
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational study on catalyst-free BCl3-promoted chloroboration of carbonyl compounds†
Abera Tadie Derese *a,
Mengistu Gemech Menkirb,
Mihret Kendie Woliec and
Desalegn Addis Yemamd
*a,
Mengistu Gemech Menkirb,
Mihret Kendie Woliec and
Desalegn Addis Yemamd
aDepartment of Chemistry, Debre Tabor University, Ethiopia. E-mail: aberatadie@gmail.com
bDepartment of Chemistry, Bahir Dar University, Ethiopia
cDepartment of Chemistry, Debark University, Ethiopia
dDepartment of Chemistry, Debre Tabor University, Ethiopia
First published on 28th January 2025
Abstract
DFT calculations were performed to investigate the possible reaction mechanisms underlying catalyst-free chloroboration reactions of carbonyl compounds with BCl3. The interaction between BCl3 and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of carbonyl compounds is a two-step reaction. In the first step, B of BCl3 forms a bond with the O of the CO moiety, followed by the 1,3-Cl migration process from BCl3 to the C of the carbonyl group. To indicate the versatility of our synthetic methodology, a catalyst-free chloroboration of a variety of aldehydes and ketones with a broad range of electron-donating and electron-withdrawing groups with BCl3 was checked. According to DFT results, BCl3-induced chloroboration of aldehydes and ketones progressed under a kinetically favorable condition with <20 kcal mol−1 of activation free energy.
O moiety of carbonyl compounds is a two-step reaction. In the first step, B of BCl3 forms a bond with the O of the CO moiety, followed by the 1,3-Cl migration process from BCl3 to the C of the carbonyl group. To indicate the versatility of our synthetic methodology, a catalyst-free chloroboration of a variety of aldehydes and ketones with a broad range of electron-donating and electron-withdrawing groups with BCl3 was checked. According to DFT results, BCl3-induced chloroboration of aldehydes and ketones progressed under a kinetically favorable condition with <20 kcal mol−1 of activation free energy.
Introduction
Boronate esters, which are produced when carbonyl compounds undergo hydroboration, are frequently employed as significant applications in biomedical and pharmaceutical research, as well as in organic synthesis.1–8 Because boronate esters are important as synthetic intermediates in a wide range of chemical processes, several techniques for producing them have been devised. Traditionally, stoichiometric additions of reactive BH3 to generate borates, which can be hydrolyzed into alcohols, or stoichiometric amounts of dangerous metal-hydrides, such as LiAlH4 and NaBH4, have been used to induce hydroboration of CO bonds.9–14 In terms of cost and atom efficiency, carbonyl reduction using transition metal-catalyzed hydrogenation procedures employing combustible and highly pressured H2 gas is also optimal.15,16 Regardless of their amazing success, these procedures exhibit low yields, poor functional group compatibility, and the need for stoichiometric reagents for large-scale applications.
The discovery of transition metal-catalyzed hydroboration, which uses pinacolborane (HBpin) or catecholborane (HBcat) to selectively reduce unsaturated carbonyl compounds, has been a significant advancement in the past years.17–22 Classically, different methods with the involvement of metals, either as catalysts or in stoichiometric proportions, have been discovered to synthesize organoboron compounds.23–26 However, the involvement of metals may render these preparative techniques less economical and environmentally benign, and it may result in heavy metal contamination of the final boron products.27
To eliminate the above-listed drawbacks, transition-metal-free catalytic methods were widely adopted over the traditional approach.28 Among these, transition-metal-free catalytic diboration of unsaturated hydrocarbons,29,30 transition-metal-free catalytic β-boration of α,β-unsaturated compounds,31 transition-metal-free catalytic borylation of allylic and propargylic alcohols,32 and transition-metal-free catalytic hydroboration of unsaturated hydrocarbons33–36 have been reported.
The use of protocols in organic transformations that do not require solvents or catalysts has gained considerable interest lately as a result of the growing need for low-cost, atom-economical, and environmentally safe synthetic processes.37,38 Recently, catalyst-free and solvent-free hydroboration of carbonyl compounds using pinacol borane has been reported. For example, in 2018, Stachowiak and coworkers studied catalyst-free and solvent-free hydroboration of aldehydes.39 In addition, Wang and colleagues experimentally and computationally investigated catalyst-free and solvent-free hydroboration of ketones40 and carboxylic acids.41 However, for the hydroboration of ketones and carboxylic acids, computational results reveal that the reactions are hard to proceed at room temperature because of the high energy barrier.
Furthermore, borylative cyclization of unsaturated compounds without any catalyst, such as intramolecular amination of alkenes and alkynes, metal-free borylative cyclization of alkynes, intramolecular aminoboration of allenes, and catalyst-free annulative thioboration of unfunctionalized olefins was demonstrated using BCl3 as a boron source.42–45 However, the action of BCl3 on catalyst-free chloroboration of carbonyl compounds has not yet studied. Inspired by the work described above, we decided to carry computational work on BCl3-induced chloroboration of carbonyl compounds in a catalyst-free manner. In this study, we performed a DFT calculation for catalyst-free chloroboration of aldehydes and ketones using BCl3 as a boron source.
Computational methodology
The Gaussian 09![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 46 computational program suite, along with its Gauss View 5.0 graphical user interface, was utilized for all calculations carried out in this work. Using the 6-31+G(d) basis set and M062X47 hybrid functional, the geometry optimizations of all the structures were performed. Frequency calculations were performed at the same theoretical level for each stationary point to classify them as minima (no imaginary frequency) or transition states (one imaginary frequency) and to derive thermodynamic energy corrections. To confirm that the transition states link two pertinent local minima along the potential energy surface, intrinsic reaction coordinate (IRC)48 computations were performed on the transition structures. To obtain more accurate energy estimates, the larger basis set 6-311++G(d,p) and M062X49 functional were used in single-point energy calculations in toluene based on gas phase optimized geometries utilizing the PCM50 model.
46 computational program suite, along with its Gauss View 5.0 graphical user interface, was utilized for all calculations carried out in this work. Using the 6-31+G(d) basis set and M062X47 hybrid functional, the geometry optimizations of all the structures were performed. Frequency calculations were performed at the same theoretical level for each stationary point to classify them as minima (no imaginary frequency) or transition states (one imaginary frequency) and to derive thermodynamic energy corrections. To confirm that the transition states link two pertinent local minima along the potential energy surface, intrinsic reaction coordinate (IRC)48 computations were performed on the transition structures. To obtain more accurate energy estimates, the larger basis set 6-311++G(d,p) and M062X49 functional were used in single-point energy calculations in toluene based on gas phase optimized geometries utilizing the PCM50 model.
Results and discussion
First, the mechanism of benzaldehyde and acetophenone and the substrates of the chloroboration model was investigated. Scheme 1 depicts the proposed reaction mechanism for BCl3-promoted chloroboration of aldehydes and ketones to produce boronate esters. The first and most common step for both carbonyl substrates is the bond formation between the B of BCl3 and O of the CO moiety. The second step involves a chloride shift from BCl3 to the carbonyl carbon to afford a chlorinated borane. DFT calculations were performed to investigate the detailed proposed mechanism. The calculated energetic profiles are shown in Fig. 1.
 | ||
| Scheme 1 Proposed mechanism for the BCl3-mediated chloroboration of aldehydes and ketones (RH for aldehydes; RCH3 for ketones). | ||
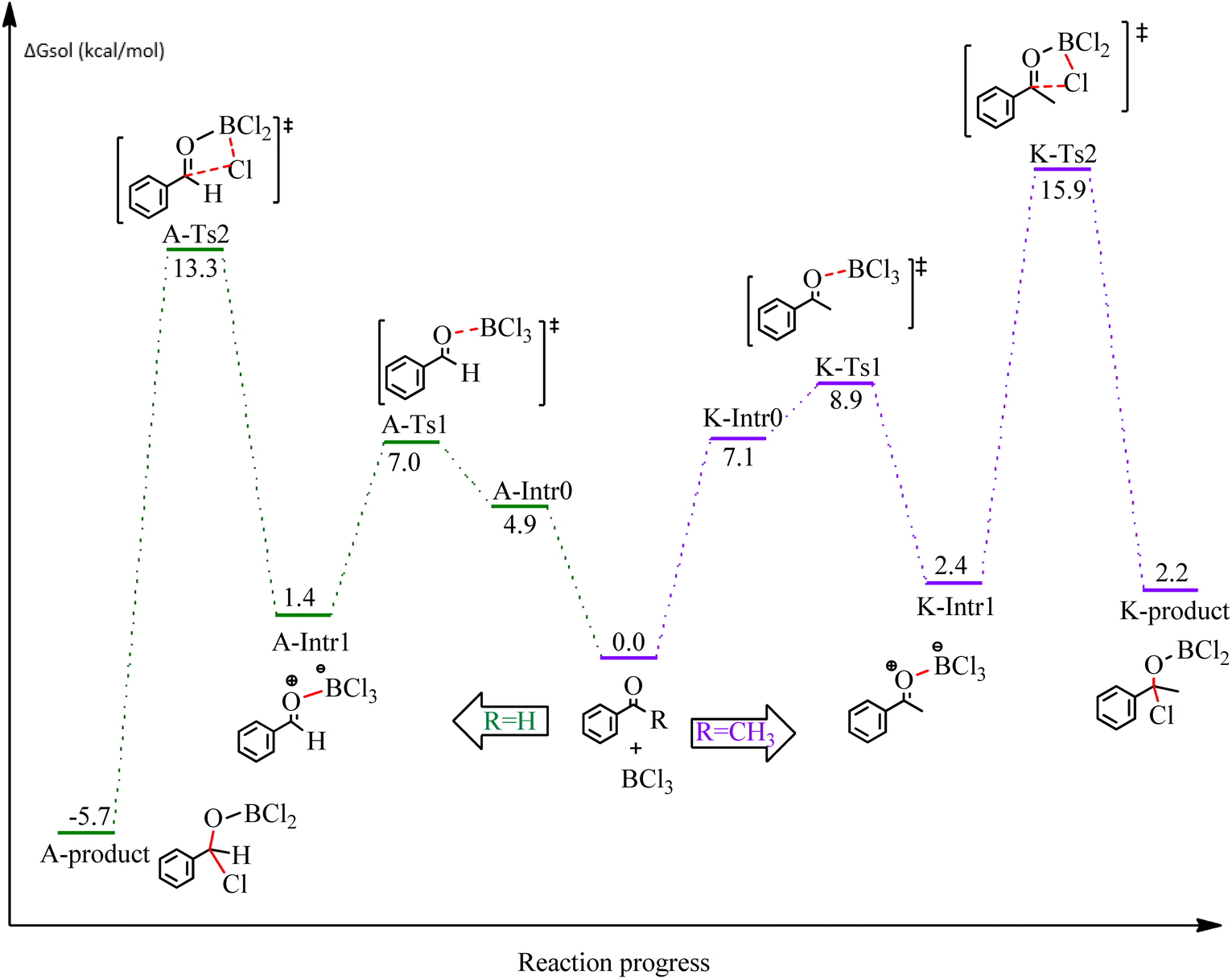 | ||
| Fig. 1 DFT-computed free energies for the chloroboration reaction of benzaldehyde and acetophenone. | ||
We started the study by reacting benzaldehyde and BCl3 without the use of a catalyst. Based on the DFT calculation results shown in Fig. 1, the production of the coordinated complex A-Intr0 between the CO group of the model reactant (benzaldehyde) and BCl3 is found to be 4.9 kcal mol−1. Through A-Ts1, A-Intr0 is subsequently transformed into the zwitterion intermediate A-Intr1 at a lower activation energy of 2.1 kcal mol−1. The generation of the borylated intermediate A-product via the four-membered ring transition state A-Ts2 requires 11.9 kcal mol−1, which corresponds to the formation of C–Cl. The breaking B–Cl distance is 2.12 Å, whereas the forming C–Cl distance is 2.18 Å.
Following the identification of the most advantageous route for the chloroboration of the aldehyde model reactant, we perform analogous DFT computations on ketones using acetophenone as a substrate. The complexation process between BCl3 and the CO moiety of the model reactant, acetophenone, initiates the reaction. Fig. 1 illustrates that the former transition state, K-Ts1, has a lower energy barrier of 1.8 kcal mol−1, making it more kinetically beneficial to afford K-Intr1. Following the formation of K-Intr1, the K-product is formed by the easy transfer of the chloride anion to the carbonyl carbon via K-Ts2, which demands an energy of 13.5 kcal mol−1 (1.6 kcal mol−1 less stable than A-Ts2). Note that 2.09 Å is the breaking B–Cl distance, while 2.17 Å is the forming C–Cl distance.
According to DFT results displayed in Fig. 1, aldehydes exhibited a higher reactivity than ketones owing to the steric hindrance effect and ketones' reduced electrophilicity. Fig. 2 shows the structure of the transition states and optimized intermediates during the chloroboration of aldehydes and ketones.
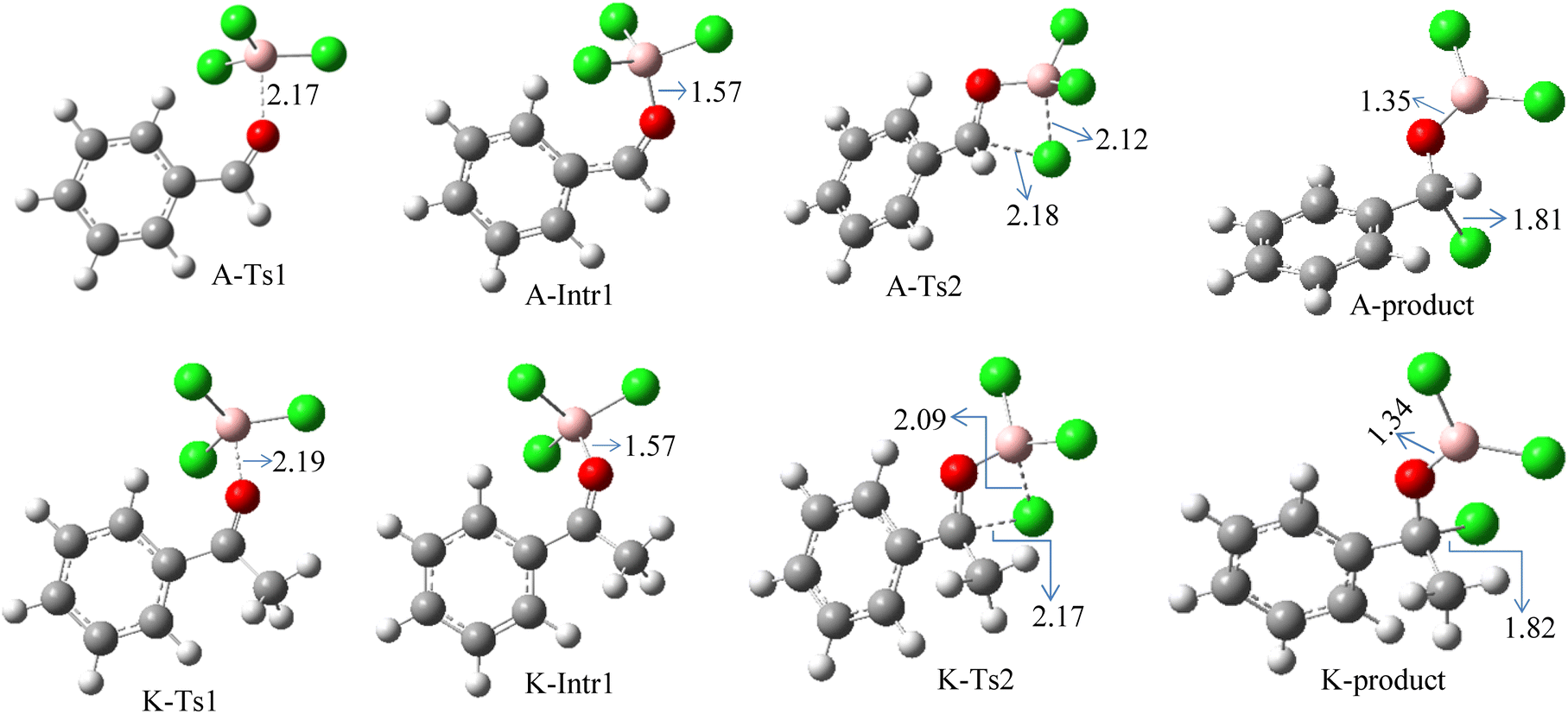 | ||
| Fig. 2 Geometric structures of intermediates and transition states in the chloroboration of benzaldehyde and acetophenone (distances are in Å, carbon: gray, hydrogen: white, oxygen: red, boron: pink, chlorine: green). | ||
After we performed DFT calculation for the model substrate, we looked at the extent of the reaction when specific substituted benzaldehydes bearing methyl, methoxy, halogens, and heterocyclic derivatives were functionalized with either electron-withdrawing or electron-donating groups Table 1. Fig. 3 presents DFT results for the BCl3-induced chloroboration reaction of substituted benzaldehydes bearing m-CH3, o-CH3, p-CF3, and p-F. In contrast to the parent model substrate benzaldehyde (Fig. 1), the chloroboration of aldehydes substituted with o-CH3 and p-CF3 proceeded with lower activation energy, yielding more stable boronate ester products (Fig. 3). The reaction proceeded with somewhat greater activation energy to afford the predicted boronate esters when the substituent groups are m-CH3 and p-F when compared with the parent substrate benzaldehyde. Fig. 5 shows the DFT results for the BCl3-induced chloroboration reaction of substituted benzaldehydes bearing p-Br and p-OMe as well as picolinbenzaldehyde and thiophen-2-carbaldehyde as a substrate. p-Br and p-OMe substituted benzaldehyde and thiophen-2-carbaldehyde demands a higher energy barrier than the model reactant. Picolinbenzaldehyde consumes energy almost equal to that consumed by benzaldehyde. Fig. 4 and 6 show the geometric structures for intermediates and transition states in the chloroboration of substituted benzaldehyde.
| Substrate | B–O bond formation | 1,3-Cl migration | Substrate | B–O bond formation | 1,3-Cl migration |
|---|---|---|---|---|---|
 |
2.1 | 11.9 |  |
2.9 | 12.8 |
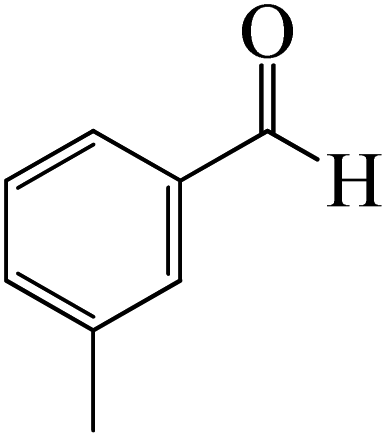 |
2.7 | 13.3 | 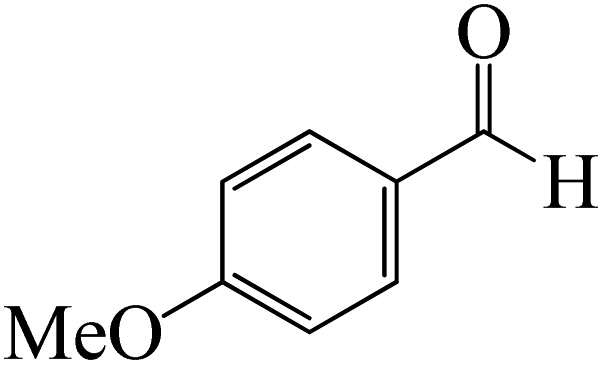 |
1.8 | 14.8 |
 |
2.3 | 10.3 | 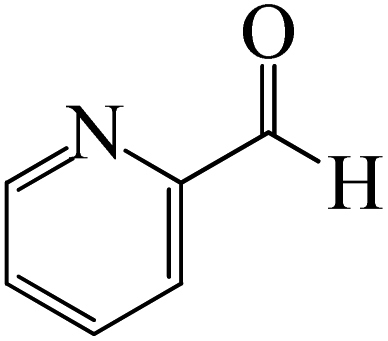 |
2.7 | 11.8 |
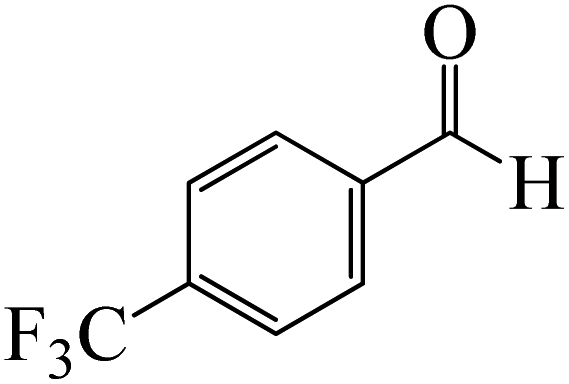 |
3.5 | 11.3 |  |
2.3 | 15.5 |
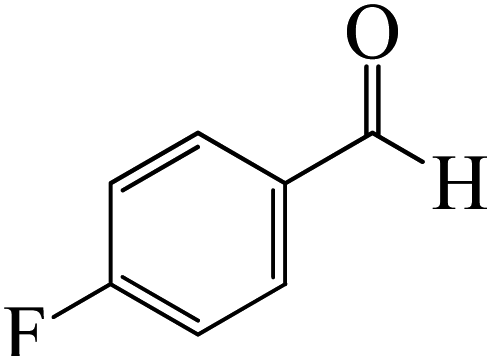 |
2.2 | 12.2 |
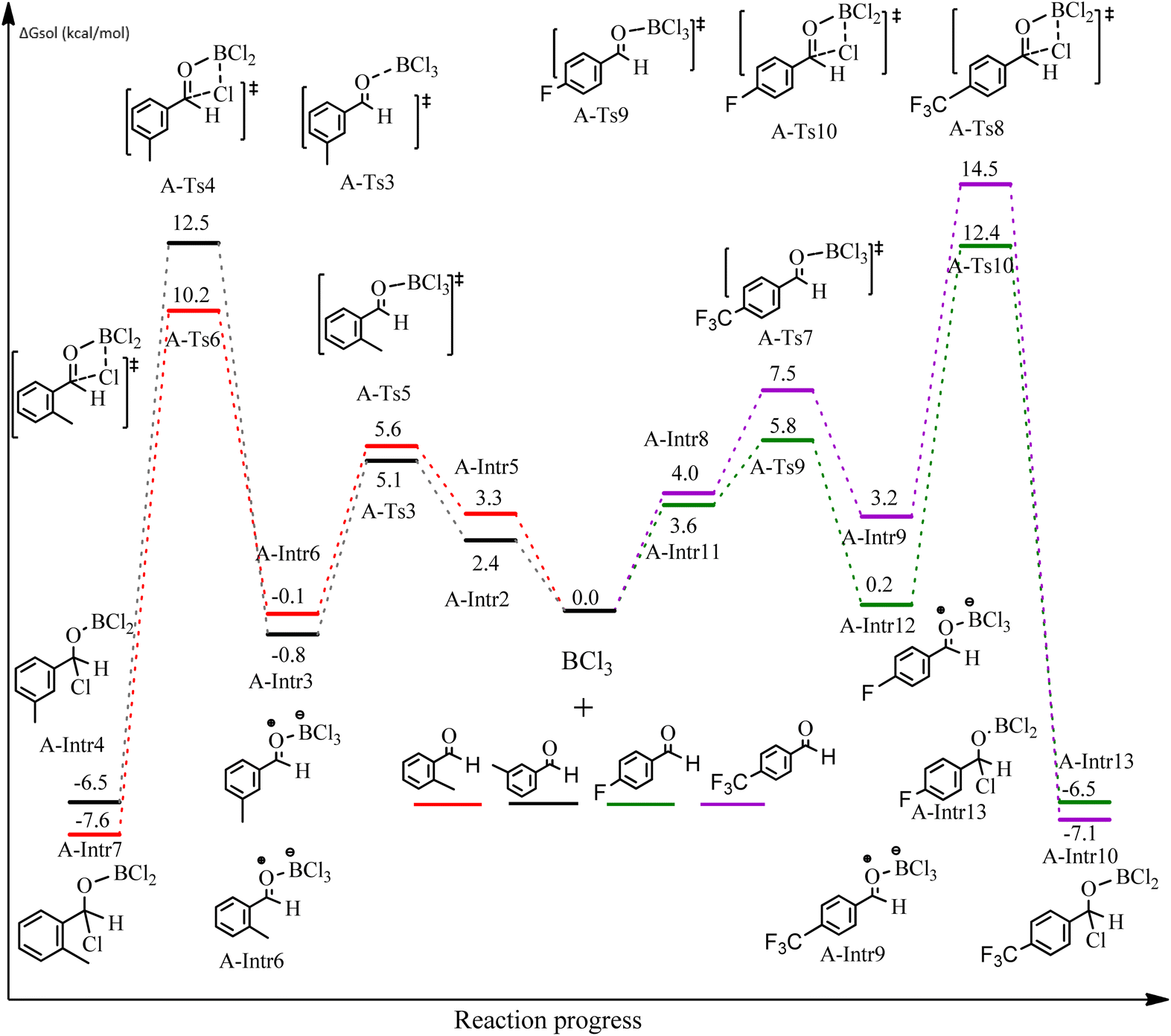 | ||
| Fig. 3 DFT-computed free energies for chloroboration reaction of substituted benzaldehyde bearing m-CH3, o-CH3, p-CF3, and p-F. | ||
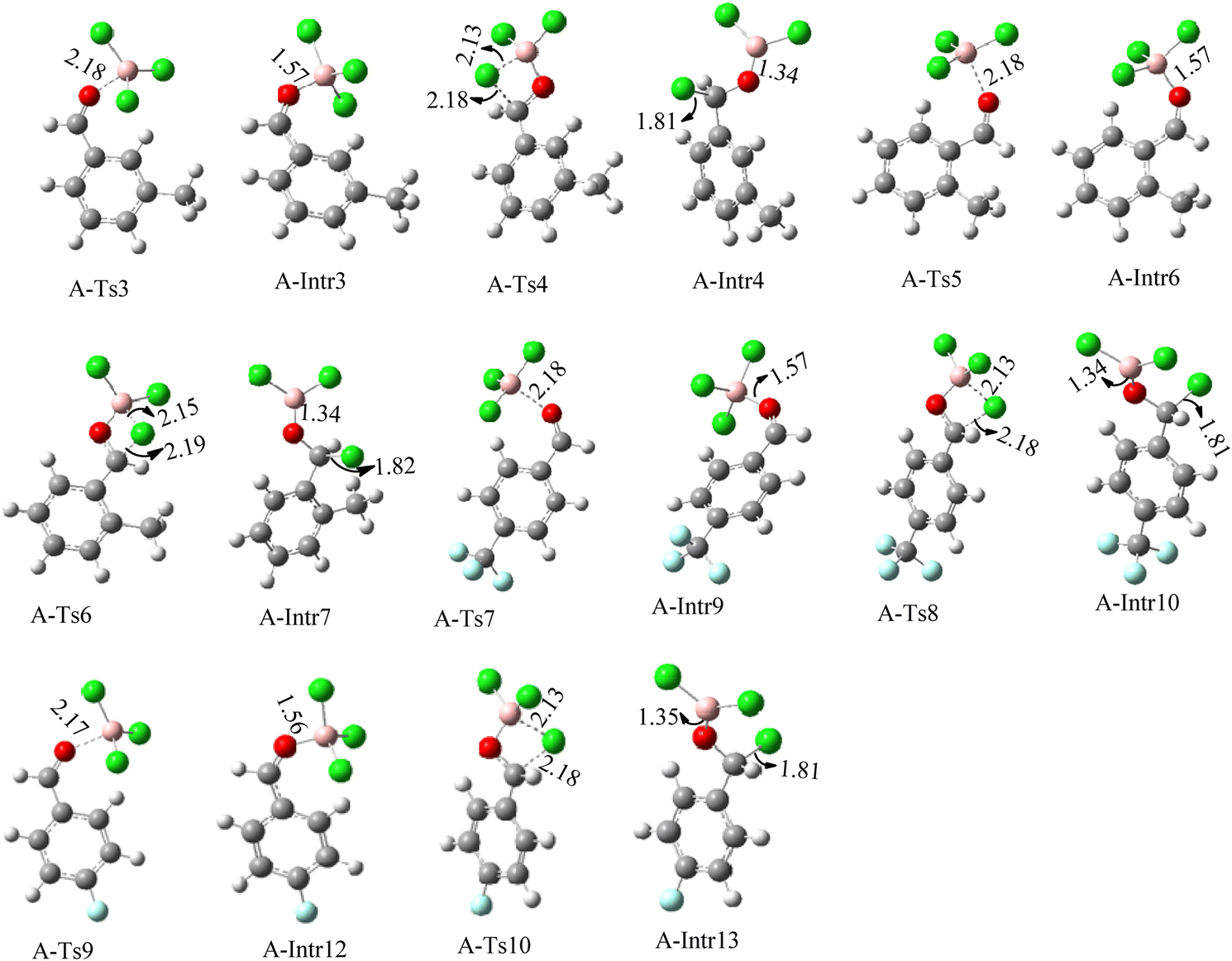 | ||
| Fig. 4 Geometric structures of intermediates and transition states in the chloroboration of substituted benzaldehyde bearing m-CH3, o-CH3, p-CF3 and p-F (distances are given in Å). | ||
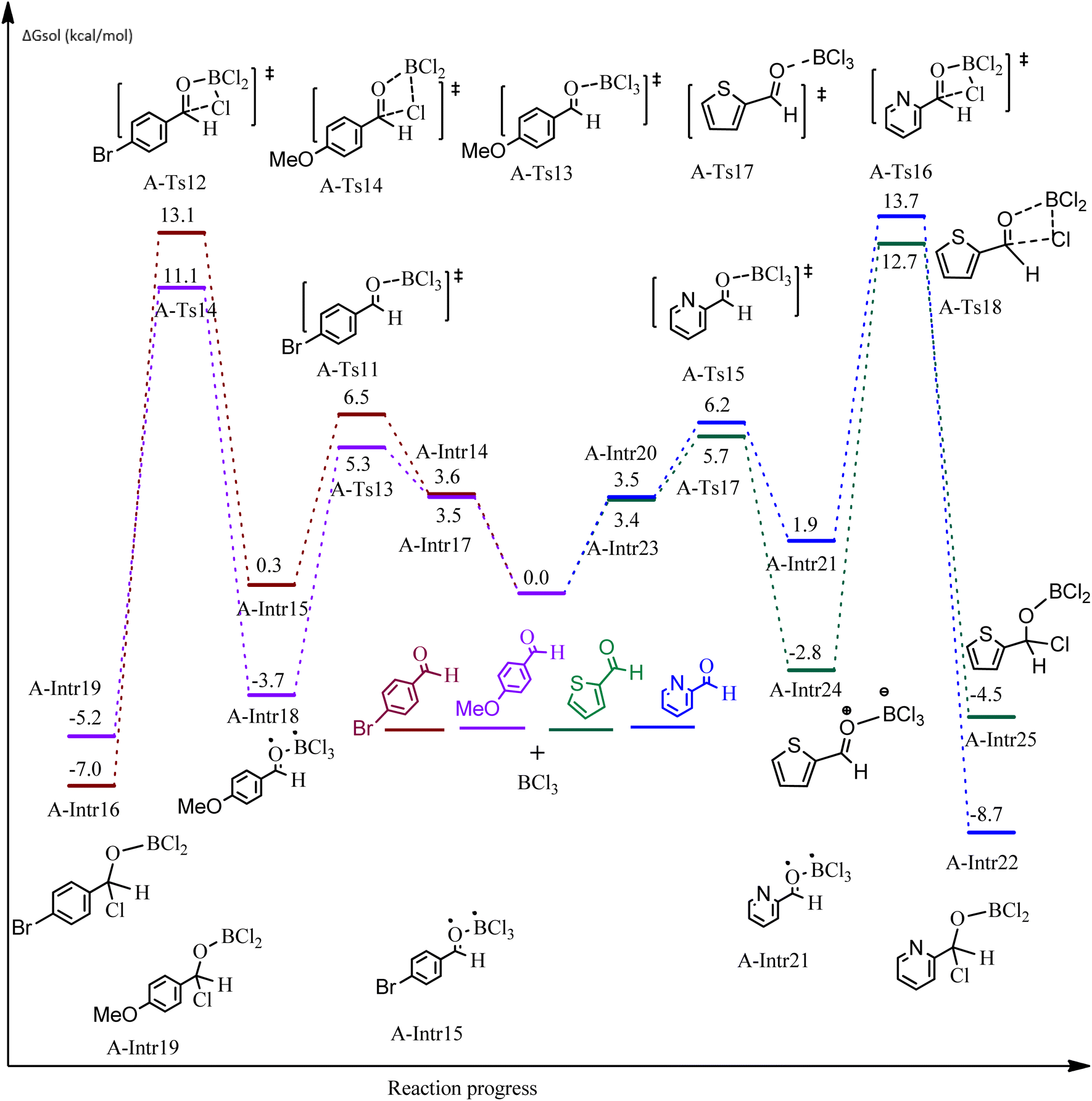 | ||
| Fig. 5 DFT-computed free energies for the chloroboration reaction of substituted benzaldehyde bearing p-Br and p-OMe, picolinbenzaldehyde and thiophen-2-carbaldehyde. | ||
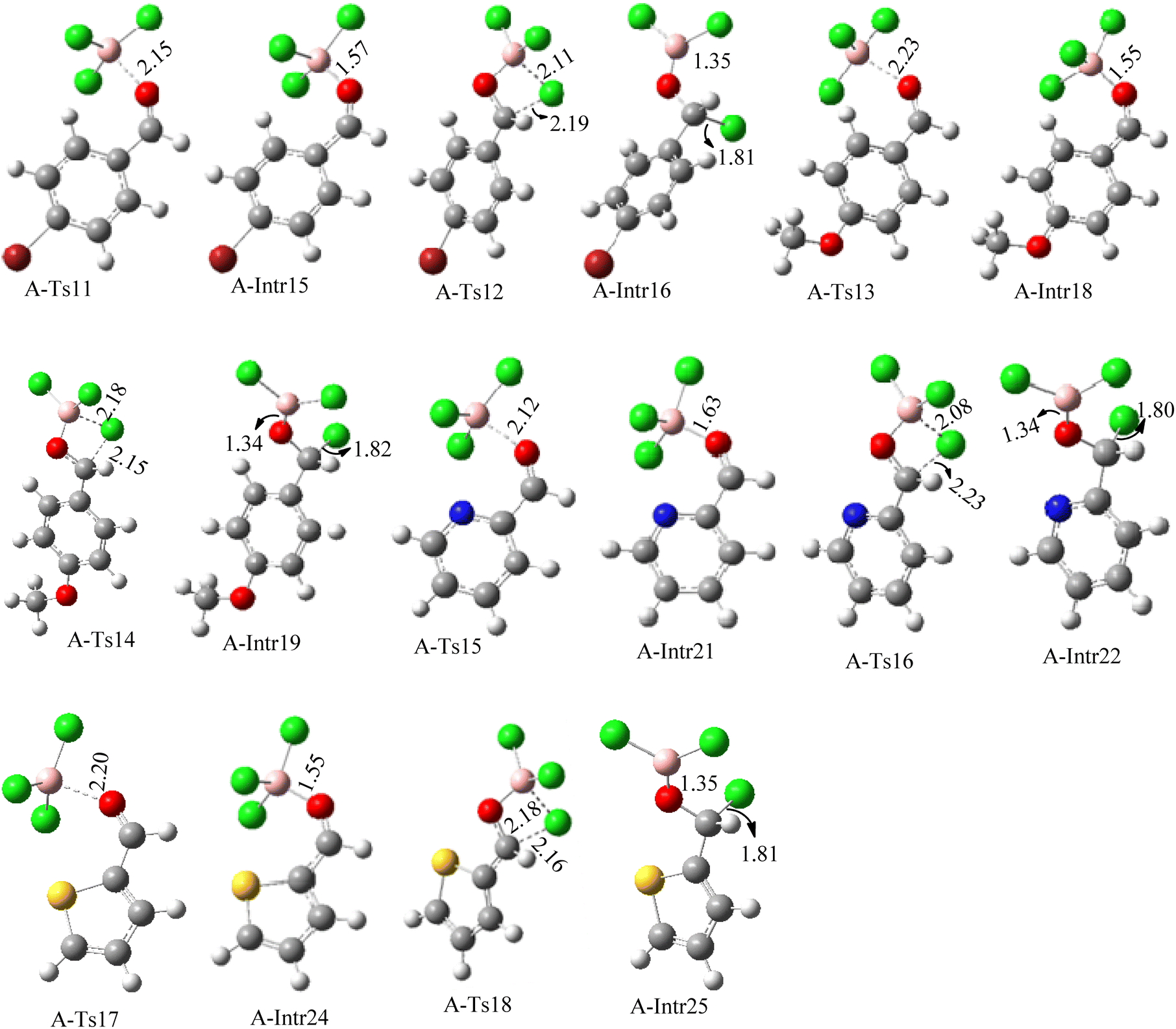 | ||
| Fig. 6 Geometric structures for intermediates and transition states in the chloroboration of substitute benzaldehyde bearing p-Br, p-OMe, picolinbenzaldehyde and thiophen-2-carbaldehyde (distances are given in Å). | ||
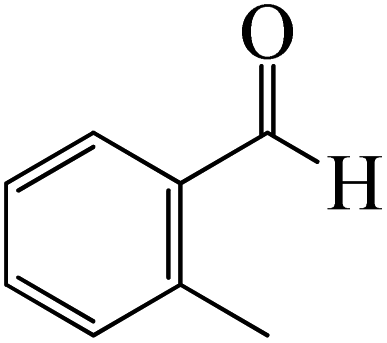
The production of the coordinated complex A-Intr2 between the CO group of the reactant (3-methylbenzaldehyde) and BCl3 is found to be 2.4 kcal mol−1 based on the calculation results given in Fig. 3. A-Intr2 is then converted via A-Ts3 into the zwitterion intermediate A-Intr3, which has a lower activation energy of 2.7 kcal mol−1. Notably, 13.3 kcal mol−1 (1.4 kcal mol−1 greater than that of the model reactant) is needed for the four-membered ring transition state A-Ts4 to form the borylated intermediate A-Intr4. The coordinated complex A-Intr5 is determined to be 3.3 kcal mol−1 when CH3 is in ortho-position (2-methylbenzaldehyde), and A-Ts5 forms A-Intr6 (requiring 2.3 kcal mol−1). A-Ts6 converts A-Intr6 into A-Intr7 at 10.3 kcal mol−1, which is 1.6 kcal mol−1 more stable than the model reactant and 3.0 kcal mol−1 from 3-methylbenzaldehyde. By increasing the electron density in the benzene ring, the CH3 group, an activating group, promotes the reaction progress.
The formation of the coordinated complex A-Intr8 between the CO group of 4-(trifluoromethyl)benzaldehyde and BCl3 is reported to be 4.0 kcal mol−1 based on the computation results displayed in Fig. 3. Next, by converting A-Intr8 via A-Ts7 (3.5 kcal mol−1), the zwitterion intermediate A-Intr9 is produced; 11.3 kcal mol−1 is required for the four-membered ring transition state A-Ts8 to produce the borylated intermediate A-Intr10. The coordinated complex A-Intr11 is determined to be 3.6 kcal mol−1 when the substituent is p-F. This is followed by the production of A-Intr12 via A-Ts9, which requires 2.2 kcal mol−1. A-Ts10 converts A-Intr12 into A-Intr13 at a cost of 12.2 kcal mol−1. This indicates that compared to 4-(fluoro)benzaldehyde, 4-(trifluoromethyl)benzaldehyde is more reactive because compared to F atoms, CF3 is a more potent electron-withdrawing group. The structure of the optimal intermediates and transition states during the chloroboration of replacement benzaldehyde is shown in Fig. 4.
ased on the computation findings shown in Fig. 5, the synthesis of the coordinated complex A-Intr14 between the CO group of the reactant (4-bromobenzaldehyde) and BCl3 is found to be 3.6 kcal mol−1. The zwitterion intermediate A-Intr15, which has lower activation energy of 2.9 kcal mol−1, is subsequently created by converting A-Intr14 via A-Ts11. The borylated intermediate A-Intr16 is obtained by converting the four-membered ring transition state A-Ts12 to 12.8 kcal mol−1. When p-OMe is the substituent, A-Intr17 is formed with 3.5 kcal mol−1 of energy, while A-Intr18 through A-Ts13 is formed with 1.8 kcal mol−1. As a result, A-Ts14 forms A-Intr19, requiring 14.8 kcal mol−1 of energy, which makes it 2.9 kcal mol−1 less stable than the model substrate.
The synthesis of the coordinated complex A-Intr20 between CO group picolinbenzaldehyde and BCl3 is found to be 3.5 kcal mol−1, based on the computation results displayed in Fig. 5. Next, using A-Ts15 with 2.7 kcal mol−1, A-Intr20 is converted into the zwitterion intermediate A-Intr21. The four-membered ring transition state A-Ts16 requires 11.8 kcal mol−1 free energy to yield the borylated intermediate A-Intr22. A-Intr23 (3.4 kcal mol−1) is produced when thiophen-2-carbaldehyde is used as the substrate in a reaction with BCl3. A-Ts17 (2.3 kcal mol−1) then converts this product into A-Intr24. A-Intr24 is converted into A-Intr25 via A-Ts18 (15.5 kcal mol−1, 3.6 kcal mol−1 less stable than the model reactant).
Following a computational study on the chloroboration of substituted benzaldehyde, we performed DFT calculations to better understand the scope of BCl3-promoted chloroboration of ketones in a catalyst-free manner when specific substituted acetophenones containing methyl, methoxy, halogens, nitro, and heterocyclic derivatives were functionalized with either electron-donating or electron-withdrawing groups Table 2. Fig. 7 and 9 show DFT-computed free energies for the chloroboration reactions of substituted acetophenone. Fig. 8 and 10 show the geometric structures for intermediates and transition states in the chloroboration of substituted acetophenone.
| Substrate | B–O bond formation | 1,3-Cl migration | Substrate | B–O bond formation | 1,3-Cl migration |
|---|---|---|---|---|---|
 |
1.8 | 13.5 |  |
2.9 | 12.6 |
 |
1.8 | 15.7 | 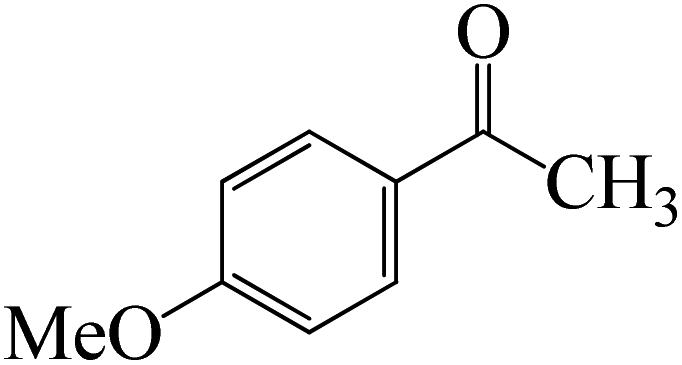 |
1.5 | 15.3 |
 |
2.6 | 13.4 | 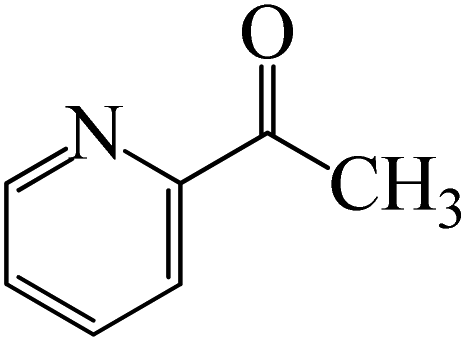 |
1.5 | 11.6 |
 |
3.3 | 12.2 |  |
1.1 | 17.0 |
 |
2.0 | 13.2 |
 | ||
| Fig. 7 DFT-computed free energies for the chloroboration reaction of substituted acetophenone bearing p-CH3, m-CH3, p-NO2 and p-Br. | ||
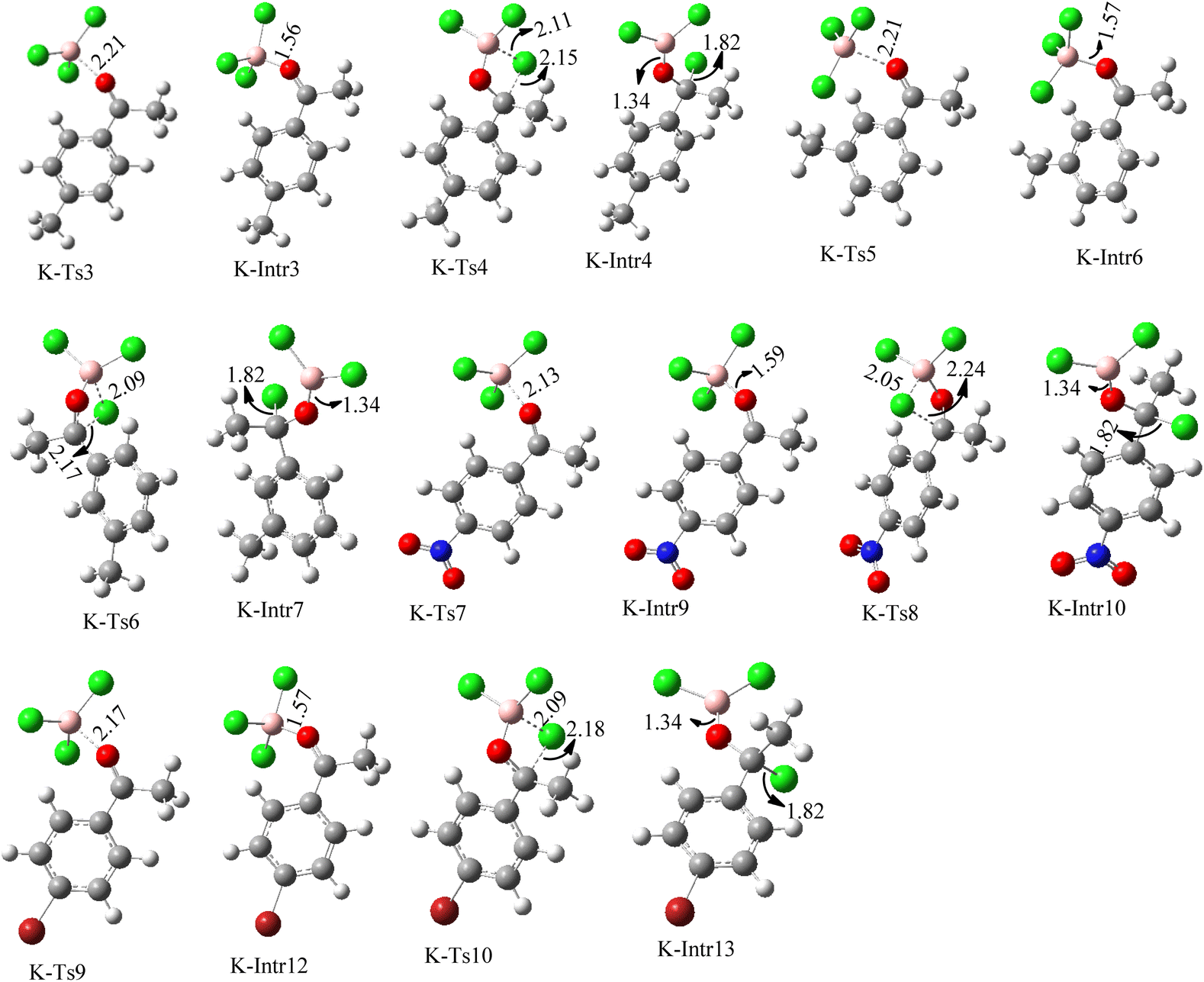 | ||
| Fig. 8 Geometric structures of intermediates and transition states in the chloroboration of substituted acetophenone bearing p-CH3, o-CH3, p-NO2 and p-Br (distances are given in Å). | ||
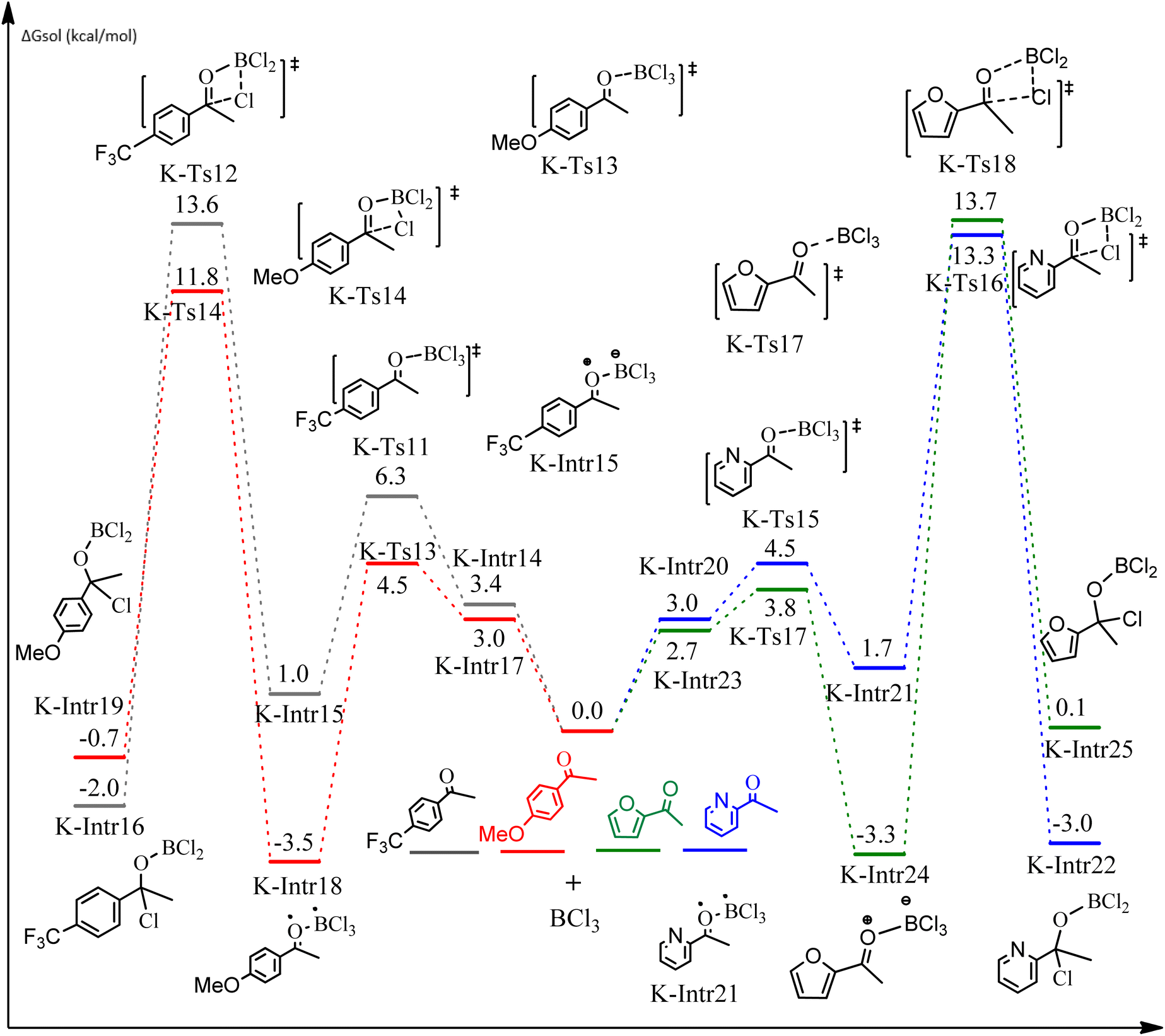 | ||
| Fig. 9 DFT-computed free energies for the chloroboration reaction of substituted acetophenone bearing p-CF3, p-OMe, 1-(pyridin-2-yl)ethanone and 1-(furan-2-yl)ethanone. | ||
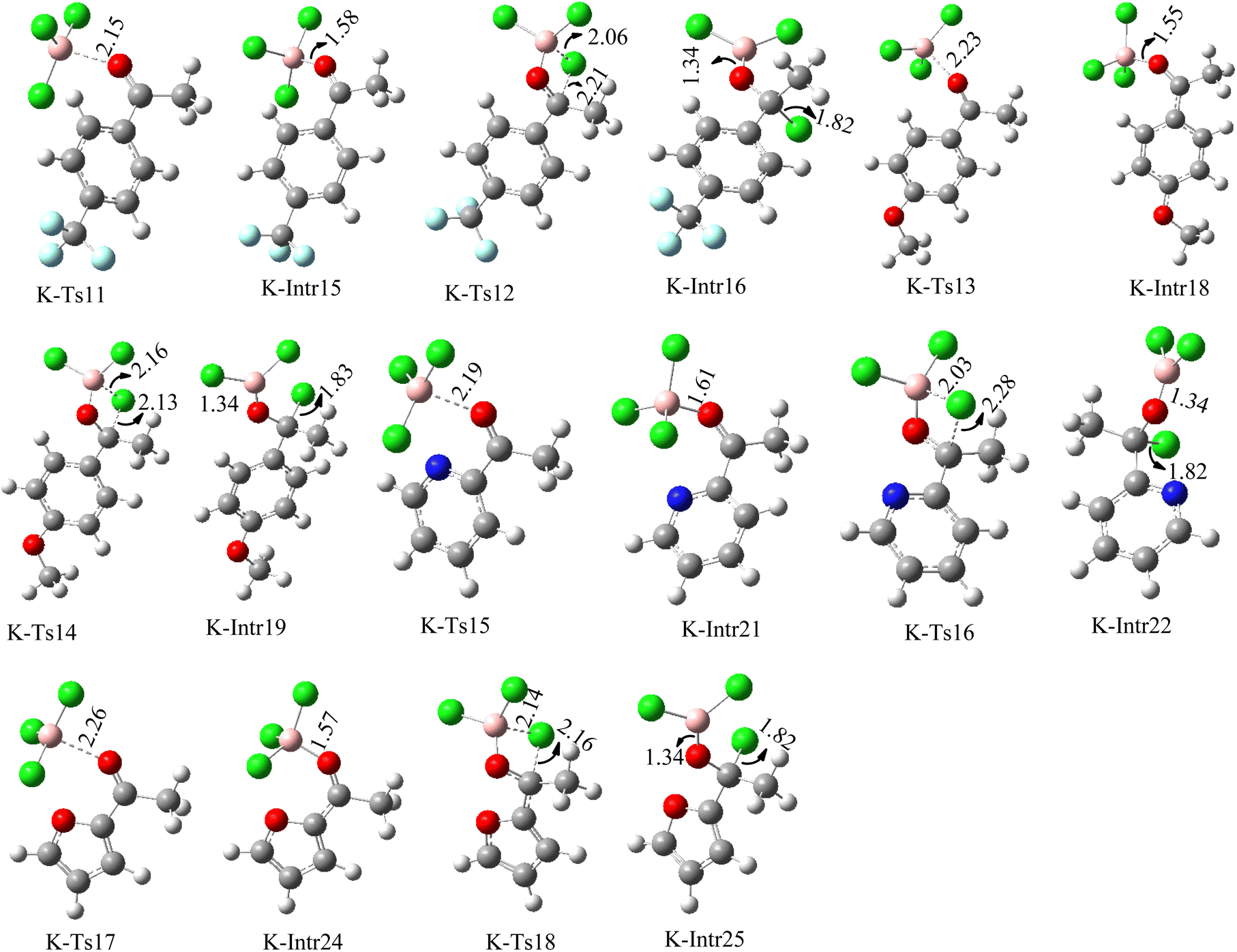 | ||
| Fig. 10 Geometric structures of intermediates and transition states in the chloroboration of substituted acetophenone bearing p-CF3, p-OMe, 1-(pyridin-2-yl)ethanone and 1-(furan-2-yl)ethanone (distances are given in Å). | ||
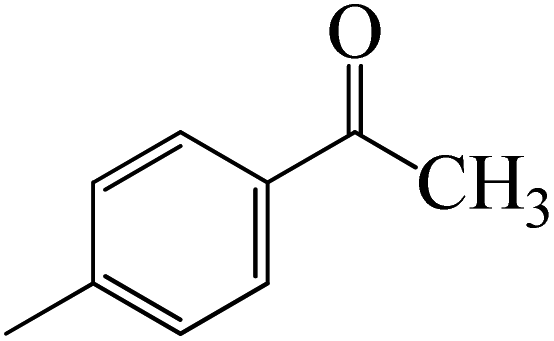
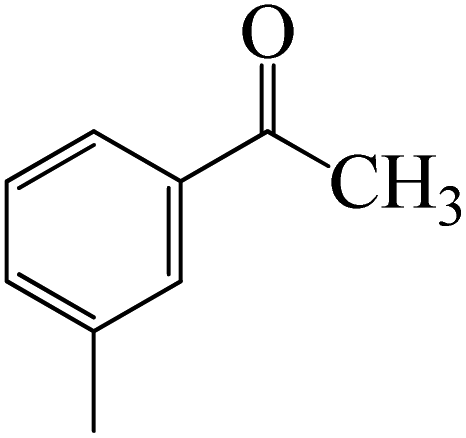
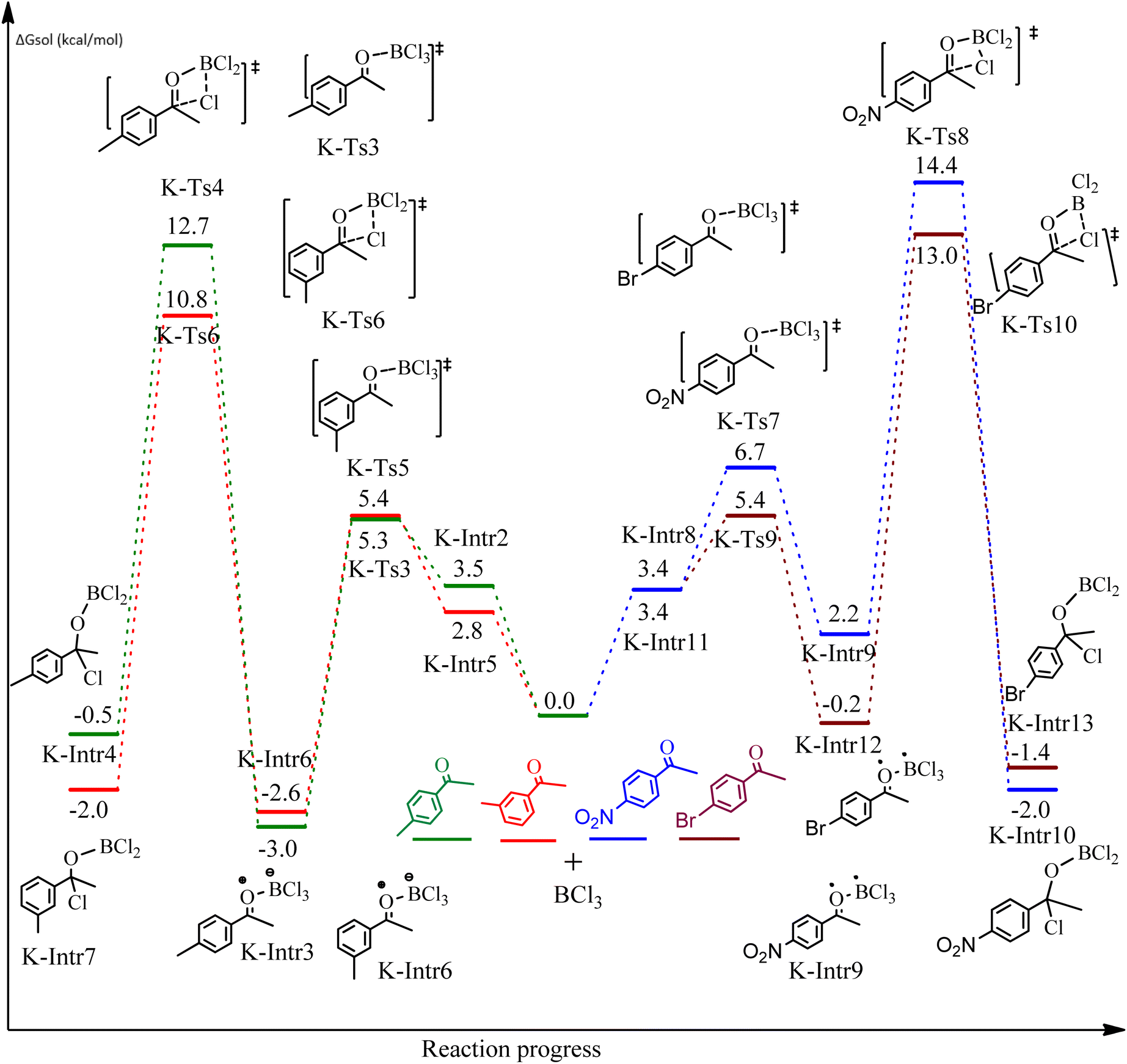
Complexation of BCl3 with 1-(p-tolyl)ethanone, which occurs kinetically at 3.5 kcal mol−1 to produce K-Intr2, is the first step. K-Intr3 is then generated through the transition state K-Ts3, which has a lower energy barrier of 1.8 kcal mol−1. After K-Intr3 (2.4 kcal mol−1) is created, K-Ts4 (15.7 kcal mol−1) facilitates the easy transfer of the chloride anion to the carbonyl carbon to generate K-Intr4. It is less stable by 2.2 kcal mol−1 compared to the model substrate. The coordinated complex A-Intr5 forms with a reduced activation energy of 2.8 kcal mol−1 when the substrate is 1-(m-tolyl)ethanone. With a lower energy barrier of 2.6 kcal mol−1, the previous transition state, K-Ts5, is more kinetically advantageous to produce K-Intr6. After K-Intr6 is produced, K-Ts6, which is 13.4 kcal mol−1, may be used to transfer the chloride anion to the carbonyl carbon with ease, yielding the desired product, K-Intr7. In comparison with K-Ts4 of 1-(p-tolyl)ethanone, K-Ts6 of 1-(m-tolyl)ethanone is stable by 2.3 kcal mol−1.
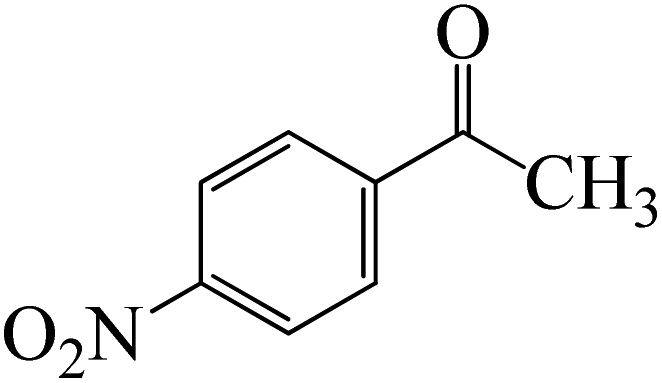
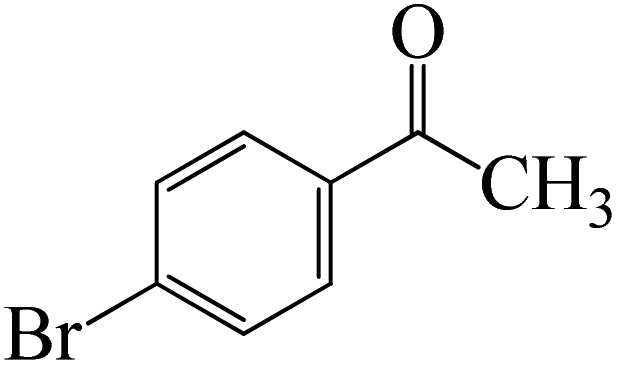
The formation of the complex intermediate K-Intr8 (3.4 kcal mol−1) occurs when 1-(4-nitrophenyl)ethanone is the substrate. K-Intr9 is generated through the transition state K-Ts7, which has a lower energy barrier of 3.3 kcal mol−1. After K-Intr9 (2.2 kcal mol−1) is created, K-Ts8, which is 12.2 kcal mol−1 (1.3 kcal mol−1 stable than the model substrate), facilitates the easy transfer of the chloride anion to the carbonyl carbon, generating K-Intr10. The coordinated complex A-Intr11 is generated with an activation energy of 3.4 kcal mol−1 when the substrate is 1-(4-bromophenyl)ethanone. With a lower energy barrier of 2.0 kcal mol−1, the previous transition state, K-Ts9, is more kinetically advantageous to produce K-Intr12. K-Ts10, which is 13.2 kcal mol−1, makes it simple to move the chloride anion to the carbonyl carbon once K-Intr12 is formed. K-Intr13 is the final product of this step.
As shown in Fig. 9, the CO moiety of the 1-(4-(trifluoromethyl)phenyl)ethanone reactant molecule combines with BCl3. K-Intr14 is kinetically advantageous since K-Ts11 has a lower energy barrier of 2.9 kcal mol−1. K-Intr16 is produced by the rapid transfer of the chloride anion to the carbonyl carbon via K-Ts12 (12.6 kcal mol−1), which occurs after K-Intr15 (1.0 kcal mol−1) is formed. When OMe is a substituent of acetophenone at para-position, 1-(4-methoxyphenyl)ethanone forms the complex K-Intr17 with BCl3 by demanding 3.0 kcal mol−1. This is followed by the synthesis of K-Intr18 through K-Ts13, which requires 1.5 kcal mol−1. Finally, using K-Ts14, K-Intro19 is generated with an activation energy of 15.3 kcal mol−1 (1.8 kcal mol−1 higher than that of the model reactant) of energy.
The CO moiety of 1-(pyridin-2-yl)ethanone reacts with BCl3 to generate K-intr20 (3.0 kcal mol−1) as the reactant. As can be observed in Fig. 9, K-Ts15, the preceding transition state, has a lower energy barrier of 1.5 kcal mol−1, making K-Intr21 kinetically favorable. When the chloride anion is shifted from K-Intr21 to the carbonyl carbon via K-Ts16 (11.6 kcal mol−1, 1.9 kcal mol−1 stable than the model reactant), K-Intr22 is formed. While 1-(furan-2-yl)ethanone serves as the substrate, K-Intr23 is created at a rate of 2.7 kcal mol−1. For K-Ts17 to convert K-Intr23 into K-Intr24, 1.1 kcal mol−1 is required. Finally, K-Ts18, which requires 17.0 kcal mol−1, transfers K-Intr24 to K-Intr24. K-Ts18 makes 1-(furan-2-yl)ethanone less stable than the model substrate by 3.5 kcal mol−1.
Conclusions
In conclusion, DFT calculations were used to carry out a mechanistic analysis of catalyst-free interactions between carbonyl compounds and BCl3. We suggest that the interaction between the CO bond and BCl3 activates the chloroboration of carbonyl compounds. The mechanism yields an overall free energy barrier of <20 kcal mol−1, which is in good agreement with the observation that reactions take place at room temperature, according to the DFT result. We reported that BCl3 facilitated catalyst-free chloroboration reactions of carbonyl compounds, supporting the universality of the finding. Therefore, our computations offer mechanistic insights into the crucial reactions involved in the synthesis of chemicals derived from organoborane derivatives, and they could be beneficial in the development and execution of novel reactions of this type.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI material.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express our gratitude to the Department of Chemistry, Debre Tabor University, for allowing us to utilize their computer facility for performing all of the DFT calculations necessary for this study.Notes and references
- S. R. Tamang, D. Bedi, S. Shafiei-Haghighi, C. R. Smith, C. Crawford and M. Findlater, Cobalt-catalyzed hydroboration of alkenes, aldehydes, and ketones, Org. Lett., 2018, 20(21), 6695–6700 CrossRef PubMed.
- Z. Zhu, X. Wu, X. Xu, Z. Wu, M. Xue, Y. Yao, Q. Shen and X. Bao, n-Butyllithium catalyzed selective hydroboration of aldehydes and ketones, J. Org. Chem., 2018, 83(17), 10677–10683 CrossRef PubMed.
- J. Wu, H. Zeng, J. Cheng, S. Zheng, J. A. Golen, D. R. Manke and G. Zhang, Cobalt (II) coordination polymer as a precatalyst for selective hydroboration of aldehydes, ketones, and imines, J. Org. Chem., 2018, 83(16), 9442–9448 CrossRef PubMed.
- M. K. Bisai, T. Das, K. Vanka and S. S. Sen, Easily accessible lithium compound catalyzed mild and facile hydroboration and cyanosilylation of aldehydes and ketones, Chem. Commun., 2018, 54(50), 6843–6846 RSC.
- U. K. Das, C. S. Higman, B. Gabidullin, J. E. Hein and R. T. Baker, Efficient and Selective Iron-Complex-Catalyzed Hydroboration of Aldehydes, ACS Catal., 2018, 8(2), 1076–1081 CrossRef.
- W. Wang, X. Shen, F. Zhao, H. Jiang, W. Yao, S. A. Pullarkat, L. Xu and M. Ma, Ytterbium-catalyzed hydroboration of aldehydes and ketones, J. Org. Chem., 2018, 83(1), 69–74 CrossRef PubMed.
- T. Ghatak, K. Makarov, N. Fridman and M. S. Eisen, Catalytic regeneration of a Th–H bond from a Th–O bond through a mild and chemoselective carbonyl hydroboration, Chem. Commun., 2018, 54(78), 11001–11004 RSC.
- D. M. Ould and R. L. Melen, Arsenic Catalysis: Hydroboration of Aldehydes Using a Benzo-Fused Diaza-benzyloxy-arsole, Chem.–Eur. J., 2018, 24(57), 15201–15204 CrossRef CAS PubMed.
- H. C. Brown and B. S. Rao, A new technique for the conversion of olefins into organoboranes and related alcohols, J. Am. Chem. Soc., 1956, 78(21), 5694–5695 CrossRef CAS.
- H. C. Brown and G. Zweifel, Hydroboration as a convenient procedure for the asymmetric synthesis of alcohols of high optical purity, J. Am. Chem. Soc., 1961, 83(2), 486–487 Search PubMed.
- H. C. Brown, N. R. Ayyangar and G. Zweifel, Hydroboration. XVIII. The Reaction of Diisopinocampheylborane with Representative cis-Acyclic, Cyclic, and Bicyclic Olefins. A Convenient Systhesis of Optically Active Alcohols and Olefins of High Optical Purity and Established Configuration, J. Am. Chem. Soc., 1964, 86(3), 397–403 CrossRef CAS.
- M. Hudlicky, Reductions in organic chemistry, 1996 Search PubMed.
- R. Noyori and S. Hashiguchi, Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes, Acc. Chem. Res., 1997, 30(2), 97–102 CrossRef CAS.
- P. Mäki-Arvela, J. Hájek, T. Salmi and D. Y. Murzin, Chemoselective hydrogenation of carbonyl compounds over heterogeneous catalysts, Appl. Catal., A, 2005, 292, 1–49 CrossRef.
- (a) L. Cervený, Catalytic hydrogenation, Elsevier, 1986 Search PubMed; (b) J. G. de Vries and C. J. Elsevier, The Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, 2007 Search PubMed.
- J.-i. Ito and H. Nishiyama, Recent topics of transfer hydrogenation, Tetrahedron Lett., 2014, 55(20), 3133–3146 CrossRef.
- D. Männig and H. Nöth, Catalytic hydroboration with rhodium complexes, Angew Chem. Int. Ed. Engl., 1985, 24(10), 878–879 CrossRef.
- A. Y. Khalimon, P. Farha, L. G. Kuzmina and G. I. Nikonov, Catalytic hydroboration by an imido-hydrido complex of Mo (IV), Chem. Commun., 2012, 48(3), 455–457 RSC.
- L. Koren-Selfridge, H. N. Londino, J. K. Vellucci, B. J. Simmons, C. P. Casey and T. B. Clark, A boron-substituted analogue of the Shvo hydrogenation catalyst: catalytic hydroboration of aldehydes, imines, and ketones, Organometallics, 2009, 28(7), 2085–2090 CrossRef.
- H.-W. Suh, L. M. Guard and N. Hazari, A mechanistic study of allene carboxylation with CO 2 resulting in the development of a Pd (ii) pincer complex for the catalytic hydroboration of CO 2, Chem. Sci., 2014, 5(10), 3859–3872 RSC.
- M. J. Sgro and D. W. Stephan, Frustrated Lewis pair inspired carbon dioxide reduction by a ruthenium tris (aminophosphine) complex, Angew. Chem., Int. Ed. Engl., 2012, 51(45), 11343–11345 CrossRef.
- S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, An efficient nickel catalyst for the reduction of carbon dioxide with a borane, J. Am. Chem. Soc., 2010, 132(26), 8872–8873 CrossRef PubMed.
- H. C. Brown and T. E. Cole, Organoboranes. 31. A simple preparation of boronic esters from organolithium reagents and selected trialkoxyboranes, Organometallics, 1983, 2(10), 1316–1319 CrossRef.
- T. Ishiyama, M. Murata and N. Miyaura, Palladium (0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: a direct procedure for arylboronic esters, J. Org. Chem., 1995, 60(23), 7508–7510 CrossRef.
- Z. Zuo, H. Wen, G. Liu and Z. Huang, Cobalt-Catalyzed Hydroboration and Borylation of Alkenes and Alkynes, Synlett, 2018, 29(11), 1421–1429 CrossRef.
- T. Ishiyama and N. Miyaura, Transition metal-catalyzed borylation of alkanes and arenes via C–H activation, J. Organomet. Chem., 2003, 680(1–2), 3–11 CrossRef.
- L. Zhu, J. Duquette and M. Zhang, An improved preparation of arylboronates: application in one-pot Suzuki biaryl synthesis, J. Org. Chem., 2003, 68(9), 3729–3732 CrossRef.
- C.-L. Sun and Z.-J. Shi, Transition-metal-free coupling reactions, Chem. Rev., 2014, 114(18), 9219–9280 CrossRef PubMed.
- A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Transition-metal-free diboration reaction by activation of diboron compounds with simple Lewis bases, Angew. Chem., Int. Ed., 2011, 50(31), 7158–7161 CrossRef PubMed.
- A. Verma, R. F. Snead, Y. Dai, C. Slebodnick, Y. Yang, H. Yu, F. Yao and W. L. Santos, Substrate-Assisted, Transition-Metal-Free Diboration of Alkynamides with Mixed Diboron: Regio-and Stereoselective Access to trans-1, 2-Vinyldiboronates, Angew. Chem., 2017, 129(18), 5193–5197 CrossRef.
- K.-s. Lee, A. R. Zhugralin and A. H. Hoveyda, Efficient C–B Bond Formation Promoted by N-Heterocyclic Carbenes: Synthesis of Tertiary and Quaternary B-Substituted Carbons through Metal-Free Catalytic Boron Conjugate Additions to Cyclic and Acyclic α, β-Unsaturated Carbonyls, J. Am. Chem. Soc., 2010, 132(36), 12766 CrossRef CAS.
- N. Miralles, R. Alam, K. J. Szabó and E. Fernández, Transition-Metal-Free Borylation of Allylic and Propargylic Alcohols, Angew. Chem., 2016, 128(13), 4375–4379 CrossRef.
- K. Yang and Q. Song, Transition-metal-free regioselective synthesis of alkylboronates from arylacetylenes and vinyl arenes, Green Chem., 2016, 18(4), 932–936 RSC.
- S. Hong, W. Zhang, M. Liu, Z.-J. Yao and W. Deng, Transition-metal-free hydroboration of terminal alkynes activated by base, Tetrahedron Lett., 2016, 57(1), 1–4 CrossRef CAS.
- Y. Wen, C. Deng, J. Xie and X. Kang, Recent synthesis developments of organoboron compounds via metal-free catalytic borylation of alkynes and alkenes, Molecules, 2018, 24(1), 101 CrossRef.
- E. Davenport and E. Fernandez, Transition-metal-free synthesis of vicinal triborated compounds and selective functionalisation of the internal C–B bond, Chem. Commun., 2018, 54(72), 10104–10107 RSC.
- M. B. Gawande, V. D. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, Solvent-free and catalysts-free chemistry: a benign pathway to sustainability, ChemSusChem, 2014, 7(1), 24–44 CrossRef CAS PubMed.
- A. Sarkar, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin, V. N. Charushin and A. Majee, A decade update on solvent and catalyst-free neat organic reactions: a step forward towards sustainability, Green Chem., 2016, 18(16), 4475–4525 RSC.
- H. Stachowiak, J. Kaźmierczak, K. Kuciński and G. Hreczycho, Catalyst-free and solvent-free hydroboration of aldehydes, Green Chem., 2018, 20(8), 1738–1742 RSC.
- W. Wang, M. Luo, W. Yao, M. Ma, S. A. Pullarkat, L. Xu and P.-H. Leung, Catalyst-free and solvent-free hydroboration of ketones, New J. Chem., 2019, 43(27), 10744–10749 RSC.
- W. Wang, M. Luo, D. Zhu, W. Yao, L. Xu and M. Ma, Green hydroboration of carboxylic acids and mechanism investigation, Org. Biomol. Chem., 2019, 17(14), 3604–3608 RSC.
- W. Liu, R. Zeng, Y. Han, Y. Wang, H. Tao, Y. Chen, F. Liu and Y. Liang, Computational and experimental investigation on the BCl 3 promoted intramolecular amination of alkenes and alkynes, Org. Biomol. Chem., 2019, 17(10), 2776–2783 RSC.
- Y. Wei, D. Liu, X. Qing and L. Xu, Mechanistic Insights on a Metal-Free Borylative Cyclization of Alkynes Using BCl3: A Theoretical Investigation, Asian J. Org. Chem., 2017, 6(11), 1575–1578 CrossRef.
- C.-H. Yang, M. Han, W. Li, N. Zhu, Z. Sun, J. Wang, Z. Yang and Y.-M. Li, Direct intramolecular aminoboration of allenes, Org. Lett., 2020, 22(13), 5090–5093 CrossRef PubMed.
- Z. Yang, C.-H. Yang, S. Chen, X. Chen, L. Zhang and H. Ren, Catalyst free annulative thioboration of unfunctionalized olefins, Chem. Commun., 2017, 53(89), 12092–12095 RSC.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian, Gaussian 09, Revision A. 1, Gaussian Inc, Wallingford (CT), 2009 Search PubMed.
- M. Frisch, G. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, and G. Petersson, Gaussian 09, Gaussian. Inc., Wallingford CT, 2009, vol. 121, pp. 150–166 Search PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, and G. Petersson, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin and VN. 50.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, and G. Petersson, Gaussian 09, EM64L-G09RevB. 01, Gaussian Inc, Wallingford CT, 2010 Search PubMed.
- G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of solvation. I. General formalism, J. Chem. Phys., 2010, 132(11), 114110 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates (Å). See DOI: https://doi.org/10.1039/d4ra06893a |
| This journal is © The Royal Society of Chemistry 2025 |