
Visible-light responsive defluorination-acyl fluoride exchange for photoclick labeling based on phenoxazine chromophores†
Lijun
Deng‡
,
Sitong
Li‡
,
Cefei
Zhang
,
Yuqiao
Zhou
 ,
Zhishan
Su
*,
Changwei
Hu
,
Xiaohu
Zhao
and
Zhipeng
Yu
*
,
Zhishan
Su
*,
Changwei
Hu
,
Xiaohu
Zhao
and
Zhipeng
Yu
*
Key Laboratory of Green Chemistry & Technology of Ministry of Education, College of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu 610064, P. R. China. E-mail: suzhishan@scu.edu.cn; zhipengy@scu.edu.cn
First published on 1st November 2024
Abstract
Photoclick chemistry represents an integration of photo- and click chemistry, enabling spatiotemporal control, high selectivity, and efficient conjugation. Photo-induced defluorination acyl fluoride exchange (photo-DAFEx), as a novel photoclick reaction, has emerged as a promising tool for the flourishing field of photoaffinity labeling (PAL) for drug discovery and the exploration of protein interactions. Currently, the first-generation photo-DAFEx reaction relies on the photo-defluorination of a monocyclic m-trifluoromethylaniline, and consequently the excitation wavelength (λex.) falls within the UV-B band (311 nm), limiting its widespread applications. Therefore, there is a high demand for the discovery of innovative cores that can expedite visible-light-induced photo-DAFEx reactions and for the exploration of their crosslinking capabilities. Herein, we report that the combination of phenoxazine chromophores with dialkylated amine auxochromes expands the excitation wavelength (λex.) of the photo-DAFEx reacion into the visible region (405 nm), enabling the multifunctional design of photoaffinity probes for in situ identification of drug–target interactions. By employing the phenoxazine-based photo-DAFEx reagent, we successfully developed potent PAL probes targeting hCA-II and BRD4, which can be activated with controllability using a 405 nm LED, thereby underscoring the potential of photo-DAFEx in advancing our understanding of protein–ligand interactions.
Introduction
Elucidating molecular interactions and dynamics of drugs with biomolecules, particularly with proteins within their native biological contexts, is crucial for comprehending their efficacy and leveraging their therapeutic potential.1 Photoaffinity labeling (PAL),2 which utilizes a photochemical probe to covalently conjugate drugs to targets in situ in response to light activation, has emerged as one of the few strategies for drug discovery, enabling identification of novel drug targets3 and direct mapping of interactions between drug molecules and proteins.4 Photoaffinity warheads serve as photo-controllable crosslinkers5 that can be transformed into highly reactive intermediates only upon exposure to light within a specific wavelength range. Therefore, a photoaffinity warhead, when incorporated with pharmacophores, remains dormant during drug delivery and dynamic distribution. Subsequently, it enables the non-invasive, covalent capture of transient interactions with biomolecules in vicinity within living systems.6 For visualization purposes7 and mass spectrometry (MS)-based profiling of the capturing event,4 the design of a PAL probe typically comprises both a photoaffinity warhead and a bioorthogonal handle,8 allowing for subsequent installation of either a fluorophore or an enrichment functionality, while being tethered onto the pharmacophore via a flexible linker (Scheme 1a). Since the PAL aims to capture non-covalent interacting targets spatiotemporally and dynamically, the advancement of photoaffinity warheads (photo-crosslinking reagents) with characteristic features has become a vital factor in adapting to diverse interaction scenarios.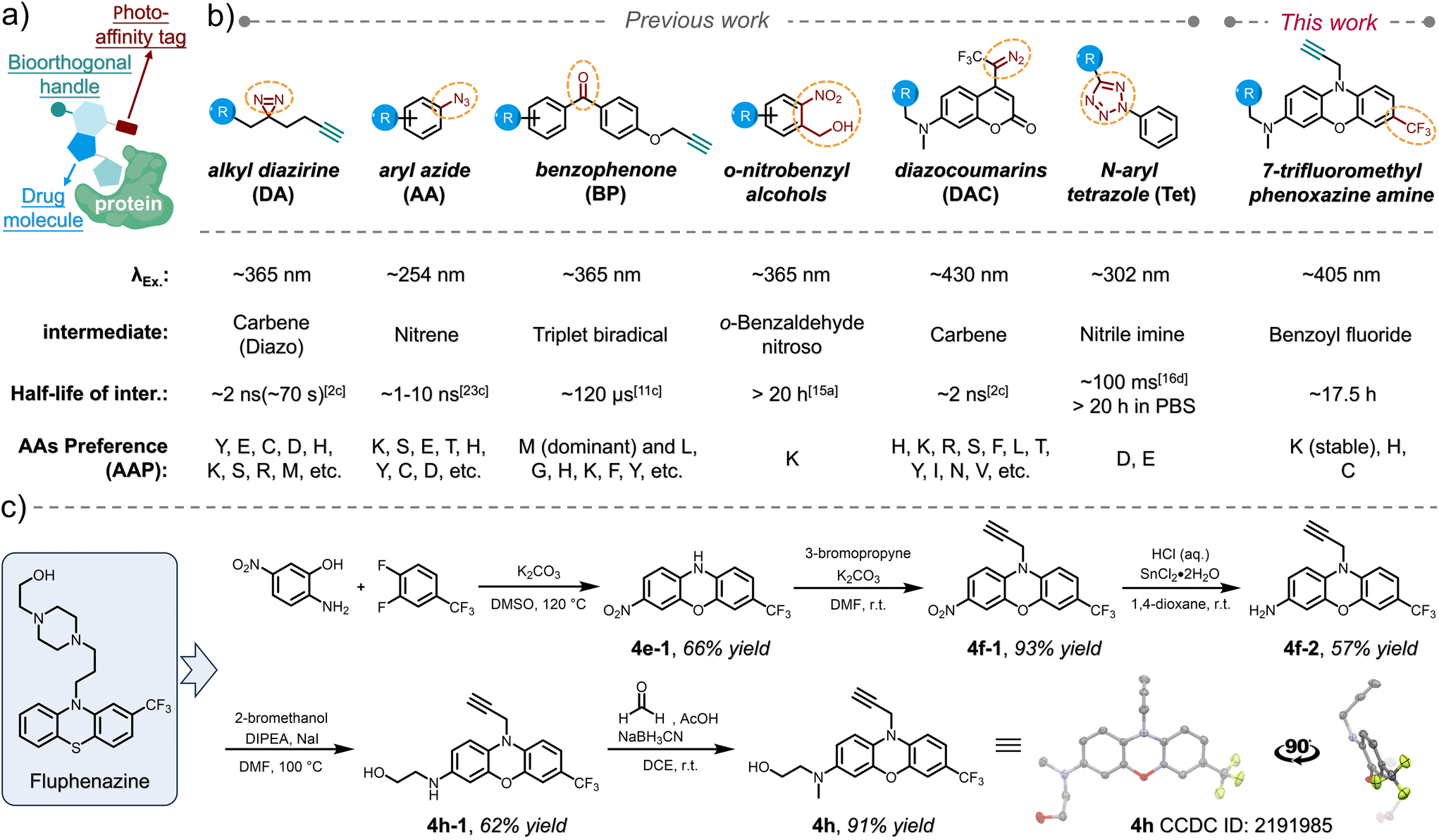 | ||
| Scheme 1 (a) Conceptual diagram of PAL probe design with a bioorthogonal handle. (b) The molecular structure of the widely used PAL warheads and the photo-DAFEx reagent in this work. Key parameters for these PAL structures are clarified. (c) The structure of fluphenazine and the synthetic route for TFMPxA and the X-ray crystal structure of 4h. | ||
Since the development of PAL by Westheimer et al. in 1962,2a a myriad of photo-crosslinker reagents have subsequently emerged, each proving unique photochemical behaviours and specific amino acid preferences (AAP) during the crosslinking process. Classical photo-crosslinkers are primarily represented by three photoreactive moieties: alkyl diazirines (DAs),9 aryl azides (AAs),10 and benzophenones (BPs)11 (Scheme 1b). Upon irradiation, these moieties are converted into highly reactive intermediates, specifically carbenes/diazos for alkyl diazirines, nitrenes for aryl azides, and triplet biradicals for benzophenones, respectively. The prevalent practice of utilizing classical photoaffinity probes in excess amounts relative to their tentative targets frequently leads to non-specific snatching of off-target biomolecules during photo-irradiation, owing to the extremely high reactivity of these intermediates during insertion as well as their wide AAP (Scheme 1b). Nevertheless, identification of potential drug interaction sites via MS/MS-based mapping is also burdened by the complexity of multiple target residues residing within a single type of biomolecule.12 Furthermore, the extremely short lifetime (ns–μs) of these intermediates (Scheme 1b) makes the spatial range of these crosslinking reactions very tight in a microenvironment.13 To overcome these challenges, photoclick14 functional groups can be regarded as valuable extensions to the photoaffinity toolbox, leveraging their superior chemoselectivity (narrow AAP) while maintaining decent reactivity. The half-life of the photoclick intermediates can also be tuned up to 20 hours, leaving plenty of temporal space for capturing the amino acid residues in a wider radius. Current advancements encompass photo-dehydration of o-nitrobenzyl alcohol, yielding o-benzaldehyde nitroso as a long-lived intermediate with exclusive affinity for lysine residues (primary amines);15 photo-induced ring-rupture of N-aryl tetrazole (Tet), generating reactive nitrile imine intermediates that react with carboxylic amino acid residues, such as glutamic acid and aspartic acid, as a unique AAP;16 photo-rearrangement of oxazole to azirine, facilitating nucleophilic additions;17 and our recent report on the photo-defluorination of m-trifluoromethylaniline (TFMA) for selective acyl fluoride exchange (photo-DAFEx) with lysine residues, whereas exchanging reactions with histidine and cysteine residues yield conjugates that are susceptible to hydrolysis.18
Given that the trifluoromethyl (–CF3) group serves as a prevalent bioisostere in drug design and is featured in numerous FDA-approved drugs,19 including in some cases being an integral part of the drug molecule itself (e.g., flufenamic acid20 and trifluoromethylphenylpiperazine21), the adoption of TFMA as a photoaffinity reagent presents significant convenience for pharmacological investigations. The photo-generated benzoyl fluoride intermediate exhibits a long half-life of approximately 13.7 hours and demonstrates hydrogen-bond activation in proximity, which is akin to that of SuFEx reagents.22
Other crucial parameters for assessing the biocompatibility of a photoaffinity reagent are the ratio of wavelength (λex.) to size and the quantum yield of photochemical transformations at λex., as it is advantageous to utilize light of longer wavelengths to initiate crosslinking under the precondition of minimizing interference with potential drug–target interaction (DTI). The utilization of longer wavelengths of light notably diminishes damage to biomolecules while minimizing background effects, thereby enhancing the feasibility of studying pharmacodynamics within living animal tissues. In fact, visible-light-induced photoaffinity labeling approaches are primarily realized through photocatalytic energy transfer7,13a,23 or the use of a limited number of specialized reagents, such as diazocoumarins (DAC, Scheme 1b).24
Results and discussion
The inaugural photo-DAFEx reaction is based on the straightforward photo-defluorination of a monoaromatic TFMA,25 with the excitation wavelength (λex.) constrained within the UV-B region (at 311 nm). As a result, its versatility across interdisciplinary biochemical applications has been limited, particularly in living tissue studies. Hence, it is a priority to seek photo-DAFEx reagents that are easily accessible while exhibiting a high wavelength-to-size efficiency (r value, Fig. 1c) and a high quantum yield (∅R, Fig. 1c). To this end, we have established a library of TFMA analogues featuring potentially extended n–π* or π–π* transition systems (Fig. 1b). Inspired by the structure of fluphenazine (a potent dopamine receptor antagonist, Scheme 1c),26 we also included a series of TFMA reagents derived from phenoxazine or phenothiazine chromophores. Notably, the 7-CF3-phenoxazine-3-amine (TFMPxA) framework can be incorporated with a propargyl moiety at the N10 position as a ‘click’ handle, and it can be easily synthesized via a scalable 5-step route, which retains an additional hydroxyl group for conjugating with drug molecules of interest (Scheme 1c). The X-ray structure analysis of 4h elucidated a non-planar structure for the TFMPxA core,27 implying that the two aromatic rings are relatively independent π-systems, which are simultaneously bridged by nitrogen and oxygen atoms to form a non-aromatic entirety.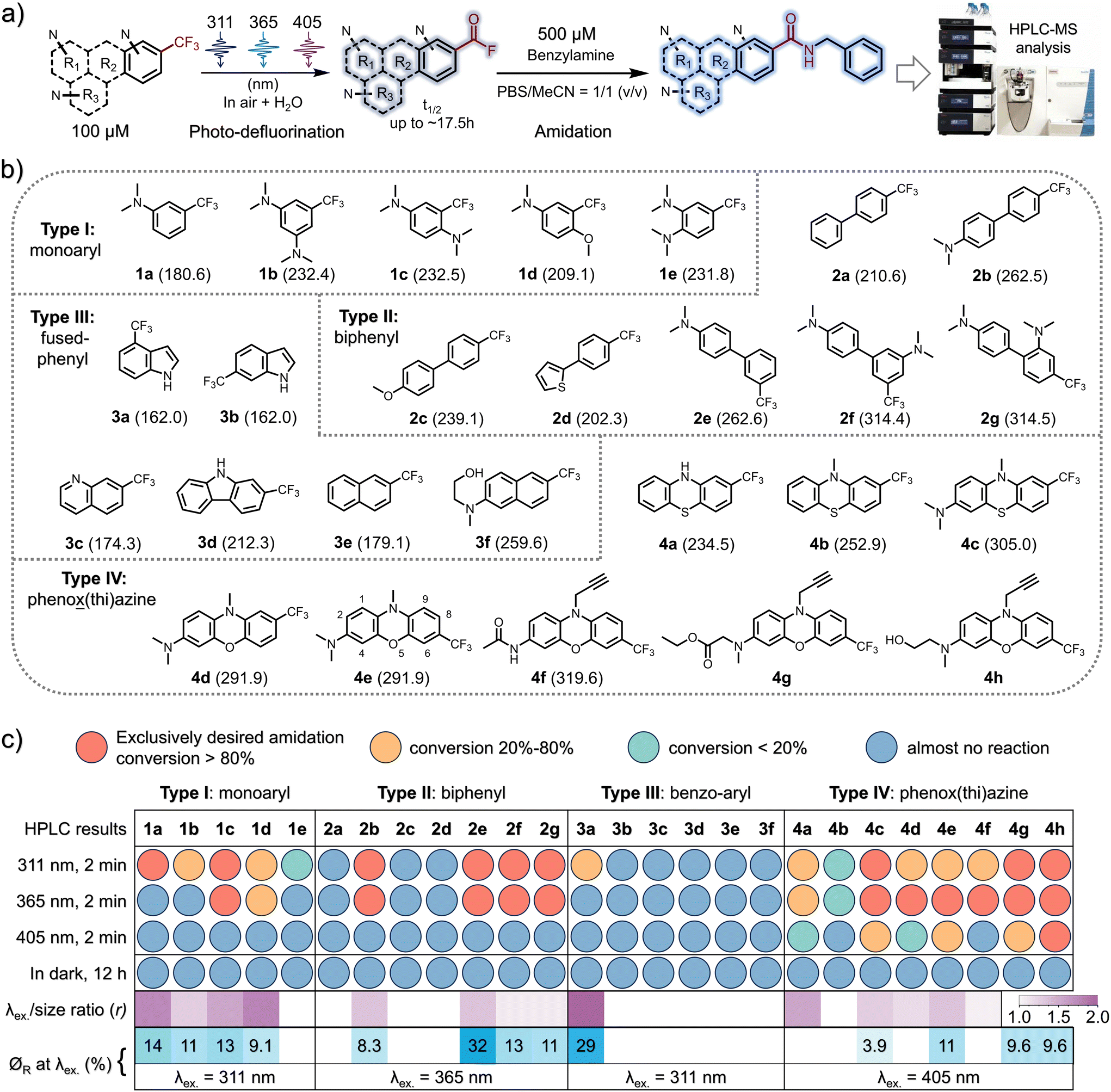 | ||
| Fig. 1 HPLC-MS screening for the photo-DAFEx reaction under various irradiation conditions. (a) The photo-DAFEx reaction scheme. (b) The structure of the TFMA analogues in the library; the molecular van der Waals (vdW) volume (Å3) is given in the parenthesis. (c) The irradiation conditions, the colour codes for the characteristics of each photo-DAFEx reaction, and the color-coded screening results via the HPLC-MS analysis. Purple colour heatmap for representing the r value (nm Å−3) and blue colour heatmap indicating the quantum efficiency of the photo-defluorination step at the longest λex. | ||
To achieve a high λex.-to-size ratio (r, Fig. 1c) in the photo-DAFEx system, the TFMA analogue library was divided into four categories (Fig. 1b). The core size was expanded from the smallest monophenyl (type-I, 1a–1e) to biphenyl (type-II, 2a–2g), fused-phenyl (type-III, 3a–3f), and phenox(thi)azine (type-IV, 4a–4h) structures, enabling us to precisely screen out highly efficient photo-defluorination structures across various excitation wavelengths (λex.).
Thus, HPLC-MS conversion screening under conditions involving exposure of 100 μM TFMA reagent to an LED array for 2 minutes at various excitation wavelengths (λex. = 311, 365 or 405 nm) or in darkness, in a solvent mixture of MeCN/PBS (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), was able to rapidly reveal which structures are crucial for photo-defluorination, accompanied by a bathochromic shift in the excitation wavelength (λex.) (Fig. 1a and b). 500 μM benzylamine was added to ensure complete conversion of the intermediates into amide ligation products, while simultaneously assessing the potential fluorescence turn-on performance of the resulting conjugates.18 Having obtained the longest λex. and the λex.-to-size ratio (r value) data, we proceeded to investigate the quantum yield (∅R) of each compound upon illumination at the possible longest λex., aiming to identify molecules capable of undergoing photo-defluorination reactions with high reactivities (Fig. 1c, heatmaps at the bottom). The screening results (Fig. S1–S26, ESI†) reveal that in the CF3-monophenyl type-I compound depicted in Fig. 1c, the incorporation of dual N,N-dimethylamine groups is capable of initiating photo-DAFEx reactions under 365 nm irradiation. However, this photo-reactivity is observed exclusively in a para-disubstituted form with a good quantum efficiency (1c, r = 1.56, ∅R311 = 13%). When the meta-disubstitution pattern was utilized, the λex. shifted back to 311 nm (1b, r = 1.34, ∅R311 = 11%), whereas in the case of ortho-disubstitution, photo-defluorination was largely inhibited, even at 311 nm (1e, r = 0, indicating very poor reactivity). The presence of both a methoxy group and a dimethylamine at the para-position was also advantageous for reactivity at λex. = 365 nm but with a slightly poorer quantum yield (1d, r = 1.75, ∅R311 = 9.1%), further emphasizing the importance of the relative position of these electronic tuning groups. Within the type-II CF3-biphenyl category (Fig. 1c), N,N-dimethylamine groups, which serve as strong electron-donating auxochromes (in contrast to electron-donating thienyl or methoxy groups), were also crucial for the success of photo-DAFEx reactions (2b, 2e–2g). Interestingly, the relative positioning of the auxochromes and the CF3 moiety on the two σ-linked arene rings was found to be sensitive for photo-defluorination efficiency. When a dimethylamine is present at the para-position of one phenyl ring, the photo-DAFEx reaction at 365 nm proves practicable, regardless of whether the CF3 group is located at the para-position (4,4′- for compound 2b, with r = 1.39) or the meta-position (4,3′- for compound 2e, also with r = 1.39) of the other phenyl. However, the ∅R365 of the meta-position 2e is the highest at 32% in comparison with 8.3% for 2b. Dual N,N-dimethylamines are of assistance in 365 nm promoted photo-DAFEx with a moderate quantum efficiency when they are located at the 4,3′- (2f, r = 1.16, ∅R365 = 13%) or 4,2′- (2g, r = 1.16, ∅R365 = 11%) positions, respectively. Unfortunately, none of the type-II TMFA reagents were able to achieve a visible-light (405 nm) photo-DAFEx reaction, perhaps due to the twisted π orbitals of the biphenyl system, which may hinder an efficient internal charge transfer. Unexpectedly, within the type-III category of CF3-containing fused-aromatics (Fig. 1c), neither naphthalene nor quinoline scaffolds, nor imidazole, displayed any observable photo-defluorination, with the exception of 4-CF3-indole (3a, r = 1.92). The photo-DAFEx reaction of 3a yielded a moderate amount of product solely upon irradiation at 311 nm with a good ∅R311 = 29%, although its absorption coefficient was low. This contrast indicates that the presence of electron-donating amino groups in the fused-aromatic systems does not facilitate the photoelectron energy transfer, which is necessary to weaken the C(sp3)–F bond in the excited state. In the final type-IV class of CF3-phenox(thi)azine chromophores (Fig. 1c), we made an exciting discovery: apart from 4b (r = 1.23), which closely resembles fluphenazine and exhibited poor defluorination under both 311 and 365 nm irradiation (∅R ≈ 0), other substitution patterns especially with a dialkylated amine on the trifluoromethylated phenox(thi)azines showed acceptable photo-DAFEx reactivities under 365 nm light. Notably, the TFMPxA frameworks (4c, r = 1.33; 4e, r = 1.39) were found to efficiently perform photo-DAFEx under 405 nm LED irradiation (∅R405 = 3.9% and 11%, respectively). Interestingly, 3-acetylamino-TFMPxA (4f, r = 1.14), despite being unable to extend the excitation wavelength (λex.) to 405 nm, demonstrated excellent fluorescence turn-on of up to 89-fold at 530 nm during the efficient photo-DAFEx process induced by 365 nm light (Fig. 2b). This feature is particularly advantageous for washing-free labeling and imaging applications.
:1), was able to rapidly reveal which structures are crucial for photo-defluorination, accompanied by a bathochromic shift in the excitation wavelength (λex.) (Fig. 1a and b). 500 μM benzylamine was added to ensure complete conversion of the intermediates into amide ligation products, while simultaneously assessing the potential fluorescence turn-on performance of the resulting conjugates.18 Having obtained the longest λex. and the λex.-to-size ratio (r value) data, we proceeded to investigate the quantum yield (∅R) of each compound upon illumination at the possible longest λex., aiming to identify molecules capable of undergoing photo-defluorination reactions with high reactivities (Fig. 1c, heatmaps at the bottom). The screening results (Fig. S1–S26, ESI†) reveal that in the CF3-monophenyl type-I compound depicted in Fig. 1c, the incorporation of dual N,N-dimethylamine groups is capable of initiating photo-DAFEx reactions under 365 nm irradiation. However, this photo-reactivity is observed exclusively in a para-disubstituted form with a good quantum efficiency (1c, r = 1.56, ∅R311 = 13%). When the meta-disubstitution pattern was utilized, the λex. shifted back to 311 nm (1b, r = 1.34, ∅R311 = 11%), whereas in the case of ortho-disubstitution, photo-defluorination was largely inhibited, even at 311 nm (1e, r = 0, indicating very poor reactivity). The presence of both a methoxy group and a dimethylamine at the para-position was also advantageous for reactivity at λex. = 365 nm but with a slightly poorer quantum yield (1d, r = 1.75, ∅R311 = 9.1%), further emphasizing the importance of the relative position of these electronic tuning groups. Within the type-II CF3-biphenyl category (Fig. 1c), N,N-dimethylamine groups, which serve as strong electron-donating auxochromes (in contrast to electron-donating thienyl or methoxy groups), were also crucial for the success of photo-DAFEx reactions (2b, 2e–2g). Interestingly, the relative positioning of the auxochromes and the CF3 moiety on the two σ-linked arene rings was found to be sensitive for photo-defluorination efficiency. When a dimethylamine is present at the para-position of one phenyl ring, the photo-DAFEx reaction at 365 nm proves practicable, regardless of whether the CF3 group is located at the para-position (4,4′- for compound 2b, with r = 1.39) or the meta-position (4,3′- for compound 2e, also with r = 1.39) of the other phenyl. However, the ∅R365 of the meta-position 2e is the highest at 32% in comparison with 8.3% for 2b. Dual N,N-dimethylamines are of assistance in 365 nm promoted photo-DAFEx with a moderate quantum efficiency when they are located at the 4,3′- (2f, r = 1.16, ∅R365 = 13%) or 4,2′- (2g, r = 1.16, ∅R365 = 11%) positions, respectively. Unfortunately, none of the type-II TMFA reagents were able to achieve a visible-light (405 nm) photo-DAFEx reaction, perhaps due to the twisted π orbitals of the biphenyl system, which may hinder an efficient internal charge transfer. Unexpectedly, within the type-III category of CF3-containing fused-aromatics (Fig. 1c), neither naphthalene nor quinoline scaffolds, nor imidazole, displayed any observable photo-defluorination, with the exception of 4-CF3-indole (3a, r = 1.92). The photo-DAFEx reaction of 3a yielded a moderate amount of product solely upon irradiation at 311 nm with a good ∅R311 = 29%, although its absorption coefficient was low. This contrast indicates that the presence of electron-donating amino groups in the fused-aromatic systems does not facilitate the photoelectron energy transfer, which is necessary to weaken the C(sp3)–F bond in the excited state. In the final type-IV class of CF3-phenox(thi)azine chromophores (Fig. 1c), we made an exciting discovery: apart from 4b (r = 1.23), which closely resembles fluphenazine and exhibited poor defluorination under both 311 and 365 nm irradiation (∅R ≈ 0), other substitution patterns especially with a dialkylated amine on the trifluoromethylated phenox(thi)azines showed acceptable photo-DAFEx reactivities under 365 nm light. Notably, the TFMPxA frameworks (4c, r = 1.33; 4e, r = 1.39) were found to efficiently perform photo-DAFEx under 405 nm LED irradiation (∅R405 = 3.9% and 11%, respectively). Interestingly, 3-acetylamino-TFMPxA (4f, r = 1.14), despite being unable to extend the excitation wavelength (λex.) to 405 nm, demonstrated excellent fluorescence turn-on of up to 89-fold at 530 nm during the efficient photo-DAFEx process induced by 365 nm light (Fig. 2b). This feature is particularly advantageous for washing-free labeling and imaging applications.
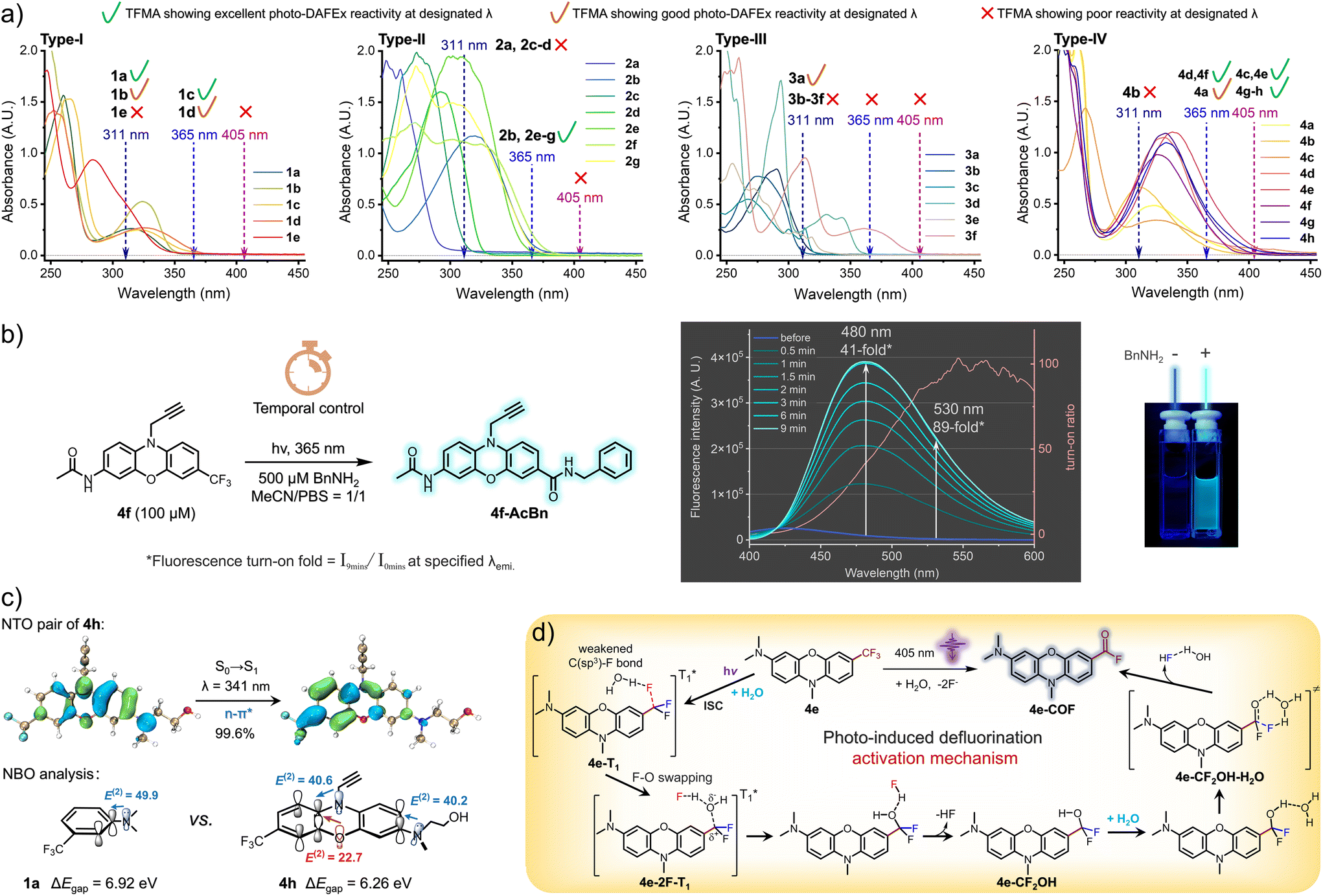 | ||
| Fig. 2 (a) The UV-Vis absorbance spectra of all the TFMA reagents, 100 μM in MeCN/PBS = 1/1, pH = 7.4. (b) Fluorescence turn-on spectra of the photo-DAFEx reaction process under the control by 365 nm irradiation, fluorescence λex. = 390 nm. (c) Top: NTO analysis of S0 → S1 transitions and the corresponding eigenvalue of NTO pair (in percentage). Bottom: The energy gap between the HOMO and LUMO (ΔEgap) and the second order stabilizing energy (E(2), in kcal mol−1) of the key NBO pairs. (d) Proposed photoactivation mechanism of the TFMA reagent for the photo-defluorination of C(sp3)–F bonds. | ||
In view of the structural sensitivity of these photo-DAFEx reagents, we next collected their ultraviolet-visible absorption spectra (UV-Vis) and displayed them in accordance with the four types, respectively (Fig. 2a, type-I). By analyzing the data, we found that among the type-I monophenyl compounds, both 1c and 1d held a certain degree of absorbance at 365 nm (extinction coefficients, ε365 = 300 and 520 cm−1 M−1, respectively). Both the compounds, featuring either two para-positioned dimethylamine groups or a para-dimethylamine–methoxy pair, underwent photo-DAFEx at 365 nm, emphasizing the crucial role of para-disubstitution in combination with strong electron-donating groups (EDGs) for extending the absorbance band and maintaining a good photo-defluorination efficiency. Interpretation regarding the spectra of the type-II compounds (Fig. 2a, type-II) also clarified the importance of the para-dimethylamine position on the σ-linked biphenyl. Reagents 2b and 2e, with a para-dimethylamine on one of the phenyls and a CF3 group located at the 4,4′- and 4,3′-positions, respectively, possessed decent absorbance at 365 nm (ε365 = 1250 and 440 cm−1 M−1, respectively) and efficiently transferred the excited-state energy for defluorination. In contrast, 2a, 2c and 2d, which do not contain a para-dimethylamine, not only displayed no absorbance at 365 nm but also failed to undergo transformation even under 311 nm irradiation despite their ε311 being up to 470, 1390, and 8940 cm−1 M−1, respectively. Additionally, the presence of dual dimethylamines on each phenyl significantly enhanced the 365 nm absorbance, only when the conformation of the biphenyl was less twisted. For instance, compound 2f with 4,3′-di(dimethylamines) exhibited an ε of 2360 cm−1 M−1 (∅R365 = 13%), in contrast to 2g with 4,2′-di(dimethylamines), which had a much lower ε of 440 cm−1 M−1 (∅R365 = 11%). Similar to the unexpected HPLC-MS findings depicted in Fig. 2a (type-III), the majority of the fused-aromatic structures, despite showing decent absorbance at either 311 or 365 nm, were unable to undergo photo-defluorination, with the exception of 3a, which demonstrated the capability for photo-induced defluorination solely at 311 nm, providing an ε311 of 1520 cm−1 M−1. In the case of the type-IV phenox(thi)azine derivatives (Fig. 2a, type-IV), we observed a prominent extension of the absorbance band, particularly when a dialkylated amine was present at the 3-position (4e, ε405 = 760 cm−1 M−1, ∅R405 = 11%). However, not all CF3-phenox(thi)azine derivatives with absorbance at 405 nm allow photo-defluorination efficiently. For instance, compounds 4a and 4d, although they represent absorbance at 405 nm (ε405 = 230 and 210 cm−1 M−1, respectively), defluorinate efficiently only when irradiated at 365 nm. Collectively, the structure of 4e emerged as the most effective visible-light-responsive photo-DAFEx core, featuring tri-alkylation on both the bridging 10-amine and the terminal 3-amine positions (also including 4g and 4h, ε405 = 130, 490 cm−1 M−1, ∅R405 = 9.6%, 9.6%, respectively). Notably, 4f, which possesses a 3-acetamide moiety, displayed a significant hypsochromic shift of the absorbance band, thereby leading to an inability to be efficiently excited by 405 nm light, yet its efficient fluorescence turn-on performance (up to 89-fold) under control of a 365 nm LED (ε365 = 3220 cm−1 M−1) merits further exploration (Fig. 2b). Collectively, a visible-light photo-DAFEx reagent depends critically on both its significant absorbance in the visible region and the nature of its chromophore core, as well as the precise positioning of dialkylated amines relative to CF3 substitutions.
To elucidate the structural uniqueness of the phenoxazine core in terms of its absorption in the visible region, (TD)DFT calculations were performed to characterize the S0 → S1 excitation of 4h (for calculation details, see the ESI†). Natural transition orbital (NTO) analyses (Fig. 2c)28,29 reveal that the S0 → S1 excitation of 4h, peaking at 341 nm, is of the n–π* type, and is accompanied by an absorption band spanning 365–405 nm. In the natural bond orbital (NBO) analysis30,31 of both 1a and 4h, significant p–π* interactions were observed between the lone pair of the nitrogen atom of the terminal amine group and the ArCF3 entity, with second order stabilizing energies (E(2)) of 49.9 kcal mol−1 for 1a and 40.2 kcal mol−1 for 4h, respectively. In contrast to 1a, there are more strong p–π* interactions between the bridging atoms (N and O) and the ArCF3 unit in 4h, with an E(2) of 40.6 and 22.7 kcal mol−1, respectively. Compared with 1a (6.92 eV), the additional conjugations in 4h decrease the energy gap between the HOMO and the LUMO (ΔEgap) to 6.26 eV, consequently leading to a red shift in the absorption spectrum. These effects also facilitate the activation of the C(sp3)–F bond by promoting the distribution of excitation electrons at the CF3 moiety.
To illustrate a possible mechanism for the photo-induced defluorination of trifluoromethyl phenoxazine, we have delineated a plausible photochemical pathway based on our previous research (Fig. 2d),18 emphasizing the significance of water molecules in the excited triplet states. Compound 4e undergoes intersystem crossing (ISC) in the excited singlet state to populate an excited triplet state (4e-T1), in which a water molecule polarizes the C(sp3)–F single bond, weakening its bond strength via hydrogen bonding. Then, a swap between the fluorine atom and the oxygen of the water molecule occurs to form an intermediate (4e-2F-T1), a hydrogen-bond-stabilized ion pair. Subsequently, it relaxes to the ground singlet state, forming a hydrogen fluoride and difluorophenylmethanol intermediate (4e-CF2OH). Then, the secondary defluorination is a thermodynamic process mediated by a water molecule through a six-membered hydrogen-bonding transition state (4e-CF2OH-H2O), which eliminates a secondary hydrogen fluoride hydrate to ultimately yield an acyl fluoride on the phenoxazine core (4e-COF, Fig. 2d). The interesting point is that the phenoxazine-amine core can transfer electrons to weaken the C(sp3)–F bond in the excited triplet state via electron delocalization, and it can be excited by 405 nm light.
Accurately identifying drug–target interactions (DTIs), affinity, and binding sites is paramount not only for drug screening,32 but also for drug repositioning33 and design.34 The PAL-based DTI identification offers a spatiotemporally controlled strategy for revealing unprecedented drug–target interactions, depending on their pharmacological affinity (Kd), as illustrated in Fig. 3a.35 Hence, diverse PAL approaches, capable of establishing covalent ligation with varying lifetimes, offer unique prospects for capturing spatiotemporally sensitive pharmacological affinities (Scheme 1b). To demonstrate the visible-light PAL capability, we firstly examined the biostability of 4e and 4h in the dark under biomimicking conditions (Fig. S36 and S37, ESI†), which clarified the high resistance to thiol addition up to 10 days. The half-lives of several acyl fluoride intermediates formed after photo-defluorination were verified (Fig. S31–S35, ESI†). Of these intermediates, 4h-AcF exhibited a notably long half-life of 17.5 hours in a mixture of MeCN/PBS = 1/1 (pH = 7.4). Then, we validated the AAP of the photo-DAFEx reaction by employing 4h as the warhead, which disclosed a high selectivity toward His, Cys and Lys residues after 405 nm LED irradiation for 3.0 min with complete defluorination of 4h (Fig. 3c and S27–S30, ESI†). However, in contrast to the stable benzoylamide conjugate formed at the Lys residue, the corresponding acylimidazole (formed upon exchange with His) and thioester (formed upon exchange with Cys) conjugates were reversibly hydrolysable in an unconstrained aqueous microenvironment.18 Subsequently, human carbonic anhydrase II (hCA-II) was chosen as the target for drug–target interaction (DTI) studies, owing to its ready availability, high stability, and well-defined structural information.36 This enzyme catalyzes the reversible hydration of CO2 and can be competitively inhibited by acetazolamide (AZA, depicted in Fig. 3a).37 Thus, two sulfonamide-4h probes, designated as P1 and P2 (Fig. 3b), were synthesized and tethered via either a flexible chain or a more rigid piperazine carbamide linker, respectively.
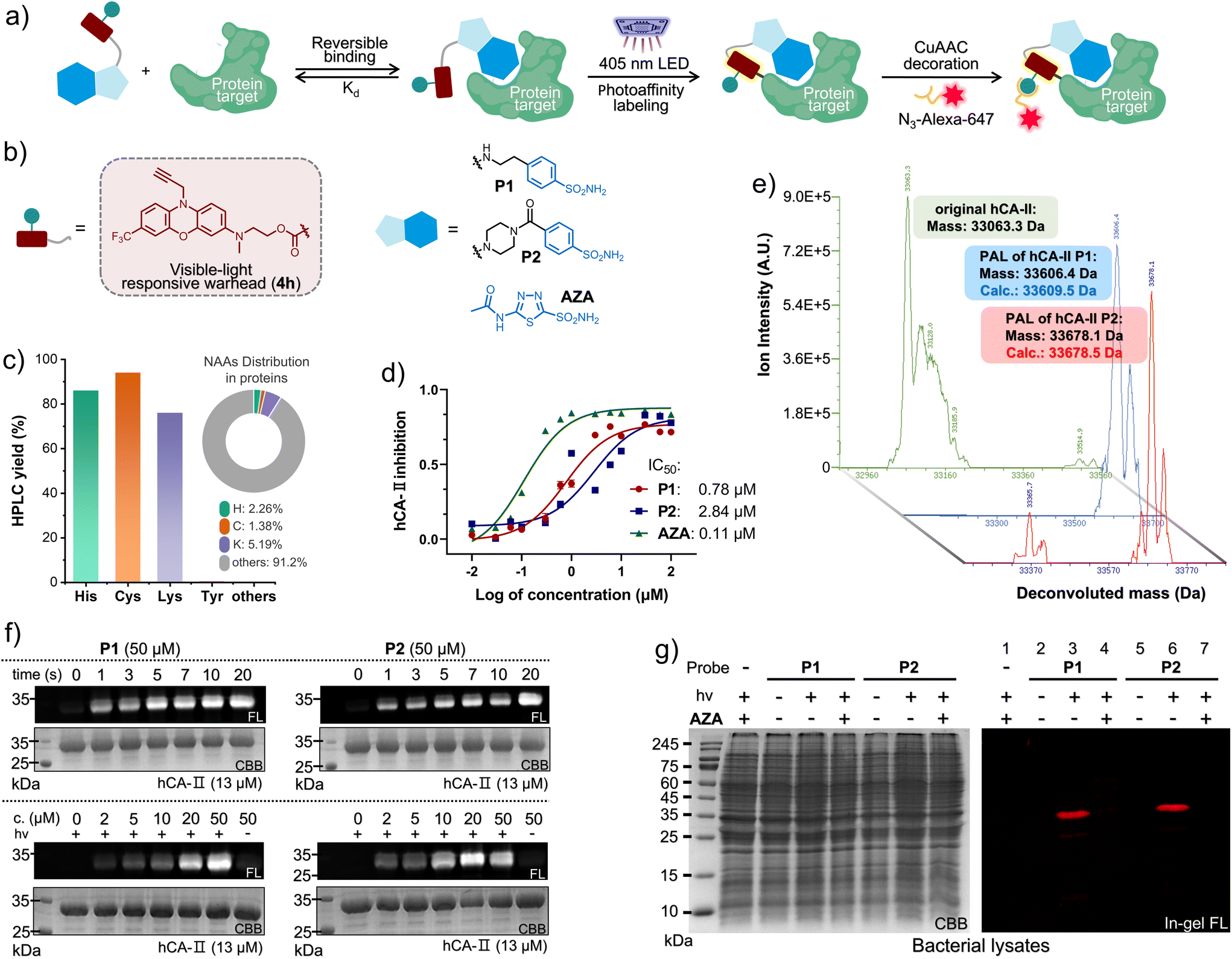 | ||
| Fig. 3 (a) Conceptual diagram of PAL toward the interacting proteins via a multifunctional probe for covalent conjugation, and subsequent “click” decoration for imaging. (b) The chemical structure of the photo-DAFEx probes targeting hCA-II. (c) The photo-conjugation yield of 4h (100 μM, 405 nm LED for 60 s) with various Nα-protected NAA esters (500 μM) as a small molecule, obtained via HPLC-MS analysis (Fig. S27–S30, ESI†) as well as the native NAA distribution. (d) The plot of the enzymatic activity of hCA-II (1.0 μM) catalysing the hydrolysis of p-nitrophenyl acetate (2.5 mM) vs. the inhibitor concentration. Error bars indicate SD, n = 3. (e) The deconvoluted HPLC-MS spectra for identifying the purified hCA-II over-expressed in E. coli cells (BL21 (DE3)), and hCA-II after photo-DAFEx with P1/P2, 100 μM hCA-II and 130 μM probe in a PBS buffer (pH = 7.4) after 405 nm LED irradiation for 20 s. (f) In-gel fluorescence and Coomassie Brilliant Blue (CBB) staining images of SDS-PAGE of the isolated hCA-II after PAL with P1 (left) and P2 (right) under 405 nm control. (g) SDS-PAGE imaging analysis of the PAL toward hCA-II (3.0 μM, [probe] = 20 μM, 405 nm for 20 s) in an E. coli lysate in the presence/absence of AZA (200 μM) as a competitive inhibitor. Fluorophore decoration was completed via CuAAc with a N3-Alexa-647 conjugate. The visible-light photo-DAFEx PAL should be performed in a strictly dark environment. | ||
Prior to the PAL assay, the inhibitory efficacy of these probes against hCA-II was estimated, with IC50 values of 0.78 μM for P1 and 2.84 μM for P2 (Fig. 3d and S38, ESI†), albeit slightly lower than that of the commercial AZA (0.11 μM).37Via deconvolution HPLC-MS analysis (Fig. 3e), we successfully identified the purified hCA-II protein (33063.3 Da, green spectrum, Fig. 3e) as well as the modified hCA-II species (33606.4 and 33678.1 Da, blue and red spectra, Fig. 3e) obtained after PAL treatment with 2 eq. of the probes (P1/P2), mediated by irradiation with a 405 nm LED for 20 s (Fig. S40, ESI†). This resulted in mass shifts of 543 Da and 615 Da plus on the hCA-II protein, which corresponded well with the calculated masses for the PAL modifications (33609.5 and 33678.5 Da). After obtaining the direct evidence from HPLC-MS, we capitalized on a CuAAC reaction to append an Alexa-647 fluorophore onto the N10-propargyl moiety of the crosslinked hCA-II protein, thereby facilitating the evaluation of the time- and dose-dependence of the photo-DAFEx PAL (Fig. 3a). Afterward, in-gel fluorescence qualification determined that 10 seconds of irradiation with 405 nm light is optimal for the PAL using 50 μM of either probe P1 or P2 (Fig. 3f, upper panel, and Fig. S41 and S43, ESI†). Additionally, a minimal concentration of 20 μM of either probe was sufficient to reach the plateau of crosslinking efficiency (Fig. 3f, lower panel, and Fig. S42 and S44, ESI†). Furthermore, the 405 nm-induced PAL reaction towards the over-expressed h-CAII in an E. coli lysate (Fig. 3g and S45, ESI†) not only validated the high affinity of our designed probes (P1/P2) for hCA-II (Fig. 3g, right panel, lanes 3/6), but also disclosed their high specificity for the binding site through competitive displacement with 10 eq. of AZA (Fig. 3g, right panel, lanes 4/7). These findings indicate that the hCA-II probes designed for PAL are compatible with complex environments and possess high target specificity.
The BRD4 protein, a member of the bromodomain and extra-terminal (BET) family, frequently participates in transcriptional and epigenetic regulation (Fig. 4a).38 It contains two consecutive bromodomains (BD1 and BD2) that recognize and bind to acetylated lysine residues on histones, playing pivotal roles in gene regulation and cell growth. Moreover, BRD4 has been associated with NUT gene translocations in midline carcinomas.39,40 BRD4 serves as a primary target for BET inhibitors, notably including (+)-JQ1 (also known as SGCBD01, Fig. 4a, left panel),41 a potent antagonist of BET bromodomains. To explore the versatility of the visible-light photo-DAFEx, we also designed two BRD4-targeted PAL probes, P3 and P4, by joining the 4h scaffold to (+)-JQ1 at the solvent-exposed terminal (specifically, the exposed tBu ester), utilizing carbamide linkers of varying lengths (depicted in Fig. 4a, right panel). Subsequently, optimal photo-DAFEx conditions for these probes were established through a series of SDS-PAGE gel-based analyses. Additionally, the N3-Alexa-647 probe was appended to the photo-crosslinked BRD4 via CuAAC chemistry. As illustrated in Fig. 4b (and Fig. S46–S49, ESI†), in the absence of 405 nm excitation, both probes failed to covalently label the BRD4 protein (evident in the first lanes of the upper panels and the last lanes of the lower panels in Fig. 4b), resulting in undetectable in-gel signals. However, upon brief 405 nm LED irradiation for as little as 1 s, the photo-DAFEx reaction produced a distinct in-gel signal corresponding to the BRD4 bands (observed in upper imaging groups, also in Fig. S46 and S48, ESI†). Extending the irradiation time to 5–7 s resulted in saturation of the in-gel fluorescence, underscoring the remarkably high photoconversion and crosslinking efficiency of these two probes, which minimizes the potential damage caused by prolonged exposure. In the concentration gradient tests, the two probes exhibited distinct behaviors: P3 at a concentration of 5 μM displayed a clearly observable labeled BRD4 band, whereas P4 required a concentration up to 20 μM to enable a noticeable signal (Fig. 4b, lower groups; Fig. S47 and S49, ESI†). To rigorously validate the affinity of the two probes for BRD4, we further conducted photo-DAFEx experiments in an E. coli lysate. As depicted in Fig. 4c (Fig. S50, ESI†), distinct Alexa-647 bands matching to the molecular weight of BRD4 were observed within the SDS-PAGE gel (lanes 3 and 6). Notably, the Alexa-647 intensity of BRD4 crosslinked viaP3 was approximately 5.7-fold higher than that of P4 (with the linker lengths of n = 2 for P3 better than the n = 4 for P4, Fig. 4a and c), implying that the crosslinked Lys residue may be in tighter proximity. Upon quenching with a 10-fold excess of the competitive inhibitor (+)-JQ1 (native form), the fluorescence signal was once again completely extinguished (lanes 2 and 5), emphasizing the site-specific nature of these probes. These findings confirm that both probes selectively bind to the acetyl-lysine recognition site41 of BRD4 and undergo covalent conjugation with either the Lys91 (distance = 11.63 Å) or Lys141 (distance = 11.01 Å) residue under 405 nm light control, while maintaining good inertness without exposure.
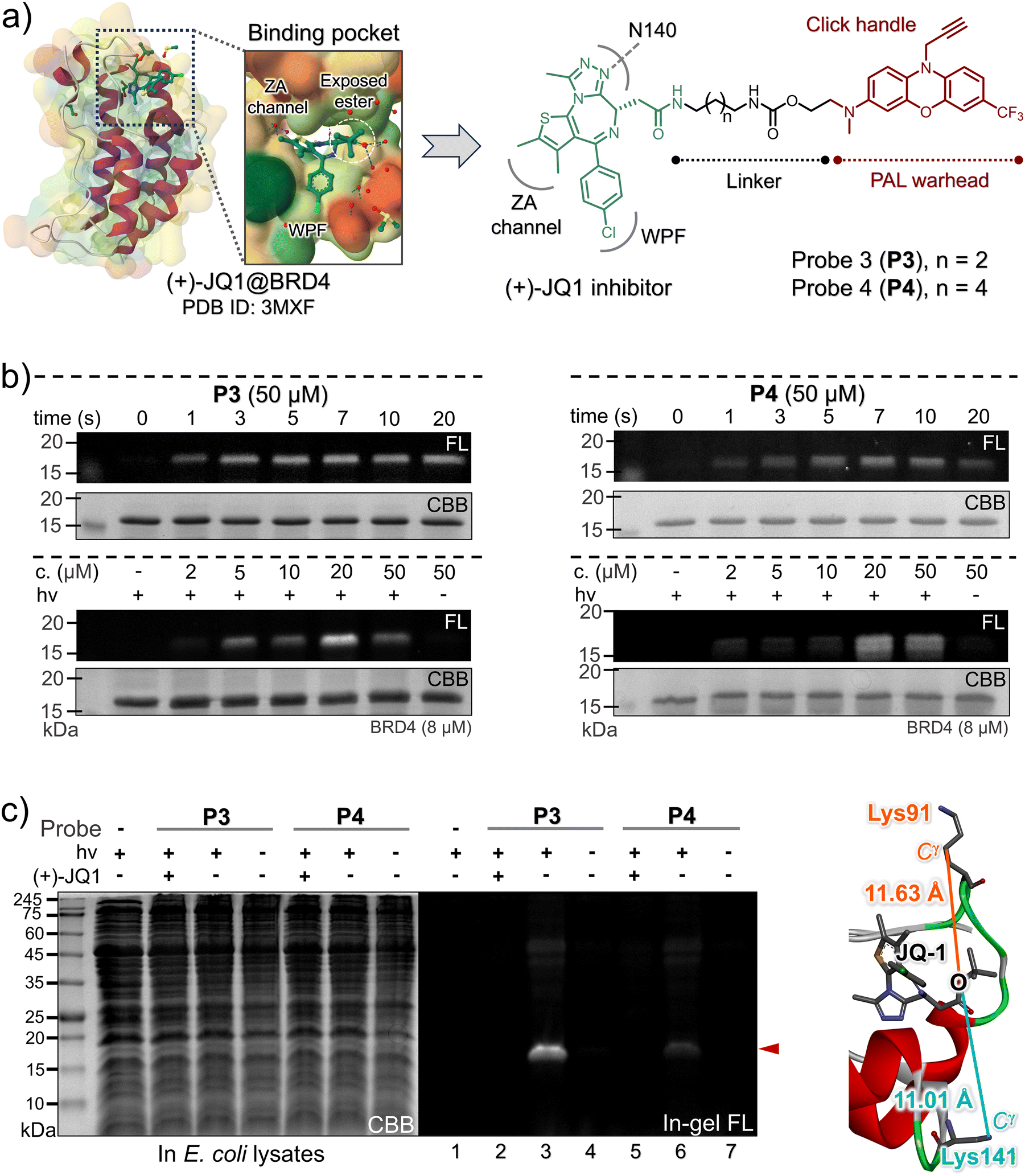 | ||
| Fig. 4 (a) The DTI model of (+)-JQ1 on BRD4 based on the co-crystal structure,41 and the design of the two (+)-JQ1-4h probes. (b) In-gel fluorescence and CBB staining images of the SDS-PAGE for the isolated BRD4 after PAL with P3 (left) and P4 (right) under 405 nm control. (c) SDS-PAGE imaging analysis of the PAL toward BRD4 (3.0 μM, [probe] = 20 μM, 405 nm for 20 s) in an E. coli lysate in the presence/absence of (+)-JQ1 (200 μM) as a competitive inhibitor. The spatial distance between the Cγ of the lysine site (Lys91 and Lys141) and the oxygen atom of the ester moiety in (+)-JQ1 obtained from the co-crystal structure. | ||
Conclusions
Collectively, we have successfully synthesized a library of tailored TFMA reagents specifically designed for visible-light-induced photo-DAFEx reactions, and subsequently examined their reactivity through UV-Vis absorption spectroscopy and HPLC-MS screening. Notably, our research uncovered that dialkylated 7-CF3-phenoxazine-3-amine (4e, 4g–4h) as a chromophore can be efficiently excited by a 405 nm LED, triggering C(sp3)–F bond cleavage to yield acyl fluorides, which function as highly reactive yet stable and long-lived intermediates. Importantly, this photo-defluorination process facilitates selective covalent ligation with nucleophilic moieties, such as lysine, cysteine, or histidine residues, leading to the formation of amide, thioester, or acyl imidazole linkages, respectively. We validated the efficacy of this photoclick reaction for visible-light proximity-dependent labeling (PAL) in two distinct drug–target interaction (DTI) models by designing and utilizing two sets of drug-4h conjugates. Specifically, in the hCA-II DTI model, the 4h-based arylsulfonamide probes achieved rapid crosslinking within seconds upon exposure to 405 nm light, as confirmed by in-gel fluorescence analysis and MS deconvolution. Furthermore, our demonstration of visible-light PAL in bacterial lysates highlights the ability of 4h-based probes to achieve high target fidelity in complex biological environments, including the (+)-JQ1@BRD4 DTI model. Our findings expand the application potential of photo-DAFEx for visible-light PAL, offering the potential to enhance light penetration in living tissues while alleviating the phototoxicity associated with irradiation. Since a photo-crosslinking core need to be as small as possible to minimize interference with the normal functions of biomolecules, further exploration of the TFMA reagents is still in demand, particularly for a superior λex.-to-size ratio, excellent quantum efficiency, and straightforward synthetic accessibility.Author contributions
L. D., S. L., Z. S., and Z. Y. initiated the idea, designed the experiments and wrote the manuscript. L. D. and S. L. synthesized the main compounds and conducted all of the experiments. C. Z., Z. S., and C. H. carried out the theoretical calculations. Y. Z. conducted the X-ray single crystal analysis and X. Z. helped with compound synthesis and in vitro experiments. Z. S. and Z. Y. conceived and directed the project and prepared the manuscript. All authors contributed to the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for compound 4h have been deposited at the CCDC under ID: 2191985 and can be obtained from https://www.ccdc.cam.ac.uk/structures/.
Additional information related to the single crystal XRD analysis is also available in Fig. S51 in the uploaded ESI pdf file.†
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (22001181, 22077090 and 22477088), the Fundamental Research Funds for the Central Universities (20826041D4117) and the Institutional Research Fund from Sichuan University (2020SCUNL105). We also express our appreciation to the Xiaoming Feng laboratory (Sichuan University) for access to equipment.References
- (a) D. M. Mitrea, M. Mittasch, B. F. Gomes, I. A. Klein and M. A. Murcko, Modulating biomolecular condensates: a novel approach to drug discovery, Nat. Rev. Drug Discovery, 2022, 21, 841–862 CrossRef CAS PubMed; (b) M. Konstantinidou and M. R. Arkin, Molecular glues for protein-protein interactions: Progressing toward a new dream, Cell Chem. Biol., 2024, 31, 1064–1088 CrossRef CAS; (c) P. Thiel, M. Kaiser and C. Ottmann, Small-Molecule Stabilization of Protein–Protein Interactions: An Underestimated Concept in Drug Discovery?, Angew. Chem., Int. Ed., 2012, 51, 2012–2018 CrossRef CAS.
- (a) A. Singh, E. R. Thornton and F. H. Westheimer, The Photolysis of Diazoacetylchymotrypsin, J. Biol. Chem., 1962, 237, 3006–3008 CrossRef CAS; (b) F. Kotzyba-Hibert, I. Kapfer and M. Goeldner, Recent Trends in Photoaffinity Labeling, Angew. Chem., Int. Ed. Engl., 1995, 34, 1296–1312 CrossRef CAS; (c) R. A. Homan, J. D. Lapek, C. M. Woo, S. Niessen, L. H. Jones and C. G. Parker, Photoaffinity labelling with small molecules, Nat. Rev. Methods Primers, 2024, 4, 30 CrossRef CAS.
- (a) E. Smith and I. Collins, Photoaffinity Labeling in Target- and Binding-Site Identification, Future Med. Chem., 2015, 7, 159–183 CrossRef CAS PubMed; (b) N. R. Burton, P. Kim and K. M. Backus, Photoaffinity labelling strategies for mapping the small molecule–protein interactome, Org. Biomol. Chem., 2021, 19, 7792–7809 RSC.
- (a) J. M. Wozniak, W. Li, P. Governa, L.-Y. Chen, A. Jadhav, A. Dongre, S. Forli and C. G. Parker, Enhanced mapping of small-molecule binding sites in cells, Nat. Chem. Biol., 2024, 20, 823–834 CrossRef CAS; (b) D. P. Murale, S. C. Hong, M. Haque and J.-S. Lee, Photo-affinity labeling (PAL) in chemical proteomics: a handy tool to investigate protein-protein interactions (PPIs), Proteome Sci., 2016, 15, 14 CrossRef.
- P. P. Geurink, L. M. Prely, G. A. van der Marel, R. Bischoff and H. S. Overkleeft, Photoaffinity Labeling in Activity-Based Protein Profiling, Topics in Current Chemistry, ed. S. Sieber, Springer, Berlin, Heidelberg, 2011, vol. 324, pp. 85–114 Search PubMed.
- C. P. Seath, A. J. Burton, X. Sun, G. Lee, R. E. Kleiner, D. W. C. MacMillan and T. W. Muir, Tracking chromatin state changes using nanoscale photo-proximity labelling, Nature, 2023, 616, 574–580 CrossRef CAS PubMed.
- N. E. S. Tay, K. A. Ryu, J. L. Weber, A. K. Olow, D. C. Cabanero, D. R. Reichman, R. C. Oslund, O. O. Fadeyi and T. Rovis, Targeted activation in localized protein environments via deep red photoredox catalysis, Nat. Chem., 2023, 15, 101–109 CrossRef CAS PubMed.
- (a) H. Guo and Z. Li, Developments of bioorthogonal handle-containing photo-crosslinkers for photoaffinity labeling, Med. Chem. Commun., 2017, 8, 1585–1591 RSC; (b) K. Kozoriz, O. Shkel, K. T. Hong, D. H. Kim, Y. K. Kim and J.-S. Lee, Multifunctional Photo-Cross-Linking Probes: From Target Protein Searching to Imaging Applications, Acc. Chem. Res., 2023, 56, 25–36 CrossRef CAS PubMed; (c) S. Pan, S.-Y. Jang, D. Wang, S. S. Liew, Z. Li, J.-S. Lee and S. Q. Yao, A Suite of “Minimalist” Photo-Crosslinkers for Live-Cell Imaging and Chemical Proteomics: Case Study with BRD4 Inhibitors, Angew. Chem., Int. Ed., 2017, 56, 11816–11821 CrossRef CAS.
- (a) R. A. G. Smith and J. R. Knowles, Aryldiazirines. Potential reagents for photolabeling of biological receptor sites, J. Am. Chem. Soc., 1973, 95, 5072–5073 CrossRef CAS PubMed; (b) A. V. West, G. Muncipinto, H.-Y. Wu, A. C. Huang, M. T. Labenski, L. H. Jones and C. M. Woo, Labeling Preferences of Diazirines with Protein Biomolecules, J. Am. Chem. Soc., 2021, 143, 6691–6700 CrossRef CAS PubMed; (c) L. P. Conway, A. M. Jadhav, R. A. Homan, W. Li, J. S. Rubiano, R. Hawkins, R. M. Lawrence and C. G. Parker, Evaluation of fully-functionalized diazirine tags for chemical proteomic applications, Chem. Sci., 2021, 12, 7839–7847 RSC.
- G. W. J. Fleet, R. R. Porter and J. R. Knowles, Affinity Labelling of Antibodies with Aryl Nitrene as Reactive Group, Nature, 1969, 224, 511–512 CrossRef CAS.
- (a) R. E. Galardy, L. C. Craig, J. D. Jamieson and M. P. Printz, Photoaffinity Labeling of Peptide Hormone Binding Sites, J. Biol. Chem., 1974, 249, 3510–3518 CrossRef CAS; (b) P. Kleiner, W. Heydenreuter, M. Stahl, V. S. Korotkov and S. A. Sieber, A Whole Proteome Inventory of Background Photocrosslinker Binding, Angew. Chem., Int. Ed., 2017, 56, 1396–1401 CrossRef CAS; (c) G. Dormán, H. Nakamura, A. Pulsipher and G. D. Prestwich, The Life of Pi Star: Exploring the Exciting and Forbidden Worlds of the Benzophenone Photophore, Chem. Rev., 2016, 116, 15284–15398 CrossRef.
- C. Iacobucci, M. Götze, C. H. Ihling, C. Piotrowski, C. Arlt, M. Schäfer, C. Hage, R. Schmidt and A. Sinz, A cross-linking/mass spectrometry workflow based on MS-cleavable cross-linkers and the MeroX software for studying protein structures and protein–protein interactions, Nat. Protoc., 2018, 13, 2864–2889 CrossRef CAS.
- (a) J. V. Oakley, B. F. Buksh, D. F. Fernández, D. G. Oblinsky, C. P. Seath, J. B. Geri, G. D. Scholes and D. W. C. MacMillan, Radius measurement via super-resolution microscopy enables the development of a variable radii proximity labeling platform, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203027119 CrossRef CAS; (b) J. B. Geri, J. V. Oakley, T. Reyes-Robles, T. Wang, S. J. McCarver, C. H. White, F. P. Rodriguez-Rivera, D. L. Parker Jr., E. C. Hett, O. O. Fadeyi, R. C. Oslund and D. W. C. MacMillan, Microenvironment mapping via Dexter energy transfer on immune cells, Science, 2020, 367, 1091–1097 CrossRef CAS PubMed; (c) M. Li, Y. Xu, H.-W. Rhee and J. S. Kim, Deep-red-light photocatalytic protein proximity labeling, Chem, 2022, 8, 3171–3174 CrossRef CAS.
- (a) B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Photoclick Chemistry: A Bright Idea, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed; (b) G. S. Kumar and Q. Lin, Light-Triggered Click Chemistry, Chem. Rev., 2021, 121, 6991–7031 CrossRef CAS.
- (a) A.-D. Guo, D. Wei, H.-J. Nie, H. Hu, C. Peng, S.-T. Li, K.-N. Yan, B.-S. Zhou, L. Feng, C. Fang, M. Tan, R. Huang and X.-H. Chen, Light-induced primary amines and o-nitrobenzyl alcohols cyclization as a versatile photoclick reaction for modular conjugation, Nat. Commun., 2020, 11, 5472 CrossRef CAS PubMed; (b) A.-D. Guo, K.-N. Yan, H. Hu, L. Zhai, T.-F. Hu, H. Su, Y. Chi, J. Zha, Y. Xu, D. Zhao, X. Lu, Y.-J. Xu, J. Zhang, M. Tan and X.-H. Chen, Spatiotemporal and global profiling of DNA–protein interactions enables discovery of low-affinity transcription factors, Nat. Chem., 2023, 15, 803–814 CrossRef CAS.
- (a) Y. Tian, M. P. Jacinto, Y. Zeng, Z. Yu, J. Qu, W. R. Liu and Q. Lin, Genetically Encoded 2-Aryl-5-carboxytetrazoles for Site-Selective Protein Photo-Cross-Linking, J. Am. Chem. Soc., 2017, 139, 6078–6081 CrossRef CAS; (b) K. Cheng, J.-S. Lee, P. Hao, S. Q. Yao, K. Ding and Z. Li, Tetrazole-Based Probes for Integrated Phenotypic Screening, Affinity-Based Proteome Profiling, and Sensitive Detection of a Cancer Biomarker, Angew. Chem., Int. Ed., 2017, 56, 15044–15048 CrossRef CAS; (c) A. Herner, J. Marjanovic, T. M. Lewandowski, V. Marin, M. Patterson, L. Miesbauer, D. Ready, J. Williams, A. Vasudevan and Q. Lin, 2-Aryl-5-carboxytetrazole as a New Photoaffinity Label for Drug Target Identification, J. Am. Chem. Soc., 2016, 138, 14609–14615 CrossRef CAS PubMed; (d) S. Flesch and P. Vöhringer, Ultrafast Dynamics of Photochemical Nitrile Imine Formation, Angew. Chem., Int. Ed., 2022, 61, e202205803 CrossRef CAS PubMed.
- K. Cheng, J. Qi, X. Ren, J. Zhang, H. Li, H. Xiao, R. Wang, Z. Liu, L. Meng, N. Ma and H. Sun, Developing Isoxazole as a Native Photo-Cross-Linker for Photoaffinity Labeling and Chemoproteomics, Angew. Chem., Int. Ed., 2022, 61, e202209947 CrossRef CAS.
- L. Deng, C. Zhang, B. Li, J. Fu, Z. Zhang, S. Li, X. Zhao, Z. Su, C. Hu and Z. Yu, Photo-induced defluorination acyl fluoride exchange as a fluorogenic photo-click reaction for photo-affinity labeling, Chem. Sci., 2023, 14, 3630–3641 RSC.
- A. S. Nair, A. K. Singh, A. Kumar, S. Kumar, S. Sukumaran, V. P. Koyiparambath, L. K. Pappachen, T. M. Rangarajan, H. Kim and B. Mathew, FDA-Approved Trifluoromethyl Group-Containing Drugs: A Review of 20 Years, Processes, 2022, 10, 2054 CrossRef CAS.
- V. López-Mejías, J. W. Kampf and A. J. Matzger, Nonamorphism in Flufenamic Acid and a New Record for a Polymorphic Compound with Solved Structures, J. Am. Chem. Soc., 2012, 134, 9872–9875 CrossRef.
- L. J. Schep, R. J. Slaughter, J. A. Vale, D. M. G. Beasley and P. Gee, The clinical toxicology of the designer “party pills” benzylpiperazine and trifluoromethylphenylpiperazine, Clin. Toxicol., 2011, 49, 131–141 CrossRef CAS.
- J. Dong, K. Krasnova, M. G. Finn and K. B. Sharpless, Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- (a) S. D. Knutson, B. F. Buksh, S. W. Huth, D. C. Morgan and D. W. C. MacMillan, Current advances in photocatalytic proximity labeling, Cell Chem. Biol., 2024, 31, 1145–1161 CrossRef CAS; (b) D. C. Cabanero, S. K. Kariofillis, A. C. Johns, J. Kim, J. Ni, S. Park, D. L. Parker Jr., C. P. Ramil, X. Roy, N. H. Shah and T. Rovis, Photocatalytic Activation of Aryl(trifluoromethyl) Diazos to Carbenes for High-Resolution Protein Labeling with Red Light, J. Am. Chem. Soc., 2024, 146, 1337–1345 CrossRef CAS; (c) Y. Zhang, J. Tan and Y. Chen, Visible-light-induced protein labeling in live cells with aryl azides, Chem. Commun., 2023, 59, 2413–2420 RSC; (d) Y. Zhang, S. Liu, F. Guo, S. Qin, N. Zhou, Z. Liu, X. Fan and P. R. Chen, Bioorthogonal Quinone Methide Decaging Enables Live-Cell Quantification of Tumor-Specific Immune Interactions, J. Am. Chem. Soc., 2024, 146, 15186–15197 CrossRef CAS.
- S.-Y. Dai and D. Yang, A Visible and Near-Infrared Light Activatable Diazocoumarin Probe for Fluorogenic Protein Labeling in Living Cells, J. Am. Chem. Soc., 2020, 142, 17156–17166 CrossRef CAS PubMed.
- J. Fu, S. Li, L. Deng, X. Zhao and Z. Yu, A genetically encodable and fluorogenic photo-crosslinker via photo-induced defluorination acyl fluoride exchange, Chem. Commun., 2023, 59, 11073–11076 RSC.
- S. Leucht, M. Tardy, K. Komossa, S. Heres, W. Kissling, G. Salanti and J. M. Davis, Antipsychotic drugs versus placebo for relapse prevention in schizophrenia: a systematic review and meta-analysis, Lancet, 2012, 379, 2063–2071 CrossRef CAS.
- Deposition number 2191985 (for compound 4h) contains the supplementary crystallographic data for this paper.†.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. P. Foster and F. Weinhold, Natural hybrid orbitals, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- A. E. Reed and F. Weinhold, Natural bond orbital analysis of near–Hartree–Fock water dimer, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS.
- Q. Ye, C.-Y. Hsieh, Z. Yang, Y. Kang, J. Chen, D. Cao, S. He and T. Hou, A unified drug–target interaction prediction framework based on knowledge graph and recommendation system, Nat. Commun., 2021, 12, 6775 CrossRef CAS PubMed.
- T. T. Ashburn and K. B. Thor, Drug repositioning: identifying and developing new uses for existing drugs, Nat. Rev. Drug Discovery, 2004, 3, 673–683 CrossRef CAS PubMed.
- (a) T. A. Baillie, Targeted Covalent Inhibitors for Drug Design, Angew. Chem., Int. Ed, 2016, 55, 13408–13421 CrossRef CAS; (b) T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Counting on natural products for drug design, Nat. Chem., 2016, 8, 531–541 CrossRef CAS.
- F. Vincent, A. Nueda, J. Lee, M. Schenone, M. Prunotto and M. Mercola, Phenotypic drug discovery: recent successes, lessons learned and new directions, Nat. Rev. Drug Discovery, 2022, 21, 899–914 CrossRef CAS PubMed.
- V. P. Mocharla, B. Colasson, L. V. Lee, S. Röper, K. B. Sharpless, C.-H. Wong and H. C. Kolb, In Situ Click Chemistry: Enzyme-Generated Inhibitors of Carbonic Anhydrase II, Angew. Chem., Int. Ed., 2004, 44, 116–120 CrossRef PubMed.
- A. A. Barrese, C. Genis, S. Z. Fisher, J. N. Orwenyo, M. T. Kumara, S. K. Dutta, E. Phillips, J. J. Kiddle, C. Tu, D. N. Silverman, L. Govindasamy, M. Agbandje-McKenna, R. McKenna and B. C. Tripp, Inhibition of Carbonic Anhydrase II by Thioxolone: A Mechanistic and Structural Study, Biochemistry, 2008, 47, 3174–3184 CrossRef CAS.
- J. Shi and C. R. Vakoc, The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition, Mol. Cell, 2014, 54, 728–736 CrossRef CAS.
- L. Zeng and M.-M. Zhou, Bromodomain: an acetyl-lysine binding domain, FEBS Lett., 2002, 513, 124–128 CrossRef CAS.
- C. A. French, Demystified molecular pathology of NUT midline carcinomas, J. Clin. Pathol., 2010, 63, 492–496 CrossRef.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Selective inhibition of BET bromodomains, Nature, 2010, 468, 1067–1073 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, HPLC-MS analysis, spectral properties, calculation details, and characterization of all new compounds. CCDC 2191985. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01870b |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |