
Synergy of prediction rule and total synthesis in solving the stereochemical puzzle of eucalactam B†
Chenqi
Wang
a,
Junyang
Liu
 *b and
Tao
Ye
*ac
*b and
Tao
Ye
*ac
aState Key Laboratory of Chemical Oncogenomics, Peking University Shenzhen Graduate School, Shenzhen 518055, China. E-mail: yet@pkusz.edu.cn
bSchool of Pharmacy and Food Engineering, Wuyi University, Jiangmen, 529020, China. E-mail: liujy@wyu.edu.cn
cQianYan (Shenzhen) Pharmatech. Ltd., Shenzhen 518172, China
First published on 24th October 2024
Abstract
The absolute configurations of the fungal-derived reduced polyketide eucalactam B were initially predicted using the “Biochemistry-based rule” and later confirmed through its first successful total synthesis. This accomplishment involved key reactions such as Brown crotylation, the Paterson anti-aldol reaction, cross-metathesis, and macrolactamization, furnishing eucalactam B in 19 linear steps, with an overall yield of 6.0%. The high degree of alignment between the synthetic compound and the natural product provided strong validation for the accuracy of the “Biochemistry-based rule” in predicting the absolute configurations of fungal-derived reduced polyketides.
Introduction
The identification of biologically active lead compounds from natural sources remains a critical component of drug discovery and development.1 A pivotal step in advancing these potential drug candidates is the comprehensive structural characterization of bioactive natural products. This involves not only determining their molecular composition but also precisely defining their stereochemical configuration and molecular conformation. Accurate structural elucidation is essential, as it provides the basis for understanding the compound's mechanism of action, enhancing its pharmacological properties, and guiding further synthetic modifications in the drug development process. Modern spectroscopic techniques have significantly improved the efficiency and precision of structural elucidation for natural products.2 However, the inherent structural complexity of many natural compounds, coupled with the often limited quantities available for study, can present substantial challenges. These difficulties can lead to ambiguities in assigning all stereogenic centers or, in some cases, result in incorrect structural interpretations. This issue is particularly pronounced with polyketides, where the lack of single-crystal X-ray structures makes it difficult to accurately assign stereogenic centers. As the number of stereocenters increases and molecular flexibility becomes more pronounced, the challenge intensifies. In such cases, total synthesis has emerged as a highly reliable and powerful tool for structural elucidation. By enabling the complete construction and comparison of proposed structures, total synthesis allows for the unambiguous verification of the compound's stereochemistry and overall structure, making it indispensable in cases where spectroscopic data alone may fall short.3Eucalactam B, a secondary metabolite first discovered in 2020 by Gao and colleagues, was isolated from the solid culture extract of the endophytic fungus Diaporthe eucalyptorum KY-95.4 This intriguing compound is characterized by its highly complex 21-membered macrocyclic structure, which consists of a unique combination of a tripeptide unit and a polyketide domain (Fig. 1). The molecule contains eight stereogenic centers embedded within its macrocyclic framework, contributing to its structural complexity. Detailed analyses using nuclear magnetic resonance (NMR) and mass spectrometry (MS) techniques revealed key structural moieties. The tripeptide moiety was identified as a Thr-Gly-Gly sequence, with the threonine residue conclusively identified to be L-threonine via Marfey's method,5 a widely used approach for determining amino acid stereochemistry. Further chemical derivatization and advanced NMR studies provided additional clarity on the relative configuration of the polyketide subunit, confirming that the hydroxyl groups at C-3 and C-5 adopt a relative syn configuration. However, determining the absolute stereochemistry of the entire polyketide region proved challenging due to the inherent conformational flexibility of the molecule. Conventional methods, such as X-ray crystallography and the crystalline sponge technique, were unable to provide a definitive stereochemical assignment. This structural ambiguity underscores the difficulty of resolving complex natural products and highlights the limitations of current crystallographic approaches in cases involving flexible, non-rigid frameworks. In 2023, Marin-Felix and collaborators also successfully isolated eucalactam B from the ethyl acetate extracts of the endophytic fungus Diaporthe africana.6 Despite extensive analyses, including the acquisition of a ROESY (Rotating-Frame Overhauser Effect Spectroscopy) spectrum, significant correlations were observed, yet the relative stereochemistry at several key positions—C-2, C-3, C-5, C-10, C-11, and C-12—remained unresolved. This left crucial gaps in understanding the complete stereochemical arrangement of eucalactam B. Given the complexity of the polyketide moiety, which contains six chiral centers, synthesizing all 64 possible stereoisomers would be highly impractical and time-consuming. Such an exhaustive approach would demand significant resources and might still fail to yield the desired stereochemical clarity due to the challenges of separating and analyzing all possible isomers. Therefore, innovative strategies, such as biochemistry-guided prediction, may play a key role in narrowing down the stereochemical possibilities. These approaches could accelerate the determination of the absolute configuration and help unlock the full structural and biological potential of eucalactam B.
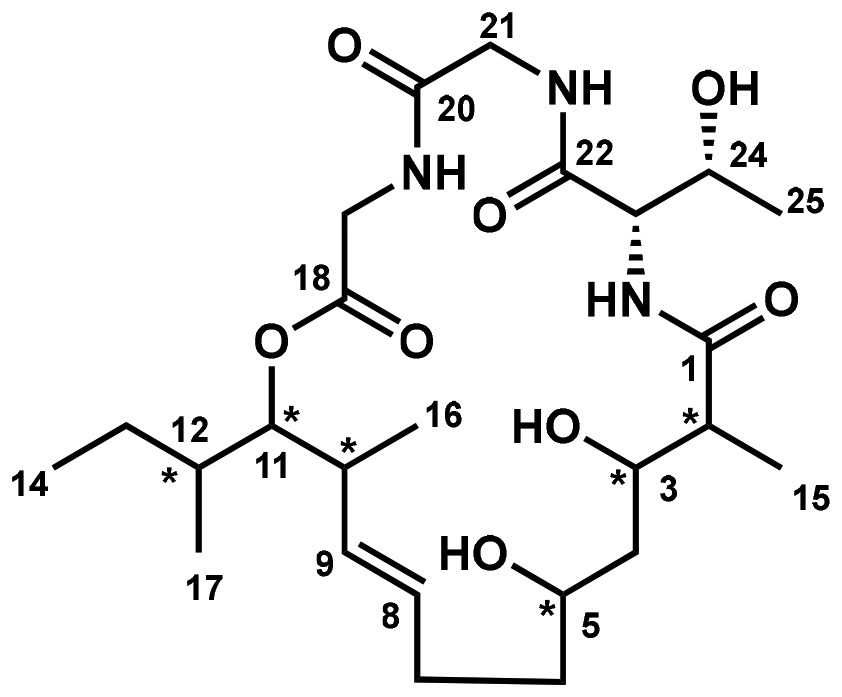 | ||
| Fig. 1 Original structure of eucalactam B. | ||
Results and discussion
Fungal highly reducing polyketide synthases (HR-PKSs) represent a distinct class of single-module enzymes that play a critical role in the biosynthesis of polyketides. These enzymes catalyze the elongation of linear fatty acyl chains, which act as the fundamental building blocks for polyketides.7 In addition to their elongation function, HR-PKSs exhibit precise stereochemical control over these chains, resulting in the formation of specific stereoisomers. Recently, in collaboration with the Oikawa group, we developed a unified stereochemical model that explains the configuration of several complex polyhydroxylated polyketides.8 These include phialotides,9 phomenoic acid,10 and ACR-toxin (acyl carrier protein toxin).11 This model led to the formulation of a biochemistry-based rule that enables the prediction of the absolute configuration of fungal-derived reduced polyketides. The reliability of this rule was demonstrated by its accurate prediction of the absolute configurations of valactamide A12 and acremolides A-B,16 which were further confirmed through their total synthesis. This rule has the potential to streamline the stereochemical determination process for future fungal-derived polyketide compounds. We hypothesized that the Biochemistry-based rule could be leveraged to predict the absolute configuration of fungal-reduced eucalactam B, potentially streamlining synthetic efforts and reducing the need for redundant synthetic work.The mechanism of action of fungal HR-PKSs involves a series of intricate reactions, including initiation, elongation, methylation, reduction, and dehydration. According to the “Biochemistry-guided Rule”,8 the keto reductase (KR) domain reduces a keto group to a °R-configuration hydroxyl group, the enoyl reductase (ER) domain reduces enoyl moieties to a °S-configuration methyl group, and the methyltransferase (MT) domain introduces a °R-configuration methyl group (Fig. 2a). It's important to note that the definitions of °R/°S configuration in the Biochemistry-based Rule slightly differ from the conventional R/S configuration. The °R-configuration can be determined by following these steps: (1) label the group on the PK chain elongation side as RC; (2) label the group on the PK chain initiation side as RM; (3) establish the priority order: OH > RC > RM > Me > H; (4) observe the stereogenic center from the opposite side of the hydrogen atom (the group with the lowest priority), and arrange the remaining three groups in descending order of priority. If this sequence forms a clockwise direction, it is defined as a °R configuration. Conversely, if the sequence forms a counterclockwise direction, it is defined as a °S configuration.
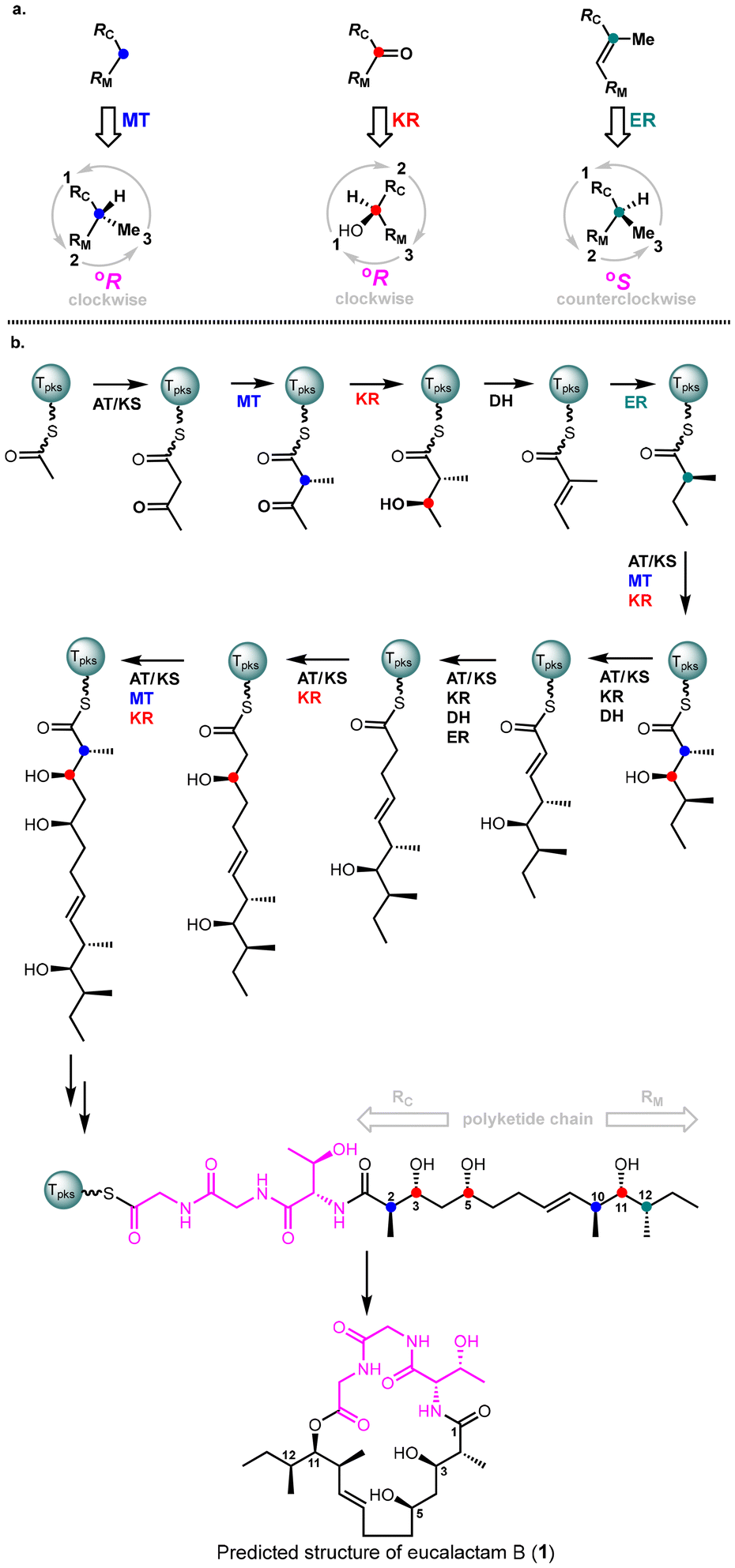 | ||
| Fig. 2 (a). Definition of °R/°S configuration. (b). Biochemistry-based rule guided prediction of the structure of eucalactam B (1). AT: acyltransferase, MT: methyltransferase, KR: keto reductase, ER: enoyl reductase, KS: keto synthase, DH: dehydratase. | ||
With this framework in mind, the absolute configuration of eucalactam B was predicted and is shown in Fig. 2b. Specifically, the ketoreductase (KR) domain of the polyketide synthase (PKS) reduces the corresponding keto groups, leading to a °R configuration for the hydroxyl groups at C3, C5, and C11 (marked in red). Additionally, the enoyl reductase (ER) domain iteratively reduces the corresponding intermediate, producing a °S configuration for the methyl group at C12 (marked in green). Meanwhile, the methyltransferase (MT) domain installs methyl groups at C2 and C10 with a °R configuration (marked in blue). Consequently, the predicted absolute stereochemistry of eucalactam B is illustrated in Fig. 2b. The next essential step is to synthesize this compound to experimentally validate our stereochemical prediction. This will not only confirm the proposed structure but also provide deeper insights into the accuracy of our biochemistry-based prediction model, enhancing its broader application to other fungal polyketide systems.
To achieve the synthesis of eucalactam B, a convergent synthetic route has been designed, as depicted in Scheme 1. The retrosynthetic analysis begins by identifying the amide bond between L-threonine and the polyketide acid as the optimal site for ring closure. This bond will serve as the key disconnection point, enabling the synthesis to proceed in a modular manner. Further disconnection of the amide bond between the two glycine residues generates two key intermediates: dipeptide fragment 3 and the long-chain polyketide fragment 2. The next step involves synthesizing compound 4 through the esterification of fragment 2 with the appropriate alcohol group. This esterification prepares the polyketide moiety for subsequent transformations. Fragment 4, representing the polyketide portion of the molecule, can then be disconnected into known precursors 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 and 6,14 both of which can be accessed via established synthetic methods. To construct the polyketide backbone, olefin cross-metathesis will be employed to couple these two precursors, forming the core carbon framework. The critical stereogenic centers within the polyketide chain will be introduced through a Paterson anti-aldol reaction,15 ensuring the correct relative and absolute configurations of the hydroxyl and methyl groups. This retrosynthetic plan is designed to streamline the overall synthetic process, enabling efficient assembly of the complex macrocyclic structure of eucalactam B. The use of a convergent synthetic approach will provide flexibility in modifying stereochemistry, which could facilitate the generation of analogs for further biological testing and structural validation in future studies.
13 and 6,14 both of which can be accessed via established synthetic methods. To construct the polyketide backbone, olefin cross-metathesis will be employed to couple these two precursors, forming the core carbon framework. The critical stereogenic centers within the polyketide chain will be introduced through a Paterson anti-aldol reaction,15 ensuring the correct relative and absolute configurations of the hydroxyl and methyl groups. This retrosynthetic plan is designed to streamline the overall synthetic process, enabling efficient assembly of the complex macrocyclic structure of eucalactam B. The use of a convergent synthetic approach will provide flexibility in modifying stereochemistry, which could facilitate the generation of analogs for further biological testing and structural validation in future studies.
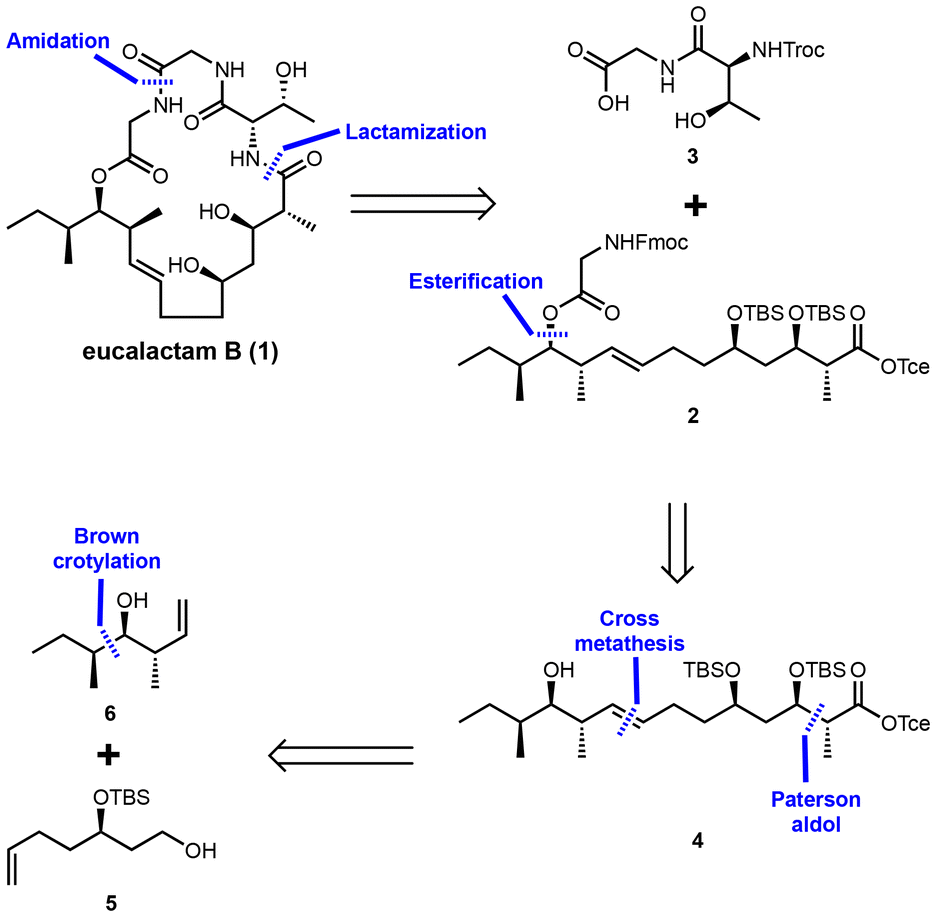 | ||
| Scheme 1 Retrosynthetic analysis of eucalactam B (1). | ||
The synthesis of fragment 12 commenced from the known precursor 5, which was prepared from D-aspartic acid in a six-step sequence.13 Oxidation of the primary alcohol 5 using Dess–Martin Periodinane16 afforded the corresponding aldehyde 7. This aldehyde then underwent a Paterson anti-aldol reaction, affording hydroxyl ketone 9 as a single diastereoisomer with an excellent 87% yield. Subsequent protection of the hydroxyl group in intermediate 9 as a tert-butyldimethylsilyl (TBS) ether gave the protected ketone 10. This ketone was then transformed into carboxylic acid 11via a three-step process with an overall yield of 78%. The transformation involved: (1) reduction of the ketone to the corresponding diol using NaBH4 and K2CO3 in methanol; (2) oxidative cleavage of the diol to provide the corresponding aldehyde; and (3) further oxidation under Pinnick conditions17 to yield carboxylic acid 11. Finally, the carboxylic acid 11 was protected as a Tce (2,2,2-trichloroethyl) ester, delivering fragment 12 in 80% yield (Scheme 2).
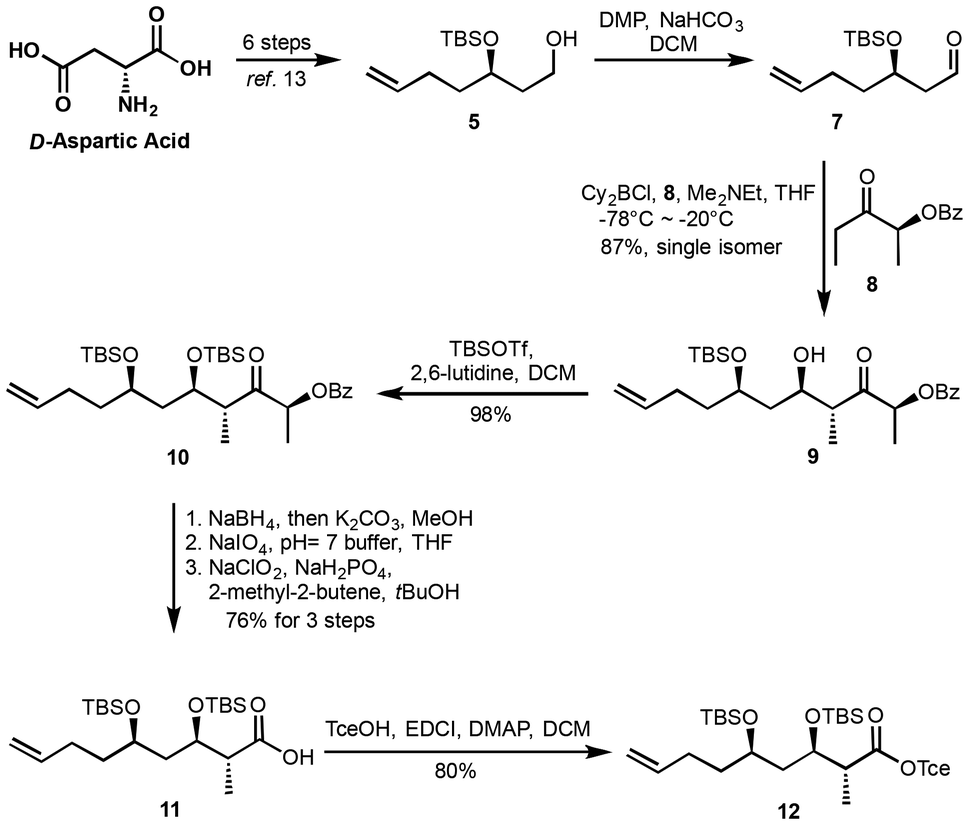 | ||
| Scheme 2 Synthesis of ester 12. | ||
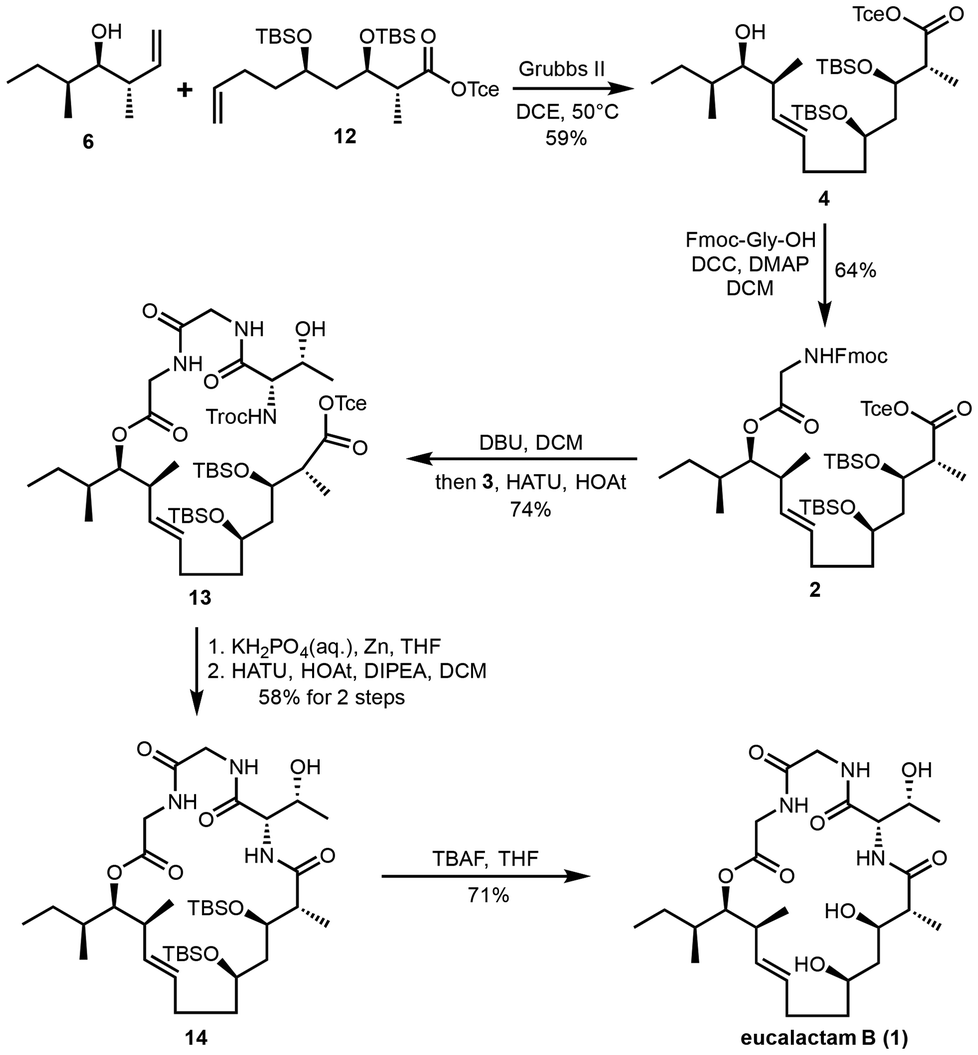
With fragment 12 in hand, we shifted our focus to the completion of the total synthesis (Scheme 3). The known homoallylic alcohol 6 was readily prepared in two steps from (S)-2-methyl-1-butanol via a Brown crotylation reaction,18 providing a reliable route to this key intermediate. The olefin cross-metathesis (CM) reaction between terminal alkenes 6 and 12 was then carried out smoothly using Grubbs II catalyst, delivering the polyketide fragment 4 in a modest 59% yield.19 Next, the secondary alcohol in fragment 4 was esterified with Fmoc-Gly-OH under Steglich conditions, utilizing DCC/DMAP as coupling agents.20 This reaction afforded ester 2 in 64% yield. In parallel, the dipeptide acid 3 was synthesized via a two-step sequence, beginning with the coupling of Troc-protected L-threonine and methyl glycinate hydrochloride, followed by the hydrolysis of the resulting methyl ester (see the ESI† for further details). To elongate ester 2 with dipeptide acid 3, we employed Boger's one-pot peptide coupling protocol,21 which successfully furnished compound 13 in 74% yield. With cyclization precursor 13 in hand, we proceeded to the crucial deprotection step. After several trials, we found that the Troc- and Tce-protecting groups could be simultaneously removed using zinc powder under mild acidic conditions. The resulting free amino acid was then subjected to a HATU-promoted macrolactamization, effectively cyclizing the structure and forming macrocycle 14 in an overall yield of 58% over two steps. The final step in the total synthesis involved global deprotection of the silyl ethers. This was achieved by treatment with TBAF, which completed the synthesis of eucalactam B (1) in 71% yield. With this, the total synthesis of eucalactam B was successfully completed.
 | ||
| Scheme 3 Completion of the total synthesis of eucalactam B (1). | ||
The structure of our synthetic eucalactam B (1) was confirmed by comprehensive spectroscopic analysis. The HRMS, 1H, and 13C NMR spectroscopic data were found to be in excellent agreement with the reported literature data for natural eucalactam B (see Fig. 3 and ESI† for details). The matching chemical shifts and coupling constants provided strong evidence for the successful synthesis. In addition, the obtained specific optical rotation value for the synthesized eucalactam B {[α]20D = −17.7 (c 0.3, MeOH)} was in excellent agreement with the reported value {[α]27D = −17 (c 0.23, MeOH)}.4 As a result, the structure of eucalactam B (1) was unambiguously assigned as depicted in Fig. 2b.
 | ||
| Fig. 3 Comparison of 13C NMR data of synthetic and natural eucalactam. | ||
Conclusions
In summary, the absolute stereochemical configuration of eucalactam B was initially predicted using biochemistry-based rules and subsequently confirmed through total synthesis. The synthesis was completed in 19 linear steps, beginning from D-aspartic acid, with an overall yield of 6.0%. Key features of the synthetic strategy included a highly efficient Paterson anti-aldol reaction, a successful olefin cross-metathesis, and Steglich esterification, all of which were pivotal in assembling the complex polyketide structure. This work highlights the effectiveness of biochemistry-based rules in accurately predicting and validating the absolute stereochemistry of fungal-derived reduced polyketides. It demonstrates their potential as a powerful tool not only for structure elucidation but also for total synthesis and structural validation in natural product chemistry.Author contributions
T. Y. and J. L. conceived and directed the project. C. W. performed the experiments and analysed the data. T. Y., J. L., and C. W. wrote the manuscript with input from all the authors. All authors have read and approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no conflict of interest to report.Acknowledgements
This research was funded by the 2022 Shenzhen Sustainable Supporting Funds for Colleges and Universities (No. 20220811103834002), the Technology & Innovation Bureau of Longgang District (RCTDPT-2019-008), the Guangdong Basic and Applied Basic Research Foundation (2024A1515010958), the Natural Science Foundation of Guangdong Province (2021A1515010344), the Department of Education of Guangdong Province (2021ZDJS097), the National Natural Science Foundation of China (22171014, 21901013 and 22371212).References
- G. M. Cragg and D. J. Newman, Natural Products: A Continuing Source of Novel Drug Leads, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, I. E. Orhan, M. Banach, J. M. Rollinger, D. Barreca, W. Weckwerth, R. Bauer, E. A. Bayer, M. Majeed, A. Bishayee, V. Bochkov, G. K. Bonn, N. Braidy, F. Bucar, A. Cifuentes, G. D'Onofrio, M. Bodkin, M. Diederich, A. T. Dinkova-Kostova, T. Efferth, K. El Bairi, N. Arkells, T.-P. Fan, B. L. Fiebich, M. Freissmuth, M. I. Georgiev, S. Gibbons, K. M. Godfrey, C. W. Gruber, J. Heer, L. A. Huber, E. Ibanez, A. Kijjoa, A. K. Kiss, A. Lu, F. A. Macias, M. J. S. Miller, A. Mocan, R. Müller, F. Nicoletti, G. Perry, V. Pittalà, L. Rastrelli, M. Ristow, G. L. Russo, A. S. Silva, D. Schuster, H. Sheridan, K. Skalicka-Woźniak, L. Skaltsounis, E. Sobarzo-Sánchez, D. S. Bredt, H. Stuppner, A. Sureda, N. T. Tzvetkov, R. A. Vacca, B. B. Aggarwal, M. Battino, F. Giampieri, M. Wink, J.-L. Wolfender, J. Xiao, A. W. K. Yeung, G. Lizard, M. A. Popp, M. Heinrich, I. Berindan-Neagoe, M. Stadler, M. Daglia, R. Verpoorte, C. T. Supuran and T. the International Natural Product Sciences, Natural Products in Drug Discovery: Advances and Opportunities, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS.
- (a) A. Dangi, B. Pande, S. Agrawal, D. Sarkar, K. R. Vamkudoth and U. K. Marelli, Total Synthesis, Structure Elucidation and Expanded Bioactivity of Icosalide A: Effect of Lipophilicity and Ester to Amide Substitution on its Bioactivity, Org. Biomol. Chem., 2023, 21, 5725–5731 RSC; (b) H. Jeon, H. Cho and S. Kim, Total Synthesis and Structural Elucidation of (−)-Cephalezomine G, Org. Lett., 2019, 21, 1121–1124 CrossRef CAS PubMed.
- Y.-Q. Gao, S.-T. Du, J. Xiao, D.-C. Wang, W.-B. Han, Q. Zhang and J.-M. Gao, Isolation and Characterization of Antifungal Metabolites from the Melia azedarach-Associated Fungus Diaporthe eucalyptorum, J. Agric. Food Chem., 2020, 68, 2418–2425 CrossRef CAS.
- P. Marfey, Determination ofD-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene, Carlsberg Res. Commun., 1984, 49, 591 CrossRef CAS.
- B. M. Kemkuignou, C. Lambert, M. Stadler, S. K. Fogue and Y. Marin-Felix, Unprecedented Antimicrobial and Cytotoxic Polyketides from Cultures of Diaporthe africana sp. nov, J. Fungi, 2023, 9, 781 CrossRef PubMed.
- (a) R. J. Cox, Curiouser and Curiouser: Progress in Understanding the Programming of Iterative Highly-reducing Polyketide Synthases, Nat. Prod. Rep., 2023, 40, 9–27 RSC; (b) X.-L. Yang, S. Friedrich, S. Yin, O. Piech, K. Williams, T. J. Simpson and R. J. Cox, Molecular Basis of Methylation and Chain-length Programming in a Fungal Iterative Highly Reducing Polyketide Synthase, Chem. Sci., 2019, 10, 8478–8489 RSC.
- J. Takino, A. Kotani, T. Ozaki, W. Peng, J. Yu, Y. Guo, S. Mochizuki, K. Akimitsu, M. Hashimoto, T. Ye, A. Minami and H. Oikawa, Biochemistry–Guided Prediction of the Absolute Configuration of Fungal Reduced Polyketides, Angew. Chem., Int. Ed., 2021, 60, 23403–23411 CrossRef CAS PubMed.
- A. Yagi, R. Uchida, K. Kobayashi and H. Tomoda, Polyketide Glycosides Phialotides A to H, New Potentiators of Amphotericin B Activity, Produced by Pseudophialophora sp. BF-0158, J. Antibiot., 2020, 73, 211–223 CrossRef CAS.
- M. Devys, J.-P. Férézou, R. S. Topgi, M. Barbier, J.-F. Bousquet and A. Kollmann, Structure and Biosynthesis of Phomenoic Acid, an Antifungal Compound Isolated from Phoma Lingam Tode, J. Chem. Soc., Perkin Trans. 1, 1984, 2133–2137 RSC.
- J. M. Gardner, Y. Kono, J. H. Tatum, Y. Suzuki and S. Takeuchi, Plant Pathotoxins from Alternaria Citri: The Major Toxin Specific for Rough Lemon Plants, Phytochemistry, 1985, 24, 2861–2867 CrossRef CAS.
- T. Zhang, J. Feng, H. Oikawa, Y. Guo and T. Ye, Synergy of Prediction Rule and Total Synthesis in Solving the Stereochemical Puzzle of Valactamides, Precis. Chem., 2024, 2, 120–126 CrossRef CAS.
- (a) D. Saha, S. Guchhait and R. K. Goswami, Total Synthesis and Stereochemical Assignment of Penicitide A, Org. Lett., 2020, 22, 745–749 CrossRef CAS PubMed; (b) J. A. Frick, J. B. Klassen, A. Bathe, J. M. Abramson and H. Rapoport, An Efficient Synthesis of Enantiomerically Pure (R)-(2-Benzyloxyethyl)oxirane from (S)-Aspartic Acid, Synthesis, 1992, 621–623 CrossRef CAS.
- H. C. Brown, K. S. Bhat and R. S. Randad, Chiral synthesis via organoboranes. 21. Allyl- and crotylboration of .alpha.-chiral aldehydes with diisopinocampheylboron as the chiral auxiliary, J. Org. Chem., 1989, 54, 1570–1576 CrossRef CAS.
- I. Paterson, Polyketide Synthesis Using the Boron-mediated, Anti-aldol Reactions of Lactate-derived Ketones: Total Synthesis of (-)-ACRL Toxin IIIB, Synthesis, 1998, 639–652 CrossRef CAS.
- D. B. Dess and J. C. Martin, Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- B. S. Bal, W. E. Childers and H. W. Pinnick, Oxidation of α,β-unsaturated Aldehydes, Tetrahedron, 1981, 37, 2091–2096 CrossRef CAS.
- H. C. Brown and K. S. Bhat, Enantiomeric Z- and E-crotyldiisopinocampheylboranes. Synthesis in High Optical Purity of All Four Possible Stereoisomers of β-methylhomoallyl Alcohols, J. Am. Chem. Soc., 1986, 108, 293–294 CrossRef CAS.
- F. C. Engelhardt, M. J. Schmitt and R. E. Taylor, Kinetically Controlled Cross-Metathesis Reactions with High E-Olefin Selectivities, Org. Lett., 2001, 3, 2209–2212 CrossRef CAS PubMed.
- B. Neises and W. Steglich, Simple Method for the Esterification of Carboxylic Acids, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- N. A. Isley, Y. Endo, Z.-C. Wu, B. C. Covington, L. B. Bushin, M. R. Seyedsayamdost and D. L. Boger, Total Synthesis and Stereochemical Assignment of Streptide, J. Am. Chem. Soc., 2019, 141, 17361–17369 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo01777c |
| This journal is © the Partner Organisations 2025 |