
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, anti-inflammatory evaluation, and molecular docking studies of novel quinazoline-4(3H)-one-2-carbothioamide derivatives†
Le Thanh Hang Nguyena,
Dinh Hoang Vua,
Minh Quan Pham bc,
Quoc Anh Ngo*d and
Ngoc Binh Vo*d
bc,
Quoc Anh Ngo*d and
Ngoc Binh Vo*d
aSchool of Chemistry and Life Sciences, Hanoi University of Science and Technology, 1 Dai Co Viet Street, Hanoi, Vietnam
bInstitute of Natural Products Chemistry (INPC), Vietnam Academy of Science and Technology (VAST), Hanoi, Vietnam
cGraduate University of Science and Technology (GUST), Vietnam Academy of Science and Technology (VAST), Hanoi, Vietnam
dInstitute of Chemistry, Vietnam Academy of Science and Technology, Hanoi, Vietnam. E-mail: binhvn.tq@gmail.com
First published on 28th January 2025
Abstract
In this paper, a series of novel quinazoline-4(3H)-one-2-carbothioamide derivatives (8a–p) were designed and synthesized via the Wilgerodt–Kindler reaction between 2-methylquinazoline-4-one 10 and amines using S8/DMSO as the oxidizing system. Their characteristics were confirmed by IR, NMR, HRMS spectra, and their melting point. These novel derivatives (8a–p) were evaluated for their anti-inflammatory activity by inhibiting NO production in lipopolysaccharide (LPS)-activated RAW 264.7 macrophage cells. Compounds 8d (IC50 = 2.99 μM), 8g (IC50 = 3.27 μM), and 8k (IC50 = 1.12 μM) exhibited potent inhibition of NO production compared to the standard drug dexamethasone (IC50 = 14.20 μM). Compound 8a (IC50 = 13.44 μM) exhibited NO inhibition comparable to dexamethasone. Structure–activity relationship (SAR) studies indicated that the presence of both the thioamide functional group (NH–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S) directly attached to the phenyl ring containing halogen substituents (4-Cl, 8d), (4-Br, 8g) and (4-CF3, 8k), is responsible for the potent anti-inflammatory activity of these novel quinazolinone derivatives. Computational modeling studies revealed that compounds 8d, 8g, and 8k are potent inhibitors of TLR4 signaling through the formation of hydrophobic interactions and are stabilized by hydrogen bonds. Replacing the thioamide (8k) with an amide (8q) resulted in an 83-fold decrease in NO inhibitory potency. This highlights the important role of H-bonding involving the thioamide group. The structural shape difference results in favorable interactions of quinazolinones containing thioamide linkers compared to amide linkers to the target receptor. Furthermore, the ADMET profiles and physicochemical properties of these three lead compounds were predicted to meet the criteria for drug-like properties. Therefore, these compounds may be potential candidates for the treatment of many inflammatory diseases associated with immune disorders.
S) directly attached to the phenyl ring containing halogen substituents (4-Cl, 8d), (4-Br, 8g) and (4-CF3, 8k), is responsible for the potent anti-inflammatory activity of these novel quinazolinone derivatives. Computational modeling studies revealed that compounds 8d, 8g, and 8k are potent inhibitors of TLR4 signaling through the formation of hydrophobic interactions and are stabilized by hydrogen bonds. Replacing the thioamide (8k) with an amide (8q) resulted in an 83-fold decrease in NO inhibitory potency. This highlights the important role of H-bonding involving the thioamide group. The structural shape difference results in favorable interactions of quinazolinones containing thioamide linkers compared to amide linkers to the target receptor. Furthermore, the ADMET profiles and physicochemical properties of these three lead compounds were predicted to meet the criteria for drug-like properties. Therefore, these compounds may be potential candidates for the treatment of many inflammatory diseases associated with immune disorders.
Introduction
Inflammation is a complex pathophysiological process caused by the overproduction of inflammatory mediators such as nitric oxide (NO), prostaglandin E2 (PGE2), as well as inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β).1 Overproduction of these inflammatory mediators is closely linked to several diseases such as atherosclerosis, diabetes, cancer, neuropsychiatric disorders, and various inflammatory disorders such as rheumatoid arthritis, inflammatory bowel disease, rheumatoid arthritis, and psoriasis.2–4 Among them, NO has emerged as a key mediator of inflammation, playing an important role in both acute and chronic inflammatory diseases.5,6 NO is produced by three isoforms of nitric oxide synthase (NOS) (iNOS, eNOS, and nNOS), under stimulation by proinflammatory cytokines, or bacterial lipopolysaccharide (LPS) in many cell types including macrophages.5,6 LPS is a major component of the membrane of Gram-negative bacteria, plays a role in stimulating the production of inflammatory mediators and is recognized by toll-like receptor 4 (TLR4).7 Overproduction of NO has been implicated in the progression of several inflammatory disorders in the joints, intestines, and lungs.8 Most of the current anti-inflammatory drugs, along with their therapeutic properties, are associated with some side effects at different levels such as toxicity to the liver, and gastrointestinal tract, risks for cardiovascular disease, kidney disease, and hypertension.9,10 Therefore, researchers today aim to design new-generation anti-inflammatory drugs with strong anti-inflammatory properties and low toxicity.10Quinazolinones have emerged as an important pharmacological scaffolds in organic chemistry, possessing a wide range of biological properties such as anti-inflammatory and analgesic, anticonvulsant, antibacterial, antifungal, antituberculosis, anticancer, anti-HIV, anti-leishmanicidal activity.11 Among them, the quinazolin-4(3H)-one scaffold (Fig. 1) is the most common and important of the quinazolinones12 that we selected for this study due to its remarkable anti-inflammatory activity.11,13 Proquazone (1) and fluproquazone (2) are well-known NSAIDs containing a quinazolinone scaffold. In addition, rutaecarpine (3) is a natural quinazolinone derivative from Evodia rutaecarpa with anti-inflammatory activity, highly selective for COX-2 inhibition,14 in contrast to synthetic COX-2 inhibitors such as etoricoxib and celecoxib, rutecarpine has beneficial effects on some cardiovascular diseases.15 In addition, several reports have shown that substitutions at position 2 or 3 of the quinazoline nucleus significantly affect anti-inflammatory activity.13,16–21 On the other hand, the molecular structure of quinazolin-4(3H)-one scaffold shows that they are lipophilic,13,22 which will facilitate the design of anti-inflammatory drugs.23 Sutthichat Kerdphon et al. synthesized and studied the molecular docking of quinazolinones with an aliphatic substitution at the C-2 position, which showed good to excellent inhibition of inflammatory gene expression, including COX-2, iNOS, and IL-1β mRNA through inhibition of nuclear factor κB (NF-κB). Molecular docking studies showed that the quinazolin-4(3H)-ones binding to the receptor was facilitated by hydrogen bonding, hydrophobic, and electrostatic interactions as well as π–π and amide–π interactions.21
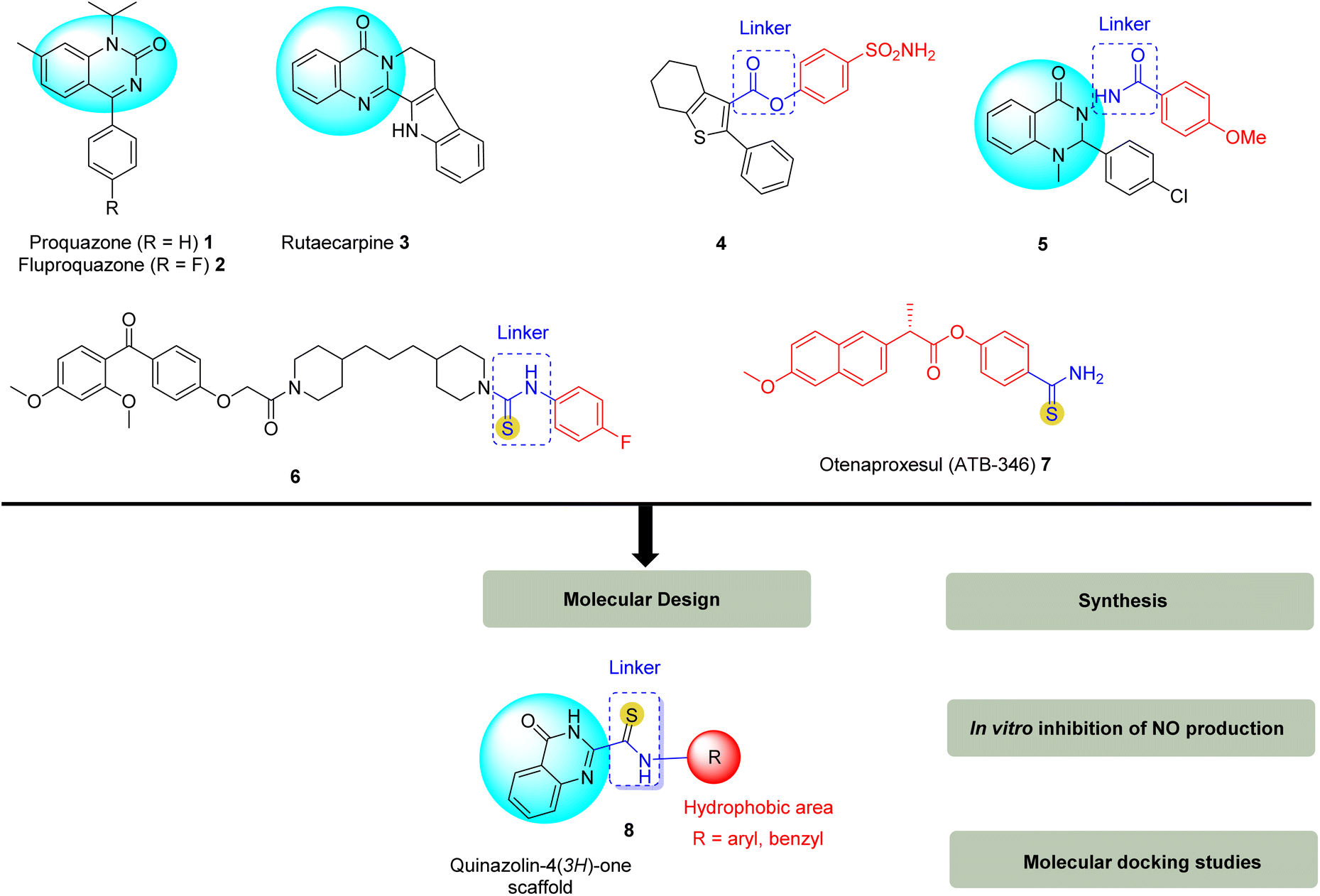 | ||
| Fig. 1 Structure of the proquazone 1, fluproquazone 2, rutaecarpine 3, some potent anti-inflammatory agents based on the ester linker 4, amide 5, thioamide 6, 7, and the designed molecule 8. | ||
Recent studies have shown that the introduction of linkers such as ester (compound 4, Fig. 1) or amide (compound 5, Fig. 1) between the aromatic/heterocyclic ring and the central heterocycle leads to improved hydrogen bond formation with the enzyme active site, resulting in compounds with potent anti-inflammatory activity and high selectivity for COX-2 inhibition.19,24–26 Kambappa Vinaya et al.27 synthesized a (4-hydroxyphenyl)(2,4-dimethoxyphenyl) methanone derivative 6 with a thioamide linker to a 4-fluorophenyl aromatic ring that exhibited potent anti-inflammatory activity at 30 mg kg−1 p.o. than the standard drug diclofenac sodium. Structural studies indicated that both the functional linkage (–CS–NH–) and the electron-withdrawing groups attached to the phenyl ring were responsible for the potent anti-inflammatory activity of the synthesized derivatives.
The thioamide functional group is a bioisostere of the amide functional group, which is believed to enhance the chemical stability and biological activity of amide-functionalized drug molecules.28 Thioamides are thought to be better hydrogen bond donors than amides.29 Isosteric substitution of an amide donor with a thioamide can increase ligand affinity due to enhanced n → π* electron transfer, as well as impart conformational stability to the thioamide.30 Furthermore, the substitution of amide with thioamide is beneficial for permeation, as well as stability under physiological conditions.31,32 In addition, compounds containing the thioamide function have attracted great attention in medicinal chemistry due to their broad spectrum of biological activity,33–35 useful synthetic substrates in the synthesis of heterocyclic rings,36 suitable stability, and low toxicity.34 A recent review of thioamides as small molecule therapeutic agents in medicinal chemistry showed that thioamides exhibit pharmacological activities and pharmacokinetic properties superior to their isosteres.35 In the field of anti-inflammatory drug design, combining NSAID pharmacological scaffolds with thioamide groups that act as H2S-releasing agents is an effective strategy to develop anti-inflammatory drugs with dual effects, i.e., increasing the efficacy of the parent compound while reducing its side effects.37–39 Among them, otenaproxesul (ATB-346) (compound 7, Fig. 1) is a new orally administered NSAID that releases hydrogen sulfide (H2S) into the gastrointestinal tract and is being developed by Antibe Therapeutics.40,41 Therefore, many new synthetic methods have been developed to access thioamide compounds.42,43 Recently, we reported a useful method for the synthesis of thioamides through the use of the S8/DMSO system that promotes the direct oxidative coupling of active methylhetarene with amines under mild conditions, with low sulfur content.44
Based on these findings, we selected quinazolin-4(3H)-one as the pharmacological scaffold for anti-inflammatory activity, the thioamide functional group attached with hydrophobic moieties such as aryl/benzyl was introduced at the C-2 position of the quinazolinone scaffold to enhance the binding affinity to the biological target. Different substituents on the phenyl ring were also introduced to study the structure–activity relationship (SAR). The novel quinazoline-4(3H)-one-2-carbothioamide derivatives were evaluated for anti-inflammatory activity through NO inhibition. Furthermore, in silico studies on physicochemical properties and ADMET of the compounds were also carried out to evaluate their potential in drug development.
Results and discussion
Chemistry
Our recent work has revealed a useful method to approach thioamide derivatives by promoting the direct oxidative coupling reaction of active methylhetarene with amine using S8/DMSO under mild temperatures.44 Following these results, we selected the molecule 2-methylquinazolin-4(3H)-one 10 as the methylhetarene moiety to apply this reaction. Firstly, compound 10 was synthesized based on a simple method previously reported by Sun et al.,45 in which 2-aminobenzamide 9 was refluxed in excess ethanol for 48 h in the presence of TBHP as an oxidant after removal of the solvent under reduced pressure and the crude product was crystallized in ethyl acetate to afford compound 10 in 65% yield (Scheme 1).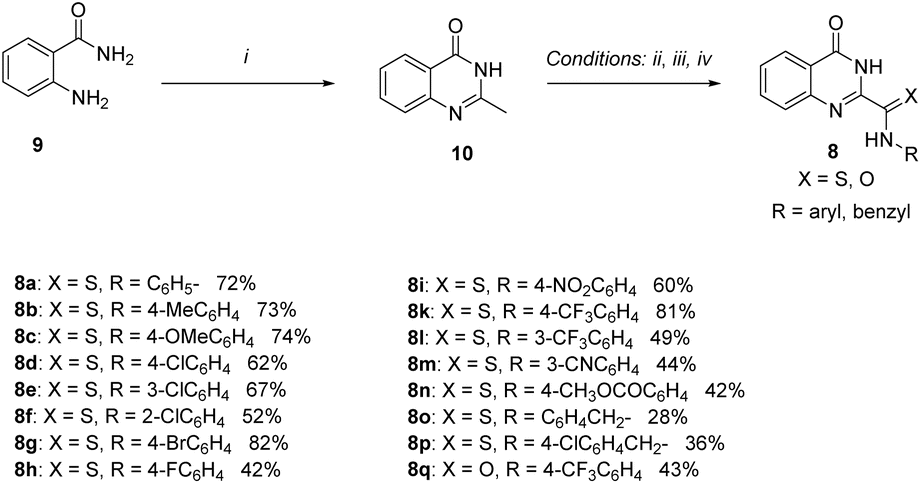 | ||
| Scheme 1 Synthesis of novel quinazoline-4(3H)-one-2-carbothioamide derivatives (8a–p) and quinazoline-4(3H)-one-2-carboxamide 8q. Reagents and conditions: (i) EtOH, tert-butyl hydroperoxide (TBHP), reflux, 48 h; (ii) for 8a–n: arylamine (11a–n), S8, DMSO, 110 °C, 16 h; (iii) for 8o, 8p: benzylamines (11o, 11p), S8, DMSO, CH3COOH, 100 °C, 16 h; (iv) for 8q: 4-(trifluoromethyl)aniline (11k), S8, DMSO, FeCl2·4H2O (5 mol%), 120 °C, 16 h. | ||
In the IR spectrum of compound 10, signals of important functional groups on the quinazolin-4(3H)-one nucleus were observed at 3446 cm−1 (N–H, amide) and 1672 cm−1 (CO, amide). In the 1H NMR spectrum of 10, two singlet signals at 12.17 ppm and 2.35 ppm were attributed to the N–H proton of the amide group and the proton of the methyl group (–CH3). In summary, the IR and NMR data of compound 10 were consistent with the published data of this compound.46,47 In the second step (Scheme 1), new quinazoline-4(3H)-one derivatives (8a–p) were synthesized via the Wilgerodt–Kindler reaction of compound 10 with arylamines 11a–n, using S8/DMSO as the oxidizing agent, at 110 °C for 16 h (ii) or with benzylamines 11o, 11p in the presence of S8/DMSO and CH3COOH, at 100 °C, 16 h (iii). Fifteen new quinazoline-4(3H)-one-2-carbothioamide derivatives (8a–p) containing the thioamide group were synthesized in yields ranging from 28% to 82%. Among them, the reaction of 10 with arylamine reagents 11a–n (from 42%–82%) gave higher yields than with benzylamines 11o, 11p (from 28–36%). Amide compound 8q was also synthesized via the reaction of 10 and arylamine 11k using S8/DMSO system as an oxidizing agent and catalyzed by FeCl2·4H2O (5 mol%), at 120 °C for 16 h.48 Amide 8q was obtained in a moderate yield of 43%. The structures of all the synthesized compounds were characterized by melting point, IR, NMR, and LC-MS spectra.
In the IR spectrum of the new quinazolin-4(3H)-ones 8a–p, in addition to the characteristic stretching bands for the amide functional group on the quinazolin-4(3H)-one nucleus such as N–H at 3481–3359 cm−1 and CO at 1709–1603 cm−1, additional stretching bands for the thioamide functional group appeared, namely N–H at 3278–3190 cm−1 and CS at 1110–1023 cm−1. In the 1H NMR spectrum of quinazolin-4(3H)-ones 8a–p, the singlet signal at 12.70–10.83 ppm was assigned to the N–H proton (amide group) on the quinazolinone nucleus, the new singlet signal appeared at 12.06–10.08 ppm demonstrating the presence of the N–H proton (thioamide functional group). The 1H NMR spectra of compounds 8o and 8p with benzylamine reagent clearly showed proton signals –CH2– at 5.00 ppm (d, J = 5.8 Hz, 2H) for 8o and 4.98 ppm (d, J = 5.9 Hz, 2H) for 8p. In the 13C NMR spectra of quinazolin-4(3H)-ones 8a–p, characteristic peaks of carbon CS (thioamide group) at 186–178 ppm and carbon CO (amide group) at 161.16–160.4 ppm were observed. Additionally, the presence of carbon signal of a methylene group (–CH2–) in compounds 8o and 8p could be observed at 50.38 ppm and 52.29 ppm, respectively. For compound 8q, the IR spectrum showed peaks at 3332 and 3188 cm−1 assigned to the N–H protons of the amide groups and 1669 cm−1 characteristic of the stretching vibration peak of the amide CO bond. In the 13C NMR spectrum of 8q, the carbon chemical shift (CO, amide) was found in the upfield region (at 161.43 and 159.14 ppm) relative to the resonance of carbon (CS, thioamide) at 180.46 ppm in 8k.29 Finally, the stereochemical configuration of the synthesized compounds 8a–p was examined through the NOESY spectrum in the CDCl3 solvent of compound 8k. In the NOESY spectrum of compound 8k, only one cross peak of the interaction of the N–H (amide) proton with H-2′ and H-6′ could be observed. Furthermore, the morphology and crystal structure of the analogous compound49 indicated that the stereochemical configuration of compounds 8a–p was the (Z) configuration. TOF MS ESI+ spectrum shows characteristic [M + H]+ peak corresponding to the molecular weight of the synthesized compound (see in ESI†).
Anti-inflammatory activity
All synthesized quinazolinone compounds 8a–q were tested for their anti-inflammatory activity by inhibiting nitric oxide (NO) production on mouse macrophages RAW 264.7, activated by lipopolysaccharide (LPS). Dexamethasone was used as the reference standard drug for our screening studies. The results of the in vitro NO production inhibition assay are presented in Table 1.| Entry | Compounds | X | R | IC50 (μM) ± SDa |
|---|---|---|---|---|
| a Data are expressed as mean IC50 ± SD (μM), n = 3, using TableCurve 2Dv4 software for data calculation. | ||||
| 1 | 8a | S | Ph | 13.44 ± 0.70 |
| 2 | 8b | S | 4-MeC6H4 | 72.89 ± 3.50 |
| 3 | 8c | S | 4-OMeC6H4 | 87.35 ± 3.07 |
| 4 | 8d | S | 4-ClC6H4 | 2.99 ± 0.19 |
| 5 | 8e | S | 3-ClC6H4 | 96.09 ± 4.93 |
| 6 | 8f | S | 2-ClC6H4 | >160.96 |
| 7 | 8g | S | 4-BrC6H4 | 3.27 ± 0.13 |
| 8 | 8h | S | 4-FC6H4 | >100 |
| 9 | 8i | S | 4-NO2C6H4 | 88.82 ± 2.56 |
| 10 | 8k | S | 4-CF3C6H4 | 1.12 ± 0.11 |
| 11 | 8l | S | 3-CF3C6H4 | >100 |
| 12 | 8m | S | 3-CNC6H4 | >100 |
| 13 | 8n | S | 4-CH3OCOC6H4 | >100 |
| 14 | 8o | S | C6H4CH2− | >100 |
| 15 | 8p | S | 4-ClC6H4CH2− | >100 |
| 16 | 8q | O | 4-CF3C6H4 | 93.32 ± 5.46 |
| 17 | Dexamethasone | 14.20 ± 0.54 | ||
The IC50 value data of the compounds in Table 1 showed that the compounds 8d, 8g, and 8k exhibited potent anti-inflammatory activity compared to the positive control drug dexamethasone (IC50 = 14.20 μM) with IC50 values of 2.99, 3.27, and 1.12 μM, respectively. Compound 8a (IC50 = 13.44 μM) exhibited anti-inflammatory activity comparable to dexamethasone. Compounds 8b, 8c, 8e, 8i, and 8q showed weak inhibitory activity on the production of inflammatory mediator NO with IC50 in the range of 72.89–96.09 μM. The remaining compounds including 8f, 8h, 8l, 8m, 8n, 8o, and 8p did not show anti-inflammatory activity.
Structure–activity relationship (SAR)
To study SAR, we introduced various substituents including both electron-withdrawing and electron-donating groups attached to the phenyl ring as substitutes linked to the thioamide group (Fig. 2). First, the simultaneous presence of both the thioamide linker and the unsubstituted phenyl ring in 8a showed good inhibition of NO production, comparable to the reference drug. The introduction of electron-withdrawing groups containing halogen elements such as –Cl (8d), –Br (8g), and –CF3 (8k) at the para-position on the phenyl ring further enhanced the in vitro anti-inflammatory activity of these derivatives, except for the case of the 4-F substituent (8h) which did not show activity. However, moving the –Cl substituent from the para- (8d) to the meta- (8e), ortho- (8f), or –CF3 to the meta- (8l) position results in decreased or loss of activity. In general, the introduction of other electron-withdrawing substituents into the phenyl ring such as (4-NO2, 8i), (3-CN, 8m), and (4-CH3OCO, 8n) all showed low or no NO inhibitory activity.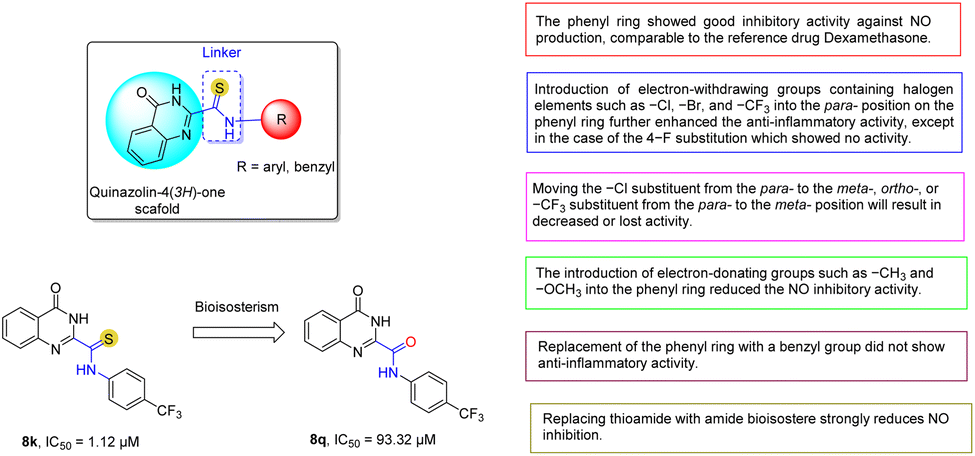 | ||
| Fig. 2 Summary of SAR studies of novel quinazoline-4(3H)-one-2-carbothioamide derivatives. | ||
The introduction of electron-donating groups such as –CH3 (8b) and –OCH3 (8c) into the phenyl ring reduced the NO inhibitory activity. Finally, the two designed compounds 8o and 8p containing the benzyl group did not show anti-inflammatory activity. This suggests that the conjugation of the thioamide linker directly attached to the aromatic ring influences the anti-inflammatory activity. In summary, the presence of both the thioamide group (NH–CS) and electron-withdrawing halogen substituents (–CF3, –Cl, –Br) at the para-position on the phenyl ring is thought to increase the lipophilicity of the molecule,10,26,50,51 which would enhance cell permeability and thereby enhance the NO production inhibitory activity of these compounds. In addition, we also attempted to explore the isosteric substitution strategy based on the 8k compound with the best NO inhibitory activity. The results showed that the amide isostere 8q had a strongly reduced potency.
Molecular docking studies
Statistical results suggest that predicting various molecular properties early in the discovery and development process is a crucial step.52 Thus, all ligands were assessed for Lipinski's Rule of Five parameters and drug-likeness properties using admetSAR1 and ProTox-III cheminformatic online web servers.53 Obtained results are presented in Table 2.| CP IDs | Dock score (kcal mol−1) | HBA (≤10) | HBD (≤5) | Log P (−4.0–5) | TPSAa (0–150) Å | HIAb (%) | LD50 (mg kg−1) | Toxic classc |
|---|---|---|---|---|---|---|---|---|
| a TPSA: molecular total polar surface area.b HIA: human intestinal absorption.c Toxicity prediction class: 1 → 6 (high toxicity to non-toxic). | ||||||||
| 8a | −7.06 | 4 | 2 | 2.899 | 83.66 | 88.67 | 200 | 3 |
| 8b | −6.72 | 4 | 2 | 3.208 | 88.40 | 93.25 | 400 | 4 |
| 8c | −6.93 | 5 | 2 | 2.908 | 90.21 | 88.67 | 200 | 3 |
| 8d | −7.71 | 4 | 2 | 3.552 | 88.67 | 89.98 | 250 | 3 |
| 8e | −6.58 | 4 | 2 | 3.552 | 88.67 | 89.98 | 580 | 4 |
| 8f | −6.02 | 4 | 2 | 3.552 | 88.67 | 89.27 | 470 | 4 |
| 8g | −7.65 | 4 | 2 | 3.661 | 91.36 | 86.26 | 200 | 3 |
| 8h | −6.51 | 4 | 2 | 3.038 | 83.62 | 90.03 | 390 | 4 |
| 8i | −7.25 | 6 | 2 | 2.807 | 90.32 | 87.25 | 200 | 3 |
| 8k | −7.84 | 4 | 2 | 3.918 | 88.66 | 96.72 | 425 | 4 |
| 8l | −6.09 | 4 | 2 | 3.918 | 88.66 | 96.72 | 2000 | 4 |
| 8m | −6.15 | 5 | 2 | 2.771 | 88.38 | 87.39 | 400 | 4 |
| 8n | −5.87 | 6 | 2 | 2.686 | 95.00 | 78.24 | 1600 | 4 |
| 8o | −6.38 | 4 | 2 | 2.577 | 86.88 | 92.49 | 900 | 4 |
| 8p | −6.14 | 4 | 2 | 3.230 | 91.89 | 93.41 | 500 | 4 |
| 8q | −7.32 | 5 | 2 | 3.117 | 81.07 | 92.69 | 200 | 3 |
| Dexamethasone | −7.10 | 5 | 3 | 1.895 | 99.94 | 99.20 | 3000 | 5 |
The results of Lipinski's Rule of Five assessment show that all studied compounds are favorable for oral drug development, each having fewer than two condition violations. Additionally, pharmacokinetic parameters and toxicity prediction results, combined with docking studies, provide valuable information for evaluating potential compounds with inhibitory abilities and drug-like properties for further development. The calculated properties showed interesting results on the toxicity scale. From Table 2, five compounds including 8a, 8c, 8d, 8g, 8i, and 8q were classified as low-toxic (rank 3). The other ten candidates were positioned at rank 4 and considered safe, including compounds 8b, 8e, 8f, 8h, 8k, 8l, 8m, 8n, 8o, and 8p. Moreover, it was reported that compounds with good oral bioavailability typically have a total polar surface area (TPSA) ranging from 70 to 140 Å2, human intestinal absorption (HIA) values above 50%, and contain 12 or fewer hydrogen bond donors (HBD) and acceptors (HBA).54 In this study, all compounds were observed to have TPSA values within the range 70–140 Å2, and the HIA percentage ranged from 78.24 to 96.72%. From physicochemical properties, these compounds are likely to be membrane permeable and easily absorbed in the human body based on satisfaction of the criteria of drug-like properties thus, demonstrating suitability in oral drug development.
AutoDock4 is among the most popular docking software, with over 6000 citations since 2010.55–57 It is a valuable tool for rapidly predicting the binding affinity of ligands to specific protein or enzyme targets. In this scenario, AutoDock4 has been utilized in searching for potential mechanisms of the bioactive compounds. The docking scores are shown in Table 2. Using Cheng–Prusoff's formula, the Ki inhibition constant is calculated as follows:
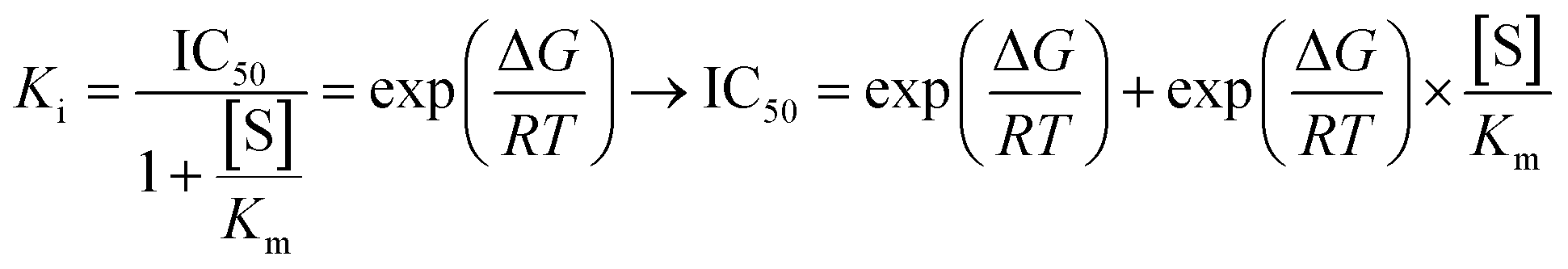 | (1) |
Assuming the IC50 value equals Ki, the experimental binding free energy could be derived from the aforementioned formula as follows: ΔGexp = RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(Ki) = RTln(IC50) where R = 1.987 × 10−3 (kcal K−1 mol−1); T = 300 (K) and inhibition constant Ki is measured in moles. Energy is measured in kilocalories per mole. Taking into account compounds that are assumed to be bioactive (IC50 less than 100 μM), the plotting of experimental binding free energies against the computed values gave a relatively high correlation coefficient of R2 = 0.79 (Fig. 3).
ln(Ki) = RTln(IC50) where R = 1.987 × 10−3 (kcal K−1 mol−1); T = 300 (K) and inhibition constant Ki is measured in moles. Energy is measured in kilocalories per mole. Taking into account compounds that are assumed to be bioactive (IC50 less than 100 μM), the plotting of experimental binding free energies against the computed values gave a relatively high correlation coefficient of R2 = 0.79 (Fig. 3).
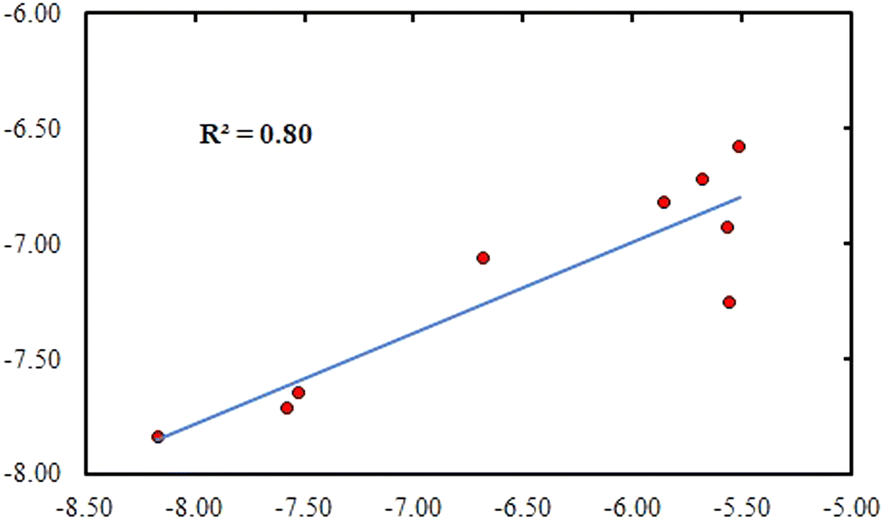 | ||
| Fig. 3 The correlation between experimental and computational binding free energies. | ||
According to the ranking criteria of Autodock4, the more negative docking energy suggests the higher binding affinity of the compound towards the targeted receptor.58 The docking score of dexamethasone was −7.10 kcal mol−1, thus, any ligand whose docking energy is close to this value or more negative would be considered a potential inhibitor. Obtained results indicate that 3 out of 15 studied compounds could be assumed as “HITs” based on their dock score, including 8d, 8g, and 8k. The stereoview of the binding mode of three HIT ligands is depicted in Fig. 4.
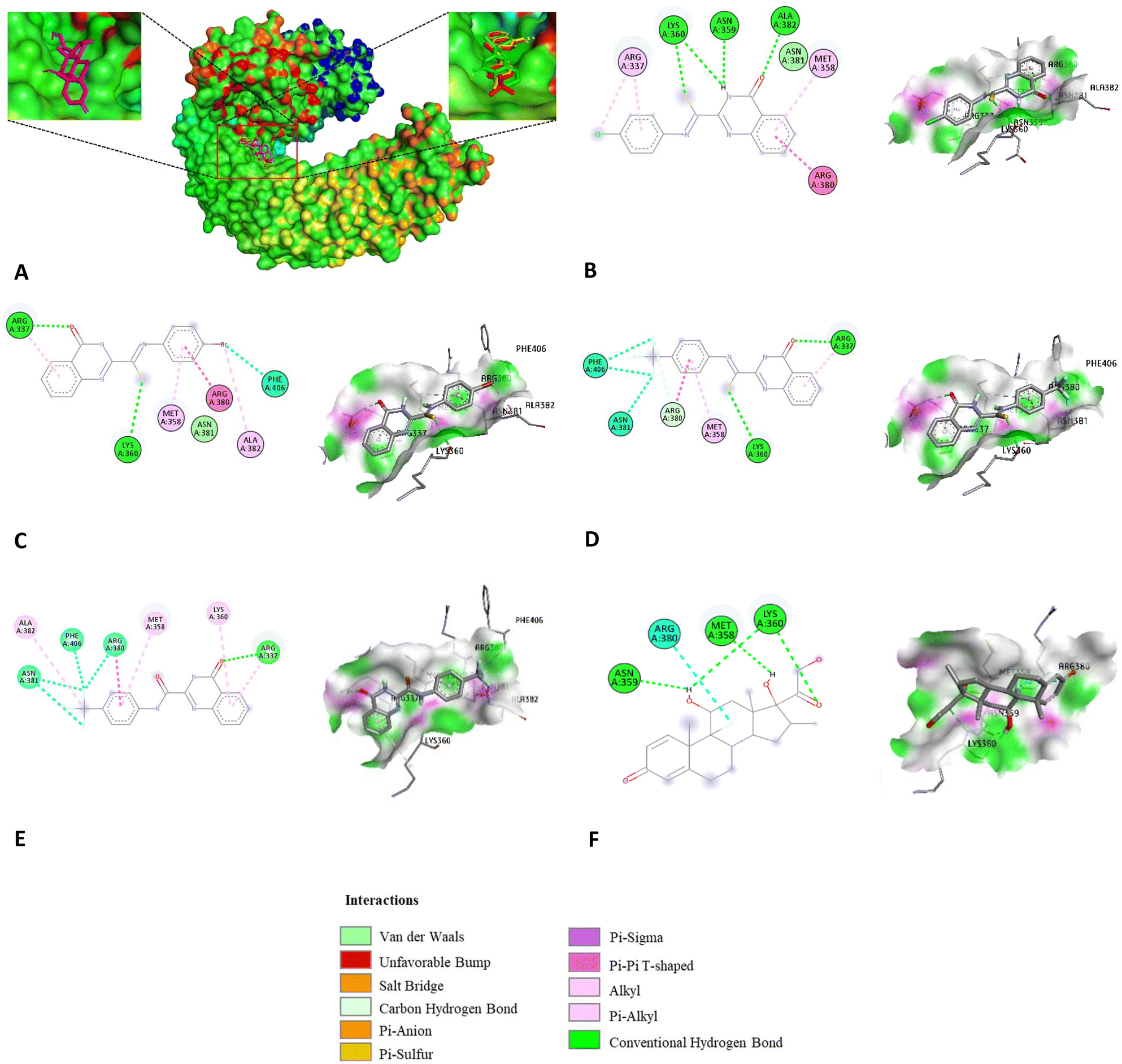 | ||
| Fig. 4 Binding conformation of studied compounds revealed by molecular docking simulations. (A) HIT compounds bind to a smooth region in TLR4, adjacent to its contact interface with MD-2: dexamethasone-magenta color, 8d-green color, 8g-yellow color, 8k-red color; (B) dock pose of 8d; (C) dock pose of 8g; (D) dock pose of 8k; (E) dock pose of 8q; (F) dock pose of dexamethasone. | ||
Previously, several studies have reported potential inhibitors of TLR4 signaling using computational tools. Thus, in this research, the molecular docking simulation was utilized to identify possible binding sites on TLR4 for potential compounds.59–61 It is observed that dexamethasone and three HIT compounds did not bind to the hydrophobic pocket in MD-2, where LPS bridges TLR4 and MD-2 to initiate signal transduction. It was bound to a cleft mainly constructed by TLR4. Fig. 3A showed that residues Arg337, Gln339, Met358, Lys360 and Arg380 from TLR4, along with His96 from MD-2, formed a smooth surface. The twisted structures of 8d, 8g, and 8k fit this shape perfectly and were closely attached to the surface.
The binding conformation of dexamethasone revealed that Met358, Asn359, and Lys360 formed three H-bonds with this ligand, the interaction was further stabilized through hydrophobic binding with Arg380. Of all the docked results, compound 8k exhibited the highest binding affinity (−7.84 kcal mol−1). Binding orientation analysis of 8k shows that Met358, Arg380, Asn381, and Phe406 were the key residues that participated in hydrophobic interaction. The interaction was further stabilized through H-bonds with Arg337 and Lys360. In the docked pose of 8d, the hydrophobic pockets formed with this ligand involving residues Arg337, Met358, Arg380, and Asn381. The interaction is further strengthened through three conventional hydrogen bonds with Asn359, Lys360, and Ala382. An array of hydrophobic interactions was observed as contributed by Met358, Arg380, Asn381, and Ala382 to the binding with compound 8g. Also, the hydrogen bonds were constituted from interaction with Arg337 and Lys360. The amide compound 8q showed a lower binding affinity (−7.32 kcal mol−1) than the corresponding thioamide isostere 8k, respectively. Binding analysis of 8q showed that it also participated in hydrophobic interactions with amino acids such as Ala382, Phe406, Arg380, Asn381, Met358, and Lys360. The carbonyl group in the quinazoline ring formed hydrogen bonds with Arg337 residues. However, the amide bond inserted into the quinazoline ring at the C-2 position did not participate in interactions with any amino acids. In contrast to the twisted conformation of 8k, amide 8q appears to have a planar conformation (Fig. 4), in which case this conformation appears not to be optimal for binding to the active site. This may explain the lower binding affinity and NO inhibitory potency compared to the 8k thioamide bioisostere.
Molecular docking simulations further revealed that HIT compounds could inhibit TLR4 signaling through direct interaction, with the twisted structure being crucial in this process. Unlike the interaction between LPS and TLR4, the three most potent compounds did not occupy the binding site in MD-2; instead, they bound to an adjacent smooth region in TLR4. This likely undermined the formation of the primary contact interface between TLR4 and MD-2 and the recognition of LPS. Given the significant role of TLR4 in inflammation and immunity, these findings suggest that they could have potent applications in treating many inflammatory diseases related to immune disorders.
Experimental
Chemistry
All chemicals and solvents used as analytical reagents were supplied by commercial chemical companies and were used directly without further purification steps. The melting temperatures of the compounds were recorded by a Krüss M5000 melting point meter at default settings. A PerkinElmer Spectrum Two FT-IR spectrometer was used to record the IR spectra, using the pellet method with KBr. 1H NMR (600 MHz) and 13C NMR (125/150 MHz) spectra were recorded using Bruker AV-600 and Bruker AV-500 spectrometers with TMS as standard, CDCl3 and DMSO-d6 as solvents. NMR data are described in terms of chemical shifts (ppm), multiplets: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), coupling constants (Hz), and integration. High-resolution mass spectral data of all compounds were collected in ESI positive mode using a SCIEX X500R QTOF high-resolution mass spectrometer. Reactions were monitored by thin-layer chromatography (TLC) using aluminum TLC plates pre-coated with silica gel 60 F254, 0.25 mm (Merck) and detected by UV light at 254 nm. The purity of HIT compounds 8d, 8g, and 8k was >94% as determined by high-performance liquid chromatography (HPLC), (LC, Agilent 1100 series; column, Agilent C18, 170 Å, 4.6 × 150 mm, 5 μm; column temperature, 25 °C; mobile phase, solvent A, methanol, solvent B, water, gradient elution, 20–99% solvent A; flow rate, 1 mL min−1; UV signals were recorded at 254 nm).Synthesis of 2-methylquinazolin-4(3H)-one (10)
Compound 10 was synthesized based on a previously reported study45 with some minor modifications. A solution of 2-aminobenzamide 9 (500 mg, 3.67 mmol), 2 mL of TBHP (70% in H2O) (14.68 mmol), and 37 mL of absolute ethanol were refluxed for 48 h. The reaction progress was monitored and checked by TLC (ethyl acetate:n-hexane = 1:1, v/v). At the end of the reaction, the reaction mixture was dried under reduced pressure to obtain the crude product. The crude product was crystallized in ethyl acetate to obtain 382 mg of 10 as a white solid. The reaction yield was 65%.
O, amide), 1467, 1382, 1340, 1292, 1250, 1145, 887, 772. 1H NMR (600 MHz, DMSO-d6) δ 12.17 (s, 1H), 8.08 (dd, J = 7.9, 1.0 Hz, 1H), 7.76 (ddd, J = 8.3, 7.1, 1.6 Hz, 1H), 7.57 (d, J = 7.5 Hz, 1H), 7.45 (ddd, J = 8.1, 7.1, 1.2 Hz, 1H), 2.35 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 162.18, 154.72, 149.44, 134.72, 127.04, 126.30, 126.13, 121.10, 21.90. HR-MS (ESI) calcd. For C9H9N2O [(M + H)+]: 161.0715; found: 161.0708.General procedure for the synthesis of 3,4-dihydroquinazoline-2-carbothioamide derivatives (8a–n)
A mixture of 10 (1.0 mmol), aryl amine 11a–n (1.0 mmol), and sulfur (40 mg, 1.25 mmol) in DMSO (0.5 mL) was heated in a 7 mL test tube sealed with a rubber stopper, at 110 °C for 16 h. The crude mixture was purified by column chromatography on silica gel (acetone:n-hexane = 3% to 5%, v/v) to obtain 8a–n compounds.44
O, amide), 1592, 1436, 1388, 1332, 1067 (CS, thioamide), 825, 766. 1H NMR (600 MHz, CDCl3) δ 11.68 (s, 1H), 10.96 (s, 1H), 8.36 (d, J = 7.8 Hz, 1H), 8.06 (d, J = 7.4 Hz, 2H), 7.84 (d, J = 3.5 Hz, 2H), 7.63–7.57 (m, 1H), 7.51–7.47 (m, 2H), 7.35 (t, J = 7.4 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 179.44, 160.51, 146.56, 144.09, 137.83, 135.06, 129.23, 128.74, 128.24, 127.47, 127.16, 122.42, 122.25. HR-MS (ESI) calcd. For C15H12N3OS [(M + H)+]: 282.0701; found: 282.0691.O, amide), 1601, 1529, 1475, 1435, 1388, 1330, 1063 (CS, thioamide), 819, 767. 1H NMR (600 MHz, CDCl3) δ 11.64 (s, 1H), 11.00 (s, 1H), 8.37 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 8.2 Hz, 2H), 7.84 (d, J = 4.1 Hz, 2H), 7.60 (dt, J = 8.1, 4.2 Hz, 1H), 7.30 (d, J = 8.1 Hz, 2H), 2.41 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 178.99, 160.57, 146.64, 144.19, 137.62, 135.39, 135.03, 129.75, 128.67, 128.21, 127.17, 122.40, 122.22, 21.26. HR-MS (ESI) calcd. For C16H14N3OS [(M + H)+]: 296.0858; found: 296.0845.O, amide), 1602, 1515, 1464, 1404, 1308, 1263, 1023 (CS, thioamide), 826, 772. 1H NMR (600 MHz, CDCl3) δ 11.61 (s, 1H), 11.00 (s, 1H), 8.37 (d, J = 7.8 Hz, 1H), 8.00 (d, J = 9.1 Hz, 2H), 7.90–7.81 (m, 2H), 7.64–7.57 (m, 1H), 7.01 (d, J = 9.0 Hz, 2H), 3.87 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 178.42, 160.58, 158.52, 146.66, 144.26, 135.05, 131.02, 128.64, 128.18, 127.17, 123.90, 122.38, 114.31, 55.59. HR-MS (ESI) calcd. For C16H14N3O2S [(M + H)+]: 312.0807; found: 312.0794.O, amide), 1602, 1517, 1491, 1437, 1397, 1326, 1177, 1123, 1099, 1067 (CS, thioamide), 1012, 824, 769, 711. 1H NMR (600 MHz, CDCl3) δ 11.66 (s, 1H), 10.89 (s, 1H), 8.35 (d, J = 7.9 Hz, 1H), 8.03 (d, J = 8.8 Hz, 2H), 7.88–7.78 (m, 2H), 7.60 (ddd, J = 8.1, 5.9, 2.3 Hz, 1H), 7.45 (d, J = 8.8 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 179.70, 160.49, 146.44, 143.94, 136.33, 135.14, 132.53, 129.34, 128.87, 128.22, 127.18, 123.50, 122.40. HR-MS (ESI) calcd. For C15H11ClN3OS [(M + H)+]: 316.0311; found: 316.0292. Purity > 95% (HPLC, see in ESI†).O, amide), 1604, 1522, 1480, 1408, 1378, 1324, 1180, 1069 (CS, thioamide), 955, 885, 748. 1H NMR (600 MHz, CDCl3) δ 11.68 (s, 1H), 10.90 (s, 1H), 8.37 (d, J = 7.8 Hz, 1H), 8.18 (t, J = 2.1 Hz, 1H), 7.94 (dd, J = 7.9, 2.2 Hz, 1H), 7.90–7.82 (m, 2H), 7.62 (ddd, J = 8.2, 5.5, 2.8 Hz, 1H), 7.43 (t, J = 8.1 Hz, 1H), 7.33 (dd, J = 8.0, 1.4 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 180.02, 160.46, 146.41, 143.86, 138.85, 135.15, 134.94, 130.24, 128.92, 128.26, 127.46, 127.21, 122.46, 122.18, 120.33. HR-MS (ESI) calcd. For C15H11ClN3OS [(M + H)+]: 316.0311; found: 316.0308.O, amide), 1602, 1526, 1468, 1438, 1407, 1324, 1127, 1070 (CS, thioamide), 735. 1H NMR (600 MHz, CDCl3) δ 12.29 (s, 1H), 10.89 (s, 1H), 9.01 (dd, J = 8.3, 1.6 Hz, 1H), 8.37 (dd, J = 7.9, 1.5 Hz, 1H), 7.94–7.81 (m, 2H), 7.62 (ddd, J = 8.2, 6.7, 1.6 Hz, 1H), 7.55 (dd, J = 8.0, 1.5 Hz, 1H), 7.42 (td, J = 7.8, 1.5 Hz, 1H), 7.29 (td, J = 7.7, 1.5 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 179.96, 160.62, 146.52, 144.10, 135.09, 134.77, 129.88, 128.89, 128.61, 127.91, 127.45, 127.12, 126.70, 122.83, 122.46. HR-MS (ESI) calcd. For C15H11ClN3OS [(M + H)+]: 316.0311; found: 316.0292.O, thioamide), 1534, 1492, 1394, 1328, 1069 (CS, thioamide), 1012, 824, 772, 714. 1H NMR (600 MHz, CDCl3) δ 11.65 (s, 1H), 10.88 (s, 1H), 8.38–8.28 (m, 1H), 8.02–7.93 (m, 2H), 7.88–7.79 (m, 2H), 7.65–7.56 (m, 3H). 13C NMR (150 MHz, CDCl3) δ 179.62, 160.42, 146.41, 143.93, 136.81, 135.12, 132.31, 128.87, 128.22, 127.19, 123.66, 122.41, 120.35. HR-MS (ESI) calcd. For C15H11BrN3OS [(M + H)+]: 359.9806; found: 359.9797. Purity > 98% (HPLC, see in ESI†).O, amide), 1602, 1523, 1478, 1435, 1410, 1382, 1327, 1239, 1177, 1124, 1067 (CS, thioamide), 829, 747. 1H NMR (600 MHz, CDCl3) δ 11.63 (s, 1H), 10.93 (s, 1H), 8.40–8.33 (m, 1H), 8.07–7.98 (m, 2H), 7.88–7.79 (m, 2H), 7.61 (ddd, J = 8.2, 6.0, 2.4 Hz, 1H), 7.21–7.17 (m, 2H). 13C NMR (150 MHz, CDCl3) δ 179.83, 160.96 (d, J = 248.85 Hz), 160.47, 146.50, 144.00, 135.10, 133.90 (d, J = 3.4 Hz), 128.81, 128.21, 127.20, 124.39 (d, J = 8.2 Hz), 122.45, 116.21, 116.06. HR-MS (ESI) calcd. For C15H11FN3OS [(M + H)+]: 300.0607; found: 300.0592.O, amide), 1595, 1549, 1509, 1481, 1446, 1375, 1334 (C–NO2), 1108, 1067 (CS, thioamide), 947, 849, 765. 1H NMR (600 MHz, CDCl3) δ 11.95 (s, 1H), 10.80 (s, 1H), 8.47–8.22 (m, 5H), 7.97–7.78 (m, 2H), 7.71–7.54 (m, 1H). 13C NMR (125 MHz, CDCl3) δ 180.79, 160.39, 146.16, 145.41, 143.71, 142.95, 135.31, 129.25, 128.33, 127.32, 125.07, 122.52, 121.87. HR-MS (ESI) calcd. For C15H11N4O3S [(M + H)+]: 327.0552; found: 327.0545.O, amide), 1602, 1535, 1479, 1437, 1386, 1315, 1241, 1167, 1111, 1071 (CS, thioamide), 840, 761, 716. 1H NMR (600 MHz, CDCl3) δ 11.78 (s, 1H), 10.84 (s, 1H), 8.34 (d, J = 7.8 Hz, 1H), 8.22 (d, J = 8.5 Hz, 2H), 7.88–7.81 (m, 2H), 7.74 (d, J = 8.4 Hz, 2H), 7.63–7.57 (m, 1H). 13C NMR (150 MHz, CDCl3) δ 180.53, 160.52, 146.42, 143.92, 140.73, 135.31, 129.13, 129.08 (q, J = 32.9 Hz), 128.40, 127.32, 126.59 (q, J = 4.1 Hz), 123.82 (q, J = 272.2 Hz), 122.56, 122.18. HR-MS (ESI) calcd. For C16H11F3N3OS [(M + H)+]: 350.0575; found: 350.0558. Purity > 94% (HPLC, see in ESI†).O, amide), 1598, 1529, 1431, 1332, 1167, 1067 (CS, thioamide), 884, 774, 696. 1H NMR (600 MHz, DMSO-d6) δ 12.70 (s, 1H), 12.06 (s, 1H), 8.41 (s, 1H), 8.22 (t, J = 8.2 Hz, 2H), 7.94 (t, J = 7.0 Hz, 1H), 7.88 (d, J = 7.9 Hz, 1H), 7.75 (t, J = 7.8 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.66 (t, J = 7.4 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ 186.14, 161.16, 149.38, 147.44, 139.72, 135.55, 130.63, 129.86 (q, J = 31.8 Hz), 128.74, 128.46, 128.03, 126.67, 124.33 (q, J = 272.3 Hz), 123.95 (d, J = 3.6 Hz), 122.41, 120.59 (q, J = 3.5 Hz). HR-MS (ESI) calcd. For C16H11F3N3OS [(M + H)+]: 350.0575; found: 350.0560.O, amide), 1604, 1534, 1481, 1432, 1380, 1318, 1070 (CS, thioamide), 878, 796, 767. 1H NMR (600 MHz, CDCl3) δ 11.75 (s, 1H), 10.82 (s, 1H), 8.51–8.49 (m, 1H), 8.40–8.33 (m, 1H), 8.28 (dt, J = 6.8, 2.4 Hz, 1H), 7.90–7.83 (m, 2H), 7.66–7.59 (m, 3H)·13C NMR (125 MHz, CDCl3) δ 180.84, 160.37, 146.26, 143.70, 138.55, 135.23, 130.56, 130.20, 129.10, 128.28, 127.27, 126.41, 125.35, 122.52, 117.81, 113.55. HR-MS (ESI) calcd. For C16H11N4OS [(M + H)+]: 307.0654; found: 307.0640.O, amide), 1599, 1534, 1436, 1391, 1281, 1110 (CS, thioamide), 849, 772, 708. 1H NMR (600 MHz, CDCl3) δ 11.83 (s, 1H), 10.88 (s, 1H), 8.36 (d, J = 7.9 Hz, 1H), 8.21 (d, J = 8.8 Hz, 2H), 8.16 (d, J = 8.8 Hz, 2H), 7.85 (d, J = 3.3 Hz, 2H), 7.65–7.58 (m, 1H), 3.95 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 179.96, 166.06, 160.44, 146.36, 143.90, 141.51, 135.16, 130.82, 128.96, 128.57, 128.28, 127.21, 122.44, 121.41, 52.29. HR-MS (ESI) calcd. For C17H14N3O3S [(M + H)+]: 340.0756; found: 340.0745.General procedure for the synthesis of 3,4-dihydroquinazoline-2-carbothioamide derivatives (8o, 8p)
A mixture of 10 (1.0 mmol), benzylamine 11o, 11p (1.0 mmol), sulfur (40 mg, 1.25 mmol), and acetic acid (60 mg, 1 mmol) in DMSO (0.5 mL) was heated in a 7 mL test tube sealed with a rubber stopper, at 100 °C for 16 h. The crude mixture was purified by column chromatography on silica gel (acetone:n-hexane = 3% to 5%, v/v) to obtain 8o, 8p compounds.44
O, amide), 1606, 1535, 1482, 1431, 1331, 1241, 1179, 1131, 1100, 1070 (CS, thioamide), 1014, 826, 756. 1H NMR (600 MHz, CDCl3) δ 10.86 (s, 1H), 10.08 (s, 1H), 8.33 (dd, J = 7.9, 1.5 Hz, 1H), 7.81–7.75 (m, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.59–7.54 (m, 1H), 7.46–7.34 (m, 5H), 5.00 (d, J = 5.8 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 183.73, 160.48, 146.83, 143.52, 135.33, 134.92, 129.11, 128.60, 128.43, 128.34, 128.26, 127.07, 122.61, 50.38. HR-MS (ESI) calcd. For C16H14N3OS [(M + H)+]: 296.0858; found: 296.0843.O, amide), 1602, 1535, 1479, 1436, 1385, 1314, 1167, 1111, 1071 (CS, thioamide), 840, 760. 1H NMR (600 MHz, CDCl3) δ 10.83 (s, 1H), 10.08 (s, 1H), 8.34 (dd, J = 7.9, 1.5 Hz, 1H), 7.82–7.76 (m, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.61–7.55 (m, 1H), 7.41–7.32 (m, 4H), 4.98 (d, J = 5.9 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ 184.02, 160.45, 146.75, 143.45, 134.97, 134.37, 133.80, 129.65, 129.25, 128.69, 128.25, 127.09, 122.62, 49.45. HR-MS (ESI) calcd. For C16H13ClN3OS [(M + H)+]: 330.0468; found: 330.0453.Synthesis of 3,4-dihydroquinazoline-2-carboxamide derivative (8q)
Compound 8q was synthesized based on a procedure previously described by Thanh Binh Nguyen et al.48 Brief, a mixture of 10 (1 mmol), 4-(trifluoromethyl)aniline 11k (1 mmol), FeCl2·4H2O (5 mol%), and sulfur (1.25 mmol, 40 mg) in DMSO (0.3 mL) was carried out in a 7 mL test tube sealed with a rubber stopper, at 120 °C for 16 h. The crude mixture was purified by column chromatography on silica gel (eluted with a mixture of EtOAc:n-hexane = 10%, v/v) to obtain compound 8q.
O, amide), 1604, 1535, 1479, 1412, 1332, 1164, 1105, 1072, 1017, 894, 843, 774. 1H NMR (600 MHz, DMSO-d6) δ 12.55 (s, 1H), 11.11 (s, 1H), 8.22 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 8.4 Hz, 2H), 7.97–7.89 (m, 2H), 7.78 (d, J = 8.5 Hz, 2H), 7.70–7.63 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ 161.43, 159.14, 147.33, 146.16, 141.79, 135.31, 128.89, 128.42, 126.71, 126.50 (q, J = 3.5 Hz), 125.64 (d, J = 271.2 Hz), 125.03 (q, J = 32.7 Hz), 123.35, 121.14. HR-MS (ESI) calcd. For C16H11F3N3O2 [(M + H)+]: 334.0803; found: 334.0796.In vitro anti-inflammatory activity
The in vitro anti-inflammatory activity of the tested compounds was evaluated by inhibiting NO production in LPS-stimulated RAW 264.7 cells following a similar procedure described by Diep Trinh Thi.63 Briefly, RAW 274.7 cells were seeded into 96-well plates at a concentration of 2 × 105 cells per well and cultured in an incubator at 37 °C and 5% CO2 for 24 h. The sample was dissolved in 100% DMSO to an initial concentration of 20 mM. The sample was diluted in a 96-well plate with cell culture medium (without FBS) into 4 concentration ranges from high to low. Cells were then incubated with different concentrations for 2 h before being stimulated to produce NO with LPS (10 μg mL−1) for 24 h. Some wells were not incubated with samples but only used sample diluent as negative controls. While the positive controls used were dexamethasone (Sigma) at concentrations of 100; 20; 4 and 0.8 μM. Nitrite (NO2−), considered an indicator for NO production, was determined using the Griess Reagent System (Promega Cooperation, WI, USA). Specifically, 100 μL of cell culture medium (incubation sample) was transferred to a new 96-well plate and 100 μL of Griess reagent was added: 50 μL of 1% (w/v) sulfanilamide in 5% (v/v) phosphoric acid and 50 μL of 0.1% (w/v) N-1-naphthylethylenediamine dihydrochloride in water. The mixture was further incubated at room temperature for 10 min and the nitrite content was measured using a microplate reader at 540 nm. DMEM medium without FBS was used as the blank well. The nitrite content of each experimental sample was determined by the NaNO2 standard curve and compared in % with the negative control sample (LPS). The corresponding NO production inhibition ability of the sample was determined by the formula: (%) inhibition = 100% − [content NOsample/content NOLPS] × 100. The test was repeated 3 times to ensure accuracy. The IC50 value (concentration that inhibits 50% of NO formation) was determined using the computer software TableCurve 2Dv4.Molecular docking studies
Since the experiments in this research were performed on RAW264.7 cells, the crystal structure of mouse TLR4 and mouse MD-2 complex were selected for docking simulation with studied compounds. The X-ray crystallographic model of the complexes was downloaded from the RCSB Protein Data Bank (PDB ID: 2Z64).64 A resolution between 1.5 and 3.0 Å is considered a good quality for docking studies.65,66 The interaction with model 2Z64, a monomeric LPS-free TLR4-MD-2 structure that acted as an inactive conformation prepared for ligand docking, was the primary basis for identifying the putative binding region. Then, the active conformation (PDB ID: 3VQ2), a dimeric TLR4/MD-2 combination with LPS, was subsequently used to confirm the results. To create a free receptor, water molecules were removed from the protein. The protein was prepared for docking simulations by assigning partial charges, solvation parameters, and hydrogens to the receptor molecule. A grid box was constructed to encompass all possible binding sites by using validated ligands as references. AutoDock 4.2 was utilized for the molecular docking simulation using Lamarckian Genetic Algorithm (LGA). The ligand conformation with the lowest binding free energy, selected from the most favored cluster, was chosen for further analysis. The results from the AutoDock modeling studies were analyzed using PyMOL67 and Discovery Studio Visualizer.68Conclusions
A library of 15 novel quinazoline-4(3H)-one derivatives functionalized at position 2nd via thioamide linkages attached to phenyl and benzyl rings was successfully designed and synthesized in two simple steps. Interestingly, compounds with potent in vitro anti-inflammatory activity were synthesized in good yields (62–82%). Such as compounds 8a, 8d, 8g and 8k. The structures of the synthesized compounds were determined by modern physicochemical analysis methods such as IR, NMR, HRMS, and melting point. These compounds were also evaluated for their anti-inflammatory activity through their ability to inhibit NO production. Compounds 8d, 8g, and 8k exhibited superior anti-inflammatory activity compared to the standard drug dexamethasone. Compound 8a inhibited the inflammatory mediator NO equivalently to dexamethasone. SAR studies demonstrated the importance of the presence of a thioamide linker with a phenyl ring attached to halogen-containing substituents at the para-position in the generation of novel quinazolinone compounds with potent anti-inflammatory activity. In silico studies showed that candidates 8d, 8g, and 8k were potent inhibitors of TLR4 signaling with significant docking scores of −7.71, −7.65, and −7.84 kcal mol−1, respectively, compared to −7.10 kcal mol−1 of dexamethasone. One of the outstanding features of our NO inhibitor essential for high potency is the presence of a thioamide group, as its replacement by an amide results in a sharp decrease in inhibitory activity. This highlights the important role of favorable sulfur interactions in protein-ligand complexes, as well as the thioamide group in drug design. Finally, physicochemical and ADMET studies of the potent compounds were also conducted, showing that they had favorable ADMET profiles, in silico studies of the lead compounds demonstrate their potential as drug candidates.Data availability
The data supporting this article are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.01-2020.49.References
- V. Ngoc Binh, S. Adorisio, D. V. Delfino and Q. A. Ngo, Prod. Commun., 2022, 17, 1934578X221105692 CAS.
- C.-L. Hsu, S.-C. Fang and G.-C. Yen, Food Funct., 2013, 4, 1216–1222 RSC.
- M. E. Bauer and A. L. Teixeira, Ann. N. Y. Acad. Sci., 2019, 1437, 57–67 CrossRef CAS.
- S. U. Rahman, Y. Li, Y. Huang, L. Zhu, S. Feng, J. Wu and X. Wang, Inflammopharmacology, 2018, 26, 319–330 CrossRef CAS.
- P. Tripathi, P. Tripathi, L. Kashyap and V. Singh, FEMS Microbiol. Immunol., 2007, 51, 443–452 CrossRef CAS.
- M. Marra, A. Mariconda, D. Iacopetta, J. Ceramella, A. D'Amato, C. Rosano, K. Tkachenko, M. Pellegrino, S. Aquaro, M. S. Sinicropi and P. Longo, Molecules, 2024, 29, 5262 CrossRef CAS.
- L.-R. Balahura Stămat and S. Dinescu, Sci. Rep., 2024, 14, 24753 CrossRef.
- J. N. Sharma, A. Al-Omran and S. S. Parvathy, Inflammopharmacology, 2007, 15, 252–259 CrossRef CAS PubMed.
- S. Sharma, D. Kumar, G. Singh, V. Monga and B. Kumar, Eur. J. Med. Chem., 2020, 200, 112438 CrossRef CAS PubMed.
- M. Bian, Q.-q. Ma, Y. Wu, H.-h. Du and G. Guo-hua, J. Enzyme Inhib. Med. Chem., 2021, 36, 2139–2159 CrossRef CAS PubMed.
- M. Bhat, S. L. Belagali, S. V. Mamatha, B. K. Sagar and E. V. Sekhar, in Studies in Natural Products Chemistry, ed. R. Atta ur, Elsevier, 2021, vol. 71, pp. 185–219 Search PubMed.
- A. R. Awwad and K. A. Fars, in Quinazolinone and Quinazoline Derivatives, ed. A.-k. Ali Gamal, IntechOpen, Rijeka, 2020, ch. 2, pp. 11–38 Search PubMed.
- M. F. Zayed, ChemEngineering, 2022, 6, 94 CrossRef CAS.
- T. C. Moon, M. Murakami, I. Kudo, K. H. Son, H. P. Kim, S. S. Kang and H. W. Chang, Inflammation Res., 1999, 48, 621–625 CrossRef CAS.
- S. Jia and C. Hu, Molecules, 2010, 15, 1873–1881 CrossRef CAS PubMed.
- K. M. Amin, M. M. Kamel, M. M. Anwar, M. Khedr and Y. M. Syam, Eur. J. Med. Chem., 2010, 45, 2117–2131 CrossRef CAS PubMed.
- A. Kumar and C. S. Rajput, Eur. J. Med. Chem., 2009, 44, 83–90 CrossRef CAS PubMed.
- B. A. Rather, T. Raj, A. Reddy, M. P. S. Ishar, S. Sivakumar and P. Paneerselvam, Arch. Pharm., 2010, 343, 108–113 CrossRef CAS PubMed.
- A. Sakr, S. Rezq, S. M. Ibrahim, E. Soliman, M. M. Baraka, D. G. Romero and H. Kothayer, J. Enzyme Inhib. Med. Chem., 2021, 36, 1810–1828 CrossRef CAS PubMed.
- S. K. Verma, R. Verma, K. P. Rakesh and D. C. Gowda, Eur. J. Med. Chem. Rep., 2022, 6, 100087 CAS.
- S. Kerdphon, N. Khamto, K. Buddhachat, J. Ngoenkam, P. Paensuwan, S. Pongcharoen, T. Singh, P. Meepowpan and J. Jongcharoenkamol, ACS Med. Chem. Lett., 2023, 14, 1167–1173 CrossRef CAS PubMed.
- A. M. Alsibaee, H. M. Al-Yousef and H. S. Al-Salem, Molecules, 2023, 28, 978 CrossRef CAS PubMed.
- S. Asirvatham, B. V. Dhokchawle and S. J. Tauro, Arabian J. Chem., 2019, 12, 3948–3962 CrossRef CAS.
- C. K. Khatri, K. S. Indalkar, C. R. Patil, S. N. Goyal and G. U. Chaturbhuj, Bioorg. Med. Chem. Lett., 2017, 27, 1721–1726 CrossRef CAS PubMed.
- M. H. Abdelrahman, B. G. M. Youssif, M. A. abdelgawad, A. H. Abdelazeem, H. M. Ibrahim, A. E. G. A. Moustafa, L. Treamblu and S. N. A. Bukhari, Eur. J. Med. Chem., 2017, 127, 972–985 CrossRef CAS PubMed.
- A. Sakr, H. Kothayer, S. M. Ibrahim, M. M. Baraka and S. Rezq, Bioorg. Chem., 2019, 84, 76–86 CrossRef CAS PubMed.
- K. Vinaya, R. Naika, C. S. A. Kumar, S. R. Ranganath, S. B. B. Prasad, V. Krishna and K. S. Rangappa, Arch. Pharm., 2009, 342, 476–483 CrossRef CAS PubMed.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS.
- N. Mahanta, D. M. Szantai-Kis, E. J. Petersson and D. A. Mitchell, ACS Chem. Biol., 2019, 14, 142–163 CrossRef CAS PubMed.
- A. Choudhary and R. T. Raines, ChemBioChem, 2011, 12, 1801–1807 CrossRef CAS PubMed.
- P. Ghosh, N. Raj, H. Verma, M. Patel, S. Chakraborti, B. Khatri, C. M. Doreswamy, S. R. Anandakumar, S. Seekallu, M. B. Dinesh, G. Jadhav, P. N. Yadav and J. Chatterjee, Nat. Commun., 2023, 14, 6050 CrossRef CAS.
- K. Tsuji, T. Ishii, T. Kobayakawa, N. Higashi-Kuwata, K. Shinohara, C. Azuma, Y. Miura, H. Nakano, N. Wada, S.-i. Hattori, H. Bulut, H. Mitsuya and H. Tamamura, J. Med. Chem., 2023, 66, 13516–13529 CrossRef CAS.
- S. Sharma, D. Singh, S. Kumar, Vaishali, R. Jamra, N. Banyal, Deepika, C. C. Malakar and V. Singh, Beilstein J. Org. Chem., 2023, 19, 231–244 CrossRef CAS PubMed.
- K. Pedrood, H. Azizian, M. N. Montazer, A. Moazzam, M. Asadi, H. Montazeri, M. Biglar, M. Zamani, B. Larijani, K. Zomorodian, M. Mohammadi-Khanaposhtani, C. Irajie, M. Amanlou, A. Iraji and M. Mahdavi, Sci. Rep., 2022, 12, 13827 CrossRef.
- G. Huang, T. Cierpicki and J. Grembecka, Eur. J. Med. Chem., 2024, 277, 116732 CrossRef CAS PubMed.
- T. S. Jagodziński, Chem. Rev., 2003, 103, 197–228 CrossRef.
- E. Korbut, M. Suski, Z. Śliwowski, D. Bakalarz, U. Głowacka, D. Wójcik-Grzybek, G. Ginter, K. Krukowska, T. Brzozowski, M. Magierowski, J. L. Wallace and K. Magierowska, Inflammopharmacology, 2024, 32, 2049–2060 CrossRef CAS.
- E. Gugliandolo, R. Fusco, R. D'Amico, A. Militi, G. Oteri, J. L. Wallace, R. Di Paola and S. Cuzzocrea, Pharmacol. Res., 2018, 132, 220–231 CrossRef CAS PubMed.
- U. Głowacka, K. Magierowska, D. Wójcik, J. Hankus, M. Szetela, J. Cieszkowski, E. Korbut, A. Danielak, M. Surmiak, A. Chmura, J. L. Wallace and M. Magierowski, Antioxid. Redox Signaling, 2021, 36, 189–210 CrossRef.
- J. L. Wallace, D. Vaughan, M. Dicay, W. K. MacNaughton and G. de Nucci, Antioxid. Redox Signaling, 2017, 28, 1533–1540 CrossRef.
- J. R. W. Glanville, P. Jalali, J. D. Flint, A. A. Patel, A. A. Maini, J. L. Wallace, A. A. Hosin and D. W. Gilroy, FASEB J., 2021, 35, e21913 CrossRef CAS PubMed.
- T. Murai, in Chemistry of Thioamides, ed. T. Murai, Springer Singapore, Singapore, 2019, pp. 45–73 Search PubMed.
- Q. Zhang, L. Soulère and Y. Queneau, Molecules, 2023, 28, 3527 CrossRef CAS.
- T. T. T. Nguyen, L. A. Nguyen, Q. A. Ngo, M. Koleski and T. B. Nguyen, Org. Chem. Front., 2021, 8, 1593–1598 RSC.
- J. Sun, T. Tao, D. Xu, H. Cao, Q. Kong, X. Wang, Y. Liu, J. Zhao, Y. Wang and Y. Pan, Tetrahedron Lett., 2018, 59, 2099–2102 CrossRef CAS.
- S. Yoshida, T. Aoyagi, S. Harada, N. Matsuda, T. Ikeda, H. Naganawa, M. Hamada and T. Takeuchi, J. Antibiot., 1991, 44, 111–112 CrossRef CAS.
- X. Yu, L. Gao, L. Jia, Y. Yamamoto and M. Bao, J. Org. Chem., 2018, 83, 10352–10358 CrossRef CAS.
- T. T. T. Nguyen, V. D. Duong, T. N. N. Pham, Q. T. Duong and T. B. Nguyen, Org. Biomol. Chem., 2022, 20, 8054–8058 RSC.
- N. Pirnazarova, U. Yakubov, S. Allabergenova, A. Tojiboev, K. Turgunov and B. Elmuradov, Acta Crystallogr., Sect. E:Crystallogr. Commun., 2022, 78, 47–50 CrossRef CAS.
- B. Khatri, S. Raghunathan, S. Chakraborti, R. Rahisuddin, S. Kumaran, R. Tadala, P. Wagh, U. D. Priyakumar and J. Chatterjee, Angew. Chem., Int. Ed., 2021, 60, 24870–24874 CrossRef CAS.
- M. Nibin Joy, M. R. Guda and G. V. Zyryanov, Pharmaceuticals, 2023, 16, 1326 CrossRef CAS PubMed.
- T. Khan, S. Dixit, R. Ahmad, S. Raza, I. Azad, S. Joshi and A. R. Khan, J. Chem. Biol., 2017, 10, 91–104 CrossRef PubMed.
- F. Cheng, W. Li, Y. Zhou, J. Shen, Z. Wu, G. Liu, P. W. Lee and Y. Tang, J. Chem. Inf. Model., 2012, 52, 3099–3105 CrossRef CAS.
- A. T. Garcia-Sosa, U. Maran and C. Hetenyi, Curr. Med. Chem., 2012, 19, 1646–1662 CrossRef CAS PubMed.
- N. T. Dan, H. D. Quang, V. Van Truong, D. Huu Nghi, N. M. Cuong, T. D. Cuong, T. Q. Toan, L. G. Bach, N. H. T. Anh, N. T. Mai, N. T. Lan, L. Van Chinh and P. M. Quan, Sci. Rep., 2020, 10, 11429 CrossRef CAS.
- S. T. Ngo, N. M. Tam, M. Q. Pham and T. H. Nguyen, J. Chem. Inf. Model., 2021, 61, 2302–2312 CrossRef CAS.
- N. T. Nguyen, T. H. Nguyen, T. N. H. Pham, N. T. Huy, M. V. Bay, M. Q. Pham, P. C. Nam, V. V. Vu and S. T. Ngo, J. Chem. Inf. Model., 2019, 60, 204–211 CrossRef PubMed.
- Q. A. Ngo, T. H. N. Thi, M. Q. Pham, D. Delfino and T. T. Do, Mol. Diversity, 2021, 25, 2307–2319 CrossRef CAS.
- X. Lin, J. Zhang, D. Fan, J. Hou, H. Wang, L. Zhu, R. Tian, X. An and M. Yan, Front. Pharmacol., 2021, 12, 643188 CrossRef CAS PubMed.
- M.-q. Tao, C.-l. Ji, Y.-j. Wu, J.-y. Dong, Y. Li, O. J. Olatunji and J. Zuo, Inflammation, 2020, 43, 1821–1831 CrossRef CAS PubMed.
- X. Gong, Y. He, D. Yang, S. Yang, J. Li, H. Zhao, Q. Chen, Q. Ren and B. Zhang, Bioorg. Chem., 2022, 127, 105939 CrossRef CAS.
- T. Chen, Y. Zhou, D. Han, L.-B. Han and S.-F. Yin, Org. Biomol. Chem., 2014, 12, 3802–3807 RSC.
- D. Trinh Thi, D. Luong Van, T. Phung Van, H. Nguyen Thi, T. Do Thi, U. Nguyen Thi To, L. Tran Thi Hoai, K. Dang Vinh, T. T. Huynh and T. Le Thi Thanh, Nat. Prod. Res., 2024, 38, 1834–1843 CrossRef CAS PubMed.
- H. M. Kim, B. S. Park, J.-I. Kim, S. E. Kim, J. Lee, S. C. Oh, P. Enkhbayar, N. Matsushima, H. Lee, O. J. Yoo and J.-O. Lee, Cell, 2007, 130, 906–917 CrossRef CAS PubMed.
- C. Didierjean and F. Tête-Favier, Acta Crystallogr., Sect. D:Struct. Biol., 2016, 72, 1308–1309 CrossRef CAS.
- C. Venugopal, C. Demos, K. Jagannatha Rao, M. Pappolla and K. Sambamurti, CNS Neurol. Disord.: Drug Targets, 2008, 7, 278–294 CrossRef CAS PubMed.
- L. Schrödinger, The PyMOL Molecular Graphics System, Version 1.3r1, 2010 Search PubMed.
- BIOVIA, Dassault Systèmes. Discovery Studio Visualizer, v21.1.0.20298, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra09094b |
| This journal is © The Royal Society of Chemistry 2025 |