
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAll-carbon supramolecular complexation of a bilayer molecular nanographene with [60] and [70]fullerenes†
Manuel
Buendía
 a,
Anton J.
Stasyuk
bc,
Salvatore
Filippone
a,
Miquel
Solà
*b and
Nazario
Martín
*ad
a,
Anton J.
Stasyuk
bc,
Salvatore
Filippone
a,
Miquel
Solà
*b and
Nazario
Martín
*ad
aDepartamento de Química Orgánica I, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Av. Complutense, S/N, 28040 Madrid, Spain. E-mail: nazmar@ucm.es
bInstitut de Quimica Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, M. Aurèlia Capmany, 69, 17003 Girona, Spain. E-mail: miquel.sola@udg.edu
cDepartament de Farmàcia i Tecnologia Farmacèutica, i Fisicoquímica, Facultat de Farmàcia i Ciències de l'Alimentació & Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona (UB), Av. Joan XXIII 27-31, Barcelona, Spain
dIMDEA-Nanociencia. C/Faraday, 9. Campus de Cantoblanco, 28049 Madrid, Spain
First published on 24th December 2024
Abstract
Supramolecular chemistry of carbon-based materials provides a variety of chemical structures with potential applications in materials science and biomedicine. Here, we explore the supramolecular complexation of fullerenes C60 and C70, highlighting the ability of molecular nanographene tweezers to capture these structures. The binding constant for the CNG-1⊃C70 complex was significantly higher than for CNG-1⊃C60, showing a clear selectivity for the more π-extended C70. DFT calculations confirmed these experimental results by showing that the interaction energy of C70 with CNG-1 is more than 5 kcal mol−1 higher than that of C60. Theoretical calculations predict that the dispersion interaction provides about 58–59% of the total interaction energy, followed by electrostatic attraction with 26% and orbital interactions, which contribute 15–16%. The racemic nanographene tweezers effectively recognize fullerene molecules and hold promise for future applications in chiral molecule recognition.
Introduction
The supramolecular chemistry of carbon-based materials has been a recurring topic in organic chemistry with the uncountable possibilities and combinations stemming from the size and shape of graphene,1,2 carbon nanotubes (CNTs),3,4 and fullerenes.5 As expected for these carbon nanostructures, non-covalent interactions such as C–H⋯π and, mostly, π⋯π interactions are those mainly controlling the formation of the resulting supramolecular ensembles.6–9 Interestingly, the non-covalent functionalization of these carbon allotropes has provided a great variety of chemical architectures, many of them showing unexpected properties with potential biomedical10 and materials science applications.11In particular, the supramolecular complexation of fullerenes has received a lot of attention due to their singular spherical shape as well as their electron-acceptor nature, affording a variety of supramolecular complexes exhibiting new structural and optoelectronic properties.6,12 Thus, a plethora of molecules of diverse structural complexity have been synthesized acting as hosts to capture guest fullerenes, especially C60 and C70. In this regard, chemical structures based on macrocycles such as calixarenes,13,14 cucurbituryls,15 cyclodextrins,16,17 molecular nanobelts,18,19 cycloparaphenylenes (CPPs),20–22 molecular cages,23,24 as well as molecular tweezers endowed with curved π-extended moieties with positive Gaussian curvature such as corannulenes,25 or with electronically complementary electron-donor molecules such as porphyrins or π-extended tetrathiafulvalenes (ex-TTFs)26,27 have extensively been reported.
More recently, molecular nanographenes have attracted the attention of the scientific community due to their remarkable chiroptical and optoelectronic properties, their quick access and tunability through benchtop synthesis and their amazing structural diversity.28–30 These nanometric fragments of graphene typically present a higher number of π–π interactions with fullerenes than other Polycyclic Aromatic Hydrocarbons (PAHs). Accordingly, molecular nanographenes have been used as hosts in host–guest chemistry with C60 and C70 species, being embedded into the aforementioned macrocycles as CPPs and nanobelts,18,31,32 and also as simple molecules with either positive or negative Gaussian curvatures.33 Interestingly, these supramolecular complexes not only have enhanced binding constants with fullerenes than their previous counterparts, but they can also showcase photoinduced electron transfer processes between the fullerene and the nanographene units.34
In this work, we report on the molecular complexation of C60 and C70 with a molecular nanographene tweezer (CNG-1) as a suitable host previously obtained by a six-step synthetic process. Furthermore, theoretical calculations using the DFT BLYP exchange–correlation functional, along with the empirical D3(BJ) dispersion correction and def2-SVP basis set, nicely underpin the experimental values, matching the difference in interaction energy with binding constant values.
Results and discussion
Racemic and chiral triindane nanographene CNG-1 was synthesized according to a procedure previously reported by our group.35 Interestingly, this bilayer molecule exhibits a suitable concave geometry for further supramolecular complexation experiments. In particular, we envisaged to seize the inner cavity of this molecular tweezer with different fullerenes, namely C60 and C70, endowed with complementary convex surfaces (Fig. 1).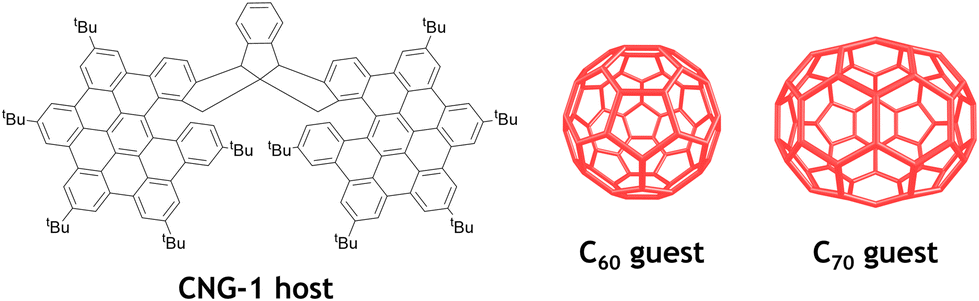 | ||
| Fig. 1 All-carbon nanostructures involved in the supramolecular complexation of CNG-1 with fullerenes C60 and C70. | ||
The X-ray crystal structure of CNG-1 revealed that both nanographene layers of the tweezer connected to the central triindane unit form an all-carbon host with an interlayer distance of around 9 Å, being a suitable host to accommodate either C60 or C70 fullerenes and, thus, affording their respective supramolecular complexes (Fig. 1). For this purpose, 1H NMR titration experiments were performed to carry out the supramolecular complexation by following the evolution of the proton signals mostly affected in the complexation process between racemic CNG-1 with fullerenes C60 and C70.
These titration experiments were performed in deuterated chlorobenzene (PhCl-d5) to ensure that both fullerene species were properly dissolved. Additionally, a complete NMR study was performed to assign all proton signals of CNG-1 and gain a deeper insight into the complexation processes, which can be found in Fig. S1–S6, ESI.† The first titration involved CNG-1 as host and fullerene C60 as guest. 1H NMR spectra of this titration showcased chemical shifts of single time-averaged signals of CNG-1 with additions up to 19 equivalents of guest C60, evidencing a fast exchange rate in solution between complex and free host and guest species as well as the supramolecular complexation process (Fig. S7†). Notable differences in chemical shifts were observed for six proton signals: two tert-butyl protons (Ha: δ0 = 1.26 ppm; Hb: δ0 = 1.58 ppm), the less shielded methylene proton of the AB system (Hc: δ0 = 2.76 ppm), the doubly benzylic protons of the stereogenic centers (Hd: δ0 = 3.79 ppm), and two notably deshielded protons from the edge of the nanographene layers (He: δ0 = 8.82 ppm; Hf: δ0 = 9.30 ppm). The highest chemical shift difference was found for signal Hc (Δδ = 0.087 ppm) which, according to the X-ray data, belongs to the proton pointing towards the inner cavity of the nanographene tweezer, nicely confirming the complexation process (Fig. 2).
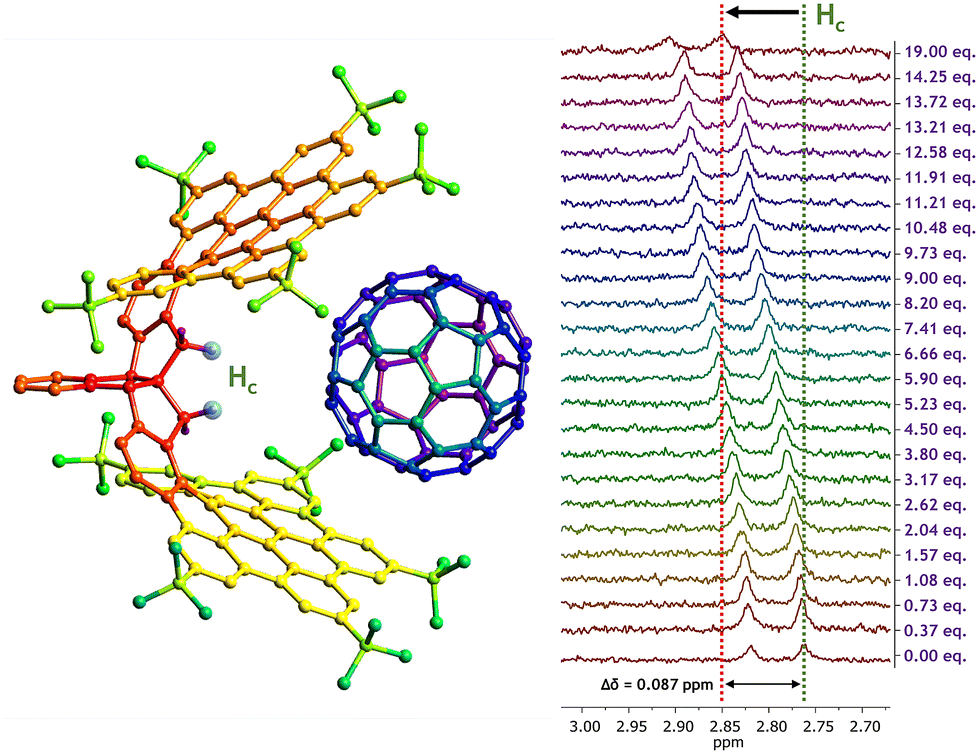 | ||
Fig. 2 Model for the supramolecular complexation of the CNG-1⊃C60 complex with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry (left), and Hc signal chemical shift variation upon addition of C60 equivalents throughout the titration in PhCl-d5 (right). :1 stoichiometry (left), and Hc signal chemical shift variation upon addition of C60 equivalents throughout the titration in PhCl-d5 (right). | ||
After plotting the titration data using BindFit fitting tool,36,37 the non-linear fit afforded a moderate binding constant of Ka = 61 ± 1 M−1 for the CNG-1⊃C60 supramolecular complex, matching a 1:1 stoichiometry model (Nelder Mead method, Fig. S8;† for molar fraction evolution of both host and complex see Fig. S9†), thus confirming the affinity of CNG-1 for fullerene C60.
The second titration involving fullerene C70 as guest revealed a similar behavior for CNG-1 host proton signals, where the same six signals from the first titration with C60 also shifted largely (Fig. S10†), with the greatest difference in chemical shift for the methylene signal Hc as well (δ0 = 2.76 ppm, Δδ = 0.049 ppm). Moreover, complete saturation of complex in solution was reached (8 equivalents of C70) as the Hc signal remained invariable, thus completing a successful titration (Fig. 3).
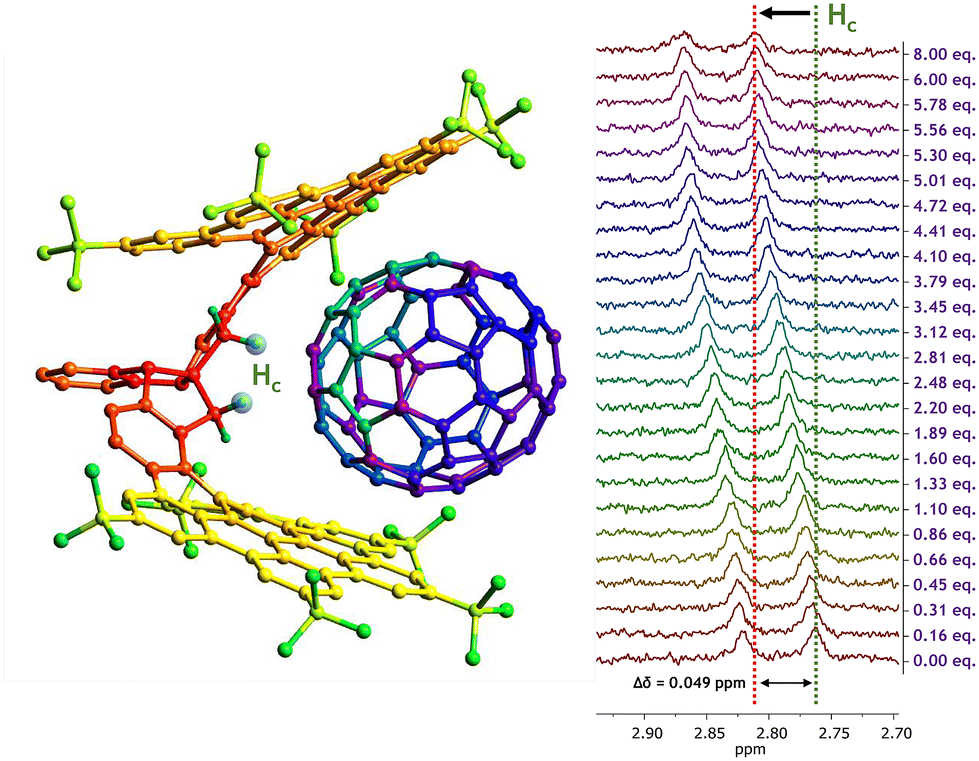 | ||
| Fig. 3 Model for the supramolecular complexation of the CNG-1⊃C70 complex with a 1:1 stoichiometry (left), and Hc signal chemical shift variation upon addition of C70 equivalents throughout the titration in PhCl-d5 (right). | ||
The non-linear adjust of these data resulted in a binding constant of Ka = 400 ± 17 M−1 for the CNG-1⊃C70 supramolecular complex, also matching with a 1:1 stoichiometry model (Nelder Mead method, Fig. S11;† for molar fraction evolution of both host and complex see Fig. S12†). This result demonstrates the higher affinity of CNG-1 C70 over C60 with a binding constant value around one order of magnitude higher. Such enhancement can be accounted for by the larger π-extended surface of the C70 guest compared to the former C60 and, hence, increasing the π–π interactions between host CNG-1 and guest C70 molecules.
To gain insight into the nature of these supramolecular complexation processes, a series of quantum chemical calculations were performed. Systems of interest were optimized utilizing the DFT BLYP38,39 exchange−correlation functional along with the empirical D3(BJ) dispersion correction40,41 and def2-SVP basis set.42,43 The calculations were performed within the ORCA 5.0.3 program44 (see ESI for more details on the computational method used†).
To assess the stability of the complexes, we calculated the interaction energy (ΔEint) between the CNG-1 tweezers and the C60/C70 units, as well as the deformation energy (ΔEdef) associated with the deformation of the units from their equilibrium geometries to the geometries they adopt in the complexes. DFT optimized structures of both complexes are provided in Fig. 4. For CNG-1⊃C60 and CNG-1⊃C70, ΔEint was found to be −61.3 and −66.1 kcal mol−1, while ΔEdef was 3.1 and 2.5 kcal mol−1, respectively (Table 1).
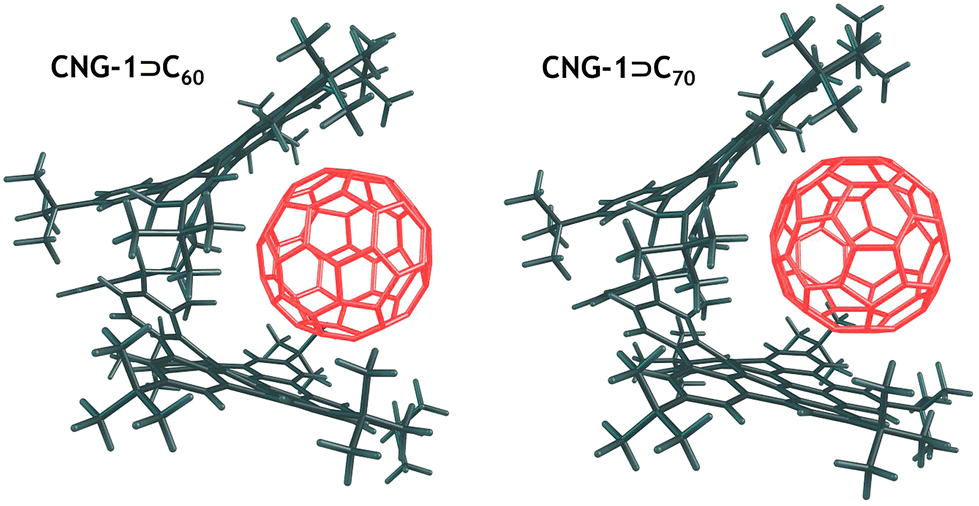 | ||
| Fig. 4 Optimized DFT structures for supramolecular complexes CNG-1⊃C60 and CNG-1⊃C70. | ||
| Complex | Energy terms | ΔEint | ΔEdef | ΔEcomplex | K a, M−1 | ||||
|---|---|---|---|---|---|---|---|---|---|
| ΔEPauli | ΔEelstat | ΔEoi | ΔEdisp | Host | C xx | ||||
| a Relative values (in parentheses) are given as a percentage and express the contribution to the sum of all attractive energy terms: ΔEelstat + ΔEoi + ΔEdisp. Complexation energy: ΔEcomplex = ΔEint + ΔEdef. | |||||||||
| CNG-1⊃C60 | 104.56 | −43.24 (26%) | −25.80 (16%) | −96.78 (58%) | −61.25 | 2.74 | 0.37 | −58.14 | 61 ± 1 |
| CNG-1⊃C70 | 116.86 | −47.36 (26%) | −28.01 (15%) | −107.57 (59%) | −66.08 | 2.18 | 0.27 | −63.63 | 400 ± 17 |
Most of the deformation energy is attributed to the CNG-1 deformation, with deformation of the fullerenes contributing only 11 to 12%. It is interesting to note that despite the significant difference in the spatial sizes of C60 and C70 fullerenes, the deformation energy of the host unit in both complexes is nearly the same. This can apparently be explained by the mutual arrangement of the fullerenes and CNG-1 units in the complexes. In particular, C70 in the CNG-1⊃C70, complex is positioned in such a way that its main axis runs along the cavity of the tweezers, and thus, the effective size of both fullerenes appears to be very similar.
To analyze the nature of the host–guest interactions, we employed the Morokuma-like interaction energy decomposition analysis (EDA)45 implemented in the ADF program.46 The EDA decomposes the interaction energy into four components: electrostatic (ΔEelstat), Pauli repulsion (ΔEPauli), orbital interactions (ΔEoi), and dispersion correction (ΔEdisp).47 This decomposition enables us to assess the role of the specific interactions in the systems. Table 1 shows the EDA analysis results for CNG-1⊃C60 and CNG-1⊃C70 complexes.
As expected for both complexes, the nature of the host–guest interactions is nearly the same. In particular, dispersion interaction provides about 58–59% of the total interaction energy, followed by electrostatic attraction with 26%, and orbital interactions which contribute 15–16%. The destabilizing term (ΔEPauli) is slightly larger for CNG-1⊃C70 than for CNG-1⊃C60, 116.9 and 104.6 kcal mol−1, respectively. However, this difference is compensated to a significant extent by a larger dispersion correction term for CNG-1⊃C70 (Table 1). Overall, based on the complexation energy values, the CNG-1⊃C70 complex is more stable than CNG-1⊃C60 by 5.5 kcal mol−1, supporting the higher binding constant for the complex with C70 fullerene observed in the titration experiment.
The analysis of the host–guest interaction topology for the complexes of interest, performed using the non-covalent interaction (NCI) index analysis,48 revealed two distinct types of areas between the fullerenes and the CNG-1 host unit. One type of these areas is located between the fullerene and the nanographene units of the tweezer.
Considering the π-conjugated nature of the fullerene and nanographene units, the interactions can be assigned as π⋯π interactions. The other type of area can be classified as C–H⋯π interactions due to its location between the fullerene and the hydrogen atoms of the central triindane fragment or the hydrogen atoms of the tert-butyl groups. The NCI isosurfaces for both complexes are presented in Fig. S13, ESI.†
Although these binding constants are lower with respect to other previously reported in literature for molecular nanographenes,49 it is important to remark that CNG-1 as a molecular receptor for fullerenes (especially C70) is endowed with an inherent chiral triindane which, eventually, could open a door for the selective supramolecular recognition of sp2 carbon-based chiral molecules.
Conclusions
The supramolecular complexation of guest fullerenes C60 and C70 with racemic molecular nanographene CNG-1 as a host is presented in this work. Relatively moderate binding constant values were afforded for CNG-1⊃C60 and larger for CNG-1⊃C70 complexes, both accommodating the respective guest fullerene inside its cavity with a 1:1 stoichiometry. Moreover, the binding constant for CNG-1⊃C70 was one order of magnitude higher than that of CNG-1⊃C60, thus revealing a clear selectivity towards the more π-extended fullerene. DFT calculations supported these experimental values, matching the difference in interaction energy with binding constant values. In this manner, we have demonstrated that racemic molecular nanographene CNG-1, can be used for the supramolecular recognition of carbon-based π-extended molecules such as fullerenes. Besides, its inherent chirality could potentially play a major role in the near future for the recognition of chiral molecules, including fullerenes.
Experimental section
Materials and methods
CNG-1 was prepared according to the procedure reported in the literature.351H NMR spectra of the titration were recorded at 300 (Bruker AVIII) MHz at 298 K using deuterated chlorobenzene (PhCl-d5) as solvent. All titrations were performed in an NMR sample tube charged with 0.5 mL of a starting CNG-1 host solution (5 × 10−4 M). Sequential aliquots of a guest stock solution were added to the initial solution of CNG-1 with a micropipette and mixing properly, recording their 1H NMR spectra afterwards. These guest fullerene stock solutions were prepared dissolving the respective fullerene with a CNG-1 solution of the same molarity as the initial to prevent dilution factors and keep a constant host concentration throughout the titration. Therefore, C60 + CNG-1 (9.5 × 10−3 M and 5 × 10−4 M, respectively) and C70 + CNG-1 (4 × 10−3 M and 5 × 10−4 M, respectively) stock solutions were afforded. The resulting titration data was fitted using BindFit fitting tool to determine the association constants.37Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. J. S. and M. S. thanks the Ministerio de Ciencia, Innovación y Universidades (MCIN/AEI/10.13039/50110001103) (Projects PID2023-147424NB-I00, PID2023-146849NB-I00, RED2022-134939-T, and FPI predoctoral grant to C. C.) and the Generalitat de Catalunya (Project 2021-SGR-623). A. J. S. gratefully acknowledge Polish high-performance computing infrastructure PLGrid (HPC Center: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2023/016841.M. B., S. F., and N. M. acknowledge financial support from the Spanish MICIN (project PID2020-114653RB-I00), they also acknowledge financial support from the ERC (SyG TOMATTO ERC-2020-951224) and from the ‘(MAD2D-CM)-UCM’ project funded by Comunidad de Madrid, by the Recovery, Transformation and Resilience Plan and by NextGenerationEU from the European Union. M. B. also acknowledges financial support from the Spanish MICIN (project CTQ2017-83531-R).
References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Electric field effect in atomically thin carbon films, Science, 2004, 306, 666–669 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, The rise of graphene, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- S. Iijima, Helical Microtubules of Graphitic Carbon, Nature, 1991, 354, 56–58 CrossRef CAS.
- D. S. Bethune, C. H. Kiang, M. S. de Vries, G. Gorman, R. Savoy, J. Vazquez and R. Beyers, Cobalt-catalysed growth of carbon nanotubes with single-atomic-layer walls, Nature, 1993, 363, 605–607 CrossRef CAS.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, C60: Buckminsterfullerene, Nature, 1985, 318, 162–163 CrossRef CAS.
- J. F. Nierengarten and N. Martín, Supramolecular Chemistry of Fullerenes and Carbon Nanotubes, Wiley, Weinheim, 2012 Search PubMed.
- E. M. Pérez and N. Martín, π-π interactions in carbon nanostructures, Chem. Soc. Rev., 2015, 44, 6425–6433 RSC.
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Noncovalent Functionalization of Graphene and Graphene Oxide for Energy Materials, Biosensing, Catalytic, and Biomedical Applications, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed.
- G. Bottari, M. A. Herranz, L. Wibmer, M. Volland, L. Rodriguez-Perez, D. M. Guldi, A. Hirsch, N. Martin, F. D'Souza and T. Torres, Chemical functionalization and characterization of graphene-based materials, Chem. Soc. Rev., 2017, 46, 4464–4500 RSC.
- Z. Liu, X. Sun, N. Nakayama-Ratchford and H. Dai, Supramolecular Chemistry on Water-Soluble Carbon Nanotubes for Drug Loading and Delivery, ACS Nano, 2007, 1, 50–56 CrossRef CAS PubMed.
- L. von Luders, R. Tilmann, K. Lee, C. Bartlam, T. Stimpel-Lindner, T. K. Nevanen, K. Iljin, K. C. Knirsch, A. Hirsch and G. S. Duesberg, Functionalisation of Graphene Sensor Surfaces for the Specific Detection of Biomarkers, Angew. Chem., Int. Ed., 2023, 62, e202219024 CrossRef.
- X. Chang, Y. Xu and M. von Delius, Recent advances in supramolecular fullerene chemistry, Chem. Soc. Rev., 2024, 53, 47–83 RSC.
- J. L. Atwood, G. A. Koutsantonis and C. L. Raston, Purification of C60 and C70 by selective complexation with calixarenes, Nature, 1994, 368, 229–231 CrossRef CAS.
- X.-S. Ke, T. Kim, V. M. Lynch, D. Kim and J. L. Sessler, Flattened Calixarene-like Cyclic BODIPY Array: A New Photosynthetic Antenna Model, J. Am. Chem. Soc., 2017, 139, 13950–13956 CrossRef CAS PubMed.
- F. Constabel and K. E. Geckeler, Solvent-free self-assembly of C60 and cucurbit[7]uril using high-speed vibration milling, Tetrahedron Lett., 2004, 45, 2071–2073 CrossRef CAS.
- T. Andersson, K. Nilsson, M. Sundahl, G. Westman and O. Wennerström, C60 embedded in γ-cyclodextrin: a water-soluble fullerene, J. Chem. Soc., Chem. Commun., 1992, 604–606 RSC.
- Z. Hu, C. Wang, F. Zhao, X. Xu, S. Wang, L. Yu, D. Zhang and Y. Huang, Fabrication of a graphene/C60 nanohybrid via gamma-cyclodextrin host-guest chemistry for photodynamic and photothermal therapy, Nanoscale, 2017, 9, 8825–8833 RSC.
- D. Lu, G. Zhuang, H. Wu, S. Wang, S. Yang and P. Du, A Large π-Extended Carbon Nanoring Based on Nanographene Units: Bottom-Up Synthesis, Photophysical Properties, and Selective Complexation with Fullerene C70, Angew. Chem., Int. Ed., 2017, 56, 158–162 CrossRef CAS.
- X. Lu, T. Y. Gopalakrishna, Y. Han, Y. Ni, Y. Zou and J. Wu, Bowl-Shaped Carbon Nanobelts Showing Size-Dependent Properties and Selective Encapsulation of C70, J. Am. Chem. Soc., 2019, 141, 5934–5941 CrossRef CAS.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Size-selective encapsulation of C60 by [10]cycloparaphenylene: formation of the shortest fullerene-peapod, Angew. Chem., Int. Ed., 2011, 50, 8342–8344 CrossRef CAS PubMed.
- J. S. Wossner, D. Wassy, A. Weber, M. Bovenkerk, M. Hermann, M. Schmidt and B. Esser, [n]Cyclodibenzopentalenes as Antiaromatic Curved Nanocarbons with High Strain and Strong Fullerene Binding, J. Am. Chem. Soc., 2021, 143, 12244–12252 CrossRef PubMed.
- M. Hermann, D. Wassy and B. Esser, Conjugated Nanohoops Incorporating Donor, Acceptor, Hetero- or Polycyclic Aromatics, Angew. Chem., Int. Ed., 2021, 60, 15743–15766 CrossRef CAS.
- Y. Shi, K. Cai, H. Xiao, Z. Liu, J. Zhou, D. Shen, Y. Qiu, Q. H. Guo, C. Stern, M. R. Wasielewski, F. Diederich, W. A. Goddard 3rd and J. F. Stoddart, Selective Extraction of C70 by a Tetragonal Prismatic Porphyrin Cage, J. Am. Chem. Soc., 2018, 140, 13835–13842 CrossRef CAS.
- C. García-Simón, M. Costas and X. Ribas, Metallosupramolecular receptors for fullerene binding and release, Chem. Soc. Rev., 2016, 45, 40–62 RSC.
- A. Sacristán-Martín, D. Miguel, A. Díez-Varga, H. Barbero and C. M. Álvarez, From Induced-Fit Assemblies to Ternary Inclusion Complexes with Fullerenes in Corannulene-Based Molecular Tweezers, J. Org. Chem., 2022, 87, 16691–16706 CrossRef.
- E. M. Pérez and N. Martín, Curves ahead: molecular receptors for fullerenes based on concave-convex complementarity, Chem. Soc. Rev., 2008, 37, 1512–1519 RSC.
- E. Huerta, H. Isla, E. M. Pérez, C. Bo, N. Martín and J. de Mendoza, Tripodal exTTF-CTV hosts for fullerenes, J. Am. Chem. Soc., 2010, 132, 5351–5353 CrossRef CAS PubMed.
- L. Chen, Y. Hernandez, X. Feng and K. Müllen, From nanographene and graphene nanoribbons to graphene sheets: chemical synthesis, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, New advances in nanographene chemistry, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- J. M. Fernández-García, P. Izquierdo-García, M. Buendía, S. Filippone and N. Martín, Synthetic chiral molecular nanographenes: the key figure of the racemization barrier, Chem. Commun., 2022, 58, 2634–2645 RSC.
- Q. Huang, G. Zhuang, H. Jia, M. Qian, S. Cui, S. Yang and P. Du, Photoconductive Curved-Nanographene/Fullerene Supramolecular Heterojunctions, Angew. Chem., Int. Ed., 2019, 58, 6244–6249 CrossRef CAS PubMed.
- J. P. Mora-Fuentes, M. D. Codesal, M. Reale, C. M. Cruz, V. G. Jiménez, A. Sciortino, M. Cannas, F. Messina, V. Blanco and A. G. Campaña, Heptagon-Containing Nanographene Embedded into [10]Cycloparaphenylene, Angew. Chem., Int. Ed., 2023, 62, e202301356 CrossRef CAS.
- S. Zank, J. M. Fernández-García, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Solà, D. M. Guldi and N. Martín, Initiating Electron Transfer in Doubly Curved Nanographene Upon Supramolecular Complexation of C60, Angew. Chem., Int. Ed., 2022, 61, e202112834 CrossRef CAS.
- G. M. Beneventi, M. Krug, D. Reger, N. Jux and D. M. Guldi, Towards understanding the competition of electron and energy transfer in “molecular” nanographenes on the example of hexa-peri-hexabenzocoronene, J. Photochem. Photobiol., C, 2023, 56, 100602 CrossRef CAS.
- M. Buendía, J. M. Fernández-García, J. Perles, S. Filippone and N. Martín, Enantioselective synthesis of a two-fold inherently chiral molecular nanographene, Nat. Synth., 2024, 3, 545–553 CrossRef.
- A. J. Lowe, F. M. Pfeffer and P. Thordarson, Determining binding constants from 1H NMR titration data using global and local methods: a case study using [n]polynorbornane-based anion hosts, Supramol. Chem., 2012, 24, 585–594 CrossRef CAS.
- D. B. Hibbert and P. Thordarson, The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis, Chem. Commun., 2016, 52, 12792–12805 RSC.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- K. Morokuma, Why do molecules interact? The origin of electron donor-acceptor complexes, hydrogen bonding and proton affinity, Acc. Chem. Res., 1977, 10, 294–300 CrossRef CAS.
- ADF, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2017, https://www.scm.com Search PubMed.
- F. M. Bickelhaupt and K. N. Houk, Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- H. He, Y. J. Lee, Z. Zong, N. Liu, V. M. Lynch, J. Kim, J. Oh, D. Kim, J. L. Sessler and X.-S. Ke, Nanographene-Fused Expanded Carbaporphyrin Tweezers, J. Am. Chem. Soc., 2024, 146, 543–551 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo02071e |
| This journal is © the Partner Organisations 2025 |