
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnticancer benzimidazole derivatives as inhibitors of epigenetic targets: a review article
Nardin Wagiha,
Islam M. Abdel-Rahmana,
Nawal A. El-Koussi ab and
Gamal El-Din A. Abuo-Rahma*ca
ab and
Gamal El-Din A. Abuo-Rahma*ca
aPharmaceutical Chemistry Department, Faculty of Pharmacy, Deraya University, New Minia 61111, Egypt. E-mail: gamalaburahma@yahoo.com
bMedicinal Chemistry Department, Faculty of Pharmacy, Assiut University, 71526, Assiut, Egypt
cMedicinal Chemistry Department, Faculty of Pharmacy, Minia University, 61519 Minia, Egypt
First published on 13th January 2025
Abstract
Cancer is one of the leading causes of morbidity and mortality worldwide. One of the primary causes of cancer development and progression is epigenetic dysregulation, which is a heritable modification that alters gene expression without changing the DNA sequence. Therefore, targeting these epigenetic changes has emerged as a promising therapeutic strategy. Benzimidazole derivatives have gained attention for their potent epigenetic modulatory effects as they interact with various epigenetic targets, including DNA methyltransferases, histone deacetylases and histone methyltransferases. This review provides a comprehensive overview of benzimidazole derivatives that inhibit different acetylation and methylation reader, writer and eraser epigenetic targets. Herein, we emphasize the therapeutic potential of these compounds in developing targeted, less toxic cancer therapies. Presently, some promising benzimidazole derivatives have entered clinical trials and shown great advancements in the fields of hematological and solid malignancy therapies. Accordingly, we highlight the recent advancements in benzimidazole research as epigenetic agents that could pave the way for designing new multi-target drugs to overcome resistance and improve clinical outcomes for cancer patients. This review can help researchers in designing new anticancer benzimidazole derivatives with better properties.
1 Introduction
Cancer is a complex disease caused by genetic and epigenetic alterations and remains a leading cause of death worldwide.1–3 Conventional cancer therapies using alkylating agents, antimetabolites, antimicrotubular agents, antibiotics, hormones, plant alkaloids, vinca alkaloids, epipodophyllotoxins, anthracyclines, taxanes, corticosteroids, and kinase inhibitors are toxic to cancer; however, they also exhibit toxicity to normal cells, leading to some serious side effects in patients and sometimes even death.4 There are different problems related to conventional cancer therapies, such as resistance to therapy, ineffectiveness, and the growth and reproliferation of cancerous cells if cytotoxic therapy is stopped.5 Therefore, epigenetics has entered the field of cancer treatment with great advancements and effectiveness. Epigenetics is the study of inheritance and reversible modes originating from chemical modifications or additions to DNA without causing any changes in the primary DNA sequence.6–8 Modifications in epigenetic regulations are the primary cause of cancer initiation, progression, and metastasis. Chromatin is a DNA and histone protein complex that is crucial in packaging DNA within the cell nucleus. Moreover, it is a dynamic structure that responds to reversible epigenetic alterations. Histones are responsible for DNA condensation and organization, making DNA accessible for replication, transcription, and repair.7 Epigenetic modifications are regulated through three types of epigenetic proteins, i.e., writers (add an epigenetic mark), readers (respond to the epigenetic mark), and erasers (remove the epigenetic mark) (Table 1).9 These modifications include histone modification, DNA methylation and non-coding RNA (ncRNA) mutation or abnormal expression. Histone modifications include histone acetylation, methylation, phosphorylation, ubiquitination, and citrullination.10,11 Histone acetylation is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), leading to chromatin opening and condensation, respectively.12 Acetylation of H3 and H4 histone proteins is responsible for the relaxed conformation of chromatin by neutralizing the lysine positive charge and preventing the electrostatic interaction of histones with DNA.13 Histone methyltransferases consist of lysine methyltransferase (KMT) and arginine methyltransferase (PRMT) families, which are responsible for various biological processes (cell cycle regulation, cell division and proliferation, survival, and genome stability) and RNA metabolism (RNA biogenesis, processing, splicing, gene expression, chromatin organization, translation, and microtubule-associated metabolic processes), respectively.14 KMT therapeutic targets include EZH2, DOT1L, and G9a.15 Lysine demethylase enzymes (KDMs) are divided into three families including lysine-specific demethylase 1 (LSD1 or KDM1A), lysine-specific demethylase 2 (LSD2) and Jumonji C domain-containing demethylases (JmjC). The JmjC members were identified as KDM 2–7.16 PRMTs are divided into type I (PRMT1, 2, 3, 4, 6, and 8), II (PRMT5 and 9), and III (PRMT7), while arginine demethylases include JMJD6 and PAD4.17 Phosphorylation, a critical histone modification, involves adding a phosphate group by kinases.18 DNA methylation is primarily mediated by DNMT1, DNMT3a, and DNMT3b, which transfer the methyl group provided by SAM10 by covalent addition of a methyl group to the fifth carbon of the cytosine base,19 and DNA demethylase enzymes (erasers) including ten-to-eleven translocation (TET) proteins.20 Non-coding RNA (ncRNA) does not encode proteins but is involved in regulating a variety of biological processes in cancer progression, such as proliferation, apoptosis, migration, and invasion.10 ncRNA includes long non-coding RNAs (lncRNA) and small non-coding RNAs such as miRNA, snoRNAs, siRNA and piRNA.21 Epigenetic dysregulation can cause developmental disorders, autoimmune diseases, neurodegenerative diseases, psychological disorders, and cancer. In cancer, it plays critical roles in cancer stages such as carcinogenesis, metastasis, and drug resistance development, and thus can be a potential preventive and diagnostic marker.2,22| Type of modification | Place | Writer | Eraser | Reader |
|---|---|---|---|---|
| Acetylation | Histone | GNAT, P300/CBP, and MYST | Class I (HDAC 1, 2, 3, and 8), Class IIa (HDAC 4, 5, 7, and 9), Class IIb (HDAC 6 and 10), Class IV (HDAC 11), and Class III or sirtuins (SIRT1 to SIRT7) | Bromodomains |
| Methylation | Lysine | DOT1L, EZH2, G9a, and MLL1 | Lysine-specific demethylase 1 (LSD1, BHC110 or KDM1), lysine-specific demethylase 2 (LSD2), and jumonji domain-containing protein (JMJD) | Plant homeodomains (PHDs), WD40 repeat (WDR) domains, royal superfamily domains |
| Arginine | Type I PRMT enzymes (PRMT1, -2, -3, -4, -6, and -8), type II enzymes (PRMT5 and PRMT9), and type III enzymes (PRMT7) | PAD4 and JMJD6 | Tudor domains, WD40 repeat (WDR) domains, and plant homeodomains (PHDs) | |
| DNA | DNMT1, DNMT3a, and DNMT3b | Ten-eleven translocation (TET) | Methyl-CpG binding domain (MBD) family, Kaiso family, and SET- and ring finger-associated (SRA) domain family |
Benzimidazole is a bicyclic aromatic organic compound consisting of a benzene ring fused to a five-membered imidazole ring at the 4- and 5-positions of the imidazole ring. Its IUPAC name is 1H-benzimidazole and it is also known as 1,3-benzodiazole, benzoglyoxaline, iminazole, and imidazole.23–25 It is a weak base with sufficient NH-acidic to be soluble in aqueous alkali and has pKa values of 5.3 and 12.3 for pKa1 and pKa2, respectively. It appears as white tabular crystals with a molecular weight of 118.14 g mol−1.23,26 The benzimidazole nucleus is a promising scaffold with different biological activities. This importance began when Woolley discovered the structural similarity of 2-aminobenzimidazole with purine27 and when 5,6-dimethylbenzimidazole was discovered as a degradation product of vitamin-B12.28 In 1872, Hoebrecker was the first to synthesize the benzimidazole ring through the reduction and dehydration of 2-nitro-4-methylacetanilide (Fig. 1), and its tautomerism.29 Nowadays, the benzimidazole ring can be synthesized from o-phenylenediamine through condensation with aldehydes, ketones, and carboxylic acids and via the rearrangement of benzodiazepinones, quinoxaline derivatives, and 1,2,3-triazole.30 The 1, 2 and/or 5(or 6) positions of the benzimidazole ring are the most important positions to be substituted to obtain different biologically active compounds.31 Substituted benzimidazole can act as antimicrobial, antituberculosis, antiviral, antiulcer, anti-inflammatory, antidiabetic, anticonvulsant, antihypertensive, antimalarial, proton pump inhibitor, anti-HIV and anticancer agents.23,24,32,33 Benzimidazole has multiple mechanisms to act as an anticancer agent through the modulation of many epigenetics targets, as summarized in Fig. 2, and many other targets such as protein kinase CK2 inhibitors, topoisomerase inhibitors, tyrosine kinase inhibitors, antiangiogenic agents, checkpoint kinase 2 inhibitors, antiproliferative inhibitors, hypoxia-inducible factor inhibitors, interleukin-2-inducible T-cell kinase inhibitors, JNK inhibitors, dual KSP and aurora-A kinase inhibitors, and thioredoxin reductase inhibitors.34 Consequently, many benzimidazole derivative drugs have been approved by the FDA, such as albendazole, mebendazole, and thiabendazole as antihelmintics, anti-psychotic drugs such as pimozide,35 omeprazole and pantoprazole as proton pump inhibitors,36 astemizole as an antihistaminic, enviradine as an antiviral, candesartan and telmisartan as antihypertensives37 and binimetinib, bendamustine, and selumetinib as anticancer agents (Fig. 3).38
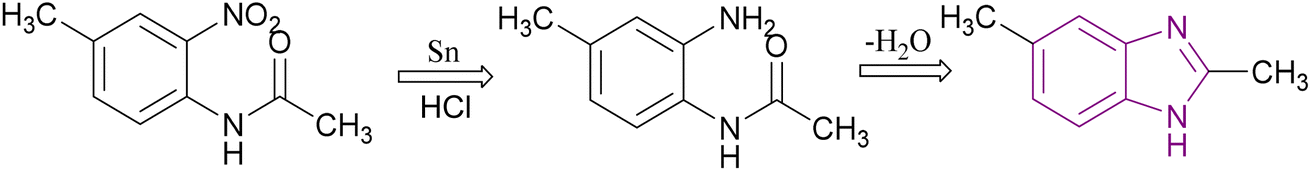 | ||
| Fig. 1 Synthesis of benzimidazole derivative by Hoebrecker. | ||
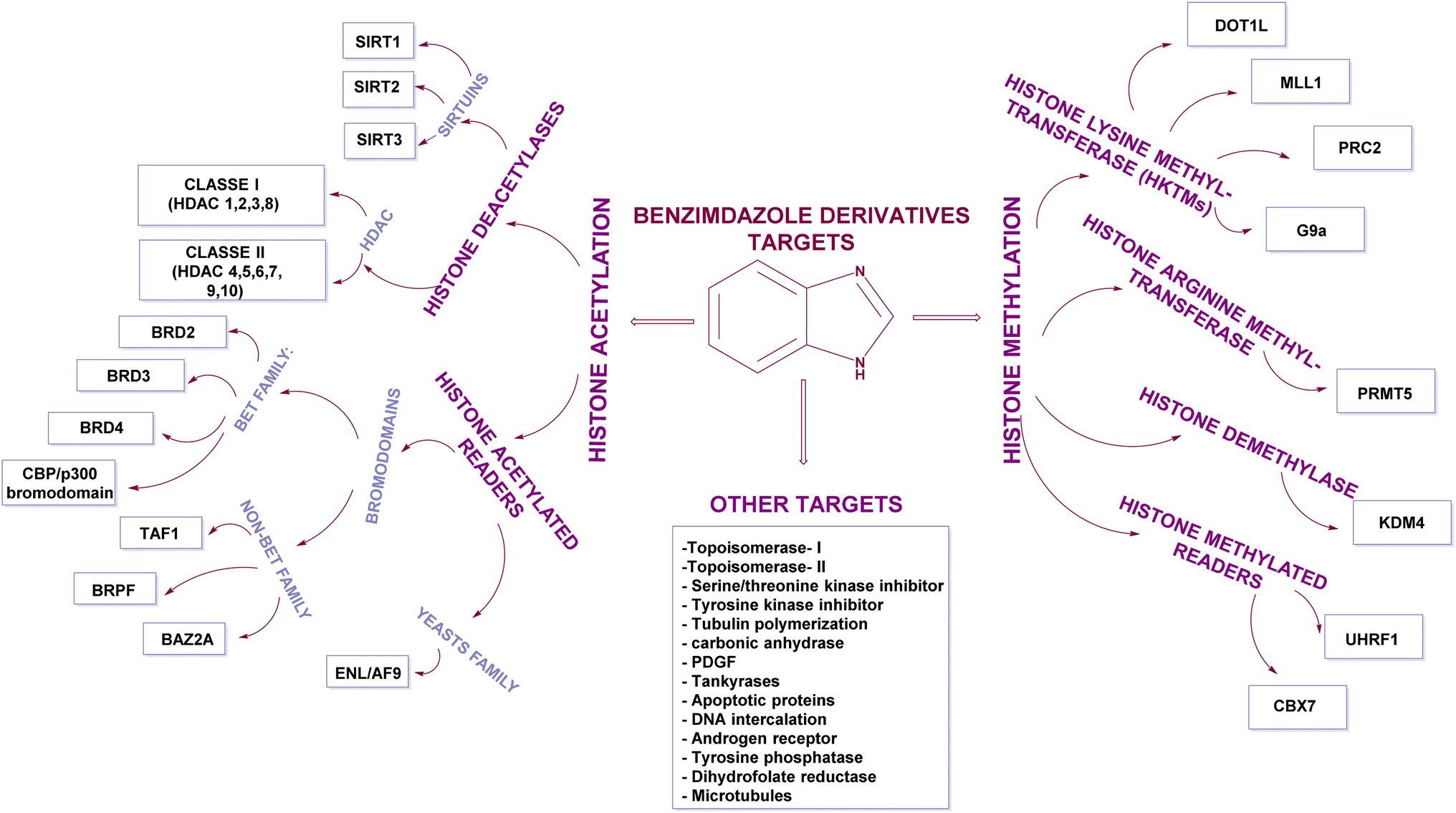 | ||
| Fig. 2 Representation of various targets of 1H-benzimidazolederivatives having anticancer effects. | ||
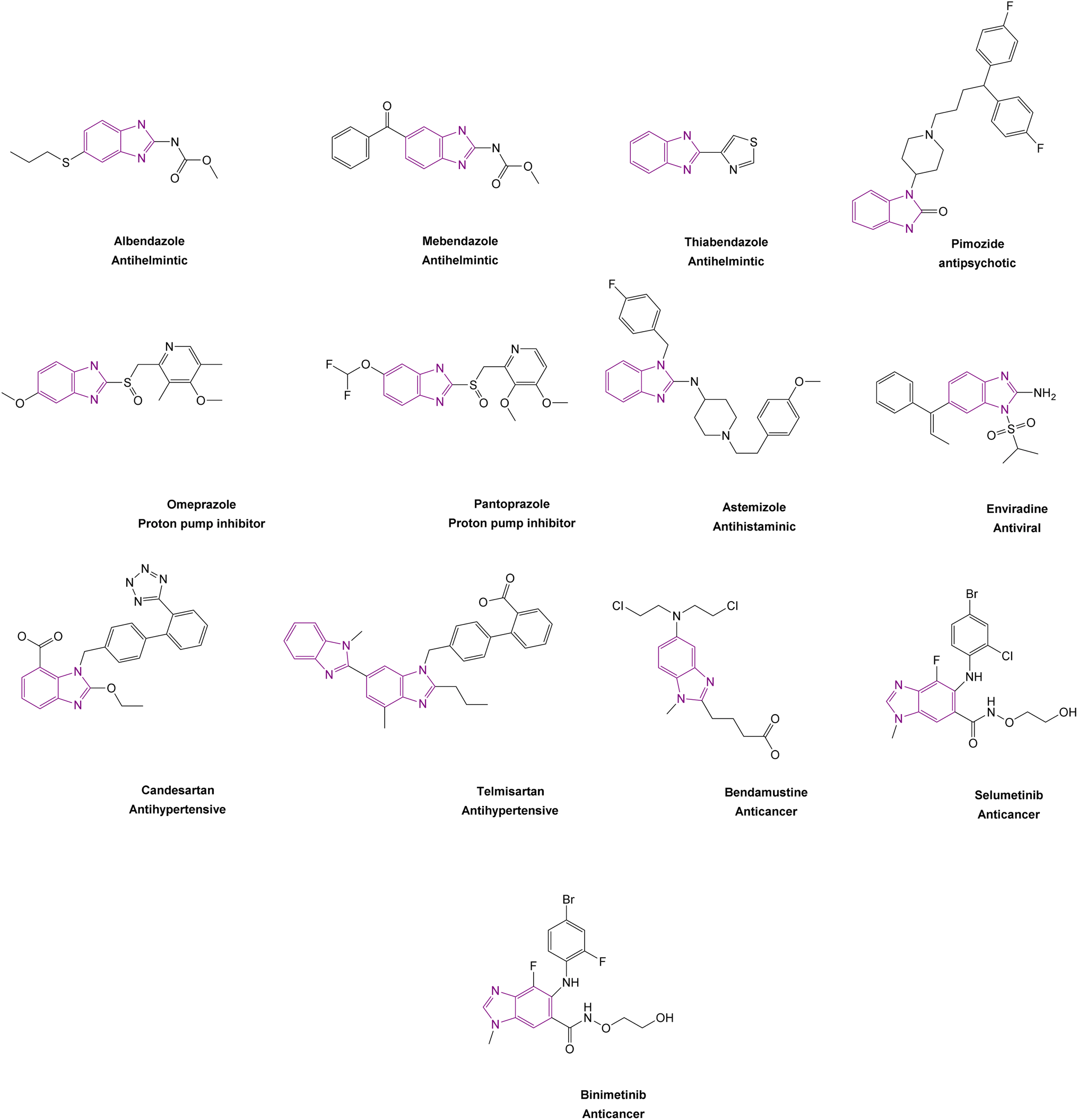 | ||
| Fig. 3 Structures of FDA-approved benzimidazole derivative drugs. | ||
There are leading review articles in the field of epigenetics and benzimidazole derivatives acting as cytotoxic agents. However, a collection of the benzimidazole derivatives that inhibit epigenetic targets is currently lacking. Thus, in this review, we aim to provide a comprehensive overview of all the benzimidazole derivatives that inhibit different epigenetic targets, as summarized in Table 2, such as histone deacetylases (HDACs), histone acetylated readers (CBP/P300 bromodomain, TAF1 bromodomain, BRPF bromodomain, and ENL), lysine methyl transferases (DOT1L, MLL1, PRC2, and G9a), arginine methyl transferases (PRMT), histone demethylases (JMJD and peptidyl arginine deiminases), histone methylated readers (UHRF1 and CBX7), and ubiquitin-specific protease 5 (USP5) and how their structure–activity relationship (SAR) affects their anti-proliferative activity.
| Benzimidazole derivative | Chemical structure | Epigenetic target | Mechanism of action | IC50/EC50/Kd/PIC50 (μM) | Tested cancer cell lines | Key findings | Ref. |
|---|---|---|---|---|---|---|---|
| a NA, not available. | |||||||
| Pracinostat (2) | 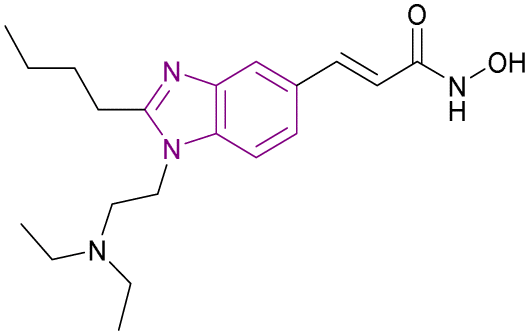 |
Pan-HDAC inhibitor | Enhances histone acetylation | HDAC1/2/3/8/4/5/7/9/6/10/11, IC50 = 0.028/0.027/0.019/0.048/0.016/0.021/0.104/0.024/0.247/0.023/0.024 | MV4-11 | It has been investigated in phase I and II clinical trials for treating hematologic malignancies and solid tumours. It has excellent oral bioavailability, safety, long duration of action, and good ADME. However, it has dose-dependent mild myelosuppression and gastrointestinal side effects | 68 and 69 |
| Ramos | |||||||
| PC-3 | |||||||
| A2780 | |||||||
| COLO 205 | |||||||
| Compound 10 | 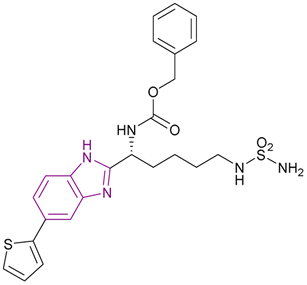 |
HDAC1 and HDAC6 | Enhances histone acetylation | HDAC1/HDAC6, IC50 = 0.20/0.48 | 293T cell line | Its PK profile showed metabolic stability and a t1/2 of 1.2 h. However, it has poor bioavailability because of the sulfamide unit | 73 |
| Compound 12 | 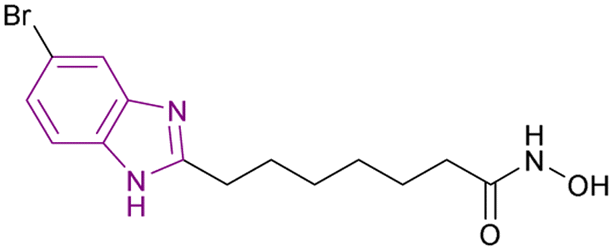 |
Pan-HDAC inhibitor | Enhances histone acetylation | (HDAC1/2/3/4/5/6/7/8/9/10/11, IC50 = 0.0202/0.0341/0.0064/0.279/0.0025/0.0019/0.313/0.282/0.0011/0.003/0.0334 | Skov-3 MiaPaCa | Less toxic side effects than vorinostat as well as its in vivo studies using the human pancreatic cancer MiaPaCa xenograft mouse model, resulted in 74% tumour growth inhibition | 74 |
| H295R | |||||||
| Mdamb231 | |||||||
| H69 | |||||||
| H23 | |||||||
| Compound 14 | 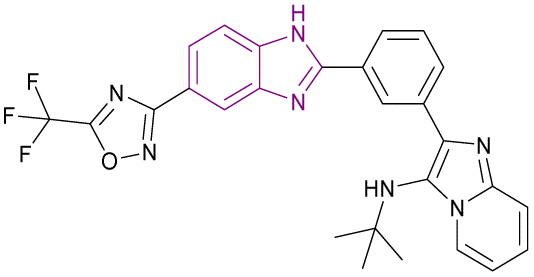 |
HDAC5 and SIRT | Enhances histone acetylation | HDAC5 IC50 = 0.31 | NAa | It can be beneficial in treating solid tumours and can be effective in treating sirtuin-associated diseases, including cancer, HIV, metabolic disorders, and neurological diseases | 75 |
| Downregulates 7 SIRT enzymes | |||||||
| Induces cell cycle arrest at G1/S phase | |||||||
| Reduces the expression of the oncogenic protein c-Myc, the anti-apoptotic genes BCL3 and BCL2, cyclin-dependent kinases (CDK2, CDK4, and CDK6), and cell cycle regulators (E2F1 and RB1 genes) but increases the expression of the proapoptotic proteins caspases 3 and 7 | |||||||
| Compound 15 | 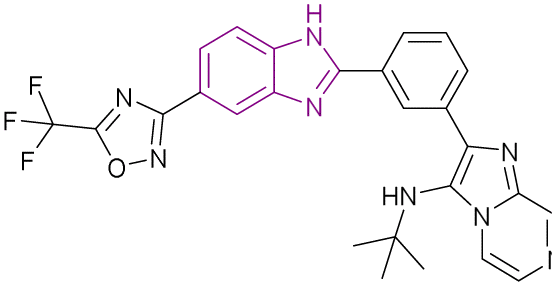 |
HDAC5 | Enhances histone acetylation | IC50 = 0.30 | NA | It can be beneficial in treating solid tumours | 75 |
| Induces cell cycle arrest at G1/S phase | |||||||
| Reduces the expression of the oncogenic protein c-Myc, the anti-apoptotic genes BCL3 and BCL2, cyclin-dependent kinases (CDK2, CDK4, and CDK6), and cell cycle regulators (E2F1 and RB1 genes) but increases the expression of the proapoptotic proteins caspases 3 and 7 | |||||||
| Compound 16 | 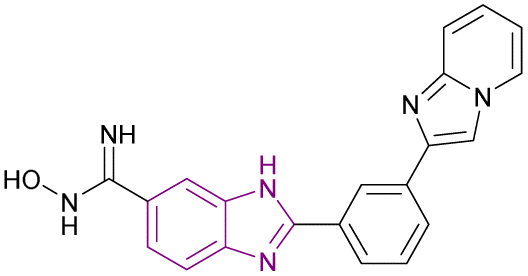 |
HDAC1, HDAC2 and SIRT | Enhances histone acetylation | HDAC1/HDAC2, IC50 = 0.55/0.4 | MCF7 breast cancer cells | It can be beneficial in treating solid tumours and can be effective in treating sirtuin-associated diseases, including cancer, HIV, metabolic disorders, and neurological diseases | 75 |
| Downregulates 7 SIRT enzymes | The non-small cell lung cancer cells A549 | ||||||
| Reduces the expression of the oncogenic protein c-Myc, the anti-apoptotic genes BCL3 and BCL2, cyclin-dependent kinases (CDK2, CDK4, and CDK6), and cell cycle regulators (E2F1 and RB1 genes) but increases the expression of the proapoptotic proteins caspases 3 and 7 | |||||||
| Ensulizole (18) | 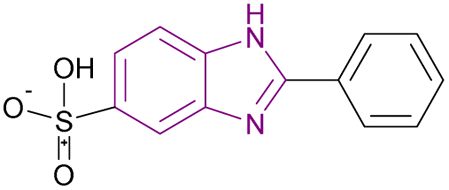 |
HDAC3 | Enhances histone acetylation | NA | HCT116 | Selective to HDAC3 | 94 |
| Compound 20 | 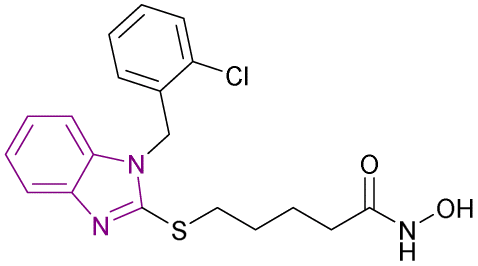 |
HDAC6 | Enhances histone acetylation and caused dose-dependent increase in the level of tubulin acetylation | IC50 = 0.09 | MCF-7 | Potent anticancer agent for selective HDAC6 inhibition and deserves further investigation | 96 |
| Compound 21 | 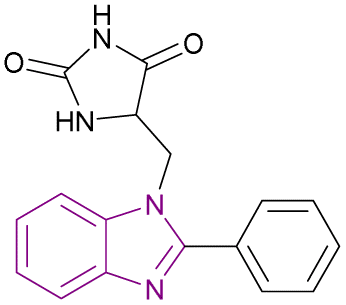 |
HDAC6 | Enhances histone acetylation | IC50 = 0.09735 | MOLT-4 | A Mannich base that fits perfectly fitting inside the binding site of HDAC6 enzyme | 97 |
| Compound 24 | 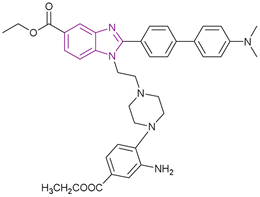 |
SIRT1, SIRT2, and SIRT3 | Inhibits SIRT1, 2 and 3 deacetylase activity | SIRT1/2/3, IC50 = 10.22/2.92/10.02 | NA | Good inhibitory activity against SIRT2 | 103 |
| BZD9L1 (31) | 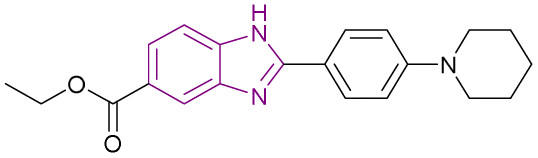 |
SIRT1 and SIRT2 | Inhibits SIRT1, and SIRT2 deacetylase activity and with a dose-dependent reduction in cell survival and migration | SIRT1/SIRT2, IC50 = 42.9/9.0) | HCT116 | It is highly selective and nongenotoxic and has no effect on cell cycle distribution or cellular senescence | 107 |
| HT-29-CRC | |||||||
| Roxyl-zhc-84 (33) | 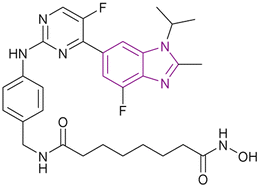 |
CDK/HDAC dual targeting | Enhances histone acetylation | (HDAC1/2/3/4/5/6/7/8/9/10/11, IC50 = 0.026/0.52/0.056/<10/<10/0.0059/<10/<10/<10/0.073/6.8) | MDA-MB-231 | It is sensitive to solid tumours with good kinase selectivity and a favourable pharmacokinetic profile. It outperformed vorinostat or abemaciclib in the in vivo tests using both the 4T1 and MDA-MB-468 mouse models | 114 |
| Inhibits cyclin-dependent kinase | MDA-MB-468 | ||||||
| Thus, it downregulates the antiapoptotic protein BCL2, upregulated p21 expression, induced caspase-3 cleavage, and caused cell cycle arrest at the G1 phase | OVCAR-5 | ||||||
| SK-OV-3 | |||||||
| Compound 36 | 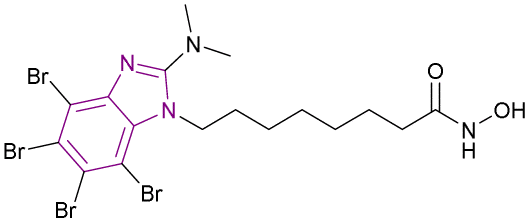 |
Multiple (HDAC1/HDAC6/CK2) | Enhances histone acetylation | HDAC1/HDAC6/CK2, IC50 = 13.7/8.98/5.89 | Jurkat | It has high cytotoxic activity, proapoptotic capability, mitochondria-targeting, and multidrug-circumventing properties | 41 |
| HCT-116 | |||||||
| MCF-7 | |||||||
| HL-60/vinc | |||||||
| HL-60/adr | |||||||
| HL-60 | |||||||
| Tinostamustine (38) | 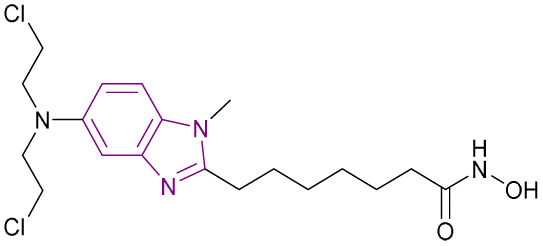 |
DNA/HDAC dual targeting | Alkylates DNA | HDAC1/2/3/4/5/6, IC50 = 0.017/0.01/0.025/0.0064/0.107/0.072 | HL60 | It was well tolerated with advanced solid tumours as had been showed in an open-label phase I/II study (NCT03345485). Also, it was well tolerated in relapsed/refractory (R/R) Hodgkin lymphoma (HL) patients as in cohort-expansion stage of a phase I study. The main side effects were haematological and thrombocytopenia, leading to treatment discontinuation | 115–119 |
| Enhances histone acetylation | Daudi cells | ||||||
| Triggers apoptosis | |||||||
| Reduces the expression of genes that control DNA repair | |||||||
| Compound 42 | 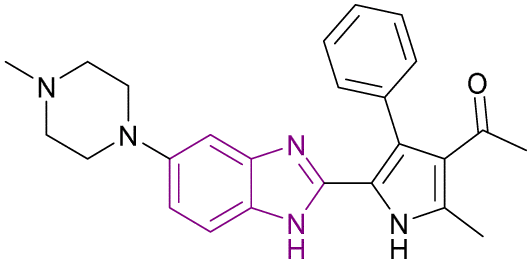 |
BRD4 (BD1) | Inhibits BET protein binding to acetylated histones | IC50 = 0.0501 | NA | It might be a potential candidate for the treatment of AML and some solid tumours. It has good metabolic stability and good oral bioavailability | 129 |
| Blocks transcriptional activity | |||||||
| Induces cell cycle arrest at G0/G1 phase through upregulation of p21 and apoptotic marker-cleaved PARP and downregulation of c-Myc | |||||||
| Inobrodib (48) | 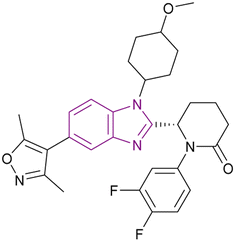 |
CBP/p300 | Blocks histone acetylation | CBP/p300, Kd = 0.0013/0.0017 | −22Rv1 | It has entered phase I/II clinical trials for treatment of AML, non-Hodgkin lymphoma, and MM (NCT04068597) and it is promising for the treatment of advanced prostate cancer | 132, 135 and 136 |
| -VCaP | |||||||
| Compound 51 | 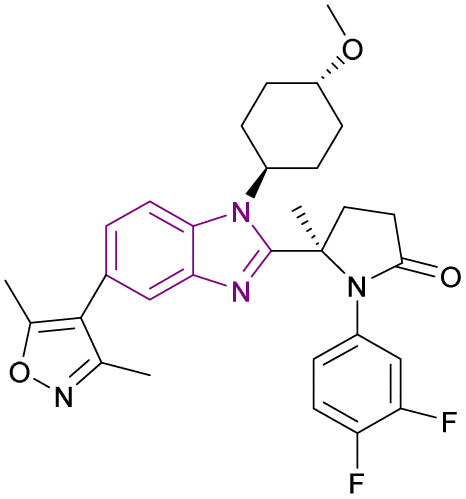 |
p300 bromodomain | Inhibits the binding to acetylated lysine residues on histone | IC50 = 0.060 | NA | It overcomes the defect of Inobrodib 48 defect of its high efflux rate and it is metabolically stable and has no hERG risk, indicating its safety for the heart | 139 |
| GNE-371 (52) | 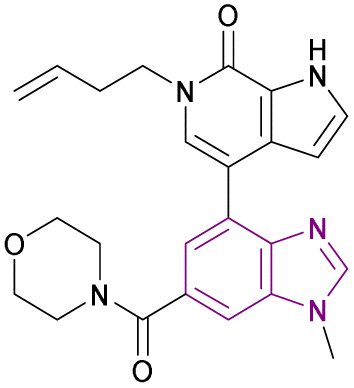 |
TAF1(2)-bromodomain | Inhibition of bromodomain-acetylated lysine binding | IC50 = 0.01 | NA | Exhibits antiproliferative synergy with JQ1 | 140 |
| GSK6853 (54) | 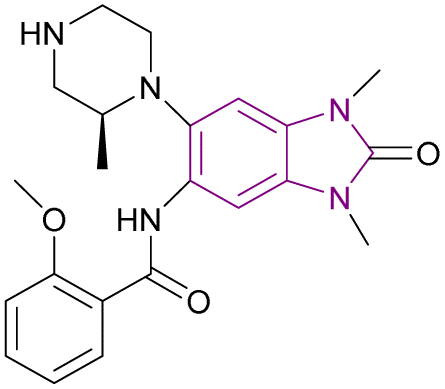 |
BRPF1 bromodomain | Inhibits BRPF1 bromodomain binding to acetylated lysine residues on histones | pIC50 = 8.1 | NA | Suitable for in vivo studies | 142 |
| Compound 58 |  |
BAZ2A bromodomain | Prevent bromodomain of BAZ2A from binding to acetylated lysine residues on histones | IC50 = 10 | NA | NA | 143 |
| Compound 60 | 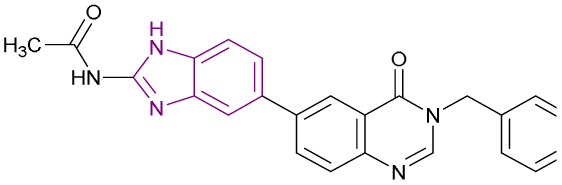 |
BRD4/PI3K/Aurora A | Inhibits BET protein binding to acetylated histones | PI3K IC50 = 0.01312 and Aurora A IC50 = 0.01019 | A549 | It has selectivity towards cancer cells superior to normal cells and provides better safety, enhanced efficacy, and delayed drug resistance | 128 |
| Blocks the transcriptional activity | H1975 | ||||||
| Upregulates the pro-apoptotic proteins cleaved-PARP and cleaved-caspase 9, while the anti-apoptotic protein Bcl-2 is downregulated | HCC827 | ||||||
| Induces cell cycle arrest in G2/M phase | |||||||
| Compound 63 | 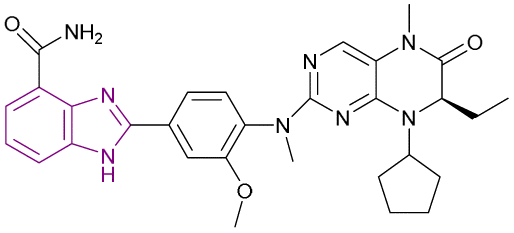 |
PARP1/BRD4/BRD4BD1/BRD4BD2 | Inhibits BET protein binding to acetylated histones | PARP1/BRD4/BRD4BD1/BRD4BD2, IC50 = 0.013/0.101/0.075/0.157 | SW1990 cells | It can be beneficial in the treatment of advanced pancreatic cancer | 144 |
| Blocks the transcriptional activity | Breast cancer MDA-MB-231 | ||||||
| Blocks Single-Strand Break (SSB) repair | |||||||
| Compound 65 | 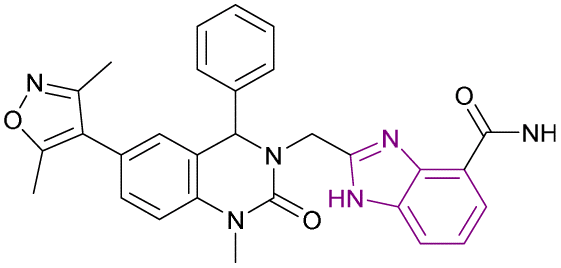 |
PARP1/BRD4 | Inhibits BET protein binding to acetylated histones | PARP1/BRD4, IC50 = 0.049/0.202 | CFPAC-1 | The inhibitory effects of compound 65 at a dose of 30 mg kg−1 on SW1990 xenograft tumour growth were more significant than that of JQ1, olaparib, and their combination | 145 |
| Blocks the transcriptional activity | Capan-1 | ||||||
| Blocks Single-Strand Break (SSB) repair | SW1990 | ||||||
| Thus, arrests the cell cycle at G0/G1 and G2/M phases, increases DNA damage, and reverses olaparib-induced adaptive resistance | MDAMB-231 | ||||||
| MDA-MB-468 | |||||||
| HCT116 | |||||||
| THP-11 | |||||||
| Compound 68 | 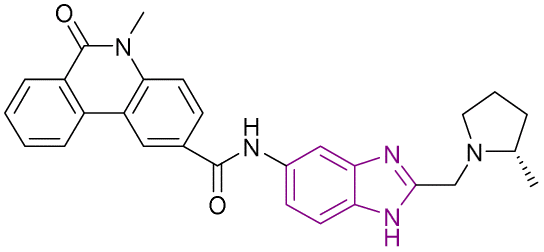 |
ENL | Blocks YEATS domain binding to acetylated lysine. Downregulates the expression of target gene MYC | IC50 = 0.0154 | MOLM-13 | It is a therapeutic target in leukemogenesis with exceptional target engagement in cellular context | 148 |
| MV4-11 | |||||||
| EPZ-5676 (70) | 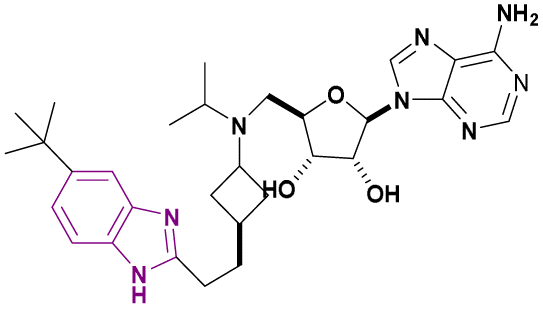 |
DOT1L | Inhibits H3K79 methylation | — | MV4-11 | It has recently entered clinical evaluation as a therapeutic agent for MLL-rearranged leukemia | 159,160 |
| Decreases HOXA9 and MEIS1 mRNA | |||||||
| Rabeprazole (71) | 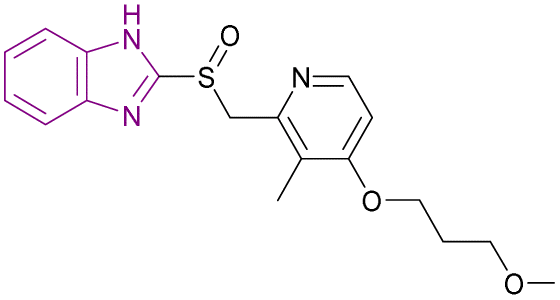 |
MLL1-WDR5 | Disrupting the MLL1-WDR5 interaction | IC50 = 0.49 | MLL-AF4 | More modifications are needed to repurpose the scaffold of substituted 2-pyridyl methyl/sulfinyl benzimidazole to eliminate proton pump inhibitory effects in the future | 161 |
| Significantly reduces MLL1's ability to methylate H3K4 | MLL-AF9 | ||||||
| Downregulates the expression of Hoxa9 and Meis1 genes | |||||||
| DC-PRC2in-01 (77) | 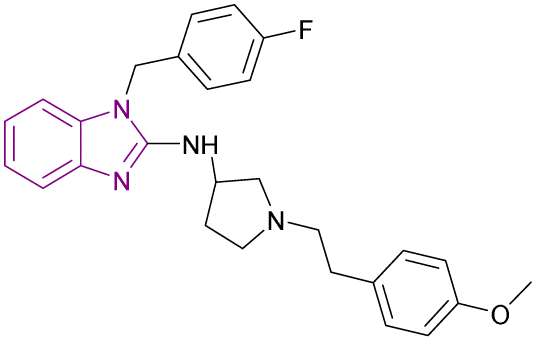 |
EZH2-EED | Inhibits the interaction of EZH2-EED by binding to EED | IC50 = 4.21 | SU-DHL-4 | A promising chemical scaffold for further development | 163 |
| Arrests cell cycle at the G0/G1 phase | Karpsa-422 | ||||||
| Pfeiffer cell lines | |||||||
| GA001 (78) | 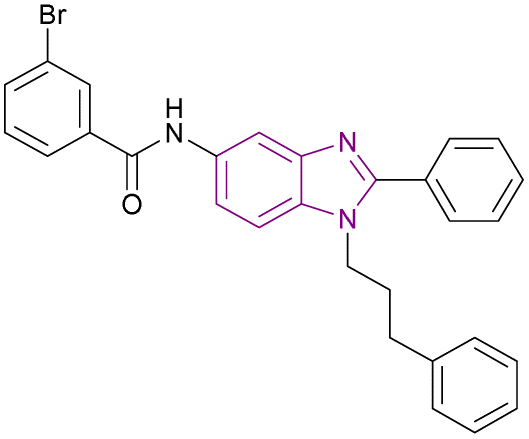 |
G9a | Blocks H3K9 methylation | IC50 = 1.32 | MCF-7 | It can help in breast cancer therapy | 164 |
| Induces autophagy and apoptosis | T-47D | ||||||
| Activates the tumour suppressor p53 by inhibiting G9a, which regulates p21 activation | MDA-MB-231 | ||||||
| MDA-MB-468 | |||||||
| MDA-MB-435 | |||||||
| Compound 83 | 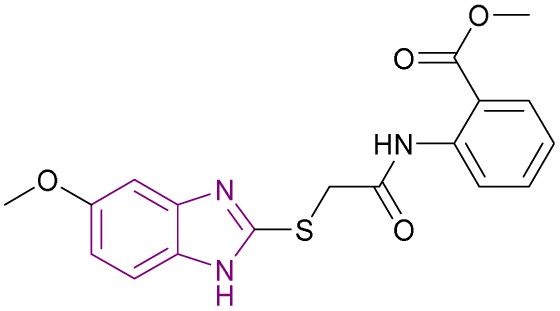 |
PRMT5 | Inhibits arginine demethylation | IC50 = 0.33 | MV4-11 | It can be beneficial in treatment of leukemia | 166 |
| Compound 84 | 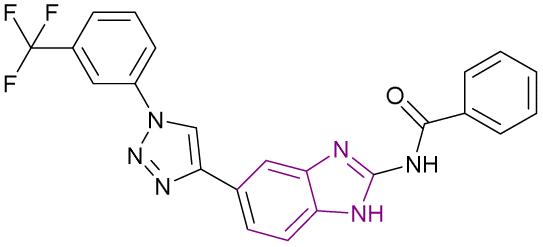 |
PRMT5 | Inhibits arginine demethylation | IC50 = 4.11 | MCF7 | It has prominent anticancer potency | 169 |
| DU145 | |||||||
| PC3 | |||||||
| HepG2 | |||||||
| Compound 88 | 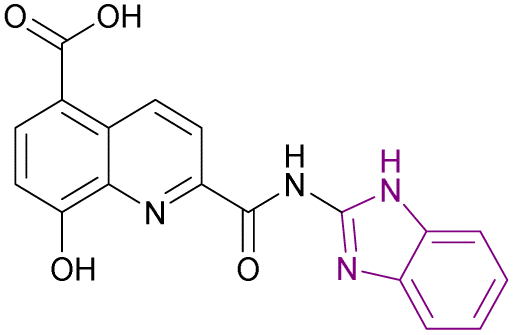 |
JMJD2A | Increases H3K9me2 and H3K9me3 levels | IC50 = 19.0 | HCT116 | A good starting point for the development of new JmjC inhibitors | 172 |
| MCF-7 | |||||||
| A549 | |||||||
| Compound 90 | 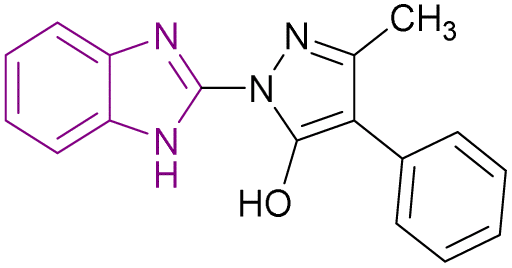 |
KDM4 | Increases H3K9 methylated levels | IC50 = 13 | LnCaP cells | It reduced the expression of genes regulated by the androgen receptor | 173 |
| -DU145 cells | |||||||
| -PC3 cells | |||||||
| Nocodazole (91) | 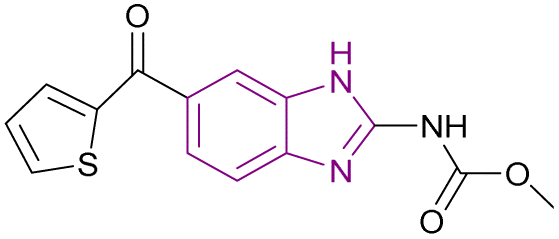 |
UHRF1 | Gradual loss of DNA methylation | NA | NA | NA | 175 |
| MS351 (93) | 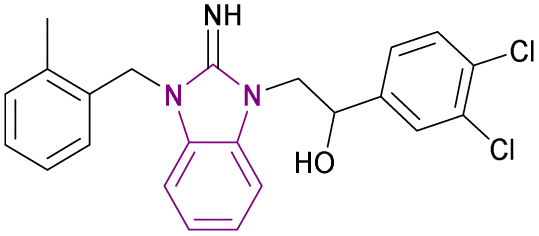 |
CBX7ChD | Disrupting recognition of H3K27me3 | Kd = 23.8 | NA | NA | 177 |
| Compound 94 | 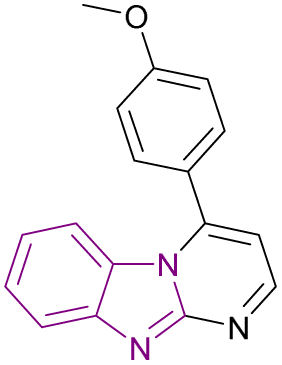 |
USP5 | Accumulating free polyubiquitin chains and disrupting ubiquitin homeostasis | Kd = 0.47 | MYCN-amplified | Selective towards cancer cell lines | 178 |
| MYCN-non-amplified neuroblastoma cells | |||||||
| Compound 96 | 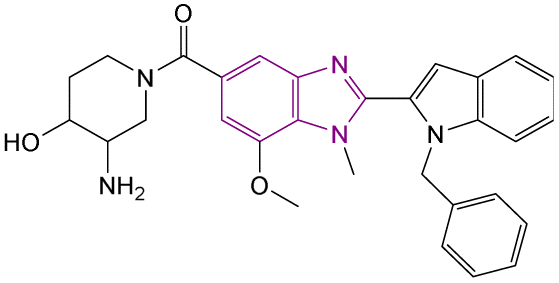 |
PAD4 | Blocking citrullination of histones | NA | 4T1 cells | The MTT assay showed potent activity | 180 |
| Compound 97 | 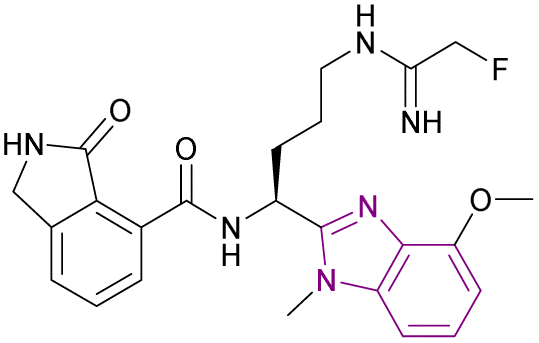 |
PAD2 | Blocking citrullination of histones | EC50 = 0.4 | NA | May be suitable for treating diseases in which PAD2 activity is dysregulated | 181 |
2 Benzimidazole compounds acting on histone modifications
2.1 Acetylation
The reversible addition and removal of the acetyl group in lysine is controlled by the activity of two characteristic enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs). Their actions lead to the formation of either the loose form of chromatin known as euchromatin or the condensed form known as heterochromatin, respectively, as shown in Fig. 4. HATs (writers) include the Gen-5-related N-acetyltransferase (GNAT) family, the p300 cAMP-responsive element-binding protein-binding protein (p300/CBP) family, and the MOZ/YBF2/SAS2/TIP60 (MYST) family. Regarding the CBP/p300 family, it consists of a HAT domain, a cysteine-rich motif, and a bromodomain, where the latter is considered one of the lysine-acetylated readers whose inhibition is involved in the treatment of cancer.39,40 The amino groups of the lysine residues in the N-terminal tails of histone and non-histone proteins, including tubulin and several transcription factors (e.g., p53, CREB, and NF-κB), are deacetylated by HDACs (erasers). The opened chromatin structure allows the cellular DNA to be more accessible to DNA-damaging agents. Accordingly, HDAC inhibitors are considered promising anticancer targets, given that the hypoacetylation of histones by HDAC leads to the development of several types of cancers.41–45 Fortunately, many benzimidazole derivatives have been found to act on most of these targets, showing great efficacy and potency when tested on cancer cell lines.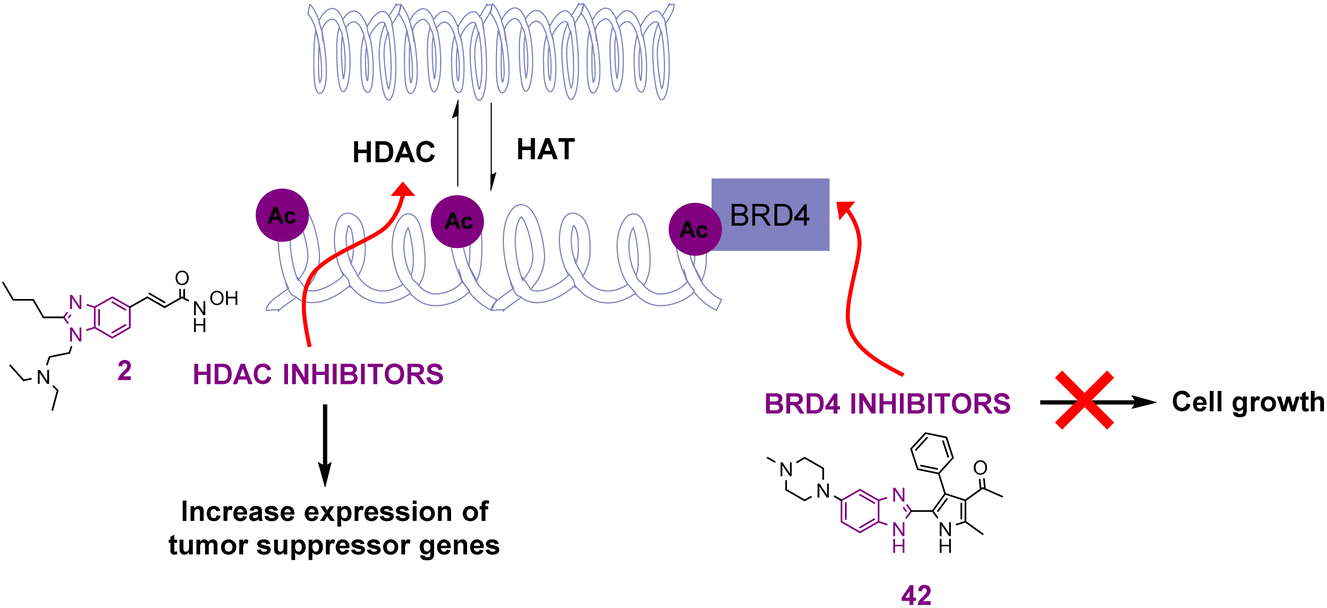 | ||
| Fig. 4 Effects of benzimidazole derivatives act as HDAC inhibitors and histone acylated reader (BRD4) on cancer cell growth and gene expression. | ||
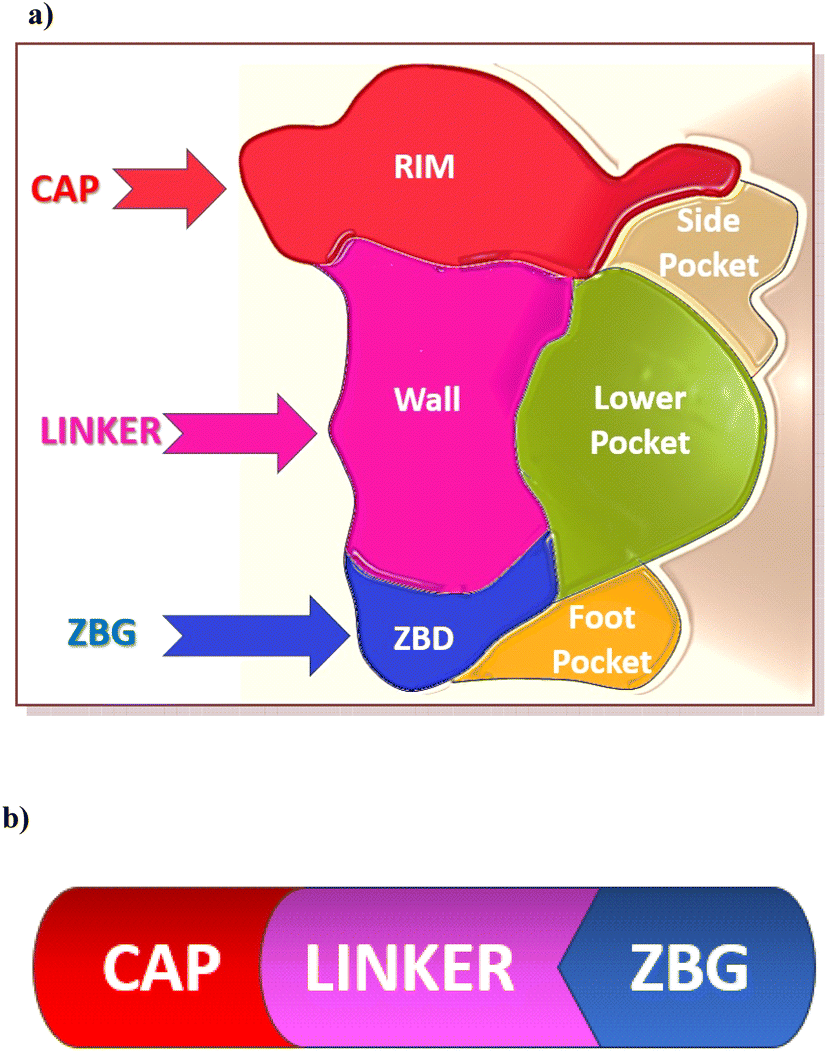 | ||
| Fig. 5 (a) HDAC isoforms structures showing rim, wall, and zinc binding groups with the additional subpockets called foot pocket in HDAC1-3, side pocket in HDAC8, and lower pocket in the HDACIIa isoforms. (b) Pharmacophore of the HDAC inhibitors. | ||
The pentacoordinate zinc ion binds to Asp178 (O δ 1), His180 (N δ 1), and Asp267 (O δ 1), whereas the acetyl moiety (carbonyl oxygen) of the substrate and a water molecule occupy the other two coordination sites. The N-acetyl group is attacked by a water molecule (a nucleophile) when HDAC is activated. The tetrahedral oxyanion intermediate, formed through the nucleophilic attack by water on the carbonyl, collapses to generate the free lysine and acetate products.51,52 Hydroxamic acid (a ZBG) forms an electrostatically favourable bidentate binding mode with two oxygen atoms to attach to the catalytic zinc cation, leaving the amide NH to create an extra H-bond connection with a histidine.53
The FDA-approved HDAC inhibitors include vorinostat (SAHA), belinostat (PXD-101), romidepsin (FK-228), panobinostat (LBH589), and chidamide (Fig. 6). However, they are all pan-HDAC inhibitors, inhibiting all the HDAC isoforms, except romidepsin and chidamide, which are HDAC 1–3 selective inhibitors. There is variation in the length of HDAC isoforms; the shortest is HDAC11, with 347 amino acids, while the longest is HDAC6, with 1215 amino acids.52 HDACs can be classified into two groups, group I (zinc-dependent amidohydrolases) and group II (NAD+-dependent hydrolases). Group I consists of classes I (isoforms 1, 2, 3, and 8), IIa (isoforms 4, 5, 7, and 9), IIb (isoforms 6 and 10), and IV (HDAC 11). Group II consists of class III or sirtuins (SIRTs), which comprise SIRT1 to SIRT7.54 In Class I HDACs, a second metal-binding site was discovered around 7 Å away from the zinc ion; depending on the salt present during crystallization, this site may be a potassium, calcium, or sodium ion.55
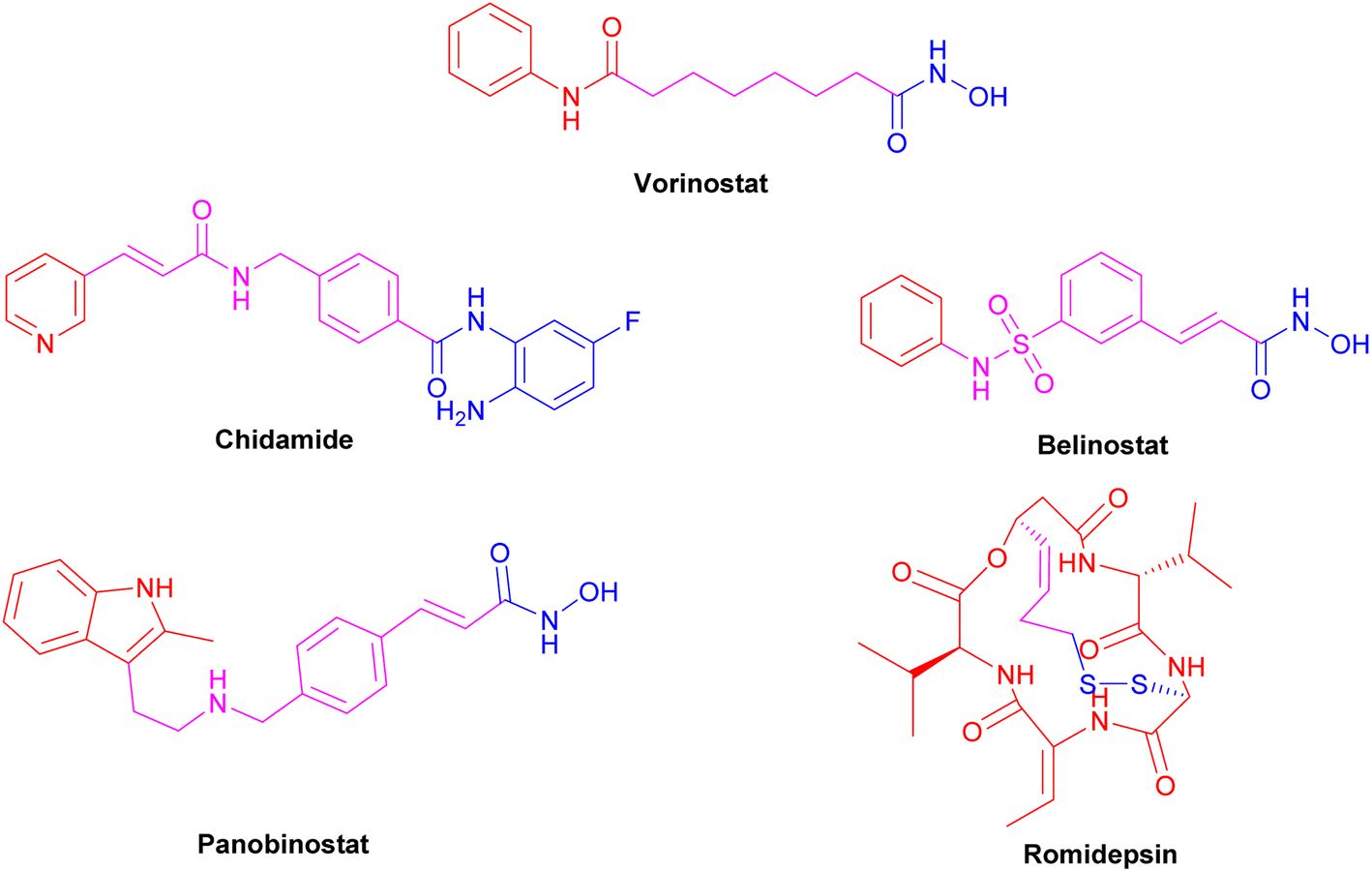 | ||
| Fig. 6 FDA-approved HDAC inhibitors. | ||
HDAC inhibitors act as anticancer agents by activating either the extrinsic pathway (death receptors and ligands up-regulation) or intrinsic apoptosis (pro-apoptotic upregulation and anti-apoptotic factors down-regulation). Consequently, HDAC inhibition leads to cell cycle arrest, inhibition of differentiation and proliferation, apoptosis, and necrosis.43,56
In addition, HDAC inhibitors target and/or modulate the expression of non-histone proteins such as transcription factors and regulators, chaperones, DNA repair enzymes, nuclear import regulators, inflammation/immune response mediators, and structural proteins.57 Thus, they play a promising role in neurodegenerative disorders,58,59 cardiovascular diseases,60 viral infections,61,62 and cancer.48,63–65
2.1.1.1 Benzimidazole derivatives as pan-HDAC inhibitors. In the following paragraphs, we elaborate on the group of benzimidazole derivatives 1–8 that function as pan-HDAC inhibitors (Fig. 7).
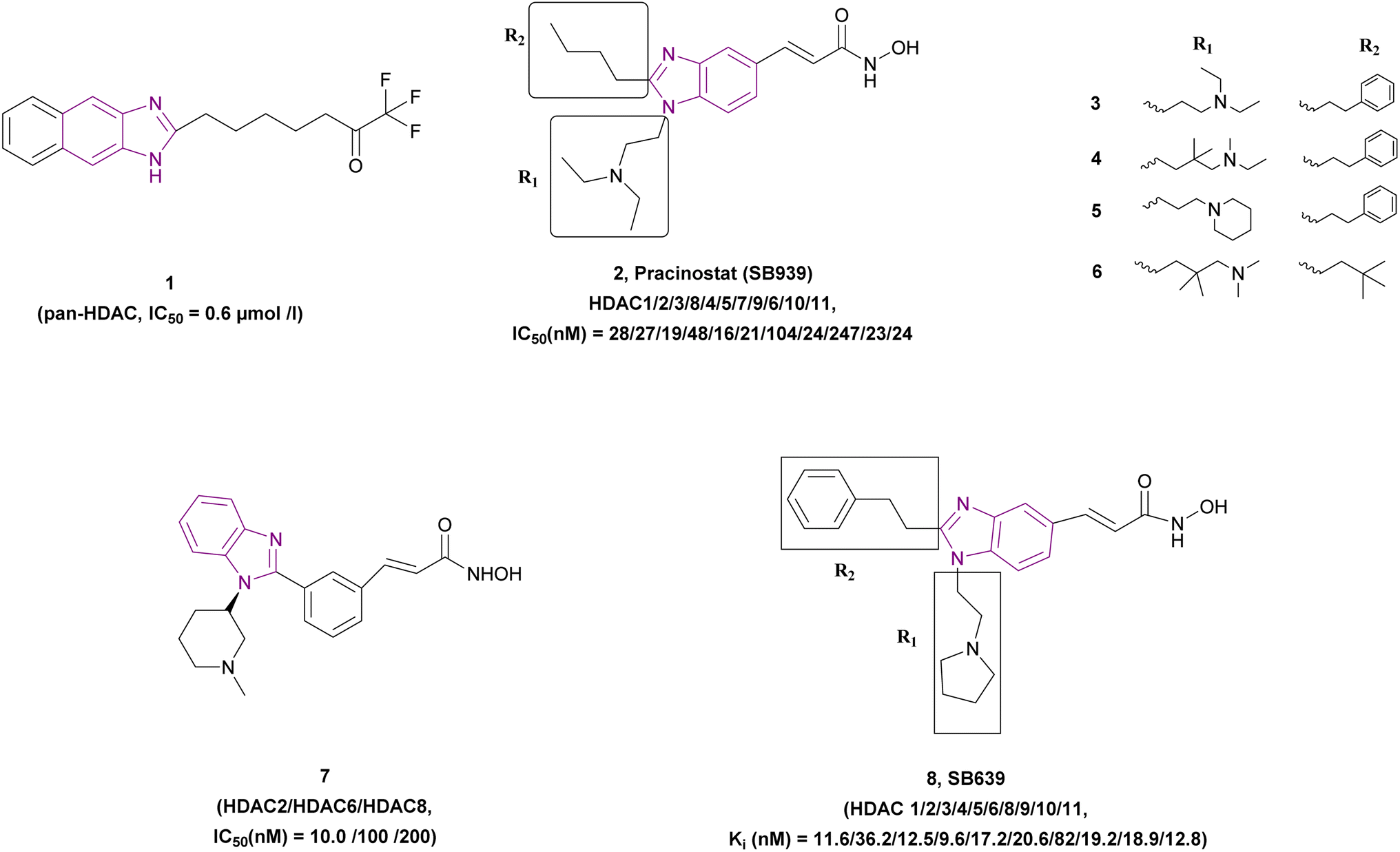 | ||
| Fig. 7 Chemical structure, pharmacophore and IC50 of pan-HDACIs 1–8. | ||
The screening of a natural compound library of 3719 compounds using a fluorescence-based HDAC activity assay showed that 1 is a potent non-hydroxamic pan-HDAC inhibitor. Wegener et al. showed that it has antiproliferative activity against embryonic childhood cancer such as neuroblastoma and medulloblastoma when tested in MYCN single-copy cells (SH-EP), MYCN stably transfected cells (WAC2), and MYCN-amplified cells [BE(2)-C, Kelly] cell lines. Also, it significantly induced the expression of the cyclin-dependent kinase inhibitor 1A gene (CDKN1A/p21WAF1/Cip1) and induced apoptosis. Moreover, it did not affect the viability of normal skin fibroblasts at 10-fold higher concentrations.66
Pracinostat 2, a dialkyl benzimidazole derivative, was a result of the optimization of compounds 3–5, which were metabolically unstable in human liver microsomal assays. With >1000-fold selectivity for HDAC Class 1 and 2 over Class 3,67 pracinostat is a very soluble, competitive, and effective pan-HDAC inhibitor,51 which formed essential interactions with HDAC1, as shown in the docking structure in Fig. 8. Additionally, it has been investigated in phase I and II clinical trials for treating hematologic malignancies and solid tumours.68 It has excellent oral bioavailability, safety, long duration of action, and good ADME. However, it has dose-dependent mild myelosuppression and gastrointestinal side effects.69 The SAR studies be Wang et al. highlighted that a basic center in the R1 group was crucial for optimal its biological activity. Additionally, the two-carbon linker of R1 has greater potency than its three-carbon counterpart. In the case of R2, a simple alkyl substitution at C-2 of the benzimidazole ring yields adequate potency and improved binding efficiency. Additionally, the cellular potency against COLO 205 would somewhat decrease if one of the ethyl groups from the R1 side chain was eliminated. Moreover, when tested on a variety of cancer cell lines, including MV4-11, Ramos, PC-3, and A2780, it exhibited antiproliferative properties, indicating that it is more effective against haematological cancers than solid tumours.68 After a pilot phase II study was carried out to investigate the efficacy and safety of a pracinostat 2 and 5-azacitidine combination in individuals with intermediate-2 or high-risk myelodysplastic syndrome (MDS), Quintás-Cardama et al. discovered that this combination was incredibly well tolerated in MDS patients.70 With respect to compound 6, the bulkiness of the tert-butylmethyl group at R2 may force it to adopt a more favourable conformation for effective interaction with Asp99, making it a competitive HDAC1 inhibitor, while the presence of a bulky gemdimethyl group in the R1 side chain increased the lipophilic interactions with HDAC1.68
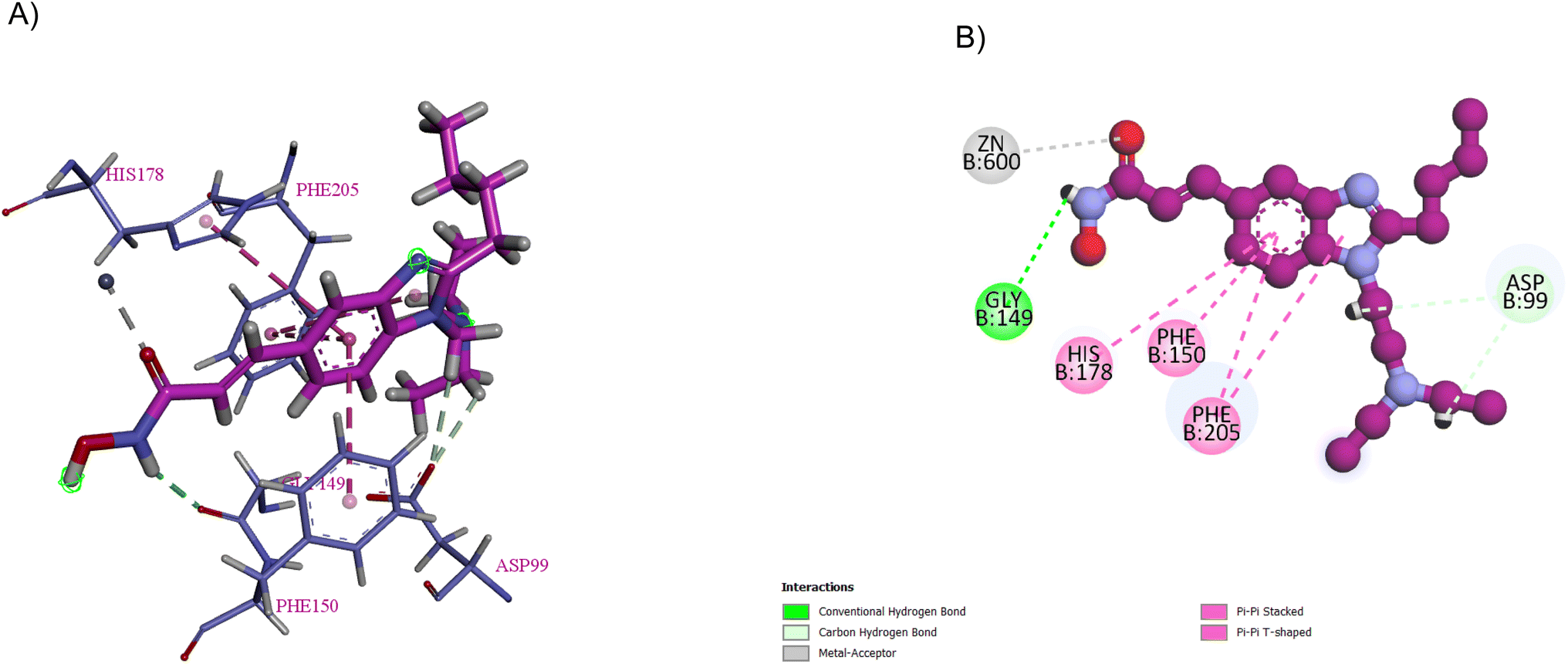 | ||
| Fig. 8 Pracinostat (in purple) interacts with HDAC1 through multiple interactions. Images were generated by Discovery Studio 4.5. (A) 3D interaction of pracinostat and HDAC1 residues. (B) 2D interaction of pracinostat and HDAC1 residues (in blue). It forms four hydrophobic interactions between the PHE205, PHE150, and HIS178 residues and the benzimidazole ring, and NH of the hydroxamic group forms hydrogen bond donor interactions with GLY149, while ASP99 forms two hydrogen bond acceptors with the alkyl groups. The carbonyl oxygen forms a metal–acceptor interaction with the zinc group. | ||
Bressi et al. synthesized compound 7, an N-hydroxy-3-[3-(1-substituted-1H-benzoimidazol-2-yl)-phenyl]-acrylamide analogue, which inhibited HDAC2 (IC50 of 10.0 nM), HDAC6 (IC50 of 100 nM), and HDAC8 (IC50 of 200 nM) with a good PK profile and p21waf induction. Its MOA was proven by H3 and H4 hyperacetylation in HL60 leukemia cells. In vitro studies showed its antiproliferative activity when tested in A549 lung (EC50 of 0.3 μM), HL60 leukemia (EC50 of 0.04 μM), and PC3 (EC50 of 0.05 μM) prostate cancer cell lines. In vivo studies using PANC-1 tumour-bearing mice, human HCT116 colon xenograft mouse model, and PC3 prostate model proved that 7 has anti-tumour activity.71
Wang et al. synthesized N-hydroxy-1,2-disubstituted-1H-benzimidazol-5-yl acrylamides, highlighting that compound 8 is a potent hydroxamic pan-HDAC inhibitor that inhibited HDAC1 with an IC50 of 0.035 ± 0.016 μM and inhibited class I, II, and IV HDAC isoenzymes in the nanomolar range, where the benzimidazole ring was a part of the cap moiety. It had antiproliferative activity when tested in HCT116, A2780, and PC3 cell lines with IC50 values (μM) of 0.20, 0.19, and 0.15, respectively. It is orally active and has good drug-like properties. According to SAR, hydrophobic and bulky groups were preferred for R1 with a short, flexible linker. Both enzymatic and cellular activities were significantly improved with a monobasic amine and a two-carbon linker compared to the compounds with longer linkers. In the case of R2, the compounds with a directly attached ring or an α-branch were not potent, whereas that with α-unsubstituted phenylalkyl chains was more potent.72
The optimization of compound 9, a lysine-derived sulfamide, resulted in compound 10, which was synthesized by Manku et al., who replaced the amide group with a benzimidazole scaffold, an isosteric heterocycle, given that compound 9 was unstable due to amidic bond cleavage (Fig. 9). Compound 10 is an HDAC1 (IC50 of 0.20 μM) and HDAC6 (IC50 of 0.48 μM) inhibitor with a sulfamide group as the ZBG and benzimidazole as its cap moiety. Interestingly, the N-carbobenzyloxy in the linker was responsible for the activity against both HDAC1 and HDAC6, given that the linear linker was selective towards HDAC6. The in vitro studies in the 293T cell line showed antiproliferative activity with an IC50 of 0.62 μM. Its PK profile showed metabolic stability and a t1/2 of 1.2 h. However, it has poor bioavailability because of the sulfamide unit.73
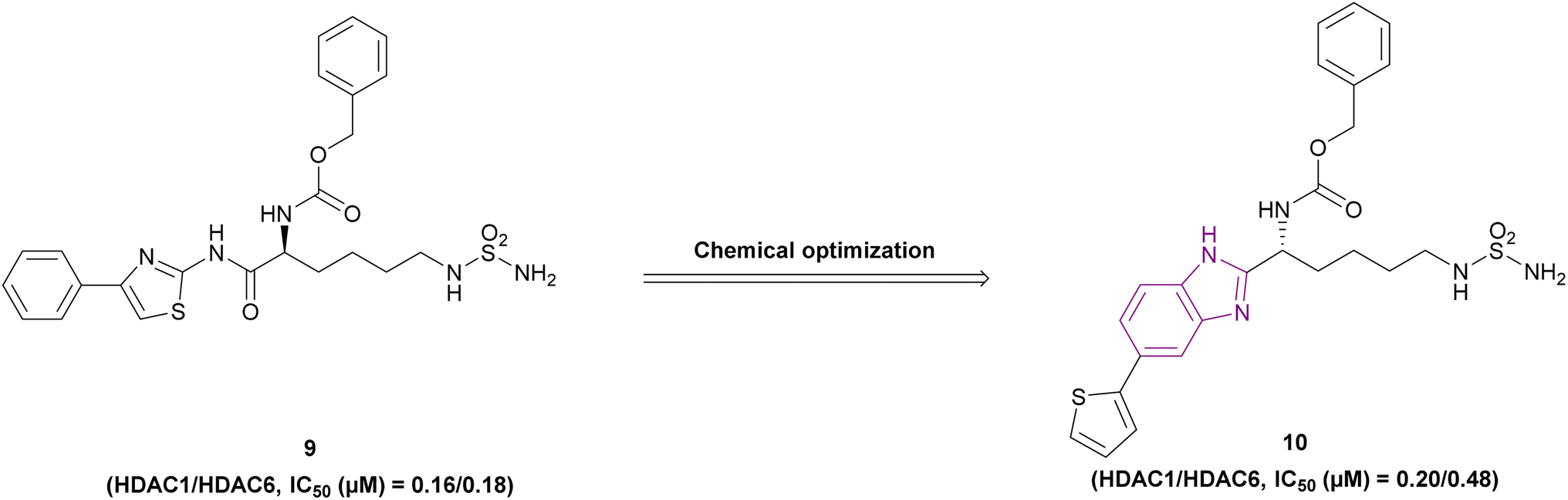 | ||
| Fig. 9 Chemical structures and IC50 of HDAC1 and 6 inhibitors 9 and 10. | ||
The metabolic studies for vorinostat 11 showed its instability due to the cleavage of its benzamide bond, generating toxic secondary metabolites. A series of benzimidazole-based HDAC inhibitors was synthesized through a series of reactions, including the Suzuki coupling reaction. Among them, compound 12, an optimized analogue of vorinostat obtained through cyclization (Fig. 10a), is considered a pan-HDAC inhibitor targeting HDAC 3, 5, 6, 9, and 10 and showed less toxic side effects than vorinostat 11 rather than exhibiting better results in in vitro studies. The SAR studies showed that a six-carbon linker was more potent than a five-carbon linker, and substitutions at the 5-position were more favorable than at the 4-position. Moreover, halogen substitutions (Cl, F, and Br) at the 5-position improved the cell activity more than electron-donating groups such as 5-methoxy and 5-dimethylamino, and the imidazole[4,5-c] pyridine system instead of the benzimidazole ring, as the cap group, eliminated the activity. Furthermore, additional groups on N-1, such as methyl, cyclopropyl, and phenyl, had deleterious effects. Compound 12 was chosen for in vitro studies instead of compound 13 (Fig. 10b) due to its better solubility, regardless of the enhanced cell activity of compound 13. As a result, compound 12 was tested in the human ovarian cancer SKOV3 cell assay (IC50 of 0.27 μM) and other different cell lines, as well as in in vivo studies using a human pancreatic cancer MiaPaCa xenograft mouse model, resulting in 74% tumour growth inhibition.74
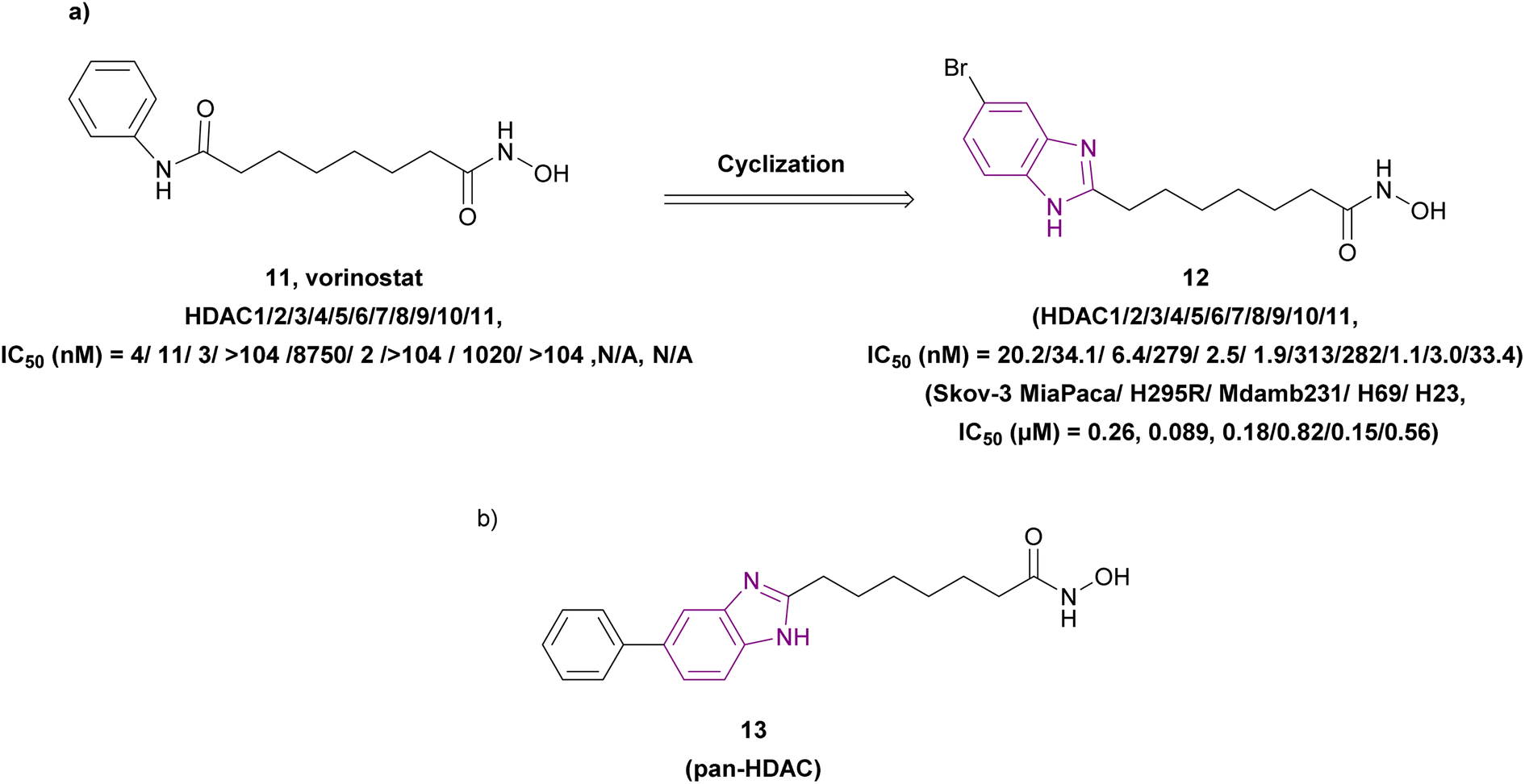 | ||
| Fig. 10 Chemical structures and IC50 of HDAC inhibitors 11–13. | ||
Rigidification of the flexible alkyl chain of vorinostat by replacing it with a benzimidazole linker enhanced its potency and selectivity. El-Awady et al. synthesized compounds 14–16, which are imidazopyridine derivatives (Fig. 11), resulted from the post-transformation of the Groebke–Blackburn–Bienaymé [4 + 1]-cycloaddition reaction products and suggested that they could be beneficial in treating solid tumours. Compounds 14 and 15 are potent HDAC-5 inhibitors (IC50 = 0.31 μM and 0.30 μM, respectively) with the trifluoromethyloxadiazole group (TFMO) as a nonchelating metal-ZBG group and induce cell cycle arrest at the G1/S phase. Compound 16 showed selectivity towards HDAC-1 and HDAC-2 (IC50 of 0.55 and 0.4 μM, respectively), showing significant anti-tumour activity when tested in MCF7 breast cancer cells and A549 non-small cell lung cancer cells. It had no activity towards HDAC-5 because it replaces the trifluoromethyloxadiazole group with N-hydroxyl amidine. Also, it did not inhibit human CYP450s 2D6 and 2C9 and had moderate kinetic aqueous solubility with lower stability in human liver microsomes. These compounds reduced the expression of the oncogenic protein c-Myc, the anti-apoptotic genes BCL3 and BCL2, cyclin-dependent kinases (CDK2, CDK4, and CDK6), and cell cycle regulators (E2F1 and RB1 genes) but increased the expression of the proapoptotic proteins caspases 3 and 7. However, compounds 14 and 16 reduced transcription factor NF-kB expression, while compound 15 enhanced its expression. Moreover, compounds 14 and 16 downregulated 7 SIRT enzymes (Class III HDACs). Thus, they can be effective in treating sirtuin-associated diseases, including cancer, HIV, metabolic disorders, and neurological diseases.75
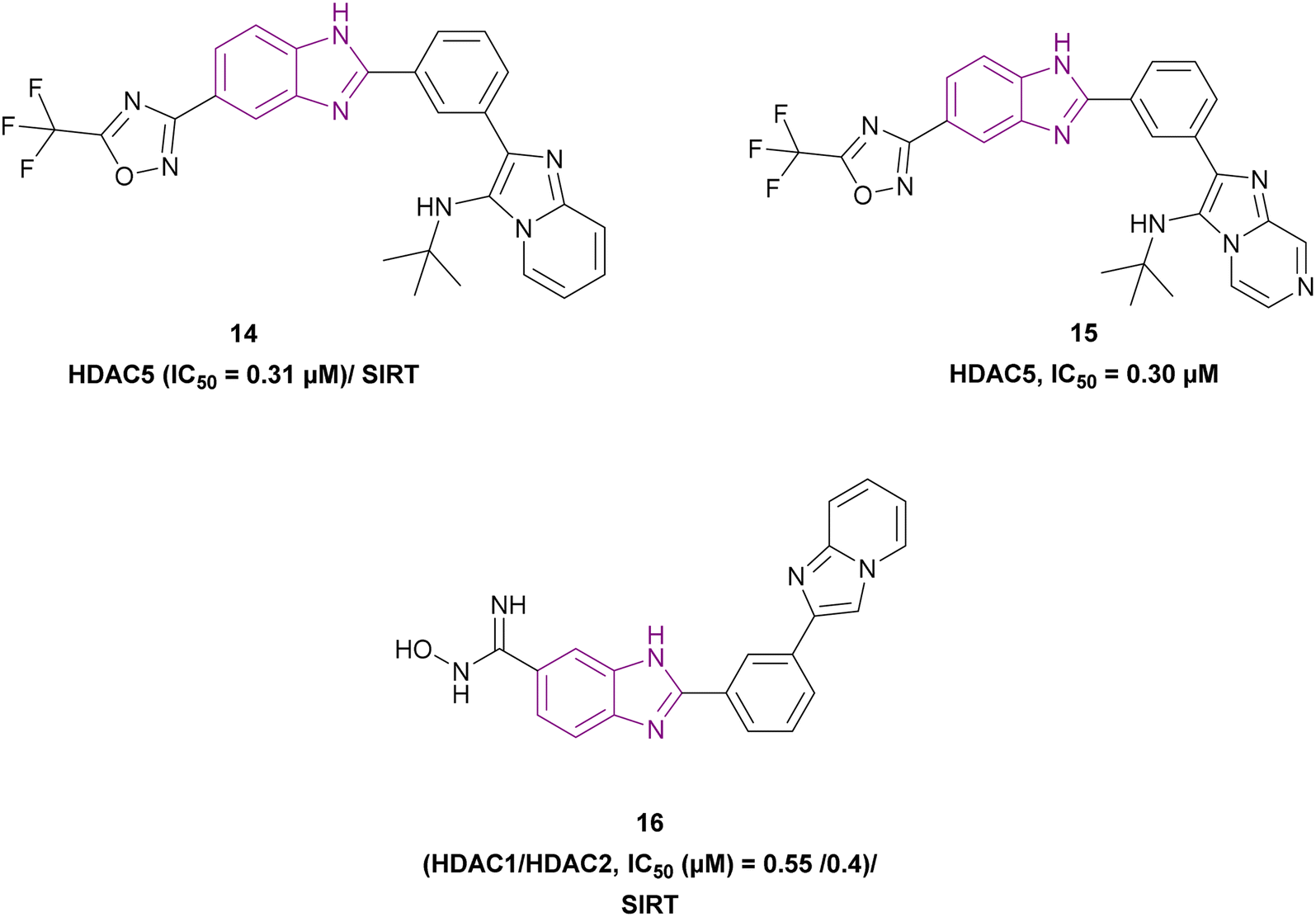 | ||
| Fig. 11 Chemical structures and IC50 of HDAC and SIRT inhibitors 14–16. | ||
2.1.1.2 Benzimidazole derivatives as selective HDAC inhibitors. HDAC-selective inhibitors could reduce side effects and be more specific, given that certain HDAC isoforms have a direct correlation with certain types of illnesses.76 Pan-HDACIs make up the majority of hydroxamic acid-based HDACIs,77 and their hydroxamic moieties are responsible for side-effects such as nausea, thrombocytopenia, anaemia, and other metabolic abnormalities due to their strong chelation with the zinc ion. Thus, researchers are trying to find different ZBGs with weaker binding and greater selectivity.53
Importantly, the foot pocket (HDAC1-3), lower pocket (HDACIIa), and side pocket (HDAC8) binding groups are essential for selectivity given that they are not present in all isoforms. The homological identities and high sequence similarity of Class I HDACs are considered a challenge in the development of HDAC1/2/3-selective compounds. Currently, most synthesized selective compounds are HDAC8 inhibitors.76
Class I HDACs produce inflammatory cytokines, which contribute to innate immunity and possess an additional internal cavity (foot pocket) supposed to be used for water entry and acetic acid removal.52 In this class, the HDAC channel is deeper and narrower than that of class IIb.78 High expression levels of HDAC1, 2, and 3 have been shown to be associated with gastric and ovarian cancers.79 HDAC1 was significantly overexpressed in various cancers, including prostate, gastric, breast, pancreatic, and colon cancers. Also, HDAC1 interacts with several non-histone protein-forming complexes. For example, HDAC1 acts as a pivotal component in its interaction with the tumour suppressor protein p53.80
HDAC2 is overexpressed in various malignancies, including prostate, gastric, non-small lung, colon, hepatocellular, and renal cancer,81 and is a crucial target for cancer and neurodegenerative diseases. The 14 Å foot pocket in HDAC2 can occupy benzamides well, such as chidamide and entinostat. Interestingly, the aromatic substitution of benzamide inhibitors at 5-position resulted in further entry into the foot pocket and a pronounced increase in the HDAC2 inhibitory potency.82
HDAC3 shares ∼50% sequence homology with HDAC1 and HDAC2. The divergence is mainly in the C-terminal region, with HDAC3 containing a domain for binding to the nuclear NCoR/SMRT complexes.83 HDAC3 inhibitors are effective for the treatment of inflammatory diseases, diabetes mellitus, cardiac diseases, HIV, different neurodegenerative disorders, rheumatoid arthritis, cancer, etc. The benzamide scaffold mainly as ZBG, has great selectivity towards HDAC3. However, synthesizing an effective compound against HDAC3 is still challenging.84
HDAC6 is the largest in size85 and the only enzyme in the HDAC family that has two deacetylase domains and a zinc finger domain. HDAC6 catalyzes the deacetylation of acetyl groups from non-histone proteins such as α-tubulin, Hsp90, and cortactin86 and is involved in the control of cell motility and VEGFR regulation in cancer cells.87 It regulates cellular viral RNA sensing and promotes the angiogenesis and migration of endothelial cells.88 Compared to the other HDACs, the rim of HDAC6 is shallower and wider. Therefore, compounds such as citarinostat, which have huge, rigid cap groups, can fit into the HDAC6 rim area easily.46 The bulky and lipophilic cap and substituted phenyl unit as a linker are selective for HDAC6 over other isozymes more than using an alkyl chain as the linker.86 It was reported that oxazoles attached to hydroxamate (ZBG) increased the potency and selectivity towards HDAC6.89 The X chromosome carries the gene for HDAC8, the only known HDAC that has the ability to function as a monomer.90 It has a larger surface opening and a wider active site pocket than HDAC1-3 (ref. 91) and is overexpressed in advanced neuroblastoma.92 In addition, HDAC11 is overexpressed mainly in rhabdomyosarcoma.93
Compounds 17–23, as shown in Fig. 12, are selective HDAC inhibitors. CID-18150975 17 was revealed to have the most selective inhibitory effect against class I HDAC inhibitors, particularly HDAC1, due to the presence of a benzamide functional group, which is a zinc binding group.80
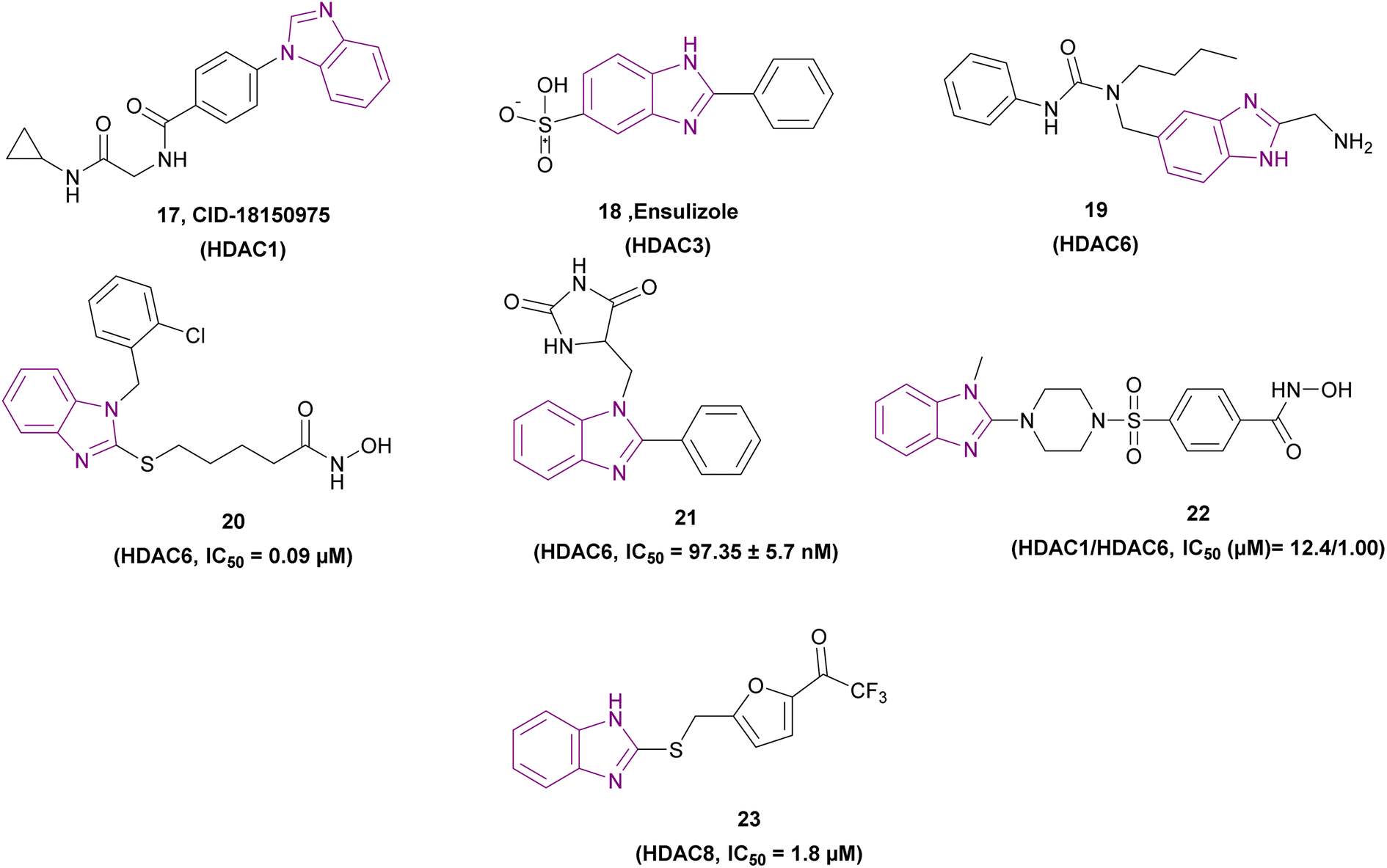 | ||
| Fig. 12 Chemical structure and IC50 of selective HDACIs 17–23. | ||
Ensulizole 18 is a selective HDAC3 inhibitor. In the HCT116 viability experiment, its IC50 was 13.39 ± 2.80 μM, while its efficacy was 70%.94
The modification of Nexturastat A by Alves Avelar et al. involved substituting a fragment of (1H-benzoimidazol-2-yl)-methylamine in place of the hydroxamic acid (a ZBG). This resulted in compound 19, which could serve as a lead structure for the synthesis of new selective non-hydroxamic HDAC6 selective inhibitors. The fragment of (1H-benzoimidazol-2-yl)-methylamine was selected as a potential ZBG through a structure-based virtual screening of designed libraries, and then evaluated with molecular docking and molecular dynamics simulations. The free amino group of the methylamino group and the non-alkylated imidazolic nitrogen, both of which demonstrated interactions with the zinc ion. Given that the HDAC6 binding site is shallow and wide, it could accommodate its cap group, but class I HDACs have a tighter hydrophobic channel.95
Compound 20 is a selective HDAC6 inhibitor with an IC50 of 0.09 μM and consists of a benzimidazole cap and four carbon-chain-containing thioether linkers, providing antiproliferative effects when tested against the MCF-7 cancer cell line and causing a dose-dependent increase in the level of tubulin acetylation.96
Mansour et al. synthesized compound 21, a Mannich base, through an aminoalkylation reaction. It is a potent HDAC6 inhibitor consisting of the benzimidazole nucleus as the capping moiety, the hydantoin moiety as the non-hydroxamate ZBG, and the CH2 group as a linker. Both CCRF-CEM and MOLT-4 cell lines were susceptible to the cytotoxic effects of compound 21. However, the MOLT-4 cell line was more susceptible, with an IC50 of 3.66 μM. The SAR studies showed that phenyl benzimidazole scaffold was more active than methyl benzimidazole, which was more active than unsubstituted benzimidazole. Also, the hydantoin derivatives were more potent than the thiohydantoin analogues.97
Compound 22, a 2-benzazolyl-4-piperazin-1-ylsulfonylbenzenecarbohydroxamic acid derivative, is a selective HDAC6 inhibitor that induces cell cycle arrest in the S-phase, has antiproliferative activity with IC50 values of 1.49 μM and 6.1 μM in HCC4017 and HCC4018, respectively, and selectivity towards cancer cells. Its structure formed of [CO(NHOH)] as a zinc-binding group, a piperazine-sulfonylbenzene moiety as the linker, and 1-methylbenzimidazole as the cap group. The catalytic pocket surface of HDAC6 and benzimidazole fitted perfectly, indicating a greater inhibitory effect for HDAC6 over HDAC1. The compounds that were meta-substituted exhibited no biological activity. The elimination of the methyl group that was substituted on the imidazole ring led to the complete elimination of cellular activity because of the reduced permeability of the cells. Less potent than the well-known HDACIs, the compounds containing a hydroxamic acid functionality in the para-position of the benzene ring demonstrated good HDAC inhibitory and antiproliferative properties. However, their HDAC6 selectivity was higher.98
The ZBG-based pharmacophore model for HDAC8 was used to filter a virtual screening of about 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 chemicals. Among the 3 HITs, only compound 23 with a trifluoroacetyl group as ZBG was chosen for further research because it selectively inhibited HDAC8 over HDAC1 and HDAC4. Compound 23 exhibited anti-proliferative action when tested in the MDA-MB231 and HCT116 cell lines.56
000 chemicals. Among the 3 HITs, only compound 23 with a trifluoroacetyl group as ZBG was chosen for further research because it selectively inhibited HDAC8 over HDAC1 and HDAC4. Compound 23 exhibited anti-proliferative action when tested in the MDA-MB231 and HCT116 cell lines.56
2.1.1.3 Benzimidazole derivatives as sirtuins inhibitors. Class III HDACs called sirtuins use NAD+ as a cofactor to remove acetylated groups from histones. By breaking the bonds that hold NAD+ and niacinamide ribosomes together, sirtuins transfer the acetylated groups from proteins to ADP-ribose, and then release the deacetylated products.99 They have been divided into seven human isoforms (SIRT1-SIRT7). Nearly all SIRT1, SIRT6, and SIRT7 are found within the nucleus. SIRT3- SIRT5 are called mitochondrial sirtuins, and SIRT2 is the main component in the cytoplasm. In the catalytic core area, SIRT2 can almost represent the structural basis of each isoform. The catalytic core of SIRT2 consists of a large and a small domain. The large domain contains an inverted Rossmann fold, which is a typical NAD+(H) binding site. The small domain contains a helical module and a zinc finger module. The two domains are joined by a lysine channel and three large-domain polypeptide chains, all of which form a large groove. This junctional groove includes the NAD+ binding site, which has three subpockets (A, B, and C). The ADP-ribose moiety binds to sub-pocket A. Nicotinamide-ribose is accommodated in the B sub-pocket, while during catalysis, it occupies the C sub-pocket, allowing the acetylated group to transfer from the Lys residue to the ribose portion of the substrate.99,100 Sirtuins regulate biological pathways, such as cell cycle and apoptosis, and are associated with a variety of age-associated diseases such as type II diabetes, obesity, and Alzheimer's disease.99,101,102
1,2,5-Trisubstituted benzimidazole derivatives 24 and 25–27 are shown in Fig. 13 and 14, respectively, which act as inhibitors of sirtuins. Compound 24 was the most potent SIRT1, SIRT2, and SIRT3 inhibitor, followed by compound 25, and then compound 26. The planar benzimidazole scaffold occupied the adenosine binding site (pocket A) and ribose binding site (pocket B) of SIRT2. The N,N-dimethylbenzylamine substituent, a potent electron-donating basic terminal group, increased the sirtuin inhibitory activity. The side chain of ethyl piperazinyl benzoate was located deep inside the nicotinamide binding site, which is important for controlling the activity of the enzyme. In a comparison study, compound 27, which has a linear octyl chain instead of ethylpiperazinyl benzoate, was less potent than compound 24.103
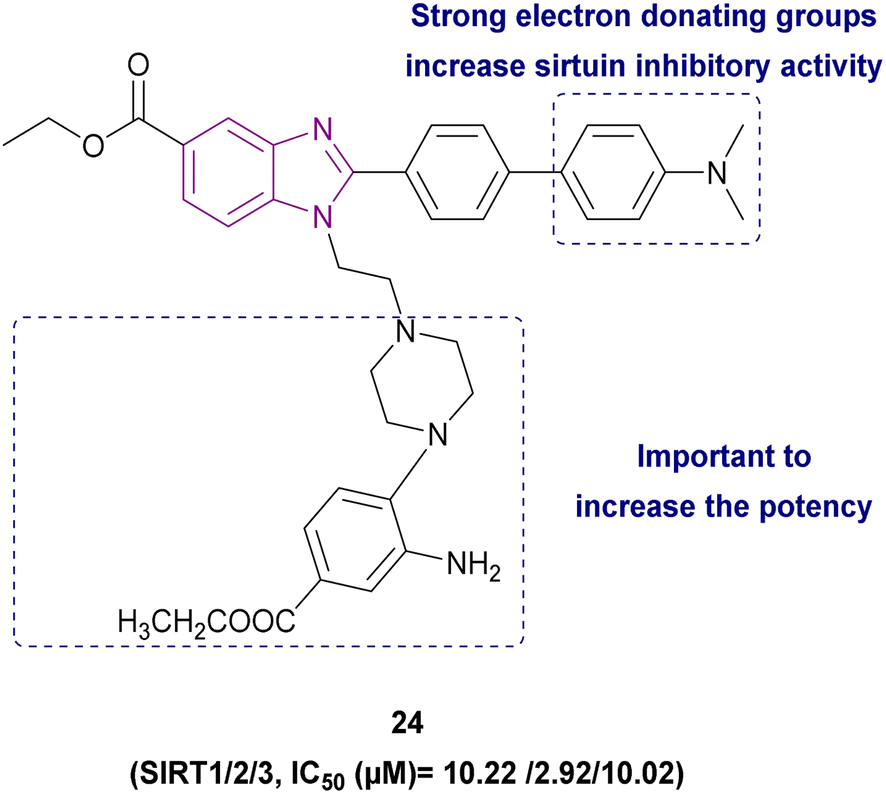 | ||
| Fig. 13 Chemical structure, SAR and IC50 of SIRT inhibitor 24. | ||
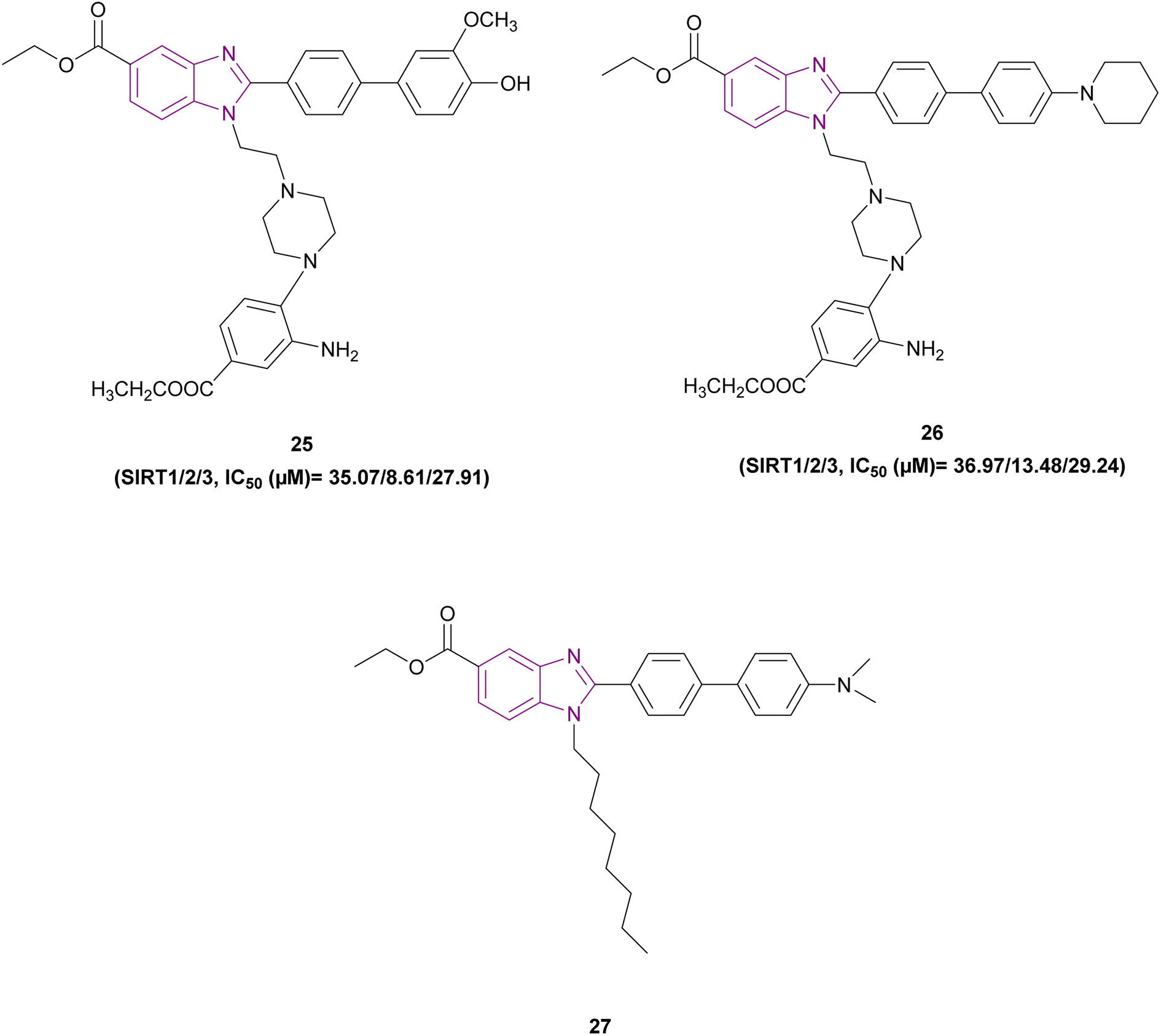 | ||
| Fig. 14 Chemical structure and IC50 of SIRT inhibitors 25–27. | ||
Compounds 28–31 are selective sirtuin inhibitors, as illustrated in Fig. 15. Yoon et al. used green chemistry to synthesize compound 28, which is a non-toxic SIRT1 and SIRT2 inhibitor. Additionally, when it was screened against several cancer cell lines derived from HCT-116, MDA-MB-468, and CCRF-CEM cell lines, it was the most effective anti-proliferative compound. Regarding the SAR of these candidates, it was found that the dimethylamino, a potent electron-donating group, has the best sirtuin inhibitory activity, and its removal significantly reduced its activity. The substituted benzene ring of the benzimidazole core was stabilized through the π–π stacking interactions between the imidazole group of His187 and the benzene ring of Phe235. The phenyl ring of the dimethylaminobenzene substituent as well as the N from the dimethylamino group with Val233 and Phe119, respectively, formed lone-pair oxygen–π interactions.104
 | ||
| Fig. 15 Chemical structure, SAR and IC50 of selective SIRT inhibitors 28–31. | ||
Compound 29, an amino acid derivative of substituted benzimidazole, is an SIRT1 inhibitor with an IC50 of 0.77 μM and % inhibition of 89.99%, together with SIRT2 and SIRT3 inhibition of 15% and 20%, respectively. It has antiproliferative activity against human cancer cell lines (HepG2 and MCF7). The benzimidazole moiety formed π–π or π–cation interactions with the binding site. The phenyl and indole groups were important for the binding affinity and the inhibitory activity, respectively. The 4-OMe function group was necessary for the inhibitory activity of SIRT1. Also, replacing it with 2-Cl or 4-F decreased the stability of the compound.105
Compound 30, a benzimidazole derivative, is an SIRT1 (IC50 of 58.43 μM) and SIRT2 (IC50 of 45.12 μM) competitive inhibitor with NAD+ cofactor. It formed H-bond, hydrophobic, and mild polar interactions with the receptor. The H-bonds between H from the CYS324 and LYS287 residues in the pocket and N from the dimethylamino group helped to stabilize the binding complex. In vitro studies in breast cancer cell lines MDA-MB-468 and MCF-7 showed its good antiproliferative activity due to its potent SIRT2 inhibition. SAR studies showed that the compounds with a 2-phenylbenzimidazole moiety and a strong electron-donating group (dimethylamino) in the para-position have more potential antitumour activity than that with weaker electron-donating groups such as methyl, hydroxyl, and methoxyl groups. The bulkier piperidine moiety instead of the dimethylamino group was unable to fit into the cavity at the end of the pocket.42
BZD9L1 31 is a potent, competitive, and reversible SIRT1/SIRT2 inhibitor with a greater affinity towards SIRT2. The strong electron-donating groups substituted at the phenyl ring, such as piperidinyl group 31 and the dimethylamino group, gave the best inhibitory activity and helped in stabilizing the benzimidazole core. In the case of replacing piperidinyl with methoxy, hydroxyl, methyl, or tert-butyl, it caused poor inhibition given that they shifted from the binding site with different binding orientations. Replacing the piperidinyl with the morpholinyl group weakened the inhibitory activity due to the importance of the basicity of the side chain.106 It had strong antiproliferative effects against HCT116 and HT-29-CRC cells, with a dose-dependent reduction in cell survival and migration. It is highly selective and nongenotoxic and has no effect on the cell cycle distribution or cellular senescence. In HCT116 cells, the proapoptotic genes BAX, BCL2, and GADD45A were upregulated, while TRAF2, a prosurvival gene, was downregulated. However, it had no effect on TRAF2 gene expression in the HT-29 cell line. At lower concentrations, cleaved PARP, a biological indicator of apoptosis, was seen in HCT-116 cells and HT-29. Only in HCT-116 cells, the p21 expression was reduced, while the PUMA protein was activated, given that both of them were not expressed in the p53 mutant cell line HT-29.107
2.1.1.4 Benzimidazole derivatives as dual-targeting HDAC inhibitors. Nowadays, multitarget-directed ligands (MTDLs) have marked importance, especially for complex multifactorial diseases such as diabetes, neurodegenerative syndromes, cardiovascular diseases, and cancer. They may be a new way to reduce drug resistance. Merged-pharmacophore mode, non-cleavable linked-pharmacophore mode, fused-pharmacophore mode, and cleavable linked-pharmacophore mode are the most common strategies for the synthesis of MTDLs.108 MTDLs are compounds consisting of more than one pharmacophore, each having a different target, which can be beneficial for cancer treatment.109 Multidrug targets are an alternative approach to combination therapy with the advantage of overcoming the problems associated with combination therapy, such as patient compliance, simplified dosing schedules, more predictable pharmacokinetics and pharmacodynamics, solubility/bioavailability issues, drug–drug interactions, and decreasing toxicity.110,111 For HDAC inhibitors to be a part of MTDLs, only their cap group could be replaced without impairing their potency due to the crucial effect of ZBG in the HDAC inhibitory activity, which could be useful in solid tumour treatment.57,78 This is because they resist treatment through some mechanisms such as signaling proteins, thioredoxin expression, cell cycle proteins, NF-kB expression, and apoptosis-related proteins.112 However, there are some challenges associated with dual inhibitors including poor pharmacokinetics due to their large molecular weight given that they are synthesized through either a pharmacophore-linked or -fused design technique. The second difficulty is in optimizing the synergistic therapeutic efficacy of the drug in vivo and maximizing its safety profile. Moreover, most of the dual epigenetic inhibitors are alkyl hydroximic acid-based HDAC inhibitors. However, the alkyl hydroximic acid scaffold has pharmacological liabilities, often resulting in short pharmacokinetic half-lives, poor oral bioavailability and toxicity.113
Huang et al. linked vorinostat 11 (an HDAC inhibitor) and abemaciclib 32 (a CDK inhibitor), resulting in compound 33, which is a CDK/HDAC dual-targeting hybrid inhibitor, and also targets JAKs (Fig. 16). To achieve HDAC inhibition, hydroxamic acid is essential for good zinc binding and activity, as in vorinostat, and the CH2–NH–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)– moiety acts as a linker. Moreover, the 2-aminopyrimidine moiety is responsible for CDK inhibition, as in abemaciclib 32. Compared to the pan-HDAC inhibitor vorinostat, compound 33 inhibited HDAC classes I, II, and IV with significantly greater selectivity. Moreover, it showed anti-tumour activity when evaluated in MDA-MB-231, MDA-MB-468, OVCAR-5, and SK-OV-3 cell lines, and considerably outperformed vorinostat or abemaciclib in the in vivo tests using both the 4T1 and MDA-MB-468 mouse models. Also, it downregulated the antiapoptotic protein BCL2, upregulated p21 expression, induced caspase-3 cleavage, and caused cell cycle arrest at the G1 phase. Interestingly, compound 33 was sensitive to solid tumours given that it inhibited the phosphorylation of JAK1 in the MDA-MB-231 and MDA-MB-468 (TNBC) cell lines, which activated STAT3-stimulated antiapoptotic signaling and limited the effect of HDAC inhibitors on solid tumours. Additionally, it had good kinase selectivity and a favourable pharmacokinetic profile.114
O)– moiety acts as a linker. Moreover, the 2-aminopyrimidine moiety is responsible for CDK inhibition, as in abemaciclib 32. Compared to the pan-HDAC inhibitor vorinostat, compound 33 inhibited HDAC classes I, II, and IV with significantly greater selectivity. Moreover, it showed anti-tumour activity when evaluated in MDA-MB-231, MDA-MB-468, OVCAR-5, and SK-OV-3 cell lines, and considerably outperformed vorinostat or abemaciclib in the in vivo tests using both the 4T1 and MDA-MB-468 mouse models. Also, it downregulated the antiapoptotic protein BCL2, upregulated p21 expression, induced caspase-3 cleavage, and caused cell cycle arrest at the G1 phase. Interestingly, compound 33 was sensitive to solid tumours given that it inhibited the phosphorylation of JAK1 in the MDA-MB-231 and MDA-MB-468 (TNBC) cell lines, which activated STAT3-stimulated antiapoptotic signaling and limited the effect of HDAC inhibitors on solid tumours. Additionally, it had good kinase selectivity and a favourable pharmacokinetic profile.114
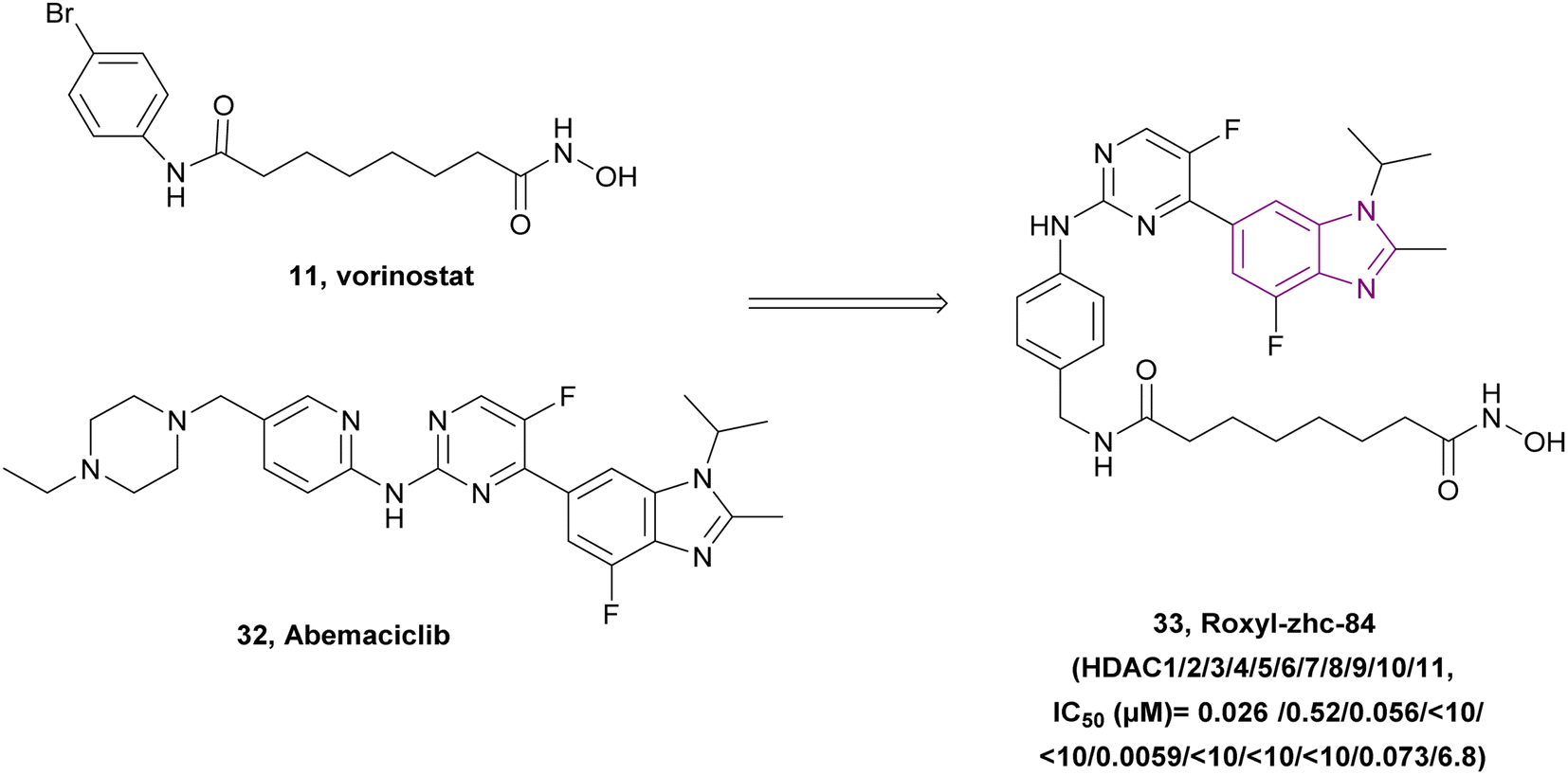 | ||
| Fig. 16 Combination between compound 11 and 32 resulted in the CDK/HDAC dual inhibitor roxyl-zhc-84 33. | ||
2-Dimethylamino-4,5,6,7-tetrabromo-benzimidazole (DMAT) scaffold 34 and hydroxamic acid (ZBG), which inhibited CK2 and HDAC, were linked, forming compounds 35 and 36 as selective dual HDAC/CK2 inhibitors, respectively (Fig. 17). Various cell lines such as Jurkat, HCT-116, MCF-7, HL-60/vinc, HL-60/adr, and HL-60 were used in the in vitro studies and demonstrated that both compounds inhibited HDAC-1, HDAC-6, and CK2. However, compound 36 showed the highest cytotoxic activity, proapoptotic capability, mitochondria-targeting, and multidrug-circumventing properties. Thus, compound 36 was employed in further in vivo studies. SAR demonstrated that the addition of the triazole ring in the linker lowered the cytotoxic and pro-apoptotic activities, while having no effect on the inhibitory action, and the 4-carbon linker was less active in both enzymes. Comparing compounds 35 and 36 to derivatives with aromatic linkers, the former had greater action against several types of cancer cells. The cytotoxic action of inhibitors 35 and 36 was influenced by their aliphatic chain length. Specifically, among the dual inhibitors that were evaluated, compound 36 was the most active.41
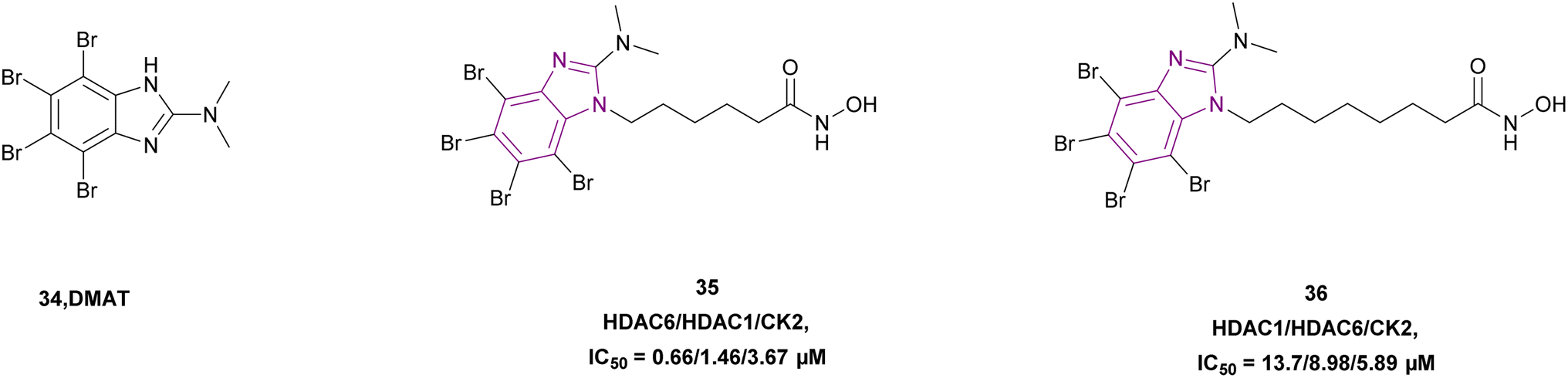 | ||
| Fig. 17 Chemical structure of CK2 inhibitor 34 and HDAC/CK2 inhibitors 35 and 36 with their IC50. | ||
Due to the critical role played by HDAC in DNA synthesis and repair in cancer cells, DNA/HDAC dual-targeting compounds have been synthesized to overcome the resistance of cancer cells to genotoxic treatments.115 Mehrling et al. fused vorinostat 11 and bendamustine 37, resulting in tinostamustine 38, which has DNA alkylating and HDAC inhibitory activity, as shown in Fig. 18. Tinostamustine inhibited HDAC1, 2, 3, 8, 6, and 10 with IC50 values of 9, 9, 25, 107, 6, and 72 nM, respectively, and formed essential interactions with HDAC1, as shown in the docking structure of Fig. 19. The inhibition of HDACs in living cells was assessed in PBMC from fifteen female rats, proving that HDAC inhibition of tinostamustine was at the maximum activity. It caused DNA cross-linking at lower concentrations than bendamustine and melphalan and had antitumour activity when evaluated in HL60 and Daudi cells. In the Daudi xenograft model, it triggered apoptosis,116 reduced the expression of genes that control DNA repair, and showed selectivity towards cancer cells.115 An open-label phase I/II study (NCT03345485) was conducted to investigate the safety, pharmacokinetics, and efficacy of tinostamustine in treating patients with advanced solid tumours, which indicated that it was well tolerated.117 Also, the cohort-expansion stage of a phase I study in relapsed/refractory (R/R) Hodgkin lymphoma (HL) patients suggested that it was well tolerated with no unexpected adverse effects.118 The same results were obtained in another R/R HL expansion cohort of a phase I study (NCT02576496). The main TEAEs were haematological and thrombocytopenia, leading to treatment discontinuation.119
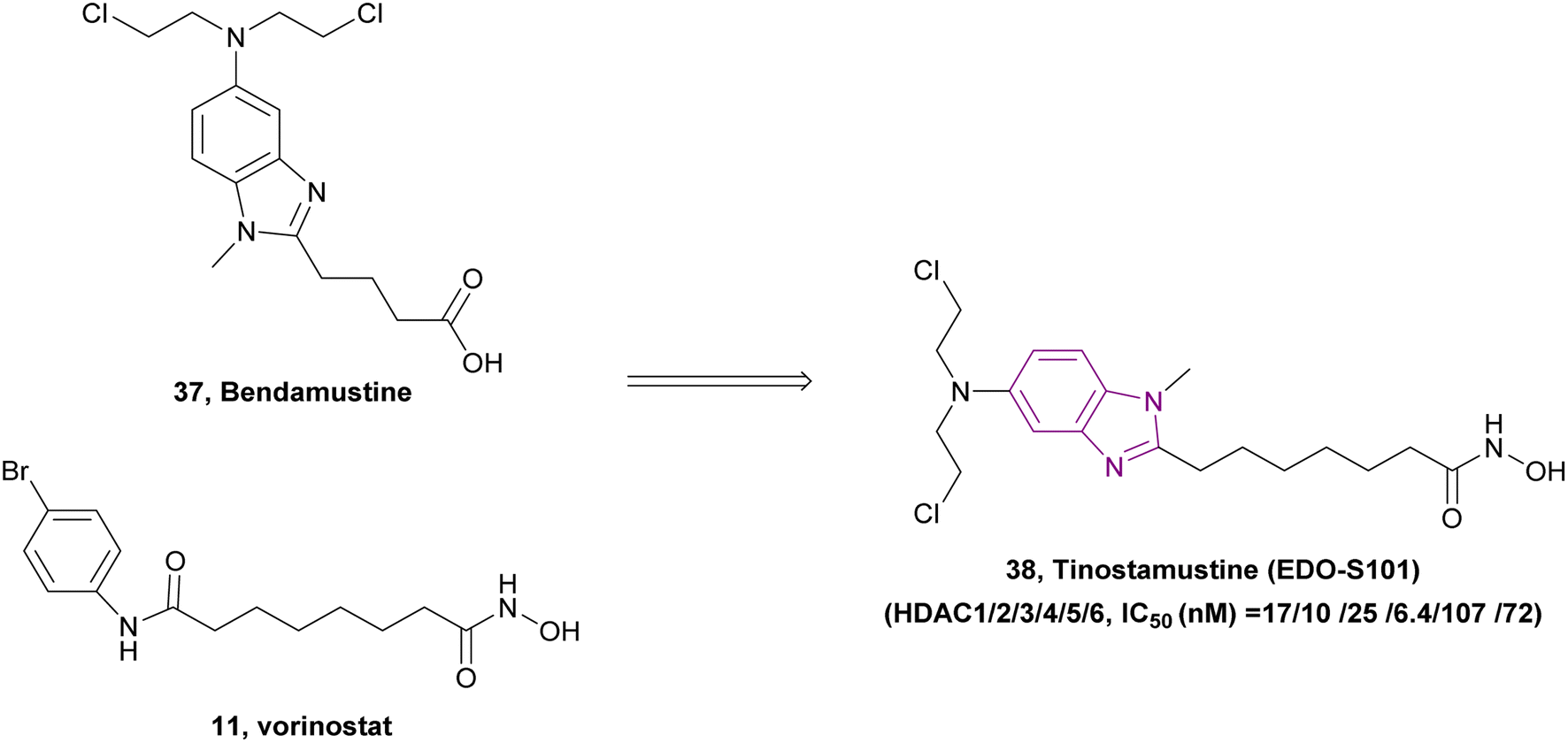 | ||
| Fig. 18 Combination of compounds 37 and 11 resulted in DNA/HDAC inhibitor 38. | ||
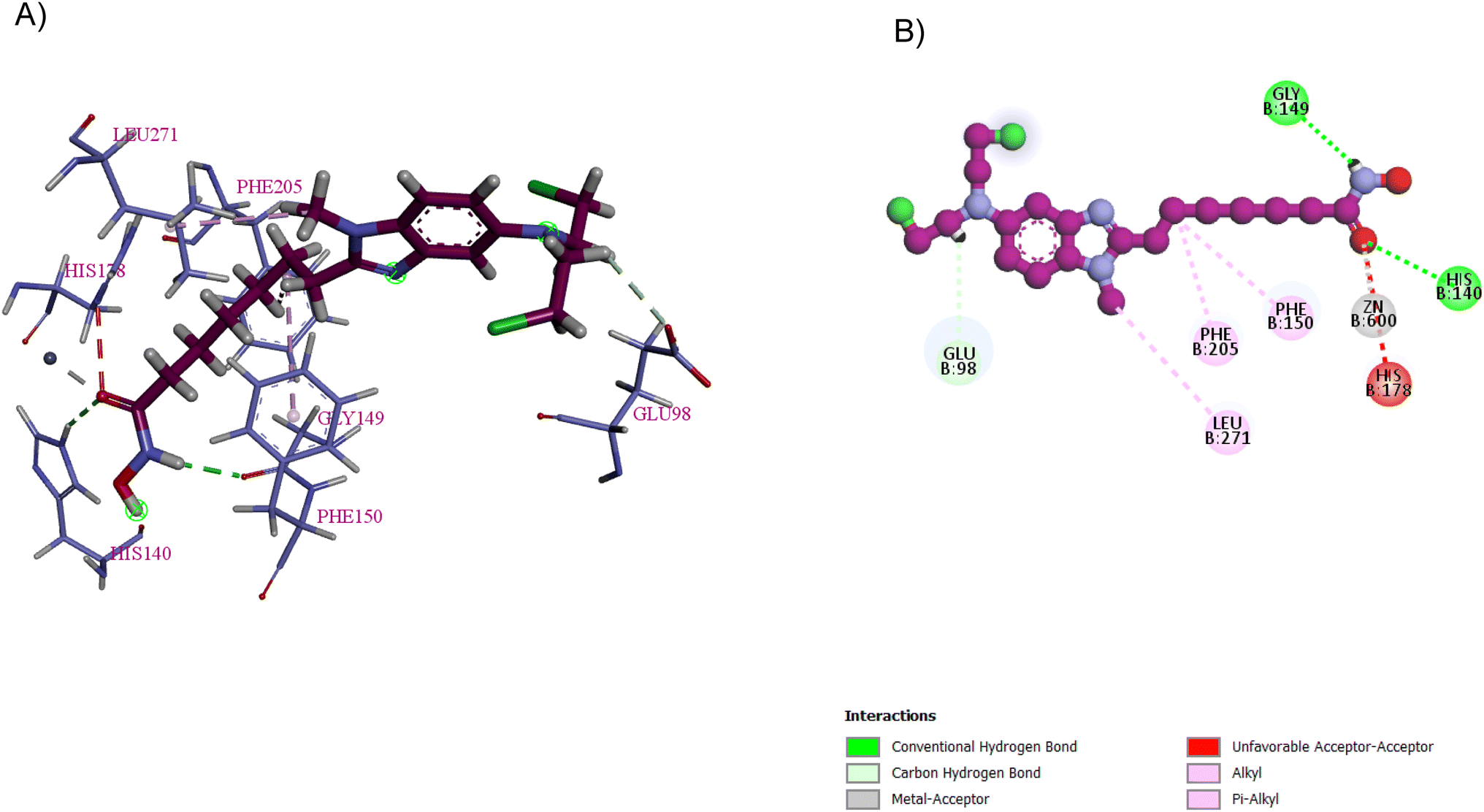 | ||
| Fig. 19 Tinostamustine (in purple) interacts with HDAC1 through multiple interactions. Images were generated by Discovery Studio 4.5. (A) 3D interaction of tinostamustine and HDAC1 residues. (B) 2D interaction of tinostamustine and HDAC1 residues (in blue). The carbonyl oxygen, amidic H, and CH2 of the alkyl group form three hydrogen bond donor interactions with the HIS140, GLY149, and GLU98 residues, respectively. The CH3 group forms hydrophobic interactions with LEU271 and the second CH2 of the linker forms two hydrophobic interactions with the PHE205 and PHE150 residues. Moreover, the carbonyl oxygen forms metal–acceptor interactions with the zinc group. | ||
2.1.2.1 Bromodomain. Bromodomains (BRDs) are acetylated histone readers. The human genome encodes 61 bromodomains in 46 proteins with different cellular functions.120,121 BRDs are comprised of the bromodomain and extra-terminal domain (BET) family and non-BET bromodomain family. The BET family members are bromodomain-containing protein 2 (BRD2), BRD3, BRD4, and BRDT localized in the nucleus and consist of two N-terminal bromodomains (BD1 and BD2), which are responsible for the process of locating and binding to Kac.122–125 BRD2, BRD3, and BRD4 are expressed in different cells, but BRDT is only expressed in tissues such as pachytene spermatocytes, diplotene spermatocytes and round spermatids.121
The bromodomain inhibitors bind to the acetyl-binding sites of bromodomains, which are deep hydrophobic binding pockets and highly selective and competitive inhibitors of Kac.126 BRD4 reads and binds to acetylated lysine in histones H3 and H4 through BD1 and BD2. Inhibition of BRD4 has been shown to decrease the expression of MYC, an oncogene,127 and Bcl-2, an antiapoptotic.121 Also, BRD4 was proven to be overexpressed in non-small cell lung cancer, promoting cancer progression.128 BRD2 is supposed to be a promising target to prevent KSVH transmission. BIC1 is a BRD2 bromodomain inhibitor given that it inhibits its binding to acetylated H4K12.122
2.1.2.1.1 Benzimidazole derivatives as BET family inhibitors. Compounds 39–43, as shown in Fig. 20, are BET family inhibitors. 3,5-Dimethylisoxazole aryl-benzimidazole derivative 39 is a BRD4 inhibitor that has anticancer activity against colorectal cancer (RKO) and selectivity towards BET bromodomains with sufficient hepatic stability. It competitively inhibited binding to BD1 (Ki of 7.5 nM) and BD2 (Ki of 3.1 nM) of BRD4. The SAR demonstrated that the cyclopropyl benzimidazole enhanced both its biochemical and cellular potency, and the two methyl groups of the isoxazole ring were crucial for its maintaining potency. When compound 39 was used to treat MM.1S tumour-bearing mice and SU-DHL-10 tumour-bearing animals, it was well tolerated and inhibited the tumour growth.127
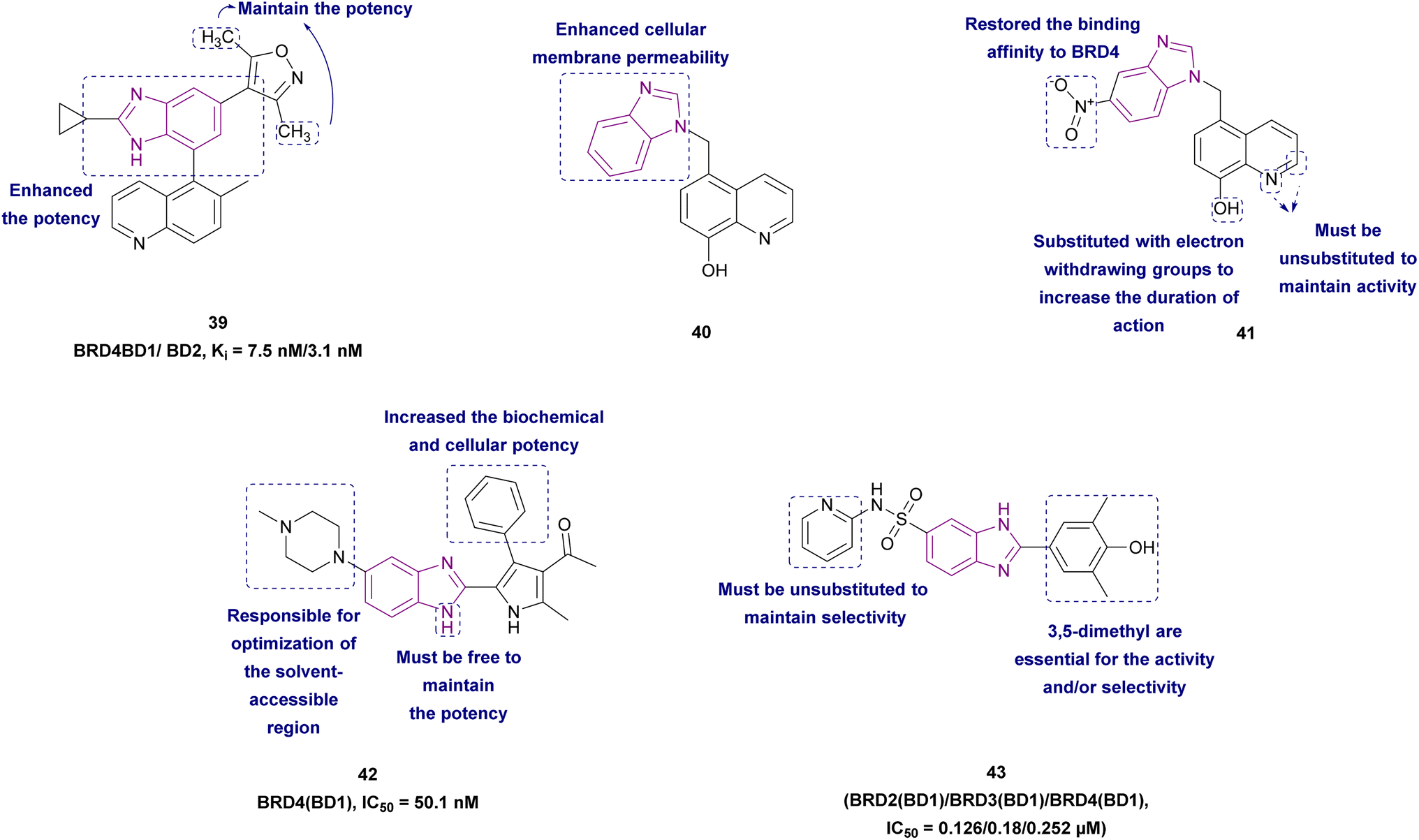 | ||
| Fig. 20 Chemical structure, SAR and IC50 of BET family inhibitors 39–43. | ||
Xing J., et al. synthesized BRD4 inhibitors 40 and 41, 5-(1H-imidazol-1-ylmethyl)-8-quinolinol analogues, and proved that they could treat acute monocytic leukaemia and basal-type triple-negative breast cancer, respectively, with little risk of hERG toxicity. 5-(1H-Imidazol-1-ylmethyl)-8-quinolinol was optimized to increase its cellular membrane permeability. As a result, the imidazole group in 5-(1H-imidazol-1-ylmethyl)-8-quinolinol was changed to 1H-benzoimidazole in 40, which made it very potent but decreased the binding affinity to BRD4. To restore the activity at both the molecular and cellular levels, a nitro group was added to the benzene ring in 40 to produce 41. According to SAR, the non-substituted nitrogen in the quinoline ring was required for activity, and the methylene linker directed the benzimidazole group to the WPF shelf of the pocket. For a longer duration effect, stronger electron-withdrawing groups may be added to the 8-hydroxyl group to reduce its phase II metabolism. Position 2 of quinoline must be unoccupied given that replacements would make the molecule less likely to act as a BRD4 inhibitor. In contrast to 41, which was sensitive to MDA-MB-231 (IC50 1.1 μM), 40 was sensitive to the MV4-11 cell line. In the MV4-11 cell line, 40 and 41 downregulated the c-Myc levels.123
Compound 42 resulted from the optimization of (1-(2,4-dimethyl-5-(thiophen-3-yl)-1H-pyrrol-3-yl)ethan-1-one). Kong et al. synthesized 42 via acetylation, bromination, Suzuki cross-coupling, and cyclization reactions. It is a well-tolerated BRD4 inhibitor with good metabolic stability and good oral bioavailability. Significantly, it may be a potential candidate for the treatment of AML and some solid tumours. It induced cell cycle arrest at the G0/G1 phase through the upregulation of p21 and apoptotic marker-cleaved PARP and downregulation of c-Myc. The introduction of N-methylpiperazine (a hydrophilic function group) at the 5-position of benzimidazole optimized the binding interactions towards the solvent-accessible region. The rigid phenyl substituent at the 4-position of the pyrrole increased the biochemical and cellular potency given that it was surrounded by a hydrophobic pocket. Substituting the ortho-, meta-, and para-position of the phenyl with –Cl or –OCH3 or the para-/meta-position with methyl, methylthio, ethoxyl, dimethylamino, 3-fluoro-4-methoxyl, 3,4-dimethoxyl, benzo[d][1,3]dioxol-5-yl and 2,3-dihydrobenzo[b][1,4]dioxin-6-yl had a negative influence on the potency.129
Compound 43, a benzoimidazole-6-sulfonamide derivative, exhibited a better selectivity profile for all BD1 BETs with features of solubility, cell permeability, and stability. It caused a decrease in the expression of c-Myc without significant cytotoxicity. The benzimidazole scaffold was responsible for acceptable cellular permeability and was used as a bioisostere for the azobenzene scaffold. SAR studies revealed that the removal of the pyridine decreased both the affinity and selectivity. The separation of the pyridine ring from the sulfonamide function by a CH2 group caused complete loss of activity. The activity shifted to non-BET proteins when the pyridine moiety was replaced with an aliphatic ring; however, when the pyridine moiety was replaced with a cyclopropyl group, there was significant off-target binding and a partial loss of affinity to BETs, with only binding to the BD2 of BRDT remaining. Its activity was attributed to the substitution of 3,5-dimethyl-4-phenol; removal of the 5-methyl or shifting the position of dimethyl to the 2,6 positions resulted in the loss of the inhibitory activity. Moreover, the activity of the unsubstituted phenol derivative was significantly less selective.124
Three HITs were chosen among 224205 compounds using the 3D-QSAR pharmacophore model and docking as BRD4 inhibitors. Subsequently, the total binding free energy was determined using the MM/PBSA method, which showed that only the 44/BRD4 complex had the best binding affinity towards BRD4 (Fig. 21) with the lowest binding free energy. The ADMET results showed that it has low aqueous solubility, good absorption, non-hepatotoxic and does not inhibit CYP2D6. In compound 44, the benzimidazole moiety formed an H-bond. Besides, the 2H-chromen-2-one moiety formed π–π T-shaped interaction and π–anion interaction. Additionally, it formed van der Waals interactions and hydrophobic interactions.121 Compound 45 is a BRD2 bromodomain inhibitor given that it inhibits its binding to acetylated H4K12.122
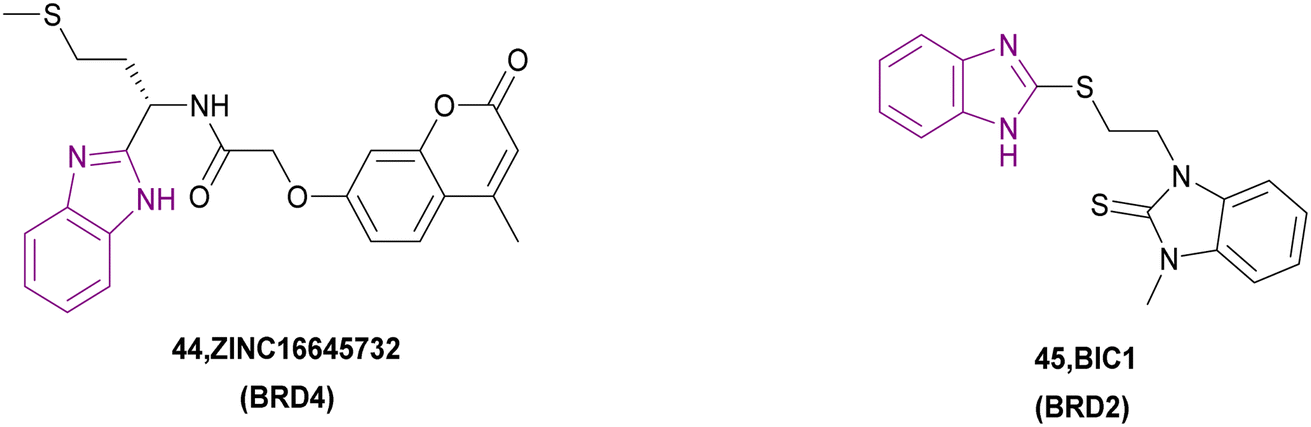 | ||
| Fig. 21 Chemical structure of BET family inhibitors 44–45. | ||
2.1.2.1.2 Benzimidazole derivatives as CBP/p300 bromodomain inhibitors. CBP and its paralog p300 are members of the lysine acetyltransferase superfamily. CBP and p300 share highly similar sequences and structural organizations, with overlapping but distinct functions, and thus they are typically referred to as CBP/p300.130 CBP and p300 are multidomain proteins containing HAT and BRD domains. The CBP/p300 bromodomain has been associated with malignancies and is considered a therapeutic agent.131 Moreover, CBP/p300 coactivates important oncogenes such as MYC and IRF4, which are significant in many types of haematological cancers.132
Compounds 46–50 are CBP/p300 bromodomain inhibitors (Fig. 22). CBP30 46 is a potent and selective CBP/p300 bromodomain inhibitor (Kd = 0.021 and 0.032 μM, respectively) with a pIC50 of 7.1 ± 0.049. Its dimethylisoxazole mimics the key Kac binding interactions of the CBP BRD.133 Z. Chen et al. optimized CBP30 46 utilizing bioisosterism and conformational restriction strategies, which resulted in compound 47, which is a potent p300 bromodomain inhibitor with a benzimidazole scaffold and a propiolactam linker, showing better antiproliferative activity against the OPM-2, 22RV1, MCF-7, and SCC-9 cell lines compared to CBP30 46. Moreover, it downregulated c-Myc expression and induced apoptosis and cell cycle arrest in the G1/G0 phase.134
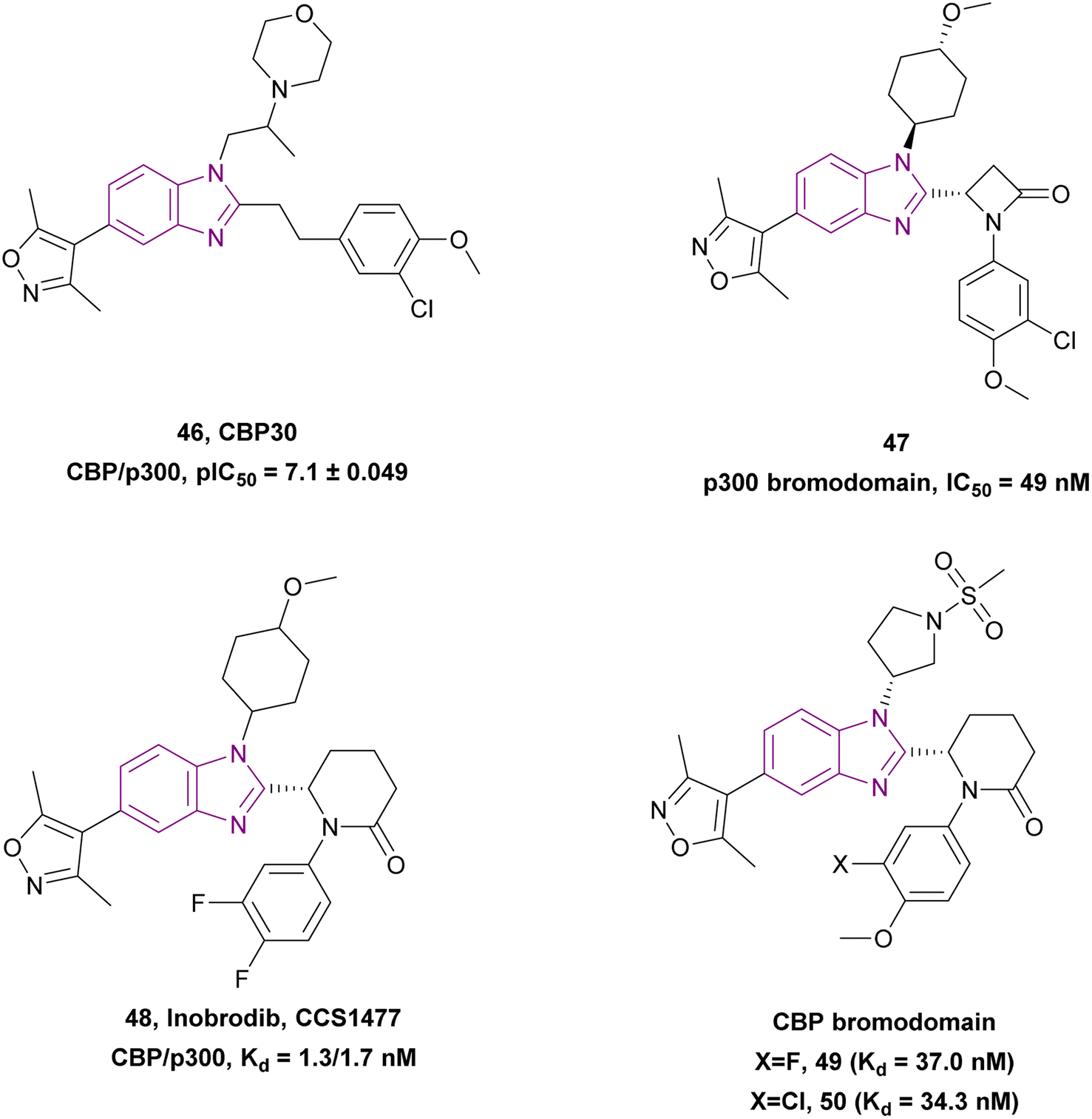 | ||
| Fig. 22 Chemical structure, pIC50, IC50 and Kd of CBP/p300 bromodomain inhibitors 46–50. | ||
CCS1477 48 is currently in phase I/II clinical trials for the treatment of AML, non-Hodgkin lymphoma, and MM (NCT04068597).132 It is a potent, selective, and orally active small molecule inhibitor of the p300/CBP bromodomain with high affinity (Kd = 1.3/1.7 nM) and selectivity. It has antiproliferative activity against prostate cell lines (22Rv1 and VCaP) with IC50 values of 96 and 49 nM, respectively. In 22Rv1 cells, p300/CBP inhibition resulted in the complete inhibition of plasma PSA. Moreover, the 22Rv1 xenograft model treated with it showed complete tumour growth inhibition.135 It is promising for the treatment of advanced prostate cancer given that it exhibited antitumour activity, regulating AR and c-Myc signalling in androgen receptor (AR) splice variant (SV)-driven models, and could regulate CRPC biopsy biomarker expression and modulate the KLK3 blood levels.136 Further studies showed that CCS1477 was effective in haematological malignancies, including MM and AML, as monotherapy or in combination with standard care agents. It arrested the cell cycle and inhibited differentiation through the upregulation of selected differentiation markers (e.g., CD11b and CD86).137 Moreover, it could be used in combination with azacitidine and/or venetoclax in AML and B-cell lymphomas. In an OPM-2 xenograft model, it had additive or synergistic inhibitory activity when combined with each of bortezomib, lenalidomide and vorinostat, suggesting its clinical utility in the treatment of MM as monotherapy and/or in combination with standard care agents.138
Compounds 49 and 50 (analogues of CCS1477 48) are CBP bromodomain inhibitors. Compound 49 showed better selectivity than CCS1477 48. Compound 49 displayed a similar binding affinity to that of CCS1477, with a Kd value of 37 nM.120
Compound 51, (R)-5-methylpyrrolidin-2-one derivative, is a structurally modified compound of CCS1477 48. Compound 51 is a p300 bromodomain inhibitor with antiproliferative activity against different cell lines. With its high cell permeability, it could overcome the defect of CCS1477 48 of high efflux rate. Moreover, it is metabolically stable and has no hERG risk, indicating its safety for the heart. Replacing the five-membered ring (pyrrolidine) with a four-membered (azetidine) or six-membered ring (piperidine) weakened its inhibitory activity. The methoxy substituted on trans-cyclohexyl group was important for its binding affinity. However, the methoxy group was a potential metabolizable site. Additionally, its potency was diminished when alternative heterocycles were used in place of 3,5-dimethylisoxazole. Generally, the meta- and para-disubstituted phenyl-containing compounds were more potent than the para- or meta-monosubstituted compounds (Fig. 23).139
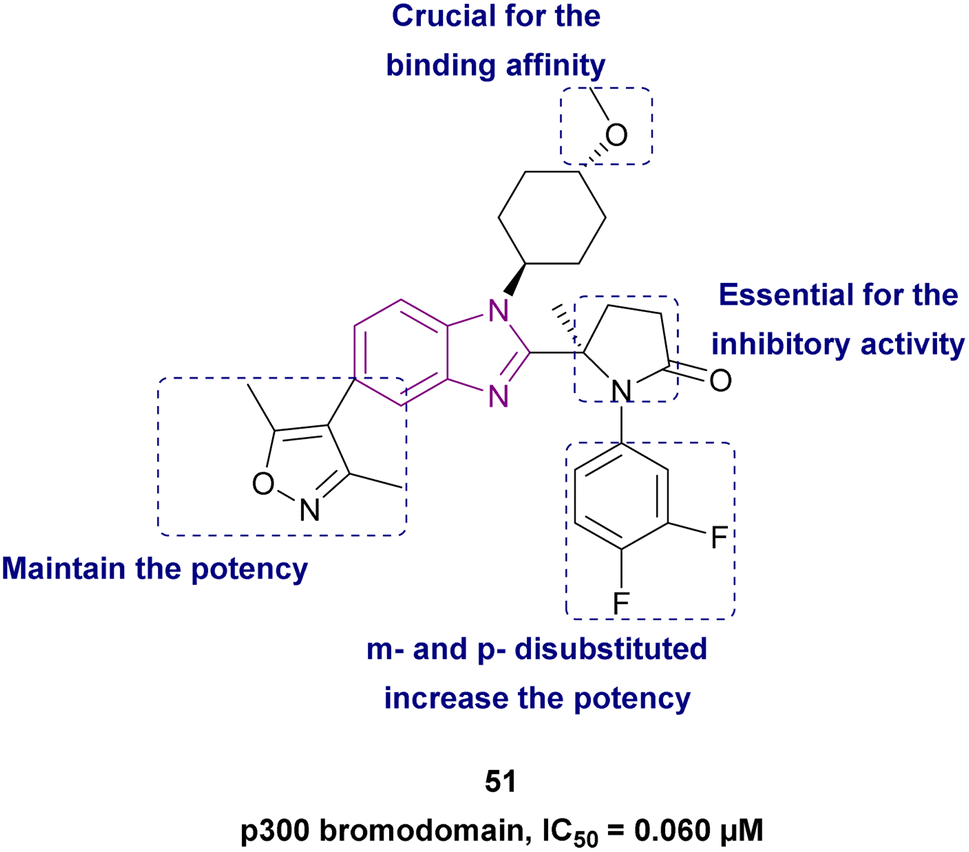 | ||
| Fig. 23 Chemical structure, SAR and IC50 of CBP/p300 bromodomain inhibitor 51. | ||
2.1.2.1.3 Benzimidazole derivatives as TAF1 bromodomain inhibitors. TAF1 is a non-BET bromodomain protein that contains a histone acetyltransferase (HAT) domain and a tandem bromodomain module (TAF1(1) and TAF1(2)). TAF1(2) could recognize acetyl lysine as well as butyryl and crotonyl lysine. Moreover, TAF1(2) synergized with BRD4 in controlling cancer cell proliferation. Compound 52 is a TAF1(2)-bromodomain inhibitor with an IC50 of 10 nM and excellent selectivity over other bromodomain-family members, showing that the benzimidazole ring is essential for improving the potency and selectivity (Fig. 24). It exhibited antiproliferative synergy with the BET inhibitor JQ1.140
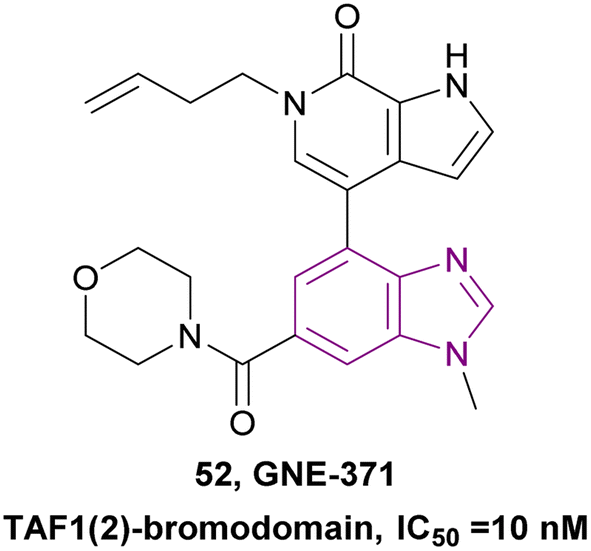 | ||
| Fig. 24 Chemical structure and IC50 of TAF1 bromodomain inhibitor 52. | ||
2.1.2.1.4 Benzimidazole derivatives as BRPF bromodomain inhibitors. The BRPF family (bromodomain and PHD finger-containing family of BRPF1, BRPF2/BRD1, and BRPF3) is non-BET bromodomain-containing proteins (1). BRPF1 contains multiple epigenetic reader modules, including a unique double PHD, zinc finger assembly (PZP), a bromodomain, and a chromo/Tudor-related Pro-Tyr-Tyr-Pro (PWWP) domain (2). Demont et al. showed that (5,6)-disubstituted benzimidazolones has greater selectivity for BRPF1 over BRD4. They synthesized compound 53 given that the amide at the 5-position interacted with a BRPF1 ZA loop and 2-methoxy phenyl is more potent than 3-methoxy phenyl carboxamide and unsubstituted phenyl.141 Bamborough et al. optimized compound 53 (pIC50 = 7.1) by replacing piperidine with piperazine, where the extra basic nitrogen formed an additional hydrogen bond and improved the solubility, resulting in compound 54, which was a potent and selective BRPF1 bromodomain inhibitor (pIC50 = 8.1) with greater solubility, potency (300 pM), and cell activity (20 nM) (Fig. 25). However, changes at the 3- and 5- positions of the benzimidazolone had detrimental effects.142
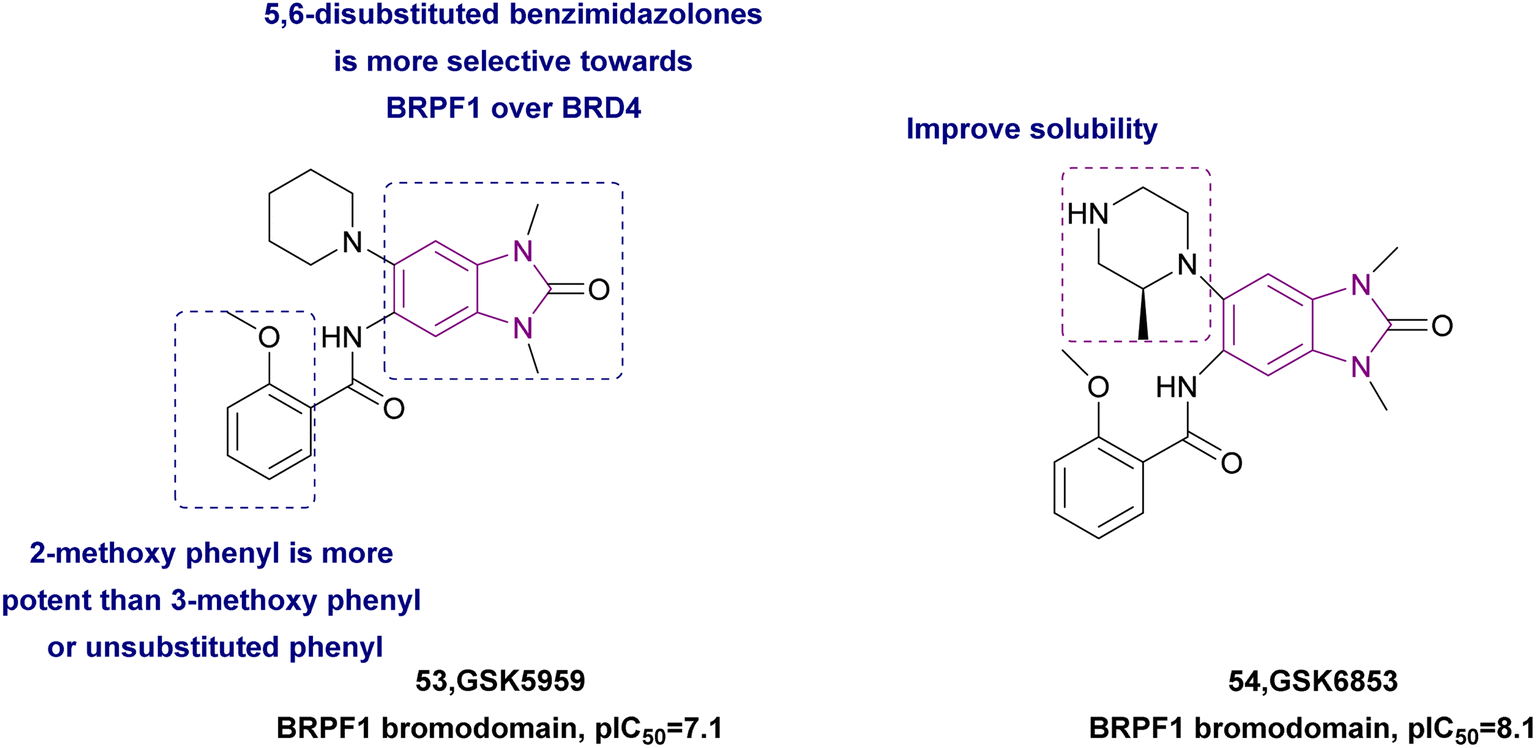 | ||
| Fig. 25 Chemical structure, SAR, and pIC50 of TAF1 bromodomain inhibitors 53 and 54. | ||
2.1.2.1.5 Benzimidazole derivatives as BAZ2A bromodomain inhibitors. BAZ2A is a non-BET bromodomain-containing protein. Acetylated Lys16 on histone H4 is recognized by the bromodomain and PHD zinc finger domain of BAZ2A, which then bind HDAC1 to create heterochromatin. The metastatic potential of prostate cancer cells has been revealed to be impacted by BAZ2A downregulation, whereas overexpression of the protein has been linked to the aggressiveness of the disease. Among 54
794 molecules from the Maybridge Screening Collection and Enamine Golden Fragment libraries docked into the crystal structure of the BAZ2A bromodomain, only compound 55 inhibited the BAZ2A bromodomain with an IC50 of 28 μM, forming 3 H-bonds by the benzimidazole nitrogen and the N2 and N3 nitrogen atoms of the triazole ring. Moreover, compounds 56, 57, and 58 from the docked compounds showed an increase in binding affinity. The potency of compound 58 could be further improved by substituting the methyl group on the triazole ring with a longer aliphatic tail (Fig. 26).143
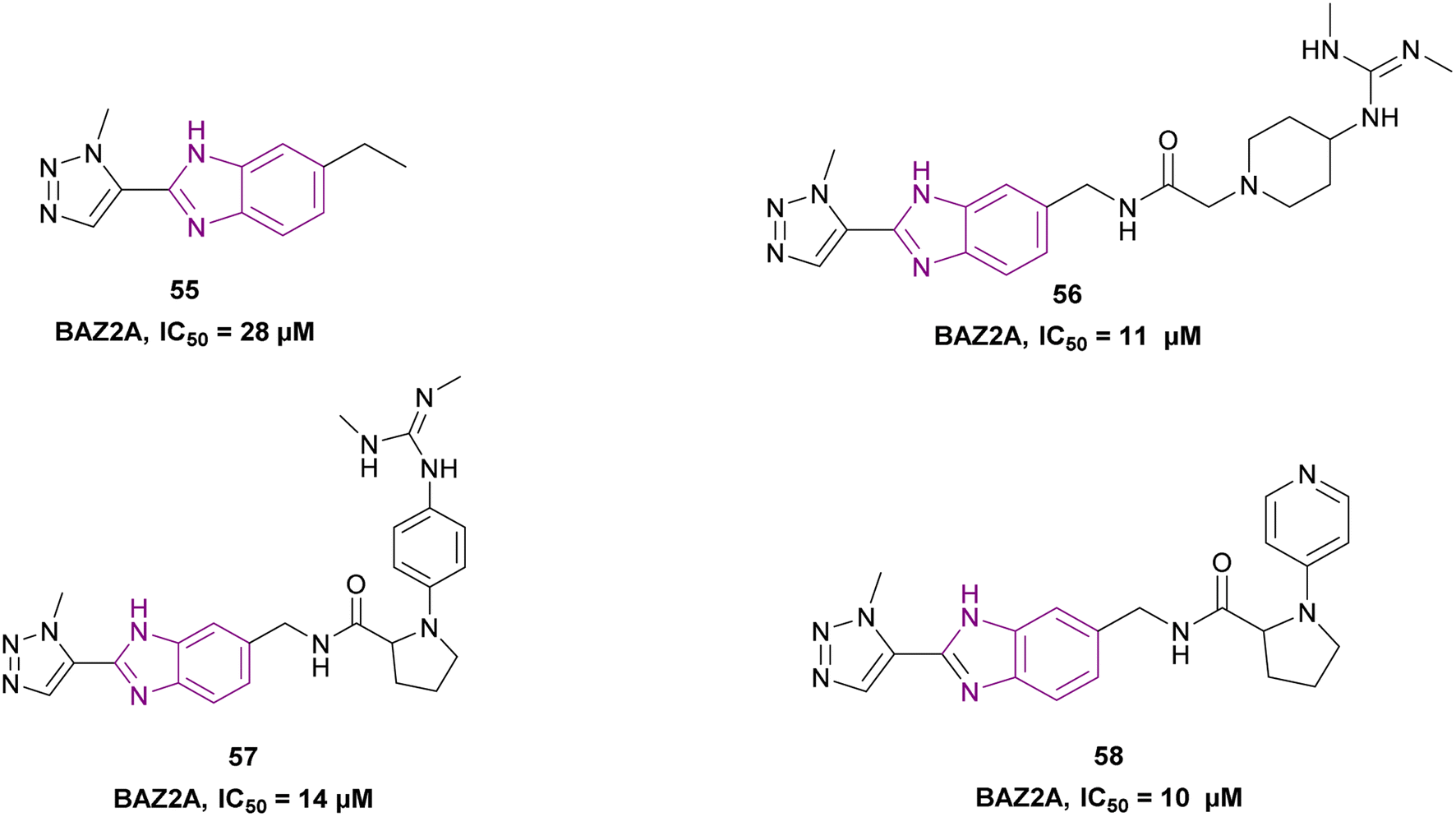 | ||
| Fig. 26 Chemical structure and IC50 of BAZ2A bromodomain inhibitors 55–58. | ||
2.1.2.1.6 Benzimidazole derivatives as dual inhibitors. Further structure optimization of compound 59 resulted in compound 60 (Fig. 27), which is a BRD4, phosphatidylinositol 3-kinase (PI3K) (IC50 = 13.12 nM) and aurora A (IC50 = 10.19 nM) multi-targeted inhibitor, providing a lower dose, better safety, enhanced efficacy, and delayed drug resistance. In compound 60, SAR studies revealed that replacing its benzyl moiety with an aromatic heterocyclic group or increasing the length of the alkyl side chain attached to its amide group may decrease its anticancer activity. It was tested in drug-resistant cell lines (A549 and H1975 cells) and non-drug-resistant cell lines (HCC827 cells), showing antiproliferation activity with IC50 values of 0.83 μM, 1.02 μM, and 0.26 μM, respectively. The pro-apoptotic proteins cleaved-PARP and cleaved-caspase 9 were upregulated by it, while the anti-apoptotic protein Bcl-2 was downregulated. Also, it showed superior selectivity towards cancer cells to normal cells and induced cell cycle arrest in the G2/M phase.128
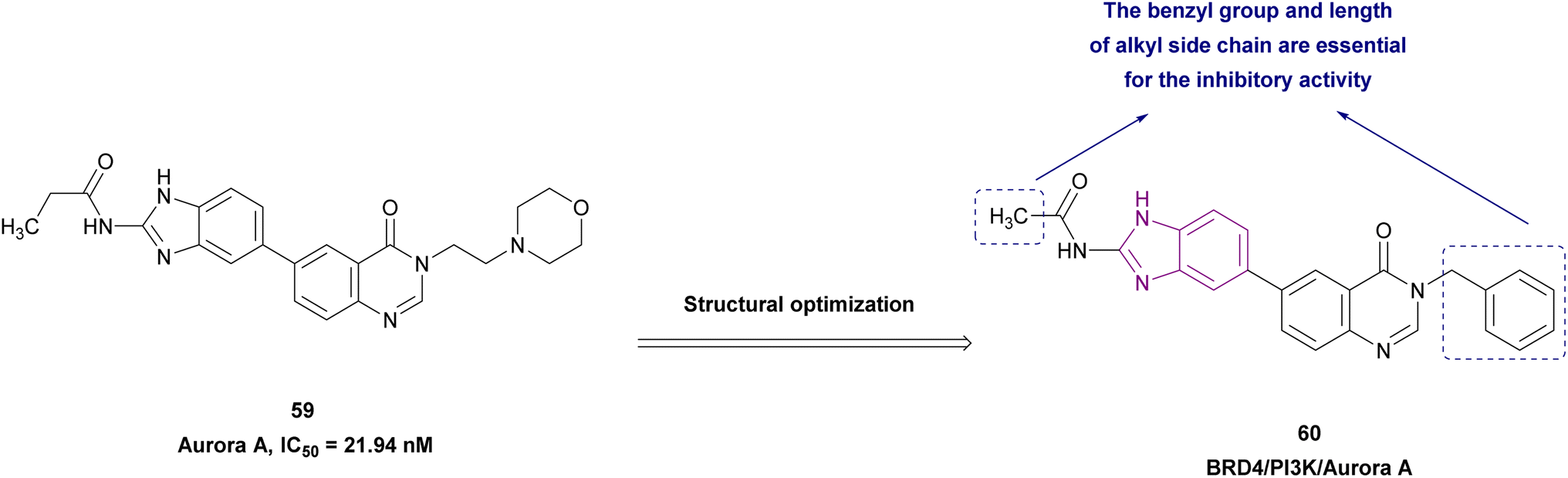 | ||
| Fig. 27 Chemical structure, IC50, and SAR of 59 and 60. | ||
BI2536 61, a BRD4 inhibitor, and veliparib 62, a PARP1/2 inhibitor, were combined to create compound 63, a dual PARP/BRD4 inhibitor (Fig. 28). Compound 63 inhibited PARP1, BRD4, BRD4 BD1, and BRD4 BD2 with IC50 of 13 ± 2.5 nM, 101 ± 11 nM, 75 ± 10 nM, and 157 ± 15 nM, respectively. It exhibited anti-tumour action in pancreatic ductal adenocarcinoma (PDAC) SW1990 cells and breast cancer MDA-MB-231 cells with IC50 of 0.98 μM and 0.34 μM, respectively. Moreover, it could reverse olaparib-induced resistance by promoting cell cycle arrest, DNA damage, and autophagy-related cell death.144
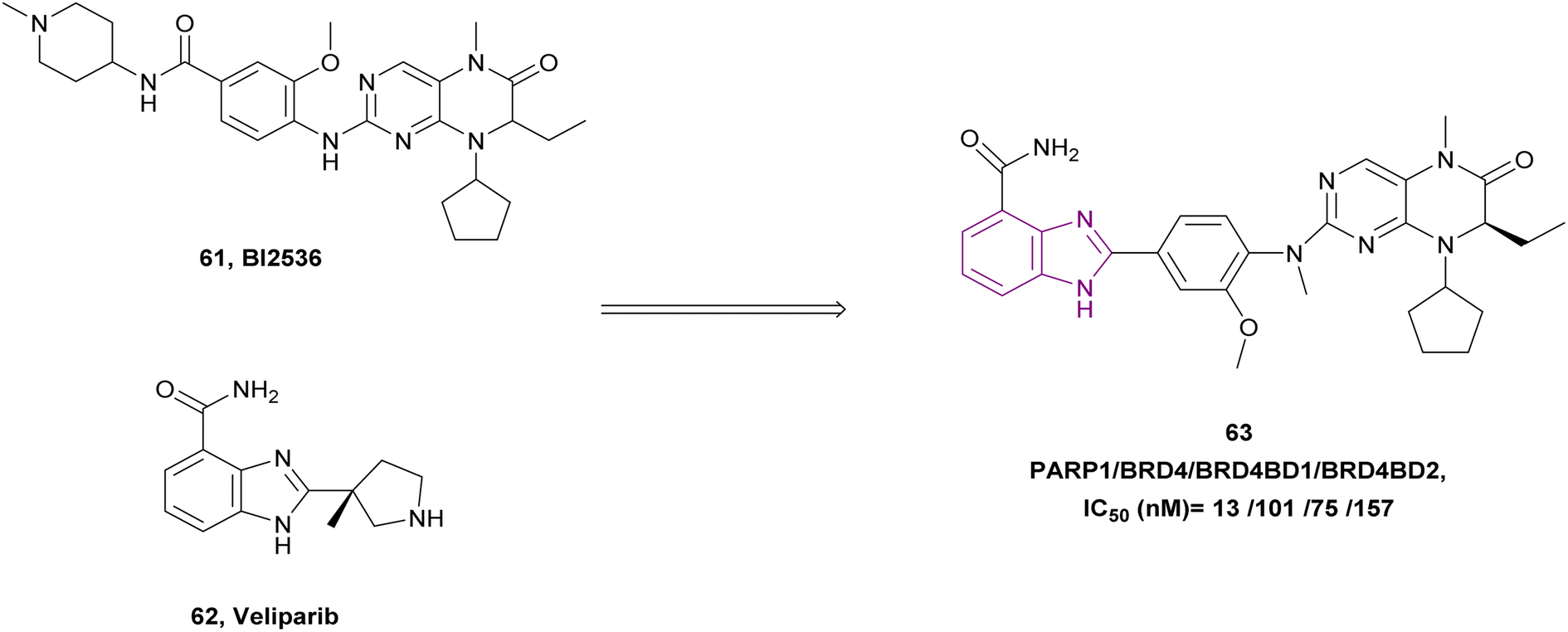 | ||
| Fig. 28 Combination of compound 61 and 62 resulted in PARP1/BRD4 inhibitor 63. | ||
Inhibition of BRD4 elevates homologous recombination (HR), the most effective repair pathway, and sensitizes cancer cells to PARP1/2 inhibitors. Huang, Cao et al. linked the 1H-benzimidazole-4-carboxamide moiety of veliparib acting as a PARP1 inhibitor with compound 64 acting as a BRD4 inhibitor to synthesize compound 65 (Fig. 29). Compound 65 is a dual PARP1/BRD4 inhibitor that arrests the cell cycle at G0/G1 and G2/M phases, increases DNA damage, and reverses olaparib-induced adaptive resistance. It was more effective than JQ-1 and olaparib in inhibiting pancreatic cancer cell (CFPAC-1, Capan-1, and SW1990) growth and had an antiproliferative effect on BRCA-proficient triple-negative breast cancer (MDAMB-231 and MDA-MB-468), BRCA2-mutated colonic cancer (HCT116), and leukaemic cancer (THP-11) cell lines. The inhibitory effects of compound 65 at a dose of 30 mg kg−1 on SW1990 xenograft tumour growth were more significant than that of JQ1, olaparib, and their combination.145
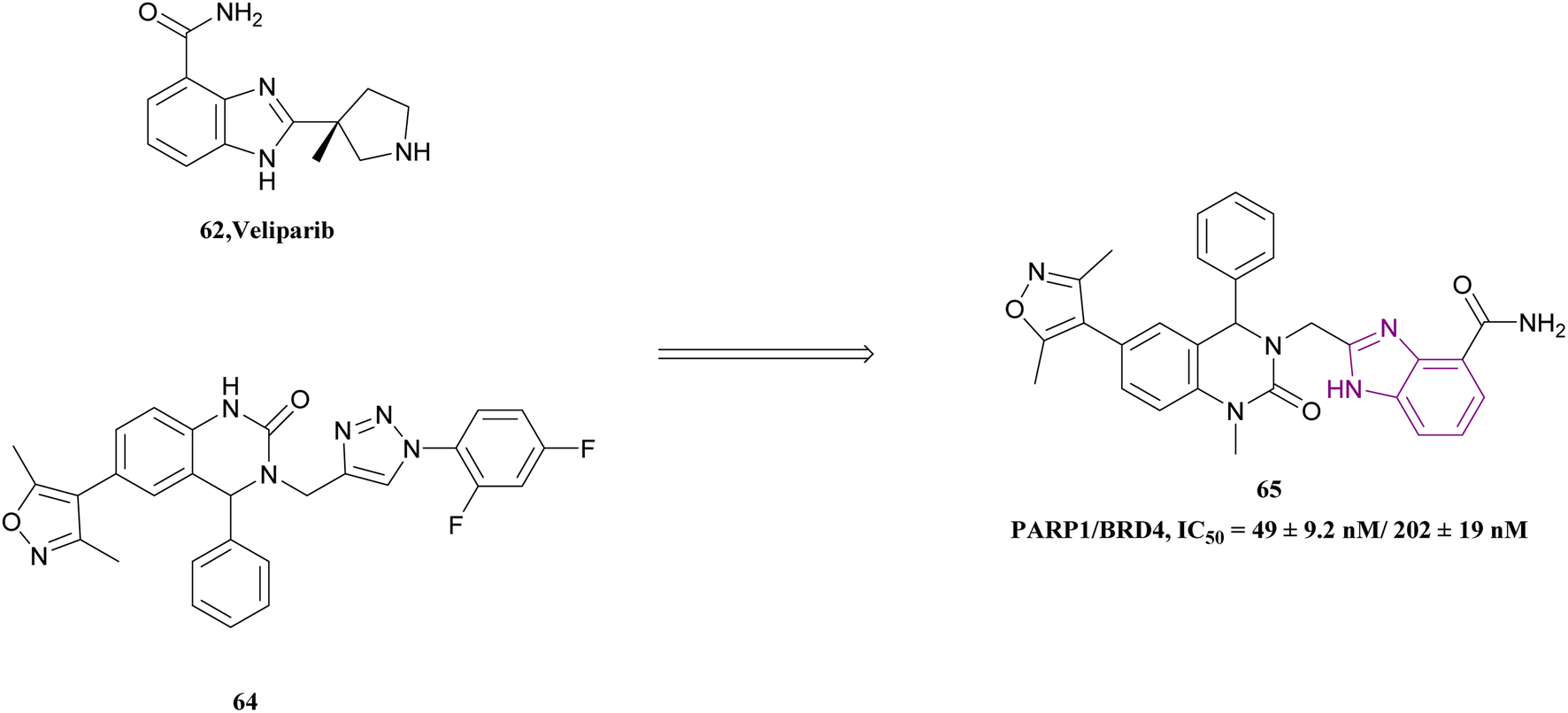 | ||
| Fig. 29 Combination of compound 62 and 64 resulted in PARP1/BRD4 inhibitor 65. | ||
2.1.2.2 Benzimidazole derivatives as YEATS family inhibitors. YEATS is named after the 5 protein members containing this conserved domain (Yaf9, ENL, AF9, Taf14, and Sas5).146 The YEATS domain is an acetylated and crotonylated lysine reader.147 Humans have four proteins that contain the YEATS domain, GAS41 (YEATS4), AF9, YEATS2, and ENL.148,149 Native ENL is necessary for leukaemia development, and thus the YEAST domain of ENL is considered as target for leukemogenesis.146,148
Compounds 66–68 inhibit ENL, as shown in Fig. 30. SGC-iMLLT 67 inhibited the YEATS domain (YD) of ENL (IC50 = 0.26 μM) and AF9. It resisted metabolism with the primary process for metabolism by N-demethylation, upregulated the expression of CD86, and downregulated the expression of MYC and DDN. Moustakim et al. optimized compound 66 due to its labile methyl ester, revealing SGC-iMLLT 67, which has more potent binding affinity. Replacement of the amide bond with sulfonamide eliminated the inhibitory activity. Incubation of cells with SGC-iMLLT in the presence of vorinostat significantly decreased t1/2 for MLLT1 and MLLT3 (MLLT1: P < 0.0001, MLLT3: P = 0.0185).150
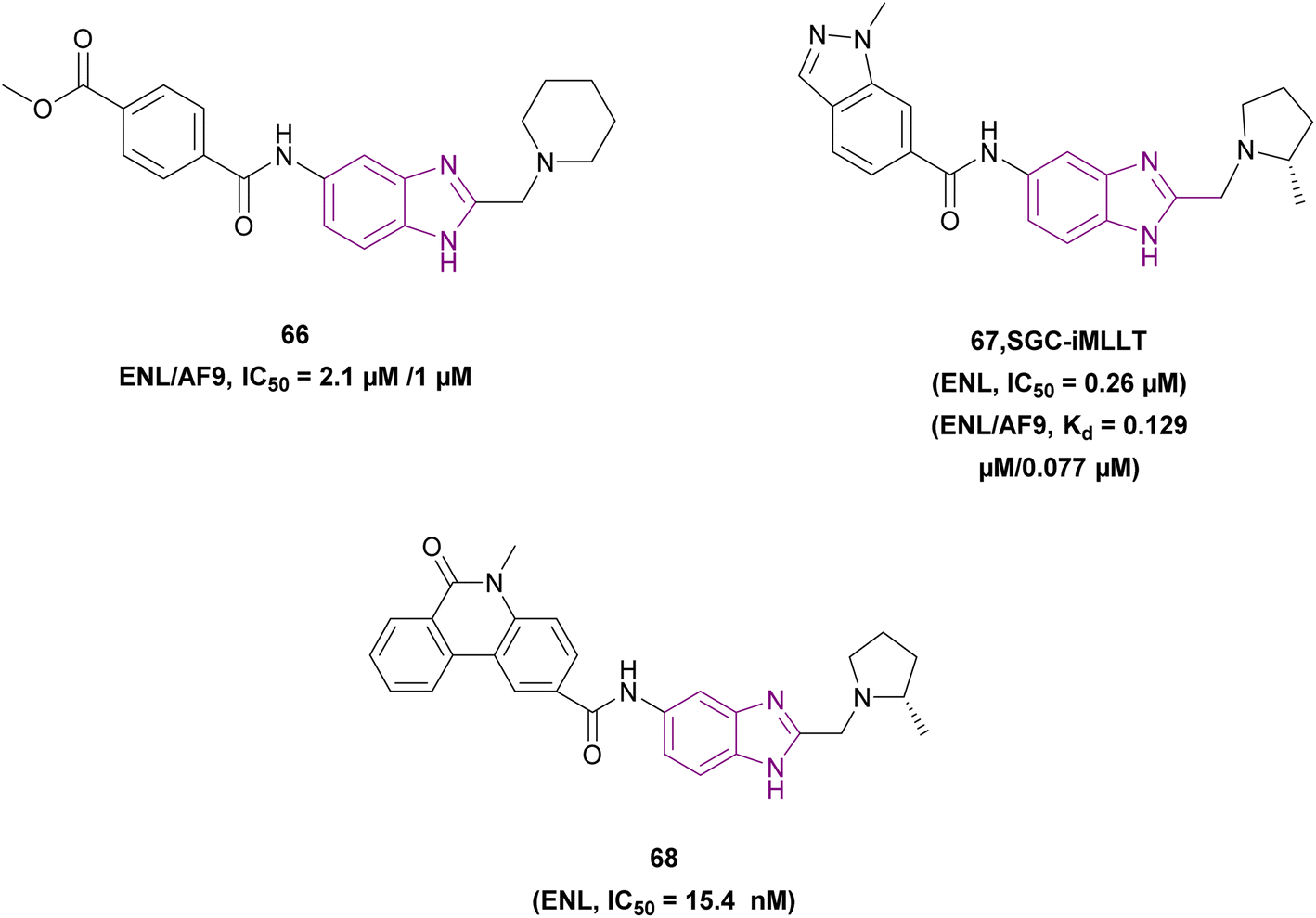 | ||
| Fig. 30 Chemical structure and IC50 of ENL inhibitors 66–68. | ||
To improve the anti-leukemic cell activities of SGC-iMLLT 67, Guo, Jia, et al. replaced the indazole ring with a larger aromatic system, such as methyl phenanthridinone in compound 68. This increased the π–π stacking interaction with residue histidine 56 (H56), which is located at the substrate entrance through which the H3Kac27 peptide entered. Compound 68, an ENL inhibitor with a Kd of 17.0 nM, is a therapeutic target in leukemogenesis, which was the most powerful SGC-iMLLT analogue because of its exceptional target engagement in the cellular context. It downregulated the expression of the target gene MYC alone or in combination with JQ-1. It had inhibitory activities towards MLL-rearranged cell lines (MOLM-13 and MV4-11) and showed the highest thermal stability for the ENL domain but did not affect the GAS41 YEATS domain. The in vivo pharmacokinetic properties of 68 showed a t1/2 of 5.2 h and Cmax of 33.6 ng mL−1 at 0.5 h with better oral exposure. The mean resident time after oral administration was 5.5 times higher than that of SGC-iMLLT. However, it needs further optimization due to its low oral bioavailability.148
2.2 Histone methylation
2.2.1.1 Histone lysine methyltransferases (HKMTs). All known lysine histone methyltransferases contain a conserved methyltransferase domain termed a SET domain. It consists of 4 families including SUV39, SET1, SET2, and RIZ.155 HKMTs catalyze the transfer of a methyl group from S-adenosylmethionine (SAM) to the ε-amino group of lysine.156 H3K4, 9, 27, 36, 79 and H4K20 are the commonly recognized lysine methylation sites.151 The six lysine methylation sites have 24 distinct HKMTs including SETD1A, SETD1B, MLL1, MLL2, MLL3, MLL4, SETD7, and PRDM9 for H3K4 methylation, EZH1 and EZH2 for H3K27 methylation, SETD2, NSD1, NSD2, NSD3, and ASH1L for H3K36 methylation, SUV39H1, SUV39H2, SETDB1, G9A/EHMT2, and GLP/EHMT1 for H3K39 methylation, DOT1L for H3K79 methylation, and SETD8, SUV420H1 and SUV420H2 for H4K20 methylation.157 Here, we discuss some benzimidazole derivatives that target lysine histone methyltransferases, MLL1 and EZH2, which belong to the SET1 family, G9a, belonging to the SUV39 family, and DOT1L, as shown in Fig. 31.
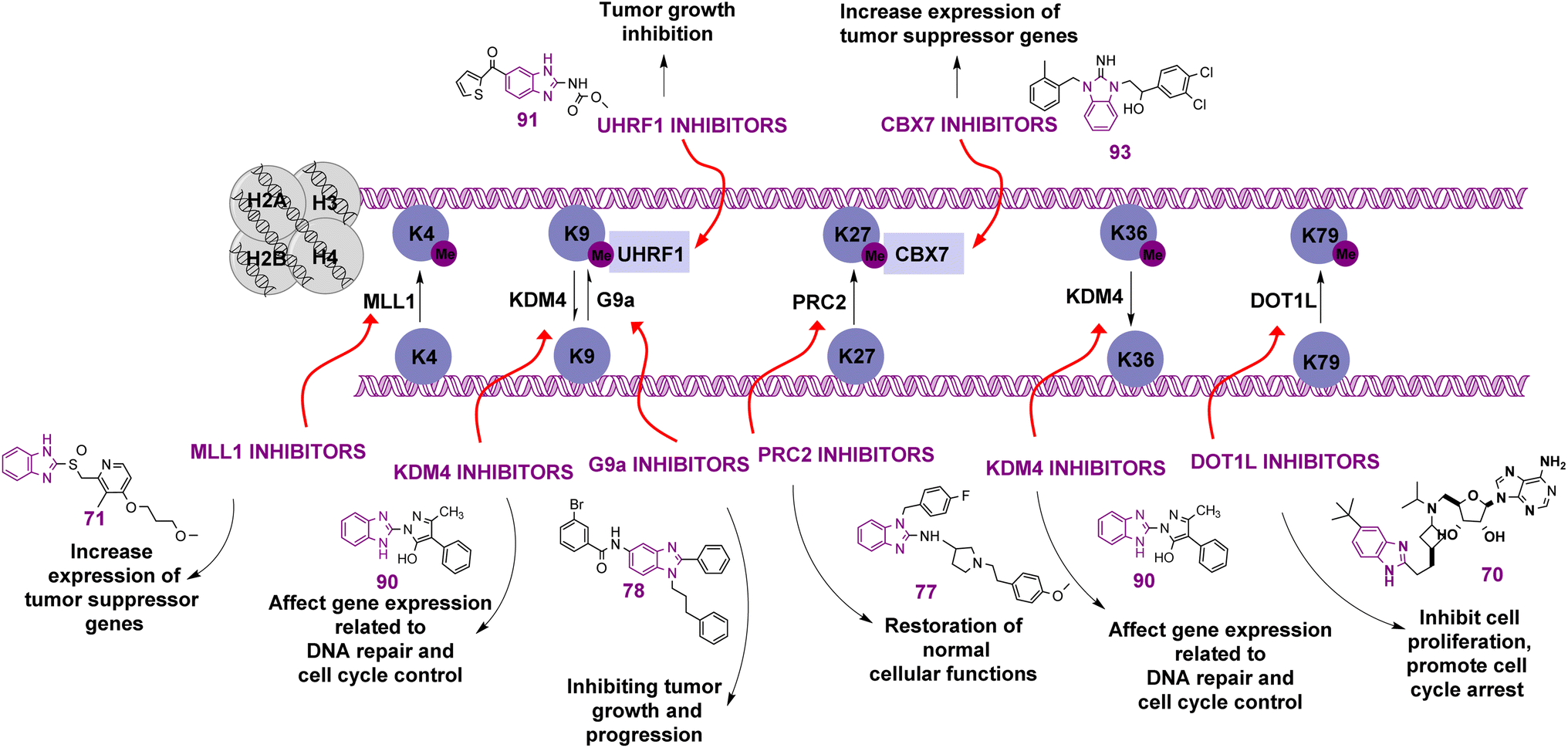 | ||
| Fig. 31 Effects of benzimidazole derivatives as histone lysine methyl transferase inhibitors, histone demethylase inhibitors, and histone methylated readers inhibitors on cancer cell growth and gene expression. | ||
2.2.1.1.1 Benzimidazole derivatives as DOT1L inhibitors. DOT1L is a histone methyltransferase enzyme that causes mono-, di-, or trimethylation of H3K79. The hypermethylation of H3K79 has been associated with the development of acute leukemias characterized by MLL (mixed-lineage leukemia) rearrangements (MLLr cells). Therefore, the growth of MLLr cells is inhibited by the suppression of H3K79 methylation.158 Optimization of EPZ004777 69 using the structure-guided design resulted in EPZ-5676 70 (Fig. 32), which is a potent and selective aminonucleoside DOT1L inhibitor. It has recently entered clinical evaluation as a therapeutic agent for MLL-rearranged leukemia because it decreased HOXA9 and MEIS1 mRNA. By occupying the SAM binding pocket, EPZ-5676 70 altered the conformation of DOT1L, which allowed additional interactions to be formed. It had an antiproliferative effect on MV4-11 cells with an IC50 of 3 nM. Also, in vivo studies using MV-411 xenograft-bearing rats showed that continuous IV infusion for 14 days or longer gave optimal activity.159,160
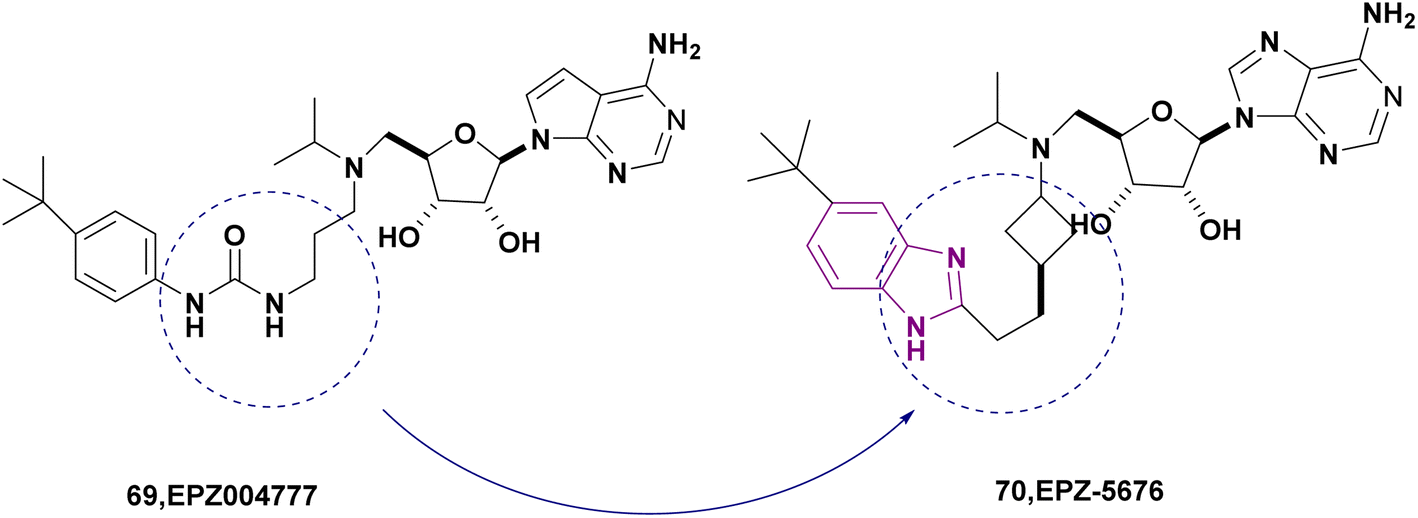 | ||
| Fig. 32 Chemical structure of DOT1L inhibitors 69–70. | ||
2.2.1.1.2 Benzimidazole derivatives as mixed lineage leukemia 1(MLL1) inhibitors. The mixed lineage leukemia 1 (MLL1) protein belongs to the SET1 family and catalyzes H3K4 methylation in all three states (mono-, di-, and tri-). Also, it is required for the maintenance of normal hematopoiesis. Dysregulation of the function of MLL1 is responsible for acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML). The MLL1 core complex consists of MLL1, WDR5, Ash2L, RBBP5, and DPY30. Hence, disturbing the MLL1-WDR5 interaction is an attractive approach for MLL-rearranged leukemias to downregulate the methylation level of H3K4 and the overexpression of Hox genes, which are known to induce leukemia.161
The outcome of cell-based screening on 1200 compounds from an in-house approved drug database was rabeprazole 71 (Fig. 33), which inhibited the MLL1-WDR5 interaction competitively. It displayed antiproliferation activity selective for cancer cell lines such as MLL-fusion leukemia cell lines MV4-11 (MLL-AF4) and MOLM-13 (MLL-AF9). However, it had no inhibitory effect on K562 (wild-type MLL1). Moreover, it downregulated the expression of the Hoxa9 and Meis1 genes.
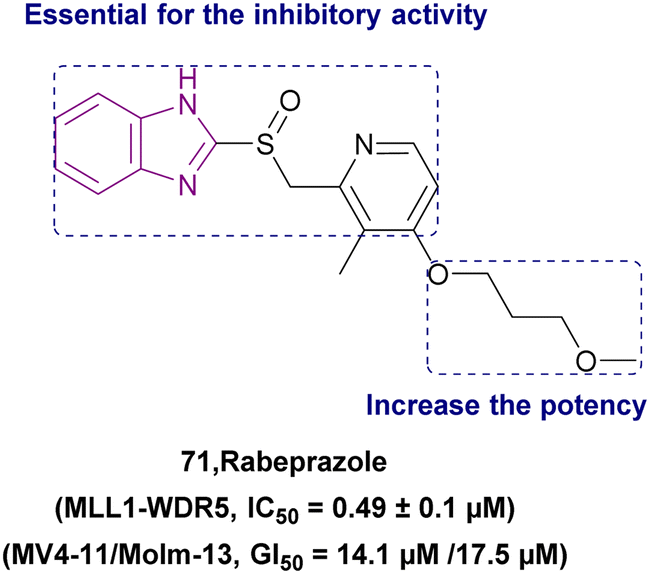 | ||
| Fig. 33 Chemical structure, IC50, SAR, and in vitro potency of MLL1 inhibitor 71. | ||
The MLL1-WDR5 complex was competitively inhibited by further proton pump inhibitors such as omeprazole 72, esomeprazole 73, lansoprazole 74, and R-lansoprazole 75, showing anti-proliferation effects against MV4-11 (MLL-AF4) and Molm-13 (MLL-AF9) (Fig. 34). The SAR of omeprazole 72 showed that removing –OCH3 from its benzimidazole ring did not affect its biochemical potency. Also, its biochemical potency was unaffected by the addition of a difluoromethoxy moiety to C-5 on the benzimidazole ring, but the MLL1-WDR5 inhibition activity decreased by the removal of the pyridine ring substituents or their replacement with a phenyl ring. In addition, the benzimidazole group was inserted into the central channel of WDR5, and the sulfoxide chain and benzimidazole ring were necessary for the inhibitory action.
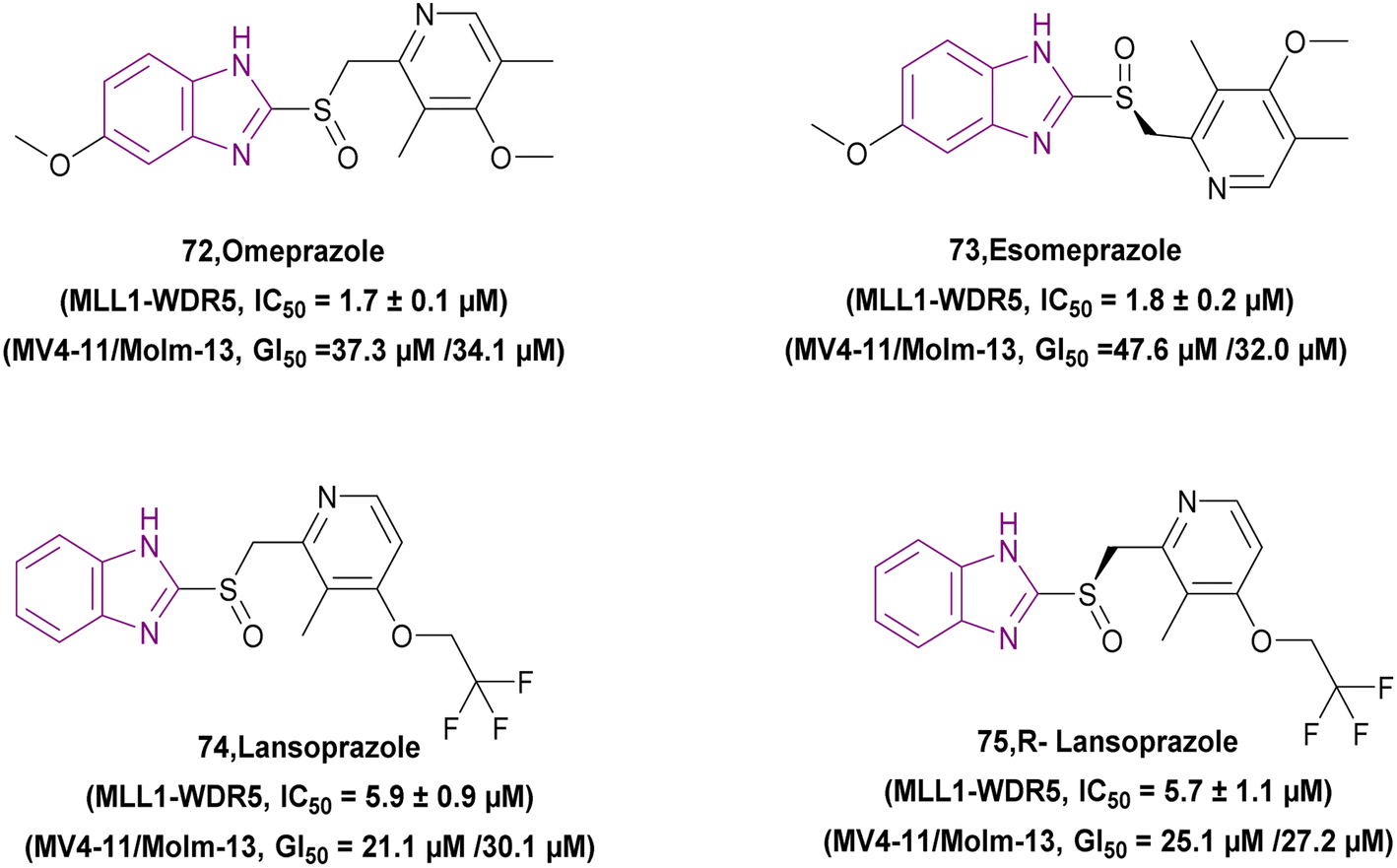 | ||
| Fig. 34 Chemical structure, IC50, and in vitro potency of MLL1 inhibitors 72–75. | ||
In the fluorescence polarization (FP) assay, lansoprazole 74 and (R)-lansoprazole 75 had similar activity, suggesting that the sulfoxide-induced conformational isomerism did not significantly impact inhibitory activity. Rabeprazole 71, carrying a hydrophobic chain, showed the most potent activity in disturbing the MLL1-WDR5 interaction. More modifications are needed to repurpose the scaffold of substituted 2-pyridyl methyl/sulfinyl benzimidazole to eliminate its proton pump inhibitory effects in the future.161
2.2.1.1.3 Benzimidazole derivatives as polycomb repressive complex 2 (PRC2) inhibitors. PRC2 is a methyltransferase that responsible for the mono/di/trimethylation of histone H3k27 and is comprised of various catalytic subunits, including EZH1/2, EED, and SUZ12.8 Heterozygous mutations in EZH2 cause hyper-trimethylation in H3K27, causing GC B-cell type diffuse large B-cell lymphoma (GCB-DLBCL) and follicular lymphoma. Importantly, the interaction between the EZH2 and EED subunits of PRC2 is crucial for the trimethylation of H3K27. Thus, destabilizing of the PRC2 complex by inhibiting the interaction of EZH2-EED subunits can be a good target in the treatment of cancer.162
Virtual screening of 1000 compounds revealed that the FDA-approved astemizole 76 drug could displace the EZH2 peptide with an IC50 of 93.80 μM by binding to EED. Thus, it competed with EZH2, causing dissociation of the PRC2 complex as the benzimidazole ring bound to the hydrophobic groove of EED. This study proved the invalidity of previous studies, which explained that the antiproliferative effect of astemizole was due to the blockage of the EAG1 ion channel. The combination of astemizole and EPZ (an EZH2 inhibitor) had a synergistic effect on DB and Toledo cells.162 Given that the distance between the two nitrogen atoms was essential for the formation of hydrogen bonds, the optimization of astemizole led to DC-PRC2in-01 77 by substituting the 4-aminopiperidine of astemizole with 3-aminopyrrolidine (Fig. 35). The methoxy group of astemizole and DC-PRC2in-01 maintained potency due to the generation of more hydrophobic contacts. DC-PRC2in-01 77 inhibited the interaction of EZH2-EED by binding to EED and arrested the cell cycle at the G0/G1 phase. The Pfeiffer cell line was the most sensitive cell line to DC-PRC2in-01 77, according to studies using the SU-DHL-4, Karpsa-422, DB, and Pfeiffer cell lines to demonstrate its antiproliferative activity.163
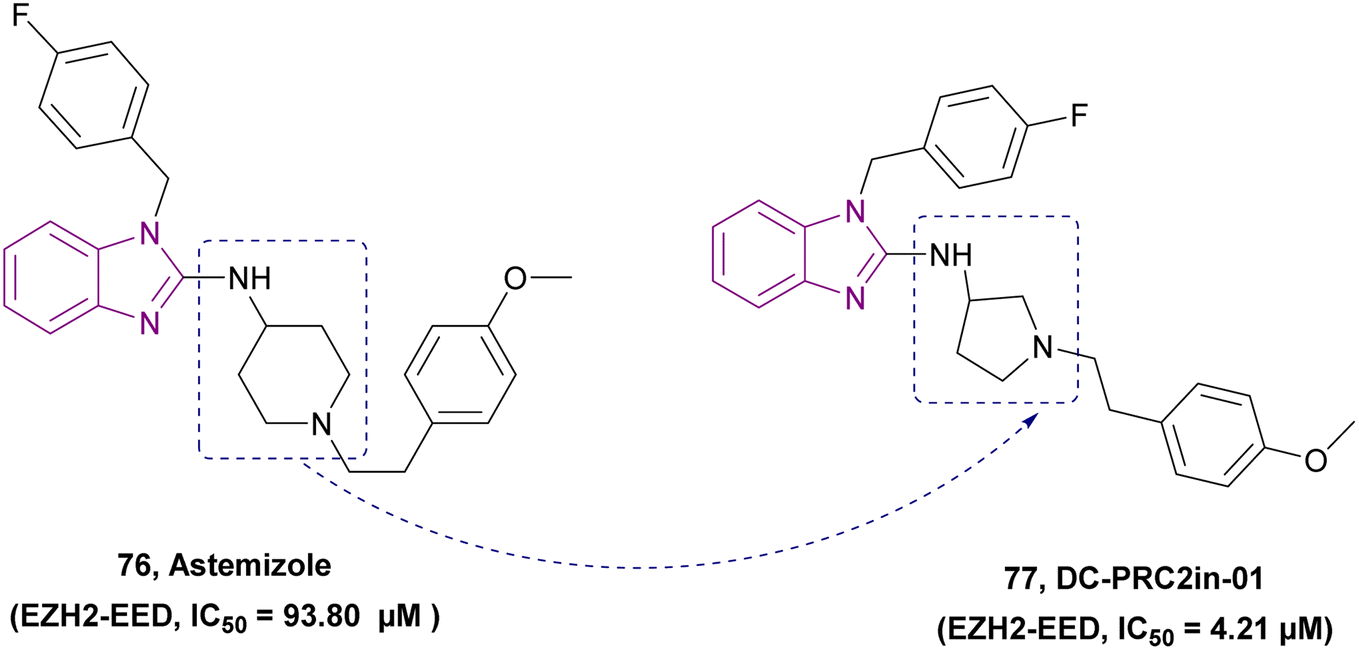 | ||
| Fig. 35 Chemical structure and IC50 of PRC2 inhibitors 76–77. | ||
2.2.1.1.4 Benzimidazole derivatives as G9a inhibitors. G9a (also known as KMT1C or EHMT2) is a histone methyltransferase of histone H3 (H3K9me1 and H3K9me2). G9a is overexpressed in various human cancers, such as prostate cancer, leukemia, hepatocellular carcinoma, pancreatic cancer, lung cancer, and breast cancer.15,164
The chemical structure of compounds 78–81, which are G9a inhibitors, is provided in Fig. 36. Virtual screening for compounds from the ChemBridge CORE library followed by in silico studies revealed 10 hits with a benzoxazole moiety in 4 of them. The benzoxazole moiety was subjected to further optimization by replacing it with a benzimidazole moiety and introducing phenylpropyl, a strong hydrophobic group, to the nitrogen atom of benzimidazole. Compound 78 was the most selective and potent inhibitor for G9a with antiproliferative activity when tested in MCF7 cells. The MTT assay showed that MCF-7 was the most sensitive cell line to it among five different cancer cell lines (MCF-7, T-47D, MDA-MB-231, MDA-MB-468, and MDA-MB-435 cells). It could induce autophagy via AMPK in MCF7 cells. In addition, high concentrations of 78 could induce apoptosis through p21-Bim signalling cascades in MCF7 cells. Thus, it activated the tumour suppressor p53 by inhibiting G9a, which regulated p21 activation.164
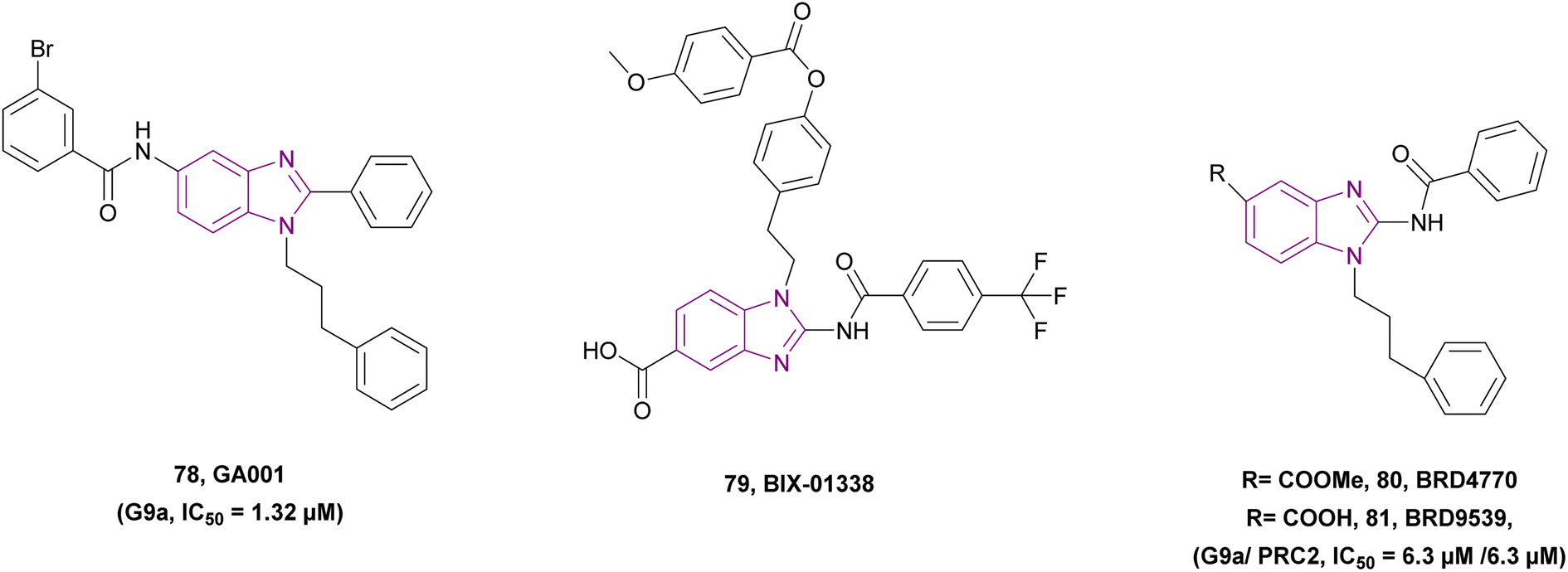 | ||
| Fig. 36 Chemical structure and IC50 of G9a inhibitors 78–81. | ||
Compound 80, an analogue of BIX-01338 79, was found to inhibit G9a more effectively in cells than the latter. 80 is a SAM mimetic prodrug, which is converted to 81 (the active form) in cells. Compound 81 was a more potent biochemical G9a inhibitor than 80, but it had no activity in cell-based assays due to its impaired cell permeability. Thus, it must be in the methyl-ester form (80). Compound 81 inhibited G9a and PRC2 activity, but there was no evidence for PRC2 inhibition by 80. Compound 80 was nontoxic and induced a senescent phenotype in a pancreatic cancer cell line, which inhibited both anchorage-independent and anchorage-dependent, and caused cell-cycle arrest at the G2/M phase. In addition, compound 80 activated the ATM pathway without inducing DNA damage, while the ATR pathway was not affected.165
2.2.1.2 Benzimidazole derivatives as arginine methyltransferases inhibitors. The post-translational methylation of arginine residues is catalyzed by protein arginine methyltransferases (PRMTs) type I–III; type I PRMT enzymes (PRMT1, -2, -3, -4, -6, and -8), type II enzymes (PRMT5 and PRMT9), and type III enzymes (PRMT7).166 These enzymes carry the methyl group from S-adenosylmethionine (AdoMet) to a terminal guanidino nitrogen of arginine, which resulted in S-adenosylhomocysteine (AdoHcy) and methylarginine.167 Importantly, PRMT inhibitors can reverse drug resistance and sensitize cancer cells to anti-cancer therapies, especially PRMT5.168
The increased PRMT5 levels in lymphomas, breast cancer, lung cancer, colorectal cancer, and glioblastoma were validated as a target in mantle cell lymphoma and glioblastoma. The optimization of compound 82, a PRMT5 inhibitor, and its SAR studies led to the development of compound 83, which selectively inhibited PRMT5 with IC50 of 0.33 μM and Kd of 0.987 μM. Compound 83 is a SAM competitive inhibitor and a peptide noncompetitive inhibitor (Fig. 37 and 38). The benzimidazole ring and the phenyl ring were crucial for maintaining activity. Substitutions at C-4 of the benzimidazole caused a loss of activity, but methoxy substitution at C-5 improved the potency. The methoxycarbonyl on the phenyl ring must be retained as the oxygen of the carbonyl forms a hydrogen bond with the L315 residues. It was sensitive only to MV4-11 cells, with an EC50 of 6.53 μM.166
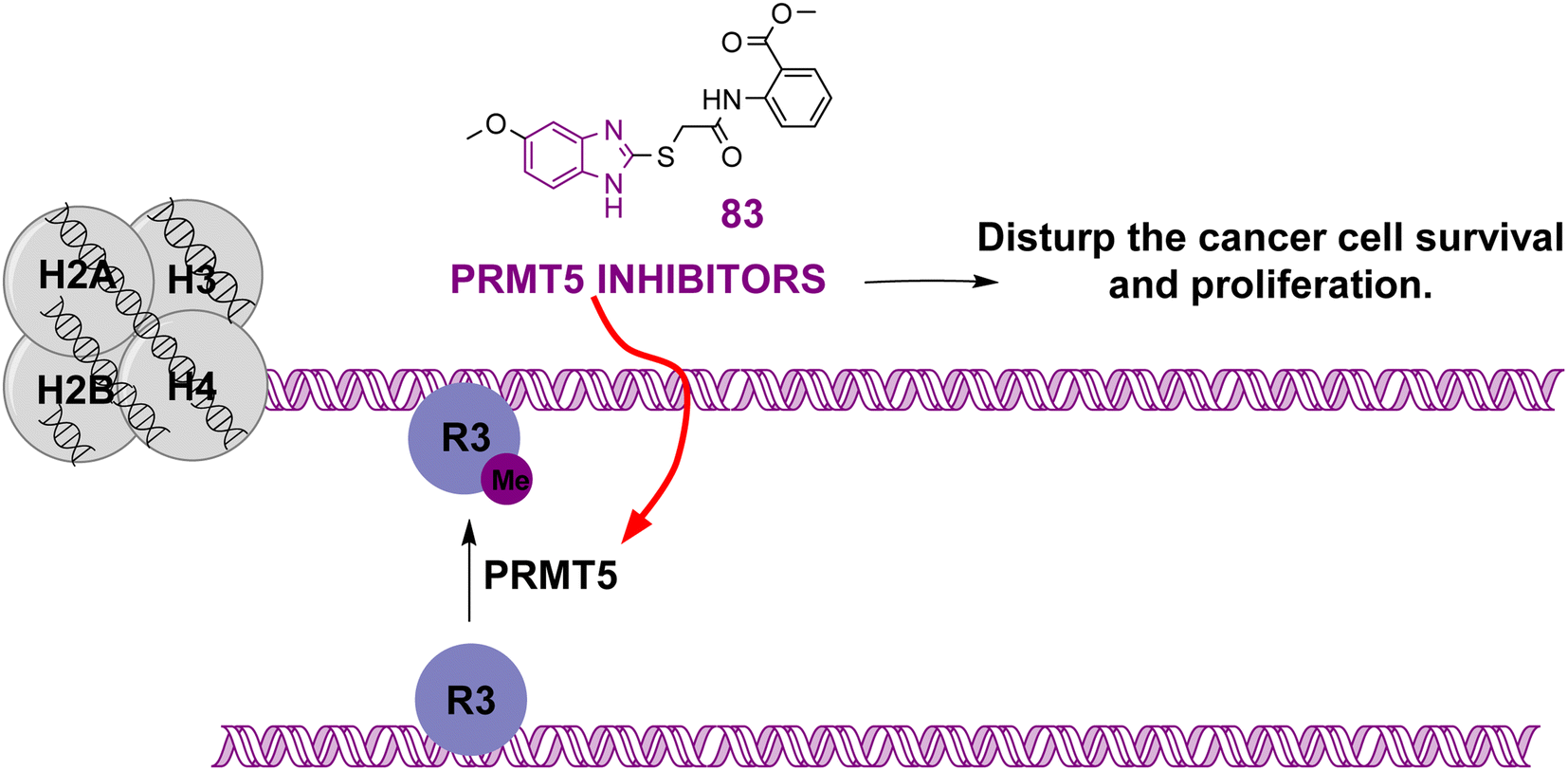 | ||
| Fig. 37 Effects of benzimidazole derivatives act as arginine methylation inhibitors on cancer cell growth and gene expression. | ||
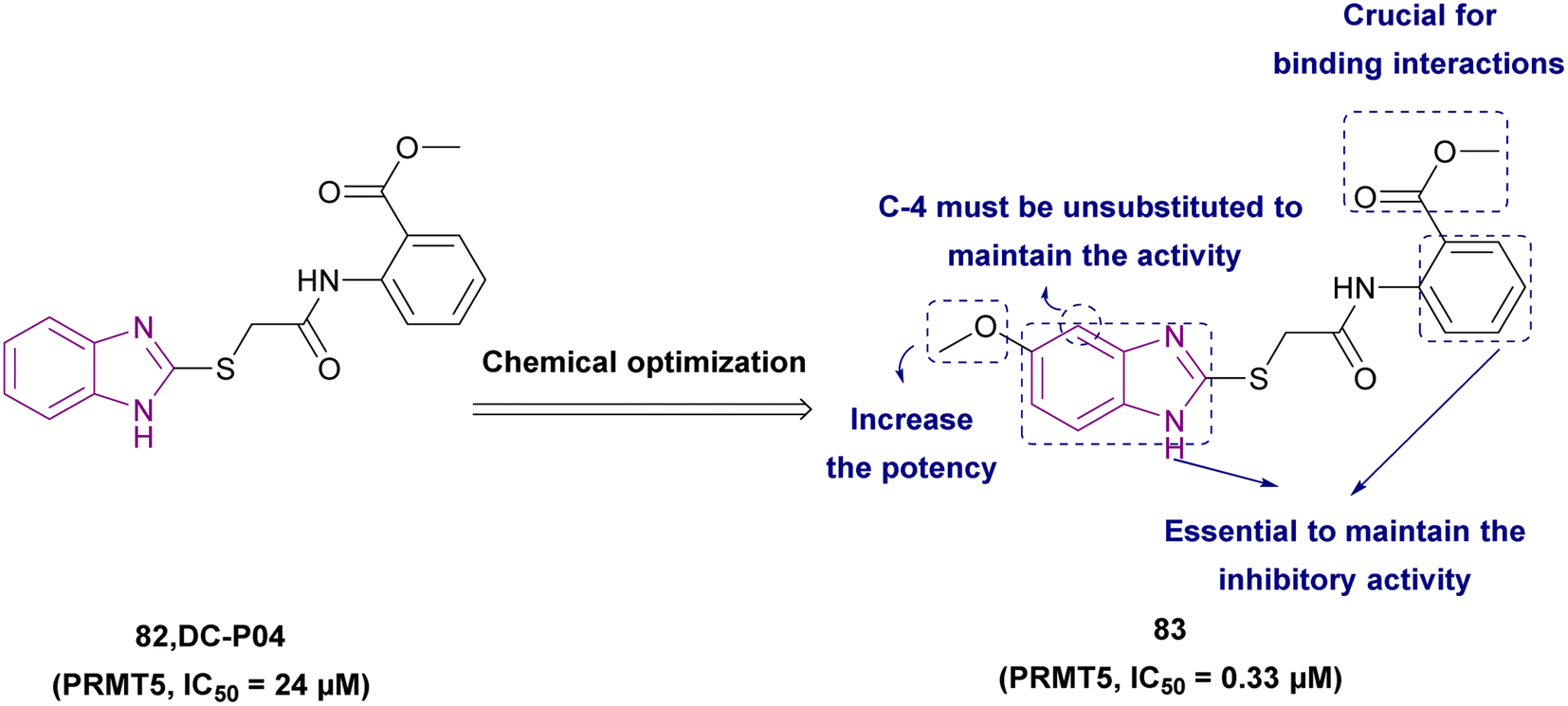 | ||
| Fig. 38 Chemical structure and IC50 of PRMT5 inhibitors 82–83. | ||
A new triazole-benzimidazole derivative, 84, is a PRMT5 inhibitor with anti-tumour activity against the MCF7, DU145, PC3, and HepG2 cell lines. The benzimidazole ring formed π–π interactions and H-bonds with the active site residue. It was synthesized through a series of reactions, including Sonogashira coupling, HATU-promoted amide coupling, and copper-catalyzed cycloaddition reaction to form 84. Also, compounds 85, 86, and 87 had inhibitory activity against cancer cells and PRMT5 inhibition, but to a lesser extent than 84 (Fig. 39).169
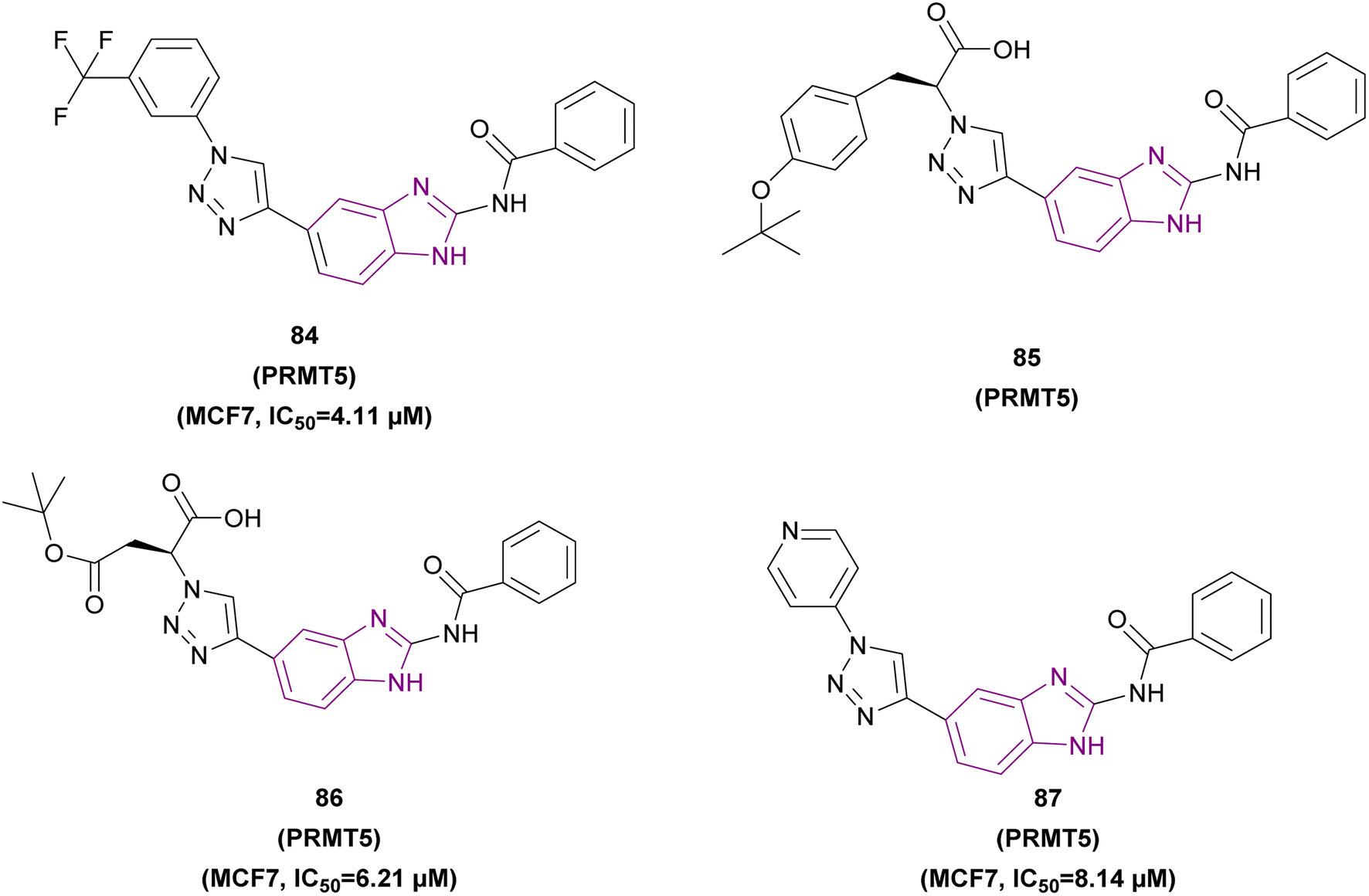 | ||
| Fig. 39 Chemical structure, IC50, and in vitro potency of PRMT5 inhibitors 84–87. | ||
2.2.2.1 Benzimidazole derivatives as jumonji domain-containing protein (JMJD) inhibitors. Jumonji domain-containing proteins (JMJDs) are a class of histone lysine demethylases.172 The KDM4 subfamily consists of KDM4A-E (JMJD2A-E). KDM4 demethylates di- and tri-methylated lysine 9 (KDM4A-E) and lysine 36 (KDM4A-C) of histone H3 and requires Fe2+ and α-ketoglutarate (α-KG) as cosubstrates.173,174
JMJD2A is a promoter of colon, lung, and breast cancer cell proliferation. An abnormality in the expression of JMJD2A can lead to colon cancer, lung cancer, and breast cancer. Due to the low cell permeability of 5-carboxy-8-hydroxyquinoline, it was optimized using structure-based design, leading to compound 88 (Fig. 40), which was synthesized through a series of reactions, including the Doebner–Miller reaction. It selectively inhibited JMJD2A with an IC50 of 19.0 μM and had anti-proliferative activity against HCT116, MCF-7, and A549 cell lines with good aqueous solubility and permeability.172
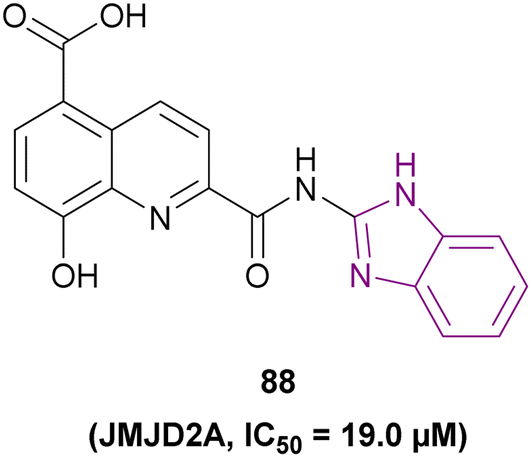 | ||
| Fig. 40 Chemical structure and IC50 of JMJD inhibitor 88. | ||
JMJD inhibitors 89 and 90 are illustrated in Fig. 41. A library of 32032 compounds was screened and filtered by fluorescence-based and FDH-mediated coupled enzyme assays. CBN-209350 89 was revealed after choosing PAINS compounds and excluding false-positive compounds. CBN-209350 had a benzimidazole pyrazolone scaffold with KDM4A-E selectivity. Also, it inhibited KDM2A and PHF-8. Higher concentrations of CBN-209350 had cytostatic effects on LnCaP, DU145, and PC-3 cell lines given that it was less permeable to the cell membrane because of its large polar surface area and low clogP value. Its safety for non-cancerous cells was established, and it produced unique biphasic inhibition curves in both the fluorescence assay and ELISA.174
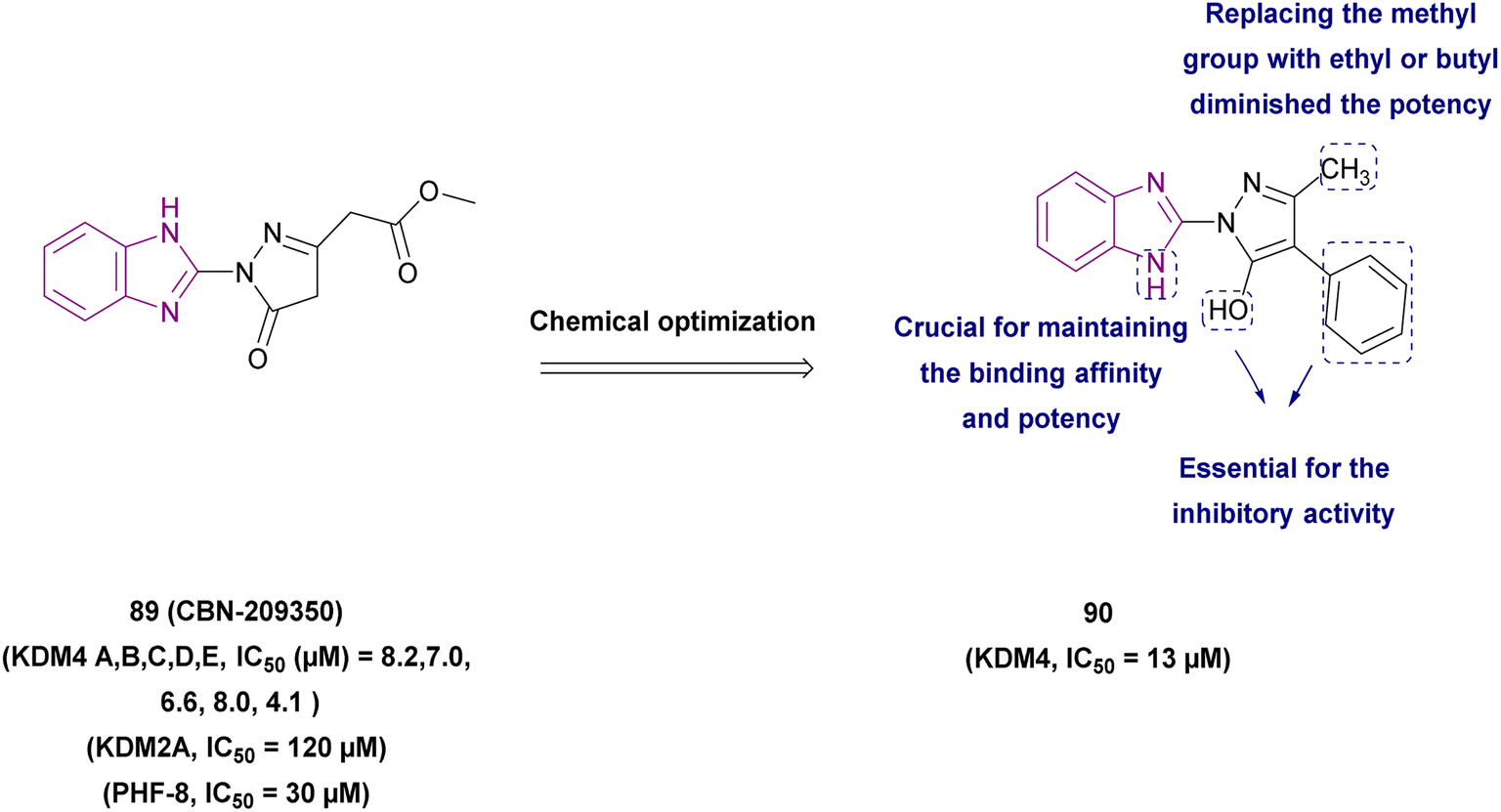 | ||
| Fig. 41 Chemical structure and IC50 of JMJD inhibitors 89 and 90. | ||
Carter et al. optimized CBN-209350 89, resulting in compound 90, given that CBN-209350 89 lacks the pyrazole-substituted sidechains that mimic the terminal ethyl carboxylate present in the α-KG and are necessary for interacting with the active site of the enzyme. Compound 90 had an uncompetitive and non-competitive mechanism of inhibition with the α-KG cofactor and peptide substrate, respectively. It downregulated AR-dependent PSA gene expression in LnCaP cells and inhibited the growth of all the PCa cell lines tested, including LnCaP cells, DU145 cells, PC3 cells, and a non-disease control cell line (HuPrEC). In compound 90, the phenyl group (nonpolar, electron-rich sidechain) and hydroxyl moiety were crucial, given that their replacement would reduce the inhibitory activity. The bidentate heterocyclic nitrogen atoms were essential for binding affinity, given that replacing the benzimidazole NH moiety with a sulfur atom or methylating the NH moiety reduced the potency. Furthermore, the potency was diminished when the methyl group was replaced with ethyl or butyl. Replacing the benzimidazole moiety with pyridine or 3-chloropyridazine decreased the potency.173
2.2.3.1 Benzimidazole derivatives as UHRF1 inhibitors. UHRF1 is a histone-methylated reader. UHRF1 upregulation resulted in silencing of tumour suppressor genes. Nocodazole 91 is a UHRF1 inhibitor (Fig. 42) that causes downregulation of UHRF1. It acquired an L-shaped conformation in the binding cavity, with the core benzimidazole ring surrounded by Leu462, Gly464, Gly465, and Tyr466, according to molecular docking studies conducted within the SRA domain of UHRF1.175
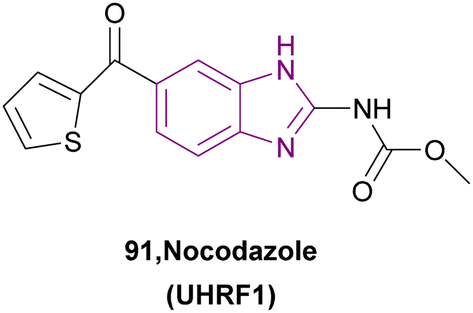 | ||
| Fig. 42 Chemical structure of UHRF1 inhibitor 91. | ||
2.2.3.2 Benzimidazole derivatives as CBX7 inhibitors. CBX7, a subunit of polycomb repressive complex 1 (PRC1), with its chromodomain (CBX7ChD) is a reader of H3K27me3 (ref. 176) and is responsible for decreasing the transcription of p16INK4a, a tumour suppressor gene, in prostate cancer cells. Hence, CBX7ChD is an important target in cancer treatment. In silico chemical screening of approximately 100
000 drug-like compounds resulted in MS452 92, which had poor cell permeability. Consequently, a ligand development strategy and cell-based SAR study discovered MS351 93, which is a chromodomain antagonist that inhibited H3K27me3 binding to CBX7ChD and selectively bound to CBX7ChD (Fig. 43). The crystal structure of the CBX7ChD/93 complex revealed that the iminobenzimidazole core interacted with the aromatic cage residues Phe11, Trp32, and Trp35, which normally house the methyl-lysine of the bound H3K27me3 peptide. It upregulated the expression of p16INK4a in human prostate cancer PC3 cells. Further chemical optimization of 93 was suggested, such as a conformationally constrained linker connecting the iminobenzimidazole and dichlorobenzene and certain substituent groups on the iminobenzimidazole might optimize its interactions with the cage residues.177
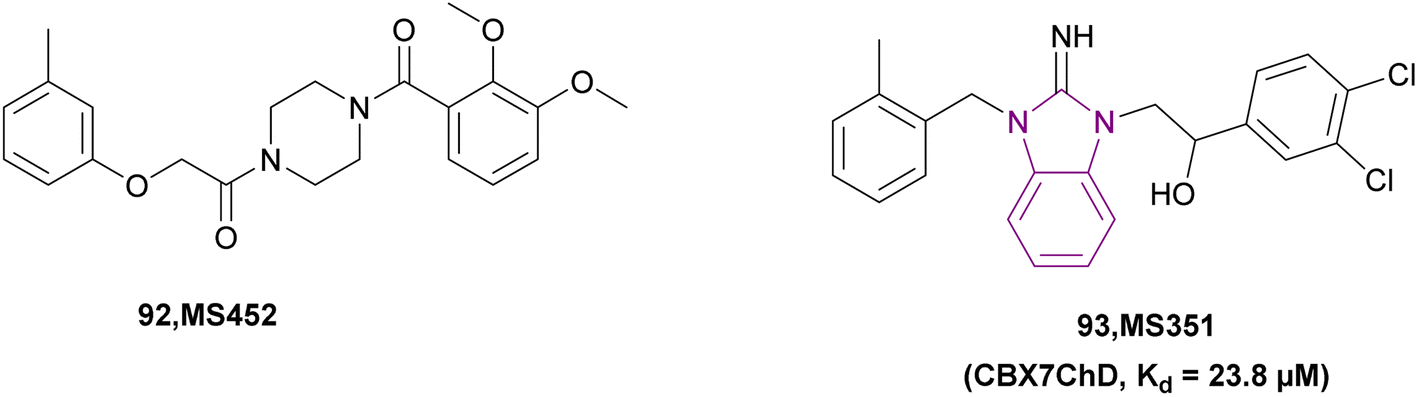 | ||
| Fig. 43 Chemical structure and Kd of CBX7 inhibitors 92 and 93. | ||
2.3 Benzimidazole derivatives inhibit other epigenetic enzymes
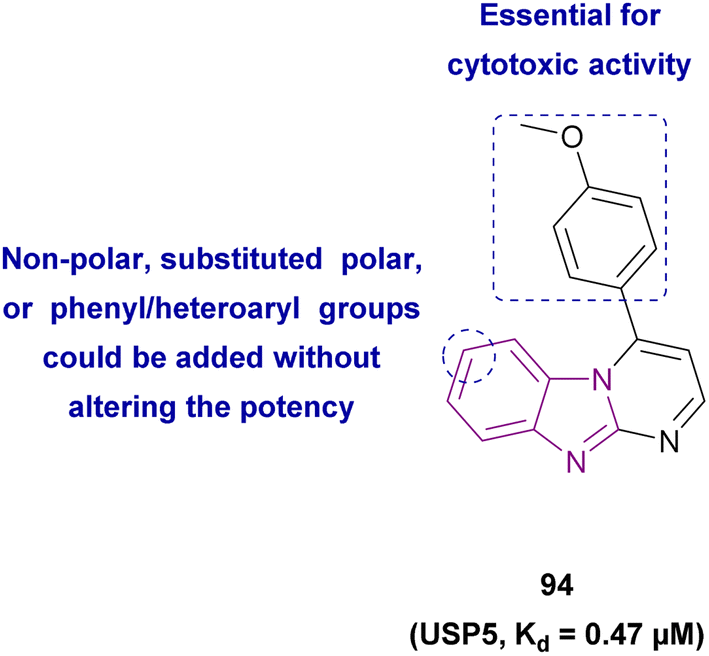 | ||
| Fig. 44 Chemical structure and Kd of USP5 inhibitor 94. | ||
GSK-484 95 is a PAD4 inhibitor, where replacing its the N-cyclopropylmethyl group on the indole ring with N-benzyl resulted in a more potent PAD4 inhibitor, compound 96 (Fig. 45). Compound 96 is a non-covalent PAD4 inhibitor, which was synthesized through the copper-catalyzed C–H arylation of benzimidazoles. The indole ring was important for the activity, given that replacing it with benzofuran, naphthalene, and pyridine diminished the activity. In vitro studies of 96 in 4T1 cells caused a marked decrease in viability, and the MTT assay showed potent activity.180
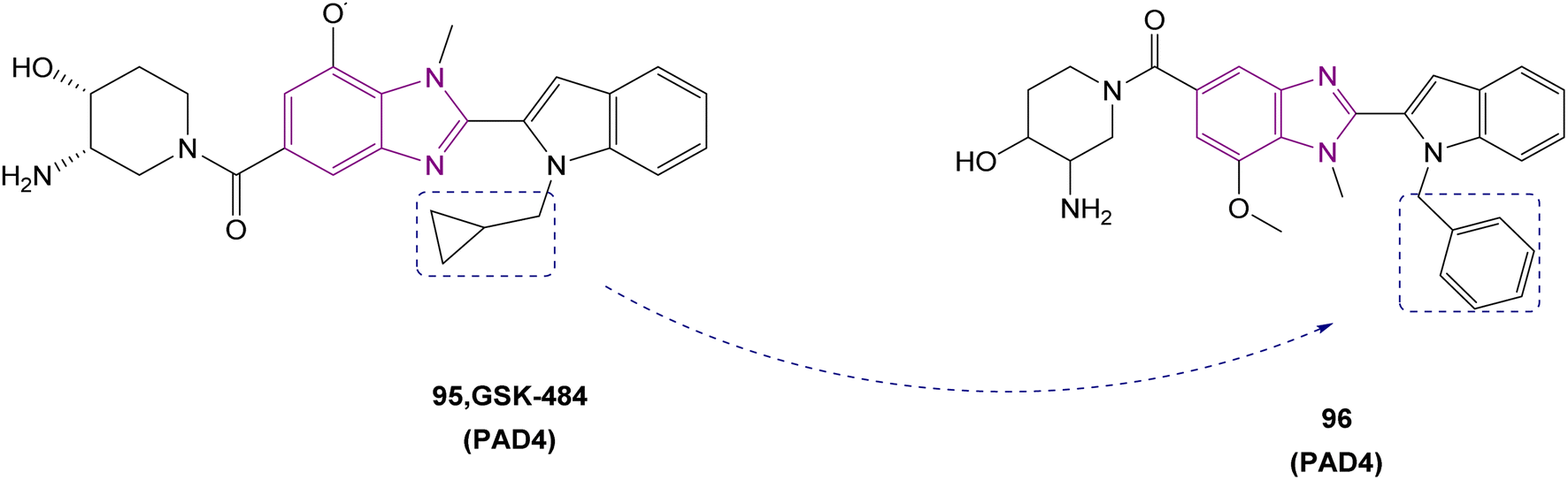 | ||
| Fig. 45 Chemical structure and EC50 of PAD inhibitors 95 and 96. | ||
PAD2 is overexpressed in luminal breast cancers. Compound 97 is a potent PAD2 inhibitor with selectivity towards PAD2 over other PADs with a large therapeutic window (Fig. 46).181
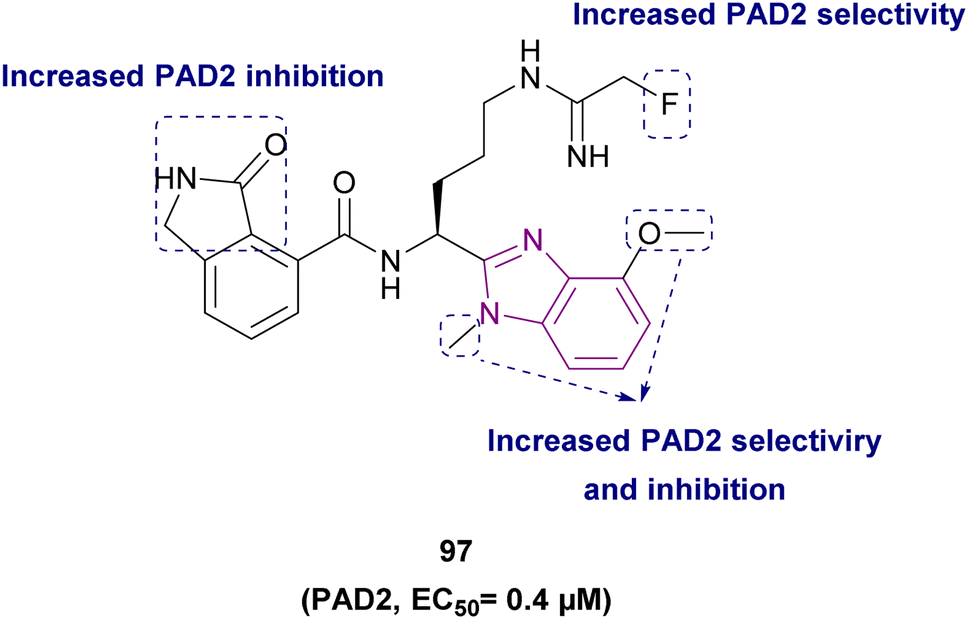 | ||
| Fig. 46 Chemical structure and EC50 of PAD inhibitor 97. | ||
3 Conclusions and future perspectives
The benzimidazole is a promising scaffold. It has many biological activities given that it mimics the purine ring. It this review, it was highlighted that the benzimidazole ring has been implicated in the structure of many epigenetic inhibitors, especially in HDACIs, and targeting different epigenetic enzymes, which plays a critical role in cancer therapy. HDAC inhibitors containing a benzimidazole ring could act as pan-inhibitors, dual inhibitors, or selective-isoforms inhibitors, where benzimidazole acts as a cap, linker, or zinc-binding group, respectively. These compounds had antiproliferative activity against many cell lines in the nanomolar range. Further research and investigations can be beneficial in synthesizing less resistant and selective HDAC inhibitors and dual acting benzimidazole derivatives, given the promising future of treating cancer with epigenetics, and especially because the dual-targeting of HDAC as a single target did not show great efficiency in cancer therapy. Alternatively, further efforts should focus on improving their pharmacokinetics and optimizing the synergistic therapeutic efficacy of the drug in vivo and maximizing its safety profile. The benzimidazole moiety plays a crucial role in inhibiting other epigenetic targets through crucial interactions with the essential residues of the target pocket. However, this area of research was not fully and precisely covered and needs further research to make the benzimidazole scaffold well established in the field of epigenetic target inhibitors. Also, benzimidazole derivatives that act as PRMT5 inhibitors are a great step in overcoming the anticancer drug resistance. However, their exact mechanism is not known yet. Synthesizing non-hydroxamic ZBG as in compound 19 and 21 was beneficial given that the hydroxamic moieties are responsible for side-effects such as nausea, thrombocytopenia, anaemia, and other metabolic abnormalities due to their strong chelation with the zinc ion. Thus, researchers are trying to find different ZBGs with weaker binding and greater selectivity. The most promising benzimidazole derivatives were pracinostat 2, a pan-HDAc inhibitor that entered phase I and phase II clinical trials for treating hematologic malignancies and solid tumours, tinostamustine 38, a DNA alkylating and HDAC inhibitor, which entered phase I/II studies in advanced solid tumours and phase I study in relapsed/refractory (R/R) Hodgkin lymphoma (HL), exhibiting haematological and thrombocytopenia adverse effects, and Inobrodib (CCS1477) 48, which inhibited the p300/CBP bromodomain in phase I/II clinical trials in non-Hodgkin lymphoma (including B-cell lymphoma), AML, and multiple myeloma. This area of research seems to be promising and requires more effort from researchers. Accordingly, this review article can help researchers design better, less resistant, and less toxic benzimidazole derivative anticancer agents.Abbreviations
| 22RV1 | Human prostate carcinoma epithelial cell line |
| A2780 | Ovarian cancer cell line |
| A549 | Human lung carcinoma cell line |
| AML | Acute myeloid leukaemia |
| Ash2L | Absent, small or homeotic 2-like protein |
| BAX | Bcl-2-associated X protein |
| BCL3 | B-cell chronic lymphatic leukemia protein 3 |
| BCL2 | Anti-apoptotic B cell lymphoma 2 |
| BRD | Bromodomain |
| BRDT | Bromodomain testis-specific protein |
| BRPF | Bromodomain and PHD finger-containing family |
| CBP | cAMP-regulated-enhancer (CRE)-binding protein (CREB)-binding protein |
| CBX7 | Chromobox protein homolog 7 |
| CCRF-CEM | Blood leukemia |
| CD86 | Cluster of differentiation 86 |
| CDK | Cyclin-dependent kinase |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A |
| CK2 | Casein kinase 2 |
| CREB | Cyclic-AMP response element binding |
| CRPC | Castration-resistant prostate cancer |
| DNMT | DNA methyltransferases |
| DOT1L | Disruptor of telomeric silencing 1 (Dot1)-like proteins |
| DU145 | Human prostate cancer cell line |
| E2F1 | E2 promoter binding factor 1 |
| EED | Embryonic ectoderm development |
| ENL | Eleven-nineteen-leukemia protein |
| EZH2 | Enhancer of zeste homolog 2 |
| G9a | Histone H3 Lys-9 methyltransferase |
| GADD45A | Growth arrest and DNA damage-inducible 45 |
| GNAT | Gen-5-related N-acetyltransferase |
| H1975 | Anlotinib-resistant lung cancer cell line |
| HATs | Histone acetyltransferases |
| HCC827 | Lung adenocarcinoma cell line |
| HCC4017 | Lung large cell carcinoma |
| HCC4018 | Small cell lung cancer |
| HCT-116 | Human colorectal carcinoma cell line |
| HDACs | Histone deacetylases |
| HepG2 | Human liver cancer cell line |
| hERG | Human ether-a-go-go-related gene |
| HKMTs | Histone lysine methyltransferases |
| HL-60 | Human Caucasian promyelocytic leukemia cell line |
| HL-60/adr | Adult acute myeloid leukemia cell line |
| HL-60/vinc | Adult acute myeloid leukemia cell line |
| HOXA9 | Homeobox A9 |
| Hsp90 | Heat shock protein 90 |
| HT-29 | Human colorectal adenocarcinoma cell line |
| IRF4 | Interferon regulatory factor 4 |
| JAK1 | Janus kinase 1 |
| JmjC | Jumonji C domain-containing demethylases |
| JNK | c-Jun N-terminal kinase |
| Kac | Acetylated lysine |
| KDMs | Lysine demethylase enzymes |
| KLK3 | Kallikrein related peptidase 3 |
| KMTs | Lysine methyltransferase |
| LSD1 | Lysine-specific demethylase 1 |
| LSD2 | Lysine-specific demethylase 2 |
| KSP | Kinesin spindle protein |
| KSVH | Kaposi's sarcoma-associated herpesvirus |
| LnCaP | Lymph node carcinoma of the prostate cell line |
| lncRNA | Long non-coding RNAs |
| MBD | Methyl-CpG binding domain family |
| MCF7 | Human breast cancer cell line |
| MDA-MB-231 | Triple-negative breast cancer(TNBC) cell line |
| MDA-MB-435 | Amelanotic melanoma cell line |
| MDA-MB-468 | Triple-negative breast cancer(TNBC) cell line |
| MEIS1 | Myeloid ecotropic viral integration site 1 |
| MLL1 | Mixed lineage leukemia 1 |
| MM | Multiple myeloma |
| MOLM-13 | Human leukemia cell line |
| MOLT-4 | T lymphoblast cell line |
| MTDLs | Multitarget-directed ligands |
| MTT | (3-(4,5-Dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) assay |
| MV4-11 | Acute monocytic leukemia cell line |
| MYC | Master regulator of cell cycle entry and proliferative metabolism |
| MYCN protein | Avian myelocytomatosis viral oncogene neuroblastoma derived homolog |
| MYST | MOZ/YBF2/SAS2/TIP60 |
| NAD+ | Nicotinamide adenine dinucleotide |
| ncRNA | Non-coding RNA |
| NF-κB | Nuclear factor kappa B |
| OPM-2 | Human myeloma cell line |
| OVCAR-5 | High-grade serous ovarian adenocarcinoma cell line |
| p16INK4a | Cyclin-dependent kinase inhibitor 2A |
| p21WAF1/Cip1 | Wildtype activating factor-1/cyclin-dependent kinase inhibitory protein-1 |
| p53 | Tumour protein P53 |
| PAD | Peptidyl arginine deiminase |
| PARP | Poly-ADP ribose polymerase |
| PBMC | Peripheral blood mononuclear cell |
| PC-3 | Human prostate cancer cell line |
| PHDs | Plant homeodomains |
| piRNA | Piwi-interacting RNA |
| PRC2 | Polycomb repressive complex 2 |
| PRMTs | Arginine methyltransferase |
| PUMA | p53 upregulated modulator of apoptosis |
| RAMOS | B lymphocyte cell line |
| RB1 | Retinoblastoma 1 |
| RBBP5 | Retinoblastoma binding protein 5 |
| SAM | S-adenosyl methionine |
| SCC-9 | Squamous carcinoma cell line |
| SET | Su(var)3–9, enhancer-of-zeste and trithorax |
| siRNA | Short interfering RNA |
| SK-OV-3 | Ovarian cancer cell line |
| snoRNAs | Small nucleolar RNAs |
| SRA | SET- and ring finger-associated domain family |
| STAT3 | Signal transducer and activator of transcription 3 |
| T-47D | Human breast cancer cell line |
| TAF1 | TATA-box binding protein associated factor 1 |
| TEAEs | Treatment-emergent adverse events |
| TET | Ten-to-eleven translocation |
| THP-11 | Leukaemic cancer cell lines |
| TNBC | Triple-negative breast cancer |
| TRAF2 | Tumour necrosis factor (TNF) receptor associated factor-2 |
| UHRF1 | Ubiquitin-like with containing PHD and RING finger domains 1 |
| USP5 | Ubiquitin-specific protease 5 |
| VCaP | Prostate cancer cell line |
| WDR | WD40 repeat domains |
| ZBD | Zinc-binding domain |
Data availability
No primary research results, software or code has been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- A. K. A. Bass, E.-S. M. Nageeb, M. S. El-Zoghbi, M. F. A. Mohamed, M. Badr and G. E.-D. A. Abuo-Rahma, Utilization of cyanopyridine in design and synthesis of first-in-class anticancer dual acting PIM-1 kinase/HDAC inhibitors, Bioorg. Chem., 2022, 119, 105564 CrossRef CAS PubMed.
- H. Yuan, Y. Lu, Y. Feng and N. Wang, Epigenetic inhibitors for cancer treatment, International Review of Cell and Molecular Biology, Academic Press, 2023 Search PubMed.
- F. Recillas-Targa, Cancer Epigenetics: An Overview, Arch. Med. Res., 2022, 53(8), 732–740 CrossRef CAS PubMed.
- S. Chakraborty and T. Rahman, The difficulties in cancer treatment, Ecancermedicalscience, 2012, 6, ed16 Search PubMed.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra and D. K. Pandey, et al., Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics, Genes Dis., 2023, 10(4), 1367–1401 CrossRef CAS PubMed.
- E. Hollwey, A. Briffa, M. Howard and D. Zilberman, Concepts, mechanisms and implications of long-term epigenetic inheritance, Curr. Opin. Genet. Dev., 2023, 81, 102087 CrossRef CAS.
- A. Farsetti, B. Illi and C. Gaetano, How epigenetics impacts on human diseases, Eur. J. Intern. Med., 2023, 114, 15–22 CrossRef CAS.
- B. Sreeshma and A. Devi, JARID2 and EZH2, the eminent epigenetic drivers in human cancer, Gene, 2023, 879, 147584 CrossRef CAS.
- S. Biswas and C. M. Rao, Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy, Eur. J. Pharmacol., 2018, 837, 8–24 CrossRef CAS PubMed.
- D. Wang, Y. Zhang, Q. Li, Y. Li, W. Li and A. Zhang, et al., Epigenetics: Mechanisms, potential roles, and therapeutic strategies in cancer progression, Genes Dis., 2023, 11, 101020 CrossRef PubMed.
- D. Zhu, Y. Zhang and S. Wang, Histone citrullination: a new target for tumors, Mol. Cancer, 2021, 20(1), 90 CrossRef CAS.
- S. Ropero and M. Esteller, The role of histone deacetylases (HDACs) in human cancer, Mol. Oncol., 2007, 1(1), 19–25 CrossRef CAS PubMed.
- J. Liu, J. Kelly, W. Yu, D. Clausen, Y. Yu and H. Kim, et al., Selective Class I HDAC Inhibitors Based on Aryl Ketone Zinc Binding Induce HIV-1 Protein for Clearance, ACS Med. Chem. Lett., 2020, 11(7), 1476–1483 CrossRef CAS.
- L. L. Marzochi, C. I. Cuzziol, C. H. V. D. Nascimento Filho, J. A. dos Santos, M. M. U. Castanhole-Nunes and É. C. Pavarino, et al., Use of histone methyltransferase inhibitors in cancer treatment: A systematic review, Eur. J. Pharmacol., 2023, 944, 175590 CrossRef CAS.
- Q. Liao, J. Yang, S. Ge, P. Chai, J. Fan and R. Jia, Novel insights into histone lysine methyltransferases in cancer therapy: From epigenetic regulation to selective drugs, J. Pharm. Anal., 2023, 13(2), 127–141 CrossRef.
- G. A. Holdgate, C. Bardelle, A. Lanne, J. Read, D. H. O'Donovan and J. M. Smith, et al., Drug discovery for epigenetics targets, Drug Discovery Today, 2022, 27(4), 1088–1098 CrossRef CAS.
- J. E. Lee and M.-Y. Kim, Cancer epigenetics: Past, present and future, Semin. Cancer Biol., 2022, 83, 4–14 CrossRef CAS PubMed.
- S. J. Nowak and V. G. Corces, Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation, Trends Genet., 2004, 20(4), 214–220 CrossRef CAS PubMed.
- T. Ozben, Basic principles of epigenetics: DNA methylation, histone modifications and non-coding RNAS, Clin. Chim. Acta, 2019, 493, S736 CrossRef.
- C. Seidel, C. Florean, M. Schnekenburger, M. Dicato and M. Diederich, Chromatin-modifying agents in anti-cancer therapy, Biochimie, 2012, 94(11), 2264–2279 CrossRef CAS PubMed.
- S. Talebian, H. Daghagh, B. Yousefi, Y. Ozkul, K. Ilkhani and F. Seif, et al., The role of epigenetics and non-coding RNAs in autophagy: A new perspective for thorough understanding, Mech. Ageing Dev., 2020, 190, 111309 CrossRef CAS.
- N. Abdelaziz, L. Therachiyil, H. Q. Sadida, A. M. Ali, O. S. Khan, M. Singh, et al., Epigenetic inhibitors and their role in cancer therapy, International Review of Cell and Molecular Biology, Academic Press, 2023 Search PubMed.
- Y. T. Lee, Y. J. Tan and C. E. Oon, Benzimidazole and its derivatives as cancer therapeutics: The potential role from traditional to precision medicine, Acta Pharm. Sin. B, 2023, 13(2), 478–497 CrossRef CAS PubMed.
- G. Satija, B. Sharma, A. Madan, A. Iqubal, M. Shaquiquzzaman and M. Akhter, et al., Benzimidazole based derivatives as anticancer agents: Structure activity relationship analysis for various targets, J. Heterocycl. Chem., 2022, 59(1), 22–66 CrossRef CAS.
- O. Ebenezer, F. Oyetunde-Joshua, O. D. Omotoso and M. Shapi, Benzimidazole and its derivatives: Recent Advances (2020–2022), Results Chem., 2023, 5, 100925 CrossRef CAS.
- P. K. Singh and O. Silakari, Chapter 2 – Benzimidazole: Journey From Single Targeting to Multitargeting Molecule, in Key Heterocycle Cores for Designing Multitargeting Molecules, ed. Silakari O., Elsevier, 2018, pp. 31–52 Search PubMed.
- D. W. Woolley, Some Biological Effects Produced By Benzimidazole And Their Reversal By Purines, J. Biol. Chem., 1944, 152(2), 225–232 CrossRef CAS.
- N. G. Brink and K. Folkers, Vitamin B12. VI. 5, 6-Dimethylbenzimidazole, a degradation product of vitamin B12, J. Am. Chem. Soc., 1949, 71(8), 2951 CrossRef CAS.
- J. B. Wright, The chemistry of the benzimidazoles, Chem. Rev., 1951, 48(3), 397–541 CrossRef CAS.
- H. E. Hashem and Y. El Bakri, An overview on novel synthetic approaches and medicinal applications of benzimidazole compounds, Arabian J. Chem., 2021, 14(11), 103418 CrossRef CAS.
- Y. Bansal and O. Silakari, The therapeutic journey of benzimidazoles: A review, Bioorg. Med. Chem., 2012, 20(21), 6208–6236 CrossRef CAS.
- B. Pathare and T. Bansode, Review-biological active benzimidazole derivatives, Results Chem., 2021, 3, 100200 CrossRef CAS.
- V. A. S. Pardeshi, N. S. Chundawat, S. I. Pathan, P. Sukhwal, T. P. S. Chundawat and G. P. Singh, A review on synthetic approaches of benzimidazoles, Synth. Commun., 2021, 51(4), 485–513 CrossRef CAS.
- A. A. El Rashedy and H. Y. Aboul-Enein, Benzimidazole derivatives as potential anticancer agents, Mini-Rev. Med. Chem., 2013, 13(3), 399–407 CAS.
- A. Sharma, V. Luxami and K. Paul, Purine-benzimidazole hybrids: Synthesis, single crystal determination and in vitro evaluation of antitumor activities, Eur. J. Med. Chem., 2015, 93, 414–422 CrossRef CAS PubMed.
- D. Sun, C. Wang, Y. Fan and J. Gu, Identification, structure elucidation and origin of a common pyridinium-thiocyanate intermediate in electrospray mass spectrometry among the benziamidazole-class proton pump inhibitors, J. Pharm. Anal., 2023, 13(6), 683–688 CrossRef PubMed.
- G. Yadav and S. Ganguly, Structure activity relationship (SAR) study of benzimidazole scaffold for different biological activities: A mini-review, Eur. J. Med. Chem., 2015, 97, 419–443 CrossRef CAS PubMed.
- K. Haider and M. Shahar Yar, Advances of Benzimidazole Derivatives as Anticancer Agents: Bench to Bedside, 2022 Search PubMed.
- B. K. Gajjela and M.-M. Zhou, Bromodomain inhibitors and therapeutic applications, Curr. Opin. Chem. Biol., 2023, 75, 102323 CrossRef CAS PubMed.
- M. Biel, V. Wascholowski and A. Giannis, Epigenetics – an epicenter of gene regulation: histones and histone-modifying enzymes, Angew Chem. Int. Ed. Engl., 2005, 44(21), 3186–3216 CrossRef CAS.
- R. Martínez, B. Di Geronimo, M. Pastor, J. M. Zapico, C. Coderch and R. Panchuk, et al., Multitarget Anticancer Agents Based on Histone Deacetylase and Protein Kinase CK2 Inhibitors, Molecules, 2020, 25(7), 1497 CrossRef.
- Y. K. Yoon, M. A. Ali, A. C. Wei, T. S. Choon, H. Osman and K. Parang, et al., Synthesis and evaluation of novel benzimidazole derivatives as sirtuin inhibitors with antitumor activities, Bioorg. Med. Chem., 2014, 22(2), 703–710 CrossRef CAS PubMed.
- S. Manku, M. Allan, N. Nguyen, A. Ajamian, J. Rodrigue and E. Therrien, et al., Synthesis and evaluation of lysine derived sulfamides as histone deacetylase inhibitors, Bioorg. Med. Chem. Lett., 2009, 19(7), 1866–1870 CrossRef CAS.
- C. W. Hsu, D. Shou, R. Huang, T. Khuc, S. Dai and W. Zheng, et al., Identification of HDAC Inhibitors Using a Cell-Based HDAC I/II Assay, J. Biomol. Screening, 2016, 21(6), 643–652 CrossRef CAS.
- S. E. Kassab, S. Mowafy, A. M. Alserw, J. A. Seliem, S. M. El-Naggar and N. N. Omar, et al., Structure-based design generated novel hydroxamic acid based preferential HDAC6 lead inhibitor with on-target cytotoxic activity against primary choroid plexus carcinoma, J. Enzyme Inhib. Med. Chem., 2019, 34(1), 1062–1077 CrossRef CAS.
- N. Cheshmazar, M. Hamzeh-Mivehroud, H. Nozad Charoudeh, S. Hemmati, J. Melesina and S. Dastmalchi, Current trends in development of HDAC-based chemotherapeutics, Life Sci., 2022, 308, 120946 CrossRef CAS.
- T. S. Ibrahim, T. A. Sheha, N. E. Abo-Dya, M. A. AlAwadh, N. A. Alhakamy and Z. K. Abdel-Samii, et al., Design, synthesis and anticancer activity of novel valproic acid conjugates with improved histone deacetylase (HDAC) inhibitory activity, Bioorg. Chem., 2020, 99, 103797 CrossRef CAS PubMed.
- G. N. Vaidya, P. Rana, A. Venkatesh, D. R. Chatterjee, D. Contractor and D. P. Satpute, et al., Paradigm shift of “classical” HDAC inhibitors to “hybrid” HDAC inhibitors in therapeutic interventions, Eur. J. Med. Chem., 2021, 209, 112844 CrossRef CAS PubMed.
- M. F. A. Mohamed, M. S. A. Shaykoon, M. H. Abdelrahman, B. E. M. Elsadek, A. S. Aboraia and G. E.-D. A. A. Abuo-Rahma, Design, synthesis, docking studies and biological evaluation of novel chalcone derivatives as potential histone deacetylase inhibitors, Bioorg. Chem., 2017, 72, 32–41 CrossRef CAS.
- J. Melesina, C. V. Simoben, L. Praetorius, E. F. Bülbül, D. Robaa and W. Sippl, Strategies To Design Selective Histone Deacetylase Inhibitors, ChemMedChem, 2021, 16(9), 1336–1359 CrossRef CAS PubMed.
- T. C. S. Ho, A. H. Y. Chan and A. Ganesan, Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight, J. Med. Chem., 2020, 63(21), 12460–12484 CrossRef CAS PubMed.
- J. Roche and P. Bertrand, Inside HDACs with more selective HDAC inhibitors, Eur. J. Med. Chem., 2016, 121, 451–483 CrossRef CAS.
- L. Whitehead, M. R. Dobler, B. Radetich, Y. Zhu, P. W. Atadja and T. Claiborne, et al., Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors, Bioorg. Med. Chem., 2011, 19(15), 4626–4634 CrossRef CAS PubMed.
- E. Seto and M. Yoshida, Erasers of histone acetylation: the histone deacetylase enzymes, Cold Spring Harbor Perspect. Biol., 2014, 6(4), a018713 CrossRef PubMed.
- C. Micelli and G. Rastelli, Histone deacetylases: structural determinants of inhibitor selectivity, Drug Discovery Today, 2015, 20(6), 718–735 CrossRef CAS PubMed.
- X. Hou, J. Du, R. Liu, Y. Zhou, M. Li and W. Xu, et al., Enhancing the Sensitivity of Pharmacophore-Based Virtual Screening by Incorporating Customized ZBG Features: A Case Study Using Histone Deacetylase 8, J. Chem. Inf. Model., 2015, 55(4), 861–871 CrossRef CAS.
- T. Qiu, L. Zhou, W. Zhu, T. Wang, J. Wang and Y. Shu, et al., Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials, Future Oncol., 2013, 9(2), 255–269 CrossRef CAS PubMed.
- D.-M. Chuang, Y. Leng, Z. Marinova, H.-J. Kim and C.-T. Chiu, Multiple roles of HDAC inhibition in neurodegenerative conditions, Trends Neurosci., 2009, 32(11), 591–601 CrossRef CAS.
- Y. Li, S. Lin, Z. Gu, L. Chen and B. He, Zinc-dependent deacetylases (HDACs) as potential targets for treating Alzheimer's disease, Bioorg. Med. Chem. Lett., 2022, 76, 129015 CrossRef CAS.
- R. A. Bagchi and K. L. Weeks, Histone deacetylases in cardiovascular and metabolic diseases, J. Mol. Cell. Cardiol., 2019, 130, 151–159 CrossRef CAS.
- B. Pitt, N. R. Sutton, Z. Wang, S. N. Goonewardena and M. Holinstat, Potential repurposing of the HDAC inhibitor valproic acid for patients with COVID-19, Eur. J. Pharmacol., 2021, 898, 173988 CrossRef CAS PubMed.
- L. Zhang, A. Ogden, R. Aneja and J. Zhou, Diverse roles of HDAC6 in viral infection: Implications for antiviral therapy, Pharmacol. Ther., 2016, 164, 120–125 CrossRef CAS.
- M. J. Ramaiah, A. D. Tangutur and R. R. Manyam, Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy, Life Sci., 2021, 277, 119504 CrossRef CAS.
- M. Huang, J. Zhang, C. Yan, X. Li, J. Zhang and R. Ling, Small molecule HDAC inhibitors: Promising agents for breast cancer treatment, Bioorg. Chem., 2019, 91, 103184 CrossRef CAS PubMed.
- A. Iglesias-Linares, R. M. Yañez-Vico and M. A. González-Moles, Potential role of HDAC inhibitors in cancer therapy: Insights into oral squamous cell carcinoma, Oral Oncol., 2010, 46(5), 323–329 CrossRef CAS PubMed.
- D. Wegener, H. E. Deubzer, I. Oehme, T. Milde, C. Hildmann and A. Schwienhorst, et al., HKI 46F08, a novel potent histone deacetylase inhibitor, exhibits antitumoral activity against embryonic childhood cancer cells, Anticancer Drugs, 2008, 19(9), 849–857 CrossRef CAS PubMed.
- A. Quintás-Cardama, H. M. Kantarjian, G. Borthakur, Z. Estrov, J. E. Cortes and S. Verstovsek, Therapy with the Histone Deacetylase Inhibitor Sb939 for Patients with Myelofibrosis, Blood, 2011, 118(21), 3863 CrossRef.
- H. Wang, N. Yu, D. Chen, K. C. L. Lee, P. L. Lye and J. W. W. Chang, et al., Discovery of (2E)-3-{2-Butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an Orally Active Histone Deacetylase Inhibitor with a Superior Preclinical Profile, J. Med. Chem., 2011, 54(13), 4694–4720 CrossRef CAS.
- H. Wang, N. Yu, D. Chen, E. T. Sun, K. Sangthongpitag and Z. Bonday, et al., 136 POSTER Discovery of SB939, an HDAC inhibitor with a superior preclinical profile, Eur. J. Cancer Suppl., 2008, 6(12), 44 CrossRef.
- A. Quintás-Cardama, H. M. Kantarjian, F. Ravandi, C. Foudray, N. Pemmaraju and T. M. Kadia, et al., Very High Rates of Clinical and Cytogenetic Response with the Combination of the Histone Deacetylase Inhibitor Pracinostat (SB939) and 5-Azacitidine in High-Risk Myelodysplastic Syndrome, Blood, 2012, 120(21), 3821 CrossRef.
- J. C. Bressi, R. Jong, Y. Wu, A. J. Jennings, J. W. Brown and S. O'Connell, et al., Benzimidazole and imidazole inhibitors of histone deacetylases: Synthesis and biological activity, Bioorg. Med. Chem. Lett., 2010, 20(10), 3138–3141 CrossRef CAS PubMed.
- H. Wang, N. Yu, H. Song, D. Chen, Y. Zou and W. Deng, et al., N-Hydroxy-1,2-disubstituted-1H-benzimidazol-5-yl acrylamides as novel histone deacetylase inhibitors: Design, synthesis, SAR studies, and in vivo antitumor activity, Bioorg. Med. Chem. Lett., 2009, 19(5), 1403–1408 CrossRef CAS.
- S. Manku, M. Allan, N. Nguyen, A. Ajamian, J. Rodrigue and E. Therrien, et al., Synthesis and evaluation of lysine derived sulfamides as histone deacetylase inhibitors, Bioorg. Med. Chem. Lett., 2009, 19(7), 1866–1870 CrossRef CAS PubMed.
- T. Wang, M. Sepulveda, P. Gonzales and S. Gately, Identification of novel HDAC inhibitors through cell based screening and their evaluation as potential anticancer agents, Bioorg. Med. Chem. Lett., 2013, 23(17), 4790–4793 CrossRef CAS PubMed.
- R. El-Awady, E. Saleh, R. Hamoudi, W. S. Ramadan, R. Mazitschek and M. A. Nael, et al., Discovery of novel class of histone deacetylase inhibitors as potential anticancer agents, Bioorg. Med. Chem., 2021, 42, 116251 CrossRef CAS PubMed.
- Y. Luo and H. Li, Structure-Based Inhibitor Discovery of Class I Histone Deacetylases (HDACs), Int. J. Mol. Sci., 2020, 21(22), 8828 CrossRef CAS PubMed.
- K. Ranganna, C. Selvam, A. Shivachar and Z. Yousefipour, Histone Deacetylase Inhibitors as Multitarget-Directed Epi-Drugs in Blocking PI3K Oncogenic Signaling: A Polypharmacology Approach, Int. J. Mol. Sci., 2020, 21(21), 8198 CrossRef CAS.
- D. A. Rodrigues, P. S. M. Pinheiro and C. A. M. Fraga, Multitarget Inhibition of Histone Deacetylase (HDAC) and Phosphatidylinositol-3-kinase (PI3K): Current and Future Prospects, ChemMedChem, 2021, 16(3), 448–457 CrossRef CAS.
- A. K. A. Bass, M. S. El-Zoghbi, E.-S. M. Nageeb, M. F. A. Mohamed, M. Badr and G. E.-D. A. Abuo-Rahma, Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors, Eur. J. Med. Chem., 2021, 209, 112904 CrossRef CAS.
- H. Sirous, G. Campiani, V. Calderone and S. Brogi, Discovery of novel hit compounds as potential HDAC1 inhibitors: The case of ligand- and structure-based virtual screening, Comput. Biol. Med., 2021, 137, 104808 CrossRef CAS PubMed.
- M. Mustafa, A. A. Abd El-Hafeez, D. Abdelhamid, G. D. Katkar, Y. A. Mostafa and P. Ghosh, et al., A first-in-class anticancer dual HDAC2/FAK inhibitors bearing hydroxamates/benzamides capped by pyridinyl-1,2,4-triazoles, Eur. J. Med. Chem., 2021, 222, 113569 CrossRef CAS.
- Q. Gao, P. Yao, Y. Wang, Q. Yao and J. Zhang, Discovery of potent HDAC2 inhibitors based on virtual screening in combination with drug repurposing, J. Mol. Struct., 2022, 1247, 131399 CrossRef CAS.
- M. J. Emmett and M. A. Lazar, Integrative regulation of physiology by histone deacetylase 3, Nat. Rev. Mol. Cell Biol., 2019, 20(2), 102–115 CrossRef CAS PubMed.
- R. Sarkar, S. Banerjee, S. A. Amin, N. Adhikari and T. Jha, Histone deacetylase 3 (HDAC3) inhibitors as anticancer agents: A review, Eur. J. Med. Chem., 2020, 192, 112171 CrossRef CAS.
- T. Li, C. Zhang, S. Hassan, X. Liu, F. Song and K. Chen, et al., Histone deacetylase 6 in cancer, J. Hematol. Oncol., 2018, 11(1), 111 CrossRef PubMed.
- D. A. Rodrigues, S. Thota and C. A. Fraga, Beyond the Selective Inhibition of Histone Deacetylase 6, Mini-Rev. Med. Chem., 2016, 16(14), 1175–1184 CrossRef CAS.
- J. H. Park, S. H. Kim, M. C. Choi, J. Lee, D. Y. Oh and S. A. Im, et al., Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors, Biochem. Biophys. Res. Commun., 2008, 368(2), 318–322 CrossRef CAS.
- D. Li, S. Xie, Y. Ren, L. Huo, J. Gao and D. Cui, et al., Microtubule-associated deacetylase HDAC6 promotes angiogenesis by regulating cell migration in an EB1-dependent manner, Protein Cell, 2011, 2(2), 150–160 CrossRef CAS.
- J. Senger, J. Melesina, M. Marek, C. Romier, I. Oehme and O. Witt, et al., Synthesis and Biological Investigation of Oxazole Hydroxamates as Highly Selective Histone Deacetylase 6 (HDAC6) Inhibitors, J. Med. Chem., 2016, 59(4), 1545–1555 CrossRef CAS PubMed.
- A. Chakrabarti, I. Oehme, O. Witt, G. Oliveira, W. Sippl and C. Romier, et al., HDAC8: a multifaceted target for therapeutic interventions, Trends Pharmacol. Sci., 2015, 36(7), 481–492 CrossRef CAS PubMed.
- J. R. Somoza, R. J. Skene, B. A. Katz, C. Mol, J. D. Ho and A. J. Jennings, et al., Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases, Structure, 2004, 12(7), 1325–1334 CrossRef CAS.
- I. Oehme, H. E. Deubzer, D. Wegener, D. Pickert, J.-P. Linke and B. Hero, et al., Histone deacetylase 8 in neuroblastoma tumorigenesis, Clin. Cancer Res., 2009, 15(1), 91–99 CrossRef CAS PubMed.
- S. A. Abbass, H. A. Hassan, M. F. A. Mohamed, G. A. I. Moustafa and G. E.-D. A. Abuo-Rahma, Recent Prospectives of Anticancer Histone Deacetylase Inhibitors, J. Adv. Biomedical Pharm. Sci., 2019, 2(4), 135–151 Search PubMed.
- C.-W. Hsu, D. Shou, R. Huang, T. Khuc, S. Dai and W. Zheng, et al., Identification of HDAC Inhibitors Using a Cell-Based HDAC I/II Assay, SLAS Discovery, 2016, 21(6), 643–652 CrossRef CAS.
- L. A. Alves Avelar, D. Ruzic, N. Djokovic, T. Kurz and K. Nikolic, Structure-based design of selective histone deacetylase 6 zinc binding groups, J. Biomol. Struct. Dyn., 2020, 38(11), 3166–3177 CrossRef CAS.
- P. H. Nguyen, B. T. B. Hue, M. Q. Pham, T. P. Hoa, Q. De Tran and H. Jung, et al., Novel histone deacetylase 6 inhibitors using benzimidazole as caps for cancer treatment, New J. Chem., 2023, 47, 7622–7631 RSC.
- R. E. S. Mansour, H. G. Abdulwahab and H. El-Sehrawi, Novel mannich bases derived from 2-substituted benzimidazole and (thio) hydantoin moieties as potent histone deacetylase 6 (hdac6) inhibitors, Al-Azhar J. Pharm. Med. Sci., 2021, 1(3), 66–74 Search PubMed.
- L. Wang, M. Kofler, G. Brosch, J. Melesina, W. Sippl and E. D. Martinez, et al., 2-Benzazolyl-4-Piperazin-1-Ylsulfonylbenzenecarbohydroxamic Acids as Novel Selective Histone Deacetylase-6 Inhibitors with Antiproliferative Activity, PLoS One, 2015, 10(12), e0134556 CrossRef PubMed.
- Y. Wang, J. He, M. Liao, M. Hu, W. Li and H. Ouyang, et al., An overview of Sirtuins as potential therapeutic target: Structure, function and modulators, Eur. J. Med. Chem., 2019, 161, 48–77 CrossRef CAS.
- L. Sacconnay, P.-A. Carrupt and A. Nurisso, Human sirtuins: Structures and flexibility, J. Biomol. Struct. Dyn., 2016, 196(3), 534–542 CAS.
- T. Kosciuk, M. Wang, J. Y. Hong and H. Lin, Updates on the epigenetic roles of sirtuins, Curr. Opin. Chem. Biol., 2019, 51, 18–29 CrossRef CAS PubMed.
- B. C. Smith, W. C. Hallows and J. M. Denu, Mechanisms and Molecular Probes of Sirtuins, Chem. Biol., 2008, 15(10), 1002–1013 CrossRef CAS.
- Y. K. Yoon, H. Osman and T. S. Choon, Potent sirtuin inhibition with 1,2,5-trisubstituted benzimidazoles, MedChemComm, 2016, 7(11), 2094–2099 RSC.
- Y. K. Yoon, M. A. Ali, A. C. Wei, A. N. Shirazi, K. Parang and T. S. Choon, Benzimidazoles as new scaffold of sirtuin inhibitors: Green synthesis, in vitro studies, molecular docking analysis and evaluation of their anti-cancer properties, Eur. J. Med. Chem., 2014, 83, 448–454 CrossRef CAS PubMed.
- N. Purushotham, M. Singh, B. Paramesha, V. Kumar, S. Wakode and S. K. Banerjee, et al., Design and synthesis of amino acid derivatives of substituted benzimidazoles and pyrazoles as Sirt1 inhibitors, RSC Adv., 2022, 12(7), 3809–3827 RSC.
- Y. K. Yoon, M. A. Ali, A. C. Wei, T. S. Choon, A. N. Shirazi and K. Parang, Discovery of a potent and highly fluorescent sirtuin inhibitor, MedChemComm, 2015, 6(10), 1857–1863 RSC.
- Y. J. Tan, Y. T. Lee, K. Y. Yeong, S. H. Petersen, K. Kono and S. C. Tan, et al., Anticancer activities of a benzimidazole compound through sirtuin inhibition in colorectal cancer, Future Med. Chem., 2018, 10(17), 2039–2057 CrossRef CAS PubMed.
- X. Li, X. Li, F. Liu, S. Li and D. Shi, Rational Multitargeted Drug Design Strategy from the Perspective of a Medicinal Chemist, J. Med. Chem., 2021, 64(15), 10581–10605 CrossRef CAS.
- M. F. A. Mohamed, B. G. M. Youssif, M. S. A. Shaykoon, M. H. Abdelrahman, B. E. M. Elsadek and A. S. Aboraia, et al., Utilization of tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidinone as a cap moiety in design of novel histone deacetylase inhibitors, Bioorg. Chem., 2019, 91, 103127 CrossRef CAS.
- C. Barbaraci, V. di Giacomo, A. Maruca, V. Patamia, R. Rocca and M. Dichiara, et al., Discovery of first novel sigma/HDACi dual-ligands with a potent in vitro antiproliferative activity, Bioorg. Chem., 2023, 140, 106794 CrossRef CAS PubMed.
- W. Zhang, J. Pei and L. Lai, Computational Multitarget Drug Design, J. Chem. Inf. Model., 2017, 57(3), 403–412 CrossRef CAS PubMed.
- A. Suraweera, K. J. O'Byrne and D. J. Richard, Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi, Front. Oncol., 2018, 8, 92 CrossRef.
- E. Shirbhate, V. Singh, V. Jahoriya, A. Mishra, R. Veerasamy and A. K. Tiwari, et al., Dual inhibitors of HDAC and other epigenetic regulators: A novel strategy for cancer treatment, Eur. J. Med. Chem., 2024, 263, 115938 CrossRef CAS PubMed.
- Z. Huang, W. Zhou, Y. Li, M. Cao, T. Wang and Y. Ma, et al., Novel hybrid molecule overcomes the limited response of solid tumours to HDAC inhibitors via suppressing JAK1-STAT3-BCL2 signalling, Theranostics, 2018, 8(18), 4995–5011 CrossRef CAS.
- C. Liu, H. Ding, X. Li, C. P. Pallasch, L. Hong and D. Guo, et al., A DNA/HDAC dual-targeting drug CY190602 with significantly enhanced anticancer potency, EMBO Mol. Med., 2015, 7(4), 438–449 CrossRef CAS PubMed.
- T. Mehrling and Y. Chen, The Alkylating-HDAC Inhibition Fusion Principle: Taking Chemotherapy to the Next Level with the First in Class Molecule EDO-S101, Anticancer Agents Med. Chem., 2016, 16(1), 20–28 CrossRef CAS PubMed.
- A. Mita, M. Loeffler, N. Bui, D. Remmy, T. Mehrling and M. Mita, et al., 470P – A phase I study of tinostamustine in patients (pts) with advanced solid tumours, Ann. Oncol., 2019, 30, v177–v8 CrossRef.
- H. Ghesquieres, A. Pinto, A. Sureda, O. Tournilhac, F. Morschhauser and T. Janik, et al., Safety and Efficacy of Tinostamustine in Heavily Pre-Treated Patients with Relapsed/Refractory (R/R) Hodgkin Lymphoma (HL), Blood, 2021, 138, 2472 CrossRef.
- A. Sureda, A. Pinto, H. Ghesquieres, F. Morschhauser, O. Tournilhac and P. Mutseaers, et al., Safety and Efficacy of Tinostamustine in Pre-Treated Patients with Relapsed/Refractory (R/R) Hodgkin Lymphoma (HL): Early Results from an Additional Expansion Cohort of a Phase I Study, Blood, 2022, 140, 3696–3697 CrossRef.
- H. Xu, G. Luo, T. Wu, J. Hu, C. Wang and X. Wu, et al., Structural insights revealed by the cocrystal structure of CCS1477 in complex with CBP bromodomain, Biochem. Biophys. Res. Commun., 2022, 623, 17–22 CrossRef CAS.
- J. Dong and X. Wang, Identification of novel BRD4 inhibitors by pharmacophore screening, molecular docking, and molecular dynamics simulation, J. Mol. Struct., 2023, 1274, 134363 CrossRef CAS.
- T. Ito, T. Umehara, K. Sasaki, Y. Nakamura, N. Nishino and T. Terada, et al., Real-Time Imaging of Histone H4K12–Specific Acetylation Determines the Modes of Action of Histone Deacetylase and Bromodomain Inhibitors, Chem. Biol., 2011, 18(4), 495–507 CrossRef CAS PubMed.
- J. Xing, R. Zhang, X. Jiang, T. Hu, X. Wang and G. Qiao, et al., Rational design of 5-((1H-imidazole-1-yl)methyl)quinolin-8-ol derivatives as novel bromodomain-containing protein 4 inhibitors, Eur. J. Med. Chem., 2019, 163, 281–294 CrossRef CAS PubMed.
- A. Cipriano, C. Milite, A. Feoli, M. Viviano, G. Pepe and P. Campiglia, et al., Discovery of Benzo[d]imidazole-6-sulfonamides as Bromodomain and Extra-Terminal Domain (BET) Inhibitors with Selectivity for the First Bromodomain, ChemMedChem, 2022, 17(20), e202200343 CrossRef CAS.
- S. Wang, V. Tsui, T. D. Crawford, J. E. Audia, D. J. Burdick and M. H. Beresini, et al., GNE-371, a Potent and Selective Chemical Probe for the Second Bromodomains of Human Transcription-Initiation-Factor TFIID Subunit 1 and Transcription-Initiation-Factor TFIID Subunit 1-like, J. Med. Chem., 2018, 61(20), 9301–9315 CrossRef CAS PubMed.
- S. Gokani and L. K. Bhatt, Bromodomains: A novel target for the anticancer therapy, Eur. J. Pharmacol., 2021, 911, 174523 CrossRef CAS PubMed.
- D. Sperandio, V. Aktoudianakis, K. Babaoglu, X. Chen, K. Elbel and G. Chin, et al., Structure-guided discovery of a novel, potent, and orally bioavailable 3,5-dimethylisoxazole aryl-benzimidazole BET bromodomain inhibitor, Bioorg. Med. Chem., 2019, 27(3), 457–469 CrossRef CAS.
- Y. Fan, F. Luo, M. Su, Q. Li, T. Zhong and L. Xiong, et al., Structure optimization, synthesis, and biological evaluation of 6-(2-amino-1H-benzo[d]imidazole-6-yl)-quinazolin-4(3H)-one derivatives as potential multi-targeted anticancer agents via Aurora A/PI3K/BRD4 inhibition, Bioorg. Chem., 2023, 132, 106352 CrossRef CAS PubMed.
- B. Kong, Z. Zhu, H. Li, Q. Hong, C. Wang and Y. Ma, et al., Discovery of 1-(5-(1H-benzo[d]imidazole-2-yl)-2,4-dimethyl-1H-pyrrol-3-yl)ethan-1-one derivatives as novel and potent bromodomain and extra-terminal (BET) inhibitors with anticancer efficacy, Eur. J. Med. Chem., 2022, 227, 113953 CrossRef CAS PubMed.
- P. Gou and W. Zhang, Protein lysine acetyltransferase CBP/p300: A promising target for small molecules in cancer treatment, Biomed. Pharmacother., 2024, 171, 116130 CrossRef CAS.
- Z.-X. He, B.-F. Wei, X. Zhang, Y.-P. Gong, L.-Y. Ma and W. Zhao, Current development of CBP/p300 inhibitors in the last decade, Eur. J. Med. Chem., 2021, 209, 112861 CrossRef CAS.
- N. Brooks, T. Knurowski, A. Hughes, K. Clegg, W. West and N. A. Pegg, et al., CCS1477, a Novel p300/CBP Bromodomain Inhibitor, Enhances Efficacy of Azacitidine and Venetoclax in Pre-Clinical Models of Acute Myeloid Leukaemia and Lymphoma, Blood, 2021, 138, 3291 CrossRef.
- D. A. Hay, O. Fedorov, S. Martin, D. C. Singleton, C. Tallant and C. Wells, et al., Discovery and Optimization of Small-Molecule Ligands for the CBP/p300 Bromodomains, J. Am. Chem. Soc., 2014, 136(26), 9308–9319 CrossRef CAS PubMed.
- Z. Chen, J. Li, H. Yang, Y. He, Q. Shi and Q. Chang, et al., Discovery of novel benzimidazole derivatives as potent p300 bromodomain inhibitors with anti-proliferative activity in multiple cancer cells, Bioorg. Med. Chem., 2022, 66, 116784 CrossRef CAS PubMed.
- N. Pegg, N. Brooks, J. Worthington, B. Young, A. Prosser and J. Lane, et al., Characterisation of CCS1477: A novel small molecule inhibitor of p300/CBP for the treatment of castration resistant prostate cancer, J. Clin. Oncol., 2017, 35(15_suppl), 11590 Search PubMed.
- J. Welti, A. Sharp, N. Brooks, W. Yuan, C. McNair and S. N. Chand, et al., Targeting the p300/CBP Axis in Lethal Prostate Cancer, Cancer Discovery, 2021, 11(5), 1118–1137 CrossRef CAS.
- N. Brooks, M. Raja, B. W. Young, G. J. Spencer, T. C. P. Somervaille and N. A. Pegg, CCS1477: A Novel Small Molecule Inhibitor of p300/CBP Bromodomain for the Treatment of Acute Myeloid Leukaemia and Multiple Myeloma, Blood, 2019, 134, 2560 CrossRef.
- L. Nicosia, N. Brooks, F. Amaral, O. Sinclair, N. A. Pegg and W. West, et al., Potent Pre-Clinical and Early Phase Clinical Activity of EP300/CBP Bromodomain Inhibitor CCS1477 in Multiple Myeloma, Blood, 2022, 140, 852–853 CrossRef.
- R. Liu, H. Yang, Z. Chen, K. Zhou, Q. Shi and J. Li, et al., Design, synthesis and biological evaluation of (R)-5-methylpyrrolidin-2-ones as p300 bromodomain inhibitors with Anti-Tumor activities in multiple tumor lines, Bioorg. Chem., 2022, 124, 105803 CrossRef CAS PubMed.
- S. Wang, V. Tsui, T. D. Crawford, J. E. Audia, D. J. Burdick and M. H. Beresini, et al., GNE-371, a Potent and Selective Chemical Probe for the Second Bromodomains of Human Transcription-Initiation-Factor TFIID Subunit 1 and Transcription-Initiation-Factor TFIID Subunit 1-like, J. Med. Chem., 2018, 61(20), 9301–9315 CrossRef CAS PubMed.
- E. H. Demont, P. Bamborough, C. W. Chung, P. D. Craggs, D. Fallon and L. J. Gordon, et al., 1,3-Dimethyl Benzimidazolones Are Potent, Selective Inhibitors of the BRPF1 Bromodomain, ACS Med. Chem. Lett., 2014, 5(11), 1190–1195 CrossRef CAS PubMed.
- P. Bamborough, H. A. Barnett, I. Becher, M. J. Bird, C.-W. Chung and P. D. Craggs, et al., GSK6853, a Chemical Probe for Inhibition of the BRPF1 Bromodomain, ACS Med. Chem. Lett., 2016, 7(6), 552–557 CrossRef CAS PubMed.
- A. Dalle Vedove, G. Cazzanelli, J. Corsi, M. Sedykh, V. G. D'Agostino and A. Caflisch, et al., Identification of a BAZ2A Bromodomain Hit Compound by Fragment Joining, ACS Bio Med Chem Au, 2021, 1(1), 5–10 CrossRef CAS PubMed.
- S.-P. Wang, Y. Li, S.-H. Huang, S.-Q. Wu, L.-L. Gao and Q. Sun, et al., Discovery of Potent and Novel Dual PARP/BRD4 Inhibitors for Efficient Treatment of Pancreatic Cancer, J. Med. Chem., 2021, 64(23), 17413–17435 CrossRef CAS.
- S.-H. Huang, R. Cao, Q.-W. Lin, S.-Q. Wu, L.-L. Gao and Q. Sun, et al., Design, synthesis and mechanism studies of novel dual PARP1/BRD4 inhibitors against pancreatic cancer, Eur. J. Med. Chem., 2022, 230, 114116 CrossRef CAS PubMed.
- H. Hu and A. G. Muntean, The YEATS domain epigenetic reader proteins ENL and AF9 and their therapeutic value in leukemia, Exp. Hematol., 2023, 124, 15–21 CrossRef CAS.
- A. Kabra and J. Bushweller, The Intrinsically Disordered Proteins MLLT3 (AF9) and MLLT1 (ENL) – Multimodal Transcriptional Switches With Roles in Normal Hematopoiesis, MLL Fusion Leukemia, and Kidney Cancer, J. Mol. Biol., 2022, 434(1), 167117 CrossRef CAS PubMed.
- S. Guo, T. Jia, X. Xu, F. Yang, S. Xiao and Z. Hou, et al., Design, synthesis of novel benzimidazole derivatives as ENL inhibitors suppressing leukemia cells viability via downregulating the expression of MYC, Eur. J. Med. Chem., 2023, 248, 115093 CrossRef CAS PubMed.
- Y. Li, H. Wen, Y. Xi, K. Tanaka, H. Wang and D. Peng, et al., AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation, Cell, 2014, 159(3), 558–571 CrossRef CAS.
- M. Moustakim, T. Christott, O. P. Monteiro, J. Bennett, C. Giroud and J. Ward, et al., Discovery of an MLLT1/3 YEATS Domain Chemical Probe, Angew Chem. Int. Ed. Engl., 2018, 57(50), 16302–16307 CrossRef CAS PubMed.
- N. C. Sutopo, J. H. Kim and J. Y. Cho, Role of histone methylation in skin cancers: Histone methylation–modifying enzymes as a new class of targets for skin cancer treatment, Biochim. Biophys. Acta, Rev. Cancer, 2023, 1878(3), 188865 CrossRef CAS.
- D. Orioli and E. Dellambra, Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases, Cells, 2018, 7(12), 268 CrossRef CAS PubMed.
- R. J. Separovich and M. R. Wilkins, Ready, SET, Go: Post-translational regulation of the histone lysine methylation network in budding yeast, J. Biol. Chem., 2021, 297(2), 100939 CrossRef CAS PubMed.
- D. Strepkos, M. Markouli, A. Klonou, A. G. Papavassiliou and C. Piperi, Histone Methyltransferase SETDB1: A Common Denominator of Tumorigenesis with Therapeutic Potential, Cancer Res., 2021, 81(3), 525–534 CrossRef CAS PubMed.
- R. Schneider, A. J. Bannister and T. Kouzarides, Unsafe SETs: histone lysine methyltransferases and cancer, Trends Biochem. Sci., 2002, 27(8), 396–402 CrossRef CAS.
- A. J. Bannister and T. Kouzarides, Regulation of chromatin by histone modifications, Cell Research, 2011, 21(3), 381–395 CrossRef CAS.
- E. K. Ko and B. C. Capell, Methyltransferases in the Pathogenesis of Keratinocyte Cancers, Cancers, 2021, 13(14), 3402 CrossRef CAS.
- C. Bon, Y. Si, M. Pernak, M. Barbachowska, E. Levi-Acobas and D. V. Cadet, et al., Synthesis and Biological Activity of a Cytostatic Inhibitor of MLLr Leukemia Targeting the DOT1L Protein, Molecules, 2021, 26(17), 5300 CrossRef CAS PubMed.
- A. Basavapathruni, L. Jin, S. R. Daigle, C. R. Majer, C. A. Therkelsen and T. J. Wigle, et al., Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L, Chem. Biol. Drug Des., 2012, 80(6), 971–980 CrossRef CAS PubMed.
- S. R. Daigle, E. J. Olhava, C. A. Therkelsen, A. Basavapathruni, L. Jin and P. A. Boriack-Sjodin, et al., Potent inhibition of DOT1L as treatment of MLL-fusion leukemia, Blood, 2013, 122(6), 1017–1025 CrossRef CAS PubMed.
- W.-L. Chen, D.-D. Li, X. Chen, Y.-Z. Wang, J.-J. Xu and Z.-Y. Jiang, et al., Proton pump inhibitors selectively suppress MLL rearranged leukemia cells via disrupting MLL1-WDR5 protein-protein interaction, Eur. J. Med. Chem., 2020, 188, 112027 CrossRef CAS.
- X. Kong, L. Chen, L. Jiao, X. Jiang, F. Lian and J. Lu, et al., Astemizole Arrests the Proliferation of Cancer Cells by Disrupting the EZH2-EED Interaction of Polycomb Repressive Complex 2, J. Med. Chem., 2014, 57(22), 9512–9521 CrossRef CAS.
- D. Du, D. Xu, L. Zhu, G. Stazi, C. Zwergel and Y. Liu, et al., Structure-Guided Development of Small-Molecule PRC2 Inhibitors Targeting EZH2–EED Interaction, J. Med. Chem., 2021, 64(12), 8194–8207 CrossRef CAS.
- J. Zhang, D. Yao, Y. Jiang, J. Huang, S. Yang and J. Wang, Synthesis and biological evaluation of benzimidazole derivatives as the G9a Histone Methyltransferase inhibitors that induce autophagy and apoptosis of breast cancer cells, Bioorg. Chem., 2017, 72, 168–181 CrossRef CAS.
- Y. Yuan, Q. Wang, J. Paulk, S. Kubicek, M. M. Kemp and D. J. Adams, et al., A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma, ACS Chem. Biol., 2012, 7(7), 1152–1157 CrossRef CAS PubMed.
- R. Mao, J. Shao, K. Zhu, Y. Zhang, H. Ding and C. Zhang, et al., Potent, Selective, and Cell Active Protein Arginine Methyltransferase 5 (PRMT5) Inhibitor Developed by Structure-Based Virtual Screening and Hit Optimization, J. Med. Chem., 2017, 60(14), 6289–6304 CrossRef CAS PubMed.
- A. Di Lorenzo and M. T. Bedford, Histone arginine methylation, FEBS Lett., 2011, 585(13), 2024–2031 CrossRef CAS PubMed.
- Y. Zhu, T. Xia, D.-Q. Chen, X. Xiong, L. Shi and Y. Zuo, et al., Promising role of protein arginine methyltransferases in overcoming anti-cancer drug resistance, Drug Resistance Updates, 2024, 72, 101016 CrossRef CAS.
- K. S. Rao, K. Nalla, C. Ramachandraiah and K. B. Chandrasekhar, et al., Design and Synthesis of New Triazole-Benzimidazole Derivatives as Potential PRMT5 Inhibitors, ChemistrySelect, 2023, 11, 1–10 Search PubMed.
- L. Zhang, Y. Chen, Z. Li, C. Lin, T. Zhang and G. Wang, Development of JmjC-domain-containing histone demethylase (KDM2-7) inhibitors for cancer therapy, Drug Discovery Today, 2023, 28(5), 103519 CrossRef CAS PubMed.
- K. M. Arcipowski, C. A. Martinez and P. Ntziachristos, Histone demethylases in physiology and cancer: a tale of two enzymes, JMJD3 and UTX, Curr. Opin. Genet. Dev., 2016, 36, 59–67 CrossRef CAS.
- T. Feng, D. Li, H. Wang, J. Zhuang, F. Liu and Q. Bao, et al., Novel 5-carboxy-8-HQ based histone demethylase JMJD2A inhibitors: Introduction of an additional carboxyl group at the C-2 position of quinoline, Eur. J. Med. Chem., 2015, 105, 145–155 CrossRef CAS PubMed.
- D. M. Carter, E. Specker, P. H. Małecki, J. Przygodda, K. Dudaniec and M. S. Weiss, et al., Enhanced properties of a benzimidazole benzylpyrazole lysine demethylase inhibitor: mechanism-of-action, binding site analysis, and activity in cellular models of prostate cancer, J. Med. Chem., 2021, 64(19), 14266–14282 CrossRef CAS PubMed.
- D. M. Carter, E. Specker, J. Przygodda, M. Neuenschwander, J. P. von Kries and U. Heinemann, et al., Identification of a Novel Benzimidazole Pyrazolone Scaffold That Inhibits KDM4 Lysine Demethylases and Reduces Proliferation of Prostate Cancer Cells, SLAS Discovery, 2017, 22(7), 801–812 CrossRef CAS.
- S. Polepalli, S. M. George, R. Valli Sri Vidya, G. S. Rodrigues, L. Ramachandra and R. Chandrashekar, et al., Role of UHRF1 in malignancy and its function as a therapeutic target for molecular docking towards the SRA domain, Int. J. Biochem. Cell Biol., 2019, 114, 105558 CrossRef CAS.
- A. Molitor and W.-H. Shen, The Polycomb Complex PRC1: Composition and Function in Plants, J. Genet. Genomics, 2013, 40(5), 231–238 CrossRef CAS.
- C. Ren, S. G. Smith, K. Yap, S. Li, J. Li and M. Mezei, et al., Structure-Guided Discovery of Selective Antagonists for the Chromodomain of Polycomb Repressive Protein CBX7, ACS Med. Chem. Lett., 2016, 7(6), 601–605 CrossRef CAS.
- S. Gadde, A. Kleynhans, J. K. Holien, M. Bhadbhade, P. L. D. Nguyen and R. Mittra, et al., Pyrimido[1,2-a]benzimidazoles as inhibitors of oncoproteins ubiquitin specific protease 5 and MYCN in the childhood cancer neuroblastoma, Bioorg. Chem., 2023, 136, 106462 CrossRef CAS.
- K. Yu and P. Proost, Insights into peptidylarginine deiminase expression and citrullination pathways, Trends Cell Biol., 2022, 32(9), 746–761 CrossRef CAS.
- Z. Guo, L. Shi, B. Wang, G. He, Y. Wang and G. Chen, Synthesis of reversible PAD4 inhibitors via copper-catalyzed C−H arylation of benzimidazole, Sci. China:Chem., 2019, 62(5), 592–596 CrossRef CAS.
- A. Muth, V. Subramanian, E. Beaumont, M. Nagar, P. Kerry and P. McEwan, et al., Development of a Selective Inhibitor of Protein Arginine Deiminase 2, J. Med. Chem., 2017, 60(7), 3198–3211 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |