
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis, and in silico studies of new quinazolinones tagged thiophene, thienopyrimidine, and thienopyridine scaffolds as antiproliferative agents with potential p38α MAPK kinase inhibitory effects†
Aisha A. Alsfouka,
Ismail M. M. Othmanb,
Manal M. Anwar *c,
Asmaa Saleha and
Eman S. Nossier*de
*c,
Asmaa Saleha and
Eman S. Nossier*de
aDepartment of Pharmaceutical Sciences, College of Pharmacy, Princess Nourah bint Abdulrahman University, P. O. Box 84428, Riyadh 11671, Saudi Arabia
bDepartment of Chemistry, Faculty of Science, Al-Azhar University, Assiut 71524, Egypt
cDepartment of Therapeutic Chemistry, Pharmaceutical and Drug Industries Research Institute, National Research Centre, El-Bohouth Street, Dokki, P. O. Box 12622, Cairo, Egypt. E-mail: manal.hasan52@live.com
dPharmaceutical Medicinal Chemistry and Drug Design Department, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo, 11754, Egypt. E-mail: dr.emannossier@gmail.com; dremannossier@azhar.edu.eg
eThe National Committee of Drugs, Academy of Scientific Research and Technology, Cairo, 11516, Egypt
First published on 16th January 2025
Abstract
The current work focuses on the creation of novel derivatives of the quinazolinone ring system, with various substituted thiophene, thienopyrimidine, and thienopyridine scaffolds 3a,b–11. Employing the standard MTT assay, every target compound's in vitro antiproliferative efficacy was evaluated in comparison with doxorubicin against both normal WI-38 cells and various cancer cell lines. Derivatives 6, 8a, and 8b demonstrated the most potent activity, alongside their safety profiles against WI-38. The in vitro enzyme assay showed that the new analogues had a better ability to inhibit p38α MAPK kinase than SB 202190 (IC50s = 0.18 ± 0.02, 0.23 ± 0.05, 0.31 ± 0.04, and 0.27 ± 0.06 μM, respectively). Additionally, apoptosis tests conducted on MCF-7 cells revealed that 6, 8a, and 8b significantly increased the levels of Bax (by approximately 7.31, 13.8, and 8.86 fold) and caspase 3 (by approximately 3.55, 4.22, and 3.87 fold), respectively, in comparison to the untreated cells. They decreased the amount of Bcl-2 by ∼1.99, 3.69, and 2.66 fold, respectively. The most powerful counterpart, 8a, underwent additional investigation of the cell cycle and apoptosis. It caused necrotic and apoptotic effects in the late stages and stopped the MCF-7 cell cycle at the G2/M phase. Based on the molecular docking study, candidates 6, 8a, and 8b all fit well within p38α MAPK kinase, with energy scores of −10.88, −11.28, and −10.96 kcal mol−1, respectively. Based on the in silico computer examination of physico-chemical and ADMET properties, the latter analogues seem to be promising candidates for further development and optimization in research.
1. Introduction
Targeted therapy, which regulates cell development and targets specific proteins, is thought to be a pinpoint cancer treatment approach. A class of serine/threonine protein kinases called mitogen-activated protein kinases (MAPKs) are widely distributed in immunological, inflammatory, and endothelial cells.1–4 They are necessary for functions within cells, including cell division, death, and inflammation, as well as regulating the channels of signaling. The MAPK family is subdivided into four groups: p38 MAPKs, extracellular signal-regulated kinases (ERK1 and 2), and c-Jun N-terminal kinase 1 (JNK1).5,6 The main differences between these kinases, aside from their activation in response to distinct stimuli, are in the size and sequencing of their activation loops. p38 protein kinases in mammals are further divided into four isoforms (p38α, p38β, p38γ, and p38δ).7 Among them, p38α has been the most thoroughly researched isoform, which is essential for controlling pro-inflammatory signaling as well as the production and function of important pro-inflammatory cytokines, including interleukin-1 beta (IL-1β) and tumor necrosis factor alpha (TNF-α).7Elevated p38α MAPK levels are associated with various cancer types, including thyroid, liver, breast, colon, and lung malignancies.8,9 There is ample evidence to suggest efficient function of excessively expressed p38α MAPK as a tumor booster, primarily through impairing control of cell cycle and promoting anti-apoptotic mechanisms.10–13 Due to its significant function, numerous cancer processes are linked to p38α MAPK, which is considered a significant therapeutic anticancer target. This has rendered the use of p38α-specific small-molecule inhibitors as pharmacologic anticancer therapies possible.1,3,14 Up to now, no FDA-approved medication for marketing has been developed using selective p38α inhibitors, despite tremendous efforts in their design and synthesis.5 Nonetheless, at various phases of assessment, a limited quantity of p38α MAPK suppressors have advanced to clinical development and research.
Based on the distinctive manner of binding within the kinase active site, p38α MAPK inhibitors are divided into different categories (Fig. 1). In a DFG-in conformation, type I inhibitors bind at the point where the ATP adenine ring occurs. Through hydrogen bonding, they engage with p38α MAPK to link the Met109 acid that is in connection with the hydrophobic area I.5
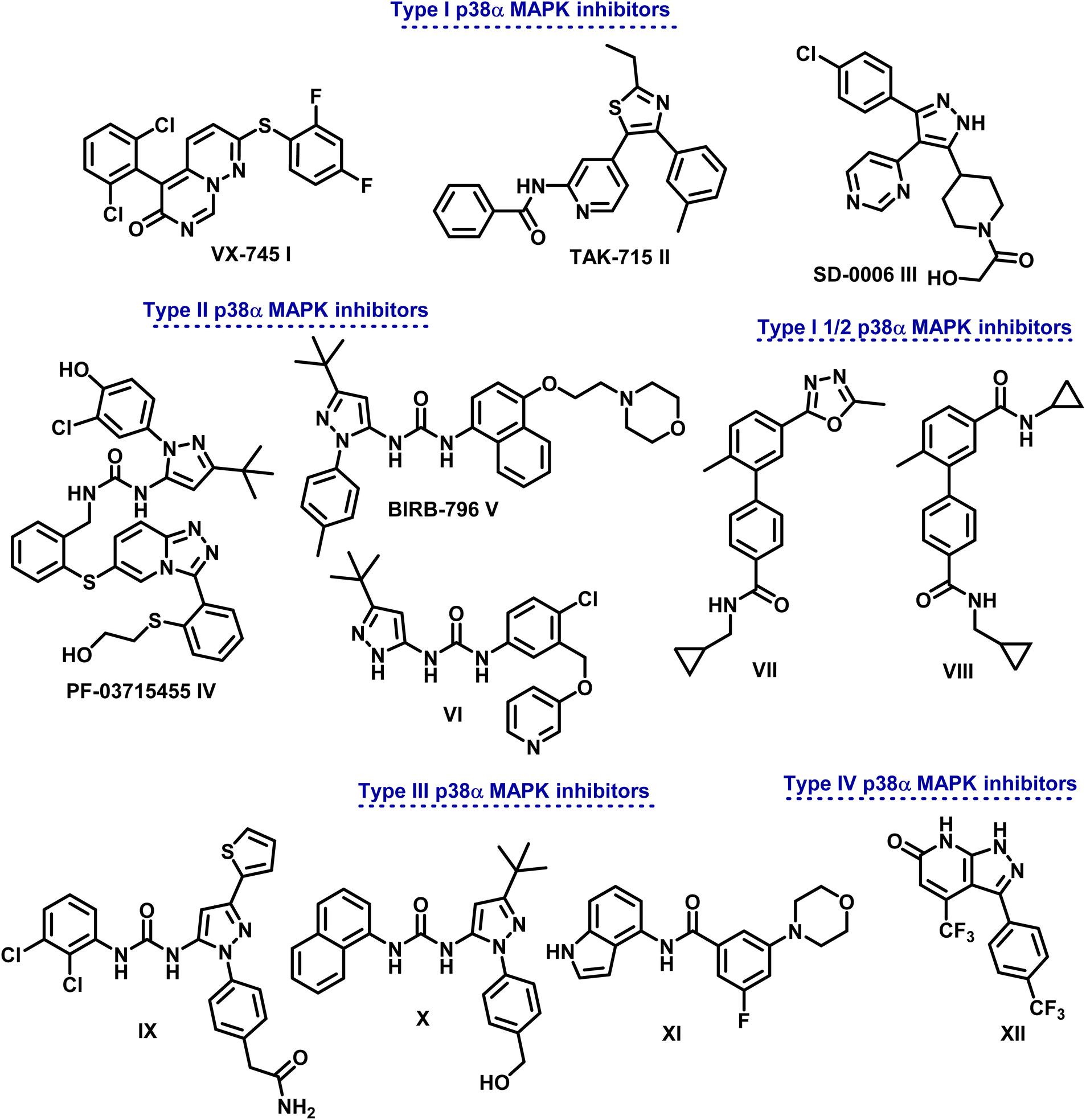 | ||
| Fig. 1 Different types of p38α MAPK inhibitors. | ||
Different types of pyrimido[1,6-b]pyridazine I, pyridinyl II, and pyrimidinyl III derivatives are class I selective ATP-competitive p38α MAPK suppressors that have been tested in both I and II phases of clinical research.15–17
Additionally, class II P38α MAPK inhibitors like IV (PF-03715455), V (BlRB-796), and VI bind in a DFG-out conformation to the hydrophobic pocket close to the ATP binding site. Phase II clinical studies are underway.18 Adhering to an allosteric site allows them to establish more hydrogen bonds with the hinge Met109 residue. On the other hand, type III inhibitors bind to an allosteric pocket next to the ATP-binding pocket.19,20
Because they are non-competitive ATP, they can attach themselves directly to the dormant protein. Molecules IX–XI serve as examples.21 In order to generate conformational changes that render a protein inactive, type IV inhibitors—ATP non-competitive suppressors—connect with the allosteric site that is separate from the ATP-binding region. One example of this class of inhibitors is compound XII (Fig. 1).22
Research has demonstrated the distinct pharmacophores of drugs that inhibit p38α MAPK, with heterocyclic scaffolds such as imidazole, thiazole, oxadiazole, pyridine, and pyrimidine serving as intriguing cores that can be fused or connected to other heterocyclic rings.23–26
In medicinal chemistry, heterocyclic scaffolds are regarded as important molecular structures. They generate a wide range of biological actions aside from being found in the framework of several biological components, including hemoglobin, hormones, vitamins, RNA, DNA, and heterocyclic compounds.27,28 As a result, heterocyclic molecules have long been desirable synthetic targets in drug design and discovery owing to their biological significance and structural diversity.
The pharmacophore quinazoline is a class of heterocyclic compounds that has recently developed into a remarkably versatile framework due to its many pharmacological characteristics, which include antiviral,29 antibacterial,30 anti-inflammatory,31,32 antihypertensive,33 antioxidant,34 and anticancer.35 Certain cancer chemotherapeutic drugs, such as quinazoline and its derivatives, have proven to be highly effective in treating solid tumors. The FDA has approved quinazoline-based anticancer medications as Gefitinib, Afatinib, Vendetanib, Erlotinib, Lapatinib, and Dacomitinib (Fig. 2).36 Furthermore, thieno[2,3-b]pyrimidines and thieno[2,3-b]pyridines constitute other two classes of heterocyclic molecules with therapeutic potential for a wide range of illnesses. Several substituted thieno[2,3-b]pyrimidines and thienopyridine compounds have been identified as potent antiproliferative agents against a variety of human cancer cell lines in recent studies.37–40 According to several investigations, hydrogen bonds between the heteroatoms of the molecules and the backbone NH group of various amino acid residues of the target enzyme are the primary mechanism through which heterocyclic rings interact.41–43 Olmutinib (Fig. 2) is an EGFR tyrosine kinase inhibitor that was recently authorized by the FDA to treat breast and non-small cell lung cancers.44
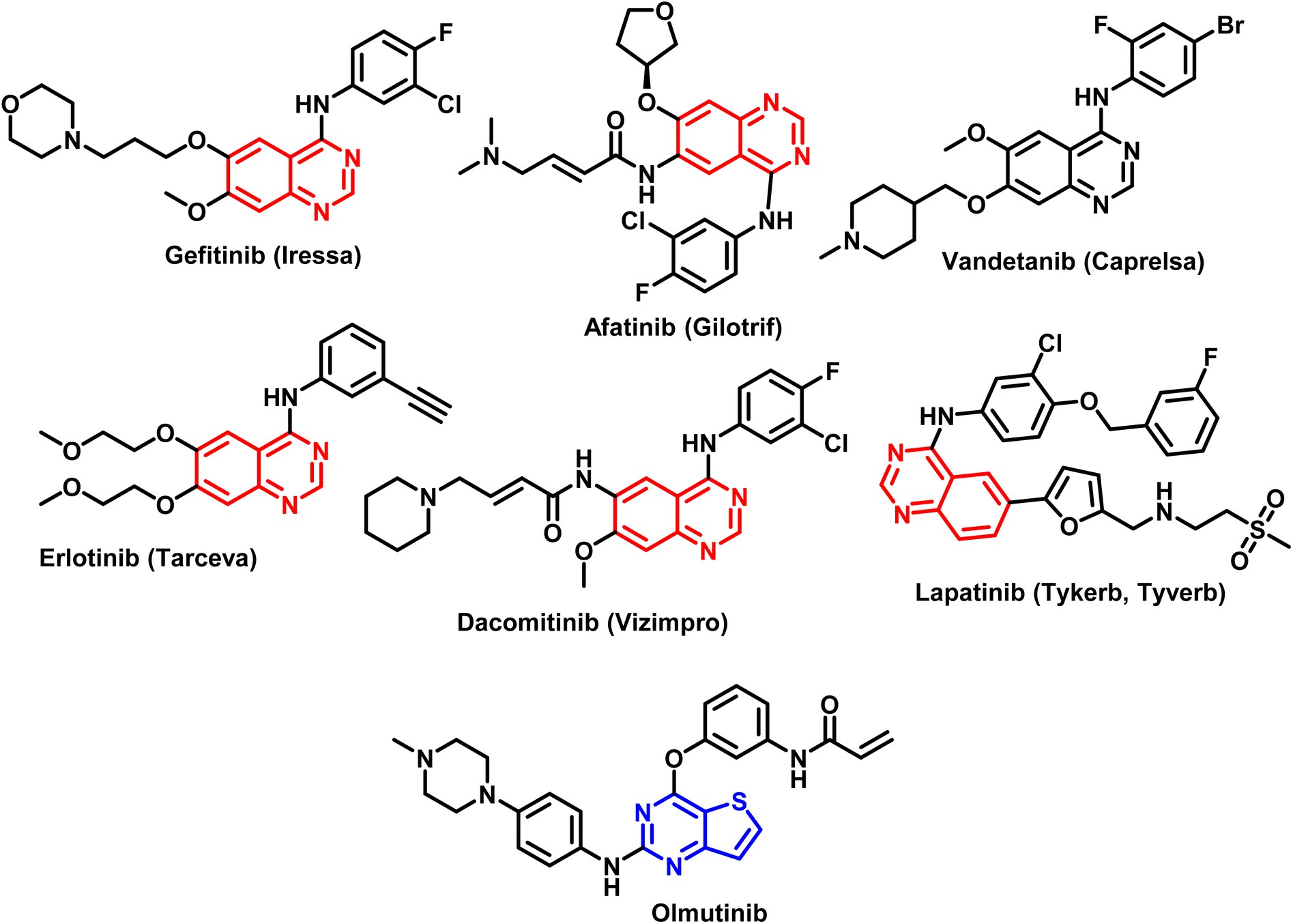 | ||
| Fig. 2 Different FDA approved drugs based on quinazoline and thienopyrimidine ring systems. | ||
Major fundamental barriers to successful clinical use of these drugs have persisted, including the high toxicity of various commonly used chemotherapeutics, the insensitivity of different tumor cells to anticancer drugs, and the incapacity of some antitumor medications to exert their effects in specific cases.
High toxicity of many frequently utilized chemotherapy remedies, inability of some antitumor medications to achieve the desired effect in particular cases, as well as insensitivity of various malignant cells to anticancer drugs are among the major fundamental obstacles that have persisted to the successful clinical use of these drugs.45,46
To enhance patient quality of life and overall survival, it is crucial to find new chemotherapeutics with clearly distinguished mechanisms of action that can be used individually or in combination. Integrating more than one scaffold into a single molecule also provides a workable hybrid pharmacophore strategy for brand-new anticancer prospects.47–49
Keeping these factors in mind and also depending upon the effect of variations in ring size, molecular orientation, and the total number of heteroatoms that could contribute additional hydrogen binding with the active ATP binding sites, the goal of the current work was to generate novel molecules based on quinazoline, thienopyrimidine, and thienopyridine rings coupled with various heterocyclic cores with documented anticancer potential, aiming to establish new anticancer agents with potential p38α MAP kinase inhibitory action. Fig. 3 illustrates the proposed layout of the new derivatives.
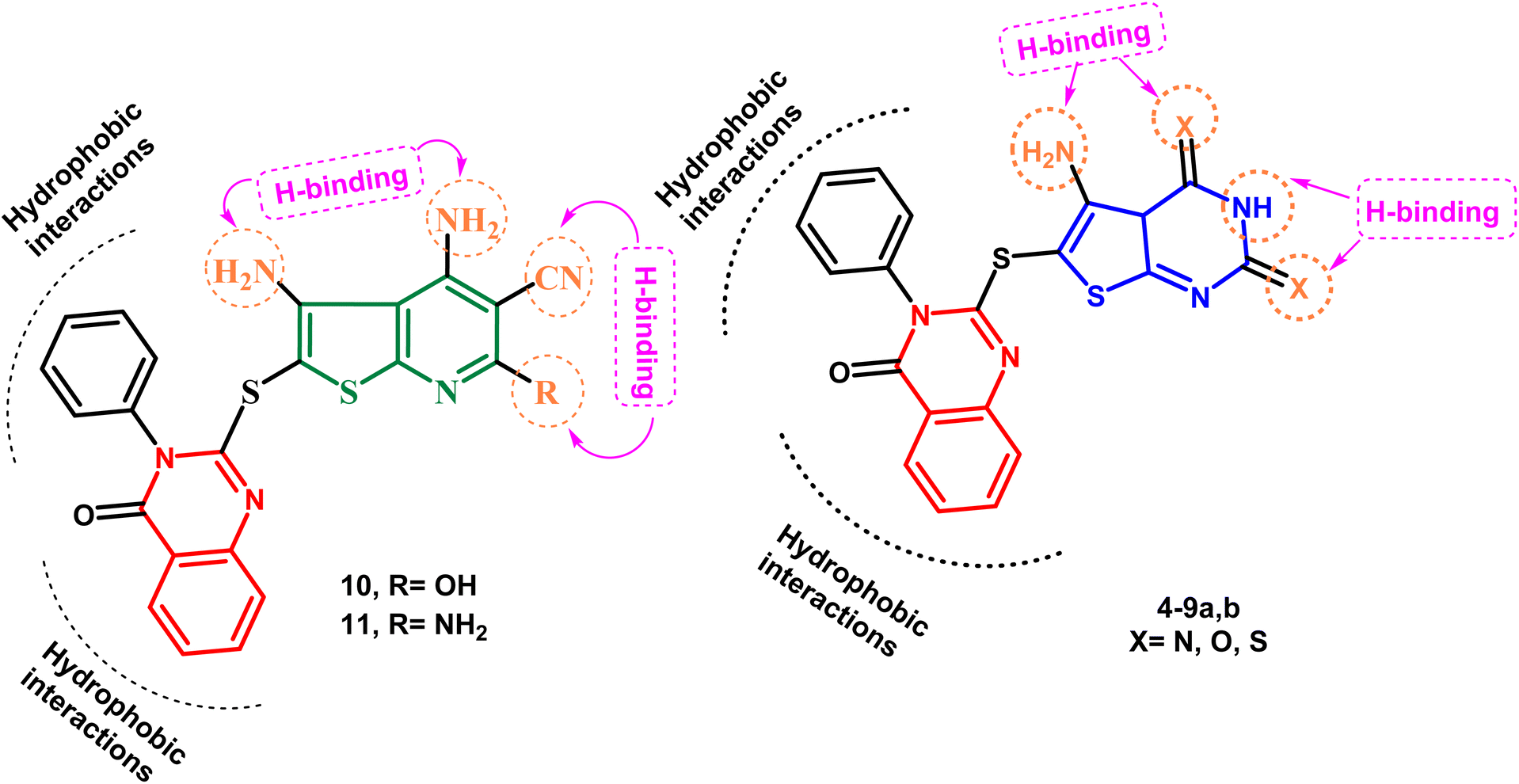 | ||
| Fig. 3 New thienopyrimidine and thienopyridine derivatives: postulated hypothesis models. | ||
2. Experimental
2.1. Chemistry
The instruments used for measuring the melting points, spectral data (IR, mass, 1H NMR and 13C NMR) and elemental analysis are provided in details in ESI material.†The ESI material† contains a detailed description of the instruments used to measure the melting points, elemental analyses and spectrum data (1H NMR, 13C NMR, IR and mass).
The starting compound 2-mercapto-3-phenylquinazolin-4-one was prepared according to the reported method.50
Yield 86%; mp 119–121 °C; IR (νmax/cm−1): 3034 (CH-rom.), 2925 (CH-aliph.), 2215 (CN), 1659 (CO); 1H NMR (400 MHz, DMSO-d6): δ 4.20 (s, 2H, CH2), 7.27–7.98 (m, 9H, Ar-H); 13C NMR (101 MHz, DMSO-d6): 33.7 (CH2), 115.2 (CN), 120.1, 126.7, 126.8, 126.9, 129.9, 130.0, 130.8, 135.2, 147.1, 156.5, 165.6 (CO). Anal. calcd for C16H11N3OS (293.34): C, 65.51; H, 3.78; N, 14.32; S, 10.93%. Found: C, 65.73; H, 3.59; N, 14.54; S, 10.71%.
2.1.2.1. 2,4-Diamino-5-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)thiophene-3-carbonitrile (3a). Yield 88%, brown crystal; mp 197–199 °C; IR (νmax/cm−1): 3223, 3190 (2NH2), 3043 (CH-arom.), 2218 (CN), 1673 (CO); 1H NMR (400 MHz, DMSO-d6) δ: 6.14 (s, 2H, NH2, D2O exchangeable), 6.61 (s, 2H, NH2, D2O exchangeable), 7.44–8.12 (m, 9H, Ar-H); 13C NMR (101 MHz, DMSO-d6): 110.6, 115.1 (CN), 120.4, 121.8, 122.2, 122.5, 124.3, 127.1, 127.3, 129.5, 129.6, 130.4, 134.2, 141.4, 150.9, 151.5, 166.2 (CO). Anal. calcd for C19H13N5OS2 (391.47): C, 58.29; H, 3.35; N, 17.89; S, 16.38%. Found: C, 58.51; H, 3.57; N, 17.66; S, 16.59%.
2.1.2.2. Ethyl 2,4-diamino-5-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)thiophene-3-carboxylate (3b). Yield 84%, brown crystal; mp 193–195 °C; IR (νmax/cm−1): 3211, 3165 (2NH2), 3057 (CH-arom.), 1725, 1686 (2CO); 1H NMR (400 MHz, DMSO-d6) δ: 1.21 (t, 3H, J = 7.2 Hz, C
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2CH3), 4.12 (q, 2H, J = 7.2 Hz, OC2CH3), 6.31 (s, 2H, NH2, D2O exchangeable), 6.84 (s, 2H, NH2, D2O exchangeable), 7.48–8.09 (m, 9H, Ar-H); 13C NMR (101 MHz, DMSO-d6): 14.5, 61.3, 120.1, 122.7, 125.6, 125.8, 126.6, 126.9, 129.8, 30.0, 130.5, 131.5, 134.7, 135.2, 148.7, 156.6, 161.1, 163.3, 166.1. Anal. calcd for C21H18N4O3S2 (438.52): C, 57.52; H, 4.14; N, 12.78; S, 14.62%. Found: C, 57.75; H, 4.36; N, 12.54; S, 14.83%.
2CH3), 4.12 (q, 2H, J = 7.2 Hz, OC2CH3), 6.31 (s, 2H, NH2, D2O exchangeable), 6.84 (s, 2H, NH2, D2O exchangeable), 7.48–8.09 (m, 9H, Ar-H); 13C NMR (101 MHz, DMSO-d6): 14.5, 61.3, 120.1, 122.7, 125.6, 125.8, 126.6, 126.9, 129.8, 30.0, 130.5, 131.5, 134.7, 135.2, 148.7, 156.6, 161.1, 163.3, 166.1. Anal. calcd for C21H18N4O3S2 (438.52): C, 57.52; H, 4.14; N, 12.78; S, 14.62%. Found: C, 57.75; H, 4.36; N, 12.54; S, 14.83%.
2.1.3.1. 5-Amino-2-methyl-6-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)thieno[2,3-d]pyrimidin-4(3H)-one (4). Yield 67%, buff crystals; mp 278–280 °C; IR (νmax/cm−1): 3352, 2277 (NH2), 2201 (NH), 3064 (CH-arom.), 2985 (CH-aliph.), 1697, 1683 (2CO); 1H NMR (400 MHz, DMSO-d6) δ: 2.13 (s, 3H, CH3), 6.71 (s, 2H, NH2, D2O exchangeable), 7.06–8.46 (m, 10H, Ar-H + NH); 13C NMR (101 MHz, DMSO-d6): 21.3, 121.1, 125.2, 126.1, 126.6, 126.9, 127.5, 128.8, 129.9, 130.0, 130.1, 130.6, 132.1, 147.6, 148.2, 156.3, 158.1, 162.9, 167.6. Anal. calcd for C21H15N5O2S2 (433.51): C, 58.18; H, 3.49; N, 16.16; S, 14.79%. Found: C, 58.39; H, 3.71; N, 16.37; S, 14.58%.
2.1.3.2. 5-Amino-6-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)thieno[2,3-d]pyrimid in-4(3H)-one (5). Yield 65%, pale brown crystals; mp 273–275 °C; IR (νmax/cm−1): 3344, 2269 (NH2), 2175 (NH), 3045 (CH-arom.), 1695, 1667 (2CO); 1H NMR (400 MHz, DMSO-d6) δ: 6.18 (s, 2H, NH2, D2O exchangeable), 6.87–8.75 (m, 10H, Ar-H + CH-pyrimidine), 8.83 (s, 1H, NH, D2O exchangeable); 13C NMR (101 MHz, DMSO-d6): 122.2, 124.3, 124.5, 125.4, 126.8, 127.9, 129.1, 130.0, 130.7, 131.0, 131.1, 138.8, 146.6, 148.2, 158.8, 159.0, 164.7, 166.2. Anal. calcd for C20H13N5O2S2 (419.48): C, 57.26; H, 3.12; N, 16.70; S, 15.29%. Found: C, 57.47; H, 3.33; N, 16.52; S, 15.49%.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 251 (NH2), 3183, 3112 (2NH), 3050 (CH-rom.), 1682 (CO); 1H NMR (400 MHz, DMSO-d6) δ: 6.20 (s, 2H, NH2, D2O exchangeable), 7.12–8.15 (m, 10H, Ar-H + NH), 12.50 (s, 1H, NH, D2O exchangeable); 13C NMR (101 MHz, DMSO-d6): 119.6, 120.1, 124.5, 126.0, 126.4, 126.7, 126.8, 129.9, 130.1, 130.5, 136.1, 136.6, 148.6, 151.1, 158.5, 164.3, 182.4. Anal. calcd for C20H13N5OS4 (467.61): C, 51.37; H, 2.80; N, 14.98; S, 27.43%. Found: C, 51.58; H, 2.60; N, 14.77; S, 27.65%.
251 (NH2), 3183, 3112 (2NH), 3050 (CH-rom.), 1682 (CO); 1H NMR (400 MHz, DMSO-d6) δ: 6.20 (s, 2H, NH2, D2O exchangeable), 7.12–8.15 (m, 10H, Ar-H + NH), 12.50 (s, 1H, NH, D2O exchangeable); 13C NMR (101 MHz, DMSO-d6): 119.6, 120.1, 124.5, 126.0, 126.4, 126.7, 126.8, 129.9, 130.1, 130.5, 136.1, 136.6, 148.6, 151.1, 158.5, 164.3, 182.4. Anal. calcd for C20H13N5OS4 (467.61): C, 51.37; H, 2.80; N, 14.98; S, 27.43%. Found: C, 51.58; H, 2.60; N, 14.77; S, 27.65%.2.1.6.1. 2-((4,5-Diamino-2-thioxo-1,2-dihydrothieno[2,3-d]pyrimidin-6-yl)thio)-3-phenyl quinazolin-4(3H)-one (8a). Yield 72%, yellow crystals; mp 292–294 °C; IR (νmax/cm−1): 3405, 3315 (2NH2), 2210 (NH), 3082 (CH-arom.), 1675 (CO); 1H NMR (400 MHz, DMSO-d6) δ: 6.28 (s, 2H, NH2, D2O exchangeable), 6.86–8.72 (m, 9H, Ar-H), 8.35 (s, 1H, NH2, D2O exchangeable), 8.81 (s, 1H, NH, D2O exchangeable); 13C NMR (101 MHz, DMSO-d6): 120.2, 120.6, 123.5, 126.4, 126.9, 127.6, 128.7, 129.3, 129.9, 130.0, 130.4, 130.9, 147.1, 152.3, 152.8, 156.5, 162.9, 181.1; MS: m/z = 450 [M+], 198 (100%). Anal. calcd for C20H14N6OS3 (450.56): C, 53.31; H, 3.13; N, 18.65; S, 21.35%. Found: C, 53.53; H, 3.35; N, 18.87; S, 21.56%.
2.1.6.2. 4,5-Diamino-6-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)thieno[2,3-d]pyrimidin-2(1H)-one (8b). Yield 70%, yellow crystals; mp 298–300 °C; IR (νmax/cm−1): 3392, 3278 (2NH2), 2196 (NH), 3059 (CH-arom.), 1689, 1664 (2CO); 1H NMR (400 MHz, DMSO-d6) δ: 6.17 (s, 2H, NH2, D2O exchangeable), 7.38–8.04 (m, 11H, Ar-H + NH2), 9.44 (s, 1H, NH, D2O exchangeable); 13C NMR (101 MHz, DMSO-d6): 110.5, 119.1, 120.0, 124.2, 126.5, 126.9, 127.2, 129.9, 130.0, 130.5, 134.1, 136.2, 147.6, 147.8, 155.2, 156.5, 163.6, 166.1; MS: m/z = 434 [M+], 182 (100%). Anal. calcd for C20H14N6O2S2 (434.49): C, 55.29; H, 3.25; N, 19.34; S, 14.76%. Found: C, 55.51; H, 3.46; N, 19.55; S, 14.54%.
2.1.7.1. 3,4-Diamino-6-hydroxy-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)thieno[2,3-b]pyridine-5-carbonitrile (9). Yield 68%, deep yellow crystals; mp 255–257 °C; IR (νmax/cm−1): 3399 (OH), 3246, 3137 (2NH2), 3035 (CH-arom.), 2221 (CN), 1698 (CO); 1H NMR (400 MHz, DMSO-d6) δ: 6.04 (s, 2H, NH2, D2O exchangeable), 6.16 (s, 2H, NH2, D2O exchangeable), 7.11–8.18 (m, 9H, Ar-H), 11.45 (s, 1H, OH, D2O exchangeable); 13C NMR (101 MHz, DMSO-d6): 109.6, 115.3, 121.1, 121.4, 123.9, 126.1, 126.6, 126.8, 126.9, 129.7, 130.0, 136.1, 136.1, 147.2, 148.6, 151.3, 157.5, 161.1, 163.7. Anal. calcd for C22H14N6O2S2 (458.52): C, 57.63; H, 3.08; N, 18.33; S, 13.99%. Found: C, 57.85; H, 3.29; N, 18.56; S, 13.77%.
2.1.7.2. 3,4,6-Triamino-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)thieno[2,3-b]pyridine-5-carbonitrile (10). Yield 74%, deep yellow crystals; mp 281–283 °C; IR (νmax/cm−1): 3267, 3220 (NH2), 3055 (CH-arom.), 2216 (CN), 1672 (CO); 1H NMR (400 MHz, DMSO-d6) δ: 6.43 (s, 2H, NH2, D2O exchangeable), 6.83, 6.85 (s, 4H, 2NH2, D2O exchangeable), 7.46–8.10 (m, 9H, Ar-H); 13C NMR (101 MHz, DMSO-d6): 108.5, 115.7, 120.0, 120.6, 121.4, 121.8, 122.5, 123.0, 127.3, 128.1, 129.7, 129.8, 131.6, 132.0, 140.3, 142.0, 156.4, 157.3, 160.8, 16.15. Anal. calcd for C22H14N6O2S2 (457.53): C, 57.75; H, 3.30; N, 21.43; S, 14.02%. Found: C, 57.53; H, 3.51; N, 21.64; S, 14.23%.
2.1.7.3. 2-((5-Acetyl-3,4-diamino-6-hydroxythieno[2,3-b]pyridin-2-yl)thio)-3-phenyl quinazolin-4(3H)-one (11). Yield 71%, deep yellow crystals; mp over 300 °C; IR (νmax/cm−1): 3492 (OH), 3351, 3312 (2NH2), 3078 (CH-arom.), 2963 (CH-aliph.), 1716, 1659 (2CO); 1H NMR (400 MHz, DMSO-d6) δ: 2.20 (CH3), 6.15 (s, 2H, NH2, D2O exchangeable), 6.58 (s, 2H, NH2, D2O exchangeable), 7.21–8.30 (m, 9H, Ar-H), 11.23 (s, 1H, OH, D2O exchangeable); 13C NMR (101 MHz, DMSO-d6): 22.2, 110.8, 119.3, 121.4, 121.9, 122.6, 124.0, 124.3, 127.7, 128.2, 129.3, 129.8, 137.9, 139.0, 147.6, 149.3, 153.6, 157.3, 158.4, 163.2, 186.4. Anal. calcd for C23H17N5O3S2 (475.54): C, 58.09; H, 3.60; N, 14.73; S, 13.49%. Found: C, 58.31; H, 3.82; N, 14.51; S, 13.72%.
2.2. Biological activity
3. Results and discussion
3.1. Chemistry
Newly targets 3a,b, 4–9a,b, and 10–12 were created as summarized in Schemes 1–4. Firstly, in Scheme 1, the key stating compound 2-mercapto-3-phenylquinazolin-4-one synthesized according to the documented method50 was permitted to react with chloroacetonitrile in dry DMF containing sodium acetate to generate the corresponding acetonitrile compound 1.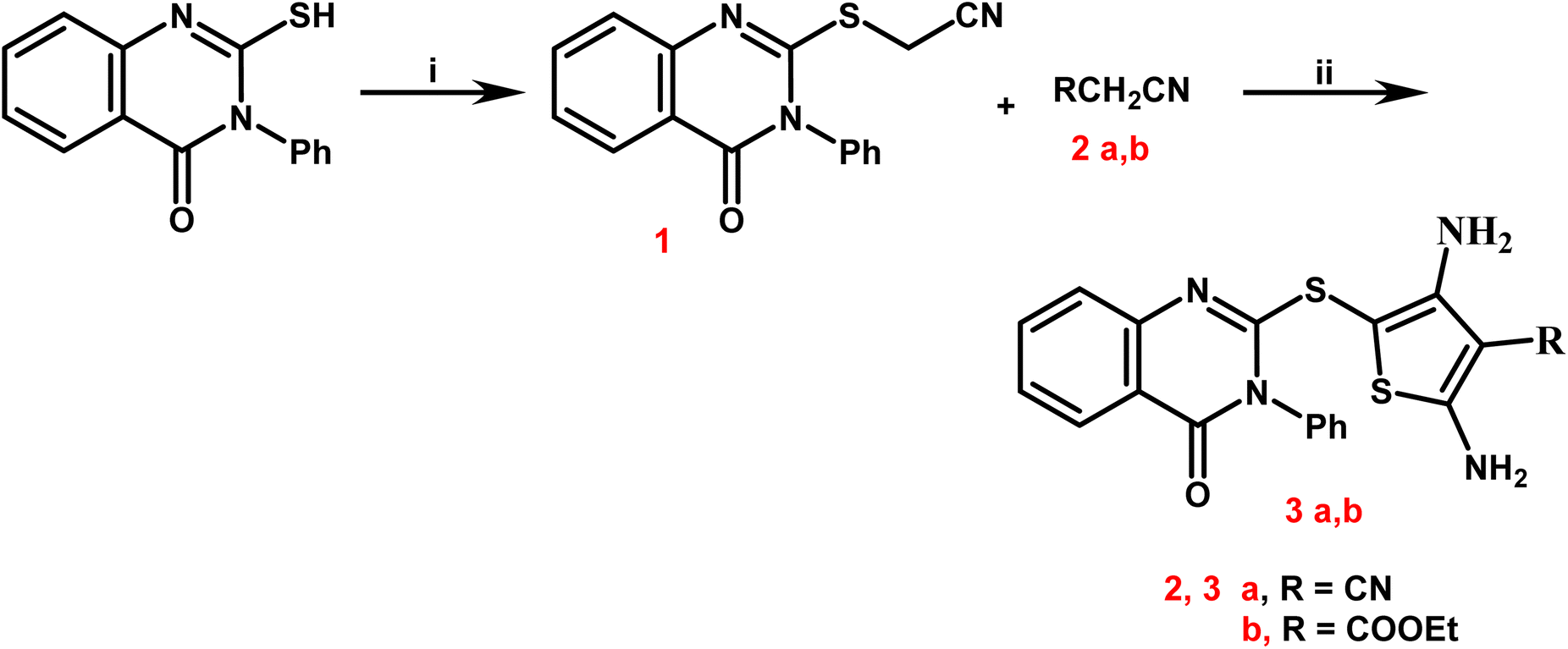 | ||
| Scheme 1 Synthetic path way for synthesis of 2,4-diamino-thiophene-3-carbonitriles 3a,b. Reagents and conditions: (i) chloro-acetonitrile, sodium acetate, dry DMF, stirring for 30 minutes, refluxing for 5 h; (ii) S, malononitrile and/or ethyl cyanoacetate, absolute ethanol, few drops of triethylamine, reflux for 7 h. | ||
The 2,4-diamino-thiophene-3-carbonitriles 3a,b were obtained via the Gewald reaction of 1 in absolute ethanol containing a catalytic amount of triethylamine with sulphur and various active methylene reagents, namely malononitrile or ethyl cyanoacetate. Characteristic bands appeared at 3223–3165 cm−1 for 2NH2, 2218 cm−1 for CN, and 1725–1686 cm−1 for CO groups, according to the IR spectra of 3a,b. 1H NMR spectra of compounds 3a,b exhibited two D2O singlet signals at δ 6.13–6.84 ppm referring to 2NH2 groups, multiplet signals in the region 7.44–8.12 corresponding to the aromatic protons. Regarding to the carboxylate protons of 3b appeared as triplet–quartet signals at δ 1.21 and 4.12 ppm and at δ 14.53 and 61.37 ppm in its 13C NMR spectrum.
In Scheme 2, compound 3a was selected as a crucial antecedent to synthesis new targets 4–8a,b. Upon treatment of 3a different reagents, namely, acetic anhydride and/or formic acid, under reflux for 24 hours furnished the corresponding thieno[2,3-d]pyrimidin-4(3H)-one 4 and 5, respectively. Also, the condensation of 3a with an equimolar amount of formamide under reflux resulted in the creation of the corresponding 4,5-diaminothieno[2,3-d]pyrimidine analogue 6.
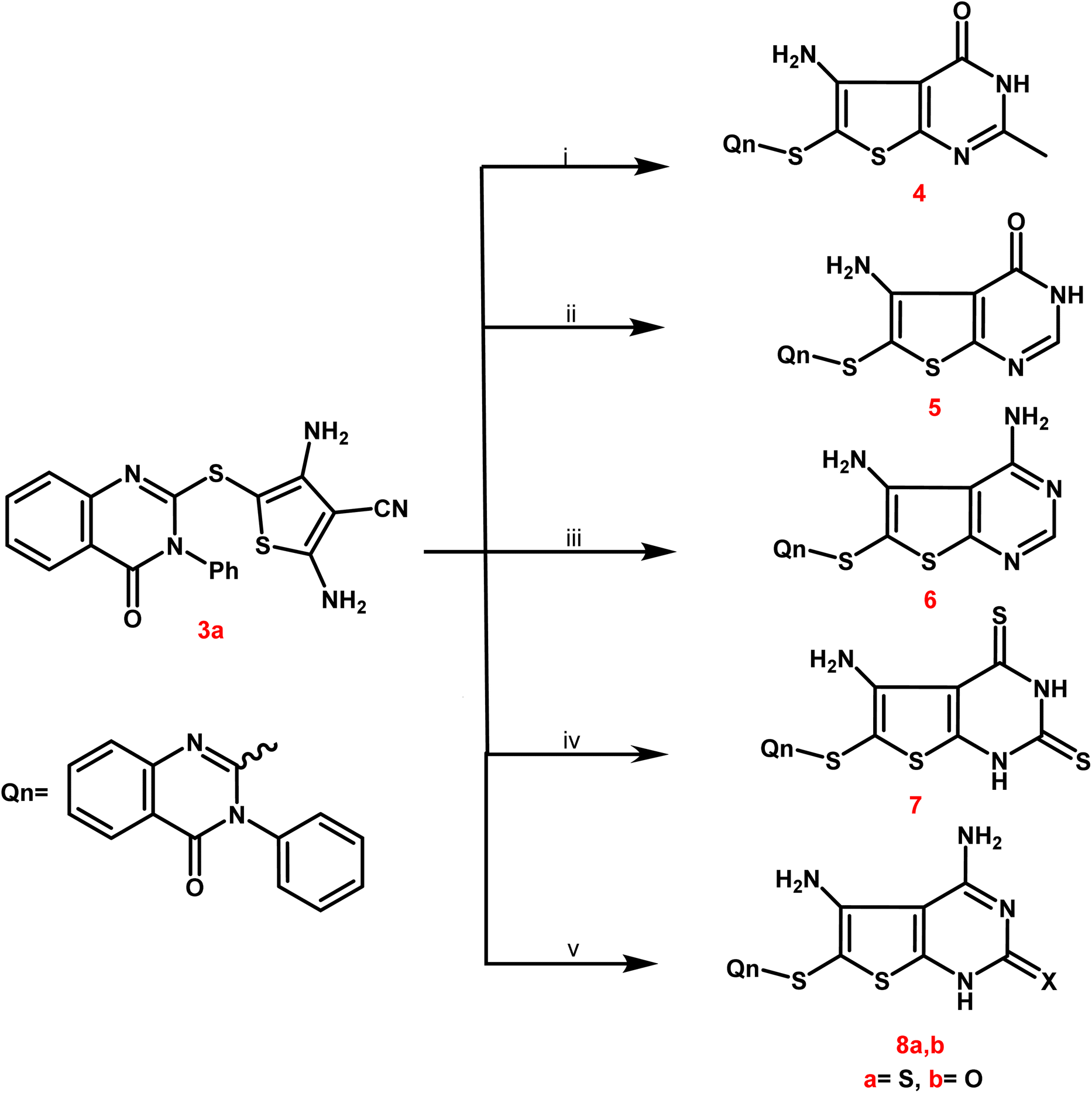 | ||
| Scheme 2 Synthetic path way for synthesis of different substituted thienopyrimidines 4–8a,b. Reagents and conditions: (i and ii) acetic anhydride and/or formic acid, reflux for 24 h; (iii) formamide, reflux for 12 h; (iv) carbon disulphide, pyridine, reflux on water bath for 10 h; (v) thiourea and/or urea, ethanolic sodium ethoxide, reflux for 9 h. | ||
IR spectra of derivatives 4 and 5 revealed two distinctive bands at 1697–1667 cm−1 assignable to CO groups. Additionally, bands of NH and NH2 groups appeared at 3352–2201 cm−1, and the 2NH2 groups of compound 6 were represented by two forked bands at 3349–3157 cm−1.
Compounds 4 and 5's 1H NMR spectra showed multiplet signals at δ 6.87–8.75 ppm caused by aromatic protons, as well as D2O exchangeable singlets at δ 6.71–6.18 ppm due to NH2 groups. Compound 4 showed a singlet signal for its 2-CH3 protons at δ 2.13 ppm and at δ 21.34 ppm in its 13C NMR. The 13C NMR spectra revealed singlet signals for the carbonyl groups of 4 and 5 at approximately δ 166.24–167.69 ppm.
Moreover, the treatment of 3a with carbon disulphide in pyridine furnished the corresponding 2,4-dithioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidine derivative 7. IR spectrum of the latter analogue exhibited different characteristic bands at 3336–3112 cm−1 due to NH2 and 2NH and at 1682 cm−1 assignable to CO group. 13C NMR spectrum of compound 7 exhibited C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CS functionalities at δ 164.35 and 182.43 ppm, respectively.
O and CS functionalities at δ 164.35 and 182.43 ppm, respectively.
Compound 3a was further subjected to the reaction with thiourea and urea in refluxing ethanolic sodium ethoxide to afford the corresponding 2-thioxo/oxo-1,2-dihydrothieno[2,3-d]pyrimidine molecules 8a,b. IR spectra of 8a,b exhibited various bands at 3405–2210 cm−1 referring to 2NH2 and NH, at 1675 cm−1 due to CO of 8a and at 1689, 1664 cm−1 due to 2CO of 8b. 13C NMR spectra of compounds 8a,b showed singlet signals at δ 162.99 and 181.14 ppm assignable to CO and CS groups.
From a mechanistic perspective, the creation of thienopyrimidine nucleus of compounds 4 and 5 as sample instances is believed to follow a multi-step route. Firstly, the carbonium ion was attacked by the lone pair of NH2 electrons through a nucleophilic reaction, resulting in the formation of the non-isolable intermediate A. The nitrile function on the enolic OH was attacked nucleophilically to produce the second non-isolable intermediate C. After that, the intermediate C underwent intramolecular rearrangement, which yielded the new thienopyrimidine derivatives 4 and 5 (Scheme 3).
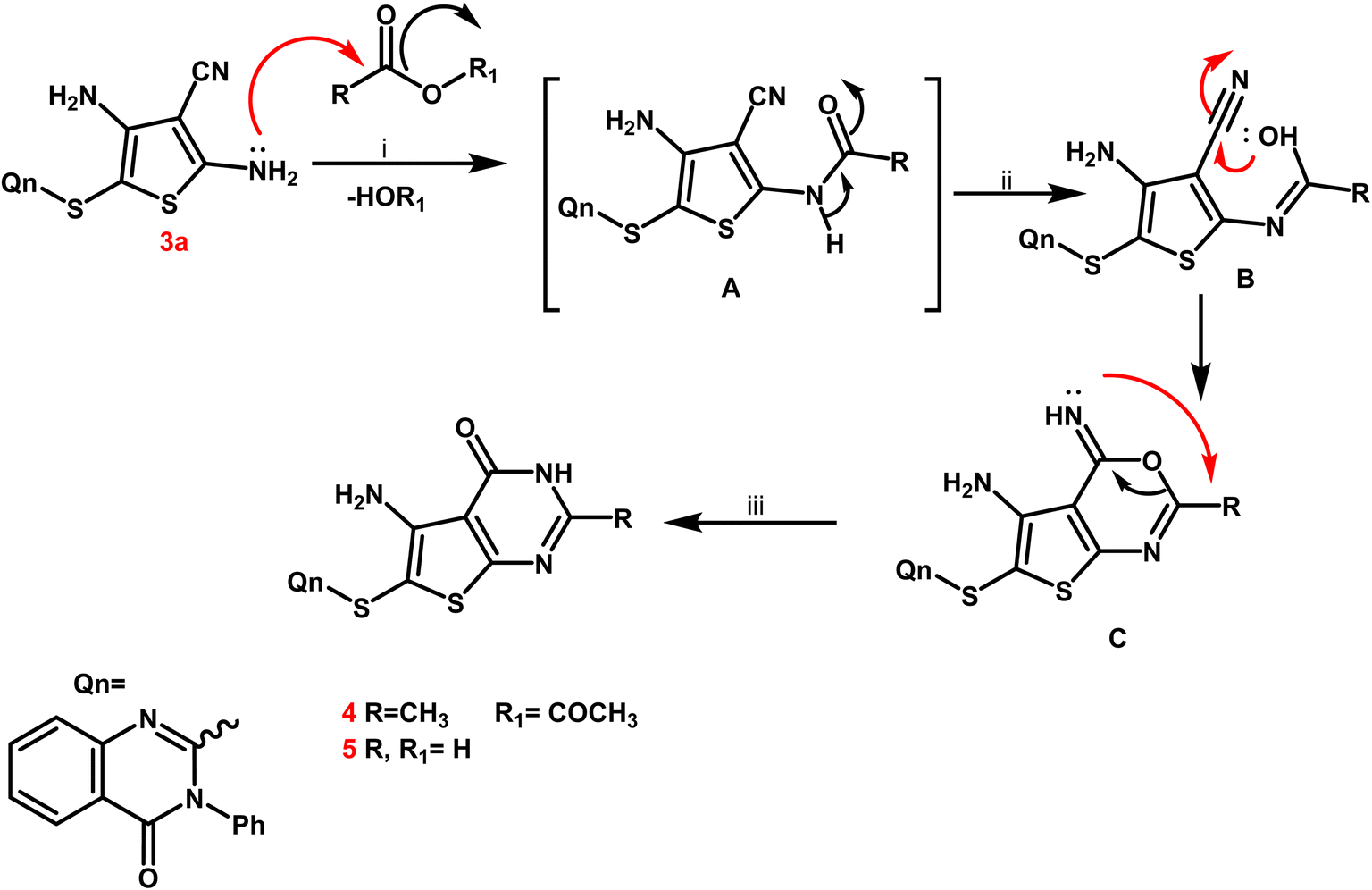 | ||
| Scheme 3 Mechanistic perspective for the synthesis of thienopyrimidine derivatives 4, 5. | ||
The third scheme (Scheme 4) involved treating 3a ethyl cyanoacetate and/or malononitrile in ethyl alcohol with a few amount of triethylamine to form the corresponding thieno[2,3-b]pyridine-5-carbonitriles 9 and 10. Furthermore, treating 3a with ethyl acetoacetate in the same way led to the production of the corresponding 5-acetyl-thieno[2,3-b]pyridine derivative 11. The IR spectrum of 9 exhibited a broad band at 3399 cm−1 corresponding to the OH group, besides the characteristic bands at 3246 and 3137 cm−1 due to NH2 groups and at 2221–2216 cm−1 assignable to the CN group. Also, two bands at 1715 and 1695 cm−1, indicating two CO functions, were observed in IR spectrum of 11. The singlet signal summed for the three protons of the 5-COCH3 group was visible in the 1H NMR spectrum of 11, and it was detected at δ 22.26 ppm in the 13C NMR spectrum, along with the expected carbons that appeared at their expected regions.
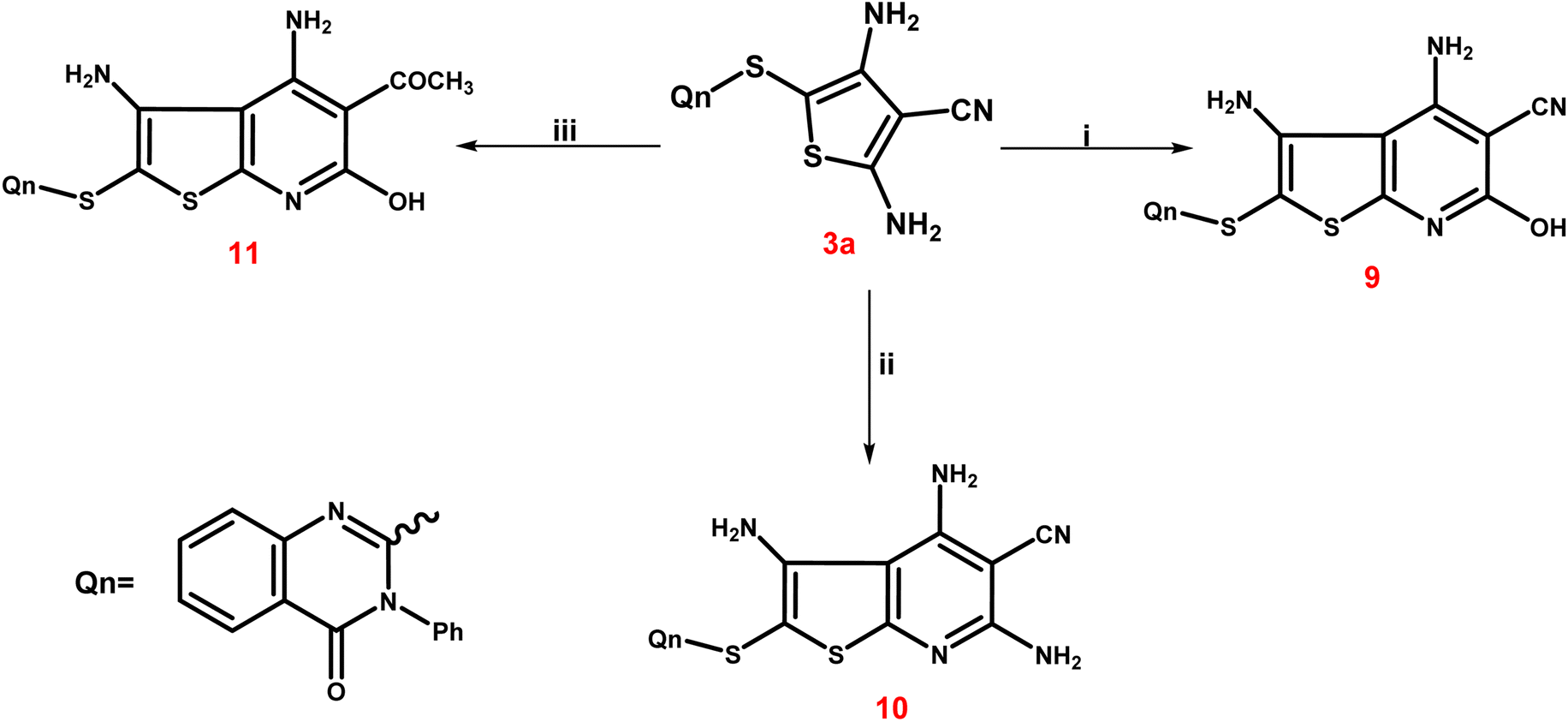 | ||
| Scheme 4 A synthetic route for creation of various substituted thienopyridines 9–11. Reagents and conditions; (i) ethyl cyanoacetate, ethanol, triethylamine, reflux for 9 h; (ii) malononitril, ethanol, triethylamine, reflux for 9 h; (iii) ethyl acetoacetate, ethanol, triethylamine, reflux for 9 h. | ||
Target compounds' mass spectra revealed molecular ion peaks that matched their molecular formulae.
Scheme 5 presents the expected multi-step molecular approach for the synthesis of the novel thienopyridine derivatives 9–11.
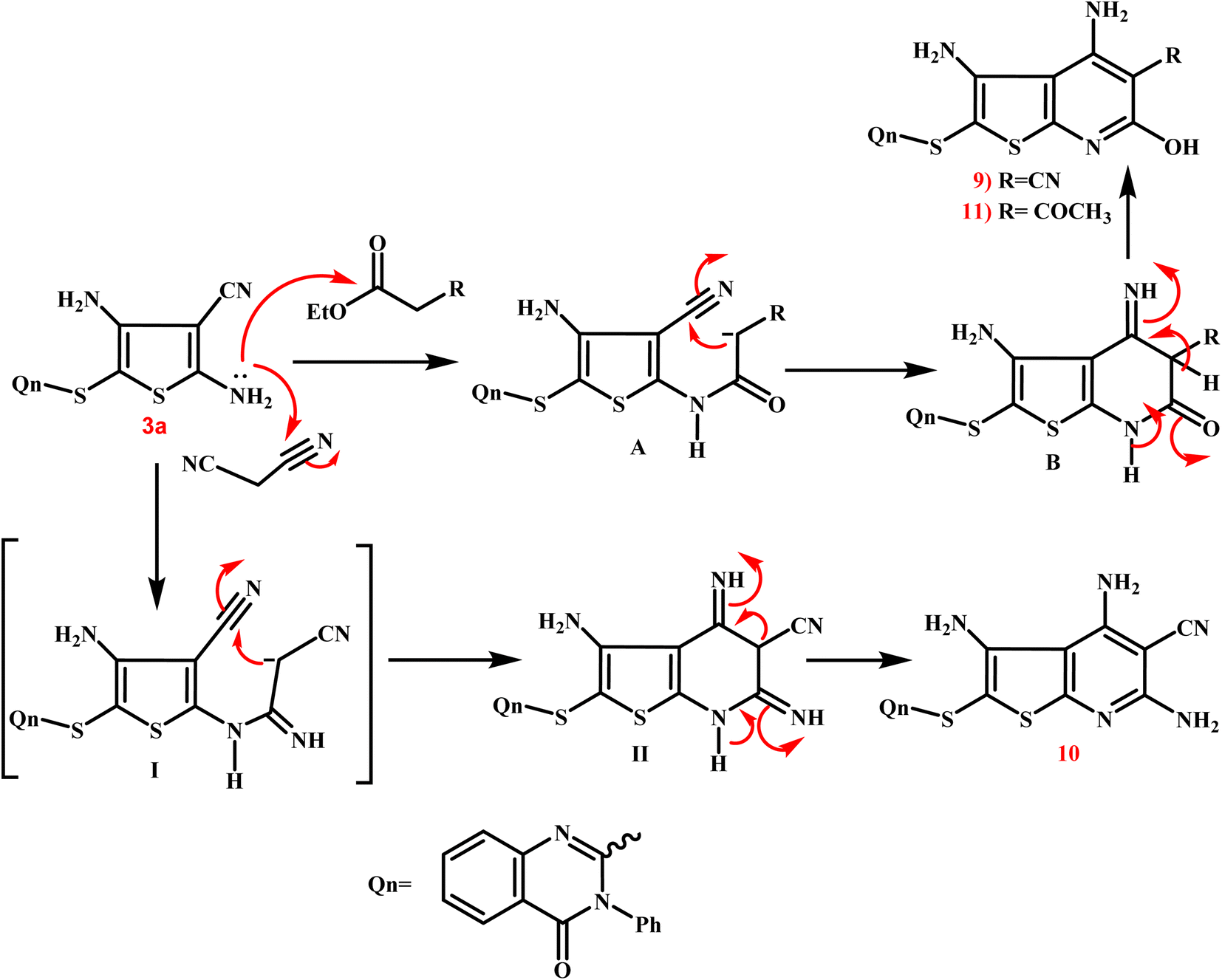 | ||
| Scheme 5 Presents the expected multi-step molecular approach for the synthesis of the novel thienopyridine derivatives 9–11. | ||
3.2. Biological evaluation
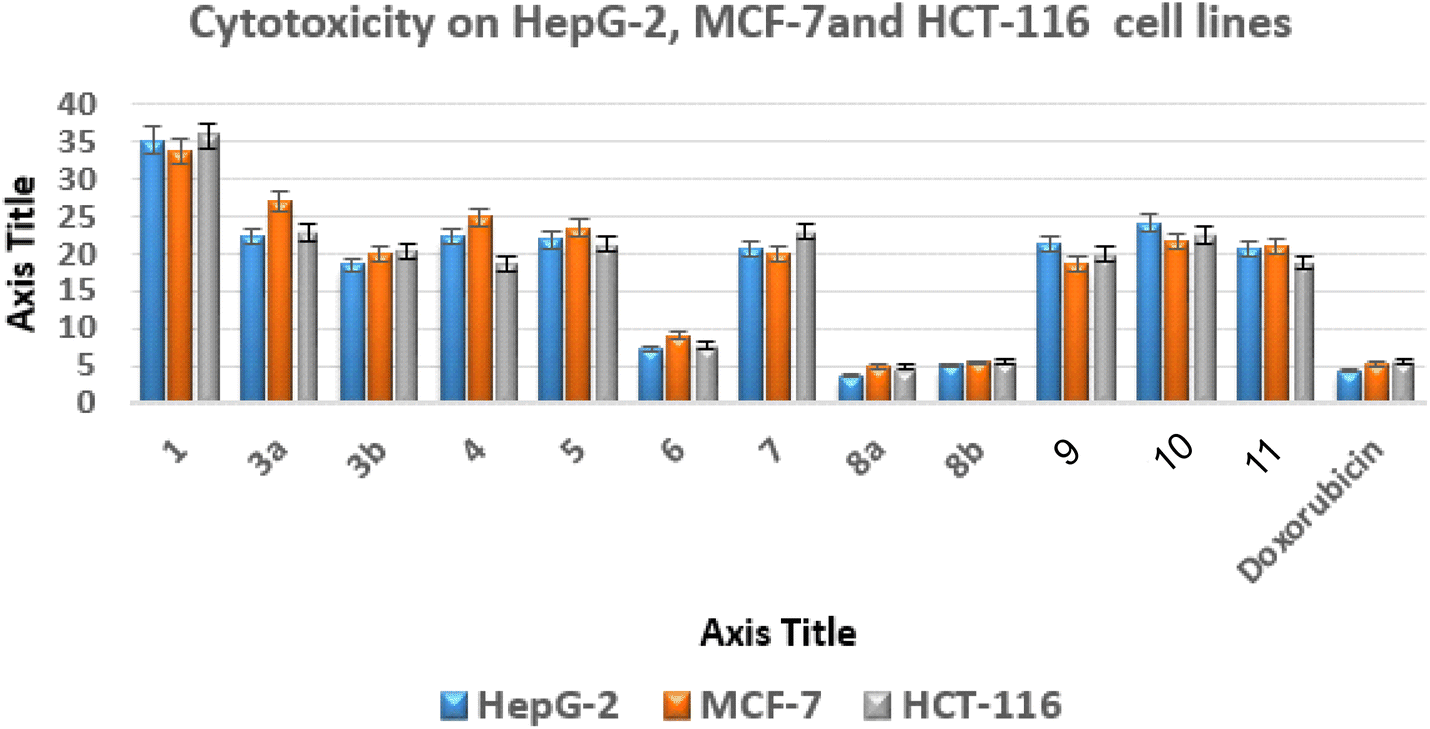 | ||
| Fig. 4 Anticancer activities of new analogues against various cell lines expressed as IC50 values. | ||
The most promising anticancer potential was demonstrated by thieno[2,3-d]pyrimidines 6, 8a, and 8b, according to the data. The 4,5-diamino-2-thioxo-1,2-dihydrothieno[2,3-d]pyrimidine derivative 8a demonstrated superior potency against the examined lines with IC50 values of 3.68 ± 0.3, 4.92 ± 0.45, and 4.88 ± 0.12 μM compared to doxorubicin with IC50 values of 4.44 ± 0.01, 5.20 ± 0.11, and 5.63 ± 0.11 μM, respectively.
Regarding the 2-oxo analogue 8b, it was approximately equivalent to doxorubicin with IC50 values of 5.13 ± 0.15, 5.44 ± 0.7, and 5.65 ± 0.37 μM. On the other hand, the 4,5-diamino-1,2-dihydrothieno[2,3-d]pyrimidine derivative 6 exhibited IC50 values of 7.22 ± 0.08, 9.14 ± 0.19, and 7.75 ± 0.3 μM against the examined cancer cells, indicating a slight decrease in activity about 2-fold less than doxorubicin.
In addition, the effects of compounds 6, 8a, and 8b on human WI-38 normal cells were evaluated, and their safety profiles were determined by calculating their selectivity indices (SI)53,54 (Table S1†).
Compound 8a had the highest level of safety and selectivity (SI > 23.12–17.29) when applied to all evaluated cancer cell lines, with compound 8b coming in second, which exhibited SI > 16.64–15.11, whereas compound 6 displayed SI > 11.12–8.80. Therefore, it is possible to draw the conclusion that compound 8a exhibits the greatest incidence of cytotoxicity among all the assessed malignant cells, with a great safety profile and excellent selectivity.55
| Compound no. | IC50 (μM) (mean ± SEM) |
|---|---|
| p38α MAPK | |
| a IC50: compound concentration needed to 50% block the activity of the enzyme, represented by the standard error of mean (SEM): each value represents the average of three readings. | |
| 6 | 0.31 ± 0.04 |
| 8a | 0.18 ± 0.02 |
| 8b | 0.23 ± 0.05 |
| SB 202190 | 0.27 ± 0.06 |
All three assessed analogues demonstrated potent p38α MAPK suppression. IC50 value of 4,5-diamino-2-thioxo derivative 8a was 0.18 ± 0.02 μM, indicating 1.5 times more powerful action than the reference medication of 0.27 ± 0.06 μM. When the 2-thioxo moiety was switched out for the 2-oxo moiety, the effect of analogue 8b was a little weaker than that of 8a, but it was still stronger than SB 202190 (IC50 = 0.23 ± 0.05 μM). Conversely, the unsubstituted thieno[2,3-d]pyrimidine ring at position 2 as 4,5-diamino-1,2-dihydrothieno[2,3-d]pyrimidine derivative 6 exhibited a further slight decrease of 1.14 fold in the inhibitory impact on p38αMAPK (IC50 = 0.31 ± 0.04 μM).
The compounds provided encouraging outcomes in enzymatic assessment as well as having the greatest antiproliferation efficacy and selectivity in the cell line studies. As a result, the thieno[2,3-d]pyrimidine fragment serves as a valuable scaffold for developing new congeners that target the p38α MAPK suppression effect as antiproliferative agents.
Also, programmed cell death (apoptosis) cannot begin until the Cysteinyl Aspartate-Specific Proteinase family, which includes caspases-3, is activated. Caspases-3 are crucial for every step of the apoptotic process, including DNA fragmentation, chromatin condensation, and cell shrinkage.58,59
Using the ELISA method, it was interesting to see how compounds 6, 8a, and 8b changed the amounts of caspase-3, Bcl-2, and Bax within MCF-7 carcinoma cells.60,61 These cells were treated with 6, 8a, and 8b at their IC50 concentrations (9.14, 4.92, and 5.44 μM, respectively) for 24 hours. The Bax level in the treated MCF-7 cells with the latter investigated drugs was measured in Pg mL−1, whereas the levels of Bcl-2 and caspase-3 were assessed in ng mL−1.62
The results showed that the three tested derivatives boosted the levels of Bax (by ∼7.31, 13.8 and 8.86 fold) and caspase 3 (by ∼3.55, 4.22 and 3.87 fold) with 6, 8a, and 8b, respectively, and decreased the level of the antiapoptotic protein Bcl-2 by ∼1.99, 3.69 and 2.66 fold, respectively in treated MCF-7 cells compared with the untreated ones. This outcome demonstrated that the compounds 6, 8a, and 8b had accelerated the cells' transition to apoptosis (Table 2).
| Compd no. | Casp-3 | Bax | Bcl-2 |
|---|---|---|---|
| Conc. ng mL−1 | Conc. Pg mL−1 | Conc. ng mL−1 | |
| 6/MCF-7 | 17.85 ± 0.22 | 172.31 ± 2.60 | 3.42 ± 0.20 |
| 8a/MCF-7 | 33.68 ± 0.42 | 205.30 ± 2.91 | 1.84 ± 0.15 |
| 8b/MCF-7 | 21.63 ± 0.15 | 188.25 ± 1.47 | 2.55 ± 0.31 |
| Cont./MCF-7 | 2.44 ± 0.05 | 48.60 ± 1.39 | 6.79 ± 0.20 |
The treatment of MCF-7 cells with 8a with its IC50 value of 4.68 μM for 24 h changes the normal cell cycle distribution of MCF-7 cells by increasing the amount of DNA in the G2/M phase by about four times with value of 32.49% and decreasing the amount of DNA in the G0–G1 48.22% and S phases 19.29% compared to MCF-7 cells that had not been treated with values 8.78%, 67.51%, 23.74%, respectively (Fig. 5 and 6). The above results suggest that compound 8a's antitumor mechanism of action could involve inducing apoptosis as a consequence of cell cycle arrest.
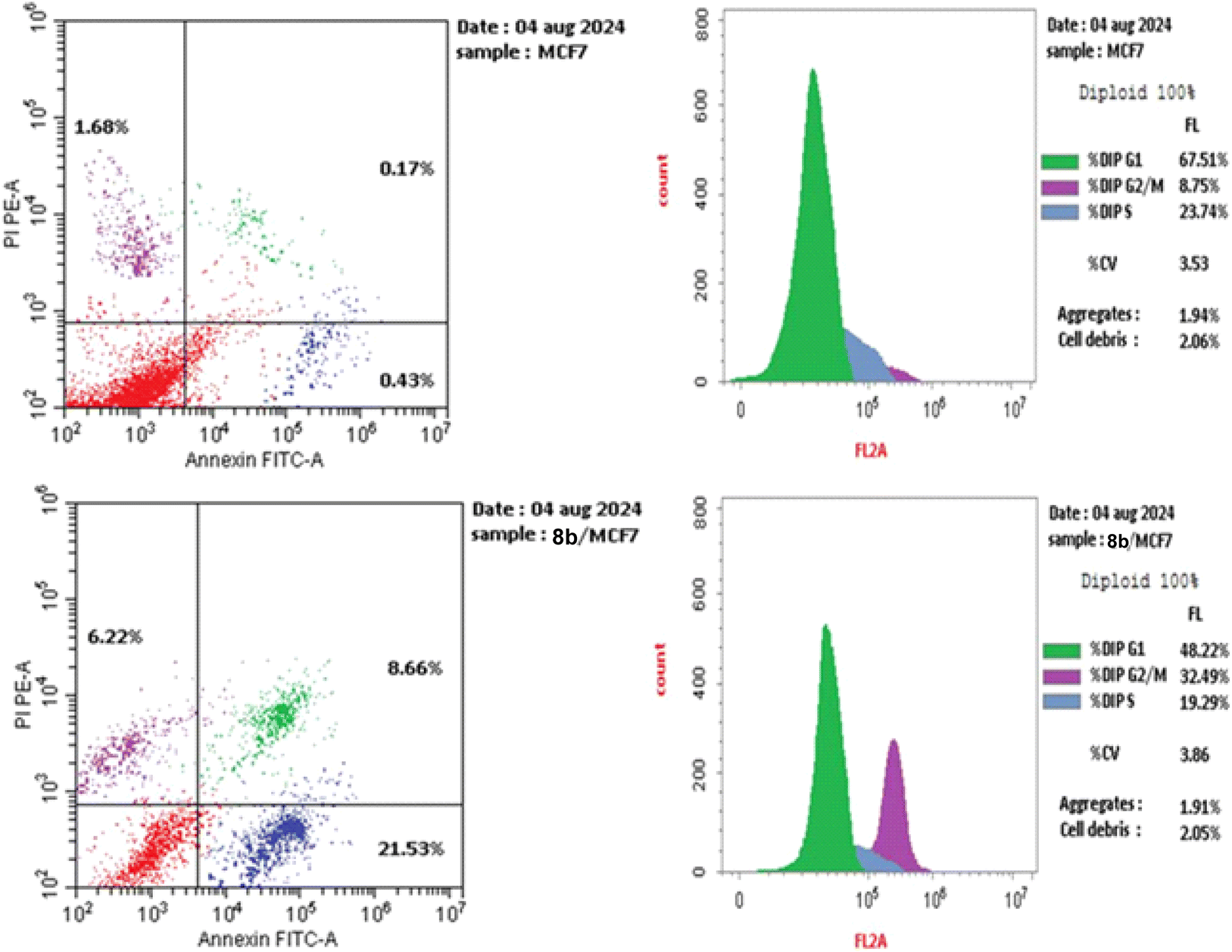 | ||
| Fig. 5 Using the annexin V/PI method, an analysis of the cell cycle and apoptosis induction of untreated control MCF-7 cell line (A) and 8a (B). | ||
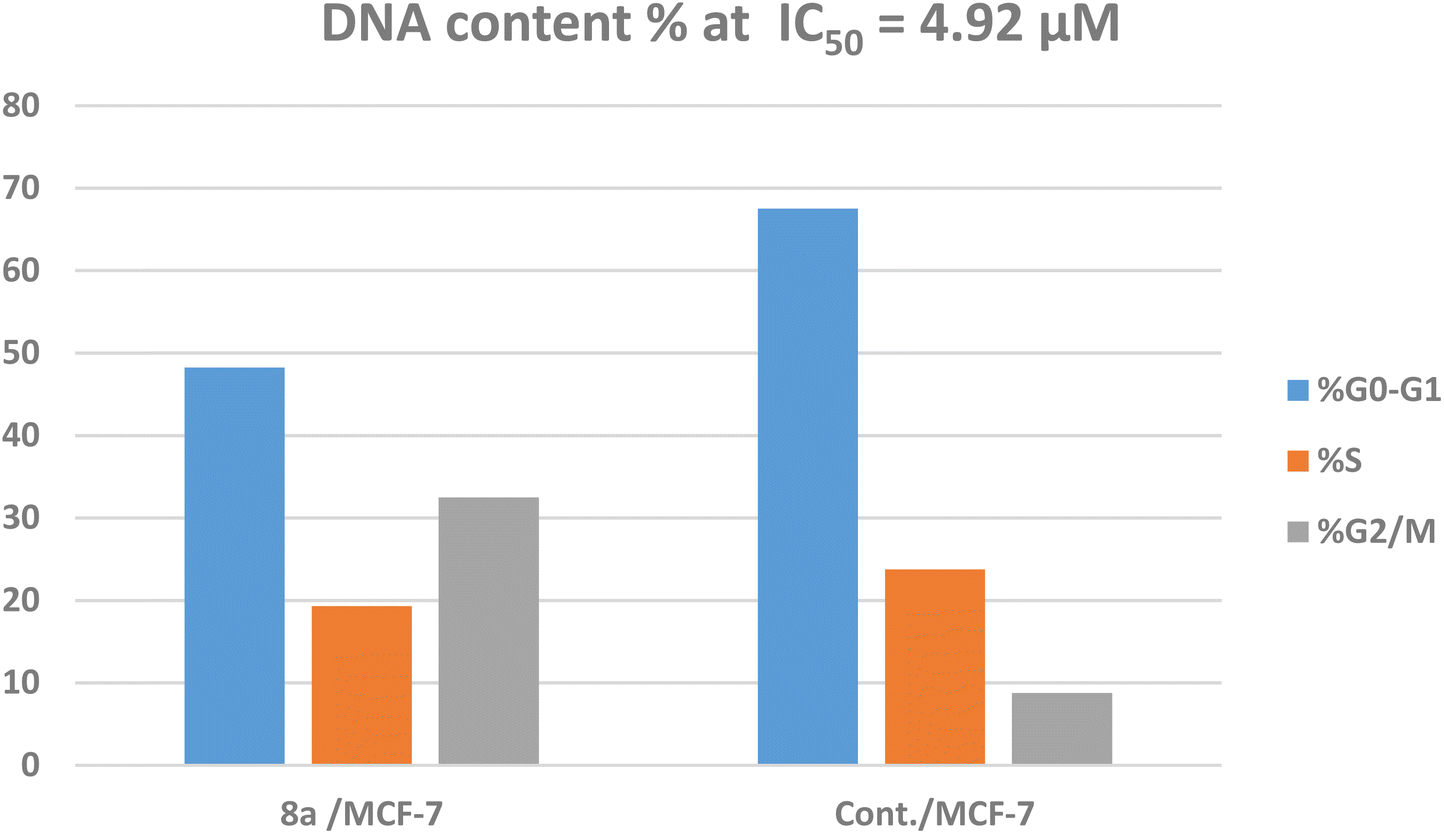 | ||
| Fig. 6 Cell cycle evaluation of the MCF-7 cell line treated with 8a. | ||
The biparametric cytofluorimetric analysis histogram shows the division of cells into four categories: necrotic (upper left side), late apoptosis (upper right side), early apoptosis (lower right side), and viable (lower left side).
It could be noticed that compound 8a enhanced the late apoptotic cell death from 0.17% for the control to 8.66% and induced necrosis of about 6.22%. Compound 8a induced a higher late apoptosis percentage than the early phase, making the recovery of apoptotic cells to safe ones more challenging. This result confirms that apoptosis is one of 8a antiproliferative activity's main modes of action (Fig. 5 and 6).
3.3. Computational studies
As shown in Fig. 7–9, all 4-oxo-3-phenylquinazoline-based candidates 6, 8a, and 8b were embedded nicely within p38α MAPK with significant energy scores of −10.88, −11.28, and −10.96 kcal mol−1, respectively. There were H-bond acceptors between N3 and the backbone of Met109 in all of the screened thieno[2,3-d]pyrimidines 6, 8a, and 8b. Also, in derivatives 6 and 8b, Thr106's side chain and the two amino groups at p-4 and p-5 formed two hydrogen bond donors. However, in 8a, the amino group at position 4 generated two hydrogen bond donors with Thr106 side chain and His107 backbone the, which were separated by 2.90 and 2.5 Å, respectively (Fig. 8 and 9). Furthermore, the amino groups at p-4 in 8b and p-5 in 8a formed ionic bonds with the water molecule HOH-42. The target 8b revealed further hydrophobic interactions between quinazoline and thieno[2,3-d]pyrimidine moieties with Lys53 and Leu108, respectively.
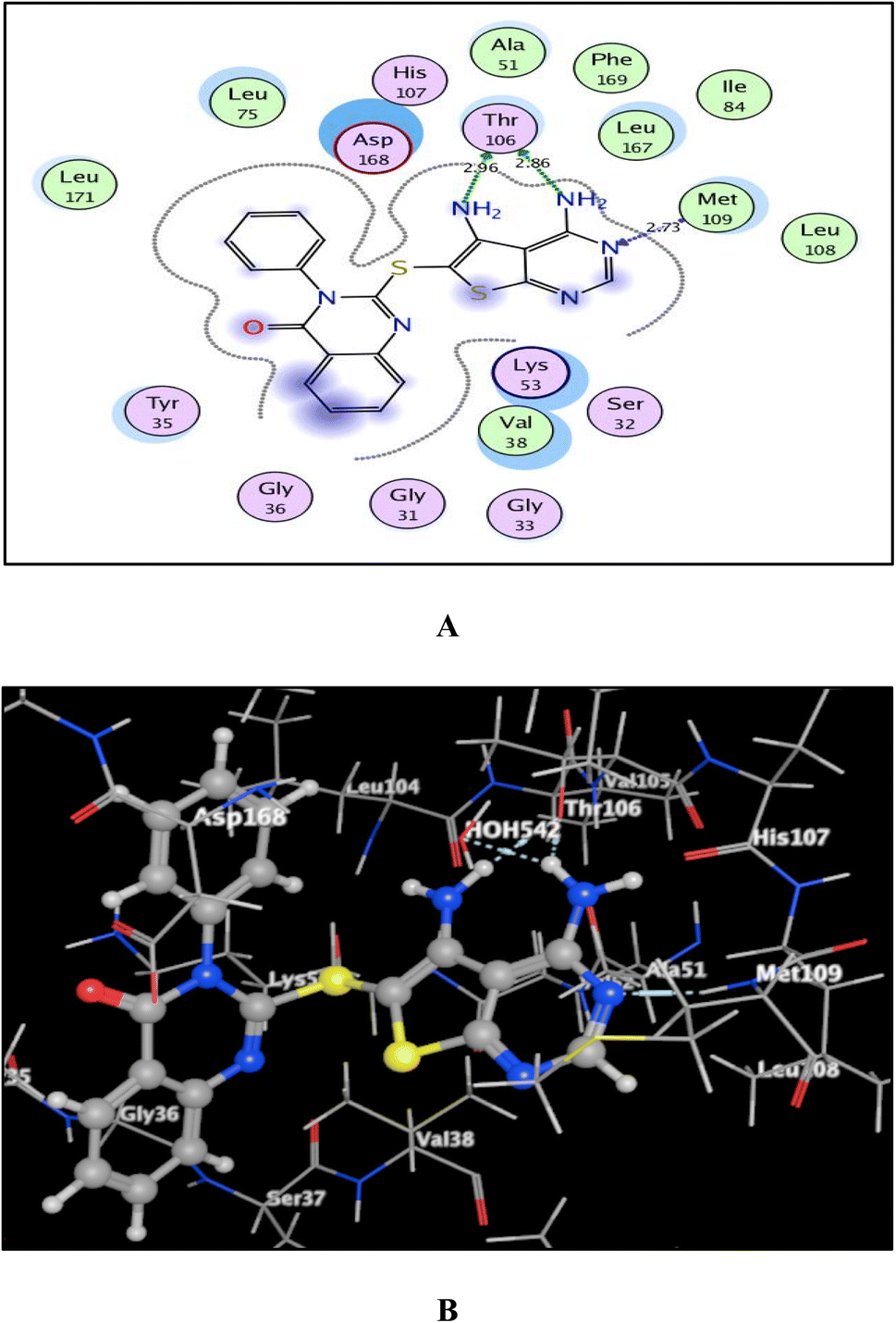 | ||
| Fig. 7 (A) and (B) patterns demonstrate two and three dimensional binding poses of the promising 4,5-diaminothieno[2,3-d]pyrimidine, 6 in the active pocket of p38α MAPK (PDB ID: 1DI9). | ||
 | ||
| Fig. 8 (A) and (B) views demonstrate two and three dimensional binding poses of promising 8a in the active pocket of p38α MAPK (PDB ID: 1DI9). | ||
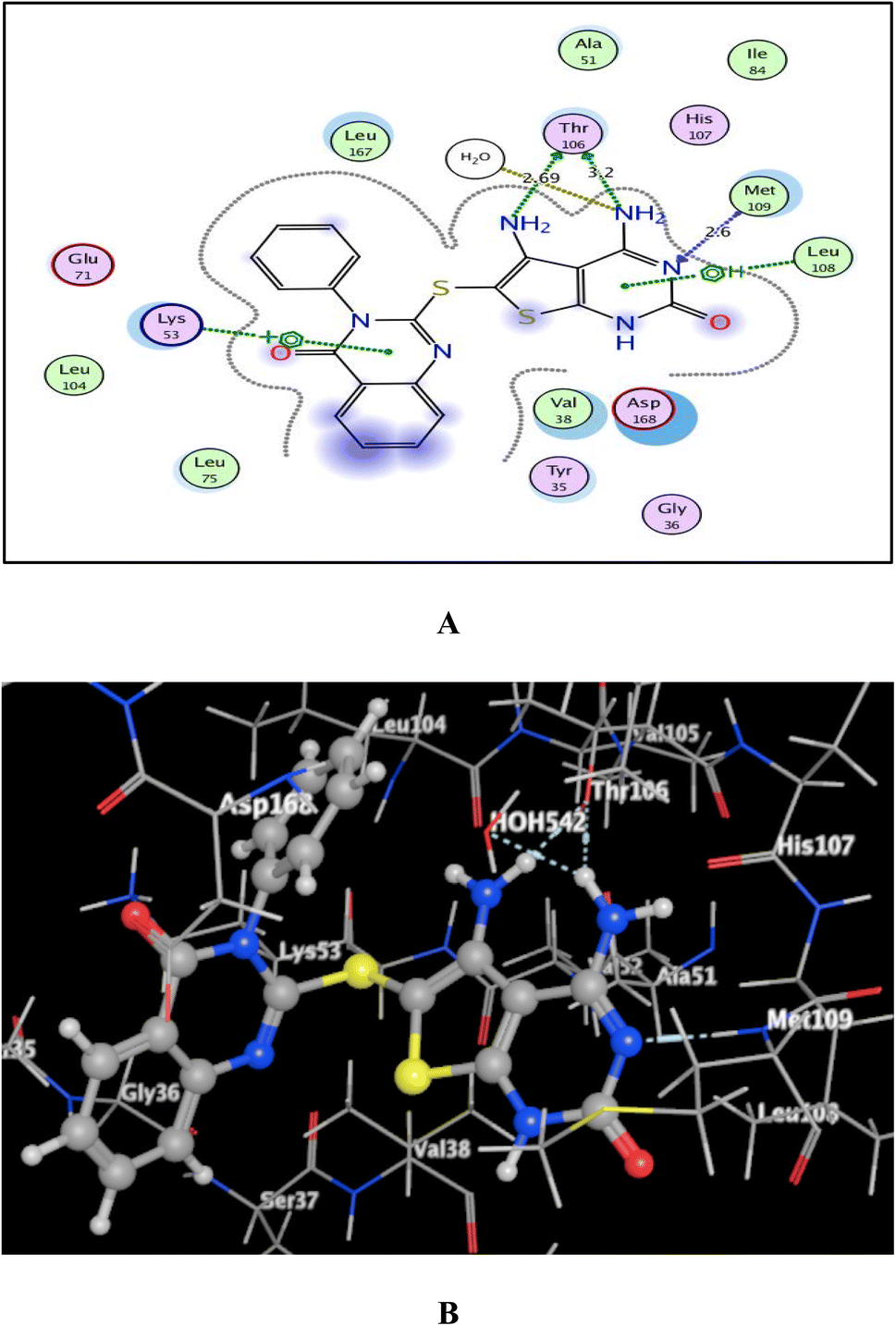 | ||
| Fig. 9 (A) and (B) views demonstrate two and three dimensional binding poses of promising 8b in the active pocket of p38α MAPK (PDB ID: 1DI9). | ||
Based upon earlier observations, the presence of a quinazoline scaffold with a thieno[2,3-d]pyrimidine core strengthened and sustained the fixation of 6, 8a and 8b within the binding pocket of p38α MAPK. The incorporation of NH2 at p-4 of thieno[2,3-d]pyrimidine was found to enhance binding with the important amino acid Thr106, resulting in enhanced inhibitory activity. Accordingly, the thieno[2,3-d]pyrimidines 6, 8a, and 8b should be looked at as possible leads for more research and design to make anticancer medications that work very well at stopping the growth of cancer cells.
Analyzing ADMET (absorption, distribution, metabolism, excretion, and toxicity) of the targeted drugs might yield crucial information on the best medication choice.
The utilization of free internet resources SwissADME and AdmetSAR 1.0 allowed for the completion of this eagerly anticipated study.69–72 Veber and Lipinski's guidelines could be used to determine which medication behaves best when taken orally. With one exception to Veber rule (TPSA > 140), it was demonstrated that thieno[2,3-d]pyrimidine-based candidates 6, 8a and 8b under examination complied with the prior regulations (Table 3).
| Compd | MWa | TPSAb (Å2) | nRBc | nHBAd | nHBDe | MlogPf |
Violationsg |
|---|---|---|---|---|---|---|---|
| a Molecular weight.b Topological polar surface area.c Number of rotatable bond.d Number of hydrogen bond acceptor.e Number of hydrogen bond donor.f Calculated lipophilicity (MlogPo/w).g Violations from Lipinski and Veber rules. |
|||||||
| Rule | ≤500 | ≤140 | ≤10 | ≤10 | ≤5 | ≤4.15 | — |
| 6 | 418.49 | 166.25 | 3 | 4 | 2 | 2.58 | 0 (Lipinski) |
| 1 (Veber) | |||||||
| TPSA > 140 | |||||||
| 8a | 450.56 | 201.24 | 3 | 3 | 3 | 2.51 | 0 (Lipinski) |
| 1 (Veber) | |||||||
| TPSA > 140 | |||||||
| 8b | 434.49 | 186.22 | 3 | 4 | 3 | 2.53 | 0 (Lipinski) |
| 1 (Veber) | |||||||
| TPSA > 140 | |||||||
The bioavailability radar map indicates that, with the exception of polarity and saturation, the evaluated thieno[2,3-d]pyrimidines 6, 8a and 8b were in the perfect range (pink region) of the critical variables (solubility, lipophilicity, flexibility and size) (Fig. 10). Thus, these studies provided substantial proof concerning the oral bioavailability of the investigated compounds.
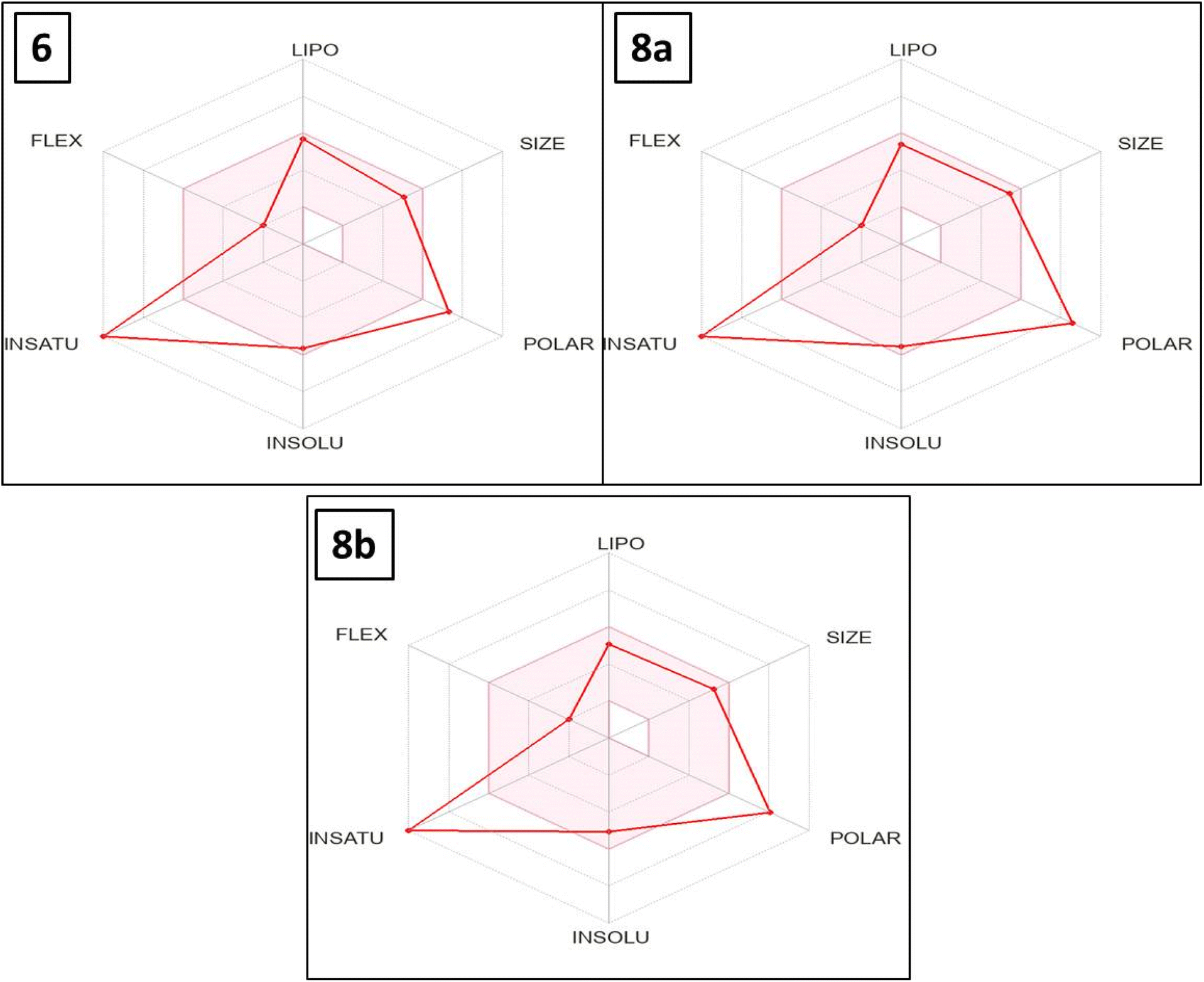 | ||
| Fig. 10 The effective thieno[2,3-d]pyrimidine-based derivatives 6, 8a and 8b's bioavailability radar map. The optimal values for each component of oral bioavailability are represented by the pink region, whereas the red lines indicate the expected values for the studied targets. | ||
Table S6† presents an admetSAR 1.0 study of ADMET linked to putative thieno[2,3-d]pyrimidine-based targets 6, 8a and 8b. With these compounds, there was a greater probability of gastrointestinal absorption without any blood–brain barrier penetration. Therefore, they might be utilized to remedy disorders at the periphery without causing central complications. Drug efflux transporter P-gp (P-glycoprotein) is responsible for removing medicines from cells and may have an impact on drug tolerance. The thieno[2,3-d]pyrimidines 6, 8a and 8b, which were screened, exhibit little discharge from cells with highest activity since they are not P-gp substrates.
Quinazoline-thieno[2,3-d]pyrimidine-based derivatives 6, 8a and 8b are distributed and concentrated within the mitochondria. Indeed, studies have indicated that inhibiting more CYP enzymes increases the possibility that a medication would interact with other active molecules in drug–drug interactions (DDI).73 As a result, it was anticipated that these drugs would have little effect on the majority of CYPs.
All screened targets did not inhibit the potassium channel related to the human ether-a-go-go gene (hERG) as predicted. This implies that there might not be much of a chance for cardiac side effects or cardiotoxicity, which are frequent worries during pharmaceutical candidate clinical trials. Thieno[2,3-d]pyrimidines 8a and 8b exhibited no Ames toxicity, implying no risk of genotoxicity, whereas 6 did not. Moreover, thieno[2,3-d]pyrimidines 6, 8a and 8b provided readings ranging from 617.6 to 688.7 mg kg−1 based on estimations of acute oral toxicity. Since these results fell into the third category (500 mg kg−1 < LD50 ≤ 5000 mg kg−1), they were categorized as harmless substances. The carcinogenicity descriptor (CARC) values of these chemicals, which varied from 424.3 to 448.5 mg per kg body weight per day, indicate that they may also be categorized as non-required and non-carcinogenic. It was expected that these derivatives would be deemed to be non-biodegradable materials when their capacity to break down in the environment was assessed.
4. Conclusion
In brief, three new series of thiophene, thieno[2,3-d]pyrimidine, and thieno[2,3-b]pyridine conjugated with 4-oxo-3-phenyl-2-thio-3,4-dihydroquinazoline scaffolds were created to evaluate their anticancer impact as p38α MAPK inhibitors. Compounds 6, 8a, and 8b exhibited the greatest anticancer efficacy against liver HepG-2, breast MCF-7, and colorectal HCT-116 carcinoma cells, which is approximately equivalent to that of doxorubicin. The latter compounds also showed encouraging safety profiles when tested against normal WI-38 cells with SI values >17.29, >15.11, and >8.80, respectively. Furthermore, compounds 6, 8a, and 8b were subjected to a p38α MAPK inhibition assay, with SB 202190 acting as the reference drug. Compounds 8a and its 2-oxo analogue 8b were more potent inhibitors than the reference drugs (IC50s = 0.18 ± 0.02, 0.23 ± 0.05 μM and 0.27 ± 0.06 μM, respectively). Derivative 6, on the other hand, had an IC50 value of 0.31 ± 0.04 μM, which was 1.14 times lower than SB 202190. Furthermore, the three promising analogues 6, 8a, and 8b boosted the levels of Bax (by ∼7.31, 13.8 and 8.86 fold, respectively) and caspase 3 (by ∼3.55, 4.22 and 3.87 fold, respectively), and decreased the level of the antiapoptotic protein Bcl-2 by ∼1.99, 2.66 and 3.69 fold, respectively in treated MCF-7 cells compared with the untreated ones. Additionally, 8a arrested MCF-7 cell cycle at G2/M phase and induced necrotic and apoptotic effects in the late stages.Molecular docking study exhibited that the candidates 6, 8a, and 8b were embedded nicely within p38α MAPK with significant energy scores of −10.88, −11.28, and −10.96 kcal mol−1, respectively. Ultimately, the physico-chemical and ADMET properties of 6, 8a, and 8b were computed in silico. According to the calculated results, these compounds have the potential to be excellent candidates for additional development and optimization in later research.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the study in this manuscript.Acknowledgements
This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R116), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.References
- M. H. El-Wakil, H. A. El-Dershaby, R. A. Ghazallah, A. F. El-Yazbi, H. A. Abd El-Razik and F. S. Soliman, Identification of new 5-(2,6-dichlorophenyl)-3-oxo-2,3-dihydro-5H-thiazolo [3,2-a]pyrimidine-7-carboxylic acids as p38α MAPK inhibitors: design, synthesis, antitumor evaluation, molecular docking and in silico studies, Bioorg. Chem., 2024, 145, 107226, DOI:10.1016/j.bioorg.2024.107226.
- O. M. Abdelhafez, E. Y. Ahmed, N. A. Abdel Latif, R. K. Arafa, Z. Y. Abd Elmageed, H. I. Ali, H. I. Abdelhafez, O. M. Ahmed, A. Y. Abdel Lalif, N. A. Arafa, R. K. Abdel Mageed and Z. Y. Ali, Design and molecular modeling of novel P38α MAPK inhibitors targeting breast cancer, synthesized from oxygen heterocyclic natural compounds, Bioorg. Med. Chem., 2019, 27, 1308–1319, DOI:10.1016/J.BMC.2019.02.027.
- K. M. Amin, Y. M. Syam, M. M. Anwar, H. I. Ali, T. M. Abdel-Ghani, A. M. Serry, A. M. Amin, K. M. Syam, Y. M. Anwar, M. M. Ali, H. I. Abdel-Ghani and T. M. Serry, Synthesis and molecular docking studies of new furochromone derivatives as p38α MAPK inhibitors targeting human breast cancer MCF-7 cells, Bioorganic Med. Chem., 2017, 25, 2423–2436, DOI:10.1016/j.bmc.2017.02.065.
- H. K. Koul, M. Pal and S. Koul, Role of p38 MAP kinase signal transduction in solid tumors, Genes Cancer, 2013, 4, 342–359, DOI:10.1177/1947601913507951.
- A. Astolfi, N. Iraci, S. Sabatini, M. L. Barreca and V. Cecchetti, p38α MAPK and type I inhibitors: binding site analysis and use of target ensembles in virtual screening, Molecules, 2015, 20, 15842–15861, DOI:10.3390/molecules200915842.
- R. J. Enslen, H. Brancho and D. M. Davis, Molecular determinants that mediate selective activation of P38 MAP kinase isoforms, EMBO J., 2000, 19, 1301–1311 CrossRef PubMed.
- P. Raubo, R. Evans and P. Willis, The discovery and evaluation of 3-amino-2(1H)-pyrazinones as a novel series of selective p38α MAP kinase inhibitors, Bioorg. Med. Chem. Lett., 2020, 30(18), 127412, DOI:10.1016/j.bmcl.2020.127412.
- M. Pomérance, J. Quillard, F. Chantoux, J. Young and J. P. Blondeau, High-level expression, activation, and subcellular localization of p38-MAP kinase in thyroid neoplasms, J. Pathol., 2006, 209, 298–306, DOI:10.1002/path.1975.
- L. Hui, L. Bakiri, E. Stepniak and E. F. Wagner, p38alpha: a suppressor of cell proliferation and tumorigenesis, Cell Cycle, 2007, 6, 2429–2433, DOI:10.4161/CC.6.20.4774.
- J. M. Olson and A. R. Hallahan, P38 MAP kinase: a convergence point in cancer therapy, Trends Mol. Med., 2004, 10, 125–129 CrossRef PubMed.
- G. Pearson, F. Robinson and T. Beers Gibson, et al., Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions, Endocr. Rev., 2001, 22(2), 153–183 Search PubMed.
- G. L. Schieven, The biology of p38 kinase: a central role in inflammation, Curr. Top. Med. Chem., 2005, 5, 921–928 CrossRef PubMed.
- T. Zarubin and J. Han, Activation and signaling of the p38 MAP kinase pathway, Cell Res., 2005, 15, 11–18 CrossRef PubMed.
- K. M. Amin, Y. M. Syam, M. M Anwar, H. I. Ali, T. M. Abdel-Ghani and A. M. Serry, Synthesis and molecular docking study of new benzofuran and furo [3,2-g]chromone-based cytotoxic agents against breast cancer and p38α MAP kinase inhibitors, Bioorg. Chem., 2018, 76, 487–500, DOI:10.1016/j.bioorg.2017.12.029.
- A. Martínez-Limón, M. Joaquin, M. Caballero, F. Posas and E. de Nadal, The p38 pathway: from biology to cancer therapy, Int. J. Mol. Sci., 2020, 21, 1–18, DOI:10.3390/ijms21061913.
- L. Xing, Clinical candidates of small molecule p38 MAPK inhibitors for inflammatory diseases, MAP Kinase, 2016, 4, 5508–5515, DOI:10.4081/mk.2015.5508.
- T. R. Machado, T. R. Machado and P. G. Pascutti, The p38 MAPK inhibitors and their role in inflammatory diseases, ChemistrySelect, 2021, 6, 5729–5742, DOI:10.1002/SLCT.202100406.
- Q. Zhang, X. Zhang and Q. You, Lead discovery of type II BRAF V600E inhibitors targeting the structurally validated DFG-out conformation based upon selected fragments, Molecules, 2016, 21, 879–889, DOI:10.3390/molecules21070879.
- M. Angiolini, Targeting the DFG-in kinase conformation: a new trend emerging from a patent analysis, Future Med. Chem., 2011, 3, 309–337, DOI:10.4155/FMC.10.294.
- R. S. K. Vijayan, P. He, V. Modi, K. C. Duong-Ly, H. Ma, J. R. Peterson, R. L. Dunbrack and R. M. Levy, Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors, J. Med. Chem., 2015, 58, 466–479, DOI:10.1021/jm501603h.
- A. Astolfi, G. Manfroni, V. Cecchetti and M. L. Barreca, A comprehensive structural overview of p38α mitogen-activated protein kinase in complex with ATP-site and non-ATP-site binders, ChemMedChem, 2018, 13, 7–14, DOI:10.1002/CMDC.201700636.
- L. K. Gavrin and E. Saiah, Approaches to discover non-ATP site kinase inhibitors, MedChemComm, 2012, 4, 41–51, 10.1039/c2md20180a.
- S. Fehr, A. b. Unger and E. Schaeffeler, Impact of p38 MAP kinase inhibitors on LPS induced release of TNF-a in whole blood and primary cells from different species, Cell. Physiol. Biochem., 2015, 36, 2237–2249 CrossRef.
- J. Regan, S. Breitfelder and P. Cirillo, et al., Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate, J. Med. Chem., 2002, 4, 2994–3008 CrossRef.
- I. Pevet, C. Brulé, A. Tizot, A. Gohier, F. Cruzalegui, J. A. Boutin and S. Goldstein, Synthesis and pharmacological evaluation of thieno[2,3-b]pyridine derivatives as novel c-Src inhibitors, Bioorg. Med. Chem., 2011, 19, 2517–2528 CrossRef.
- X. D. Yang, W. C. Wan, X. Y. Deng, Y. Li, L. J. Yang, L. Li and H. B. Zhang, Design, synthesis and cytotoxic activities of novel hybrid compounds between 2-phenylbenzofuran and imidazole, Bioorg. Med. Chem. Lett., 2012, 22, 2726–2729 CrossRef.
- P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. U. S. Kumar, FDA Approved Drugs from 2015–June 2020: A Perspective, J. Med. Chem., 2021, 64, 2339–2381 CrossRef.
- Z. M. Alamshany, N. Y. Tashkandi, I. M. Othman, M. M. Anwar and E. S. Nossier, New thiophene, thienopyridine and thiazoline-based derivatives: design, synthesis and biological evaluation as antiproliferative agents and multitargeting kinase inhibitors, Bioorg. Chem., 2022, 127, 105964, DOI:10.1016/j.bioorg.2022.105964.
- S. Zhou, K. Wang, Z. Hu, T. Chen, Y. Dong, R. Gao, M. Wu, Y. Li and X. Ji, Design, synthesis, and structure–activity relationships of a novel class of quinazoline derivatives as coronavirus inhibitors, Eur. J. Med. Chem., 2023, 261, 115831 CrossRef.
- S. Malasala, M. N. Ahmad, R. Akunuri, M. Shukla, G. Kaul, A. Dasgupta, Y. V. Madhavi, S. Chopra and S. Nanduri, Synthesis and evaluation of new quinazoline-benzimidazole hybrids as potent anti-microbial agents against multidrug resistant Staphylococcus aureus and Mycobacterium tuberculosis, Eur. J. Med. Chem., 2021, 212, 112996 CrossRef.
- N. Krasovska, G. Berest, I. Belenichev, H. Severina, I. Nosulenko, O. Voskoboinik, S. Okovytyy and S. Kovalenko, 5+1-Heterocyclization as preparative approach for carboxy-containing triazolo[1,5-c]quinazolines with anti-inflammatory activity, Eur. J. Med. Chem., 2024, 266, 116137 CrossRef PubMed.
- R. Teixeira, T. Menengat, G. Andrade, B. Cotrim, C. Ponte, W. C. Santos and G. Resende, Microwave Assisted Synthesis of 4-Phenylquinazolin-2(1H)-one Derivatives that Inhibit Vasopressor Tonus in Rat Thoracic Aorta, Molecules, 2020, 25, 1467 CrossRef PubMed.
- A. M. Soliman, H. M. Karam, M. H. Mekkawy and M. M. Ghorab, Antioxidant activity of novel quinazolinones bearing sulfonamide: potential radiomodulatory effects on liver tissues via NF-κB/PON1 pathway, Eur. J. Med. Chem., 2020, 197, 112333 CrossRef.
- M. F. Zayed, Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases, Sci. Pharm., 2023, 91, 18 CrossRef CAS.
- N. F. El Hamaky, A. Hamdi, W. A. Bayoumi, A. A. Elgazar and M. N. Nasr, Novel quinazolin-2-yl 1,2,3-triazole hybrids as promising multi-target anticancer agents: design, synthesis, and molecular docking study, Bioorg. Chem., 2024, 148, 107437, DOI:10.1016/j.bioorg.2024.107437.
- M. E. Abdelaziz, M. M. M. El-Miligy, S. M. Fahmy, M. A. Mahran and A. A. Hazzaa, Design, synthesis and docking study of pyridine and thieno[2,3-b]pyridine derivatives anticancer PIM-1 kinase inhibitors, Bioorg. Chem., 2018, 80, 674–692, DOI:10.1016/j.bioorg.2018.07.024.
- S. A. Al-Trawneh, A. H. Tarawneh, A. V. Gadetskaya, E. Seo, M. R. Al-Ta’ani, S. A. Al-Taweel and M. M. El-Abadelah, Synthesis and cytotoxicity of thieno[2,3-b]pyridine derivatives toward sensitive and multidrug-resistant leukemia cells, Acta Chim. Slov., 2021, 68(2), 458–465, DOI:10.17344/acsi.2020.6609.
- C. Eurtivong, V. Semenov, M. Semenova, L. Konyushkin, O. Atamanenko, J. Reynisson and A. Kiselyov, Identification of anticancer agents based on the thieno[2,3-b]pyridine and 1H pyrazole molecular scaffolds, Bioorg. Med. Chem., 2017, 25(2), 658–664, DOI:10.1016/j.bmc.2016.11.041.
- S. N. Sirakanyan, D. Spinelli, A. Geronikaki, E. K. Hakobyan, H. Sahakyan, E. Arabyan, H. Zakaryan, L. E. Nersesyan, A. S. Aharonyan and I. S. Danielyan, et al., Synthesis, Antitumor Activity, and Docking Analysis of New Pyrido[30,20:4,5]furo(thieno) [3,2-d]pyrimidin-8-amines, Molecules, 2019, 24, 3952 CrossRef.
- M. A. Kang, M. Kim, J. Y. Kim, Y. Shin, J. Song and J. Jeong, A novel pyrido-thieno-pyrimidine derivative activates p53 through induction of phosphorylation and acetylation in colorectal cancer cells, Int. J. Oncol., 2015, 46, 342–350 Search PubMed.
- R. Callagham, F. Luk and M. Bebawy, Inhibition of the Multidrug Resistance PGlycoprotein: Time for a Change of Strategy?, Drug Metab. Dispos., 2014, 42, 623–631 CrossRef.
- R. Bansal and A. Malhotra, Therapeutic progression of quinazolines as targeted chemotherapeutic agents, Eur. J. Med. Chem., 2021, 211, 113016 CrossRef PubMed.
- Z. M. Alamshany, N. Y. Tashkandi, I. M. Othman, M. M. Anwar and E. S. Nossier, New thiophene, thienopyridine and thiazoline-based derivatives: design, synthesis and biological evaluation as antiproliferative agents and multitargeting kinase inhibitors, Bioorg. Chem., 2022, 127, 105964, DOI:10.1016/j.bioorg.2022.105964.
- B. C. Liao, C. C. Lin, J. H. Lee and J. C. Yang, Update on recent preclinical and clinical studies of T790M mutant-specific irreversible epidermal growth factor receptor tyrosine kinase inhibitors, J. Biomed. Sci., 2016, 23(1), 86, DOI:10.1186/s12929-016-0305-9.
- K. Nurgali, R. T. Jagoe and R. Abalo, Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae?, Front. Pharmacol, 2018, 9, 245–247, DOI:10.3389/fphar.2018.00245.
- X.-J. Liu, H.-C. Zhao, S.-J. Hou, H.-J. Zhang, L. Cheng, S. Yuan, L.-R. Zhang, J. Song, S.-Y. Zhang and S.-W. Chen, Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy, Bioorg. Chem., 2023, 133, 106425, DOI:10.1016/j.bioorg.2023.106425.
- R. Z. Batran, E. Y. Ahmed, E. S. Nossier, H. M. Awad and N. A. Latif, Anticancer activity of new triazolopyrimidine linked coumarin and quinolone hybrids: Synthesis, molecular modeling, TrkA, PI3K/AKT and EGFR inhibition, J. Mol. Struct., 2024, 1305, 137790, DOI:10.1016/j.molstruc.2024.137790.
- N. H. Amin, M. T. El-Saadi, M. M. Abdel-Fattah, A. A. Mohammed and E. G. Said, Development of certain aminoquinazoline scaffolds as potential multitarget anticancer agents with apoptotic and anti-proliferative effects: design, synthesis and biological evaluation, Bioorg. Chem., 2023, 135, 106496, DOI:10.1016/j.bioorg.2023.106496.
- A. A. Alsfouk, I. M. Othman, M. M. Anwar, W. A. Alshareef, A. Saleh and E. S. Nossier, Synthesis and computational studies of new pyridine, pyrazole, pyran, and pyranopyrimidine-based derivatives of potential antimicrobial activity as DNA gyrase and topoisomerase IV inhibitors, J. Mol. Struct., 2025, 1319, 139528, DOI:10.1016/j.molstruc.2024.139528.
- A. I. Khodair, M. A. Alsafi and M. S. Nafie, Synthesis, molecular modeling and anti-cancer evaluation of a series of quinazoline derivatives, Carbohydr. Res., 2019, 486, 107832, DOI:10.1016/j.carres.2019.107832.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. I. Warren, H. Bokesch, S. Kenney and M. R. Boyd, New colorimetric cytotoxicity assay for anticancer-drug screening, J. Natl. Cancer Inst., 1990, 82, 1107–1112, DOI:10.1093/jnci/82.13.1107.
- A. E. Amr, E. A. Elsayed, M. A. Al-Omar, H. O. Badr Eldin, E. S. Nossier and M. M. Abdallah, Design, synthesis, anticancer evaluation and molecular modeling of novel estrogen derivatives, Molecules, 2019, 24, 416, DOI:10.3390/molecules24030416.
- K. Singh, A. Gangrade, A. Jana, B. B. Mandal and N. Das, Design, synthesis, characterization, and antiproliferative activity of organoplatinum compounds bearing a 1,2,3-triazole ring, ACS Omega, 2019, 4, 835–841, DOI:10.1021/ACSOMEGA.8B02849.
- M. A. Sabry, M. A. Ghaly, A. R. Maarouf and H. I. El-Subbagh, New thiazole-based derivatives as EGFR/HER2 and DHFR inhibitors: synthesis, molecular modeling simulations and anticancer activity, Eur. J. Med. Chem., 2022, 241, 114661, DOI:10.1016/J.EJMECH.2022.114661.
- A. E. G. E. Amr, E. A. Elsayed, M. A. Al-Omar, H. O. Eldin Badr, E. S. Nossier and M. M. Abdallah, Design, synthesis, anticancer evaluation and molecular modeling of novel estrogen derivatives, Molecules, 2019, 24(3), 416, DOI:10.3390/molecules24030416.
- A. S. Hassan, G. O. Moustafa, H. M. Awad, E. S. Nossier and M. F. Mady, Design, synthe-sis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole–indole hybrids, ACS Omega, 2021, 6, 12361–12374 CrossRef CAS.
- C. M. Pfeffer and A. T. K. Singh, Apoptosis: a target for anticancer therapy, Int. J. Mol. Sci., 2018, 19, 448 CrossRef.
- S. Zaman, R. Wang and V. Gandhi, Targeting the apoptosis pathway in hematologic malignancies, Leuk. Lymphoma, 2014, 55, 1980–1992 CrossRef CAS PubMed.
- D. J. Lukin, L. A. Carvajal, W. J. Liu, L. Resnick-Silverman and J. J. Manfredi, p53 pro-motes cell survival due to the reversibility of its cell-cycle checkpoints, Mol. Cancer Res., 2014, 13, 16–28 CrossRef.
- M. Barbareschi, O. Caffo, S. M. Veronese, R. D. Leek, P. Fina, S. Fox, M. Bonzanini, S. Girlando, L. Morelli, C. Eccher and F. Pezzella, Bcl-2 and p53 expression in node-negative breast carcinoma: a study with long-term follow-up, Hum. Pathol., 1996, 27, 1149–1155 CrossRef CAS.
- R. Onur, A. Semerciöz, I. Orhan and H. Yekeler, The effects of melatonin and the antioxidant defense system on apoptosis regulator proteins (Bax and Bcl-2) in experimentally induced varicocele, Urol. Res., 2004, 32, 204–208 CrossRef CAS.
- P. Pozarowski and Z. Darzynkiewicz, Analysis of cell cycle by flow cytometry methods, Methods Mol. Biol., 2004, 281, 301–311 CAS.
- F. Farmani, M. Moein, A. Amanzadeh, H. M. Kandelous, Z. Ehsanpour and M. Salimi, Antiproliferative evaluation and apoptosis induction in MCF- 7 cells by Ziziphus spina christi leaf extracts, asian Pacific, J. Cancer Prev., 2016, 17, 315–321, DOI:10.7314/APJCP.2016.17.1.315.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutellingsperger, A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled Annexin V, J. Immunol. Methods, 1995, 184, 39–51, DOI:10.1016/0022-1759(95)00072-I.
- G. O. Moustafa, A. Shalaby, A. M. Naglah, M. M. Mounier, H. El-Sayed, M. M. Anwar and E. S. Nossier, Synthesis, characterization, in vitro anticancer potentiality, and antimicrobial activities of novel peptide–glycyrrhetinic-acid-based derivatives, Molecules, 2021, 26(15), 4573, DOI:10.3390/molecules26154573.
- A. E. G. E. Amr, R. E. A. Mageid, M. M. El-Naggar, A. S. Naglah, E. Nossier and E. A. Elsayed, Chiral Pyridine-3,5-bis-(L-phenylalaninyl-L-leucinyl) Schiff Base Peptides as Potential Anticancer Agents: Design, synthesis, and molecular docking studies targeting lactate dehydrogenase-A, Molecules, 2020, 25(5), 1096, DOI:10.3390/molecules25051096.
- A. A. El-Sayed, E. S. Nossier, A. A. Almehizia and A. E. G. E. Amr, Design, synthesis, anticancer evaluation and molecular docking study of novel 2,4-dichlorophenoxymethyl-based derivatives linked to nitrogenous heterocyclic ring systems as potential CDK-2 inhibitors, J. Mol. Struct., 2022, 1247, 131285, DOI:10.1016/j.molstruc.2021.131285.
- L. Shewchuk, A. Hassell, B. Wisely, W. Rocque, W. Holmes, J. Veal and L. F. Kuyper, Binding mode of the 4-anilinoquinazoline class of protein kinase inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-dependent kinase 2 and p38 kinase, J. Med. Chem., 2000, 43(1), 133–138, DOI:10.1021/jm990401t.
- E. M. Mohi El-Deen, E. S. Nossier and E. A. Karam, New Quinazolin-4 (3H)-one Derivatives Incorporating Hydrazone and Pyrazole Scaffolds as Antimicrobial Agents Targeting DNA Gyraze Enzyme, Sci. Pharm., 2022, 90, 52, DOI:10.3390/scipharm90030052.
- H. E. Hashem, A. E. G. E. Amr, A. A. Almehizia, A. M. Naglah, B. M. Kariuki, H. A. Eassa and E. S. Nossier, Nanoparticles of a pyrazolo-pyridazine derivative as potential EGFR and CDK-2 inhibitors: design, structure determination, anticancer evaluation and in silico studies, Molecules, 2023, 28(21), 7252, DOI:10.3390/molecules28217252.
- D. H. Dawood, M. M. Sayed, S. T. Tohamy and E. S. Nossier, New Thiophenyl-pyrazolyl-thiazole Hybrids as DHFR Inhibitors: Design, Synthesis, Antimicrobial Evaluation, Molecular Modeling, and Biodistribution Studies, ACS Omega, 2023, 8(42), 39250–39268, DOI:10.1021/acsomega.3c04736.
- Z. M. Alamshany, E. M. Algamdi, I. M. Othman, M. M. Anwar and E. S. Nossier, New pyrazolopyridine and pyrazolothiazole-based compounds as anti-proliferative agents targeting c-Met kinase inhibition: design, synthesis, biological evaluation, and computational studies, RSC Adv., 2023, 13(19), 12889–12905, 10.1039/d3ra01931d.
- F. Cheng, Y. Yu, J. Shen, L. Yang, W. Li, G. Liu, P. W. Lee and Y. Tang, Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers, J. Chem. Inf. Model., 2011, 51(5), 996–1011, DOI:10.1021/ci200028n.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06744d |
| This journal is © The Royal Society of Chemistry 2025 |