
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceS-Alkylation of sulfinamides with Zn-carbenoids: expanding stereoselective sulfoximine synthesis beyond NH derivatives†
Glebs
Jersovs
 ab,
Dzonatans
Melgalvis
ab,
Artis
Kinens
ab,
Pavel A.
Donets
a and
Edgars
Suna
*ab
ab,
Dzonatans
Melgalvis
ab,
Artis
Kinens
ab,
Pavel A.
Donets
a and
Edgars
Suna
*ab
aLatvian Institute of Organic Synthesis, Aizkraukles 21, LV-1006, Riga, Latvia. E-mail: edgars@osi.lv
bFaculty of Medicine and Life Sciences, Department of Chemistry, University of Latvia, Jelgavas 1, Riga LV-1004, Latvia
First published on 22nd November 2024
Abstract
Sulfoximines are experiencing steadily increasing use in the development of pharmaceuticals and agrochemicals. Although recently a number of synthetic methods to access this versatile motif have been disclosed, only NH-sulfoximines have been considered as the ultimate targets. Here, we report an approach toward enantiopure N-substituted sulfoximines via direct stereoretentive S-alkylation of parent sulfinamides with zinc carbenoids. Mechanistically, a carbon–sulfur bond is formed in the course of 1,2-metallate rearrangement featuring an unusual migration of the S-atom in the transient zincate complex. The approach accommodates a large variety of differently substituted sulfinamides and features excellent functional group compatibility.
Introduction
Owing to their chemical stability and ease of synthesis, sulfonamides and sulfones are widely employed motifs in drug discovery, agrochemistry and materials science. Closely structurally related sulfoximines retain the beneficial properties of sulfonamides and sulfones while offering the additional advantage of three diversity vectors combined with a configurationally stable sulfur stereocentre. The enriched structural variability allows accessing 3D chemical space and renders sulfoximines particularly suitable for modular molecular design that is difficult to achieve with sulfonamides and sulfones.Not surprisingly, sulfoximines have recently1 gained recognition in asymmetric synthesis,2 the development of insecticides3 and small-molecule drug discovery4 (Fig. 1). In the latter field, however, all recent clinical candidates exclusively feature NH-structures and the full substitution potential of the sulfoximine motif remains underutilized. On the other hand, a study conducted at Bayer demonstrated that N-alkylated sulfoximines indeed show promise5 in terms of favorable physicochemical and pharmacokinetic properties. Thus, cyclo-roniciclib features improved permeability, reduced efflux and lipophilicity while displaying potency, metabolic stability and solubility comparable to those of roniciclib (Fig. 1).
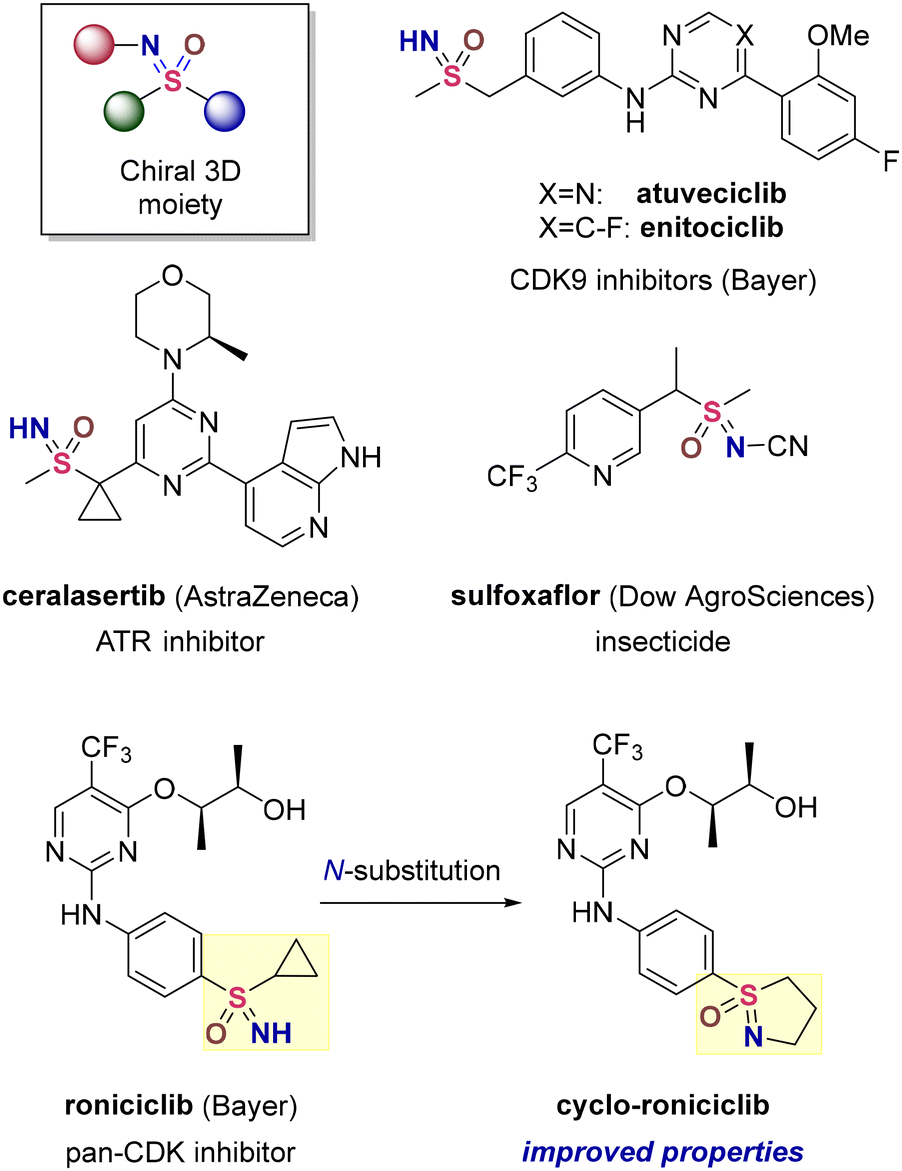 | ||
| Fig. 1 Bioactive sulfoximines. | ||
The surge of recent applications has vigorously spurred interest in the synthetic community and a stream of novel methodologies has started to fill the previously underexplored chemical space surrounding sulfoximines.6 Especially, remarkable progress has been achieved in the development of the corresponding stereoselective approaches. Thus, advances in S-imidation7 methods have allowed the direct synthesis of sulfoximines from sulfoxides.8 Moreover, asymmetric imidation of thioethers affords enantiopure sulfimides,6b,9 which in turn may be converted to sulfoximines via stereospecific oxidation. Enantiopure products are also accessible via desymmetrization10 and kinetic resolution11 of racemic NH-sulfoximines. The pioneering work of Jonson12 on nucleophilic substitution at the S-atom in sulfonimidoyl derivatives has evolved13 into another class of powerful methodologies. Despite all these advances, most methodologies focus primarily on the synthesis of NH-sulfoximines, completely neglecting possible substitution at the N-atom.
Likewise, only NH-sulfoximines are ultimately targeted by several general methods that borrow the stereogenic SNO-fragment from widely available Ellman's sulfinamide (Scheme 1). The common strategy of these approaches involves the consecutive introduction of substituents at the S-atom owing to the susceptibility of intermediate t-Bu-sulfoximines to t-Bu-cleavage (Scheme 1A). Thus, the addition of nucleophiles to sulfinamide derived S-electrophilic sulfonimidoyl fluorides allows access to a wide range of protected sulfoximines (Scheme 1B).13c Importantly, the carbamoyl protecting group plays a pivotal role as no addition occurs with other protecting groups. The inherent nucleophilic properties of sulfinamide derivatives have been exploited in another type of S-functionalization approach (Scheme 1C). Thus, both S-alkylation14a,b and S-arylation14c–e have been reported despite sulfinamides being ambident nucleophiles that predominantly exhibit N-specific reactivity (Scheme 1D).15 The desired S-selective functionalization has required the introduction of an appropriate N-protecting group such as pivaloyl for the atypical S-nucleophilicity to manifest.
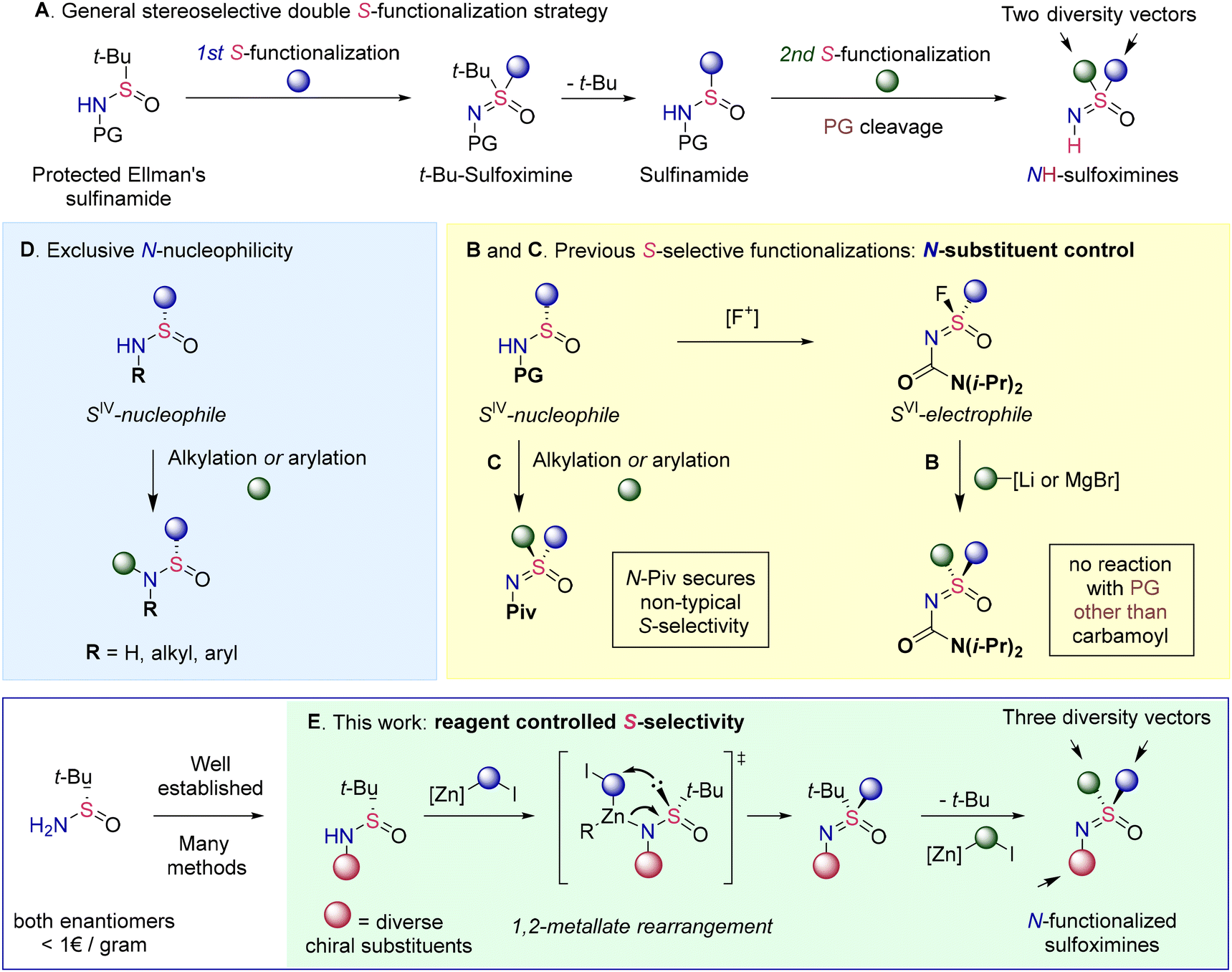 | ||
| Scheme 1 Stereoselective approaches to sulfoximines from Ellman's sulfinamide. | ||
The necessity for N-protection limits the role of Ellman's sulfinamide simply to a convenient source of S-stereogenicity. However, the chemistry developed over the years around this versatile chiral auxiliary offers innumerable opportunities for N-functionalization.16 Therefore, we realized that N-functionalized Ellman's sulfinamide derivatives could serve as excellent substrates for the modular synthesis of tri-substituted enantiopure sulfoximines. Such an approach would address the overlooked N-substitution and, considering the emerging usefulness of N-alkylated sulfoximines,4a,5 would significantly expand the diversity of accessible structures.
Herein, we report the development of a general approach for chemo- and stereospecific S-alkylation of various N-substituted sulfinamides with Zn-carbenoids (Scheme 1E). Control experiments and DFT calculations provide strong evidence that the S-selective alkylation proceeds through stereospecific 1,2-metallate rearrangement. Overall, the developed modular synthesis of tri-substituted sulfoximines allows for the reliable introduction of a broad range of N-substituents and tolerates a wide range of functional groups.
Results and discussion
In 2022, we accidentally discovered17 that the treatment of sulfinamide 1a′ with excess diethylzinc and diiodomethane delivers sulfoximine 2a′ rather than the anticipated cyclopropanation (Scheme 2). While S-alkylation of thioethers with Zn-carbenoids is precedented,18 comparable reactivity of sulfinamides has been reported only once by Zercher et al. and was primarily regarded as a synthetic obstacle.19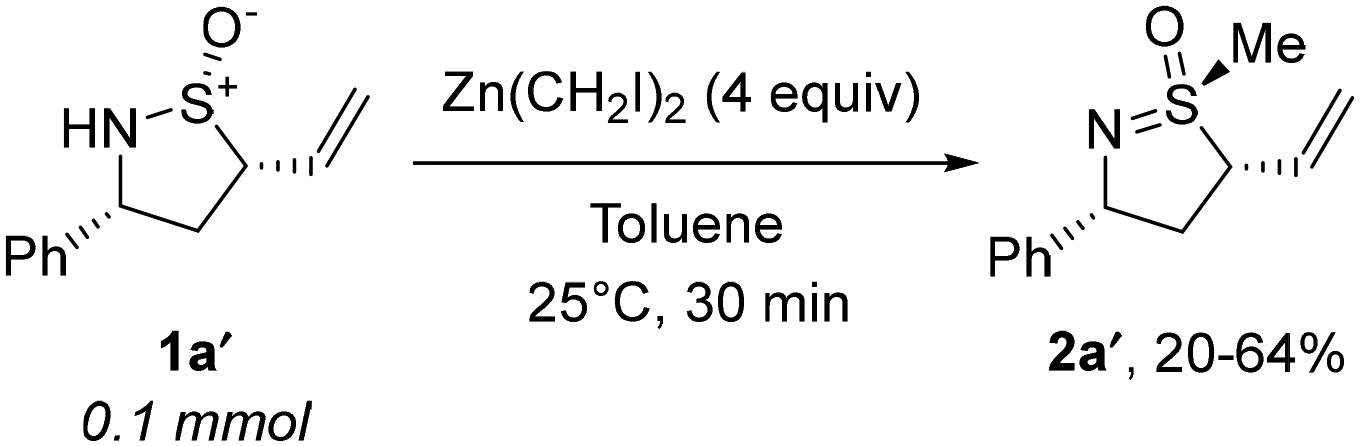 | ||
| Scheme 2 Serendipitous discovery. | ||
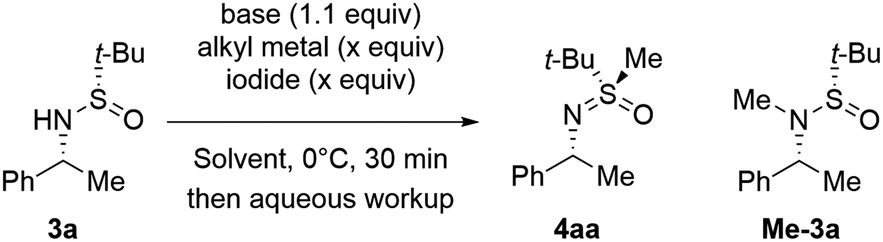
Unfortunately, the depicted transformation of 1a′ suffered from reproducibility issues. Consequently, we undertook comprehensive optimization of conditions20 using simplified tert-butyl sulfinamide 3a as a model substrate (Table 1). When sequentially treated with excess diethylzinc and diiodomethane, 3a failed to react and was quantitatively recovered. Gratifyingly, increasing the nucleophilicity of 3a by deprotonation restored the reactivity and S-methylated derivative 4aa was obtained in high yield. Irrespective of the nature of the base employed (entries 2 vs. 3), the Li-salt of 3a afforded the best results in terms of both conversion and yield (entries 2, 4 and 5). Importantly, the treatment of lithiated 3a with MeI produced N-methylation product Me-3a exclusively irrespective of the presence or absence of ZnEt2 (entries 6 and 7). The determined optimal reagent system was efficient enough to conduct the transformation under slightly over-stoichiometric conditions additionally boosting the yield of 4aa (entry 8).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Base | Carbenoid formation | Yieldb, % | ||||
| Metal alkyl | Iodide | x | 3a | 4aa | Me-3a | ||
| a Sulfinamide 3a (0.1 mmol) was deprotonated using a base (0.11 mmol) in THF (1 mL) for 15 min at 0 °C and then sequentially treated with an alkyl metal and alkyl iodide. b 1H NMR yield was measured against mesitylene as an internal standard. c No reaction with CH2Br2 instead of CH2I2 (see ref. 20). | |||||||
| 1 | — | ZnEt2 | CH2I2 | 2.5 | 100 | — | — |
| 2 | LiHMDS | ZnEt2 | CH2I2c | 2.5 | 3 | 86 | — |
| 3 | n-BuLi | ZnEt2 | CH2I2 | 2.5 | 4 | 82 | — |
| 4 | NaHMDS | ZnEt2 | CH2I2 | 2.5 | 15 | 74 | — |
| 5 | KHMDS | ZnEt2 | CH2I2 | 2.5 | 8 | 75 | — |
| 6 | LiHMDS | ZnEt2 | MeI | 2.5 | 5 | — | 89 |
| 7 | LiHMDS | — | MeI | 2.5 | — | — | 95 |
| 8 | LiHMDS | ZnEt2 | CH2I2 | 1.2 | 0 | 95 | — |
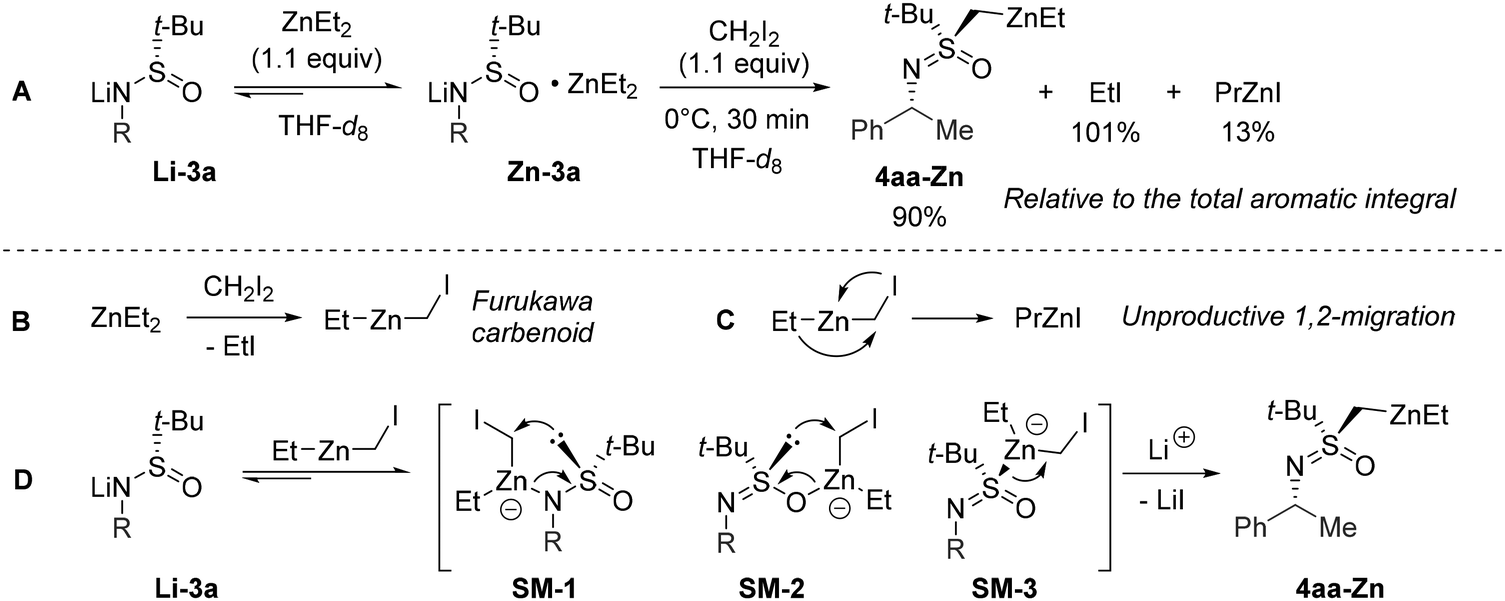
To gain insight into the mechanistic aspects of the transformation, we performed a brief NMR investigation using a nearly stoichiometric variant of the identified conditions (Scheme 3A). To begin with it was established that separately prepared lithium salt Li-3a does not interact with CH2I2. However, the treatment of Li-3a with ZnEt2 results in the reversible formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 zincate complex Zn-3a.
:1 zincate complex Zn-3a.
 | ||
| Scheme 3 NMR experiment and mechanistic hypothesis. | ||
Due to the known21 dynamic nature of ZnEt2 complexation, we were unable to determine the exact binding mode of the Zn-atom in Zn-3a using NMR spectroscopy. On the other hand, attempts to crystallize Zn-3a resulted only in the formation of Li-3a crystals.22 Nonetheless, upon the addition of CH2I2 to Zn-3a, a rapid reaction occurred. The obtained mixture contained the expected amount of EtI corresponding to quantitative I-Zn exchange leading to the formation of the Furukawa carbenoid (Scheme 3B). Next, the minor amount of PrZnI detected matched the decomposition of the excess of the formed carbenoid (Scheme 3C). The respective 1,2-metalate rearrangement is recognized as the major cause of instability in related species in coordinating media.23 Finally, the major product resulting from Li-3a was determined to be dialkylzinc 4aa-Zn.
The combined results of the NMR experiment and the optimization study allowed us to formulate a mechanistic hypothesis. Once formed in the reaction mixture, the Furukawa carbenoid will either undergo an irreversible 1,2-metalate rearrangement or engage in rapid complexation with Li-3a similarly to ZnEt2 (Scheme 3Cvs. 3D). The latter scenario should give rise to transient zincates SM-1–3 analogous to Zn-3a. Irrespective of the realized Zn-binding mode24 in SM-1–3, the anionic character of the ensuing 1,2-rearrangement renders21a the formation of 4aa-Zn faster compared to the unproductive decomposition pathway (Scheme 3C). On the other hand, the migratory aptitude of the S-atom in SM-1–3 apparently must exceed that of the Et substituent. We cannot rule out the possibility that I-Zn exchange may occur directly between the zincate Zn-3a and CH2I2; however, the same zincates SM-1–3 would arise in this case.
To gain a deeper insight into the mechanism of the transformation, a computational study at the DFT level was performed.25 Analysis of possible zincate complexes between Li-3a and the Furukawa carbenoid identified the N,O-bound adduct SM-4 as the most thermodynamically stable configuration (Fig. 2). In view of the minimal (<0.5 kcal mol−1) energy difference between the two diastereomers of SM-4, it is represented by a single structure.
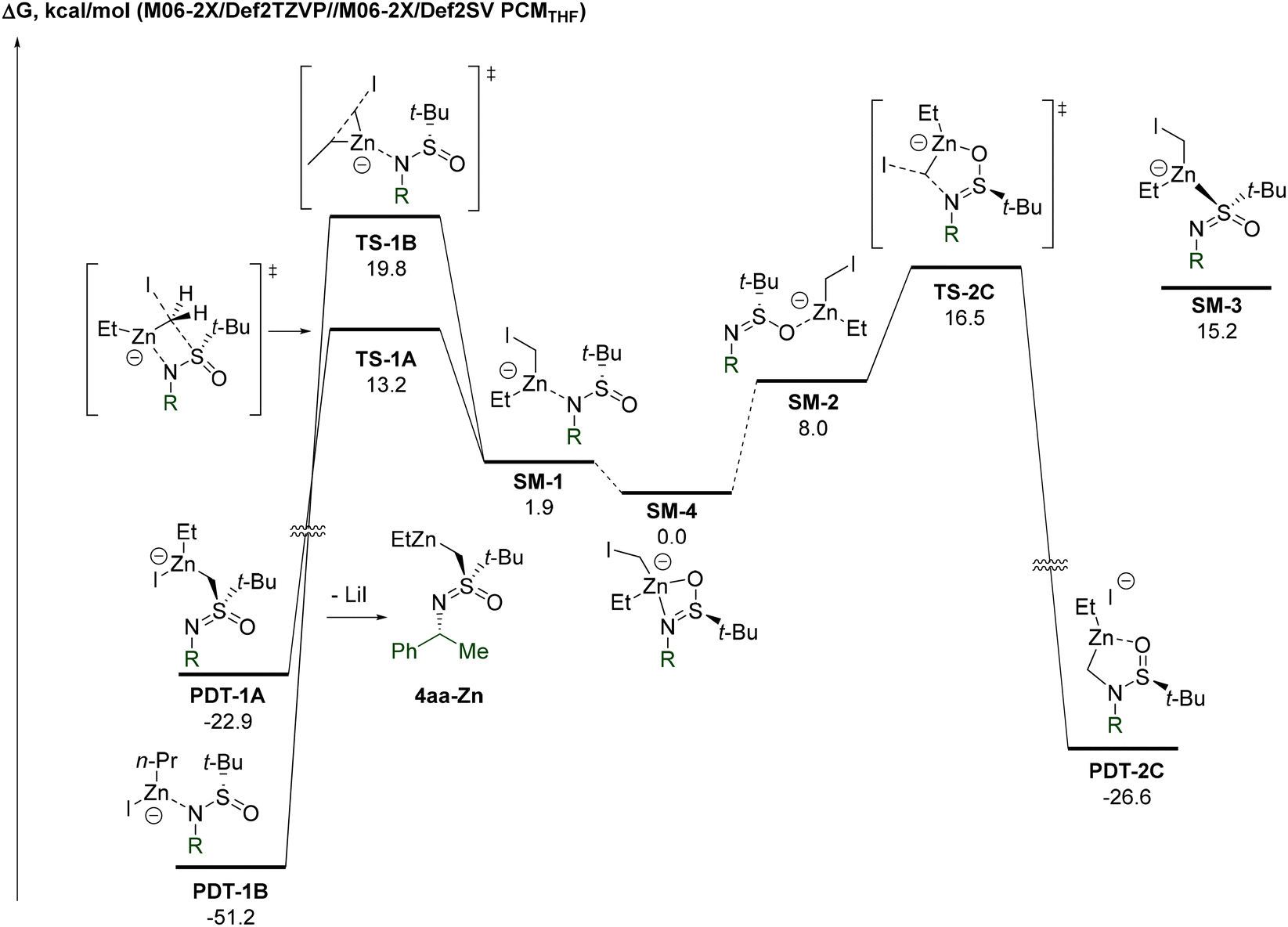 | ||
| Fig. 2 The most kinetically favored transitions of the EtZnCH2I–Li-3a complex. | ||
N-Bound SM-1 was determined to be the second most populated isomer, whereas O- and S-bound SM-2 and SM-3 are far less energetically favored. Four-membered TS-1A, which ultimately leads to the observed product 4aa-ZnviaPDT-1A, possesses the lowest energy (13.2 kcal mol−1) of all calculated transition states. The incorporation of an explicit Zn-bound THF molecule in SM-1 and TS-1A results in only a minor decrease (9.9 vs. 11.3 kcal mol−1) in the respective energy gap. Therefore, we assume that the omission of Zn-bound THF should not have a decisive impact on the overall calculated profile of the potential energy surface. The second lowest energy transition corresponds to the formation of N-alkylated PDT-2CviaTS-2C, which is 3.3 kcal mol−1 higher compared to the observed S-alkylation TS-1A. Possible competitive Et-migration stands third in the order of increase in transition state energy. The corresponding TS-1B exceeds TS-1A by 6.6 kcal mol−1. Thus, the computational study indeed confirms a significant kinetic preference for the observed S-atom migration.
The synthetic utility of dialkylzinc intermediate 4aa-Zn was probed through a series of experiments (Scheme 4). Thus, deuteration and iodination expectedly afforded the corresponding 4aa-D and 4aa-I. Moreover, transmetalation to [(trimethylsilyl)methyl]copper afforded mixed cuprates,26 which smoothly delivered homoallylic derivatives 4aa-1–4via coupling with the respective allylic bromides.
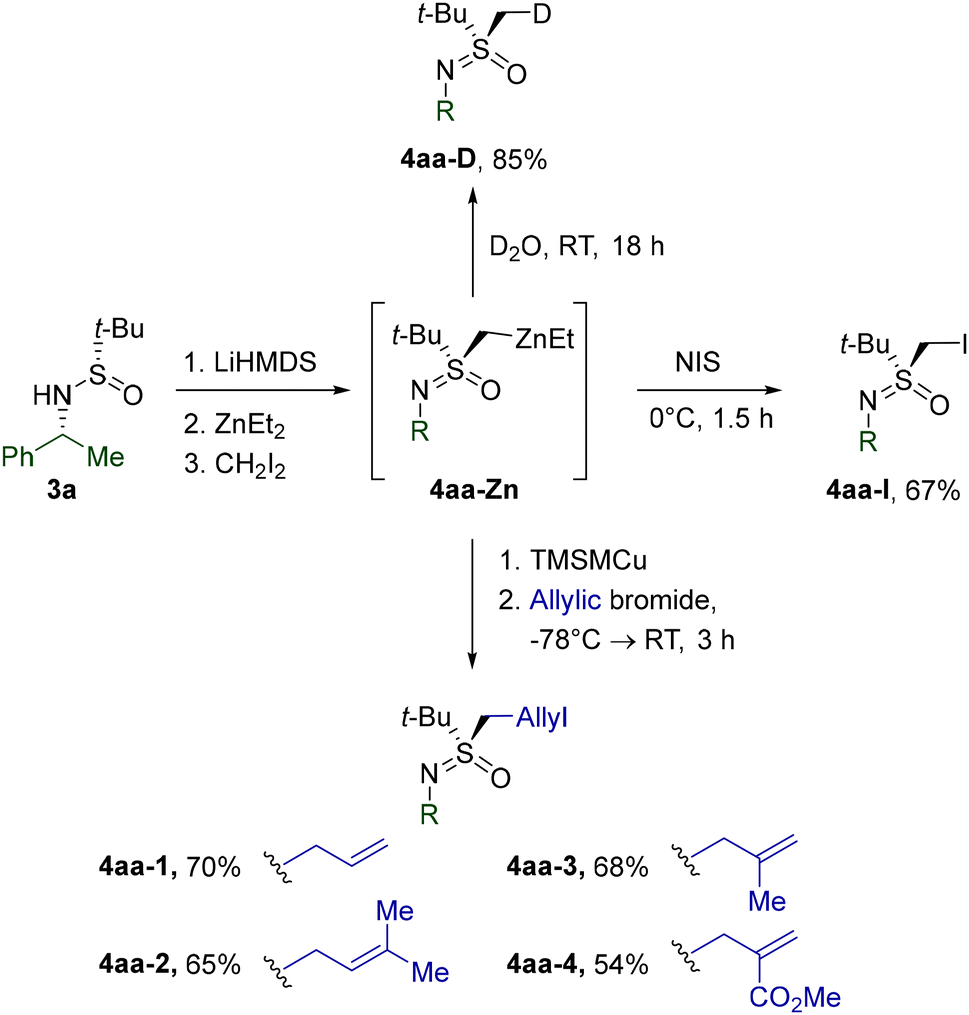 | ||
| Scheme 4 Synthetic utility of dialkylzinc intermediate 4aa-Zn. | ||
Next, we set out to explore the scope of the discovered sulfinamide alkylation with respect to geminal diiodides (Table 2). To begin with, the reliability of S-methylation with CH2I2 (5a) was confirmed in a scaled-up synthesis of 4aa. Gratifyingly, non-functionalized CH2I2 homologues 5b–g also readily engaged in the reaction. Steric crowding around the gem-diiodo carbon definitely suppressed the alkylation, as exemplified by the decreased yield of neopentylic 4ad. Despite the presence of a double bond favorably aligned for intramolecular cyclopropanation,275f afforded the expected sulfoximine 4af. The low yield of 4ag most probably arises from the acute susceptibility of the 5g-derived benzylic carbenoid toward unproductive 1,2-metalate rearrangement.
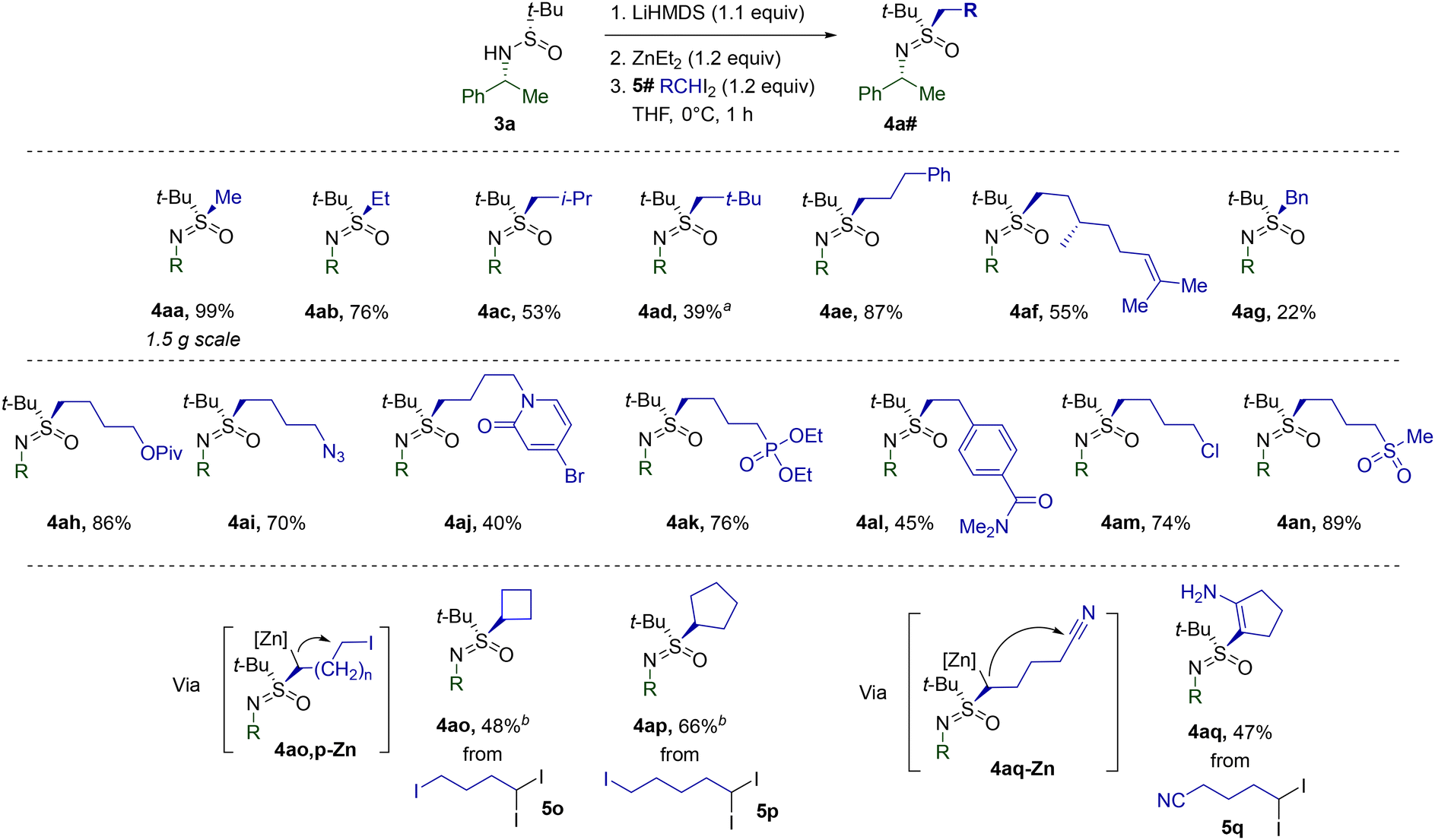
The merits of the current methodology were most vividly revealed in reactions of 3a with functionalized diiodides 5h–n. The low basicity and nucleophilicity of the employed organozinc species allow for the introduction of a variety of functional groups. Additionally, the diiodides 5o–q were found to react in a cascade manner via the dialkylzinc intermediates. While the corresponding 4ao-Zn and 4ap-Zn undergo intramolecular alkylation delivering alicyclic 4ao and 4ap, 4aq-Zn engages in Blaise-type cyclization to 4aq.
The high compatibility of the transformation with functional groups was further exploited during the investigation of possible patterns of α-N-substitution (Table 3). Sulfinamides 3b–p were prepared using Ellman's auxiliary and methylated analogously to 3a. In most cases, the optimized conditions performed adequately without the need for additional modifications. However, a lower reaction temperature was found to be beneficial for several substrates in view of the limited stability of the corresponding Li-salts at 0 °C. The presence of a hydroxyl group in 3m was successfully mitigated by higher loading of reagents. The configuration of the α-N-stereocenter apparently does not play a critical role in reaction performance, since both 4fa and 4ga were isolated with high yields. Moreover, the crystal structure obtained for 4fa unambiguously confirmed the stereoretentive character of the alkylation. Primary alkyl groups at the N-atom of the starting sulfinamides are also tolerated, as evidenced by the reaction of 3n. Notably, the corresponding 4na was formed without any loss of enantiopurity. Less nucleophilic N-arylated 3o and N-acylated 3p displayed a noticeable drop in reactivity, whereas a number of sulfinamides28 encumbered with tertiary N-alkyl substituents failed to deliver the expected products. In agreement with the computational analysis, the latter result suggests that the formation of N-bound zincate SM-1 (Fig. 2) is pivotal for successful S-alkylation.
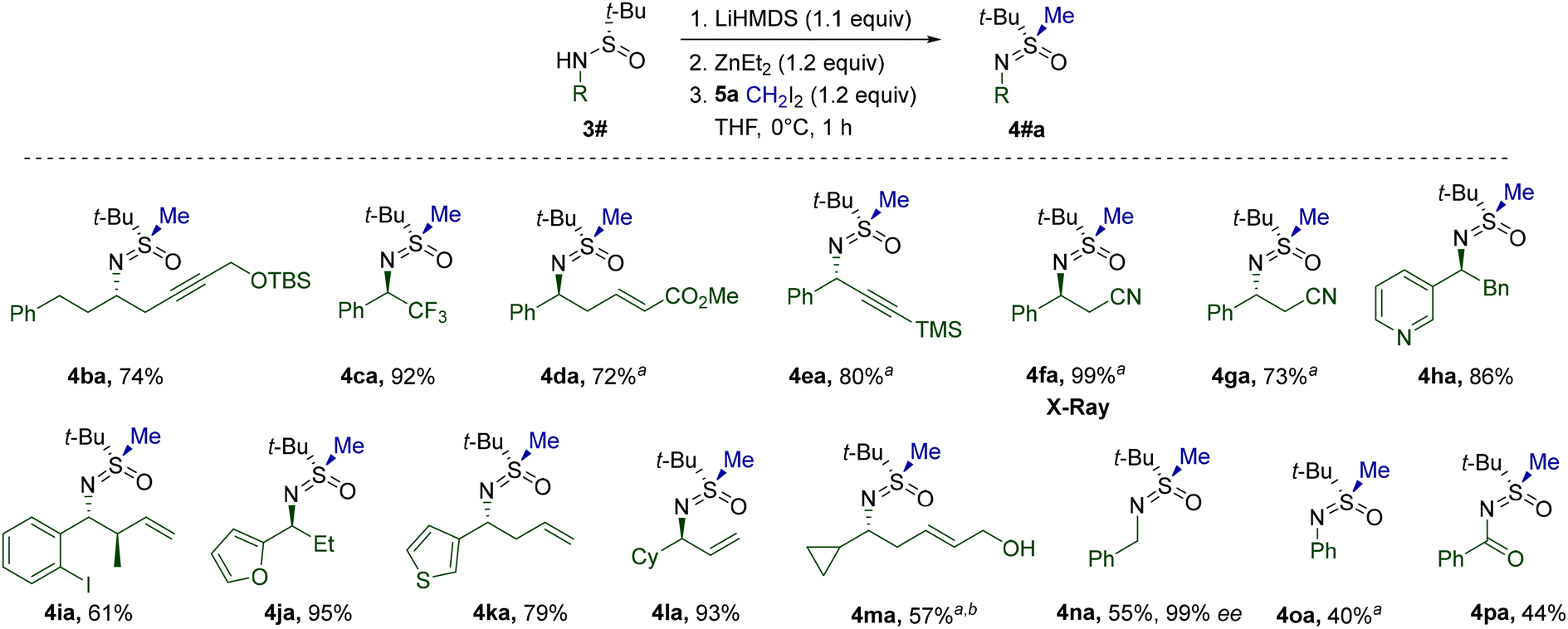
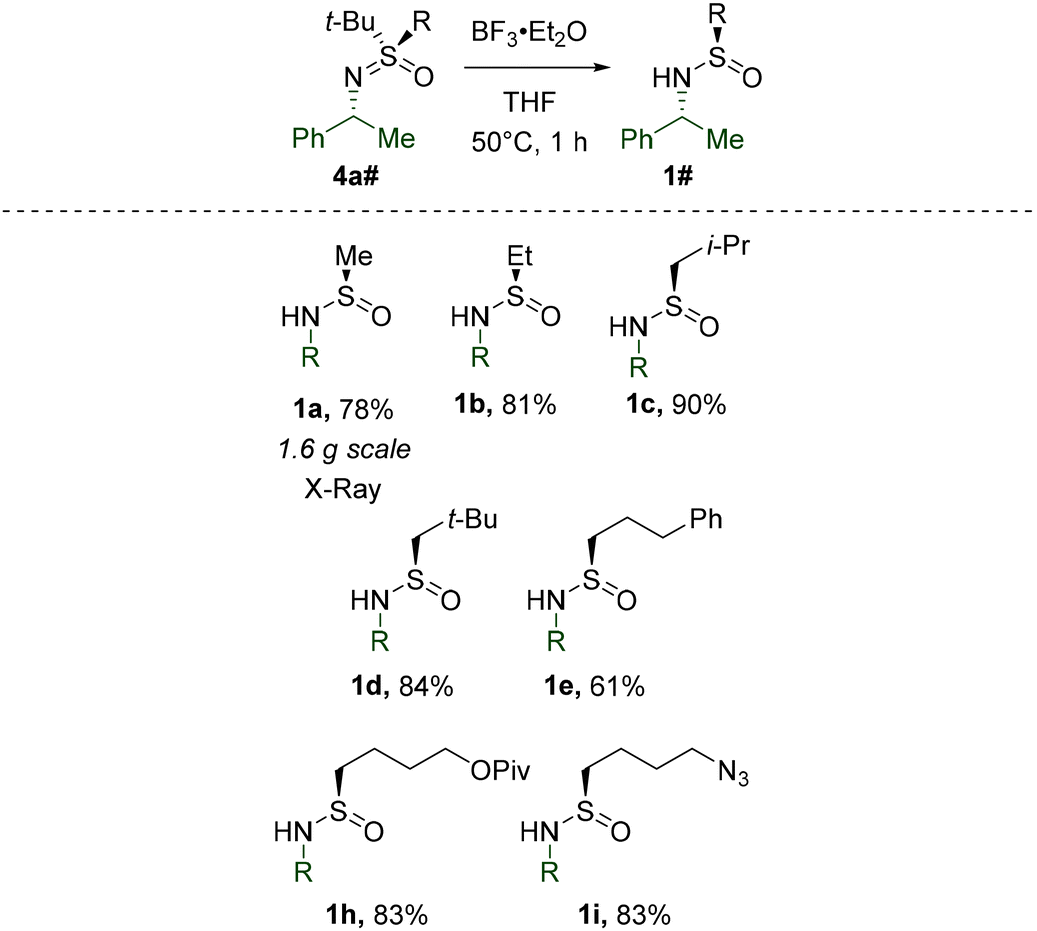
Having investigated the reactivity of S-tert-Bu-sulfinamides, we turned our attention to substrates with other substituents at the S-atom. To this end, a number of corresponding derivatives were prepared by literature known29t-Bu-cleavage (Table 4). The stereoretentive character of this transformation was confirmed by X-ray crystallography of 1a.
|
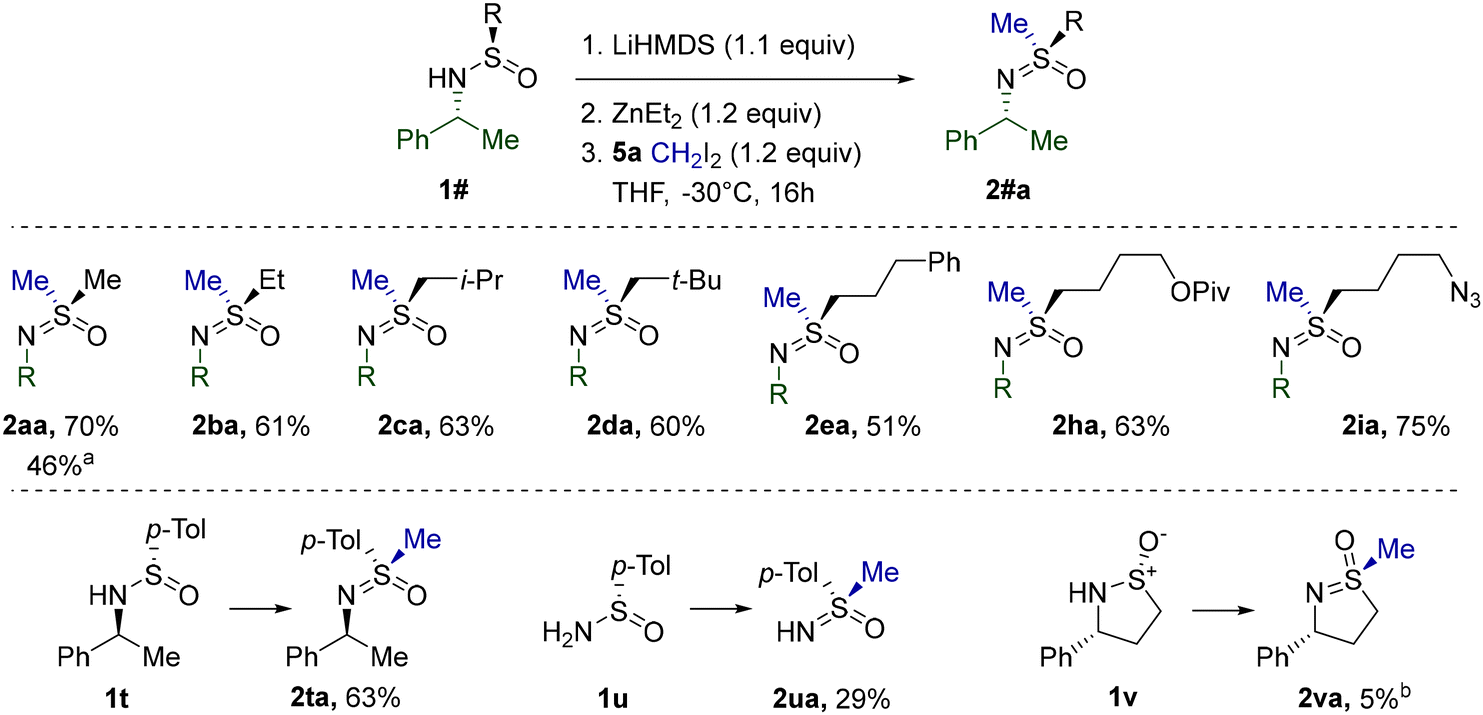
As before, S-methylation of the obtained substrates was performed first. The simplest Me-sulfinamide in the series, 1a, was found to be moderately reactive toward the CH2I2 derived Furukawa reagent under standard conditions at 0 °C (Table 5). Apparently at this temperature, the rate of carbenoid decomposition is comparable with the rate of the requisite methylation. However, at −30 °C the stability of the carbenoid is improved sufficiently in order to participate predominantly in a productive interaction with 1a. Therefore, homologues 1b–e, h, andi were methylated at −30 °C and the corresponding sulfoximines were isolated in good yields. Additionally, the reaction was determined not to be limited to S-alkyl substrates, since S-arylated 1t, derived from Davis’ sulfinamide 1u, performed equally well. However, the problematic methylation of 1u and the exceedingly low reactivity of cyclic sulfinamide 1v clearly denoted the restraints of the standard reagent system.
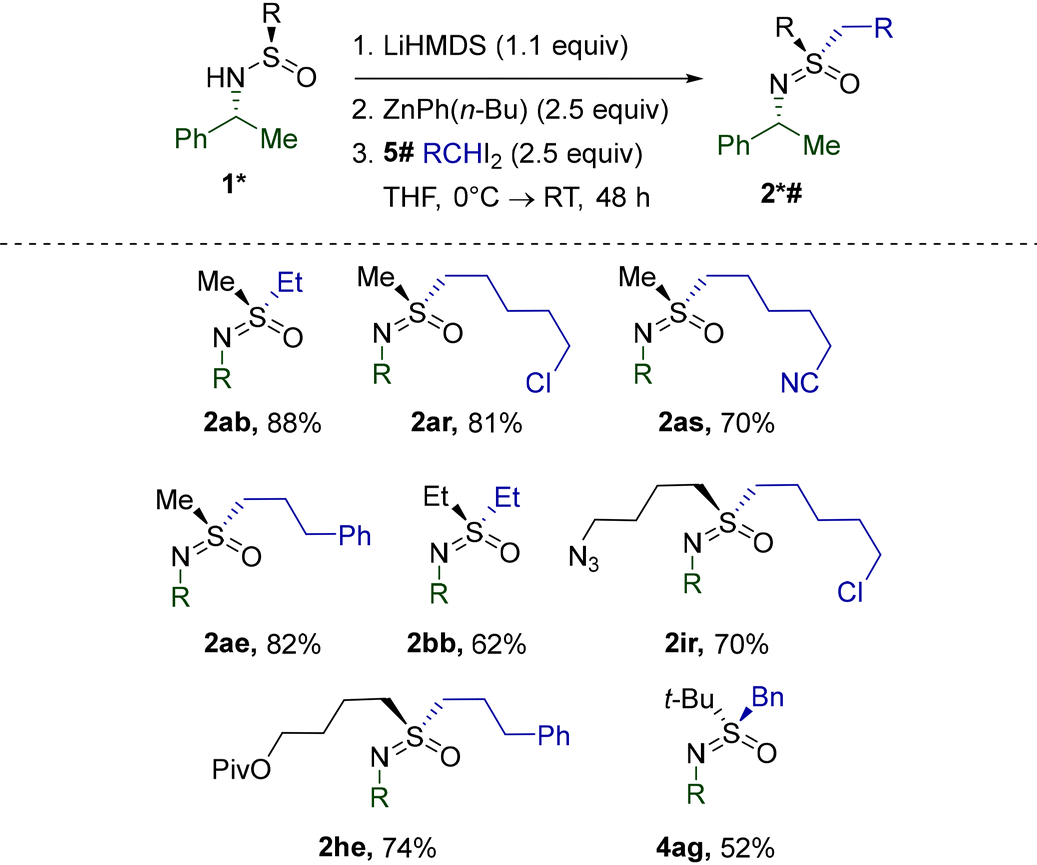
Our focus then shifted to S-alkylation with other substituted diiodides. Using the ethylation of sulfinamide 1a as a model reaction, we evaluated30 the efficacy of several diorganozinc reagents. While ZnEt2 still delivered the ethylsulfoximine 2ab in a fair yield, significant improvement was achieved using the unsymmetrical diorganozinc reagent ZnPh(n-Bu). As confirmed by a separate investigation,31 ZnPh(n-Bu) combined the high reactivity of Zn-alkyls in I-Zn exchange with the low propensity of the Ph-substituent for migration, thus increasing the stability of intermediate carbenoid species.
The discovered efficiency of ZnPh(n-Bu) encouraged us to explore the alkylation of 2a with homologues of 5b employed previously (Table 6). Importantly, the excellent functional group compatibility of the transformation was completely retained. Alkylsulfinamides 1b, i, and h with S-substituents besides methyl were also found to be competent substrates. Gratifyingly, the reaction of 3a with benzal iodide 5g mediated by ZnPh(n-Bu) afforded the corresponding 4ag with a yield significantly superior to that obtained under the standard conditions (Table 2).
|
Disappointingly, the use of ZnPh(n-Bu) did not alleviate the unusually low reactivity of cyclic sulfinamides. Nevertheless, replacing the Ph group with the apparently completely non-migratory32 dimethylsulfone fragment in diorganozinc 6 further reduced unproductive 1,2-migration and improved the performance of several cyclic sulfinamides and other previously challenging substrates (Table 7). Pleasingly, 5- and 6-membered 1v, a′, and w reacted equally smoothly. Full retention of potentially epimerizable stereocenters in 2a′ and 2wa clearly stresses the mildness of the procedure. As opposed to the initial conditions discovered for the methylation of 1a′ (Scheme 2), the current protocol afforded 2a′ reproducibly and with a better yield. Alkylation with a homologue of CH2I2 was also successfully realized in the reaction of 1v with diiodide 5r. Importantly, none of the currently existing synthetic methods (Scheme 1) possess the capacity for S-alkylation of cyclic sulfinamides. Unexpectedly, the diorganozinc 6 performed inferiorly to ZnPh(n-Bu) in the case of linear alkylsulfinamides (Table 6) and thus could not replace ZnPh(n-Bu) in this case.
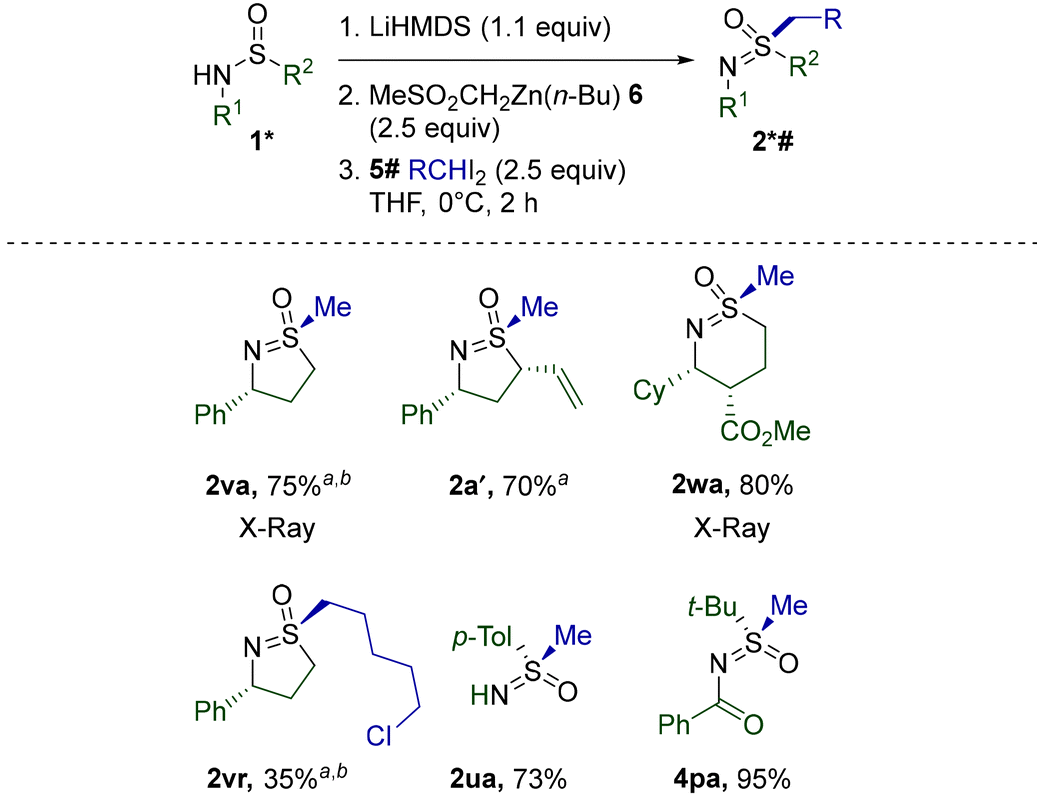
Finally, methylation mediated by 6 was probed on the so far poorly reactive Davis’ sulfinamide 1u (Table 5) and N-acylated 3p (Table 3). Gratifyingly, the corresponding sulfoximines 2vr and 4pa were obtained with much better yields. The particularly remarkable enhancement in the case of 4pa is noteworthy.
Conclusions
In summary, we have developed a new synthesis of sulfoximines based on a hitherto unknown alkylation of sulfinamide salts with Furukawa-type carbenoids. This stereospecific transformation involves an anionic 1,2-metalate rearrangement of the corresponding zincate complex, which competes with mechanistically related unproductive carbenoid decomposition. Operating under a simple reagent system based on commercially available ZnEt2, t-Bu-sulfinamides were found to be excellent substrates. Moreover, we have demonstrated the synthetic utility of the resulting organozinc intermediates. Coupled with mild t-Bu-cleavage, the transformation provides a new entry to variously N-substituted S-alkyl sulfinamides. Subsequent iterative application, on the other hand, seamlessly joins the chemistry around Ellman's auxiliary with S,S-dialkyl sulfoximines. The method may also be extended to S-arylated substrates, as illustrated by the use of Davis’ sulfinamide derivatives. While the sulfinamide structure–reactivity relationship is still not fully understood, a solution for substrates that are resilient to alkylation under standard conditions has been successfully identified. The discovered modifications of the dialkylzinc precursor effectively suppress the unproductive carbenoid decomposition, hence enforcing the requisite alkylation. It is noteworthy that structures obtained via the corresponding transformation of cyclic sulfinamides are virtually inaccessible by other contemporary methods.Data availability
Crystallographic data for 1a, 2va, 2wa, 3a-Li, and 4fa have been deposited at the Cambridge Crystallographic Data Centre under 2374035–2374039.†Experimental details, characterization data, DFT calculations, and NMR spectra have been included in the ESI.†
Author contributions
Conceptualization: G. J., P. A. D., and E. S.; investigation: G. J., D. M., and A. K.; supervision: P. A. D.; writing – original draft: G. J. and P. A. D.; writing – review & editing: G. J., P. A. D., and E. S.; funding acquisition – E. S. All authors have given approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Latvian Science Council Grant LZP-2021/1-0578. The authors thank Dr Sergey Belyakov from the Latvian Institute of Organic Synthesis for X-ray crystallographic analysis.References
- M. Reggelin and C. Zur, Sulfoximines: structures, properties and synthetic applications, Synthesis, 2000, 1–64 CrossRef CAS.
- (a) C. Worch, A. C. Mayer and C. Bolm, Synthesis and Use of Chiral Sulfoximines, in Organosulfur Chemistry in Asymmetric Synthesis, ed. T. Toru and C. Bolm, Wiley-VCH, Weinheim, 2008, ch. 6, pp. 209–232 Search PubMed; (b) H. Okamura and C. Bolm, Sulfoximines: synthesis and catalytic applications, Chem. Lett., 2004, 33, 482–487 CrossRef CAS.
- (a) G. B. Watson, M. W. Siebert, N. X. Wang, M. R. Loso and T. C. Sparks, Sulfoxaflor – a sulfoximine insecticide: review and analysis of mode of action, resistance and cross-resistance, Pestic. Biochem. Physiol., 2021, 178, 104924 CrossRef CAS PubMed; (b) T. C. Sparks, G. B. Watson, M. R. Loso, C. Geng, J. M. Babcock and J. D. Thomas, Sulfoxaflor and the sulfoximine insecticides: chemistry, mode of action and basis for efficacy on resistant insects, Pestic. Biochem. Physiol., 2013, 1, 1–7 CrossRef PubMed.
- (a) U. Lücking, New opportunities for the utilization of the sulfoximine group in medicinal chemistry from the drug designer's perspective, Chem. – Eur. J., 2022, 28, e202201993 CrossRef PubMed; (b) Y. Han, K. Xing, J. Zhang, T. Tong, Y. Shi, H. Cao, H. Yu, Y. Zhang, D. Liu and L. Zhao, Application of sulfoximines in medicinal chemistry from 2013 to 2020, Eur. J. Med. Chem., 2021, 209, 112885 CrossRef CAS PubMed; (c) P. Mäder and L. Kattner, Sulfoximines as rising stars in modern drug discovery? Current status and perspective on an emerging functional group in medicinal chemistry, J. Med. Chem., 2020, 63, 14243–14275 CrossRef PubMed; (d) U. Lücking, Neglected sulfur(VI) pharmacophores in drug discovery: exploration of novel chemical space by the interplay of drug design and method development, Org. Chem. Front., 2019, 6, 1319–1324 RSC; (e) M. Frings, C. Bolm, A. Blum and C. Gnamm, Sulfoximines from a medicinal chemist's perspective: physicochemical and in vitro parameters relevant for drug discovery, Eur. J. Med. Chem., 2017, 126, 225–245 CrossRef CAS PubMed; (f) Y. Kim, J. Kim, B. Kim, R. Kim, H. J. Kim, E. H. Lee, J. Kim, J. Park, Y. Jeong, S. I. Park, H. Kim, M. Kang, J. Lee, Y.-S. Bahn, J. W. Choi, J.-H. Park and K. D. Park, Discovery and optimization of a series of vinyl sulfoximine-based analogues as potent Nrf2 activators for the treatment of multiple sclerosis, J. Med. Chem., 2024, 67, 17866–17892 CrossRef CAS PubMed.
- (a) E. Boulard, V. Zibulski, L. Oertel, P. Lienau, M. Schäfer, U. Ganzer and U. Lücking, Increasing complexity: a practical synthetic approach to three-dimensional, cyclic sulfoximines and first insights into their in vitro properties, Chem. – Eur. J., 2020, 26, 4378–4388 CrossRef CAS PubMed; (b) B. Altenburg, M. Frings, J.-H. Schöbel, J. Goßen, K. Pannen, K. Vanderliek, G. Rossetti, S. Koschmieder, N. Chatain and C. Bolm, Chiral Analogues of PFI-1 as BET Inhibitors and Their Functional Role in Myeloid Malignancies, ACS Med. Chem. Lett., 2020, 11, 1928–1934 CrossRef CAS PubMed; (c) N. Thota, P. Makam, K. K. Rajbongshi, S. Nagiah, N. S. Abdul, A. A. Chuturgoon, A. Kaushik, G. Lamichhane, A. M. Somboro, H. G. Kruger, T. Govender, T. Naicker and P. I. Arvidsson, N-Trifluoromethylthiolated Sulfonimidamides and Sulfoximines: Anti-microbial, Anti-mycobacterial, and Cytotoxic Activity, ACS Med. Chem. Lett., 2019, 10, 1457–1461 CrossRef CAS PubMed; (d) M. Chizema, T. F. Mabasa, H. C. Hoppe and H. H. Kinfe, Design, synthesis, and antiplasmodial evaluation of a series of novel sulfoximine analogues of carbohydrate-based thiochromans, Chem. Biol. Drug Des., 2019, 93, 254–261 CrossRef CAS PubMed.
- (a) X. Zhang, F. Wang and C.-H. Tan, Asymmetric synthesis of S(IV) and S(VI) stereogenic centers, JACS Au, 2023, 3, 700–714 CrossRef CAS PubMed; (b) E. Wojaczyńska and J. Wojaczyński, Modern stereoselective synthesis of chiral sulfinyl compounds, Chem. Rev., 2020, 120, 4578–4611 CrossRef PubMed.
- (a) M. Andresini, A. Tota, L. Degennaro, J. A. Bull and R. Luisi, Synthesis and transformations of NH–sulfoximines, Chem. – Eur. J., 2021, 27, 17293–17321 CrossRef CAS PubMed; (b) V. Bizet, C. M. M. Hendriks and C. Bolm, Sulfur imidations: access to sulfimides and sulfoximines, Chem. Soc. Rev., 2015, 44, 3378–3390 RSC; (c) J. Wang, M. Frings and C. Bolm, Iron–catalyzed imidative kinetic resolution of racemic sulfoxides, Chem. – Eur. J., 2014, 20, 966–969 CrossRef CAS PubMed.
- Stereoselective synthesis of sulfoxides: (a) L. Gao, Y. Wang, Y. Zhang, Y. Fu, Y. Liu and Q. Zhang, Nickel–catalyzed enantioselective synthesis of dienyl sulfoxide, Angew. Chem., Int. Ed., 2024, 63, e202317626 CrossRef CAS PubMed; (b) J. Han, V. A. Soloshonok, K. D. Klika, J. Drabowicz and A. Wzorek, Chiral sulfoxides: advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome, Chem. Soc. Rev., 2018, 47, 1307–1350 RSC; (c) E. Wojaczyńska and J. Wojaczyński, Enantioselective synthesis of sulfoxides: 2000–2009, Chem. Rev., 2010, 110, 4303–4356 CrossRef PubMed.
- (a) Z. Liu, H. Wu, H. Zhang, F. Wang, X. Liu, S. Dong, X. Hong and X. Feng, Iron-catalyzed asymmetric imidation of sulfides via sterically biased nitrene transfer, J. Am. Chem. Soc., 2024, 146, 18050–18060 CrossRef CAS PubMed. Synthesis of sulfimides besides imidation of thioethers: (b) S. Tsuzuki and T. Kano, Asymmetric synthesis of chiral sulfimides through the O–alkylation of enantioenriched sulfinamides and addition of carbon nucleophiles, Angew. Chem., Int. Ed., 2023, 62, e202300637 CrossRef CAS PubMed; (c) N. S. Greenwood, A. T. Champlin and J. A. Ellman, Catalytic enantioselective sulfur alkylation of sulfenamides for the asymmetric synthesis of sulfoximines, J. Am. Chem. Soc., 2022, 144, 17808–17814 CrossRef CAS PubMed; (d) A. T. Champlin, N. Y. Kwon and J. A. Ellman, Enantioselective S-alkylation of sulfenamides by phase-transfer catalysis, Angew. Chem., Int. Ed., 2024, 63, e202408820 CAS.
- (a) G. Zhou, T. Zhou, A.-L. Jiang, P.-F. Qian, J.-Y. Li, B.-Y. Jiang, Z.-J. Chen and B.-F. Shi, Electrooxidative Rhodium(III)/chiral carboxylic acid-catalyzed enantioselective C−H annulation of sulfoximines with alkynes, Angew. Chem., Int. Ed., 2024, 63, e202319871 CrossRef CAS PubMed; (b) B. Wang, X. Liang and Q. Zeng, Recent advances in the synthesis of cyclic sulfoximines via C–H bond activation, Molecules, 2023, 28, 1367 CrossRef CAS PubMed; (c) S.-Y. Song, X. Zhou, Z. Ke and S. Xu, Synthesis of chiral sulfoximines via iridium–catalyzed regio– and enantioselective C–H borylation: a remarkable sidearm effect of ligand, Angew. Chem., Int. Ed., 2023, 62, e202217130 CrossRef CAS PubMed.
- (a) M. Tang, M. Yuan, S. Hong, Q. Jiang, H. Gu and X. Yang, Kinetic resolution of sulfoximines via asymmetric organocatalyzed formation of benzothiadiazine-1-oxides, Org. Lett., 2024, 26, 1914–1919 CrossRef CAS PubMed; (b) M. Brauns and N. Cramer, Efficient kinetic resolution of sulfur–stereogenic sulfoximines by exploiting CpXRhIII–catalyzed C−H functionalization, Angew. Chem., Int. Ed., 2019, 58, 8902–8906 CrossRef CAS PubMed; (c) S. Dong, M. Frings, H. Cheng, J. Wen, D. Zhang, G. Raabe and C. Bolm, Organocatalytic kinetic resolution of sulfoximines, J. Am. Chem. Soc., 2016, 138, 2166–2169 CrossRef CAS PubMed.
- (a) C. R. Johnson, K. G. Bis, J. H. Cantillo, N. A. Meanwell, M. F. D. Reinhard, J. R. Zeller and G. P. Vonk, Preparation and reactions of sulfonimidoyl fluorides, J. Org. Chem., 1983, 48, 1–3 CrossRef CAS; (b) C. R. Johnson, E. U. Jonsson and A. Wambsgans, Nucleophilic substitution at sulfur in sulfonimidoyl compounds: synthesis of sulfoximines, J. Org. Chem., 1979, 44, 2061–2065 CrossRef CAS.
- (a) S. Greed, O. Symes and J. A. Bull, Stereospecific reaction of sulfonimidoyl fluorides with Grignard reagents for the synthesis of enantioenriched sulfoximines, Chem. Commun., 2022, 58, 5387–5390 RSC; (b) G. Yang, Y. Yuan, Y. Tian, S. Zhang, X. Cui, B. Xia, G. Li and Z. Tang, Synthesis of chiral sulfonimidoyl chloride via desymmetrizing enantioselective hydrolysis, J. Am. Chem. Soc., 2023, 145, 5439–5446 CrossRef CAS PubMed; (c) S. Teng, Z. P. Shultz, C. Shan, L. Wojtas and J. M. Lopchuk, Asymmetric synthesis of sulfoximines, sulfonimidoyl fluorides and sulfonimidamides enabled by an enantiopure bifunctional S(VI) reagent, Nat. Chem., 2024, 16, 183–192 CrossRef CAS PubMed; (d) P. Mendonca, M. W. Lewis, S. P. Argent, J. C. Moore and R. A. Stockman, General method for the asymmetric synthesis of N–H sulfoximines via C–S bond formation, Org. Lett., 2020, 22, 2776–2780 CrossRef PubMed.
- (a) Y. Maeda, S. Hamada, Y. Aota, K. Otsubo, T. Kano and K. Maruoka, Practical asymmetric synthesis of chiral sulfoximines via sulfur-selective alkylation, J. Org. Chem., 2022, 87, 3652–3660 CrossRef CAS PubMed; (b) Y. Aota, T. Kano and K. Maruoka, Asymmetric synthesis of chiral sulfoximines through the s–alkylation of sulfinamides, Angew. Chem., Int. Ed., 2019, 58, 17661–17665 CrossRef CAS PubMed; (c) X. Zou, B. Shen, G. Li, Q. Liang, Y. Ouyang, B. Yang, P. Yu and B. Gao, Strain-promoted S-arylation and alkenylation of sulfinamides using arynes and cyclic alkynes, Sci. China: Chem., 2024, 67, 928–935 CrossRef CAS; (d) X. Zou, H. Wang and B. Gao, Synthesis of sulfoximines by copper-catalyzed oxidative coupling of sulfinamides and aryl boronic acids, Org. Lett., 2023, 25, 7656–7660 CrossRef CAS PubMed; (e) Y. Aota, T. Kano and K. Maruoka, Asymmetric synthesis of chiral sulfoximines via the S-arylation of sulfinamides, J. Am. Chem. Soc., 2019, 141, 19263–19268 CrossRef CAS PubMed.
- (a) Y. Liu, Z. Wang, B. Guo and Q. Cai, Asymmetric synthesis of N-aryl sulfinamides: copper(I)-catalyzed coupling of sulfinamides with aryl iodides via kinetic resolution, Tetrahedron Lett., 2016, 57, 2379–2381 CrossRef CAS; (b) X. Sun, X. Tu, C. Dai, X. Zhang, B. Zhang and Q. Zeng, Palladium-catalyzed C–N cross coupling of sulfinamides and aryl halides, J. Org. Chem., 2012, 77, 4454–4459 CrossRef CAS PubMed; (c) C. M. Simon, K. N. Robertson, P. L. DeRoy, A. A. Yadav, E. R. Johnson and M. Stradiotto, Nickel-catalyzed N-arylation of sulfinamides: a comparative study versus analogous sulfonamide cross-couplings, Organometallics, 2023, 42, 1704–1710 CrossRef CAS; (d) C. Li, K. Zhang, H. Ma, S. Wu, Y. Huang, Y. Duan, Y. Luo, J. Yan and G. Yang, An optimized Ni-catalyzed Chan-Lam type coupling: enantioretentive access to chiral N-aryl sulfinamides, Chem. – Eur. J., 2022, 28, e202202190 CrossRef CAS PubMed.
- M. T. Robak, M. A. Herbage and J. A. Ellman, Synthesis and applications of tert-butanesulfinamide, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS PubMed.
- G. Jersovs, M. Bojars, P. A. Donets and E. Suna, Synthetic approach toward enantiopure cyclic sulfinamides, Org. Lett., 2022, 24, 4625–4629 CrossRef CAS PubMed.
- S-Alkylation leading to sulfur ylides: (a) B. R. Belleniea and J. M. Goodman, Sulfonium ylide epoxidation reactions: methylene transfer, Chem. Commun., 2004, 1076–1077 RSC; (b) Z. Kosarych and T. Cohen, Method for methylidenation and ethylidenation of an allylic thioether leading to a 2,3-sigmatropic rearrangement. Failure of the Simmons-Smith reaction in the presence of thioethers, Tetrahedron Lett., 1982, 23, 3019–3022 CrossRef CAS.
- A. M. Jacobine, A. L. A. Puchlopek, C. K. Zercher, J. B. Briggs, J. P. Jasinski and R. J. Butcher, Tandem chain extension–Mannich reaction: an approach to β-proline derivatives, Tetrahedron, 2012, 68, 7799–7805 CrossRef CAS PubMed.
- For details, see Table S1 in the ESI (page S17).†.
- NMR studies on the formation and stability of carbenoids: (a) A. B. Charette and J.-F. Marcoux, Spectroscopic characterization of (iodomethyl)zinc reagents involved in stereoselective reactions: spectroscopic evidence that IZnCH2I is not Zn(CH2I)2 + ZnI2 in the presence of an ether, J. Am. Chem. Soc., 1996, 118, 4539–4549 CrossRef CAS; (b) S. E. Denmark, J. P. Edwards and S. R. Wilson, Solution and solid-state structure of the “Wittig-Furukawa” cyclopropanation reagent, J. Am. Chem. Soc., 1991, 113, 723–725 CrossRef CAS.
- See ESI, page S109† for details: Li-3a crystallizes in polymeric chains containing N- and O-bound Li-atom.
- (a) V. K. Aggarwal and K. Sommer, Rearrangements of Organozinc Compounds, in The Chemistry of Organozinc Compounds, ed. Z. Z. Rappoport and I. Marek, John Wiley & Sons, 2006, ch. 13, pp. 595–639 Search PubMed; (b) I. Marek, Sp3 Organozinc carbenoid homologation in organic synthesis, Tetrahedron, 2002, 58, 9463–9475 CrossRef CAS.
- To the best of our knowledge structures of sulfinamide zincates akin to Zn-3a have not been described. On the other hand, complexes of sulfinamides may feature a metal bound to N-, O- and S-atoms: (a) L. Wang, M. Chen, P. Zhang, W. Li and J. Zhang, Palladium/PC-Phos-catalyzed enantioselective arylation of general sulfenate anions: scope and synthetic applications, J. Am. Chem. Soc., 2018, 140, 3467–3473 CrossRef CAS PubMed; (b) T. Achard, J. Benet-Buchholz, E. C. Escudero-Adan, A. Riera and X. Verdaguer, N-Benzyl-N-phosphino-tert-butylsulfinamide and its coordination modes with Ir(I), Cu(I), Pd(II), and Pt(II): P,S or P,O?, Organometallics, 2011, 30, 3119–3130 CrossRef CAS; (c) A. J. Souers, T. D. Owens, A. G. Oliver, F. J. Hollander and J. A. Ellman, Synthesis and crystal structure of a unique and homochiral N,S-bonded N,N‘-bis(tert-butanesulfinyl)amidinate rhodium(I) complex, Inorg. Chem., 2001, 40, 5299–5301 CrossRef CAS PubMed; (d) U. Dembowski, M. Noltemeyer, J. W. Gilje and H. W. Roesky, Synthese und strukturen von metallhaltigen achtgliedrigen N-S-O–heterocyclen, Chem. Ber., 1991, 124, 1917–1921 CrossRef CAS.
- See the ESI, page S85 for details.†.
- S. H. Bertz, M. Eriksson, G. Miao and J. P. Snyder, β-Silyl organocuprates: the grail of organocuprate chemistry?, J. Am. Chem. Soc., 1996, 118, 10906–10907 CrossRef CAS.
- J. A. Bull and A. B. Charette, Intramolecular Simmons−Smith cyclopropanation. studies into the reactivity of alkyl-substituted zinc carbenoids, effect of directing groups and synthesis of bicyclo[n.,1.0]alkanes, J. Am. Chem. Soc., 2010, 132, 1895–1902 CrossRef CAS PubMed.
- See the ESI, page S38 for details.†.
- Y. Aota, Y. Maeda, T. Kano and K. Maruoka, Efficient synthesis of cyclic sulfoximines from n–propargylsulfinamides through sulfur–carbon bond formation, Chem. – Eur. J., 2019, 25, 15755–15758 CrossRef CAS PubMed.
- See the ESI, page S67 for details.†.
- See the ESI, page S81 for details.†.
- See the ESI, pages S75 and S81 for details.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2374035–2374039. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01931h |
| This journal is © the Partner Organisations 2025 |