core motif. The proliferation of skeletal editing methods aimed at manipulating molecular backbones could significantly accelerate drug discovery.24–26 In this context, 3-substituted benzo[b]thiophene 4a was used as dienes in a cycloaddition reaction with dimethyl acetylenedicarboxylate,27 after oxidation to benzo[b]thiophene S,S-dioxide, dimethyl 4-phenylnaphthalene-1,2-dicarboxylate 7a was obtained in a moderate yield of 45% (Scheme 4, part D).
Mechanism proposal
Next, we focused on establishing the mechanism of the annulative transformation. We conducted a series of control experiments (Scheme 5). Initially, we investigated the reaction's viability using the 2-(ethylsulfanyl)styrene derivative 8a under our standard conditions, which resulted in the complete conversion of 8a to 3-arylbenzo[b]thiophenes 4a, with a 67% isolated yield. This demonstrates that the reaction is not restricted to the SMe group.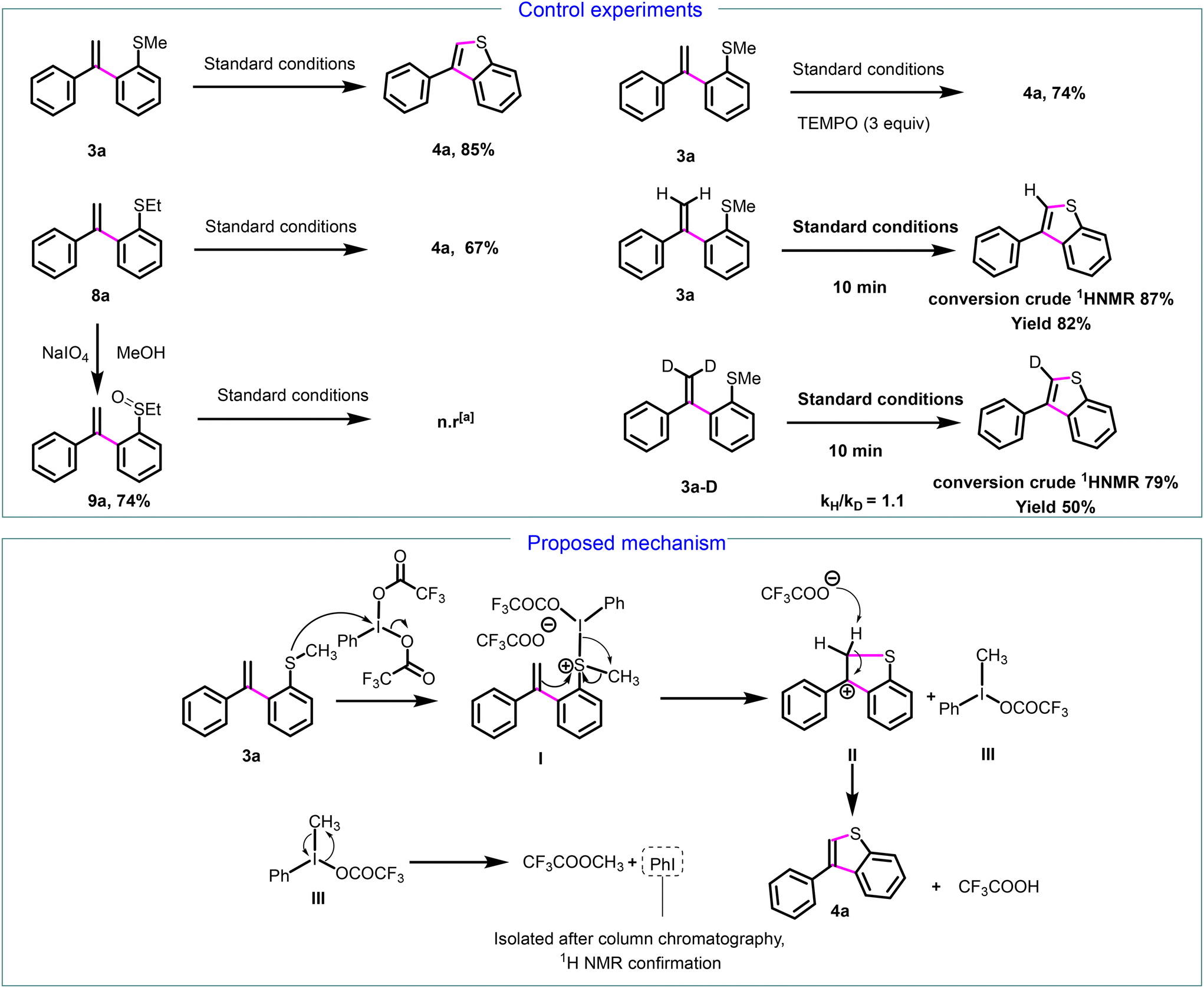 | ||
| Scheme 5 Control experiments and proposed mechanism for the formation of 3-phenylbenzo[b]thiophene 4a. an.r = no reaction. | ||
To determine if the mechanism involves the formation of a sulfoxide intermediate 9a, as observed in interrupted Pummerer reactions,20 we synthesized the sulfoxide 9a from 8a in the presence of sodium metaperiodate. No reaction was detected following the application of our standard conditions to 9a, and neither the formation of 4a nor the sulfone derivative was noted. These experiments demonstrate that, unlike interrupted Pummerer reactions, the reaction mechanism does not involve the formation of sulfoxide intermediate 9a. Next, to investigate whether this mechanism pathway entails radical formation, we conducted another control experiment under our standard conditions with the addition of TEMPO (5 equiv.). Product 4a was obtained with a 74% yield, thereby ruling out the possibility of a radical pathway (Scheme 5). Further investigations into the PIFA-mediated cyclization process were carried out through kinetic isotope effect (KIE) experiments employing 3a and deuterated 3a-D. the observed primary KIE value (for a parallel reaction kH/kD = 1.1) suggests that the deprotonation step from carbocation to 4a (see infra) is involved in the rate-determining step.
Based on the result of the control experiment and previous report on the cleavage of benzyl ethers with PIFA,28 we proposed a plausible mechanism outlined in Scheme 5 (bottom part). The reaction proceeds by the nucleophilic attack of the sulfur atom on the iodine atom of PIFA, leading to the formation of thionium complex I. Subsequently, the double bond attacks the sulfur atom of thionium salt I, resulting in the formation of carbocation II and trivalent iodine intermediate III. Trifluoroacetate anion deprotonate II, leading to 3-phenylbenzo[b]thiophene 4a. Then, intermediate III decomposes into methyl trifluoroacetate and iodobenzene which was isolated and identified by 1H NMR.
Conclusions
In conclusion, we have successfully developed a concise and flexible de novo synthesis of C3-arylated benzo[b]thiophenes involving the PIFA-mediated cyclization of methyl(2-(1-phenylvinyl)phenyl)sulfane intermediates derived from N-tosylhydrazone and 2-bromothioanisole partners. Key reaction parameters were optimized, employing PhI(OTFA)2 in CH2Cl2 at room temperature for only 10 minutes. This method demonstrates significant advantages in terms of efficiency, substrate diversity, and scalability, enabling the use of various N-tosylhydrazone derivatives and 2-bromothioanisole reagents, regardless of their electronic nature. In this study, a gram-scale reaction was successfully conducted, leading to the desired product with high efficiency, affirming the scalability of this approach. Additionally, we demonstrated the potential for expanding molecular diversity by achieving a key C2-arylation step. Our work also highlights the emerging field of ‘skeletal editing’ in synthetic chemistry, which holds promise for accelerating drug discovery. This method offers an alternative to traditional cross-coupling methods, surmounting challenges in the C3-selective functionalization of benzothiophenes while demonstrating compatibility with diverse functional groups. The synthesis of disubstituted 2,3-benzothiophenes and bis-heterocyclic compounds underscores its broad synthetic potential. Unlike interrupted Pummerer reactions, our mechanism bypasses sulfoxide intermediates, instead forming a thionium complex that is attacked by the double bond, resulting in benzo[b]thiophenes. This streamlined approach offers a valuable tool for diverse medicinal chemistry and pharmaceutical research applications. It demonstrated flexibility, efficiency, and scalability, enhancing its significance for researchers engaged in synthesizing molecular diversity for structure–activity relationship studies and pharmaceutical applications.Author contributions
The authors confirm contribution to the paper as follows: study conception and design: X. L., A. H; methodology and experiments: X. L. C. T. and O. P.; mechanism study: X. L., C. T., A. H. and J-F. S; draft manuscript preparation: X. L., O. P., C. T., J-F. S., A. H.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of this project by CNRS, Paris-Saclay University, and La Fondation ARC pour la recherche sur le cancer ARCPJA2022060005209. The authors also thank the China Scholarship Council for a fellowship (CSC) to X. L.References
- R. S. Keri, K. Chand, S. Budagumpi, S. Balappa Somappa, S. A. Patil and B. M. Nagaraja, An overview of benzo[b]thiophene-based medicinal chemistry, Eur. J. Med. Chem., 2017, 138, 1002–1033 CrossRef CAS PubMed.
- J. D. Croxtall and G. L. Plosker, Sertaconazole, Drugs, 2009, 69, 339–359 CrossRef CAS PubMed.
- A. L. Crombie, T. M. Antrilli, B. A. Campbell, D. L. Crandall, A. A. Failli, Y. He, J. C. Kern, W. J. Moore, L. M. Nogle and E. J. Trybulski, Synthesis and evaluation of azabicyclo[3.2.1]octane derivatives as potent mixed vasopressin antagonists, Bioorg. Med. Chem. Lett., 2010, 20, 3742–3745 CrossRef CAS PubMed.
- K. Liu, L. Xu, D. Szalkowski, Z. Li, V. Ding, G. Kwei, S. Huskey, D. E. Moller, J. V. Heck, B. B. Zhang and A. B. Jones, Discovery of a Potent, Highly Selective, and Orally Efficacious Small-Molecule Activator of the Insulin Receptor, J. Med. Chem., 2000, 43, 3487–3494 CrossRef CAS PubMed.
- O. Corminboeuf, S. Diethelm, C. Zumbrunn, I. Lyothier, N. Niggli, C. Gnerre, S. Jeay, F. Lehembre and C. Boss, Design of Dual EP2/EP4 Antagonists through Scaffold Merging of Selective Inhibitors, ChemMedChem, 2024, 19, e202300606 CrossRef CAS PubMed.
- R. Xiong, H. K. Patel, L. M. Gutgesell, J. Zhao, L. Delgado-Rivera, T. N. D. Pham, H. Zhao, K. Carlson, T. Martin, J. A. Katzenellenbogen, T. W. Moore, D. A. Tonetti and G. R. J. Thatcher, Selective Human Estrogen Receptor Partial Agonists (ShERPAs) for Tamoxifen-Resistant Breast Cancer, J. Med. Chem., 2016, 59, 219–237 CrossRef CAS PubMed.
- Z. He, H. J. Shrives, J. A. Fernández-Salas, A. Abengózar, J. Neufeld, K. Yang, A. P. Pulis and D. J. Procter, Synthesis of C2 Substituted Benzothiophenes via an Interrupted Pummerer/[3,3]-Sigmatropic/1,2-Migration Cascade of Benzothiophene S-Oxides, Angew. Chem., Int. Ed., 2018, 57, 5759–5764 CrossRef CAS PubMed.
- H. J. Shrives, J. A. Fernández-Salas, C. Hedtke, A. P. Pulis and D. J. Procter, Regioselective synthesis of C3 alkylated and arylated benzothiophenes, Nat. Commun., 2017, 8, 14801 CrossRef PubMed.
- R. Sang, A. Noble and V. K. Aggarwal, Chiral Benzothiophene Synthesis via Enantiospecific Coupling of Benzothiophene S-Oxides with Boronic Esters, Angew. Chem., Int. Ed., 2021, 60, 25313–25317 CrossRef CAS.
- K. Ueda, S. Yanagisawa, J. Yamaguchi and K. Itami, A General Catalyst for the β-Selective C–H Bond Arylation of Thiophenes with Iodoarenes, Angew. Chem., Int. Ed., 2010, 49, 8946–8949 CrossRef CAS.
- C. Colletto, S. Islam, F. Juliá-Hernández and I. Larrosa, Room-Temperature Direct β-Arylation of Thiophenes and Benzo[b]thiophenes and Kinetic Evidence for a Heck-type Pathway, J. Am. Chem. Soc., 2016, 138, 1677–1683 CrossRef CAS.
- D. Heijnen, J.-B. Gualtierotti, V. Hornillos and B. L. Feringa, Nickel-Catalyzed Cross-Coupling of Organolithium Reagents with (Hetero)Aryl Electrophiles, Chem. – Eur. J., 2016, 22, 3991–3995 CrossRef CAS PubMed.
- Y. Zou, G. Yue, J. Xu and J. Zhou, General Suzuki Coupling of Heteroaryl Bromides by Using Tri-tert-butylphosphine as a Supporting Ligand, Eur. J. Org. Chem., 2014, 5901–5905 CrossRef CAS.
- A. K. Sahoo, T. Oda, Y. Nakao and T. Hiyama, Cross-Coupling of Triallyl(aryl)silanes with Aryl Bromides and Chlorides: An Alternative Convenient Biaryl Synthesis, Adv. Synth. Catal., 2004, 346, 1715–1727 CrossRef CAS.
- S. Kirchberg, S. Tani, K. Ueda, J. Yamaguchi, A. Studer and K. Itami, Oxidative Biaryl Coupling of Thiophenes and Thiazoles with Arylboronic Acids through Palladium Catalysis: Otherwise Difficult C4-Selective C-H Arylation Enabled by Boronic Acids, Angew. Chem., Int. Ed., 2011, 50, 2387–2391 CrossRef CAS PubMed.
- D.-T. D. Tang, K. D. Collins and F. Glorius, Completely Regioselective Direct C–H Functionalization of Benzo[b]thiophenes Using a Simple Heterogeneous Catalyst, J. Am. Chem. Soc., 2013, 135, 7450–7453 CrossRef CAS.
- K. Funaki, T. Sato and S. Oi, Pd-Catalyzed β-Selective Direct C–H Bond Arylation of Thiophenes with Aryltrimethylsilanes, Org. Lett., 2012, 14, 6186–6189 CrossRef CAS PubMed.
- J. Wu, Y. Zong, C. Zhao, Q. Yan, L. Sun, Y. Li, J. Zhao, Y. Ge and Z. Li, Silver or cerium-promoted free radical cascade difunctionalization of o-vinylanilides with sodium aryl- or alkylsulfinates, Org. Biomol. Chem., 2019, 17, 794–797 RSC.
- K. Yuan and H. Doucet, Benzenesulfonyl chlorides: new reagents for access to alternative regioisomers in palladium-catalysed direct arylations of thiophenes, Chem. Sci., 2014, 5, 392–396 RSC.
- K. Kobayashi, M. Horiuchi, S. Fukamachi and H. Konishi, A new synthesis of benzo[b]thiophenes utilizing an interrupted Pummerer reaction, Tetrahedron, 2009, 65, 2430–2435 CrossRef CAS.
- K. Kobayashi, D. Nakamura, K. Miyamoto, O. Morikawa and H. Konishi, Convenient Synthesis of 3-Substituted Benzo[b]thiophenes by Iodine-Mediated Cyclization of α-Substituted 2-(1-Phenylethylthio)styrenes, Bull. Chem. Soc. Jpn., 2007, 80, 1780–1784 CrossRef CAS.
- T. Naret, P. Retailleau, J. Bignon, J.-D. Brion, M. Alami and A. Hamze, Palladium-Catalyzed One-Pot Synthesis of 5-(1-Arylvinyl)-1H-benzimidazoles: Overcoming the Limitation of Acetamide Partners, Adv. Synth. Catal., 2016, 358, 1833–1847 CrossRef CAS.
- C. Colletto, A. Panigrahi, J. Fernández-Casado and I. Larrosa, Ag(I)–C–H Activation Enables Near-Room-Temperature Direct α-Arylation of Benzo[b]thiophenes, J. Am. Chem. Soc., 2018, 140, 9638–9643 CrossRef CAS.
- P. Finkelstein, J. C. Reisenbauer, B. B. Botlik, O. Green, A. Florin and B. Morandi, Nitrogen atom insertion into indenes to access isoquinolines, Chem. Sci., 2023, 14, 2954–2959 RSC.
- J. Woo, A. H. Christian, S. A. Burgess, Y. Jiang, U. F. Mansoor and M. D. Levin, Scaffold hopping by net photochemical carbon deletion of azaarenes, Science, 2022, 376, 527–532 CrossRef CAS.
- S. C. Patel and N. Z. Burns, Conversion of Aryl Azides to Aminopyridines, J. Am. Chem. Soc., 2022, 144, 17797–17802 CrossRef CAS PubMed.
- T. Thiemann, Cycloaddition reactions of tetracyclones, benzo[b]thiophene Soxides, and benzo[b]thiophene S,S-dioxides with alkynes, IOSR J. Appl. Chem., 2019, 12, 24–42 CAS.
- S. Spyroudis and A. Varvoglis, Cleavage of benzyl ethers with [bis(trifluoroacetoxy)iodo]benzene, J. Chem. Soc., Chem. Commun., 1979, 615–616, 10.1039/C39790000615.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo01589d |
| This journal is © the Partner Organisations 2025 |