
Lewis acid-controlled Pd-catalyzed chemodivergent hydrocyanation of cyclopropenes†
Rongrong
Yu‡
b,
Song-Zhou
Cai‡
ab and
Xianjie
Fang
 *ab
*ab
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Key Laboratory of Organosilicon Material Technology of Zhejiang Province, College of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, 2318 Yuhangtang Road, Hangzhou 311121, China. E-mail: fangxj@hznu.edu.cn
bShanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China
First published on 22nd October 2024
Abstract
Due to the multifaceted reactivities of cyclopropenes, the divergent hydrofunctionalization of these compounds has recently attracted significant attention. Herein, we present a Pd-catalyzed hydrocyanation of cyclopropenes via aluminum Lewis acid-controlled divergent chemoselectivity. In this study, the presence of aluminum Lewis acid plays a pivotal role in the reaction pathway. In the absence of aluminum Lewis acid, the reaction predominantly yields ring-opening hydrocyanation products, whereas the addition of Lewis acid directs the formation of ring-retentive products.
Cyclopropenes have attracted substantial interest from synthetic organic chemists due to their uniquely strained structures and diverse reactivity.1 As the smallest unsaturated carbocycle, cyclopropene exhibits a high strain energy of 54.1 kcal mol−1, which allows for easier hydrofunctionalizations that are generally challenging for other unsaturated systems such as allenes, alkynes, and alkenes.2 Therefore, cyclopropenes could serve as a promising platform for the development of novel divergent hydrofunctionalization methodologies.3 Notably, in 2021, the Dong group introduced a Rh-catalyzed divergent hydrothiolation of cyclopropenes, demonstrating that the judicious choice of bisphosphine ligands plays a pivotal role in determining the reaction pathway—either ring-opening or ring-retentive mechanisms (Scheme 1a).3b More recently, Sureshkumar and co-workers reported a photocatalyzed sulfonylation and alkylation of cyclopropenes, revealing that the equilibrium between ring-opening and ring-retentive pathways was influenced by both reaction temperature and solvents (Scheme 1b).3c Despite these advancements, the development of new divergent hydrofunctionalization methods for the transformation of cyclopropenes remains highly desirable.
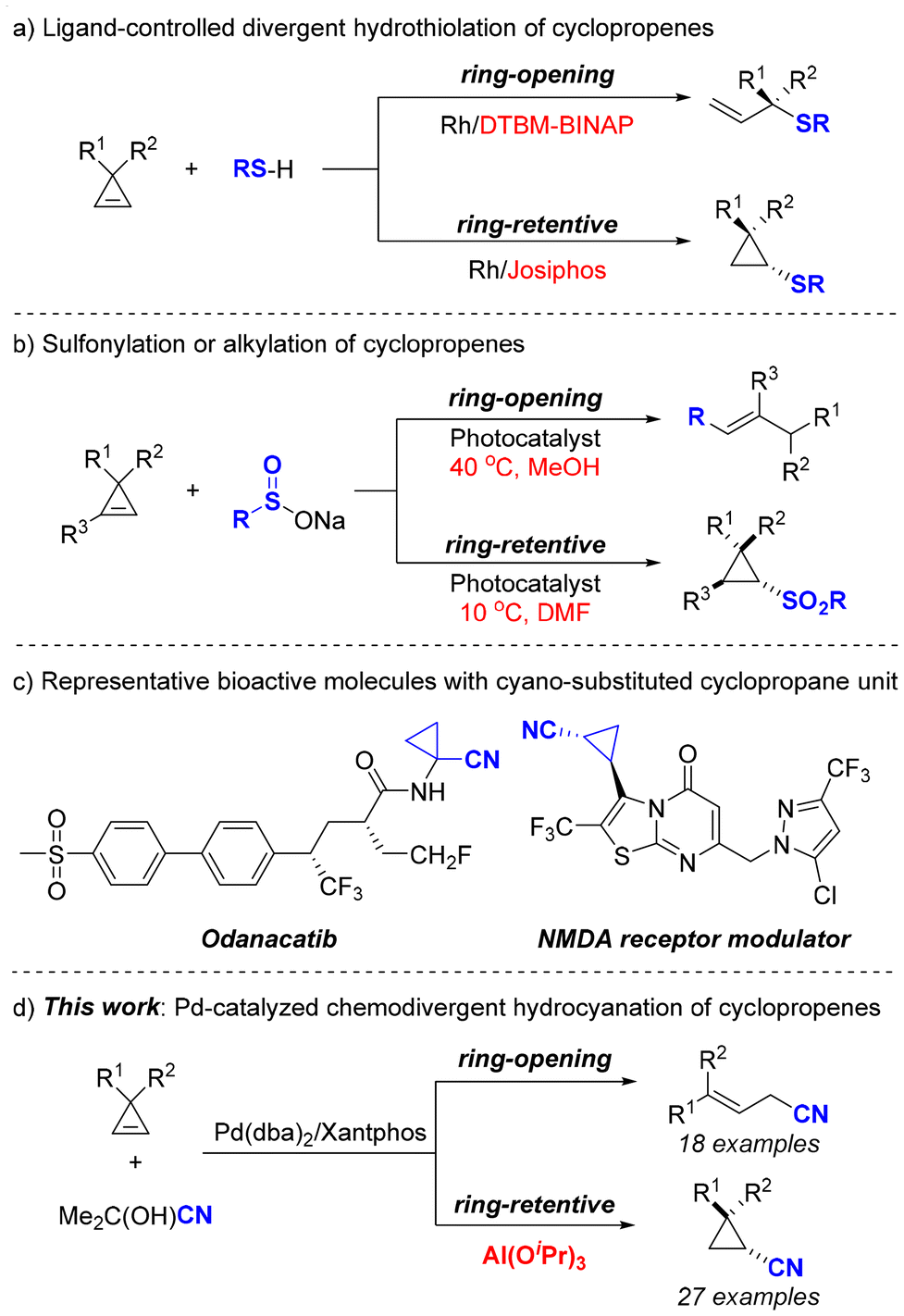 | ||
| Scheme 1 Background for the development of the current work. | ||
Nitriles are not only used as versatile synthons in organic chemistry but also widely occur in bioactive molecules, pharmaceuticals, and catalysts/ligands.4 Among various approaches, transition-metal-catalyzed hydrocyanation of unsaturated hydrocarbons represents the most atom-economical and straightforward method for the preparation of nitriles.5 Expanding the scope of unsaturated substrates and exploring the different reactivities of similar substrates are pivotal topics in this field.6 To our current knowledge, no literature has documented the hydrocyanation of cyclopropenes. Moreover, this reaction is of particular importance due to the potential formation of cyano-substituted cyclopropane motifs, which are prevalent in a wide array of natural products and bioactive molecules (Scheme 1c).4c Driven by our continuous interest in hydrocyanations and cyclopropene functionalizations,7,8 we herein disclosed a Pd-catalyzed hydrocyanation of cyclopropenes via aluminum Lewis acid-controlled divergent chemoselectivity (Scheme 1d). In this reaction, the presence of aluminum Lewis acid plays a crucial role in determining the reaction pathway. In the presence of aluminum Lewis acid, the reaction predominantly yields ring-retentive products, while the omission of Lewis acid would favor the formation of ring-opening products.
At the outset of the investigation, we employed cyclopropene 1a as the model substrate, Pd(dba)2 as the precatalyst (see Table S1† for the screening of precatalysts), and acetone cyanohydrin as the HCN source (see Table S4† for the screening of HCN sources). We first studied the ligand effects on the model reaction. The use of the most common monophosphine ligand PPh3 furnished the ring-opening allylic nitrile 2a with 20% yield (Table 1, entry 1). In contrast to monophosphine ligands, bidentate ligands exhibit superior behavior in facilitating the ring-opening hydrocyanation. When dppp and dppf were utilized as the ligands, the yields of 2a were 81% and 86%, respectively (Table 1, entries 2 and 3). However, the use of rac-BINAP resulted in a moderate yield of 45% (Table 1, entry 4). Gratifyingly, the employment of XantPhos as a ligand afforded 2a in a high yield of 92%, with an 85% isolated yield (Table 1, entry 5). Remarkably, no ring-retentive products were detected in the above reactions. In previous investigations on transition-metal-catalyzed hydrocyanation and C–CN activation, Lewis acids were frequently employed as additives to enhance reactivity and facilitate metal-mediated oxidative addition and reductive elimination processes.9,10 Accordingly, our focus shifted towards exploring Lewis acid additives and their potential impact on the hydrocyanation of cyclopropenes (see Table S2† for the screening of additives). To our delight, upon the introduction of 20 mol% BPh3 into the reaction, an 11% yield of the ring-retentive cyano-substituted cyclopropane 3a was observed, albeit with the predominant formation of the ring-opening product 2a (Table 1, entry 6). No ring-retentive product was detected when Ti(OiPr)4, LiCl and Zn(OTf)2 were used as Lewis acids (Table 1, entries 7–9). Surprisingly, the chemoselectivity was reversed when Al(OEt)3 was used, resulting in a 75% yield of 3a and only a 20% yield of the ring-opening product 2a (Table 1, entry 10). Subsequently, a series of aluminum Lewis acids was screened (Table 1, entries 11–13). Using Al(OiPr)3 as the Lewis acid, 3a was obtained as the major product with a chemoselectivity ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 11). Further investigation into solvents revealed that dimethoxyethane (DME) provided the best results in terms of both chemoselectivity and yield for ring-retentive hydrocyanation (Table 1, entry 16). It is worth mentioning that reducing the equivalent of Lewis acid would dramatically deplete the chemoselectivity, whereas increasing its equivalent showed no significant influence (Table 1, entries 18 and 19). Remarkably, the diastereoselectivity of the ring-retentive hydrocyanation was excellent, and no other diastereomer was detected. Preliminary studies on asymmetric catalysis using chiral ligands under the ring-retentive hydrocyanation conditions were performed (see Table S3† for details). Unfortunately, only DIOP achieved the ring-retentive product (16% GC yield), albeit with 0% ee. Thus, the conditions of entries 5 and 16 were selected as the optimized reaction conditions, respectively.
:1 (Table 1, entry 11). Further investigation into solvents revealed that dimethoxyethane (DME) provided the best results in terms of both chemoselectivity and yield for ring-retentive hydrocyanation (Table 1, entry 16). It is worth mentioning that reducing the equivalent of Lewis acid would dramatically deplete the chemoselectivity, whereas increasing its equivalent showed no significant influence (Table 1, entries 18 and 19). Remarkably, the diastereoselectivity of the ring-retentive hydrocyanation was excellent, and no other diastereomer was detected. Preliminary studies on asymmetric catalysis using chiral ligands under the ring-retentive hydrocyanation conditions were performed (see Table S3† for details). Unfortunately, only DIOP achieved the ring-retentive product (16% GC yield), albeit with 0% ee. Thus, the conditions of entries 5 and 16 were selected as the optimized reaction conditions, respectively.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Additive | Solvent | 2a yieldb/% | 3a yieldb/% |
| a Unless otherwise noted, all reactions were carried with 1a (0.2 mmol), Me2C(OH)CN (0.6 mmol), Pd(dba)2 (5 mol%), bidentate ligand (5 mol%) or monodentate ligand (10 mol%), additive (20 mol%), solvent (0.3 mL), 12 h. b Yield was determined via gas chromatography analysis using n-dodecane as an internal standard. c Isolated yield. d 30 mol% of additive. e 10 mol% of additive. | |||||
| 1 | PPh3 | — | Toluene | 20 | ND |
| 2 | dppp | — | Toluene | 81 | ND |
| 3 | dppf | — | Toluene | 86 | ND |
| 4 | BINAP | — | Toluene | 45 | ND |
| 5 | Xantphos | — | Toluene | 92(85) | ND |
| 6 | Xantphos | BPh3 | Toluene | 65 | 11 |
| 7 | Xantphos | Ti(OiPr)4 | Toluene | 35 | ND |
| 8 | Xantphos | LiCl | Toluene | 89 | ND |
| 9 | Xantphos | Zn(OTf)2 | Toluene | 80 | ND |
| 10 | Xantphos | Al(OEt)3 | Toluene | 20 | 75 |
| 11 | Xantphos | Al(OiPr)3 | Toluene | 12 | 72 |
| 12 | Xantphos | Al(acac)3 | Toluene | 25 | 53 |
| 13 | Xantphos | Al(OTf)3 | Toluene | 35 | 52 |
| 14 | Xantphos | Al(OiPr)3 | THF | 13 | 75 |
| 15 | Xantphos | Al(OiPr)3 | Dioxane | 13 | 77 |
| 16 | Xantphos | Al(O i Pr) 3 | DME | 10 | 83(78) |
| 17 | Xantphos | Al(OiPr)3 | DCE | 15 | 60 |
| 18d | Xantphos | Al(OiPr)3 | DME | 11 | 80 |
| 19e | Xantphos | Al(OiPr)3 | DME | 31 | 58 |
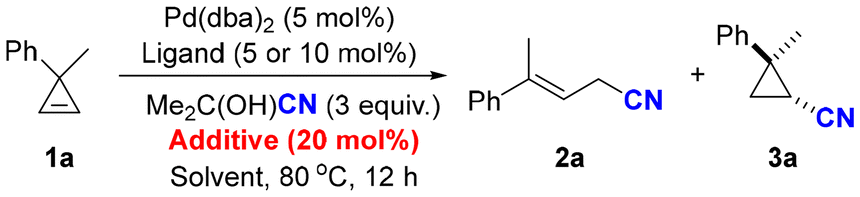
Having identified the optimal reaction conditions, the substrate scope of the ring-opening hydrocyanation was first explored. As shown in Scheme 2, a series of cyclopropenes were smoothly converted into the corresponding allylic nitriles with moderate to good yields. When para-substituents were introduced onto the phenyl rings, the ring-opening reaction was found to be compatible with both electron-donating groups, such as methoxyl (2b), t-butyl (2f), and methylthiol (2g), and electron-withdrawing groups, such as fluorine (2d) and trifluoromethyl (2e). Various functional groups, including halogens (2c, 2h) and thiophene (2n), were well tolerated. In addition, spirocyclic cyclopropene (1o) could also deliver the corresponding allylic nitriles with good yields. Interestingly, when R1 was phenyl and R2 was cyclopropyl, the corresponding allyl cyanide (2r) was successfully obtained with 50% yield, wherein the cyclopropyl group was retained and no secondary ring-opening products were detected. Unfortunately, dialkyl-substituted cyclopropene (1s) could not undergo ring-opening hydrocyanation, and cyclopropene 1s was nearly quantitatively recovered.
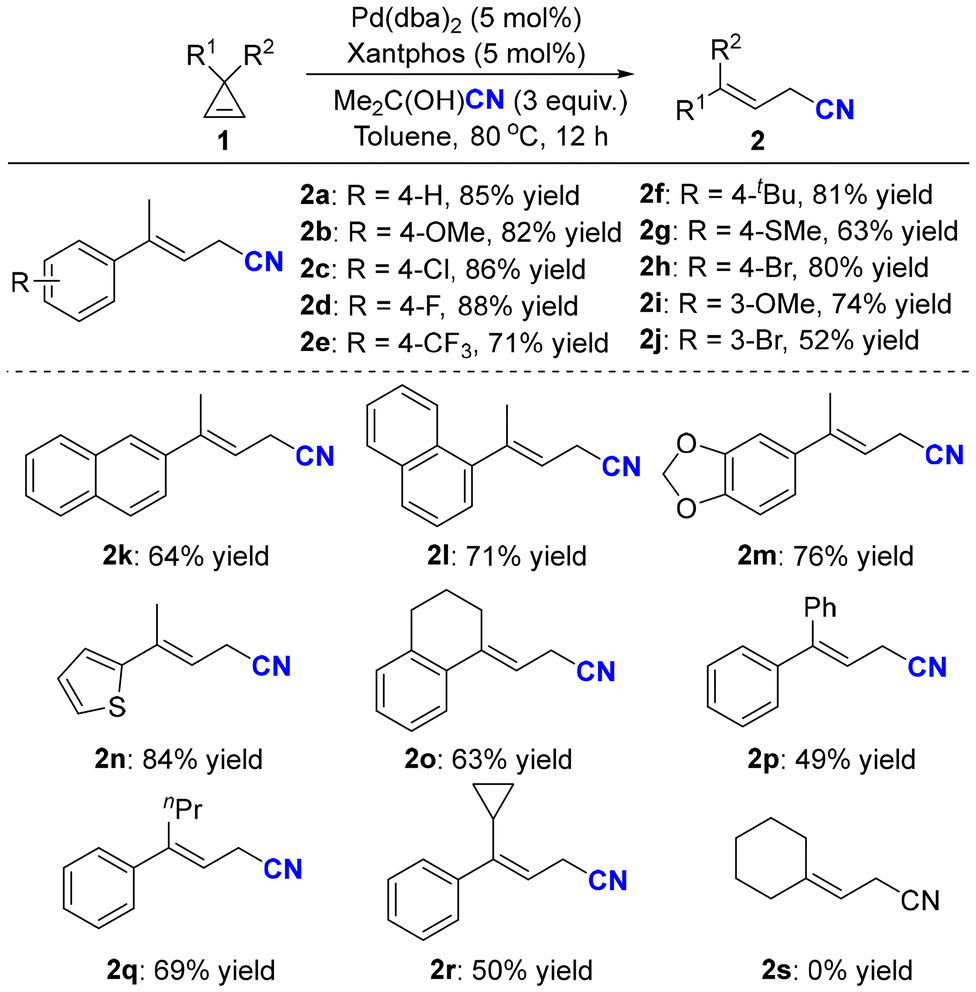 | ||
| Scheme 2 The substrate scope of the ring-opening hydrocyanation of cyclopropenes. Reactions were performed on a 0.2 mmol scale under the standard reaction conditions (Table 1, entry 5). Percentages represent isolated yields. | ||
Subsequently, we proceeded to examine the substrate scope of ring-retentive hydrocyanation (Scheme 3). Generally, cyclopropenes bearing various aryl groups proved suitable for this reaction, yielding the desired cyano-substituted cyclopropane derivatives in moderate to good yields and chemoselectivity. Multiple functional groups, such as methoxyl (3b, 3k, 3m), trifluoromethyl (3e), methylthiol (3g), bromide (3h, 3l), thiophene (3r) and phenyl (3i), were compatible under the optimized reaction conditions. However, this transformation was sensitive to the steric hindrance of the substrates—the ortho-substituted substrates (3m, 3n) gave relatively low yields and poor chemoselectivity. Apart from methyl substituents (R2 = Me), hydrogen and a variety of alkyl substituents were all suitable for this reaction (3y, 3v–3x). Spirocyclic cyclopropenes efficiently underwent ring-retentive hydrocyanation, affording the corresponding spirocyclic cyano-substituted cyclopropanes in satisfactory yields and chemoselectivity (3s, 3t). Furthermore, this reaction also exhibited good tolerance towards dialkyl-substituted substrates, furnishing products 3z and 3a-1 in 55% and 49% yields, respectively, without the formation of any ring-opening product.
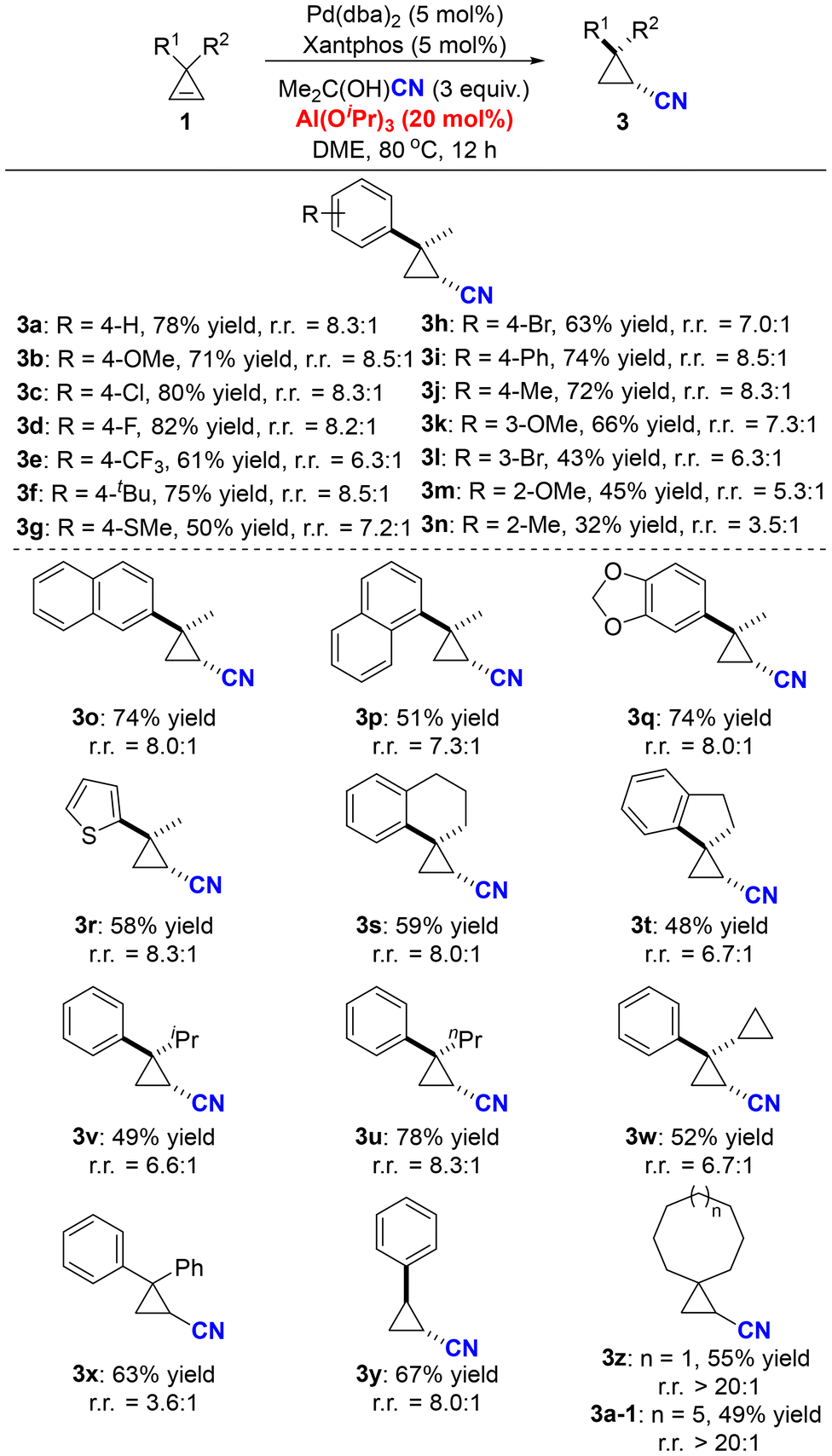 | ||
| Scheme 3 The substrate scope of the ring-retentive hydrocyanation of cyclopropenes. Reactions were performed on a 0.2 mmol scale under the standard reaction conditions (Table 1, entry 16). Percentages represent isolated yields. r.r. = the ratio of the ring-retentive product to the ring-opening product, and it was determined by GC analysis. | ||
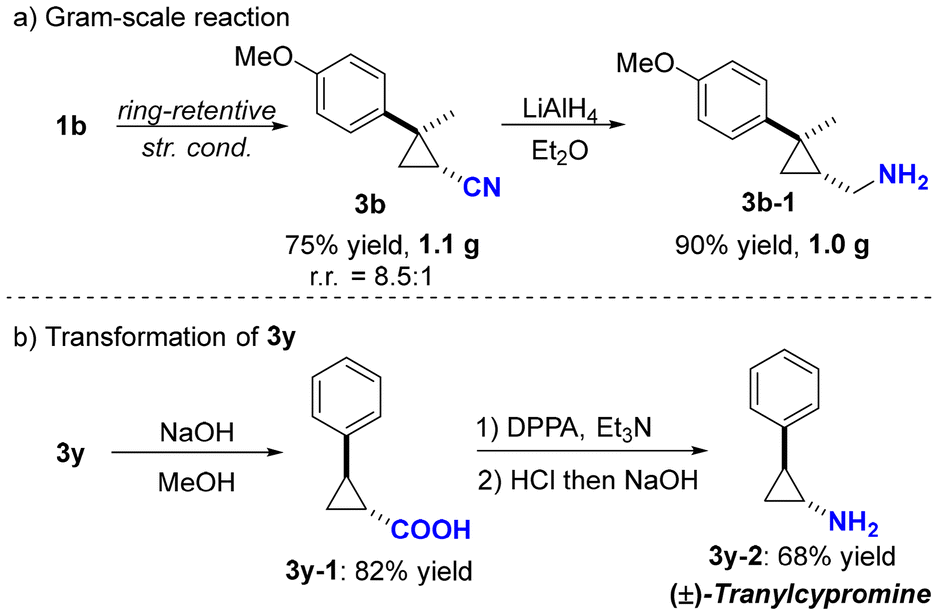
To demonstrate the practicality of this transformation, a gram-scale reaction and product derivatization were conducted (Scheme 4). The ring-retentive hydrocyanation of compound 1b was successfully performed on a gram scale, yielding 1.1 g of 3b with 75% yield and 8.5:1 chemoselectivity (Scheme 4a). Subsequent treatment of 3a with LiAlH4 proceeded efficiently, resulting in the production of 3b-1 with high yield (Scheme 4a). Additionally, (±)-tranylcypromine (3y-2) was synthesized smoothly via hydrolysis followed by the Curtius rearrangement from 3y (Scheme 4b).11
 | ||
| Scheme 4 Gram-scale reaction and product transformation. | ||
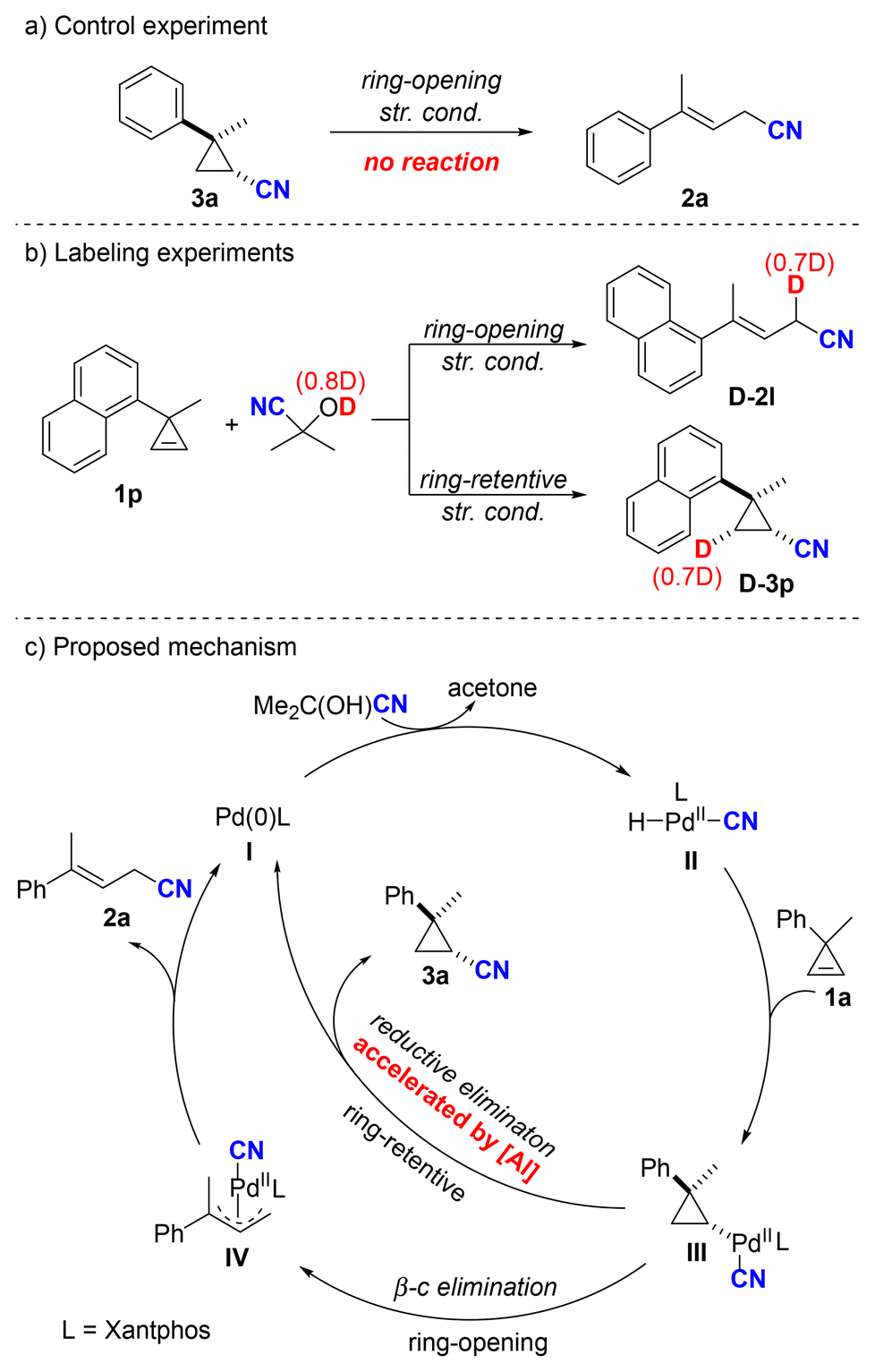
To further understand the mechanism, a control experiment was performed with 3a under the standard ring-opening reaction conditions to determine if 3a was an intermediate (Scheme 5a). However, no 2a was formed and 3a was quantitatively recovered. This result indicated that 3a was not involved as an intermediate thereof. In addition, (CH3)2C(OD)CN was utilized as the HCN source for the hydrocyanation of 1p under both ring-opening and ring-retentive standard conditions (Scheme 5b). Deuterium incorporation at the allylic position (0.7D) was observed in the ring-opening product D-2l. Under ring-retentive conditions, the corresponding product D-3p was obtained, with deuterium incorporated exclusively at the methylene position (0.7D). This deuterium labeling experiment suggests an irreversible syn-hydropalladation process involving cyclopropene. Based on the aforementioned results and tracking time course study (see the ESI† for details), we propose a plausible mechanism to elucidate the two reaction pathways (Scheme 5c). The reaction initiates with palladium(0) species, which undergoes oxidative addition with acetone cyanohydrin to give a palladium(II) intermediate II. Intermediate II subsequently undergoes hydropalladation with cyclopropene 1a to give palladium cyclopropane complex III. This pivotal intermediate III can further react via two potential pathways: β-carbon elimination or reductive elimination. According to the investigations by Nolan, Moloy, and co-workers, the coordination of Lewis acids to metal cyanides can induce partial positive charge formation at the nitrogen and carbon atoms, thereby making the metal center more electron-deficient.10a This electron deficiency accelerates the reductive elimination process, which accounts for the predominant formation of the ring-retentive product in the presence of Lewis acid. Conversely, if intermediate III undergoes β-carbon elimination, it forms the ring-opening π-allyl palladium intermediate IV. Ultimately, allylic nitrile 2a is generated by reductive elimination from IV.
 | ||
| Scheme 5 Control and labeling experiments, and the proposed mechanism. | ||
We have successfully developed a Lewis acid-controlled Pd-catalyzed chemodivergent hydrocyanation of cyclopropenes. In this study, the presence of aluminum Lewis acid plays a pivotal role in determining the reaction pathway. In the absence of aluminum Lewis acid, the reaction predominantly yields ring-opening allylic nitrile products. Conversely, the addition of Lewis acid favors the formation of ring-retentive cyano-substituted cyclopropane products. Further mechanistic studies and investigations into enantioselective ring-retentive hydrocyanation are currently underway in our laboratory.
Author contributions
R. Y., S.-Z. C. and X. F. conceived the project and wrote the manuscript. R. Y. and S.-Z. C. performed the experiments and analysed the data.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (no. 22278265 and U22B20137), the Zhejiang Provincial Natural Science Foundation of China (no. LR24B020002), the start-up funding from Hangzhou Normal University (no. 4095C50222204165), and the Interdisciplinary Research Project of Hangzhou Normal University (2024JCXK04) for financial support.References
- For selected reviews, see: (a) M. S. Baird, Thermally induced cyclopropene-carbene rearrangements: an overview, Chem. Rev., 2003, 103, 1271–1294 CrossRef CAS PubMed; (b) R. Walsh, The cyclopropenepyrolysis story, Chem. Soc. Rev., 2005, 34, 714–732 RSC; (c) M. Rubin, M. Rubina and V. Gevorgyan, Transition metal chemistry of cyclopropenes and cyclopropanes, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS; (d) Z.-B. Zhu, Y. Wei and M. Shi, Recent developments of cyclopropene chemistry, Chem. Soc. Rev., 2011, 40, 5534–5563 RSC; (e) R. Vicente, Recent progresses towards the strengthening of cyclopropene chemistry, Synthesis, 2016, 2343–2360 CrossRef CAS; (f) B. P. Raiguru, S. Nayak, D. R. Mishra, T. Das, S. Mohapatra and N. Mishra, Synthetic applications of cyclopropene and cyclopropenone: recent progress and developments, Asian J. Org. Chem., 2020, 9, 1088–1132 CrossRef; (g) P. Li, X. Zhang and M. Shi, Recent developments in cyclopropene chemistry, Chem. Commun., 2020, 56, 5457–5471 RSC; (h) R. Vicente, C–C Bond cleavages of cyclopropenes: operating for selective ring-opening reactions, Chem. Rev., 2021, 121, 162–226 CrossRef CAS.
- (a) P. V. R. Schleyer, J. E. William and K. R. Blanchard, Evaluation of strain in hydrocarbons. The strain in adamantane and its origin, J. Am. Chem. Soc., 1970, 92, 2377–2386 CrossRef CAS; (b) R. Srinivasan, Kinetics of the thermal isomerization of cyclopropene and 1-methylcyclopropene, J. Am. Chem. Soc., 1969, 91, 6250–6253 CrossRef CAS; (c) R. D. Bach and O. Dmitrenko, Strain energy of small ring hydrocarbons. Influence of C–H bond dissociation energies, J. Am. Chem. Soc., 2004, 126, 4444–4452 CrossRef CAS PubMed.
- For a selected review, see: (a) L. Dian and I. Marek, Asymmetric preparation of polysubstituted cyclopropanes based on direct functionalization of achiral three-membered carbocycles, Chem. Rev., 2018, 118, 8415–8434 CrossRef CAS PubMed. For selected examples, see: (b) S. Nie, A. Lu, E. L. Kuler and V. M. Dong, Enantioselective hydrothiolation: diverging cyclopropenes through ligand control, J. Am. Chem. Soc., 2021, 143, 6176–6184 CrossRef CAS PubMed; (c) P. Chandu, S. Biswas, K. Pal and D. Sureshkumar, Organophotoredox catalysis: switchable radical generation from alkyl sodium sulfinates for sulfonylation and alkylative activation of C–C bonds of cyclopropenes, J. Org. Chem., 2024, 89, 3912–3925 CrossRef CAS PubMed; (d) R. J. Mudd, P. C. Young, J. A. Jordan-Hore, G. M. Rosair and A.-L. Lee, Gold(I)-catalyzed addition of thiols and thioacids to 3,3-disubstituted cyclopropenes, J. Org. Chem., 2012, 77, 7633–7639 CrossRef CAS.
- (a) K. Friedrich and K. Wallenfels, The chemistry of the cyano group, Wiley-Interscience, New York, 1970 Search PubMed; (b) Y. Wang, Y. Du and N. Huang, A survey of the role of nitrile groups in protein–ligand interactions, Future Med. Chem., 2018, 10, 2713–2728 CrossRef CAS PubMed; (c) F. F. Fleming, Nitrile-containing natural products, Nat. Prod. Rep., 1999, 16, 597–606 RSC; (d) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS.
- For selected reviews, see: (a) L. Bini, C. Müller and D. Vogt, Mechanistic studies on hydrocyanation reactions, ChemCatChem, 2010, 2, 590–608 CrossRef CAS; (b) N. Kurono and T. Ohkuma, Catalytic asymmetric cyanation reactions, ACS Catal., 2016, 6, 989–1023 CrossRef CAS; (c) W.-B. Wu, J.-S. Yu and J. Zhou, Catalytic enantioselective cyanation: recent advances and perspectives, ACS Catal., 2020, 10, 7668–7690 CrossRef CAS; (d) H. Zhang, X. Su and K. Dong, Recent progress in transition-metal-catalyzed hydrocyanation of nonpolar alkenes and alkynes, Org. Biomol. Chem., 2020, 18, 391–399 RSC.
- For selected examples, see: (a) X. Zhang, X. Xie and Y. Liu, Nickel-catalyzed highly regioselective hydrocyanation of terminal alkynes with Zn(CN)2 using water as the hydrogen source, J. Am. Chem. Soc., 2018, 140, 7385–7389 CrossRef CAS; (b) F. Ye, J. Chen and T. Ritter, Rh-catalyzed anti-markovnikov hydrocyanation of terminal alkynes, J. Am. Chem. Soc., 2017, 139, 7184–7187 CrossRef CAS; (c) K. Nemoto, T. Nagafuchi, K. Tominaga and K. Sato, Efficient nickel-catalyzed hydrocyanation of alkenes using acetone cyanohydrin as a safer cyano source, Tetrahedron Lett., 2016, 57, 3199–3203 CrossRef CAS; (d) X. Shu, Y.-Y. Jiang, L. Kang and L. Yang, Ni-catalyzed hydrocyanation of alkenes with formamide as the cyano source, Green Chem., 2020, 22, 2734–2738 RSC; (e) J. P. Strache, L. Münzer, A. Adler, D. Blunk and H.-G. Schmalz, Enantioselective nickel-catalyzed hydrocyanation of homostilbenes, Eur. J. Org. Chem., 2023, e202300050 CrossRef CAS; (f) J. Gao, M. Jiao, J. Nie, R. Yu, G.-J. Chen and X. Fang, Nickel-catalyzed migratory hydrocyanation of internal alkenes: unexpected diastereomeric-ligand-controlled regiodivergence, Angew. Chem., Int. Ed., 2021, 60, 1883–1890 CrossRef CAS; (g) J. Long, S. Xia, T. Wang, G.-J. Cheng and X. Fang, Nickel-catalyzed regiodivergent cyanation of allylic alcohols: scope, mechanism, and application to the synthesis of 1,n-dinitriles, ACS Catal., 2021, 11, 13880–13890 CrossRef CAS; (h) F. Sun, C. Yang, J. Nie, G.-J. Cheng and X. Fang, Ligand-controlled regiodivergent nickel-catalyzed hydrocyanation of silyl-substituted 1,3-diynes, Org. Lett., 2021, 23, 4045–4050 CrossRef CAS PubMed; (i) J. Wilting, M. Janssen, C. Müller and D. Vogt, The enantioselective step in the nickel-catalyzed hydrocyanation of 1,3-cyclohexadiene, J. Am. Chem. Soc., 2006, 128, 11374–11375 CrossRef CAS PubMed; (j) B. Saha and T. V. RajanBabu, Nickel(0)-catalyzed asymmetric hydrocyanation of 1,3-dienes, Org. Lett., 2006, 8, 4657–4659 CrossRef CAS; (k) B. N. Bhawal, G. C. Reisenbauer, C. Ehinger and B. Morandi, Overcoming selectivity issues in reversible catalysis: a transfer hydrocyanation exhibiting high kinetic control, J. Am. Chem. Soc., 2020, 142, 10914–10920 CrossRef CAS; (l) T. Burry, S. Kullmann and B. Breit, Nickel(0)-catalyzed hydrocyanation of terminal allenes: a regio- and enantioselective approach to branched allylic nitriles, Adv. Synth. Catal., 2023, 365, 335–341 CrossRef; (m) C. Fan and Q.-L. Zhou, Nickel-catalyzed group transfer of radicals enables hydrocyanation of alkenes and alkynes, Chem. Catal., 2021, 1, 117–128 CrossRef CAS; (n) L. Song, N. Fu, B. G. Ernst, W. H. Lee, M. O. Frederick, R. A. DiStasio and S. Lin, Dual electrocatalysis enables enantioselective hydrocyanation of conjugated alkenes, Nat. Chem., 2020, 12, 747–754 CrossRef CAS PubMed; (o) S. Arai, K. Nakazawa, X.-F. Yang, M. Nakajima, S. Harada and A. Nishida, Nickel-catalysed regio- and stereoselective hydrocyanation of alkynoates and its mechanistic insights proposed by DFT calculations, Org. Biomol. Chem., 2024, 22, 3606–3610 RSC.
- (a) R. Yu, Y. Xing and X. Fang, Regio-, chemo-, and enantioselective Ni-catalyzed hydrocyanation of 1,3-dienes, Org. Lett., 2021, 23, 930–935 CrossRef CAS PubMed; (b) R. Yu, S. Rajasekar and X. Fang, Enantioselective nickel-catalyzed migratory hydrocyanation of nonconjugated dienes, Angew. Chem., Int. Ed., 2020, 59, 21436–21441 CrossRef CAS PubMed; (c) J. Long, R. Yu, J. Gao and X. Fang, Access to 1,3-dinitriles by enantioselective auto-tandem catalysis: merging allylic cyanation with asymmetric hydrocyanation, Angew. Chem., Int. Ed., 2020, 59, 6785–6789 CrossRef CAS PubMed.
- (a) R. Yu, S.-Z. Cai, C. Li and X. Fang, Nickel-catalyzed asymmetric hydroaryloxy- and hydroalkoxycarbonylation of cyclopropenes, Angew. Chem., Int. Ed., 2022, 61, e202200733 CrossRef CAS PubMed; (b) C. Li, R. Yu, S.-Z. Cai and X. Fang, Highly diastereoselective synthesis of polysubstituted cyclopropanecarbonitriles via palladium-catalyzed cyanoesterification of cyclopropenes, Org. Lett., 2023, 25, 5128–5133 CrossRef CAS.
- For selected examples of Lewis acid-promoted transition-metal-catalyzed hydrocyanation, see: (a) X. Fang, P. Yu and B. Morandi, Catalytic reversible alkene-nitrile interconversion through controllable transfer hydrocyanation, Science, 2016, 351, 832–836 CrossRef CAS; (b) A. Bhunia, K. Bergander and A. Studer, Cooperative palladium/Lewis acid-catalyzed transfer hydrocyanation of alkenes and alkynes using 1-methylcyclohexa-2,5-diene-1-carbonitrile, J. Am. Chem. Soc., 2018, 140, 16353–16359 CrossRef CAS PubMed; (c) G. Wang, X. Xie, W. Xu and Y. Liu, Nickel-catalyzed highly regioselective hydrocyanation of alkenes with Zn(CN)2, Org. Chem. Front., 2019, 6, 2037–2042 RSC.
- For selected examples of Lewis acid-promoted C–CN bond activation, see: (a) J. Huang, C. M. Haar, S. P. Nolan, J. E. Marcone and K. G. Moloy, Lewis acids accelerate reductive elimination of RCN from P2Pd(R)(CN), Organometallics, 1999, 18, 297–299 CrossRef CAS; (b) C.-S. Wang, Y. Yu, Y. Sunada, C. Wang and N. Yoshikai, Cobalt-catalyzed carbo- and hydrocyanation of alkynes via C–CN bond activation, ACS Catal., 2022, 12, 4054–4066 CrossRef CAS; (c) T. Zhang, Y.-X. Luan, S.-J. Zheng, Q. Peng and M. Ye, Chiral aluminum complex controls enantioselective nickel-catalyzed synthesis of indenes: C–CN bond activation, Angew. Chem., Int. Ed., 2020, 59, 7439–7443 CrossRef CAS; (d) D. Jiang, X. Li, J. Cai, Y. Bai, L. Zhang and L. Zhao, Mechanistic study of the cooperative palladium/Lewis acid-catalyzed transfer hydrocyanation reaction: the origin of the regioselectivity, Dalton Trans., 2021, 50, 1233–1238 RSC; (e) P. Orecchia, W. Yu and M. Oestreich, Transfer hydrocyanation of α- and α,β-substituted styrenes catalyzed by boron Lewis acids, Angew. Chem., Int. Ed., 2019, 58, 3579–3583 CrossRef CAS.
- (a) R. Csuk, M. J. Schabel and Y. Scholz, Synthesis of the enantiomer of the antidepressant tranylcypromine, Tetrahedron: Asymmetry, 1996, 7, 3505–3512 CrossRef CAS; (b) F.-C. Shu and Q.-L. Zhou, A catalytic enantioselective synthesis of antidepressant tranylcypromine, Synth. Commun., 1999, 29, 567–572 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR spectra of compounds. See DOI: https://doi.org/10.1039/d4qo01609b |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2025 |