
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyurethane-grafted graphene oxide from repurposed foam mattress waste†
Walker M. Vickery,
Juhi Singh,
Jason D. Orlando,
Ting-Chih Lin,
Julia Wang and
Stefanie A. Sydlik *
*
Department of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, PA 15213, USA. E-mail: ssydlik@andrew.cmu.edu
First published on 27th January 2025
Abstract
Polyurethanes (PU) make up a large portion of commodity plastics appearing in applications including insulation, footwear, and memory foam mattresses. Unfortunately, as thermoset polymers, polyurethanes lack a clear path for recycling and repurposing, creating a sustainability issue. Herein, using dynamic depolymerization, we demonstrate a simple one-pot synthesis for preparation of an upcycled polyurethane grafted graphene material (PU–GO). Through this dynamic depolymerization using green conditions, PU–GO nanofillers with tunable PU to GO ratios were synthesized. Chemical analysis revealed that the polyurethane graphenic materials primarily contained the polycarbamate hard-segment of polyurethane while the soft polyol component was removed in washes. PU–GOs were incorporated into bulk polyurethane foam to create composites as a filler at 0.25, 0.5, 1.0, and 2.0 weight percent filler and the thermal and mechanical properties of the resulting foams were analyzed. All PU–GO fillers were shown to improve thermal insulation up to a filler content of 0.5%, with all but 2 of the fillers demonstrating improvements up to 2% of filler content. The greatest decrease in thermal conductivity was 38.5% compared to neat PU foam, observed with the composites containing 0.5% of PU10–GO1 and 1.0% of PU3–GO1. Mechanical performance was tested for each foam and showed that lower polyurethane content graphenic composites produced foams that were less susceptible to fatiguing and more durable over cyclic loading, while higher polyurethane content graphenic composites had mechanical stability similar to neat PU but initially had greater impact resistance. Taken together, these novel PU–GO fillers prepared from repurposed PU mattress show promise as a sustainable additive to improve PU performance.
1 Introduction
Polyurethane (PU) foams are a group of complex polymers used for a wide variety of applications, due to their tunable chemical precursors and manufacturing processes. The modifiable nature of PU foams are a product of their structure, consisting of hard-segments containing diisocyanate components, and soft-segments which typically consist of polyester polyols or polyether polyols.1,2 Additionally, crosslinking agents, fillers, and blowing agents/conditions may be altered to result in foams with a wide array of properties.1,3,4 The high degree of tailorability of its component parts allows for the use of PU foams in a wide variety of applications including mechanical cushioning,4,5 sound dampening,3,6,7 and thermal insulation.8,9 The broad range of applications for PU foams has made them one of the most widely produced commodity plastic materials with ∼7746 million pounds produced in 2021 in the United States alone.10Unfortunately, the variable structure of PUs makes it difficult to utilize and process the post-consumer plastic waste in any useable form without significant purification and isolation processes. Removing or isolating undesirable components either from fillers, the soft-segment, or hard-segment fragments is often required for the chemical depolymerization, fragmentation, and upcycling of PU foams.11–13 Methods that are capable of utilizing more complex PU foam waste using simple chemical synthesis and purification would be desirable for real-world applications.
One possible avenue for the simplified upcycling of PU waste would be modifying it for use as a hybrid filler material with graphene. Thus, graphenic materials have been used to improve the morphological and physical properties of PU foams, but its benefits begin to have diminishing returns at higher loading amounts due to the lack of miscibility of the graphenic material. The miscibility of a graphenic filler in a PU foam could be improved through the covalent attachment of PU to the surface of the carbon material. This would allow improved integration of the PU modified graphene into the bulk foam composite. While traditional polymerization methods are achievable for the grafting of polymer from graphene oxide (GO), this would require the generation of new plastic waste. Ideally, post-consumer plastic waste would be used as a feedstock to prepare these plastic–graphene hybrid fillers. Previously, in an effort to make direct use of post-consumer plastic waste, we have reported a simple method, “dynamic depolymerization”, for the one-pot fragmentation and covalent attachment of polyethylene terephthalate to GO through a transesterification reaction to produce an upcycled plastic–graphene hybrid.14 The dynamic depolymerization technique efficiently and directly makes use of post-consumer condensation polymer plastic waste by utilizing chemical fragmentation to promote plastic breakdown and covalent attachment onto GO in a one-pot reaction. Using similar techniques to our previous study on polyethylene terephthalate, we were able to expand the scope of post-consumer polymers that can be covalently attached to GO by preparing a new PU–graphene hybrid material (PU–GO) for use as an upcycled PU foam filler.
Herein, the two-step, one-pot dynamic depolymerization of PU onto GO is achieved through a base catalyzed transcarbamoylation (chemical exchange of an alcohol on a carbamate). The transcarbamoylation of PU in the presence of graphene oxide (GO) was achieved through the fragmentation and covalent attachment of PU. Fragmentation and breakdown of PU carbamate units has repeatedly been achieved in green conditions through simple hydrolysis or transcarbamoylation using sodium hydroxide in the presence of water or an alcohol.12 Under these same conditions, the PU fragments can undergo chemical exchange with the alcohols on the surface of GO allowing for the subsequent covalent attachment of fragments in a one-pot reaction. We report the successful covalent attachment of PU fragments to GO in ethanol and sodium hydroxide.
2 Materials and methods
2.1 Materials
Sodium hydroxide pellets (VWR chemicals), ethanol (200 Proof, anhydrous, Decon Labs, Inc), trifluoroacetic acid (99%, Beantown Chemical), graphite flakes (99.8% metals basis, 325 mesh, Alfa Aesar), sulfuric acid (technical grade, fisher chemical), potassium permanganate (Supelco), hydrogen peroxide (30% aqueous, VWR chemicals) were procured. Polyurethane was sourced from a single-type post-consumer waste mattress that was originally purchased from Symbol Mattress company prior to use in school housing. FTIR, TGA, and NMR characterization revealed the mattress to be a single-type toluene diisocyanate (TDI)/polyether polyol slabstock foam. The mattress was cut into pieces no longer than 0.5 inches to a side and flash frozen in liquid nitrogen prior to being mechanically broken down into fragments no larger than 2 cubic mm in size using an electric blender. Polyurethane composites were prepared from Expanding Pour Foam 2 part polyurethane closed cell liquid foam (Fiberglass Supply Depot Inc). Crystal clear lacquer for DSC pan coatings was purchased from Watco. Gallium metal was purchased from Amazon.2.2 Graphene oxide synthesis
GO was prepared using a modified Hummers' method15 as previously reported.16 The reaction was run open to air in a 2 L Erlenmeyer flask, which was charged with graphite flakes (5 g) and H2SO4 (125 mL). The mixture was stirred over ice for 30 minutes before adding KMnO4 (10 g). The reaction mixture was allowed to warm to room temperature while stirring for 2 hours, then heated to 35 °C and stirred for an additional 2 hours. A blast shield was set up while the mixture was cooled on ice to at least room temperature. While cooling the flask on ice, DI water (750 mL) was added quickly with stirring. After waiting a couple of minutes, H2O2 (10 mL, 30% aq.) was then added followed by more DI water (225 mL). The mixture was stirred overnight and vacuum filtered the next day to collect the GO. GO was placed in dialysis tubing (3500 molecular weight cutoff, Snakeskin™ dialysis tubing; Thermo Scientific, Waltham, MA, USA) with DI water and dialyzed for at least 5 days changing the water every day. The resulting product was collected, frozen at −80 °C and lyophilized for 3 days to dryness.2.3 Polyurethane-grafted graphene oxide synthesis
PU–GO powder was prepared by adding GO (140 mg) and shredded PU waste (amount varies depending on weight ratio: 140 mg, 420 mg, 700 mg, and 1400 mg for PU1–GO1, PU3–GO1, PU5–GO1, and PU10–GO1, respectively) into a round bottom flask equipped with a magnetic stir bar. A 35 mL of a 0.256 M solution of NaOH in ethanol was added to the flask. The mixture was stirred briefly then sonicated for 30 minutes to disperse the GO. The mixture was then covered with a rubber septum, equipped with a needle to allow for head-room during heating which was removed after temperature was reached, and heated at 70 °C with magnetic stirring for 24 hours.The reaction mixture was collected in a centrifuge tube and the reaction vessel was rinsed out with DI water into the centrifuge tube. The reaction mixture was spun down and the supernatant discarded. The pellet was washed and centrifuged down with DI water (3 × 40 mL), THF (2 × 40 mL), and acetone (2 × 40 mL). The washed pellet was placed under vacuum overnight before grinding the resulting hard pellet into a powder. Powders were weighed and a mass yield was found for each respective material after the washes. The mass yield of powder from PU1–GO1, PU3–GO1, PU5–GO1, and PU10–GO1 were as follows: 136.22 mg, 214.99 mg, 310.41 mg, and 420.89 mg, respectively.
Several controls were also prepared using the same basic methods while excluding certain components. A graphenic control (GONP) was prepared by subjecting GO to the same reaction conditions outlined above, but in the absence of PU in order to assess changes to the graphenic structure caused by the basic conditions and mild temperatures. A degraded PU control (PUD) was prepared by subjecting shredded PU (700 mg) to the same reaction conditions in the absence of GO. During PUD preparation, the supernatant from all centrifugation steps was kept in order to assess which components were removed from the PU during the degradation and purification, and to demonstrate the ability to isolate other degraded fragments. Supernatent was collected together and any residual particulate was allowed to crash out overnight and was filtered to remove the solids. Then the solvent from the supernatant was removed through lyophilization and the obtained residue was characterized. Residue was found to have no traces of toluene diisocyanate (TDI) components and only contained polyol component (ESI Fig. S10†).17
2.4 Polyurethane-grafted graphene oxide characterization
The chemical composition of the PU–GO materials were thoroughly characterized through a series of analytical techniques as follows: ATR-FTIR, TGA, SEM, DLS, and XRD.A PerkinElmer Frontier FTIR Spectrometer with an attenuated total reflectance attachment containing a germanium crystal was used to perform FTIR spectroscopy. Raw spectra were obtained over a range of 4000–700 cm−1 with 0.4 cm−1 resolution and averaged over 16 scans. The spectra were normalized with respect to the transmittance value at 3294 cm−1.
A PerkinElmer TGA 4000 was used to analyze the thermal degradation under N2 from 50–800 °C with a ramp rate of 10 °C min−1. A ceramic crucible was cleaned using a butane torch to burn any residual organic material to ash. The mass of the crucible was tared and between 4 and 10 mg of sample was loaded into the sample holder. Onset temperature for degradation events was analyzed by taking the x value at any local minimums for the first derivative of the thermograms. Char weight percent is defined as the final char mass percent present at 800 °C.
NMR samples of PUD and residue from the supernatant of PUD washes were taken to analyze their composition. PUD and residue from the washes were dissolved in deuterated DMSO and filtered to remove sediment prior to analysis. A 500 MHz NMR (Bruker Avance™ 500) was used to analyze 1H-NMR. Data was analyzed using Topspin Software (version 4.1.4).
Powdered, as synthesized materials were spread on carbon tape with loose material being blown off via N2 gas. These materials were then loaded into a FEI Quanta 600 FEG Scanning Electron Microscope. To determine how varying amounts of PU altered the electrical conductivity of the material, the instrument's T images from secondary electrons were generated at constant brightness and contrast.
DLS was taken on a Malvern Panalytical Zetasizer Ultra taking a 60 second temperature equilibration time at 25 °C prior to each run which consisted of 5 scans. Trifluoroacetic acid was used as the dispersion and solution media. Samples were sonicated for 10 minutes prior to DLS scans. Refractive indexes of 1.28, 1.96 and 1.4 for TFA, graphenic materials, and PU/PUD materials, respectively.
As synthesized powder samples were loaded into a rectangular powder holder and flattened prior to loading into a PANalytical Empyrean X-ray diffractometer. Scans were taken with a 2θ° between 5 and 80 using 1Der FASS(Short) instrument optics, a 10 mm incident beam mask, a 1/2° divergence slit, and a 1° anti-scatter slit. Scans were taken with a scan step size of 0.05 degrees. A Cu probe, with a tension of 45 kV and a current of 40 mA, provided the incident beam. Bragg's law was used to find the interlayer spacing modes present in the sample.
2.5 Polyurethane composite preparation
1 g of the polyol solution (part B of the polyurethane foam precursors purchased from Fiberglass Supply Depot Inc.) was added to a 20 mL scintillation vial with the appropriate amount and type of filler: 5, 10, 20, 40 mg of filler for 0.25, 0.5, 1.0, and 2.0 weight% of graphenic filler, respectively. The graphenic filler mixtures were stirred mechanically with a copper wire to help mix the graphenic materials in the viscous polyol solution, and the mixture was subject to bath ultrasonication for 30 minutes to promote further dispersion and exfoliation of the graphenic materials. 1 g of the isocyanate solution (part A of the polyurethane foam precursors purchased from Fiberglass Supply Depot Inc.) was added to the polyol graphene mixture and mechanically stirred with a copper wire for 45 seconds to mix the polyurethane components. The reaction mixture began to foam and the foam was left to expand and cure overnight. The glass scintillation vials were then broken and the foams were retrieved for further processing into discs for DMA testing. Discs were prepared by cutting samples with a razor roughly 5–7 mm in height and sanding the discs down to ensure a level testing surface.2.6 Polyurethane composite thermal and mechanical characterization
The thermal and physical characteristics of the PU composites were assessed through DSC for thermal conductivity, DMA for mechanical properties, and optical spectroscopy and micro-CT for morphological analysis.A PerkinElmer DSC 4000 was used to analyze thermal conductivity in a range of 150–160 °C for indium samples with a ramp rate of 0.5 °C min−1. Thermal history of each sample was erased by a preliminary heating and cooling cycle within the respective samples ranges prior to analysis of the second heating step. PU–GO foam samples with diameter of 3.3–3.8 mm and height of 1.8–3.0 mm were placed in an aluminum pan and run as a blank to subtract any background from the sample, and then a second run was performed in which an aluminum pan with ∼5 mg of indium was placed flat on top of the foam sample. The slope of the melting isotherm for the metal was taken and used to calculate the thermal conductivity of the foams at ∼156 °C. The error reported for each point is the experimental error found by running the neat PU foam sample in triplicate and reporting the standard error for the three samples that were run. Thermal conductivity was calculated based using the following equation:18

DMA was measured on a Discovery Hybrid Rheometer 2 (TA Instruments, New Castle, DE). Compressive DMA testing was performed at room temperature in several parts. Frequency sweeps were taken of PU foam discs by applying a preforce of 1 N to the samples prior to a 1.0% strain over a range of 1–15 Hz. A perforce of 1.5 N was reapplied to account for any change in sample height during testing and the new initial height was taken from the gap size measured by the DMA prior to cyclic loading tests. Cyclic loading/deloading was applied three times for 1%, 2%, and 3% strain, each at a strain rate of 0.001 mm s−1. The final deloading was stopped when a force of <0 N was detected and this was taken as the final height of the relaxed sample after mechanical strain. Finally, frequency sweeps were once again taken of PU foam discs by applying a preforce of 1 N to the samples prior to a 1.0% strain over a range of 1–15 Hz. Triplicates of each material was run on 3 separate foam disks and experimental values were averaged and standard error was reported for each.
The foams were imaged using an optical microscope. The images were cropped, converted to 8 bit image and adjusted for brightness/contrast and threshold to develop a good differentiation between the foam and the pores using ImageJ. Pore area and Feret diameter of pores was determined as a function of pore frequency using the ImageJ software.
Cylindrical specimens of PU foams with variety of fillers at 0.5% and 1% loading percentage, with a diameter of 8.66 mm and a height ranging from 0.5 to 1.5 mm, were prepared for micro-CT scanning. The scans were conducted using a ZEISS Xradia Crystal CT® micro-CT scanner at a resolution of 3.5 μm. The current and voltage settings were maintained at 100 μA and 60 kV, respectively. A threshold intensity of greater than 5000 was employed to distinguish between the foam material and pores. The 3D reconstruction of the scans was performed, generating tiff files using Scout and Scan software. These files were subsequently analyzed with Dragonfly Pro software, where a region of interest (ROI) was cropped to eliminate edge effects and cylinder ends. The images were analyzed to determine the percentage pore volume for each foam sample.
3 Results and discussion
Powdered materials were characterized to assess both the polymer loading content for each graphenic material as well as for polymer composition and covalent attachment. PU–GO materials were prepared using several weight ratios of PU to GO during synthesis to determine if attachment amount could be varied in the preparation conditions. Weight ratios of PU to GO of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3:1, 5:1, and 10:1 were prepared and are referred to as PU1–GO1, PU3–GO1, PU5–GO1, and PU10–GO1, respectively throughout this paper. Additionally, several controls were prepared to compare to the covalently grafted products. PU was subject to the same reaction conditions as the PU–GO materials, but in the absence of GO to determine the chemical composition of the resulting polymer fragments and find approximations for the grafted polymer molecular weight. The resulting degraded polymer materials is referred to as degraded PU (PUD). Graphenic controls were prepared in a similar manner in the absence of PU (GONP), in order to find any changes in the graphenic structure that could be due to the basic conditions of the reaction which have been known to alter the graphenic structures.19 Mild temperatures can also reduce graphenic materials in suspension, so another control was prepared in which GO was heated to 70 °C in ethanol for 24 hours without any added base (GONB).
:1, 3:1, 5:1, and 10:1 were prepared and are referred to as PU1–GO1, PU3–GO1, PU5–GO1, and PU10–GO1, respectively throughout this paper. Additionally, several controls were prepared to compare to the covalently grafted products. PU was subject to the same reaction conditions as the PU–GO materials, but in the absence of GO to determine the chemical composition of the resulting polymer fragments and find approximations for the grafted polymer molecular weight. The resulting degraded polymer materials is referred to as degraded PU (PUD). Graphenic controls were prepared in a similar manner in the absence of PU (GONP), in order to find any changes in the graphenic structure that could be due to the basic conditions of the reaction which have been known to alter the graphenic structures.19 Mild temperatures can also reduce graphenic materials in suspension, so another control was prepared in which GO was heated to 70 °C in ethanol for 24 hours without any added base (GONB).
3.1 Proton nuclear magnetic resonance spectroscopy (1H-NMR)
PUD was first assessed by 1H-NMR to determine which components were left after the fragmentation reaction and which components were removed during the washes. 1H-NMR of PUD in DMSO-d6 exhibited aromatic protons (7.4–9.0 ppm, 3H), N–H protons (7.0–7.4 ppm, 2H), and aliphatic protons (2.25 ppm, 3H) in ratios and shifts that are in good agreement with literature values for toluene diisocyanate (TDI)-based PUs rather than methylene diphenyl diisocyanate (MDI) which are also commonly used for slabstock PU foam (ESI Fig. S10†).17 There is also the presence of a peak shift for etheric protons (3.5 ppm) indicative of polyethylene glycol which is a common soft-segment.20 Polyethylene glycol is present in low amounts in the PUD sample, and the washed residue obtained from the supernatant of the PUD washes contained only polyethylene glycol peaks (ESI Fig. S10†). Thus, the valuable polyol component can be isolated and retrieved through simple centrifugation using this technique for further chemical processing. The findings that the PUD component mainly contained residual TDI hard-segment polycarbamates and that the polyether soft-segments are removed during washes, are further corroborated using Fourier-transform infrared spectroscopy and thermogravimetric analysis.3.2 Fourier-transform infrared spectroscopy (FTIR)
Composition of the PU–GO materials were then assessed by FTIR to determine the chemical functional groups present on the surface of the graphenic flakes after dynamic depolymerization and to estimate the polymer content. All FTIR spectra were normalized with respect to the peak at 3294 cm−1. Characteristic vibrational modes of PU were assigned and compared to the resulting PU–GO materials to assess which chemical groups were present in the materials and what the relative abundance of PU was in each sample.All FTIR peaks for PU mattress were in good agreement with those typically observed in TDI or MDI-based slabstock foams used for foam mattresses.21,22 Peaks characteristic of the urethane component were identified at 3288 cm−1 (N–H stretching), 1538 cm−1 (N–H bending), and 1730 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbamate stretch). The waste PU was determined to contain aromatic carbamate moieties due to the presence of aromatic absorption modes (1601, 1508, and 814 cm−1). Using the relative intensities of these peaks compared to what is observed in literature for both TDI and MDI-based PU it is most likely that the waste PU sourced for this study was TDI-based.21,22 Polyether polyol soft-segments were identified based on the strong C–O absorptions at 1227 cm−1 (C–O–C asymmetric stretch) and 1110 cm−1 (C–O–C symmetric stretch) and the absence of an ester CO stretch (Fig. 1A). There are also aliphatic peaks at 2972 cm−1 (C–H asymmetric stretch), 2892 cm−1 (C–H symmetric stretch), and 1373 cm−1 (C–H bending).
O carbamate stretch). The waste PU was determined to contain aromatic carbamate moieties due to the presence of aromatic absorption modes (1601, 1508, and 814 cm−1). Using the relative intensities of these peaks compared to what is observed in literature for both TDI and MDI-based PU it is most likely that the waste PU sourced for this study was TDI-based.21,22 Polyether polyol soft-segments were identified based on the strong C–O absorptions at 1227 cm−1 (C–O–C asymmetric stretch) and 1110 cm−1 (C–O–C symmetric stretch) and the absence of an ester CO stretch (Fig. 1A). There are also aliphatic peaks at 2972 cm−1 (C–H asymmetric stretch), 2892 cm−1 (C–H symmetric stretch), and 1373 cm−1 (C–H bending).
 | ||
| Fig. 1 (A) Attenuated total reflectance FTIR of PU–GO samples with N–H, C–H, and carbonyl CO stretching modes highlighted in purple. (B) XRD of PU–GO samples. (C) TGA of PU–GO samples with first derivative in black. (D) SEM of PU–GO samples, yellow arrows are pointing to areas of brighter contrast and raster lines caused by localized charging. | ||
As the ratio of PU to GO increases during synthesis, the intensity of the carbamate peaks increases in the resulting PU–GO. FTIR spectra of PU and PUD revealed the presence of N–H stretching (3288 cm−1), N–H bending (1538 cm−1), and C–N stretching (1224 cm−1) modes. These same carbamate peaks are observed in successively increasing peak intensity in the higher weight ratio PU–GOs. Additionally, C–H asymmetric (2980 cm−1) and symmetric (2892 cm−1) stretches that are present in PU are also observable in PU–GO materials and increase in intensity with higher polymer amount used in synthesis. C–H stretches are attributed to aromatic moieties in the carbamate. Interestingly, the C–H stretch present in PU at 2921 cm−1 is not present in the PU–GO materials suggesting that this aliphatic moiety was fragmented and removed with the washes. Due to the emergence of the CC stretching modes from the PU aromatic moieties in each of the PU–GO materials, the C–H stretch at 2921 cm−1 is likely from the aliphatic component of the polyethers. The PU carbamate peak at 1730 cm−1 (CO) shifts to a lower energy stretching mode at 1702 cm−1 in PUD and PU–GO materials, which is a common indicator of hydrogen bond formation between urethane carbonyl groups, likely caused by the increased mobility of the fragmented polymers compared to the bulk plastic construct.23 FTIR shows that increased loading of the carbamate containing hard-segment onto GO was achieved while polyether segments were removed in the washes.
3.3 X-ray diffraction (XRD)
Evidence for covalent attachment of PU to GO can also be observed in XRD diffractograms of the representative materials and controls (Fig. 1B). A new interlayer spacing emerges at 7.53 Å (2θ of 11.64) for PU–GO materials which is not present in the graphenic or PU controls. This new interlayer spacing is different from that of GO which is typically 6.8 Å (2θ of 12.9°) and corresponds to a larger interlayer spacing attributed to polymer functionalization and intercalation between graphenic sheets.24 Additionally, there is a peak broadening of the new interlayer spacing, suggesting the formation of a more amorphous material.3.4 Thermogravimetric analysis (TGA)
TGA was utilized to determine the amount of PU by mass contained in each PU-GO material. Char weight percents for PU1–GO1, PU3–GO1, PU5–GO1, and PU10–GO1, were 62.6, 53.8, 33.9, and 32.7 percent, respectively. Increased polymer conjugation results in a lower char weight percentage. This suggests that more polymer is successfully attached to the graphenic basal plane as the polymer combusts to completion once 800 °C is reached, whereas graphenic materials do not (Fig. 1C). Thermograms of the graphenic and polymer controls show that GONP, GONB, PU, and PUD have char weights of 78.3, 57.9, 1.0, and 15.2 percent. By comparing the char weights of the PU-GO materials to the char weights of PUD and GONP, an approximation of the weight ratios of PU to GO in the final product can be produced (ESI eqn (S4)†). This approach yielded weight percents of polymer in PU1–GO1, PU3–GO1, PU5–GO1, and PU10–GO1, of 24.8, 38.8, 70.4, and 72.3 percent by weight compared to the graphenic component. Thus PU5–GO1 and PU10–GO1 have similar PU loading amounts, suggesting that an upper threshold to the amount of polymer that can be attached with this method is close to being achieved. First derivative for PU1–GO1, PU3–GO1, PU5–GO1, and PU10–GO1, were 301 °C, 317 °C, 330 °C, and 329 °C which is closer to the first degradation event of PU at 290 °C than the second one at 393 °C. This along with the FTIR data suggests that the first degradation event of PU consists of the hard-segment polyurethane and the second degradation event consists of the polyether polyol. Furthermore, it is well accepted that thermal degradation of TDI-based PU foam takes place in 2 distinct degradation events as the TDI hard-segment degrades around 287 °C in an inert atmosphere.21 This value is in good agreement with the first degradation event of the PU mattress used in this study further corroborating that it is a TDI-based PU foam. No significant thermal transitions were apparent in any of the materials when analyzed with DSC over a broad region of −50 °C to 200 °C (ESI Fig. S9†).3.5 Scanning electron microscopy (SEM)
Evidence for the increased loading and conjugation of PU to GO is seen in SEM images taken at constant brightness and contrast (Fig. 1D). When electrically insulating materials such as polyurethane are subjected to an electron beam, the electrons will be retained by the sample rather than ejected and detected by the secondary electron detector. By keeping brightness and contrast of the instrument consistent through each sample, the contrast between samples can be used to determine their relative electronic insulation and thus higher plastic loading in PU–GO can be determined by a darker contrast.25,26 PU3–GO1, PU5–GO1, and PU10–GO1 were darker than PU1–GO1 which was darker than GONP and GONB flakes. Additionally, bright areas on PU3–GO1, PU5–GO1, and PU10–GO1 can be observed that rapidly flare up and blur due to charging with the raster lines during scanning which is a strong indication that the PU–GO materials are charging and that the contrast changes are due to insulation rather than changes in topography or shading.263.6 Dynamic light scattering (DLS)
Relative sizes of each PU–GO material, as well as the shredded PU and PUD, were compared using DLS as we have previously done for graphenic materials.16 For 2D materials with larger flake size, DLS can be used to approximate relative size through a measurement of the hydrodynamic radius.27–31 Hydrodynamic radius can provide a useful assessment of the relative size of multilayer graphenic materials. The multilayer GO produced using a modified Hummer's method has a Z-average size of 4765.6 nm. As expected for a polymer coated graphene flake, PU–GO materials generally increased in Z-average size (Fig. 2). One notable exception was that PU1–GO1 had a similar Z-average size to PU10–GO1. PU3–GO1 and PU5–GO1 are shown to have a smaller hydrodynamic radius than GO. This can be explained by the enhanced solubility of the PU grafted GO and the sonication required for PU–GO synthesis aids in dispersing the GO. PU3–GO1 and PU5–GO1 are more dispersible in the TFA used to disperse the graphenic samples for DLS. This can be attributed to the reduced solubility of PU1–GO1 in TFA due to low grafting density, resulting in greater aggregation of the graphenic flakes. | ||
| Fig. 2 Dynamic light scattering (DLS) of PU–GO materials taken in trifluoracetic acid. DLS provides insight into the relative sizes of each graphenic material through observing light scattering effects and assumes a spherical structure. | ||
4 Material properties
4.1 Thermal conductivity
PU foams are useful for thermal insulation and are often used in refrigeration applications. PU foams are highly porous materials which is what allows them to be highly thermally insulating as the major mechanism of heat transfer is through convection of trapped gasses in the foam.32 Thermal conductivity is a measurement of how much heat is conducted through a material of a known thickness in W m−1 K−1. The thermal insulating capabilities of a material are inversely proportional to their thermal conductivity, thus a less conducting material is more thermally insulating. Thermal conductivity for insulating materials can be found utilizing differential scanning calorimetry (DSC) of a pure metal.18 This is performed by analyzing the slope of the onset of a metals melting point isotherm as heat is applied through a sample with thermal resistance, in our case a PU foam sample.To test the thermal conductivity of each PU–GO as a filler, the graphenic materials were used to prepare PU foam composites at different weight percentages. Foams were prepared by adding 0.0, 0.25, 0.5, 1.0, and 2.0 weight percent each of GO, GONP, PU1–GO1, PU3–GO1, PU5–GO1, and PU10–GO1 to a polyol solution, followed by stirring and sonication to disperse the graphenic material, and the subsequent addition of a diisocyanate solution with stirring to prepare a self-foaming composite foam material. Thermal conductivity of each composite was then tested in the DSC on samples taken using a biopsy punch to prepare foam samples with similar dimensions. These composite samples were taken and placed on the DSC between the heating element and a sample of pure indium to collect the slope of the melting point isotherm during a heating sweep.
At elevated temperatures (156 °C), nearly all foams with ≤1.0 weight percent of graphenic fillers are more insulating compared to PU. The only exception occurs with 1.0% PU1–GO1, which has the same thermal conductivity as PU (Fig. 3). Further, nearly all fillers at 2.0% enhance the insulating properties of PU, except for PU1–GO1 and GO. This demonstrates that each filler could be used in a range of quantities to improve insulation.
 | ||
| Fig. 3 Thermal conductivity of PU foams with varying weight percents of each filler. It is important to note that lower thermal conductivity indicates a greater capacity for thermal insulation. A purple box is drawn around PU3–GO1 1.0%, the only sample where the lowest thermal conductivity did not occur at 0.5% filler content. | ||
The greatest increase in thermal insulation occurs at 0.5% of PU10–GO1 and 1.0% PU3–GO1, both with a 38.5% reduction in thermal conductivity. Alterations in the insulating properties of PU foams were attributed to the surfactant and cell nucleation capabilities of the graphenic fillers during foam preparation, because the gaseous content and architecture of PU foams is primarily responsible for any thermal conductivity of a PU foam.3,32 The superior insulation capabilities of PU3–GO1 at relatively high loading amounts is attributed to smaller PU cells. Further, this is due to the enhanced miscibility of PU3–GO1 in the polymer matrix and the surfactant properties of the graphenic material. For high temperature applications, PU3–GO1 allows for the greatest insulation capabilities over the largest range of filler used.
4.2 Pore size
Smaller graphenic flakes tend to be more dispersible in a polymer medium and well dispersed graphenic materials are known to act as surfactants in PU foams.3,33 Surfactants help to nucleate pore formation by reducing surface tension in the curing foam, resulting in smaller pore sizes.3 At higher loadings of graphene, the filler aggregates, which reduces its miscibility and surfactant properties resulting in larger pore sizes with a wider distribution.3 The pore morphology of the 0.5 and 1.0 weight percent loading nanocomposite foams was assessed through optical microscopy and micro-computed tomography (micro-CT) (Fig. 4A and S8†). PU-GO materials used as additives visually appeared to have smaller pores in both the optical microscopy and micro-CT images. Utilizing the images and 3D renderings, pore areas and pore volumes could be determined for each of the composite samples tested. Optical microscopy images were used to map pore area frequencies (Fig. 4B). All graphenic composites exhibited a shift in pore area to favor a smaller size distribution than neat PU foam, suggesting surface tension reduction by all graphenic fillers during foam formation. Additionally, PU10–GO1, PU5–GO1, and PU3–GO1 had a narrower distribution of pore area while PU1–GO1, GO, and GONP had a broader distribution, which suggests less uniform pore formation. Relative pore volume changes within each PU composite were calculated from micro-CT images (Fig. 4C). Relative decreases in pore volume based on graphenic loading correlates to corresponding decreases in thermal conductivity, suggesting that pore size is a major mechanism responsible for changes in insulation capabilities of PU composites. | ||
| Fig. 4 Various pore size characterization methods for PU foam composites including: (A) optical microscopy images of PU composites, (B) pore area distribution charts showing the relative frequency of different pore areas and revealing a shift towards lower pore areas for graphenic fillers, (C) pore volumes as a percentage of the total volume of a PU foam, values were obtained from micro-CT scans which can be found in Fig. S8.† Relative changes in pore volumes correlate well with relative changes observed in thermal conductivity such that relative decreases in pore volume in samples also exhibit relative decreases in thermal conductivity. | ||
4.3 Compressive frequency sweeps (1–15 Hz)
Mechanical properties of PU foams with various fillers were tested for changes to relative impact resistance, stiffness, and fatigue. In neat PU foam, when 0.5% strain is applied and the frequency is increased from 1 Hz to 15 Hz, there is a 22.8% increase in the storage modulus and an 8.3% decrease in the loss modulus. This indicates a greater energy of deformation for an applied stress at higher frequencies and a reduced loss of energy due to permanent deformation. In general, PU10–GO1, PU5–GO1, and PU3–GO1 composites exhibited storage and loss moduli close to that of PU, whereas PU1–GO1, GO, and GONP composites have increased storage and loss moduli compared to neat PU (Fig. 5A and B). An increase in both storage and loss moduli in these samples indicates a higher energy of elastic deformation and a greater loss of energy during relaxation. This increase in moduli in the elastic region could indicate better cushioning capabilities for PU1–GO1, GO, and GONP composite foams. Neat foams did not exhibit statistically different changes in storage or loss moduli due to the frequency of the applied force. Thus, neat foams do not react differently to applied stress within the elastic region under different impact rates.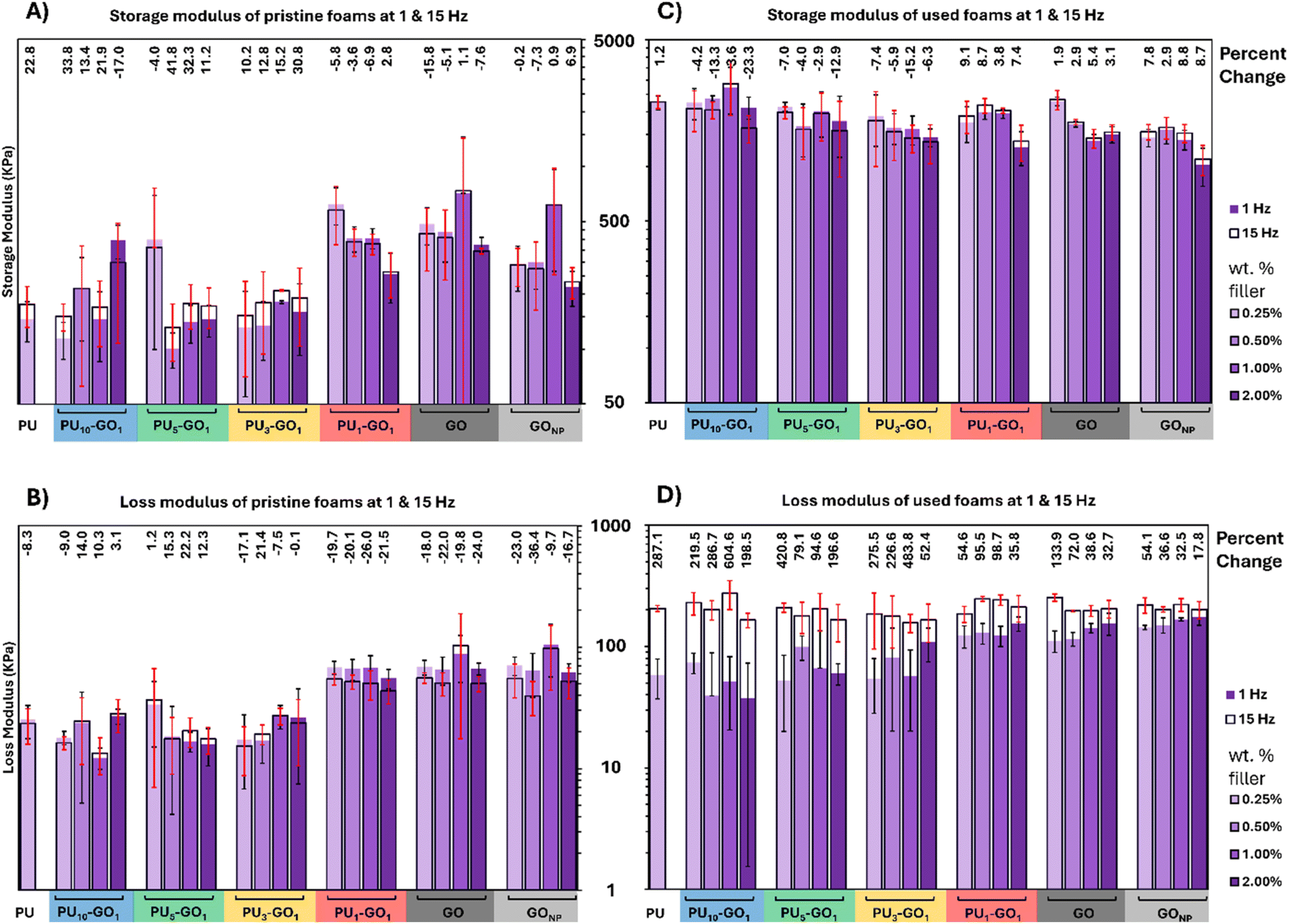 | ||
| Fig. 5 Frequency response was analyzed and compared before (A) and (B) and after (C) and (D) cyclic loading sweeps. Changes in moduli are reported as percent changes of storage (A) and (C) and loss (B) and (D) moduli between a 1 Hz run and a 15 Hz run at 0.5% strain. Storage moduli increases across all samples after cyclic loading due to compaction of the foam samples. Whereas loss moduli after cyclic loading are the only measurements that have a frequency response, with 15 Hz application of strain having higher loss moduli due to microfracturing caused by cyclic loading. | ||
After cyclic loading, the frequency response to stress for each material reveals interesting features regarding polymer foam stability (Fig. 5C and D). In all cases, the net storage and loss modulus of each foam material increases, often by at least one order of magnitude. This change is due to fragmenting and compacting of foams. For storage modulus, there is again no statistical difference between the 1 Hz and 15 Hz sweeps. PU10–GO1 and PU5–GO1 containing foams have storage moduli similar to PU while the storage modulus begins to decrease somewhat for PU3–GO1 and PU1–GO1 with the GO and GONP controls having the lowest storage moduli after cyclic loading. Thus we can conclude that there is no dependence on frequency for energy of deformation for the composites. However, loss moduli for these samples drastically increases at 15 Hz, showing increasing fracturing, friction, and fragility of the sample after cyclic loading. In PU, the loss modulus increases by 2.87 times its original value demonstrating significant damage to the neat foam due to cyclic loading. Generally, samples containing PU-GO materials showed a narrowing of this loss modulus increase indicating less damage to the samples. This narrowing is generally inversely proportional to polymer loading such that PU1–GO1 composites exhibited the least amount of damage from cyclic loading. GO and GONP also showed reduced damage from the cyclic loading suggesting that these materials can be used to tailor and decrease fracturing and permanent deformation of PU foams.
4.4 Tailorable stiffness seen in cyclic loading
Lower polymer attachment to GO (e.g. PU1–GO1), resulted in a decreased stiffness for the resulting foam. At every weight percentage, foams containing PU10–GO1 retain similar stiffness to neat PU (Fig. 6A and S6†). All other fillers, including GO and GONP, generally reduce the stiffness. GO composites had the lowest stiffness throughout all cyclic loading tests. In general, PU–GO fillers with a higher PU to GO ratio (PUX–GOX) are stiffer and have a stiffening response to loading similar to neat PU. GO and GONP have a more mixed response, generally exhibiting some stiffness at low weight percent composites but soften at higher weight percent loadings. From this experiment we conclude that PU–GO materials with higher PU content used during synthesis (e.g. PU10–GO1) retains properties close to that of neat PU due to improved composite incorporation and entanglement, while PU–GO materials with lower PU content tend to be less stiff and have a reduced stiffening effect with cyclic loading. The lower stiffness and stiffening response to stimulus in PU3–GO1 and PU1–GO1 may provide more cushioning with less wear over time, as compaction of foams is the primary mechanism of stiffening in PU foams.5 Further evidence to support the durability of these foams can be seen in the changes in plastic strain energy and fatiguing.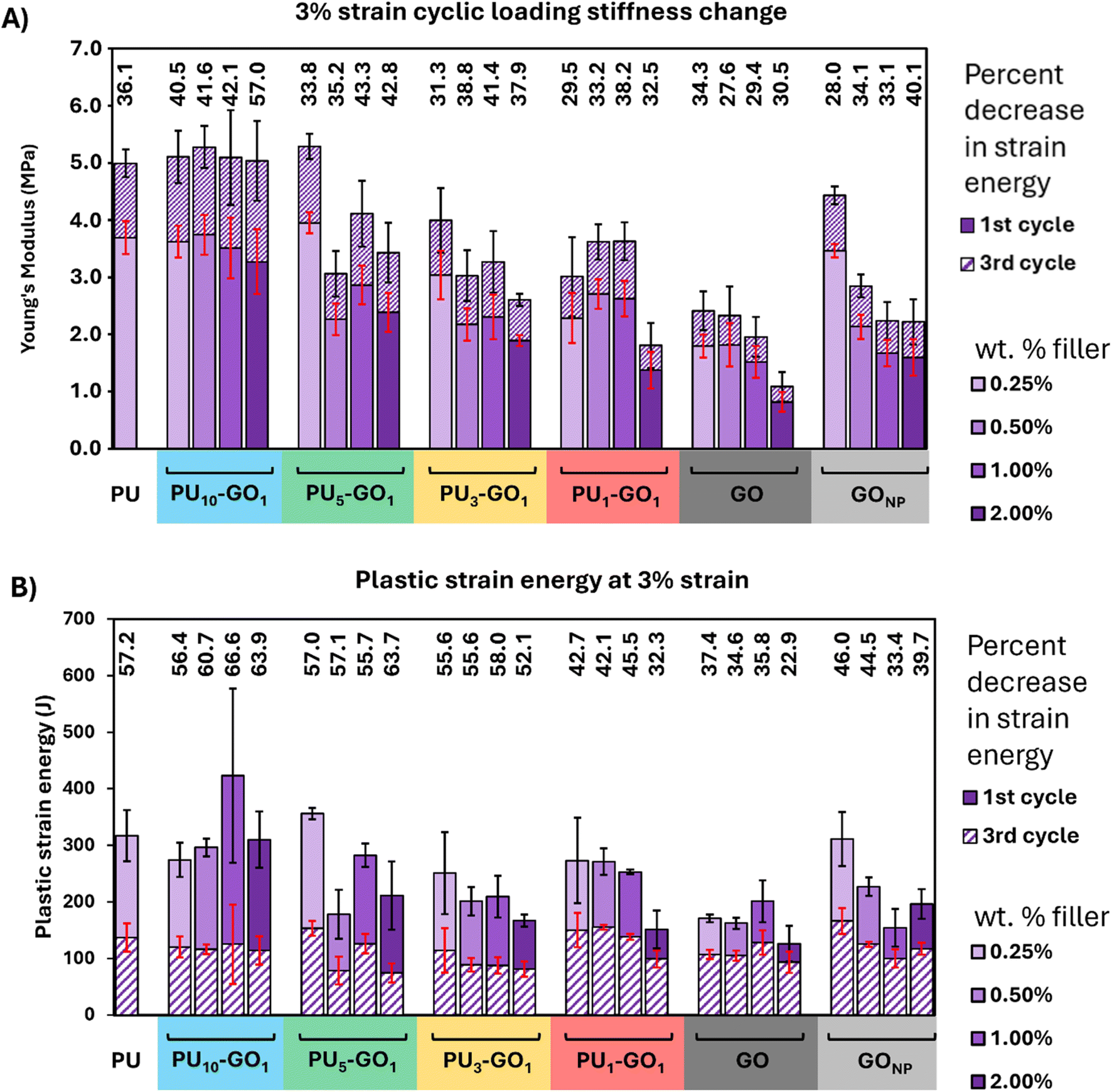 | ||
| Fig. 6 (A) Stiffening of foam composites demonstrated by Young's modulus change with cyclic loading at 3% strain. Stiffening response at 1% and 2% strain can be found in Fig. S6.† Young's modulus was taken from the slope of the second half of each loading curve to account for changes in the material due to fracturing. Stiffening occurs with cyclic loading due to fracturing and compacting of the foams over time. (B) Plastic strain energy loss for each material as a percentage of the initial plastic strain energy at 3% strain. 1% and 2% strain runs are available in Fig. S7.† Runs 1–3 are at 1% strain, runs 4–6 are at 2% strain, and runs 7–9 are at 3% strain. | ||
4.5 Fatiguing response to cyclic loading
Cyclic loading of PU foams was conducted at 1%, 2%, and 3% strain for 3 cycles, each starting with lower strain percents and ending with higher strain (Fig. 6B and S7†). Fatigue is a measure of loss of plastic strain energy, which is the energy loss in a cyclic loading sample found by taking the difference in area between a loading/deloading curve. Fatigue was analyzed by observing the change of plastic strain energy relative to the first loading/deloading cycle of a given strain percent. Fatiguing is observed in compression of PU foams due to microfracturing of the foam structural components and as microfractures occur the remaining plastic strain energy is due to friction between foam components and the plastic strain energy plateaus.34PU–GO fillers with lower PU to GO ratios (higher graphenic component) reduce fatiguing in PU foam composites. The initial plastic strain energy in each new strain region for PU3–GO1 and PU1–GO1 containing foams is lower than neat PU, exhibiting less energy loss from deformation than neat PU, PU10–GO1, and PU5–GO1 containing foams. Interestingly, PU10–GO1 and PU5–GO1 tend to fatigue to a similar degree to neat PU, which is an indication of a similar degree of microfracture formation. This is in good agreement with the trends observed in the loss modulus change seen in the frequency sweeps of used composite foams. PU1–GO1, GONP, and GO all demonstrate a decrease in fatiguing in 1%, 2%, and 3% strain, in particular at high filler content of 1 and 2 weight percent. PU1–GO1 decreases fatiguing at all weight ratios ranging from 15–25% reduction. Thus, if minimal fatigue and low stiffness is desirable for more durable and softer PU foams, PU3–GO1, PU1–GO1, GONP, or GO are the filler of choice. On the other hand, if improved thermal properties are desired for a PU foam without any change in the mechanical properties, the use of a PU10–GO1 or PU5–GO1 composite may be more desirable.
5 Conclusions
A dynamic depolymerization technique was successfully developed to covalently attach the hard-segment of polyurethane (PU) foam, sourced from post-consumer materials, to graphene oxide (GO). The attachment was accomplished using waste PU foam from a mattress and tailorable loading amounts of the PU hard-segment was demonstrated through simple adjustment of the weight ratio of PU to GO used in the synthesis. The tailorable loading has an upper limit between a ratio of PU to GO of 5:1 and 10:1. The graphenic filler materials were processed and formulated into PU foam composites with 0.25, 0.5, 1.0, and 2.0 weight percent content and tested for changes in mechanical and thermal properties.
Based on the findings from the manufactured foams, we can outline which of these fillers is ideal for specific applications. General trends that were observed suggest that greater PU to GO ratio (e.g. PU10–GO1) in composites results in foams with superior thermal insulation with retention of neat PU mechanical properties. Conversely, PU–GO materials with a lower PU to GO ratio (e.g. PU1–GO1) exhibit improved thermal insulation along with improved cushioning through reduced stiffness and improved durability from reduced fatiguing and microfracture formation. At high temperature conditions, thermal insulation is significantly improved through the addition of all graphenic fillers, including controls, at 0.25 and 0.5 weight percent filler. PU10–GO1, PU5–GO1, and PU3–GO1 also demonstrated improved insulating capabilities at higher weight percent loadings like 1% and 2%. This is atypical for most graphenic fillers, which tend to aggregate at higher filler loading percents. For applications in which an insulating foam is desired, the best fillers were 0.5% PU10–GO1 and 1.0% PU5–GO1, which both demonstrated a 38.5% reduction in thermal conductivity.
When analyzing the mechanical properties of the PU foams with graphenic fillers, it was found that PU10–GO1 generally had similar mechanical properties to PU. PU5–GO1, PU3–GO1, and PU1–GO1 also exhibited improved cushioning capabilities, and some reduction in fatiguing while having superior thermal insulating properties, making them desirable for more durable thermal insulating materials. PU1–GO1, GONP, and GO fillers were less brittle and tended to have a much lower fatiguing response to sustained stress application than the other fillers or neat PU, making them desirable for long term cushioning applications. PU1–GO1 had reductions in fatiguing ranging from 15–25% over all filler contents and strain percents. This suggests that the dynamic depolymerization provides a promising method for upcycling PU by creating PU–GO filler materials that provide enhanced properties tailorable to many traditional applications of PU.
The future prospects of this subsequent depolymerization and attachment technique for common commodity plastics highlights the potential for attachment of a number of other condensation polymers with chemically accessible backbones. For example, different hard and soft-segment polyurethanes may provide very different properties when incorporated into a hybrid graphenic material and used to make composites. While we have demonstrated the depolymerization of a polyester (previous work) and now polyurethane (this work) there are still other condensation polymers that could be attached using similar methods, such as polyamides, polyethers, polyimides, and more. Additionally, more work could be conducted to see if this method of simultaneous polymer fragmentation and attachment could be performed with other oxidized carbon nanomaterials. Up to this point this has been attempted with only graphene oxide, but it is feasible that these strategies would work with other oxidized carbon substrates such as carbon nanotubes, graphitic oxide, and oxidized fullerenes. The expansion of the various polymers and substrates that can be utilized with this type of attachment technique could expand the scope of utility for post-consumer plastic waste and the technological applications that the hybrid materials could be used in.
Data availability
Data for this article, including TGA, FTIR, XRD, SEM, thermal conductivity, micro-CT scans, microscopy images, DLS, and DMA are available at Open Science Framework at https://osf.io/6vkwy/?view_only=b4c23895436b43eb99240d1383706cc0.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the National Science Foundation under Grant No. CBET 2318652, award from the Engineering and Collaborative Research (ECR) program within the Division of Chemical, Bioengineering, Environmental, and Transport Systems (CBET). We thank J. Gillespie for providing training and use of the XPS (Materials Characterization Laboratory at the University of Pittsburgh). We thank the Wilton E. Scott Institute for Energy Innovation for funding for this project. The authors acknowledge use of the Materials Characterization Facility at Carnegie Mellon University supported by grant MCF-677785. A portion of this research was sponsored by the Army Research Laboratory and was accomplished under Cooperative Agreement Number W911NF-20-2-0175. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Army Research Laboratory or the U.S. Government. The U.S. Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation herein.References
- A. S. Dutta, Polyurethane Foam Chemistry, in Recycling of Polyurethane Foams, Elsevier, 2018, pp. 17–27, DOI:10.1016/B978-0-323-51133-9.00002-4.
- X. Jin, N. Guo, Z. You and Y. Tan, Design and Performance of Polyurethane Elastomers Composed with Different Soft Segments, Materials, 2020, 13(21), 4991, DOI:10.3390/ma13214991.
- J. M. Kim, D. H. Kim, J. Kim, J. W. Lee and W. N. Kim, Effect of Graphene on the Sound Damping Properties of Flexible Polyurethane Foams, Macromol. Res., 2017, 25(2), 190–196, DOI:10.1007/s13233-017-5017-9.
- N. Yousefi, M. M. Gudarzi, Q. Zheng, X. Lin, X. Shen, J. Jia, F. Sharif and J.-K. Kim, Highly Aligned, Ultralarge-Size Reduced Graphene Oxide/Polyurethane Nanocomposites: Mechanical Properties and Moisture Permeability, Composites, Part A, 2013, 49, 42–50, DOI:10.1016/j.compositesa.2013.02.005.
- Y. Xiao, J. Yin, X. Zhang, X. An, Y. Xiong and Y. Sun, Mechanical Performance and Cushioning Energy Absorption Characteristics of Rigid Polyurethane Foam at Low and High Strain Rates, Polym. Test., 2022, 109, 107531, DOI:10.1016/j.polymertesting.2022.107531.
- J.-H. Oh, J. Kim, H. Lee, Y. Kang and I.-K. Oh, Directionally Antagonistic Graphene Oxide-Polyurethane Hybrid Aerogel as a Sound Absorber, ACS Appl. Mater. Interfaces, 2018, 10(26), 22650–22660, DOI:10.1021/acsami.8b06361.
- T. Shou, S. Hu, Y. Wu, X. Tang, G. Fu, X. Zhao and L. Zhang, Biobased and Recyclable Polyurethane for Room-Temperature Damping and Three-Dimensional Printing, ACS Omega, 2021, 6(44), 30003–30011, DOI:10.1021/acsomega.1c04650.
- N. V. Gama, B. Soares, C. S. R. Freire, R. Silva, C. P. Neto, A. Barros-Timmons and A. Ferreira, Bio-Based Polyurethane Foams toward Applications beyond Thermal Insulation, Mater. Des., 2015, 76, 77–85, DOI:10.1016/j.matdes.2015.03.032.
- A. Demharter, Polyurethane Rigid Foam, a Proven Thermal Insulating Material for Applications between +130°C and −196°C, Cryogenics, 1998, 38(1), 113–117, DOI:10.1016/S0011-2275(97)00120-3.
- M. G. Moore, K. Belton, H. Rose-Glowacki, D. Lan and E. Sanchez, The Economic Benefits of the U.S. Polyurethanes Industry, American Chemistry Council, 2022 Search PubMed.
- C.-H. Wu, C.-Y. Chang, C.-M. Cheng and H.-C. Huang, Glycolysis of Waste Flexible Polyurethane Foam, Polym. Degrad. Stab., 2003, 80(1), 103–111, DOI:10.1016/S0141-3910(02)00390-7.
- L. Zhao and V. Semetey, Recycling Polyurethanes through Transcarbamoylation, ACS Omega, 2021, 6(6), 4175–4183, DOI:10.1021/acsomega.0c04855.
- L. Polo Fonseca, A. Duval, E. Luna, M. Ximenis, S. De Meester, L. Avérous and H. Sardon, Reducing the Carbon Footprint of Polyurethanes by Chemical and Biological Depolymerization: Fact or Fiction?, Curr. Opin. Green Sustainable Chem., 2023, 41, 100802, DOI:10.1016/j.cogsc.2023.100802.
- W. M. Vickery, K. Lee, S. M. Lee, J. D. Orlando and S. A. Sydlik, Plastic Composites from Repurposed Poly(Ethylene Terephthalate) Wasted Functionalized Graphene Oxide through Dynamic Depolymerization, ACS Appl. Nano Mater., 2024, acsanm.3c05131, DOI:10.1021/acsanm.3c05131.
- W. S. Hummers and R. E. Offeman, Preparation of Graphitic Oxide, J. Am. Chem. Soc., 1958, 80(6), 1339, DOI:10.1021/ja01539a017.
- B. D. Holt, A. M. Arnold and S. A. Sydlik, In It for the Long Haul: The Cytocompatibility of Aged Graphene Oxide and Its Degradation Products, Adv. Healthcare Mater., 2016, 5(23), 3056–3066, DOI:10.1002/adhm.201600745.
- S. Jana, D. Samanta, M. M. Fahad, S. N. Jaisankar and H. Kim, Blocking and Deblocking of Diisocyanate to Synthesize Polyurethanes, Polymers, 2021, 13(17), 2875, DOI:10.3390/polym13172875.
- Mettler Toledo, Simple Determination of the Thermal Conductivity of Polymers by DSC, Thermal Analysis UserCom 22, METTLER TOLEDO Search PubMed.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, Graphene Oxide. Origin of Acidity, Its Instability in Water, and a New Dynamic Structural Model, ACS Nano, 2013, 7(1), 576–588, DOI:10.1021/nn3047378.
- J. L. Pasek-Allen, R. K. Wilharm, J. C. Bischof and V. C. Pierre, NMR Characterization of Polyethylene Glycol Conjugates for Nanoparticle Functionalization, ACS Omega, 2023, 8(4), 4331–4336, DOI:10.1021/acsomega.2c07669.
- X. Zhao, J. Wang, J. Li and Q. Yu, Kinetic Modeling of Thermal Degradation of TDI/MDI-Based Flexible Polyurethane Foam under Nitrogen and Air Atmospheres with Shuffled Complex Evolution Algorithm: Insights from TG-FTIR Analysis, J. Anal. Appl. Pyrolysis, 2024, 177, 106279, DOI:10.1016/j.jaap.2023.106279.
- M. A. Garrido and R. Font, Pyrolysis and Combustion Study of Flexible Polyurethane Foam, J. Anal. Appl. Pyrolysis, 2015, 113, 202–215, DOI:10.1016/j.jaap.2014.12.017.
- A. Reghunadhan; S. Thomas, in Polyurethane Polymers, Elsevier, 2017, pp. 1–16, DOI:10.1016/B978-0-12-804039-3.00001-4.
- K. E. Eckhart, B. D. Holt, M. G. Laurencin and S. A. Sydlik, Covalent Conjugation of Bioactive Peptides to Graphene Oxide for Biomedical Applications, Biomater. Sci., 2019, 7(9), 3876–3885, 10.1039/c9bm00867e.
- H.-B. Zhang, R.-J. Feng and K. Ura, Utilizing the Charging Effect in Scanning Electron Microscopy, Sci. Prog., 2004, 87(4), 249–268, DOI:10.3184/003685004783238490.
- K. Ura, Contrast Mechanism of Negatively Charged Insulators in Scanning Electron Microscope, J. Electron Microsc., 1998, 47(2), 143–147, DOI:10.1093/oxfordjournals.jmicro.a023571.
- B. Gürünlü, Ç. Taşdelen Yücedağ and M. R. Bayramoğlu, Green Synthesis of Graphene from Graphite in Molten Salt Medium, J. Nanomater., 2020, 2020, 1–12, DOI:10.1155/2020/7029601.
- B. Dabrowski, A. Zuchowska, A. Kasprzak, G. Z. Zukowska and Z. Brzozka, Cellular Uptake of Biotransformed Graphene Oxide into Lung Cells, Chem.-Biol. Interact., 2023, 376, 110444, DOI:10.1016/j.cbi.2023.110444.
- W. M. Vickery, H. B. Wood, J. D. Orlando, J. Singh, C. Deng, L. Li, J.-Y. Zhou, F. Lanni, A. W. Porter and S. A. Sydlik, Environmental and Health Impacts of Functional Graphenic Materials and Their Ultrasonically Altered Products, NanoImpact, 2023, 31, 100471, DOI:10.1016/j.impact.2023.100471.
- D. L. White, L. Lystrom, X. He, S. C. Burkert, D. S. Kilin, S. Kilina and A. Star, Synthesis of Holey Graphene Nanoparticle Compounds, ACS Appl. Mater. Interfaces, 2020, 12(32), 36513–36522, DOI:10.1021/acsami.0c09394.
- A. P. Najafabadi, M. Pourmadadi, F. Yazdian, H. Rashedi, A. Rahdar and A. M. Díez-Pascual, pH-Sensitive Ameliorated Quercetin Delivery Using Graphene Oxide Nanocarriers Coated with Potential Anticancer Gelatin-Polyvinylpyrrolidone Nanoemulsion with Bitter Almond Oil, J. Drug Delivery Sci. Technol., 2023, 82, 104339, DOI:10.1016/j.jddst.2023.104339.
- H. Zhang, W.-Z. Fang, Y.-M. Li and W.-Q. Tao, Experimental Study of the Thermal Conductivity of Polyurethane Foams, Appl. Therm. Eng., 2017, 115, 528–538, DOI:10.1016/j.applthermaleng.2016.12.057.
- S. Zhang, Y. Cheng, W. Xu, J. Li, J. Sun, J. Wang, C. Qin and L. Dai, Dispersibility of Different Sized Graphene Oxide Sheets and Their Reinforcement on Polyamide 6 Fibers, RSC Adv., 2017, 7(89), 56682–56690, 10.1039/c7ra12261f.
- K. Onoue, Energy Consumption Characteristics of Concrete Using Granulated Blast-Furnace Slag Sand Related to Nucleation and Propagation of Microcracks, Constr. Build. Mater., 2019, 218, 404–412, DOI:10.1016/j.conbuildmat.2019.05.141.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06691j |
| This journal is © The Royal Society of Chemistry 2025 |