
Stereocontrolled synthesis of heterocycles from unactivated alkynes by photoredox/nickel dual-catalyzed cyclization†
Bo-Rong
Leng‡
ab,
Feng
Yang‡
a,
Jin-Lian
Bai
a,
Yu-Wen
Huang
a,
Qing-Quan
Liu
*a,
Ping
Wei
a,
De-Cai
Wang
*a and
Yi-Long
Zhu
 *a
*a
aInstitute of Materia Medica, School of Pharmaceutical Sciences, Nanjing Tech University, Nanjing 211816, P. R. China
bCollege of Life and Health, Nanjing Polytechnic Institute, Nanjing 211816, P. R. China
First published on 6th November 2024
Abstract
We introduce an innovative nickel/photoredox dual catalytic sulfonyl-arylation method for synthesizing heterocyclic compounds directly from unactivated alkynes and aryl iodides. This green chemistry approach sidesteps traditional Heck reactions, eliminating reliance on excess metal catalysts and reagents. The method ensures high chemical selectivity, curbing side reactions and facilitating the stereoselective 6-exo-dig cyclization. It simplifies the production of nitrogen- or oxygen-containing heterocyclics, such as 3,4-dihydroisoquinolin-1(2H)-ones, tetrahydroisoquinolines, and isochroman derivatives, with potential applications in medicinal chemistry. The dual catalytic system enhances reactivity with unactivated substrates, marking a significant step in synthetic chemistry.
Innovative synthetic routes for constructing heterocycles, known for their atom economy and step efficiency, have garnered significant interest from academia and industry due to their potential to enhance drug optimization, including biological activity and pharmacokinetics.1,2 The dehalogenation carbon cyclization reaction, which fuses aromatic halides with unsaturated carbon–carbon bonds, is particularly effective for creating monocyclic and polycyclic systems.3 This strategy has been adeptly applied to aromatic halide dehalogenation carbon cyclization with alkenes or alkynes through cascade reactions initiated by Heck-type reactions (Scheme 1a).4 The advent of visible-light-mediated photochemistry has transformed the field, with photoredox reagents acting as synergistic catalysts in transition-metal catalytic cycles, expanding the range of accessible reaction pathways.5 Notably, the metal/photoredox dual catalytic system has been successfully developed for aryl halide dehalogenation carbon cyclization with olefins, combining high efficiency with mild reaction conditions and aligning with green chemistry principles. Xia's group has notably pioneered a nickel/photoredox dual catalytic cyclization process for the aryl alkylation of aryl halides with non-activated olefins, yielding valuable indanethylamine derivatives, marking a significant advancement in catalytic technology (Scheme 1b).6 Despite breakthroughs in the cyclization and difunctionalization of unactivated alkynes with aryl halides, these processes still predominantly rely on Heck-type reactions, necessitating metal catalysts and stoichiometric oxidants or reducing agents.7
 | ||
| Scheme 1 The catalyzed cyclization/difunctionalization of unsaturated carbon–carbon bonds. | ||
Drawing inspiration from recent advancements, we propose a nickel/photocatalytic intramolecular cyclization strategy as an efficient and direct alternative for synthesizing heterocyclic frameworks with nitrogen or oxygen (Scheme 1c). This innovative method promises a one-step synthesis under mild conditions. However, foreseeable competitive side reactions pose a challenge in the intramolecular cyclization of unactivated alkynes catalyzed by nickel/photoredox dual catalysts. The lower affinity and reactivity of these alkynes towards nickel catalysts could impede the migration–insertion process, potentially leading to the reduction hydrogenation of halogenated benzenes8 or direct cross-coupling of sulfonyl radicals with aryl halides.9 Furthermore, in the photosensitizer-catalyzed dehalogenation cyclization of aryl halides and alkynes, the formation of undesired cyclization hydrogenation byproducts is a common issue.8,10 Additionally, the hydrosulfonation reaction between sulfonyl radicals and alkynes presents an alternative, competing pathway.11 In this research, we introduce an innovative nickel/photoredox dual catalytic sulfonyl-arylation method for the cross-coupling cyclization of aryl iodides with unactivated alkynes. This method excels in chemical selectivity, adeptly curbing side reactions and facilitating the stereoselective 6-exo-dig cyclization. It provides a potent synthetic strategy for the preparation of biologically significant heterocyclic compounds, such as 3,4-dihydroisoquinolin-1(2H)-ones, tetrahydroisoquinolines, and isochroman derivatives.
Our study began by selecting 2-iodo-N-methyl-N-(2-methylbut-3-yn-2-yl)benzamide (1a) and sodium p-tolylsulfinate (2a) as model substrates, as detailed in Table 1. Through a systematic experimental approach, we discovered a potent photocatalytic system that includes fac-Ir(ppy)3 at 5 mol% to serve as the photocatalyst, NiCl2·6H2O at 10 mol% as the nickel catalyst, and 4,4′-di-tert-butyl-2,2′-dipyridyl (L1) at 10 mol% as the ligand. Applying this system and irradiating with 465 nm blue LEDs in a DMSO solvent, we successfully synthesized 3,4-dihydroisoquinolin-1(2H)-one, achieving a yield of 35% (entry 1). Building on this initial success, we proceeded to scrutinize a range of alternative photosensitizers, such as Ru(bpy)3Cl2·6H2O, 4-CzIPN, Eosin Y, and Na2-Eosin Y, in our quest to improve the reaction's conversion rate (entries 2–5). The implementation of Na2-Eosin Y as the photocatalyst significantly enhanced the yield of our target product to 70% (entry 5). An exhaustive evaluation of various nickel catalysts and ligands underscored that the pairing of NiCl2·6H2O with L1 was optimal for the stereoselective transformation of the substrates (entries 6–11). In our solvent screening, DMSO proved to be more effective than DMA or DMF, offering higher reaction efficiency (entries 12 and 13). Control experiments were instrumental in confirming the essential roles of both the nickel catalyst and the photoredox catalyst under visible light irradiation, which were found to be crucial for the reaction's progress (entries 15 and 16).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Variations from conditions | Yieldc (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol, 2.0 equiv.), fac-Ir(ppy)3 (5 mol%), NiCl2·6H2O (10 mol%), and L1 (10 mol%) in DMSO (2.0 mL) at room temperature under N2 with 465 nm blue LEDs for 12 h. b Na2-Eosin Y instead of fac-Ir(ppy)3. c Yield of the isolated product. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | None | 35 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Ru(bpy)3Cl2·6H2O instead of fac-Ir(ppy)3 | 43 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 4-CzIPN instead of fac-Ir(ppy)3 | 50 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Eosin Y instead of fac-Ir(ppy)3 | 41 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Na2-Eosin Y instead of fac-Ir(ppy)3 | 70 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6b | Ni(OAc)2·4H2O instead of NiCl2·6H2O | 31 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7b | NiBr2·DME instead of NiCl2·6H2O | 44 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8b | Ni(PPh3)2Cl2 instead of NiCl2·6H2O | 67 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9b | L2 instead of L1 | 39 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10b | L3 instead of L1 | 46 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11b | L4 instead of L1 | 37 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12b | DMF instead of DMSO | 59 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13b | DMA instead of DMSO | 65 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14b | Without light | N.D. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15b | Without Na2-Eosin Y | N.D. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16b | Without [Ni] and ligand | N.D. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

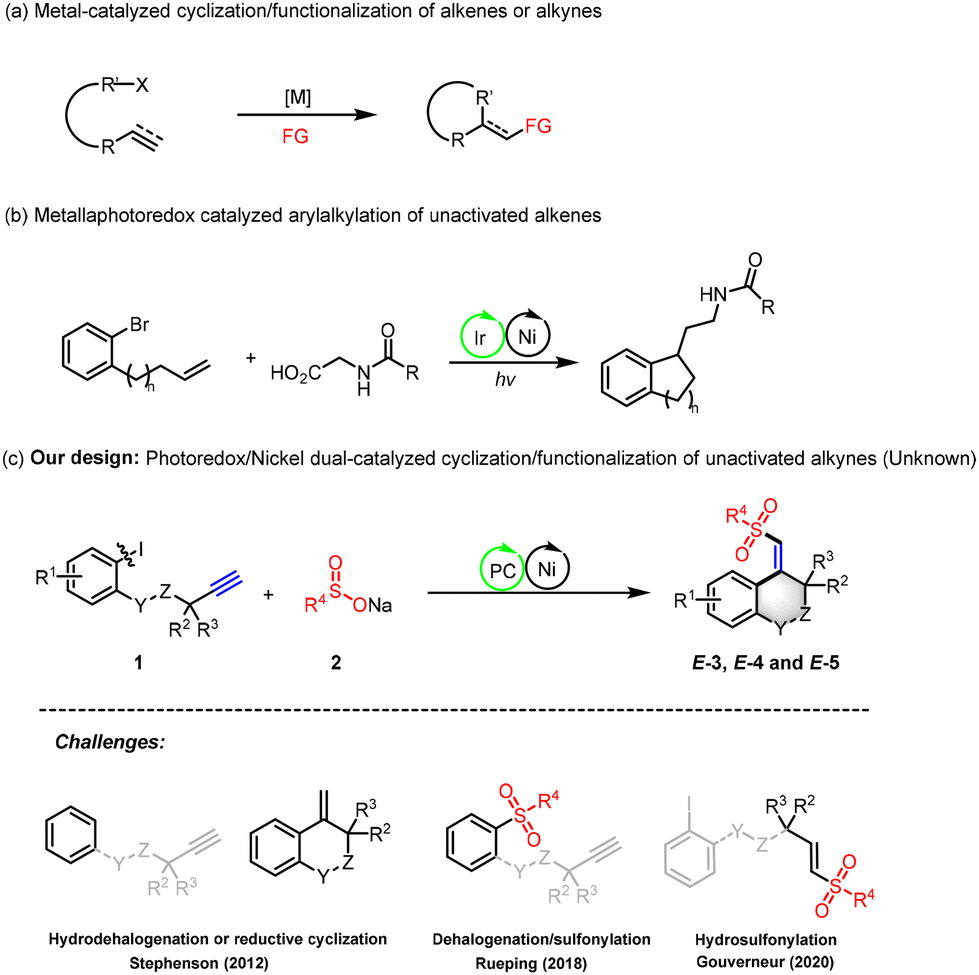
With the optimal reaction conditions determined, we explored the versatility of our nickel/photoredox dual catalytic sulfonyl-arylation method for the synthesis of 3,4-dihydroisoquinolin-1(2H)-one from nonactivated alkynes (Scheme 2). A diverse set of substituents, including electron-withdrawing (F, Cl, Br) and electron-donating (Me) groups at the 4-, 5-, and 6-positions, were well-tolerated, affording sulfonylated products with excellent stereoselectivity (3a–3i, yields: 34%–79%). Notably, electron-withdrawing substitutions at the 5-position on the arene led to higher yields than those with electron-donating groups. The method's applicability was further expanded using N-alkyl-protected substrates, such as ethyl and n-pentyl, yielding the corresponding products with 34%–48% yields (3j–3k). Subsequently, a survey of sodium aryl sulfinate (2) was conducted with 1a, revealing that a variety of sodium aryl sulfinate, irrespective of the electronic nature of their substituents (methoxy, tert-butyl, fluoro, chloro, trifluoromethyl), successfully participated in the photocatalytic process, yielding 3,4-dihydroisoquinolin-1(2H)-ones (3l–3q) with yields ranging from 35–75%. Sodium aryl sulfinates bearing ortho or meta substituents also underwent smooth photochemical transformation, producing the corresponding products (3r–3s) with yields of 53% to 83%. Additionally, sodium 2-thiophene sulfinate emerged as a viable coupling partner, providing the target product 3t in a 34% yield. Lastly, we investigated the influence of gem-dimethyl groups on the reactivity of substrates within our reaction system. We found that these groups are not strictly required for achieving successful reaction outcomes. The product 3u, which lacks gem-dimethyl groups, can be synthesized with moderate yields under standard conditions. This finding indicates that our strategy may be more flexible than initially presumed. However, internal alkynes with terminal phenyl substitutions are not compatible with this approach.
 | ||
| Scheme 2 Substrate scope of 3,4-dihydroisoquinolin-1(2H)-ones. Reaction conditions: 1 (0.2 mmol), 2 (2.0 equiv.), Na2-Eosin Y (5 mol%), NiCl2·6H2O (10 mol%), and L1 (10 mol%) in DMSO (2.0 mL) at room temperature under N2 with 465 nm blue LEDs for 12 h. Isolated yields based on 1. | ||
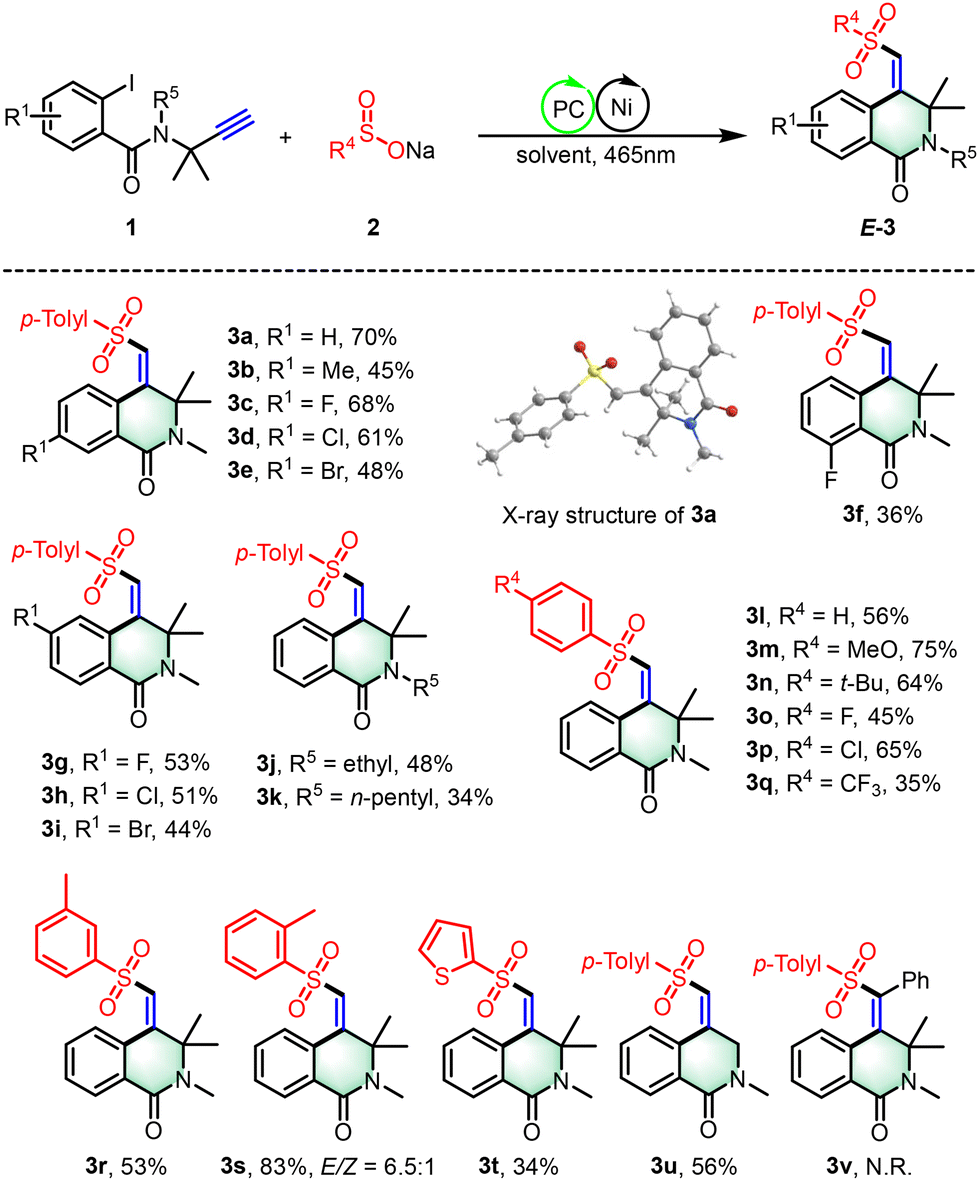
In our quest to enhance the practical applicability of our photoredox/nickel dual-catalyzed cross-coupling cyclization approach, we conducted a thorough assessment to determine the feasibility of stereoselectively constructing 4-(arylsulfonyl methylene)-1,2,3,4-tetrahydroisoquinolines using unactivated alkynes and sodium sulfinate as starting materials (Scheme 3). A diverse series of structurally distinct sodium aryl sulfinates, including those with electron-donating and electron-withdrawing functional groups, underwent the reaction smoothly, yielding products 4a–4d. Notably, substrates adorned with adjacent methyl groups encountered significant steric hindrance, potentially leading to a substantial decrease in reaction yield, as exemplified by compound 4e. Furthermore, sodium alkyl sulfinates, with methyl and ethyl substituents, integrated well within the photoredox/nickel dual-catalyzed system, displaying commendable reactivity and affording the desired products 4f–4g with yields ranging from 53% to 75%. Additionally, both N-sulfonyl protected and N-unprotected substrates have been shown to be compatible, expanding the synthetic repertoire with yields ranging from 48% to 58% for compounds 4h and 4i. It is also noteworthy that while substrate 1j′ proved to be a viable substrate, it displayed reduced stereoselectivity for the desired product 4j. This reduction in stereoselectivity is likely attributable to the diminished influence of the Thorpe–Ingold effect.
 | ||
| Scheme 3 Substrate scope of tetrahydroisoquinolines. Reaction conditions: 1 (0.2 mmol), 2 (2.0 equiv.), Ru(bpy)3Cl2·6H2O (5 mol%), NiCl2·6H2O (10 mol%), and L1 (10 mol%) in DMF (2.0 mL) at room temperature under N2 with 465 nm blue LEDs for 12 h. Isolated yields based on 1. | ||
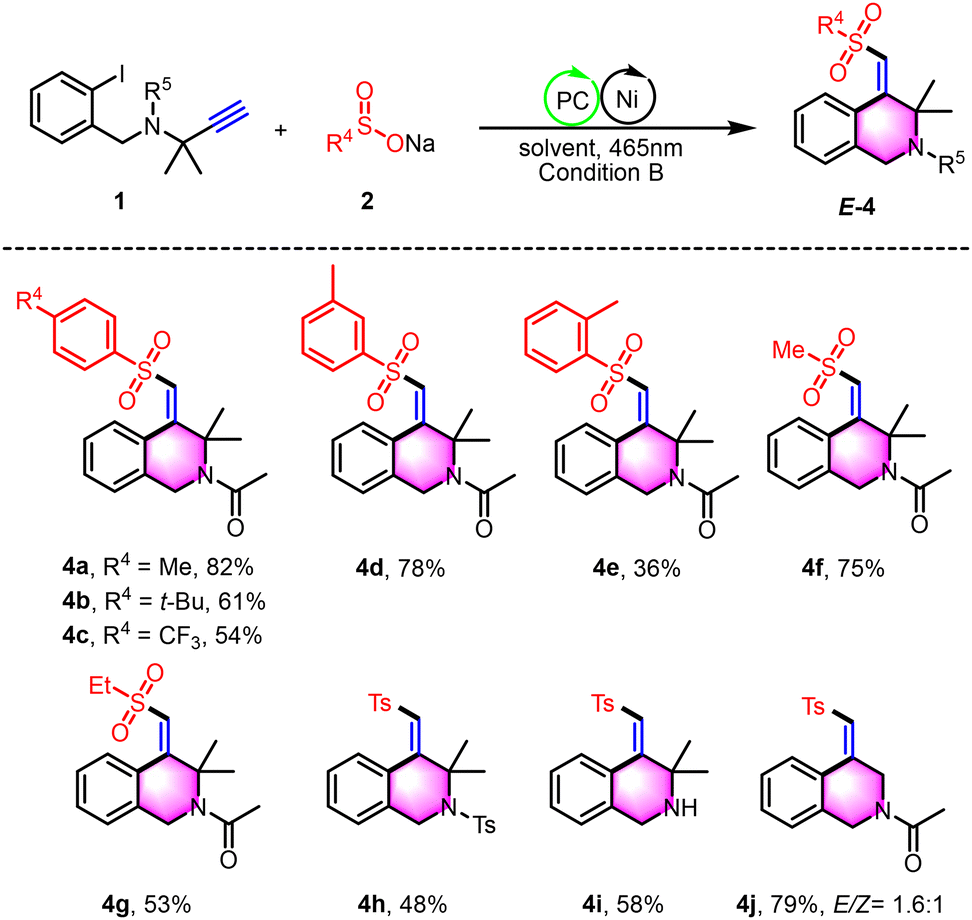
The isochromane motif, characterized by the 3,4-dihydro-1H-2-benzopyran core, is a prevalent structural element in numerous natural products and synthetic bioactive molecules.12 These compounds are celebrated for their diverse pharmacological profiles and are extensively utilized as therapeutic agents, particularly for central nervous system disorders, antitumor treatments, antibacterial applications, and anti-inflammatory therapies. In this study, we introduce an innovative photoredox/nickel dual-catalyzed approach for the synthesis of polysubstituted isochromane derivatives (Scheme 4). This method employs aliphatic terminal alkynes and sodium sulfinates as starting materials, offering a novel pathway to access these biologically relevant scaffolds. Under the photo-catalytic conditions, the reaction yielded polysubstituted isochromanes with satisfactory results, with yields ranging from 36% to 83% (5a–5m). We successfully applied a range of aryl sodium sulfinates, bearing both electron-donating groups (e.g., methyl, tert-butyl) and electron-withdrawing groups (e.g., fluorine, chlorine, bromine, trifluoromethyl), in the cross-coupling cyclization reaction with unactivated alkynes. This resulted in the formation of the corresponding isochromanes with yields between 37% and 83% (5a–5h). Notably, the method showed tolerance to steric hindrance with substrates bearing adjacent methyl groups, although this modification led to a modest reduction in yield (5i). Furthermore, we expanded the substrate scope to include sodium thiophene-2-sulfinate and sodium alkyl sulfinates, which also provided access to 4-(sulfonylmethylene) isochromanes with acceptable yields (5j–5l). Moreover, the α-methyl-substituted ether substrate was found to be readily compatible with this transformation, under scoring the versatility of our approach (5m). Additionally, this strategy is also applicable to the synthesis of product 5n, which lacks geminal dimethyl groups. However, it encounters challenges with the reaction of internal alkynes that have terminal phenyl groups. Notably, when the terminal position of the alkyne is substituted with a methyl group, the formation of trace product 5p can be observed. Moreover, the resulting sulfonyl heterocycles serve as valuable building blocks in organic synthesis (Scheme 5). We successfully alkylated the synthesized product 5a with N,N-dimethylaniline, yielding alkyl-substituted isochromanes (5aa). Furthermore, the oxidation of 5a with KMnO4 led to the formation of ketone 5ab. Additionally, product 3a can be readily converted into an alkoxy-substituted heterocyclic ring (3aa) through a simple substitution reaction with 2-iodobenzyl alcohol, highlighting its synthetic versatility.
 | ||
| Scheme 4 Substrate scope of isochromanes. Reaction conditions: 1 (0.2 mmol), 2 (2.0 equiv.), Ru(bpy)3Cl2·6H2O (5 mol%), NiCl2·6H2O (10 mol%), and L1 (10 mol%) in DMF (2.0 mL) at room temperature under N2 with 465 nm blue LEDs for 12 h. Isolated yields based on 1. Optimization of the reaction conditions see ESI.† | ||
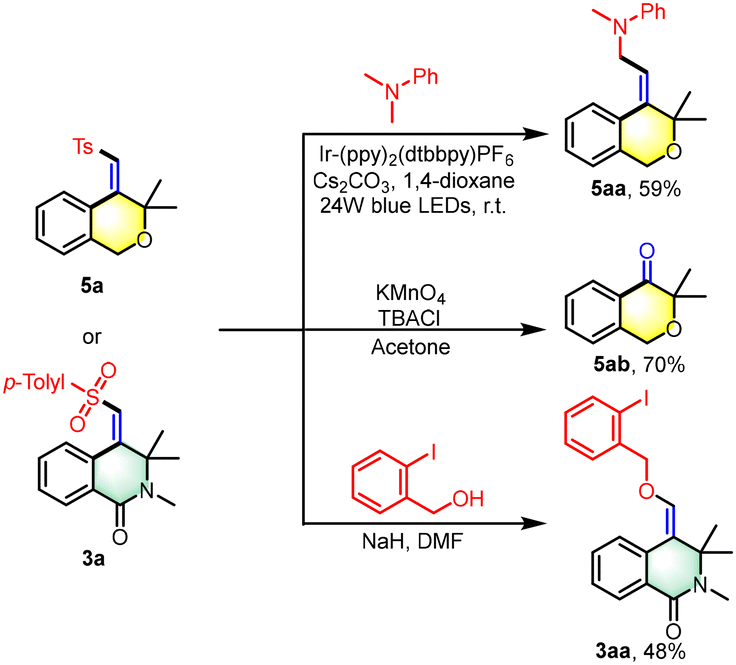 | ||
| Scheme 5 Synthetic applications. | ||
To clarify the mechanistic pathway of our catalytic protocol, we conducted a thorough series of control experiments with precision (Scheme 6). Initially, we aimed to confirm the sequence of radical addition by subjecting compound 1a to standard reaction conditions in the absence of reagent 2a. The absence of product 3a′ formation highlights the essential role of reagent 2a in this process (Scheme 6a). Subsequently, we placed compounds 1aa′ and 2a under standard reaction conditions, omitting NiCl2·6H2O and L1, to capture the addition products of sulfonyl radicals to alkynes. The failure to form products 3a or 3aa′, with the majority of starting material 1aa′ remaining unreacted, underscores the pivotal function of the Ni catalyst in facilitating the addition reaction of alkynes through highly active sulfonyl free radical nickel intermediates. Further exploration of the radical-mediated process included the strategic use of radical inhibitors and intermediate trapping agents. The introduction of 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) in a 3.0 equivalent ratio to the reaction mixture resulted in a complete suppression of the reaction. High-resolution mass spectrometry (HRMS) analysis confirmed the formation of TEMPO adducts, specifically identifying compound 4, which corroborates the involvement of a radical mechanism.
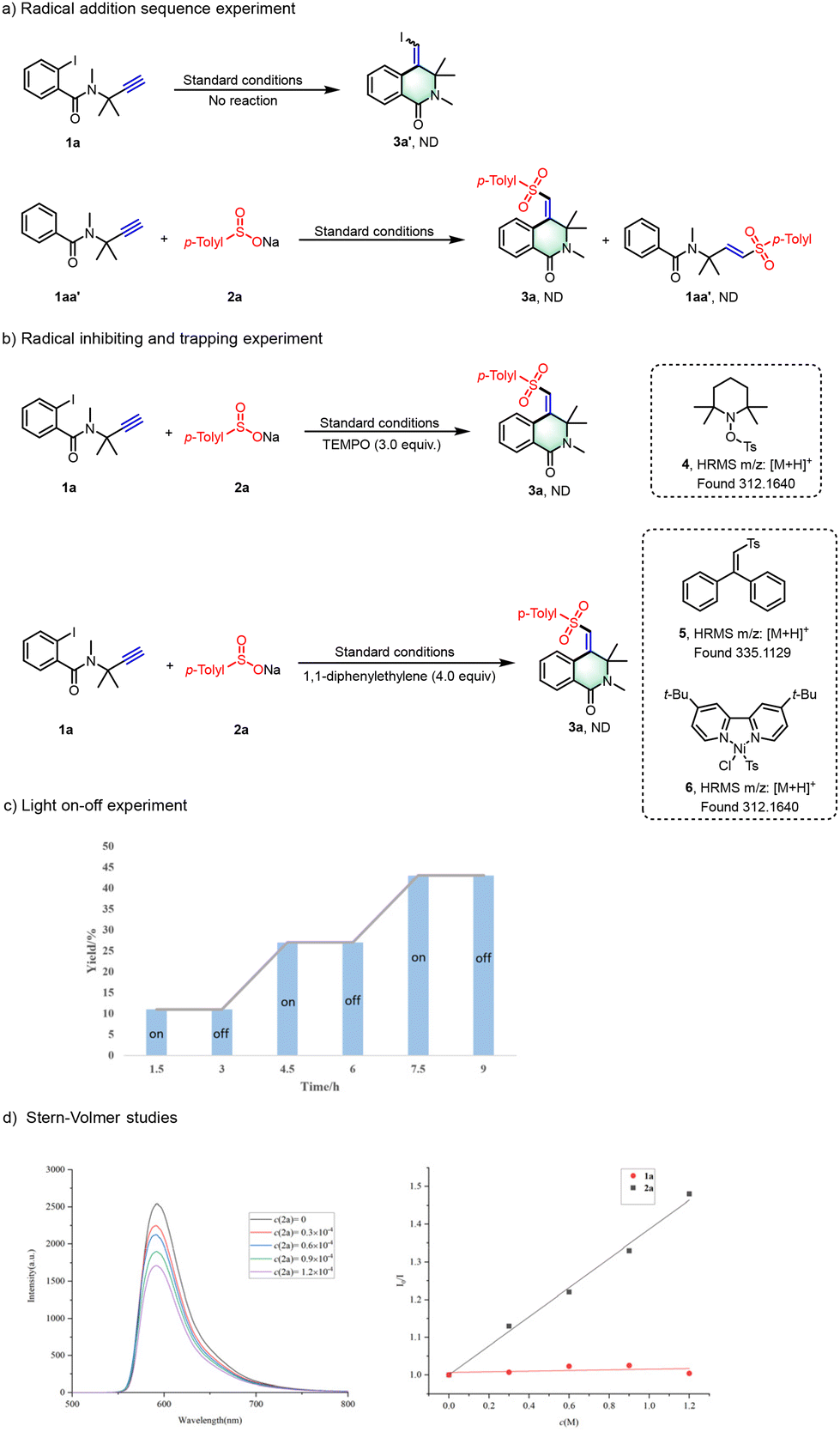 | ||
| Scheme 6 Control experiments. | ||
Additionally, employing 1,1-diphenylethylene as an intermediate trapping reagent under standard conditions, HRMS analysis enabled the identification of vinyl-trapped products 5 and the radical addition complex 6. These findings substantiate the proposed radical pathway and the sequence of addition, as illustrated in Scheme 6b. Concurrently, the photocatalytic aspect was examined through a series of visible-light irradiation on/off experiments, which decisively demonstrated the reaction's reliance on light (Scheme 6c). The reaction ceased in the absence of illumination and proceeded significantly faster under continuous visible light, underscoring the indispensable role of photocatalysis. Furthermore, a Stern–Volmer quenching experiment was performed, showing that the fluorescence intensity of Na2-Eosin Y was efficiently quenched with an increasing concentration of 2a, as depicted in Scheme 6d. In stark contrast, compound 1a showed no quenching effect on the excited state of the photosensitizer. This observation further elucidates the differential interactions between the photosensitizer and the substrates within the photocatalytic process.
Drawing from our mechanistic investigations and relevant literature,5–11 we propose a plausible mechanism for the photoredox/nickel dual-catalyzed cross-coupling cyclization, as depicted in Scheme 7. The photocatalytic cycle commences with the absorption of visible light by Na2-Eosin Y, leading to the formation of a long-lived EY* triplet excited state. This state is instrumental in oxidizing sodium p-tolylsulfinate (2a) to the sulfonyl radical A, while concurrently generating the Eosin Y radical anion, which acts as a reductant. The sulfonyl radical A is then incorporated into the nickel catalytic cycle, where it adds to a NiI complex B, yielding the Ts–NiII intermediate C. A single electron transfer (SET) event between the NiII intermediate C and the Eosin Y radical anion ensues, effectively regenerating the EY photoredox catalyst and restoring the Ts–NiI intermediate D. Following this, a 1,2 migratory insertion involving the unactivated alkyne and intermediate D takes place, culminating in the formation of intermediate E. This intermediate then undergoes an intramolecular oxidative addition cyclization with the aryl halide, succeeded by a reductive elimination step, ultimately affording the cyclization product 3a.
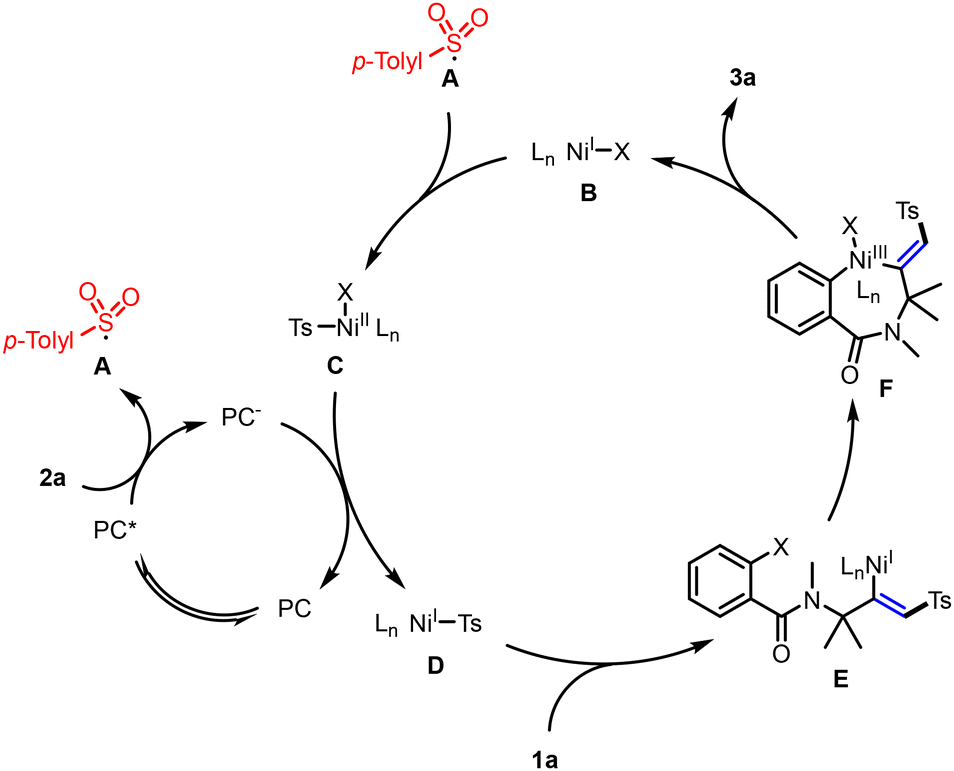 | ||
| Scheme 7 Proposed mechanistic pathway. | ||
Conclusions
In summary, we introduce a novel and environmentally friendly nickel/photoredox dual catalytic sulfonyl-arylation method for the synthesis of heterocyclic compounds. The method's innovative approach allows for the direct transformation of unactivated alkynes and aryl iodides into a variety of biologically significant heterocyclics, such as 3,4-dihydroisoquinolin-1(2H)-ones, tetrahydroisoquinolines, and isochroman derivatives. By bypassing traditional Heck reactions, this green chemistry technique minimizes the use of excess metal catalysts and reagents, enhancing chemical selectivity and curbing side reactions. The dual catalytic system's high reactivity with unactivated substrates and its ability to forge both C(sp2)–C(sp2) and C(sp2)–S bonds highlight its potential to advance synthetic chemistry. The method's operational simplicity, mild reaction conditions, and use of visible light as an energy source further underscore its ecofriendly and efficient nature, making it a promising tool for constructing heterocyclic frameworks in pharmaceuticals and biologically active compounds.Author contributions
In our collaborative research endeavor, every team member played an essential role in achieving the success of our project. B.-R. Leng and F. Yang were instrumental in optimizing the reaction conditions and expanding the scope of the reaction, ensuring the robustness of our synthetic methodology. J.-L. Bai, Y.-W. Huang, and P. Wei were responsible for experimental management, a task crucial for evaluating the reproducibility and reliability of our synthesis method. Y.-L. Zhu and D.-C. Wang provided expert guidance and strategic direction throughout the project, ensuring that our research remained on track and aligned with our scientific objectives. Y.-L. Zhu and Q.-Q. Liu were the intellectual architects of the project's conception and were also responsible for drafting the manuscript. All authors have contributed insightful comments on the manuscript and have approved the final version submitted for consideration.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 3a has been deposited at the CCDC under 2368219† and can be obtained from The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/structures/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21801130), and the Discipline Fund of the School of Pharmaceutical Sciences, Nanjing Tech University (Natural Science).References
- (a) A. Dufert and D. B. Werz, Carbopalladation Cascades Using Carbon-Carbon Triple Bonds: Recent Advances to Access Complex Scaffolds, Chemistry, 2016, 22, 16718–16732 CrossRef PubMed; (b) Y. Ke, W. Li, W. Liu and W. Kong, Ni-catalyzed ligand-controlled divergent and selective synthesis, Sci. China: Chem., 2023, 66, 2951–2976 CrossRef CAS.
- (a) D. A. Dirocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Late-stage functionalization of biologically active heterocycles through photoredox catalysis, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS; (b) A. Mermer, T. Keles and Y. Sirin, Recent studies of nitrogen containing heterocyclic compounds as novel antiviral agents: A review, Bioorg. Chem., 2021, 114, 105076 CrossRef CAS; (c) B. S. Matada, R. Pattanashettar and N. G. Yernale, A comprehensive review on the biological interest of quinoline and its derivatives, Bioorg. Med. Chem., 2021, 32, 115973 CrossRef CAS; (d) P. N. Kalaria, S. C. Karad and D. K. Raval, A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery, Eur. J. Med. Chem., 2018, 158, 917–936 CrossRef CAS; (e) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Organic synthesis provides opportunities to transform drug discovery, Nat. Chem., 2018, 10, 383–394 CrossRef CAS.
- (a) R. Arora and M. Lautens, Photoexcited Nickel-Catalyzed Carbohalogenation, ACS Catal., 2024, 14, 1970–1975 CrossRef CAS; (b) R. Arora, J. F. Rodriguez, A. Whyte and M. Lautens, Accessing Unsymmetrically Linked Heterocycles through Stereoselective Palladium-Catalyzed Domino Cyclization, Angew. Chem., Int. Ed., 2022, 61, e202112288 CrossRef CAS; (c) Y. Jin and C. Wang, Nickel-Catalyzed Asymmetric Reductive Arylalkylation of Unactivated Alkenes, Angew. Chem., Int. Ed., 2019, 58, 6722–6726 CrossRef CAS; (d) L. Li, Q. Yang, Y. Wang and Y. Jia, Catalytic asymmetric total synthesis of (-)-galanthamine and (-)-lycoramine, Angew. Chem., Int. Ed., 2015, 54, 6255–6259 CrossRef CAS; (e) A. Reding, P. G. Jones and D. B. Werz, trans-Carbocarbonation of Internal Alkynes through a Formal anti-Carbopalladation/C-H Activation Cascade, Angew. Chem., Int. Ed., 2018, 57, 10610–10614 CrossRef CAS; (f) J. Wu, L. Li, M. Liu, L. Bai and X. Luan, Selective C(sp3)-N Bond Cleavage of N,N-Dialkyl Tertiary Amines with the Loss of a Large Alkyl Group via an SN1 Pathway, Angew. Chem., Int. Ed., 2022, 61, e202113820 CrossRef CAS; (g) A. D. Marchese, A. G. Durant, C. M. Reid, C. Jans, R. Arora and M. Lautens, Pd(0)/Blue Light Promoted Carboiodination Reaction - Evidence for Reversible C-I Bond Formation via a Radical Pathway, J. Am. Chem. Soc., 2022, 144, 20554–20560 CrossRef CAS PubMed; (h) L. Zhang, X. Si, F. Rominger and A. S. K. Hashmi, Visible-Light-Induced Radical Carbo-Cyclization/gem-Diborylation through Triplet Energy Transfer between a Gold Catalyst and Aryl Iodides, J. Am. Chem. Soc., 2020, 142, 10485–10493 CrossRef CAS; (i) Y. Jin, H. Yang and C. Wang, Nickel-Catalyzed Reductive Arylalkylation via a Migratory Insertion/Decarboxylative Cross-Coupling Cascade, Org. Lett., 2019, 21, 7602–7608 CrossRef CAS.
- (a) X. W. Chen, J. P. Yue, K. Wang, Y. Y. Gui, Y. N. Niu, J. Liu, C. K. Ran, W. Kong, W. J. Zhou and D. G. Yu, Nickel-Catalyzed Asymmetric Reductive Carbo-Carboxylation of Alkenes with CO(2), Angew. Chem., Int. Ed., 2021, 60, 14068–14075 CrossRef CAS; (b) Y. Ping, Y. Li, J. Zhu and W. Kong, Construction of Quaternary Stereocenters by Palladium-Catalyzed Carbopalladation-Initiated Cascade Reactions, Angew. Chem., Int. Ed., 2019, 58, 1562–1573 CrossRef CAS; (c) U. Anwar, R. Grigg and V. Sridharan, Palladium catalysed cyclisation-Barbier-type allylation cascades, Chem. Commun., 2000, 933–934, 10.1039/b002713h; (d) M. Borjesson, D. Janssen-Muller, B. Sahoo, Y. Duan, X. Wang and R. Martin, Remote sp(2) C-H Carboxylation via Catalytic 1,4-Ni Migration with CO(2), J. Am. Chem. Soc., 2020, 142, 16234–16239 CrossRef CAS PubMed; (e) P. Fan, Y. Lan, C. Zhang and C. Wang, Nickel/Photo-Cocatalyzed Asymmetric Acyl-Carbamoylation of Alkenes, J. Am. Chem. Soc., 2020, 142, 2180–2186 CrossRef CAS; (f) Z. X. Tian, J. B. Qiao, G. L. Xu, X. Pang, L. Qi, W. Y. Ma, Z. Z. Zhao, J. Duan, Y. F. Du, P. Su, X. Y. Liu and X. Z. Shu, Highly Enantioselective Cross-Electrophile Aryl-Alkenylation of Unactivated Alkenes, J. Am. Chem. Soc., 2019, 141, 7637–7643 CrossRef CAS PubMed; (g) X. Wang, Y. Liu and R. Martin, Ni-Catalyzed Divergent Cyclization/Carboxylation of Unactivated Primary and Secondary Alkyl Halides with CO2, J. Am. Chem. Soc., 2015, 137, 6476–6479 CrossRef CAS PubMed; (h) Y. Li, K. Wang, Y. Ping, Y. Wang and W. Kong, Nickel-Catalyzed Domino Heck Cyclization/Suzuki Coupling for the Synthesis of 3,3-Disubstituted Oxindoles, Org. Lett., 2018, 20, 921–924 CrossRef CAS; (i) P.-X. Zhou, X. Yang, X. Du, S. Zhao, H. Wang, X. Tan, J. Wang and Y.-M. Liang, Pd-Catalyzed alkynyl aryl iodide cyclization/alkylation with cyclobutanols, Org. Chem. Front., 2022, 9, 2606–2611 RSC.
- (a) M. Claros, F. Ungeheuer, F. Franco, V. Martin-Diaconescu, A. Casitas and J. Lloret-Fillol, Reductive Cyclization of Unactivated Alkyl Chlorides with Tethered Alkenes under Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2019, 58, 4869–4874 CrossRef CAS; (b) A. Garcia-Dominguez, R. Mondal and C. Nevado, Dual Photoredox/Nickel-Catalyzed Three-Component Carbofunctionalization of Alkenes, Angew. Chem., Int. Ed., 2019, 58, 12286–12290 CrossRef CAS PubMed; (c) H. Jiang, X. Yu, C. G. Daniliuc and A. Studer, Three-Component Aminoarylation of Electron-Rich Alkenes by Merging Photoredox with Nickel Catalysis, Angew. Chem., Int. Ed., 2021, 60, 14399–14404 CrossRef CAS; (d) N. A. Weires, Y. Slutskyy and L. E. Overman, Facile Preparation of Spirolactones by an Alkoxycarbonyl Radical Cyclization-Cross-Coupling Cascade, Angew. Chem., Int. Ed., 2019, 58, 8561–8565 CrossRef CAS; (e) C. Zhu, H. Yue, L. Chu and M. Rueping, Recent advances in photoredox and nickel dual-catalyzed cascade reactions: pushing the boundaries of complexity, Chem. Sci., 2020, 11, 4051–4064 RSC; (f) L. Guo, F. Song, S. Zhu, H. Li and L. Chu, syn-Selective alkylarylation of terminal alkynes via the combination of photoredox and nickel catalysis, Nat. Commun., 2018, 9, 4543 CrossRef PubMed; (g) T. Long, C. Zhu, L. Li, L. Shao, S. Zhu, M. Rueping and L. Chu, Ligand-controlled stereodivergent alkenylation of alkynes to access functionalized trans- and cis-1,3-dienes, Nat. Commun., 2023, 14, 55 CrossRef CAS PubMed; (h) C. Zhu, H. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, A multicomponent synthesis of stereodefined olefins via nickel catalysis and single electron/triplet energy transfer, Nat. Catal., 2019, 2, 678–687 CrossRef CAS.
- Y. Gao, L. Gao, E. Zhu, Y. Yang, M. Jie, J. Zhang, Z. Pan and C. Xia, Nickel/photoredox dual catalyzed arylalkylation of nonactivated alkenes, Nat. Commun., 2023, 14, 7917 CrossRef CAS.
- (a) C. M. Le, P. J. Menzies, D. A. Petrone and M. Lautens, Synergistic steric effects in the development of a palladium-catalyzed alkyne carbohalogenation: stereodivergent synthesis of vinyl halides, Angew. Chem., Int. Ed., 2015, 54, 254–257 CrossRef CAS; (b) L. Fan, J. Hao, J. Yu, X. Ma, J. Liu and X. Luan, Hydroxylamines As Bifunctional Single-Nitrogen Sources for the Rapid Assembly of Diverse Tricyclic Indole Scaffolds, J. Am. Chem. Soc., 2020, 142, 6698–6707 CrossRef CAS; (c) T. Castanheiro, M. Donnard, M. Gulea and J. Suffert, Cyclocarbopalladation/cross-coupling cascade reactions in sulfide series: access to sulfur heterocycles, Org. Lett., 2014, 16, 3060–3063 CrossRef CAS PubMed; (d) C. Cheng and Y. Zhang, Palladium-Catalyzed anti-Carbosilylation of Alkynes to Access Isoquinolinone-Containing Exocyclic Vinylsilanes, Org. Lett., 2021, 23, 5772–5776 CrossRef CAS; (e) D. J. Wilger, S. E. Bottcher and L. E. Hutchinson, Nickel-Catalyzed anti-Selective Alkyne Functionalization Reactions, Synthesis, 2020, 2807–2820 CrossRef.
- J. D. Nguyen, E. M. D'Amato, J. M. Narayanam and C. R. Stephenson, Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- H. Yue, C. Zhu and M. Rueping, Cross-Coupling of Sodium Sulfinates with Aryl, Heteroaryl, and Vinyl Halides by Nickel/Photoredox Dual Catalysis, Angew. Chem., Int. Ed., 2018, 57, 1371–1375 CrossRef CAS.
- H. Kim and C. Lee, Visible-light-induced photocatalytic reductive transformations of organohalides, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 CrossRef CAS PubMed.
- (a) S. M. Hell, C. F. Meyer, A. Misale, J. B. I. Sap, K. E. Christensen, M. C. Willis, A. A. Trabanco and V. Gouverneur, Hydrosulfonylation of Alkenes with Sulfonyl Chlorides under Visible Light Activation, Angew. Chem., Int. Ed., 2020, 59, 11620–11626 CrossRef CAS; (b) X. Dong, W. Jiang, D. Hua, X. Wang, L. Xu and X. Wu, Radical-mediated vicinal addition of alkoxysulfonyl/fluorosulfonyl and trifluoromethyl groups to aryl alkyl alkynes, Chem. Sci., 2021, 12, 11762–11768 RSC.
- (a) Z. Zhao, K. Kang, J. Yue, X. Ji, H. Qiao, P. Fan and X. Zheng, Research progress in biological activities of isochroman derivatives, Eur. J. Med. Chem., 2021, 210, 113073 CrossRef CAS; (b) W. Li, C. Lee, S. H. Bang, J. Y. Ma, S. Kim, Y. S. Koh and S. H. Shim, Isochromans and Related Constituents from the Endophytic Fungus Annulohypoxylon truncatum of Zizania caduciflora and Their Anti-Inflammatory Effects, J. Nat. Prod., 2017, 80, 205–209 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2368219. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qo01627k |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2025 |