
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing intersystem crossing in multi-brominated eumelanin through transient absorption and surface hopping dynamics†
Kavya
Vinod‡
 ,
Lukhmanul
Hakeem K.‡
,
Diana
Thomas
,
Pallavi Panthakkal
Das
and
Mahesh
Hariharan
*
,
Lukhmanul
Hakeem K.‡
,
Diana
Thomas
,
Pallavi Panthakkal
Das
and
Mahesh
Hariharan
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram (IISER TVM), Maruthamala P.O., Vithura, Thiruvananthapuram 695551, Kerala, India. E-mail: mahesh@iisertvm.ac.in
First published on 8th November 2024
Abstract
Achieving intersystem crossing (ISC) through structural tuning in biological systems is an evolving area for therapeutic and materials research. Eumelanin, a natural pigment, offers huge potential for bio-inspired material design, yet remains underexplored in this regard. Herein, we report the ultrafast intersystem crossing in di-brominated (DMICE-Br2) and tri-brominated (DMICE-Br3) eumelanin model monomers through transient absorption spectroscopy and surface hopping dynamics. Femtosecond and nanosecond transient absorption experiments suggest triplet excited state populations in DMICE-Br2 and DMICE-Br3 with triplet quantum yields and rates of ISC as 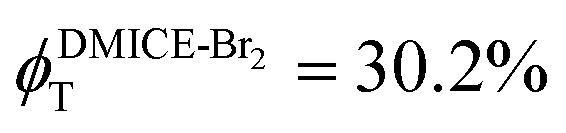 ,
, 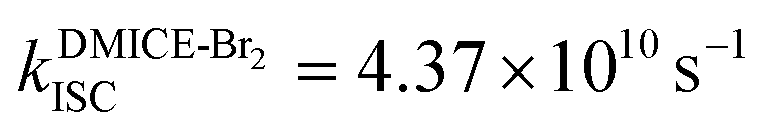 and
and 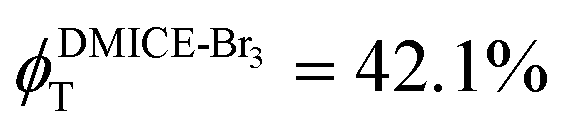 ,
, 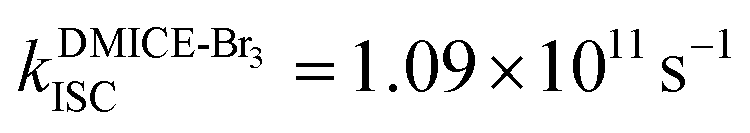 respectively. Theoretical insights into ISC were obtained with nonadiabatic dynamics simulations using the surface hopping including arbitrary couplings method coupled to potential energy surfaces, modelled by linear vibronic coupling (SHARC/LVC). The results show that for both DMICE-Br2 and DMICE-Br3, the initial S1 population decays to the T2 and T3 states in the picosecond timescale to further undergo internal conversion to T1 within sub-ns for DMICE-Br2 and sub-ps for DMICE-Br3. The simulated
respectively. Theoretical insights into ISC were obtained with nonadiabatic dynamics simulations using the surface hopping including arbitrary couplings method coupled to potential energy surfaces, modelled by linear vibronic coupling (SHARC/LVC). The results show that for both DMICE-Br2 and DMICE-Br3, the initial S1 population decays to the T2 and T3 states in the picosecond timescale to further undergo internal conversion to T1 within sub-ns for DMICE-Br2 and sub-ps for DMICE-Br3. The simulated 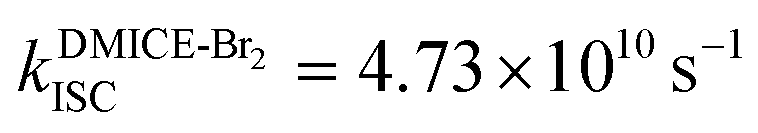 and
and 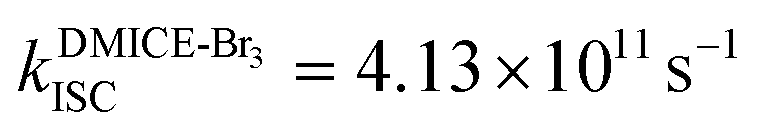 corroborate to the assignment of the ultrafast triplet excited state population observed in the experiments. The increased triplet yields and ISC rates in DMICE-Br2 and DMICE-Br3 are attributed to the enhanced heavy atom effect from additional bromine atoms. This work presents the experimental and computational evidence for ultrafast ISC in multi-brominated eumelanin monomers, with promising implications for eumelanin-inspired material design and photodynamic applications.
corroborate to the assignment of the ultrafast triplet excited state population observed in the experiments. The increased triplet yields and ISC rates in DMICE-Br2 and DMICE-Br3 are attributed to the enhanced heavy atom effect from additional bromine atoms. This work presents the experimental and computational evidence for ultrafast ISC in multi-brominated eumelanin monomers, with promising implications for eumelanin-inspired material design and photodynamic applications.
Introduction
Intersystem crossing (ISC) is the non-radiative, spin-forbidden transition between two electronic states of different multiplicities.1,2 Recently, ISC in biological molecules and the resultant photochemistry have drawn increased attention due to their potential use in clinical research.3,4 In photoactive bio-inspired molecules, ISC can be involved in the generation of reactive oxygen species (ROS) which contributes to photodamage in cells, enabling its use in photodynamic therapy.5,6 For such applications, structural modifications of biomolecules especially by heavy atom incorporation can induce ISC with good yields.7,8 Tracking the excited state dynamics of synthetic bio-inspired photosensitizers is a necessity to guarantee their efficiency, especially since numerous photophysical and photochemical transformations may occur within ultrafast timescales upon photoexcitation.9 The high degrees of freedom and the large number of electronic excited states in these molecules contribute to the availability of different decay pathways.10 In this regard, a combination of ultrafast spectroscopy and theoretical simulations is ideal to investigate the excited state species and the intricate decay modes in small bio-organic molecules. The exploration of potential energy surfaces (PESs) of organic molecules with stationary quantum chemistry is a well-established research field,11–13 but transient absorption experiments coupled with nonadiabatic dynamics simulations are relatively rarer.14–16 Recently, the ultrafast excited-state deactivation mechanism of 6-selenoguanine in water was investigated using femtosecond transient absorption and nonadiabatic trajectory surface-hopping dynamics to conclude that ISC and internal conversion (IC) channels are involved in the energy deactivation of the selenium incorporated nucleobase.17,18Eumelanin, a natural bio-pigment, has intrigued researchers due to its unique structural and optoelectronic properties.19,20 Two key monomers of eumelanin, 5,6-dihydroxyindole (DHI) and 5,6-dihydroxyindole-2-carboxylic acid (DHICA), play a pivotal role in its structure.21–23 Recent research on eumelanin and eumelanin-inspired materials positions them as a novel and versatile class with potential applications in both medical and materials science.24,25 The photochemistry of biologically relevant molecules such as DNA nucleobases upon heavy atom substitution enables their use in photodynamic therapy due to the high triplet yields.17,26 Eumelanin monomers have been underexplored in this context, though halogenation can improve their triplet quantum yields, positioning them as potential candidates for photodynamic therapy alongside DNA monomers and chlorophyll analogues.17,26,27 Our recent work on halogenated eumelanin model monomers revealed ultrafast ISC for mono-brominated (DMICE-Br) and mono-iodinated eumelanin monomers in solution and room temperature phosphorescence in the mono-iodinated monomer in the crystalline state.23 Our ongoing efforts to monitor the photogenerated excitons23,28–30 and delve deeper into ISC in eumelanin monomers prompted us to introduce multiple bromine atoms into the protected eumelanin monomer, ethyl 5,6-dimethoxyindole-2-carboxylate (DMICE), to yield DMICE-Br2 and DMICE-Br3. The experimental and theoretical results indicate that increasing the number of bromine substitutions in DMICE leads to higher rates of intersystem crossing (ISC) and greater triplet quantum yields, thereby enhancing the overall ISC efficiency in the order of DMICE-Br3 > DMICE-Br2 > DMICE-Br. The observed photophysics of the multi-brominated eumelanin monomers has broader implications in prospective photomedicine and materials science research.
Results and discussion
The brominated derivatives of DMICE, that is, DMICE-Br2 and DMICE-Br3, were synthesized according to previously reported procedures and characterized as per standard analytical techniques.31 Bromination of DMICE using N-bromosuccinimide (NBS) at 65 °C in tetrahydrofuran for 2 hours and in acetonitrile for 24 hours resulted in DMICE-Br2 and DMICE-Br3, respectively, in good yields (Fig. 1 and Schemes S1, S2†). Slow evaporation of DMICE-Br2 and DMICE-Br3 solutions separately in an ethyl acetate/hexane solvent mixture at room temperature yielded good quality colourless single crystals for X-ray crystallography. DMICE-Br2 and DMICE-Br3 crystallized in monoclinic crystal systems with P21/c and C2/c space groups respectively (Fig. S1–S3 and Table S1†). On analysing the crystal packing of DMICE-Br2 and DMICE-Br3, hydrogen bonding, π–π interactions and bromine interactions were identified. The Hirshfeld surface analyses performed on the crystal structures quantified the major stabilizing non-covalent interactions to be H⋯H (35.6% in DMICE-Br2 and 29.7% in DMICE-Br3), Br⋯H (23.3% in DMICE-Br2 and 30.5% in DMICE-Br3), O⋯H (16.8% in DMICE-Br2 and 15.5% in DMICE-Br3), C⋯C (4.9% in DMICE-Br2 and 7.6% in DMICE-Br3), Br⋯Br (4.5% in DMICE-Br3) and C⋯H (7.9% in DMICE-Br2) (Fig. S4, S5 and Table S2†).32,33 Symmetry-adapted perturbation theory (SAPT (0)) was performed on the identified dimer orientations of DMICE-Br2 and DMICE-Br3 to quantitatively elucidate the different non-covalent interactions (electrostatic, exchange, induction, and dispersion) stabilizing the crystal packing (Tables S3 and S4†).34 Co-facial orientations of DMICE-Br2 and DMICE-Br3 were identified as the most stable molecular orientations within the respective crystal packings due to the prominent dispersion interactions within the assemblies. Non-covalent interactions (NCI) index analysis also revealed that the green iso-surfaces were prominent for the co-facial orientations of DMICE-Br2 and DMICE-Br3, signifying energy stabilization within these orientations in the crystal packings (Fig. S6 and S7†).35,36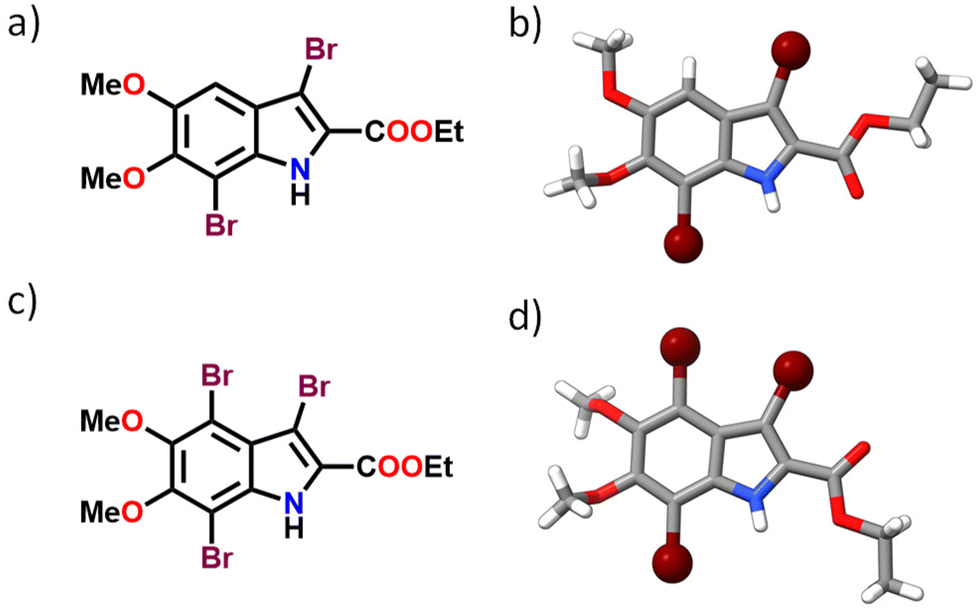 | ||
| Fig. 1 (a) Molecular structure of DMICE-Br2, (b) crystal structure of DMICE-Br2, (c) molecular structure of DMICE-Br3 and (d) crystal structure of DMICE-Br3. | ||
The primary photophysical properties of DMICE-Br2 and DMICE-Br3 were investigated using steady-state UV-vis absorption and fluorescence emission spectroscopy in toluene at room temperature (Fig. 2). The UV-vis absorption spectra of DMICE-Br2 and DMICE-Br3 displayed minimal differences with respect to each other with absorption maxima at 307 nm and 308 nm respectively. The fluorescence emission maximum of DMICE-Br2 was observed at 378 nm and that for DMICE-Br3 at 390 nm, with a red-shift of Δλ = 12 nm compared to DMICE-Br2. The non-brominated eumelanin monomer DMICE exhibited UV-vis absorption maximum at 325 nm and fluorescence emission maximum at 365 nm as reported earlier from our group.23 The fluorescence quantum yields estimated through a relative method in toluene drastically decreased with the increase in the number of bromine atoms from Φf = 49.3% in non-brominated DMICE to Φf = 3.5% in DMICE-Br to Φf < 1% in both DMICE-Br2 and DMICE-Br3.23 The significant reduction of the fluorescence emission with the incorporation of a greater number of bromine atoms in the eumelanin monomer indicates that upon multi-bromination, alternate non-radiative decay pathways become more efficient rather than fluorescence.2,23,37,38
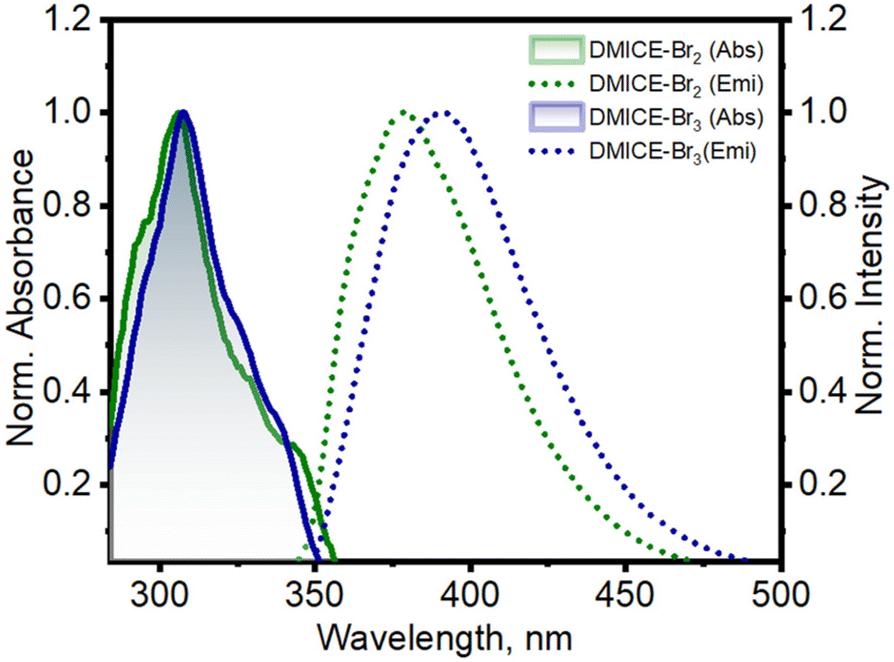 | ||
| Fig. 2 Normalized UV-vis absorption and fluorescence emission spectra of DMICE-Br2 and DMICE-Br3 in toluene. | ||
To examine the excited-state relaxation mechanisms leading to efficient non-radiative decay in multi-brominated eumelanin monomers, femtosecond transient absorption (fsTA) measurements were performed. A Spectra-Physics Tsunami oscillator (80 MHz, 800 nm) served as the seed for a Spectra-Physics Spitfire regenerative amplifier (1 kHz, 4 mJ). A portion of the amplified output was utilized to produce a 400 nm pump pulse, while the remaining 800 nm pulse was directed through a delay line within an ExciPro pump–probe spectrometer. To generate the white light continuum from the delayed 800 nm pulses, a rotating 2 mm thick CaF2 plate was used. Fig. 3 displays the fsTA spectra and the corresponding deconvoluted spectra for DMICE-Br2 and DMICE-Br3 in toluene. Following photoexcitation of DMICE-Br2 at λexc = 325 nm (O.D. at λexc = 0.3) with a 100 fs pump pulse, a broad positive absorption feature appears between 470 and 770 nm, similar to the broad singlet absorption previously observed in DHICA as well as DMICE.23,39 As the broad singlet excited state decays, DMICE-Br2 reveals a sharp additional peak around ∼520 nm within a few tens of picoseconds (Fig. 3a). The new spectral signature in DMICE-Br2 at the later time delays resembles that of the triplet excited species previously reported for DMICE-Br.23 The evolution-associated spectra (EAS) for DMICE-Br2 and DMICE-Br3 were generated using global fitting of the fsTA data in a time versus wavelength framework, modelled as an A → B → GS (GS = ground state) sequential process. Selected kinetic traces with global analysis fitted curves at different wavelengths are shown in Fig. S8† to illustrate the quality of the fits.
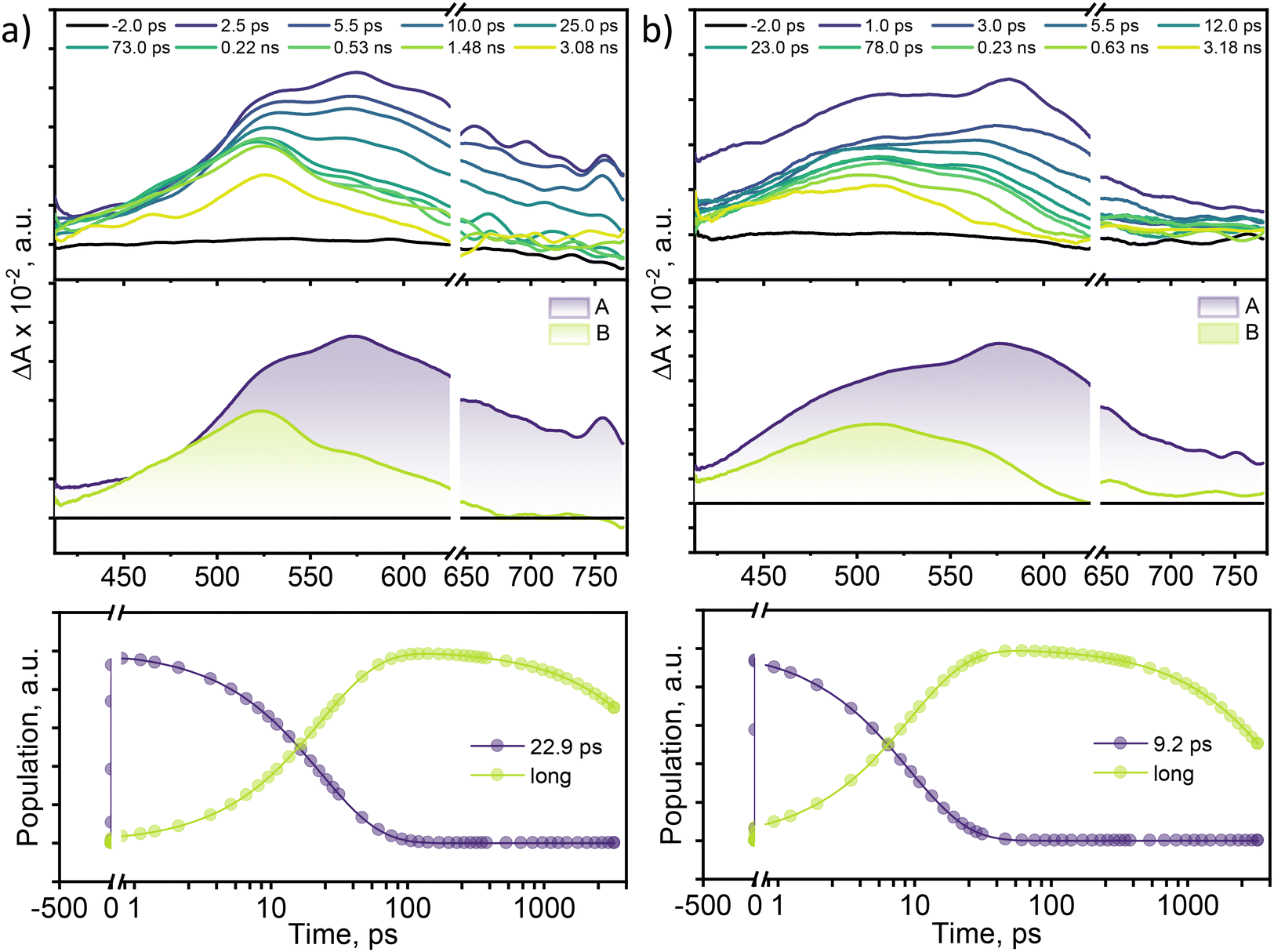 | ||
| Fig. 3 (Top) Femtosecond transient absorption spectra of (a) DMICE-Br2 and (b) DMICE-Br3 in toluene; (middle) evolution-associated spectra (EAS) obtained from the deconvolution of the total spectra through global analysis; (bottom) kinetic profiles of the deconvoluted plots fitted through global analysis using the A → B → GS sequential model. | ||
Upon spectral deconvolution, two distinct components emerge from the fsTA data for DMICE-Br2. The first component (A) is attributed to the singlet state, with a decay time constant of τA→B = 22.9 ps. The second component (B), which is blue-shifted by 50 nm relative to component A, is long-lived and persists beyond the 3.5 ns experimental window. The fsTA spectra for DMICE-Br3 in toluene also revealed the initial broad spectra upon photoexcitation, ranging from 420–750 nm. The initial broad species decay in a few picoseconds to populate another species with a spectral signature of around 420–600 nm and blue-shifted by 69 nm with respect to the initial spectral traces (Fig. 3b). The lack of an isosbestic point in the fsTA spectral traces of DMICE-Br2 and DMICE-Br3 may be attributed to the overlap between the singlet and triplet signatures in the brominated eumelanin counterparts. The deconvolution of the total spectra reveals that component A (singlet species) decays within τA→B = 9.2 ps to populate component B which is a long-lived species and persists across the experimental delay line. The rate of formation of the non-decaying species in DMICE-Br2 and DMICE-Br3 is different suggesting the impact of the number of bromine atoms in determining the rate of the singlet decay in the respective molecule. In our previous work on the mono-brominated eumelanin monomer (DMICE-Br), we reported the singlet state decay followed by triplet excited state formation in DMICE-Br within 0.13 ns.23 On the other hand, the non-brominated DMICE revealed singlet decay within 3.62 ns with negligible ISC.23 The long-lived species observed in the multi-brominated eumelanin monomers could be triplet manifolds formed from intersystem crossing.
To investigate the nature of long-lived species observed in the femtosecond transient absorption (fsTA) experiments of DMICE-Br2 and DMICE-Br3, we performed nanosecond transient absorption measurements (nsTA) in toluene. The photoexcitation of DMICE-Br2 and DMICE-Br3 at 355 nm (pulse width ≈ 8 ns) produces a positive excited state absorption (ESA) band in the nitrogen-purged solution, ranging from 440–550 nm in DMICE-Br2 and 380–600 nm in DMICE-Br3 (Fig. S9 and S10†). To confirm and quantify the formation of the triplet excited state, we performed triplet–triplet energy transfer experiments from DMICE-Br2 and DMICE-Br3 to beta-carotene as the triplet acceptor, using [Ru(bpy)3]Cl2 as the reference.23 The decay rates of the triplet excited state in DMICE-Br2, DMICE-Br3, and [Ru(bpy)3]Cl2 without the addition of beta-carotene were measured. Subsequently, after the addition of beta-carotene, the triplet excited state growth rates at 540 nm were monitored in DMICE-Br2 and DMICE-Br3 relative to [Ru(bpy)3]Cl2 (Fig. S11 and S12†). The triplet quantum yields measured in toluene show the increase in triplet population from ΦT = 30.2% in DMICE-Br2 to ΦT = 42.1% in DMICE-Br3.23 Experimental results show that DMICE-Br2 exhibits a rate of ISC of kISC = 4.37 × 1010 s−1 while DMICE-Br3 shows an increase in the rate of ISC with kISC = 1.09 × 1011 s−1. Previously, we reported the triplet quantum yield and rate of ISC (kISC) of DMICE-Br to be ΦT = 25.4% and kISC = 1.95 × 109 s−1, respectively, while for DMICE, ΦT ≈ 0.23 This trend of ISC rates and triplet quantum yields of DMICE ≪ DMICE-Br < DMICE-Br2 < DMICE-Br3 could be due to the enhanced heavy atom effect, caused by the increase in the number of bromine atoms from DMICE to DMICE-Br3.
Having established the intersystem crossing channels for DMICE-Br2 and DMICE-Br3, we performed delayed emission experiments for the multi-brominated compounds to examine the non-radiative/radiative nature of the triplet excited states in solution. Delayed emission experiments were performed at room temperature as well as 77 K at a delay time of 0.05 ms upon excitation at λexc = 310 nm in toluene. At room temperature, delayed emission was not observed in either DMICE-Br2 or DMICE-Br3. On the other hand, at 77 K, vibronically resolved delayed emission spectra were observed for both DMICE-Br2 and DMICE-Br3 (Fig. S13a†). The delayed emission spectrum of DMICE-Br2 ranges from 460–630 nm and that of DMICE-Br3 ranges from 477–650 nm. A red-shift of Δλ = 19 nm in the gated emission spectra of DMICE-Br3 with respect to DMICE-Br2 was observed. Also, the gated emission spectrum of DMICE-Br2 demonstrates a bathochromic shift of Δλ = 105 nm with respect to the prompt fluorescence. Similarly, DMICE-Br3 displays a bathochromic shift of Δλ = 112 nm between the gated emission and prompt fluorescence. The difference in the emission maxima between the prompt and delayed emission suggests that phosphorescence is activated within the multi-brominated molecules at low temperatures.40 The phosphorescence lifetimes of DMICE-Br2 and DMICE-Br3 in de-aerated toluene at 77 K were estimated to be τP = 16.27 ± 0.30 ms and 7.65 ± 0.12 ms respectively (Fig. S13b†). Generally, non-radiative transitions are repressed at low temperatures, thus rigidifying molecular conformations and reducing vibrational energy dissipations.41 Hence, phosphorescence is activated within the multi-brominated chromophores at low temperatures.
To inspect the role of bromine atoms in populating the triplet excited states in the eumelanin monomers, S–T energy gaps and spin–orbit coupling matrix elements were computed, providing critical insights into the ISC mechanism observed experimentally in DMICE-Br2 and DMICE-Br3 (Table S5†). The S1 → T2 transition appears to be more favorable, attributed to the relatively smaller S–T energy gap (ΔES1–T2 = 0.68 eV for DMICE-Br2 and ΔES1–T2 = 0.38 eV for DMICE-Br3). In contrast, the larger energy gap between the S1 and T1 states (ΔES1–T1 ∼ 1 eV) likely diminishes the probability of a direct S1 → T1 transition. This conclusion is further corroborated by a detailed investigation of the NTOs and electron density difference plots for the S1 and T2 states, which validates the ISC mechanism in both compounds (Fig. S14–S17†). In the S1 state, the Br-centered transition densities exhibit moderate rotational displacement from the indole plane, while the T2 state predominantly features an indole-centered π–π* character, which likely facilitates the necessary orbital angular momentum change for efficient ISC.
With the aim of further elucidating the origin of the experimental time constants associated with ISC and definitively identifying the dominant relaxation pathways in DMICE-Br2 and DMICE-Br3, we performed surface hopping dynamics on DMICE-Br2 and DMICE-Br3 using the SHARC/LVC method as developed by Plasser, González and co-workers (Fig. 4).42–46 An analytical LVC model was applied to depict the excited-state potential energy surfaces (PESs) of DMICE-Br2 and DMICE-Br3. LVC models utilized in SHARC were parametrized as per literature reports.42 The structures of the multi-brominated compounds were optimized in the singlet ground state, and the subsequent frequency calculations confirmed that all vibrational frequencies were positive. The calculations were performed at the B3LYP/def2-TZVP level of theory and dispersion was incorporated via the D3 Becke–Johnson scheme.47 The impact of solvent was approximately treated with the conductor-like polarizable continuum model (CPCM) with toluene (ε = 2.4, n = 1.497), and all initial calculations were performed using ORCA 5.0.4.48,49 The normal mode information for both the compounds was extracted from the frequency calculations and transformed into mass-weighted normal modes. To prevent the spurious coupling between the states, the normal modes with ωi < 300 cm−1 were excluded.50 Among the 93 normal modes, 73 normal modes were considered in order to parametrize the LVC model.50
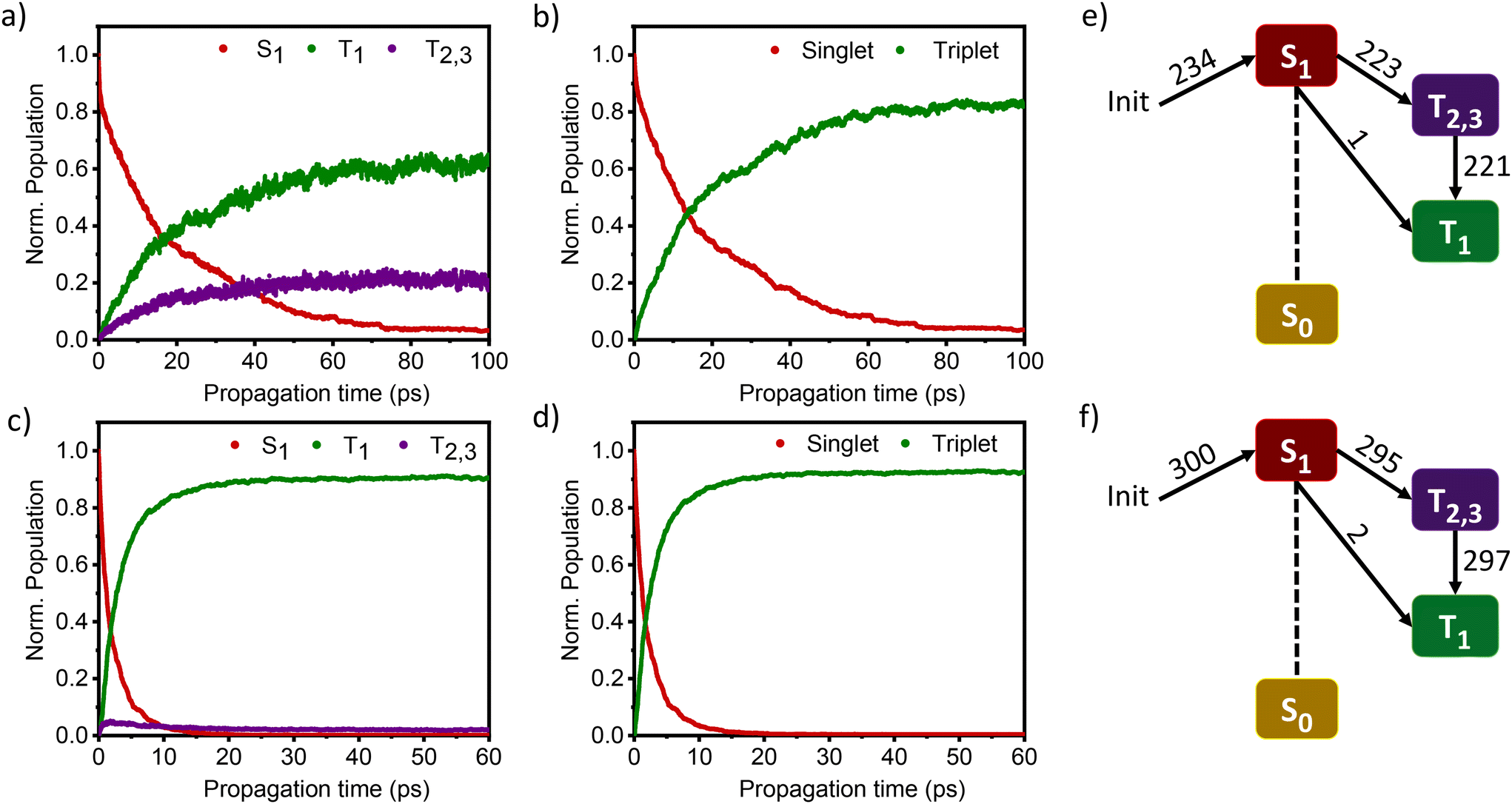 | ||
| Fig. 4 (a) Time evolution of the excited-state populations of DMICE-Br2 from an ensemble of 234 trajectories, (b) total singlet and triplet populations in DMICE-Br2, (c) time evolution of the excited-state populations of DMICE-Br3 from an ensemble of 300 trajectories, (d) total singlet and triplet populations in DMICE-Br3, (e) number of net hops in DMICE-Br2, and (f) number of net hops in DMICE-Br3. Few trajectory hops were also observed from the S1 state to the higher triplet excited states, which then underwent internal conversion to the T2,3 states (note: Init = initial; Norm. = normalized). | ||
For DMICE-Br2 and DMICE-Br3, vertical excitation time-dependent density functional theory (TDDFT) calculations with the TDA-B3LYP functional and def2-TZVP basis set were performed through the SHARC–ORCA interface at the reference geometry and along the selected normal mode coordinates. Dispersion correction was applied using the D3 Becke–Johnson scheme and solvent effects were approximately treated with the CPCM method (toluene, ε = 2.4 and n = 1.497).51 10 singlet states (ground state plus 9 excited states) and 10 triplet states were computed. The computed vertical excitation energies in eV and the respective oscillator strengths are given in the ESI (Tables S6 and S7†). For the SHARC/LVC simulations of DMICE-Br2 and DMICE-Br3, 3000 initial conditions were derived from the Wigner distribution of the harmonic oscillator of the ground state, based on normal modes and frequencies obtained at the B3LYP/def2-TZVP level of theory.42,43,52,53 At each of these initial conditions, single-point vertical excitation calculations were carried out using the SHARC/LVC interface. The optical UV-vis absorption spectra of DMICE-Br2 and DMICE-Br3 were computed from 3000 initial conditions (Fig. S18†).
The nonadiabatic dynamics simulations of DMICE-Br2 and DMICE-Br3 were carried out in the framework of the surface hopping method using the SHARC 3.0 suite of programs, which can include both nonadiabatic coupling and spin–orbit coupling (SOC) simultaneously.42,43,52,54–56 DMICE-Br2 and DMICE-Br3 were excited in the range of 3.50–3.70 eV where the dynamics predominantly started from the S1 state. For DMICE-Br2, a total of 234 trajectories were propagated for 100 ps with a time step length of 0.5 fs, and the wavefunctions were propagated by the locally diabatic method.52,57–59 The simulated excited-state dynamics for DMICE-Br2 are shown in Fig. 4a, which indicates that the S1 state is mainly populated at the initial stage. Fig. 4a further shows that the ultrafast decay of the singlet excited state population leads to the population of the triplet excited state on a picosecond timescale. The relaxation time of the singlet to the triplet for DMICE-Br2 was derived by fitting a sequential first-order kinetics model to the population data, resulting in a decay time of 21.14 ps, in good agreement with the experimental result.42Fig. 4e illustrates that out of the 234 trajectories of DMICE-Br2, a majority of 223 net hops originate from the S1 state to the T2,3 states. Subsequently, only one net hop occurs from the S1 state directly to the T1 state while 221 net hops occur from the T2,3 states to the T1 state. This strongly suggests that DMICE-Br2 follows S1 → T2,3 → T1 relaxation as the major energy decay pathway (Table S8†).
The excited state dynamics for DMICE-Br3 depicted in Fig. 4c also show that the S1 state is predominantly populated at the initial stage. The ultrafast decay of the singlet excited state results in the rapid growth of the triplet excited states in DMICE-Br3, when compared to DMICE-Br2. The faster growth kinetics of the triplet excited state of DMICE-Br3 is attributed to the enhanced heavy atom effect caused by the higher number of bromine atoms in DMICE-Br3 as compared to DMICE-Br2. A total of 300 trajectories were propagated for DMICE-Br3 with 60 ps duration and 0.5 fs time step length, and the wavefunctions were propagated by the locally diabatic method. The decay time of the singlet to the triplet for DMICE-Br3 was obtained by fitting a sequential first-order kinetics model to the population data, yielding a value of 2.42 ps, in agreement with the experimental decay time. In the case of DMICE-Br3, Fig. 4f reveals that out of the hopping dynamics across all 300 trajectories, 295 net hops occur from the S1 state to the T2,3 states, and 2 net hops occur from the S1 state directly to the T1 state. Thus DMICE-Br3 also follows S1 → T2,3 → T1 deactivation as the major energy relaxation channel (Table S8†). In DMICE-Br3, the S1–T2 crossing geometry exhibits a pronounced non-planar distortion compared to DMICE-Br2, which may facilitate a more rapid radiationless decay toward the nearest minimum energy conical intersection, which is the T2–T1 crossing geometry (Fig. S19 and S20†).60 The computed kISC for DMICE-Br2 and DMICE-Br3 follows the increasing trend of kISC = 4.73 × 1010 s−1 in DMICE-Br2 to kISC = 4.13 × 1011 s−1 in DMICE-Br3 and these values are in good agreement with the experimental results. A difference of nearly one order of magnitude is observed between the kISC of DMICE-Br2 and DMICE-Br3 both experimentally and theoretically. This is due to the additional bromine atom in DMICE-Br3 compared to DMICE-Br2 causing an increased heavy atom effect.61
Conclusions
In this work, we examine the ultrafast intersystem crossing dynamics of DMICE-Br2 and DMICE-Br3 by employing transient absorption spectroscopy and surface hopping simulations. The formation of long-lived species in both DMICE-Br2 and DMICE-Br3 was monitored through femtosecond transient absorption experiments. Nanosecond transient absorption measurements further confirmed the long-lived species to be triplet excited states. The increase in the number of bromine atoms from DMICE-Br2 to DMICE-Br3 induces a stronger heavy atom effect in DMICE-Br3 as compared with DMICE-Br2, which significantly enhances the intersystem crossing (ISC) rate by nearly one order of magnitude from kISC = 4.37 × 1010 s−1 in DMICE-Br2 to kISC = 1.09 × 1011 s−1 in DMICE-Br3. Consequently, the singlet state lifetime reduces from τs = 22.9 ps in DMICE-Br2 to τs = 9.2 ps in DMICE-Br3. The increase in kISC is also accompanied by an increase in triplet quantum yields as the number of bromine atoms increases (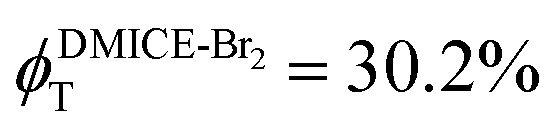 and
and 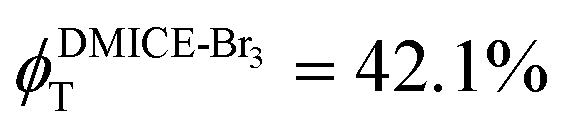 ). The experimental results align with the heavy-atom effect previously observed for DMICE-Br and are now also evident in the current molecules of interest, DMICE-Br2 and DMICE-Br3. Surface hopping dynamics simulations using the SHARC/LVC method further reveal the ultrafast ISC in both DMICE-Br2 and DMICE-Br3, in agreement with the experimental data. The SHARC/LVC method successfully reproduces the observed trend in the reduction of singlet lifetimes from τs = 21.14 ps in DMICE-Br2 to τs = 2.42 ps in DMICE-Br3. The calculated rates of ISC reflect a similar trend as obtained in experiments, with theoretical kISC = 4.73 × 1010 s−1 in DMICE-Br2 and 4.13 × 1011 s−1 in DMICE-Br3. Trajectory analysis from surface hopping simulations also reveals that the population follows the S1 → T2,3 → T1 pathway for DMICE-Br2 as well as DMICE-Br3. In conclusion, this work highlights the pivotal role of the number of bromine atoms in tuning the intersystem crossing dynamics in multi-brominated eumelanin derivatives, potentially paving the way for eumelanin-inspired photosensitizers for practical applications.
). The experimental results align with the heavy-atom effect previously observed for DMICE-Br and are now also evident in the current molecules of interest, DMICE-Br2 and DMICE-Br3. Surface hopping dynamics simulations using the SHARC/LVC method further reveal the ultrafast ISC in both DMICE-Br2 and DMICE-Br3, in agreement with the experimental data. The SHARC/LVC method successfully reproduces the observed trend in the reduction of singlet lifetimes from τs = 21.14 ps in DMICE-Br2 to τs = 2.42 ps in DMICE-Br3. The calculated rates of ISC reflect a similar trend as obtained in experiments, with theoretical kISC = 4.73 × 1010 s−1 in DMICE-Br2 and 4.13 × 1011 s−1 in DMICE-Br3. Trajectory analysis from surface hopping simulations also reveals that the population follows the S1 → T2,3 → T1 pathway for DMICE-Br2 as well as DMICE-Br3. In conclusion, this work highlights the pivotal role of the number of bromine atoms in tuning the intersystem crossing dynamics in multi-brominated eumelanin derivatives, potentially paving the way for eumelanin-inspired photosensitizers for practical applications.
Data availability
All experimental and theoretical procedures, along with the supporting data are available in the ESI.†Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
M. H. acknowledges the Science and Engineering Research Board (CRG/2023/005859), Department of Science and Technology, Government of India, for financial support. K. V. acknowledges CSIR for financial assistance. D. T. acknowledges INSPIRE for financial support. We thank Mr Alex P. Andrews for the X-ray diffraction analyses. We thank Dr Sebastian Mai and Mr Sreekumar P. Y. for their support on the calculations with SHARC. We thank the Padmanabha HPC cluster at IISER TVM for the computing time.References
- C. M. Marian, Understanding and Controlling Intersystem Crossing in Molecules, Annu. Rev. Phys. Chem., 2021, 72, 617–640 CrossRef CAS PubMed.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Access to the triplet excited states of organic chromophores, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- M. R. Detty, S. L. Gibson and S. J. Wagner, Current clinical and preclinical photosensitizers for use in photodynamic therapy, J. Med. Chem., 2004, 47, 3897–3915 CrossRef CAS.
- M. Richter, S. Mai, P. Marquetand and L. González, Ultrafast intersystem crossing dynamics in uracil unravelled by ab initio molecular dynamics, Phys. Chem. Chem. Phys., 2014, 16, 24423–24436 RSC.
- Y. Harada, C. Okabe, T. Kobayashi, T. Suzuki, T. Ichimura, N. Nishi and Y.-Z. Xu, Triplet Formation of 4-Thiothymidine and Its Photosensitization to Oxygen Studied by Time-Resolved Thermal Lensing Technique, J. Phys. Chem. Lett., 2010, 1, 480–484 CrossRef CAS.
- H. Kuramochi, T. Kobayashi, T. Suzuki and T. Ichimura, Photophysical properties of 5-substituted 2-thiopyrimidines, J. Phys. Chem. B, 2010, 114, 8782–8789 CrossRef CAS PubMed.
- S. Mai, M. Pollum, L. Martínez-Fernández, N. Dunn, P. Marquetand, I. Corral, C. E. Crespo-Hernández and L. González, The origin of efficient triplet state population in sulfur-substituted nucleobases, Nat. Commun., 2016, 7, 13077 CrossRef CAS.
- F. Peccati, S. Mai and L. González, Insights into the deactivation of 5-bromouracil after ultraviolet excitation, Philos. Trans. R. Soc., A, 2017, 375, 20160202 CrossRef.
- M. Richter, P. Marquetand, J. González-Vázquez, I. Sola and L. González, Femtosecond Intersystem Crossing in the DNA Nucleobase Cytosine, J. Phys. Chem. Lett., 2012, 3, 3090–3095 CrossRef CAS.
- M. Barbatti, A. J. Aquino, J. J. Szymczak, D. Nachtigallová, P. Hobza and H. Lischka, Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21453–21458 CrossRef CAS.
- D. Valverde, S. Mai, A. V. Sanches de Araújo, S. Canuto, L. González and A. C. Borin, On the population of triplet states of 2-seleno-thymine, Phys. Chem. Chem. Phys., 2021, 23, 5447–5454 RSC.
- Y.-H. Zhu, X.-F. Tang, X.-P. Chang, T.-S. Zhang, B.-B. Xie and G. Cui, Mechanistic Photophysics of Tellurium-Substituted Uracils: Insights from Multistate Complete-Active-Space Second-Order Perturbation Calculations, J. Phys. Chem. A, 2021, 125, 8816–8826 CrossRef CAS.
- E. Vos, T. R. Scott, J. González-Vázquez, I. Corral, D. G. Truhlar and L. Gagliardi, Intrastrand Photolesion Formation in Thio-Substituted DNA: A Case Study Including Single-Reference and Multireference Methods, J. Phys. Chem. A, 2020, 124, 10422–10433 CrossRef CAS.
- Z. Lan, Y. Lu, E. Fabiano and W. Thiel, QM/MM Nonadiabatic Decay Dynamics of 9H-Adenine in Aqueous Solution, ChemPhysChem, 2011, 12, 1989–1998 CrossRef CAS PubMed.
- X. Guo, Y. Zhao and Z. Cao, A QM/MM MD insight into photodynamics of hypoxanthine: distinct nonadiabatic decay behaviors between keto-N7H and keto-N9H tautomers in aqueous solution, Phys. Chem. Chem. Phys., 2014, 16, 15381–15388 RSC.
- R. Borrego-Varillas, D. C. Teles-Ferreira, A. Nenov, I. Conti, L. Ganzer, C. Manzoni, M. Garavelli, A. Maria de Paula and G. Cerullo, Observation of the Sub-100 Femtosecond Population of a Dark State in a Thiobase Mediating Intersystem Crossing, J. Am. Chem. Soc., 2018, 140, 16087–16093 CrossRef CAS.
- K. M. Farrell, M. M. Brister, M. Pittelkow, T. I. Sølling and C. E. Crespo-Hernández, Heavy-Atom-Substituted Nucleobases in Photodynamic Applications: Substitution of Sulfur with Selenium in 6-Thioguanine Induces a Remarkable Increase in the Rate of Triplet Decay in 6-Selenoguanine, J. Am. Chem. Soc., 2018, 140, 11214–11218 CrossRef CAS PubMed.
- D. Valverde, S. Mai, S. Canuto, A. C. Borin and L. González, Ultrafast Intersystem Crossing Dynamics of 6-Selenoguanine in Water, JACS Au, 2022, 2, 1699–1711 CrossRef CAS.
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and M. Buehler, Melanin Biopolymers: Tailoring Chemical Complexity for Materials Design, Angew. Chem., Int. Ed., 2020, 59, 11196–11205 CrossRef PubMed.
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and T. Sarna, Chemical and Structural Diversity in Eumelanins: Unexplored Bio-Optoelectronic Materials, Angew. Chem., Int. Ed., 2009, 48, 3914–3921 CrossRef PubMed.
- D. Sasikumar, K. Vinod, J. Sunny and M. Hariharan, Exciton interactions in helical crystals of a hydrogen-bonded eumelanin monomer, Chem. Sci., 2022, 13, 2331–2338 RSC.
- K. Vinod, R. Mathew, C. Jandl, B. Thomas and M. Hariharan, Electron diffraction and solid-state NMR reveal the structure and exciton coupling in a eumelanin precursor, Chem. Sci., 2024, 15, 16015–16024 RSC.
- K. Vinod, S. D. Jadhav and M. Hariharan, Room Temperature Phosphorescence in Crystalline Iodinated Eumelanin Monomer, Chem. – Eur. J., 2024, 30, e202400499 CrossRef CAS PubMed.
- W. Cao, N. C. McCallum, Q. Z. Ni, W. Li, H. Boyce, H. Mao, X. Zhou, H. Sun, M. P. Thompson, C. Battistella, M. R. Wasielewski, A. Dhinojwala, M. D. Shawkey, M. D. Burkart, Z. Wang and N. C. Gianneschi, An Abiotic Selenium Analogue of Pheomelanin, J. Am. Chem. Soc., 2020, 142, 12802–12810 CrossRef CAS.
- Z. E. Siwicka, F. A. Son, C. Battistella, M. H. Moore, J. Korpanty, N. C. McCallum, Z. Wang, B. J. Johnson, O. K. Farha and N. C. Gianneschi, Synthetic Porous Melanin, J. Am. Chem. Soc., 2021, 143, 3094–3103 CrossRef CAS.
- B. Ashwood, M. Pollum and C. E. Crespo-Hernández, Photochemical and Photodynamical Properties of Sulfur-Substituted Nucleic Acid Bases, Photochem. Photobiol., 2019, 95, 33–58 CrossRef CAS PubMed.
- M. Zhu, H. Zhang, G. Ran, Y. Yao, Z.-S. Yang, Y. Ning, Y. Yu, R. Zhang, X.-X. Peng, J. Wu, Z. Jiang, W. Zhang, B.-W. Wang, S. Gao and J.-L. Zhang, Bioinspired Design of seco-Chlorin Photosensitizers to Overcome Phototoxic Effects in Photodynamic Therapy, Angew. Chem., Int. Ed., 2022, 61, e202204330 CrossRef CAS PubMed.
- J. Sivanarayanan, E. Sebastian, K. Vinod, F. Würthner and M. Hariharan, Ultrafast Intersystem Crossing in Selenium-Annulated Perylene Bisimide, J. Phys. Chem. C, 2022, 126, 13319–13326 CrossRef CAS.
- E. Sebastian, A. M. Philip, A. Benny and M. Hariharan, Null Exciton Splitting in Chromophoric Greek Cross (+) Aggregate, Angew. Chem., Int. Ed., 2018, 57, 15696–15701 CrossRef CAS.
- A. Mohan, E. Sebastian, M. Gudem and M. Hariharan, Near-Quantitative Triplet State Population via Ultrafast Intersystem Crossing in Perbromoperylenediimide, J. Phys. Chem. B, 2020, 124, 6867–6874 CrossRef CAS PubMed.
- A. H. Aebly, J. N. Levy, B. J. Steger, J. C. Quirke and J. M. Belitsky, Expedient synthesis of eumelanin-inspired 5,6-dihydroxyindole-2-carboxylate ethyl ester derivatives, RSC Adv., 2018, 8, 28323–28328 RSC.
- M. A. Spackman and D. Jayatilaka, Hirshfeld surface analysis, CrystEngComm, 2009, 11, 19–32 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS.
- R. M. Parrish, L. A. Burns, D. G. A. Smith, A. C. Simmonett, A. E. DePrince, E. G. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. Di Remigio, R. M. Richard, J. F. Gonthier, A. M. James, H. R. McAlexander, A. Kumar, M. Saitow, X. Wang, B. P. Pritchard, P. Verma, H. F. Schaefer, K. Patkowski, R. A. King, E. F. Valeev, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS PubMed.
- P. Hobza and J. Řezáč, Introduction: Noncovalent Interactions, Chem. Rev., 2016, 116, 4911–4912 CrossRef CAS PubMed.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. M. Philip, M. Gudem, E. Sebastian and M. Hariharan, Decoding the Curious Tale of Atypical Intersystem Crossing Dynamics in Regioisomeric Acetylanthracenes, J. Phys. Chem. A, 2019, 123, 6105–6112 CrossRef CAS.
- S. Sarkar, H. P. Hendrickson, D. Lee, F. DeVine, J. Jung, E. Geva, J. Kim and B. D. Dunietz, Phosphorescence in Bromobenzaldehyde Can Be Enhanced through Intramolecular Heavy Atom Effect, J. Phys. Chem. C, 2017, 121, 3771–3777 CrossRef CAS.
- A. Ilina, K. E. Thorn, P. A. Hume, I. Wagner, R. R. Tamming, J. J. Sutton, K. C. Gordon, S. K. Andreassend, K. Chen and J. M. Hodgkiss, The photoprotection mechanism in the black-brown pigment eumelanin, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2212343119 CrossRef CAS PubMed.
- S. Freed and W. Salmre, Phosphorescence Spectra and Analyses of Some Indole Derivatives, Science, 1958, 128, 1341–1342 CrossRef CAS.
- M. A. Niyas, S. Garain, K. Shoyama and F. Würthner, Room-Temperature Near-Infrared Phosphorescence from C64 Nanographene Tetraimide by π-Stacking Complexation with Platinum Porphyrin, Angew. Chem., Int. Ed., 2024, 63, e202406353 CrossRef CAS.
- F. Plasser, S. Gómez, M. F. S. J. Menger, S. Mai and L. González, Highly efficient surface hopping dynamics using a linear vibronic coupling model, Phys. Chem. Chem. Phys., 2019, 21, 57–69 RSC.
- S. Mai, D. Avagliano, M. Heindl, P. Marquetand, M. F. S. J. Menger, M. Oppel, F. Plasser, S. Polonius, M. Ruckenbauer, Y. Shu, D. G. Truhlar, L. Zhang, P. Zobel and L. González, SHARC3.0: Surface Hopping Including Arbitrary Couplings – Program Package for Non-Adiabatic Dynamics, 2023, https://sharc-md.org/ Search PubMed.
- M. Richter, P. Marquetand, J. González-Vázquez, I. Sola and L. González, SHARC: ab Initio Molecular Dynamics with Surface Hopping in the Adiabatic Representation Including Arbitrary Couplings, J. Chem. Theory Comput., 2011, 7, 1253–1258 CrossRef CAS.
- J. P. Zobel, M. Heindl, F. Plasser, S. Mai and L. González, Surface Hopping Dynamics on Vibronic Coupling Models, Acc. Chem. Res., 2021, 54, 3760–3771 CrossRef CAS PubMed.
- A. J. Atkins and L. González, Trajectory Surface-Hopping Dynamics Including Intersystem Crossing in [Ru(bpy)3]2+, J. Phys. Chem. Lett., 2017, 8, 3840–3845 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- Y. Takano and K. N. Houk, Benchmarking the Conductor-like Polarizable Continuum Model (CPCM) for Aqueous Solvation Free Energies of Neutral and Ionic Organic Molecules, J. Chem. Theory Comput., 2005, 1, 70–77 CrossRef PubMed.
- S. Mai, M. Menger, M. Marazzi, D. L. Stolba, A. Monari and L. González, Competing ultrafast photoinduced electron transfer and intersystem crossing of [Re(CO)3(Dmp)(His124)(Trp122)]+ in Pseudomonas aeruginosa azurin: a nonadiabatic dynamics study, Theor. Chem. Acc., 2020, 139, 65 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- S. Mai, P. Marquetand and L. González, Nonadiabatic dynamics: The SHARC approach, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1370 Search PubMed.
- J. P. Dahl and M. Springborg, The Morse oscillator in position space, momentum space, and phase space, J. Chem. Phys., 1988, 88, 4535–4547 CrossRef CAS.
- J. C. Tully and R. K. Preston, Trajectory Surface Hopping Approach to Nonadiabatic Molecular Collisions: The Reaction of H+ with D2, J. Chem. Phys., 1971, 55, 562–572 CrossRef CAS.
- S. Mai, P. Marquetand and L. González, A general method to describe intersystem crossing dynamics in trajectory surface hopping, Int. J. Quantum Chem., 2015, 115, 1215–1231 CrossRef CAS.
- M. Fumanal, F. Plasser, S. Mai, C. Daniel and E. Gindensperger, Interstate vibronic coupling constants between electronic excited states for complex molecules, J. Chem. Phys., 2018, 148, 124119 CrossRef.
- G. Granucci, M. Persico and A. Toniolo, Direct semiclassical simulation of photochemical processes with semiempirical wave functions, J. Chem. Phys., 2001, 114, 10608–10615 CrossRef CAS.
- F. Plasser, M. Ruckenbauer, S. Mai, M. Oppel, P. Marquetand and L. González, Efficient and Flexible Computation of Many-Electron Wave Function Overlaps, J. Chem. Theory Comput., 2016, 12, 1207–1219 CrossRef CAS.
- A. Jain, E. Alguire and J. E. Subotnik, An Efficient, Augmented Surface Hopping Algorithm That Includes Decoherence for Use in Large-Scale Simulations, J. Chem. Theory Comput., 2016, 12, 5256–5268 CrossRef CAS.
- J. Cao and Z.-Z. Xie, Internal conversion and intersystem crossing in α,β-enones: a combination of electronic structure calculations and dynamics simulations, Phys. Chem. Chem. Phys., 2016, 18, 6931–6945 RSC.
- A. Malinge, S. Kumar, D. Chen, E. Zysman-Colman and S. Kéna-Cohen, Heavy Atom Effect in Halogenated mCP and Its Influence on the Efficiency of the Thermally Activated Delayed Fluorescence of Dopant Molecules, J. Phys. Chem. C, 2024, 128, 1122–1130 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2387375 and 2387376. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01832j |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2025 |