
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploration of novel triazolo-thiadiazine hybrids of deferasirox as multi-target-directed anti-neuroinflammatory agents with structure–activity relationship (SAR): a new treatment opportunity for Alzheimer's disease†
Syed Ahmed Shakira,
Umer Rashid *a,
Marryuma,
Nighat Fatimab,
Syeda Abida Ejazc,
Ammara Fayyazc,
Muhammad Zahid Ullaha,
Aamer Saeedd,
Ajmal Khanef,
Ahmed Al Harrasie and
Amara Mumtaz*a
*a,
Marryuma,
Nighat Fatimab,
Syeda Abida Ejazc,
Ammara Fayyazc,
Muhammad Zahid Ullaha,
Aamer Saeedd,
Ajmal Khanef,
Ahmed Al Harrasie and
Amara Mumtaz*a
aDepartment of Chemistry, COMSATS University Islamabad, Abbottabad Campus, 22060, Pakistan. E-mail: umerrashid@cuiatd.edu.pk; amaramumtaz@cuiatd.edu.pk; Tel: +92334517999 Tel: +923005316570
bDepartment of Pharmacy, COMSATS University Islamabad, Abbottabad Campus, 22060, Pakistan
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, The Islamia University of Bahawalpur, Bahawalpur 63100, Pakistan
dDepartment of Chemistry, Quaid-i-Azam University, Islamabad, 45320, Pakistan
eNatural and Medical Sciences Research Center, University of Nizwa, 616, Nizwa, Oman
fDepartment of Chemical and Biological Engineering, College of Engineering, Korea University, 145 Anan-RO, Seongbuk-Gu, Seoul, 02841, Korea
First published on 2nd January 2025
Abstract
It is believed that inflammation influences several physiological processes, including the function of the central nervous system. Moreover, the impairment of lipid mechanisms/pathways is associated with neurodegenerative disorders and onset of Alzheimer's disease (AD). AD is a chronic neurodegenerative disease representing the major cause of dementia worldwide. In this case, the overexpression of different pharmacological targets has been confirmed to address neuronal inflammation and AD, with acetylcholinesterase (AChE), monoamine oxidase-B (MAO-B), cyclooxygenase-2 (COX-2) and 5-lipoxygenase (LOX-5) being the most explored targets. Currently, the available treatments are only capable of alleviating the symptoms and not capable of delivering disease-modifying effects. Thus, the current research objective is to synthesize triazolo-thiadiazine derivatives of the deferasirox drug as multi-target compounds that could concurrently inhibit ChEs, MAOs, LOX-5 and COX-2. The synthesized derivatives were confirmed by FTIR, 1H NMR, 13C NMR and DEPT-135 spectroscopic techniques. During in vitro investigations, compound 11 was found to be the most potent inhibitor of all the targeted enzymes. Briefly, this compound exhibited inhibitory values (IC50 ± SEM) of 0.31 ± 0.16, 0.13 ± 0.16 and 0.94 ± 0.16 μM against AChE, MAO-B and COX-2, respectively, suggesting that it is a lead molecule for the synthesis of more potential multi-targeted inhibitors. Several compounds, such as compound 9 and 13, showed dual inhibition potential in comparison to standard drugs. Furthermore, molecular docking analysis was performed to validate the in vitro results, where the potent compounds showed some significant interactions with the key amino acids present in the active site of the targeted enzymes. Furthermore, molecular dynamics (MD) simulation data and physicochemical properties supported deferasirox-substituted triazolo-thiadiazine as a promising horizon for the discovery and development of new molecules to treat multifactorial diseases associated with neuro-inflammation, such as AD.
1 Introduction
Cognitive dysfunctions, such as low information processing speed, deficits in learning, persistent attention-deficit dysfunctions, poor visual perception, low mental flexibility, and memory loss, are the major symptoms of Alzheimer's disease (AD).1 In this case, acetylcholine (ACh) helps in regulating cognitive functions, particularly memory and learning, via neurotransmission. Acetyl co-enzyme A, along with choline, is the primary source for ACh synthesis in presynaptic neurons, from where Ach is released into the synaptic gaps.2 Acetylcholinesterase (AChE) disrupts ACh-mediated neurotransmission, which initially originates in neurons. AChE inhibitors are administered in AD therapy as they have the ability to increase synaptic ACh levels in the cerebral cortex and recover cholinergic transmission.3 AD is associated with a reduction in the Ach level, leading to central nervous system (CNS) disorders and, consequently, a gradual decline in cognitive function, memory, and cognition.4 At present, FDA-approved AChE inhibitors for the treatment of AD are galantamine, rivastigmine and donepezil.5 Similar to AChE, butyrylcholinesterase (BChE) significantly contributes to cholinergic neurotransmission. BChE is present in the hippocampus, temporal nerve cortex, and glial cells and is associated with cognitive function.6 In comparison to AChE, BChE has lesser substrate specificity, but both enzymes effectively hydrolyze AChE.7 In contrast, monoamine oxidases (MAOs) are considered responsible for the inactivation of catecholamine and 5-hydroxytryptamine, and therefore, they are targeted in diseases where they are considered to be involved. Additionally, selective MAO-A inhibitors possess anti-depressant activity. Thus, selective MAO-B inhibitors are employed as progressive targets for the treatment of AD and Parkinson's disease (PD).8Although the key roots of neurodegenerative diseases are diverse, microglial activation and neuroinflammation are common features in most of these diseases and the adoption of anti-inflammatory approaches to avoid neurodegeneration has been studied intensively and endorsed as a promising approach.9 Consequently, COX-1 and COX-2 have attracted significant attention, and their inhibitors are considered neuroprotective in different settings of neurotoxicity and neurodegeneration.10 COX-2 inhibitors have been uncovered as neuroprotective in several ischemia models, as well as in vitro and in vivo models of diseases such as Alzheimer's, Parkinson's and amyotrophic lateral sclerosis.11 Similarly, significant results show that 5-lipoxygenase (5-LOX), a proinflammatory protein enzyme, is a prominent modulator in the onset of age-related neuropathology. It is further believed that age-related brain diseases can be delayed or slowed down just by the inhibition of 5-LOX activation.12
Triazolo-thiadiazines are a class of compounds formed by the fusion of two active pharmacophore moieties, i.e., triazole and thiadiazine. Triazolo-thiadiazines have been found to have promising activity against different targets such as anticancer, antimicrobial,13 anti-Alzheimer's, anti-and inflammatory.14 Triazolothiadiazines show specific interactions with many different receptors due to their hydrogen bond-accepting and donating capability, making them significant pharmacophores with excellent bioactive profiles.15 Triazolothiadiazines can be synthesized by condensing the triazole amine with substituted phenacyl bromides in absolute ethanol.16 Here, we present some of the reported triazolo-thiadiazine derivatives possessing cholinesterase and monoamine oxidase inhibition potential17–19 (Fig. 1).
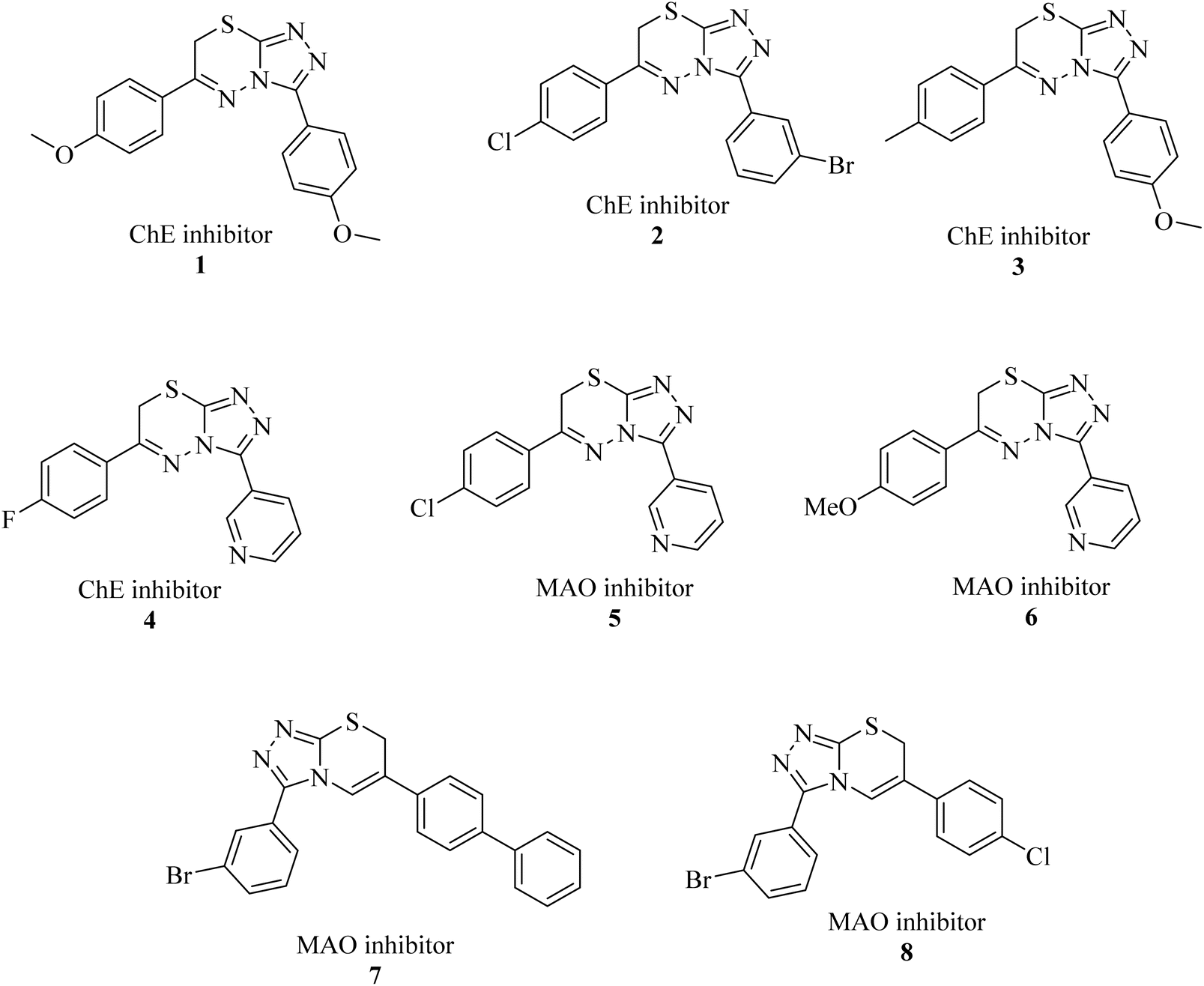 | ||
| Fig. 1 Previously reported triazolo-thiadiazine derivatives as cholinesterase and monoamine oxidase inhibitors.17–19 | ||
The multi-target-directed ligand (MTDL) strategy is an effective method for discovering novel medications because of the complex nature of the causes of AD.20 In the field of contemporary drug development, the MTDL technique has gained considerable interest and is emerging as a viable therapy option for the intricate AD. This strategy involves the fusion of more than two biologically active pharmacophores into a single molecule. Most of the newly identified diverse multitarget-directed ligands (MTDLs) function as inhibitors for many enzymes, such as acetylcholinesterase (AChE), monoamine oxidase (MAO), beta-secretase 1 (BACE1), and phosphodiesterase 4D (PDE4D). Furthermore, MTDLs that target AD exhibit the characteristics of regulators of serotonin receptors (5-HT), metal chelators, antiaggregant, neuroprotective and antioxidants agents.15,17–19 Herein, we presents a thorough evaluation of the recently discovered deferasirox derivative drugs that can interact with many targets by utilizing a mix of several pharmacophores for Alzheimer's treatment. These inhibitors have the potential to be effective therapies for AD.
2 Results and discussion
2.1. Instrumentation
Melting points were recorded in capillary tubes using an electrothermal melting point instrument, model Stuart digital melting point (SMP 10) apparatus with the specification of OE\Digital (08-09), 169/1 DMPA, 09-01. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III HD 400, 100 MHz spectrometer at the Department of Chemistry, COMSATS University Islamabad, Abbottabad Campus and Bruker 600, 150 MHz spectrometer at the Natural and Medical Sciences Research Center, University of Nizwa, respectively, utilizing DMSO-d6 (deuterated) as the solvent and TMS (tetramethylsilane) as the internal reference. High-resolution electrospray ionization mass spectra (HR-ESI-MS) were recorded on an Agilent 6530 LCQ-ToF instrument.2.2. Chemistry
The synthesis of deferasirox-substituted triazole amine 13 was carried out by using the following steps, as reported in Scheme 1.21 The first deferasirox derivative 10 was synthesised by refluxing the drug in excess methanol in the presence of sulfuric acid as a catalyst. The structure of 10 was confirmed by 1H-NMR. In the 1H-NMR analysis, the appearance of characteristic signals at 3.86 ppm as a singlet for 3 methoxy protons, in addition to the deferasirox signals confirmed the formation of an ester. Subsequently, ester 10 was reacted with hydrazine monohydrate in excess, in a methanolic solution to acquire the hydrazide derivative of deferasirox 11, whose structure was confirmed by 1H NMR with the disappearance of the methoxy signal of ester and appearance of two broad signals of NH and NHCO groups at 4.55 and 10.08, respectively. 11 was converted to compound 13 by first converting to derivative and intermediate deferasirox-substituted oxadiazole 12. The synthesis of oxadiazole derivative 12 was carried out by refluxing 11 with CS2 and KOH in ethanol for 12–15 h, and then the white precipitate obtained were washed with diethyl ether, and upon refluxing with the hydrochloric acid compound, 13 was obtained in good yield. The structure of compound 13 was confirmed by 1H NMR and 13C NMR.21 The 1H NMR data confirmed the formation of a ring after the appearance of two characteristic signals at 14.02 and 5.80 ppm for the NH and NH2 groups, respectively. The 13C NMR data further supported this data with the appearance of signals at 167.3 and at 156.8 ppm supporting ring closure21 (Scheme 1). The physical data of compounds 10–13 is presented in Table S1.†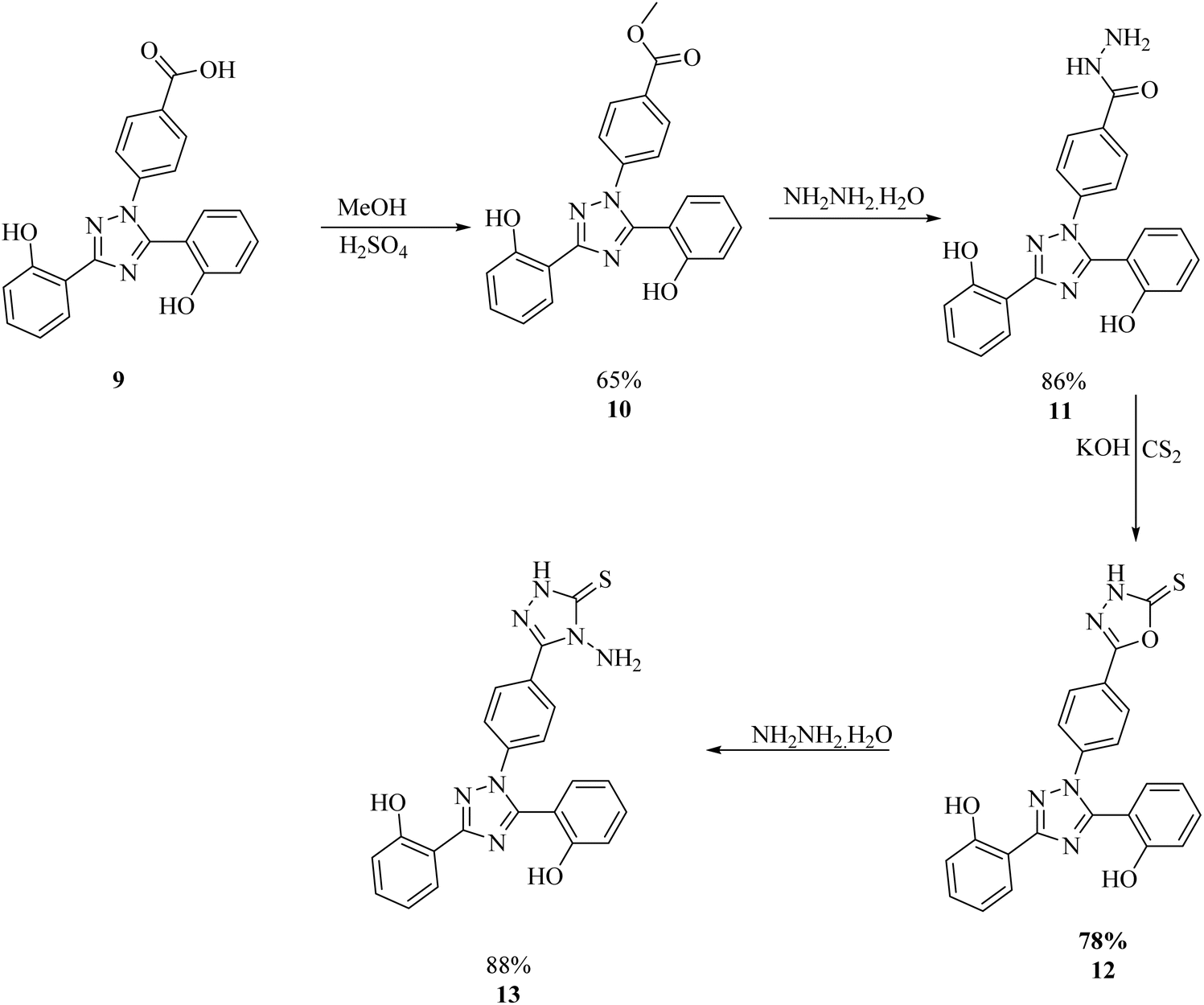 | ||
| Scheme 1 General procedure for the synthesis of the 1,3,4-triazole amine.21 | ||
For the synthesis of the deferasirox-substituted triazolo-thiadiazine derivatives (15–25), α-bromoacetophenones (14a–k) were synthesised via the reaction of substituted acetophenones with bromine in acetic acid (Scheme 2), respectively. The synthesis of the α-bromoacetophenones was confirmed by their melting points and comparison with the literature. Table S2† shows the details of the synthesized α-bromoacetophenones and their melting points (Scheme 2).
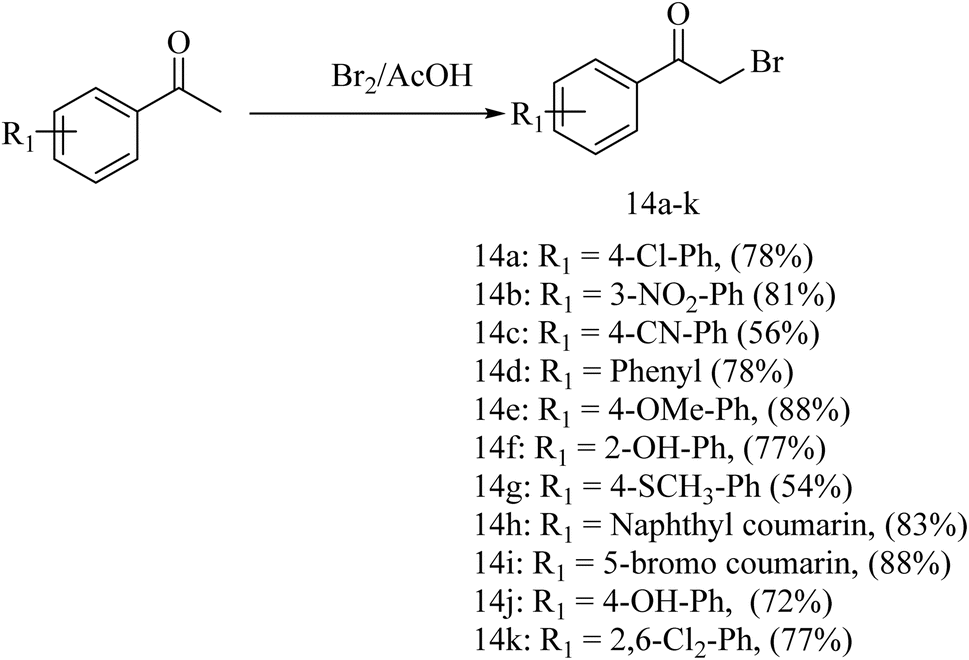 | ||
| Scheme 2 General procedure for the synthesis of α-bromoacetophenones. | ||
Our targeted compounds, triazolo-thiadiazine derivatives of deferasirox (15–25), were synthesized via the condensation of an alcoholic solution of triazole amine (13) with the substituted alpha bromo-acetophenones (14a–k) in the presence of potassium carbonate. Our targeted molecules were obtained in a good yield ranging from 65–88% (Table S3†). Structural confirmation was carried out based on the disappearance of the NH2 singlet of triazole amine derivative 13 and appearance of a characteristic singlet at 4.42–4.55 ppm for the CH2 group of triazolo-thiadiazine, confirming the ring closure. Further confirmation was carried out by 13C NMR analysis. In the 13C NMR spectra, the appearance of the characteristic signal of the CH2 group at 23.2–23.4 ppm confirmed the ring closure. DEPT-135 further confirmed the ring closure with the appearance of an inverted signal for the CH2 group in the same region (Scheme 3).
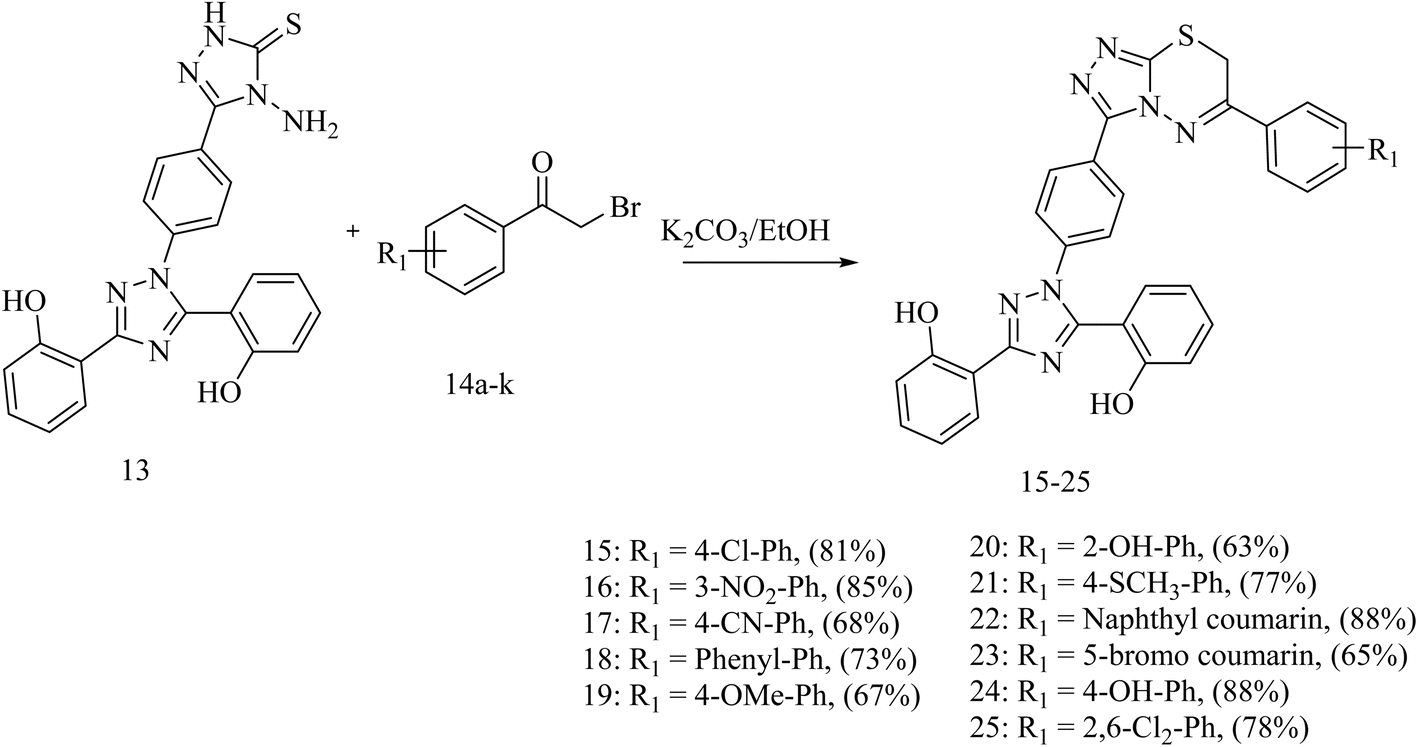 | ||
| Scheme 3 General procedure for the synthesis of triazolo-thiadiazine (15–25). | ||
2.3. Biological activities
| Code | Structure | IC50 (μM) ± SEM | |||||
|---|---|---|---|---|---|---|---|
| AChE | BChE | MAO-A | MAO-B | COX-2 | 5-LOX | ||
| 09 | 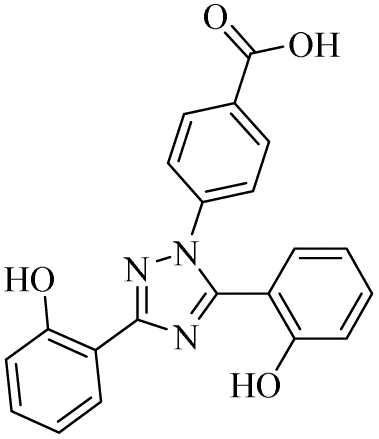 |
0.43 ± 0.05 | 0.11 ± 0.005 | 10.05 ± 1.78 | 0.18 ± 0.01 | 4.93 ± 0.38 | 1.28 ± 0.13 |
| 10 | 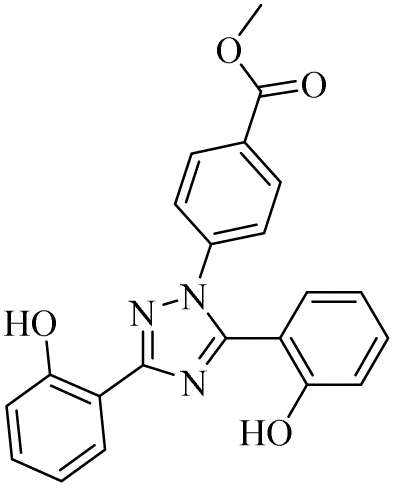 |
6.24 ± 0.16 | 0.45 ± 0.02 | 33.59 ± 3.09 | 19.55 ± 1.94 | 1.12 ± 0.14 | 26.94 ± 3.54 |
| 11 | 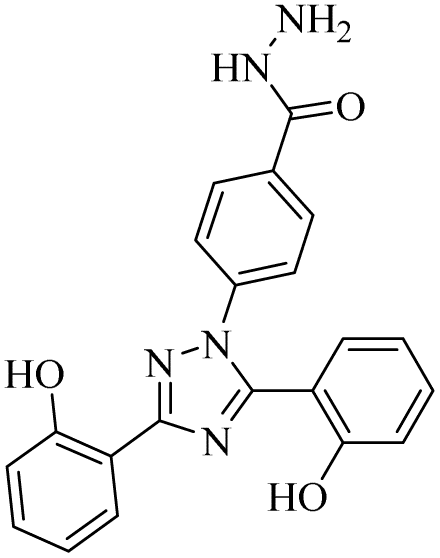 |
0.31 ± 0.02 | 0.094 ± 0.001 | 10.15 ± 1.21 | 0.13 ± 0.02 | 0.94 ± 0.11 | 12.73 ± 2.09 |
| 13 | 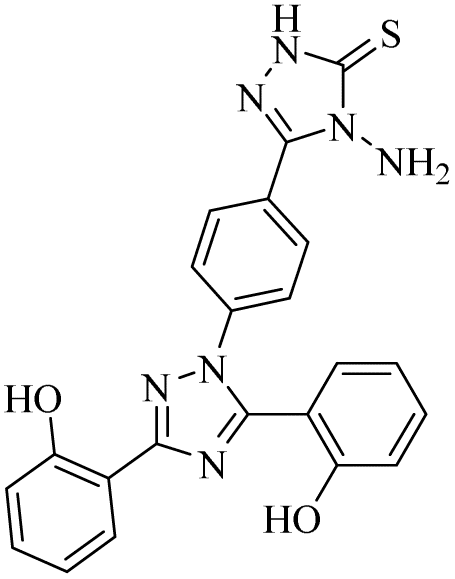 |
0.71 ± 0.16 | 0.18 ± 0.02 | 19.04 ± 2.44 | 0.41 ± 0.04 | 0.07 ± 0.001 | 0.84 ± 0.05 |
| 15 | 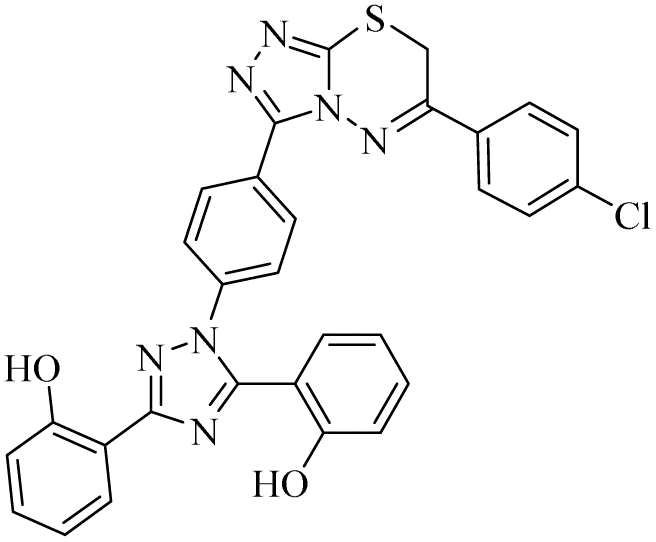 |
n.a | 3.96 ± 0.22 | 3.58 ± 0.56 | n.a | 75.29 ± 3.56 | n.a |
| 16 | 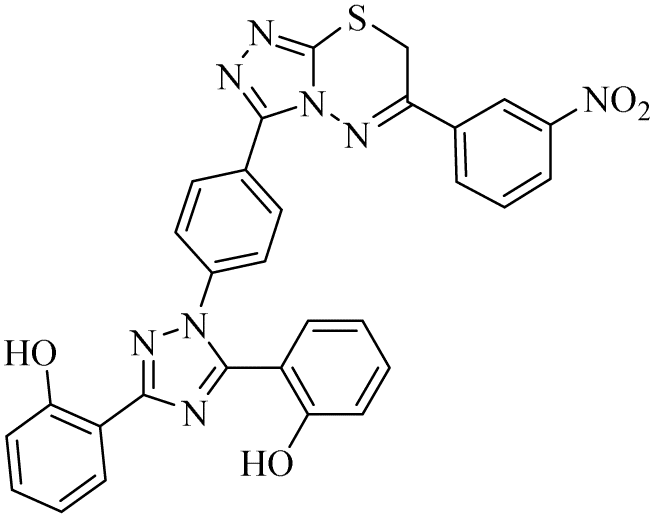 |
n.a | 6.88 ± 0.39 | 6.67 ± 0.78 | n.a | 79.55 ± 3.09 | na |
| 17 | 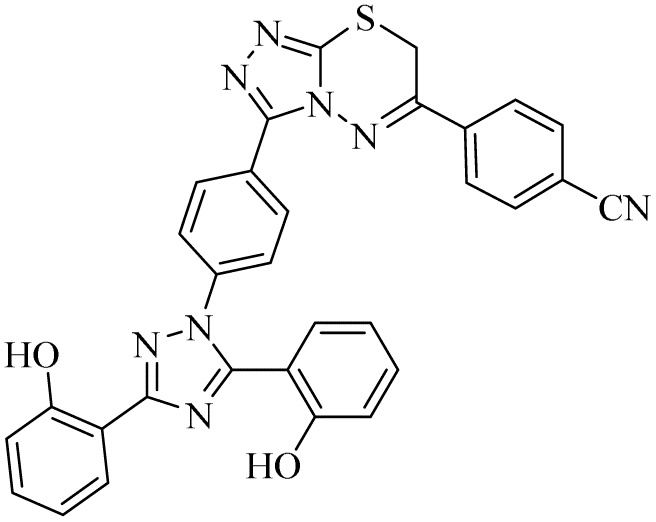 |
n.a | 0.28 ± 0.03 | 55.91 ± 5.08 | n.a | n.a | na |
| 18 | 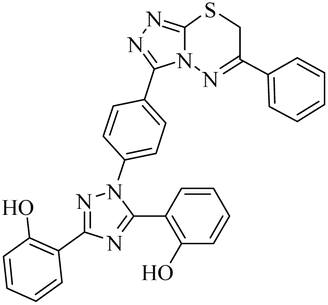 |
n.a | 3.66 ± 0.89 | 0.63 ± 0.01 | n.a | n.a | na |
| 19 | 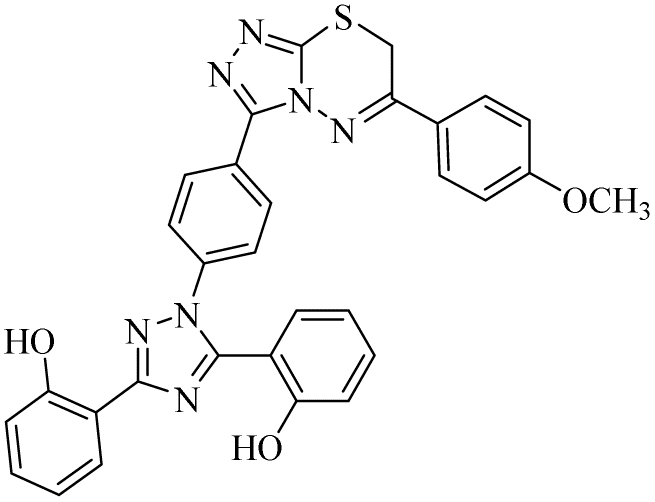 |
n.a | 0.13 ± 0.02 | 2.91 ± 0.02 | n.a | n.a | na |
| 20 | 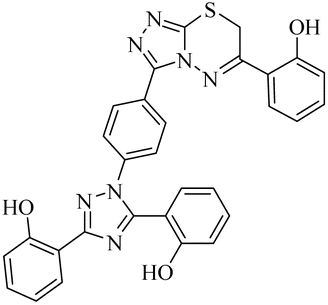 |
n.a | 0.47 ± 0.05 | 4.71 ± 0.07 | n.a | n.a | na |
| 21 | 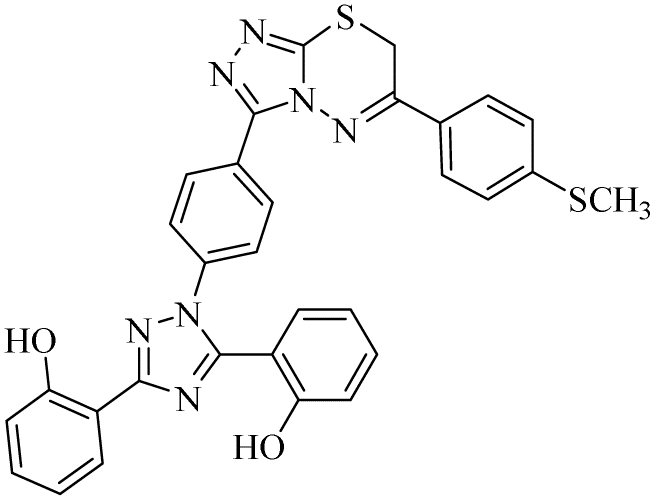 |
n.a | 0.89 ± 0.06 | 1.52 ± 0.08 | n.a | n.a | n.a |
| 22 | 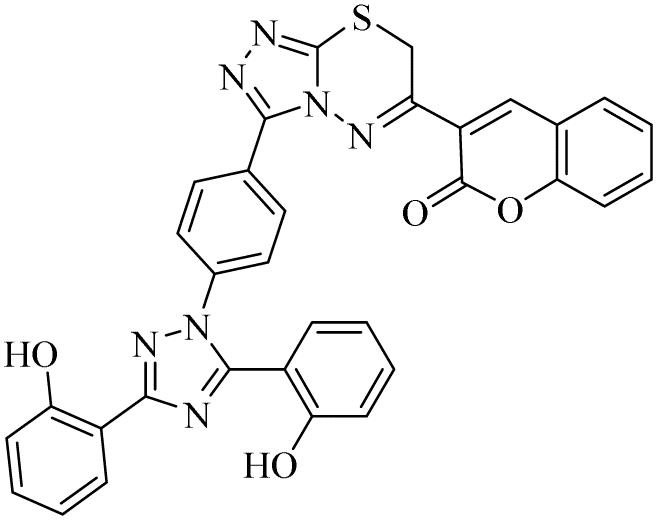 |
n.a | 10.84 ± 0.97 | 0.61 ± 0.07 | n.a | n.a | n.a |
| 23 | 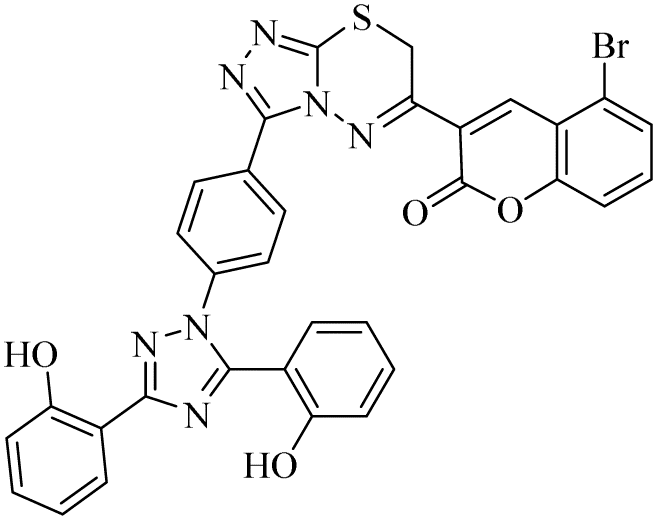 |
n.a | 13.99 ± 1.11 | 1.32 ± 0.11 | n.a | n.a | n.a |
| 24 | 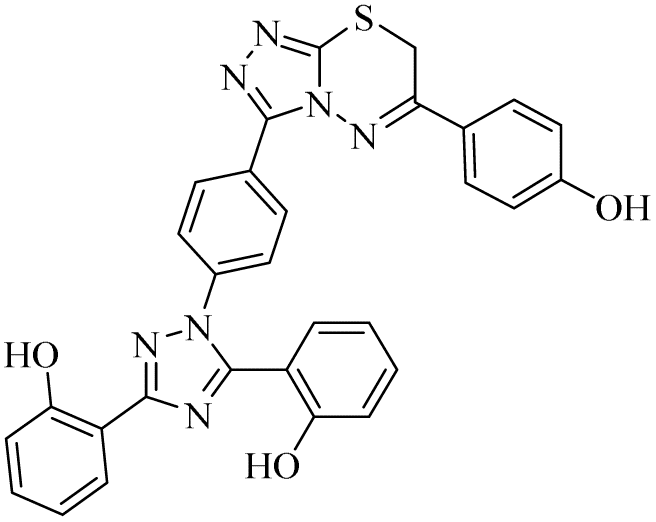 |
n.a | 0.17 ± 0.02 | 23.44 ± 1.67 | n.a | n.a | n.a |
| 25 | 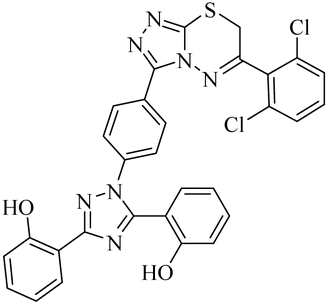 |
n.a | 1.09 ± 0.11 | 0.81 ± 0.02 | n.a | n.a | n.a |
| Donepezil | 0.047 ± 0.001 | 5.72 ± 0.43 | — | — | — | — | |
| Safinamide | — | — | 6.71 ± 0.76 | 0.027 ± 0.001 | — | — | |
| Celecoxib | — | — | — | — | 0.055 ± 0.002 | — | |
| Zileuton | — | — | — | — | — | 0.68 ± 0.009 | |
Zone 1 and zone 2 were similar in all inhibitors, while the only difference in the structures was in zone 3 due to the presence of different substituents at the aromatic ring, which are responsible for the different activities. According to the activity results shown in Table 1, the deferasirox derivatives (10–13 and 15–25) showed reasonable activity against different targets. Most of the compounds showed selective inhibition against different enzymes. Table 1 indicates their greater selectivity for BChE and MAO-A. Also, the activity of deferasirox was tested against all the enzymes in comparison to its derivatives.
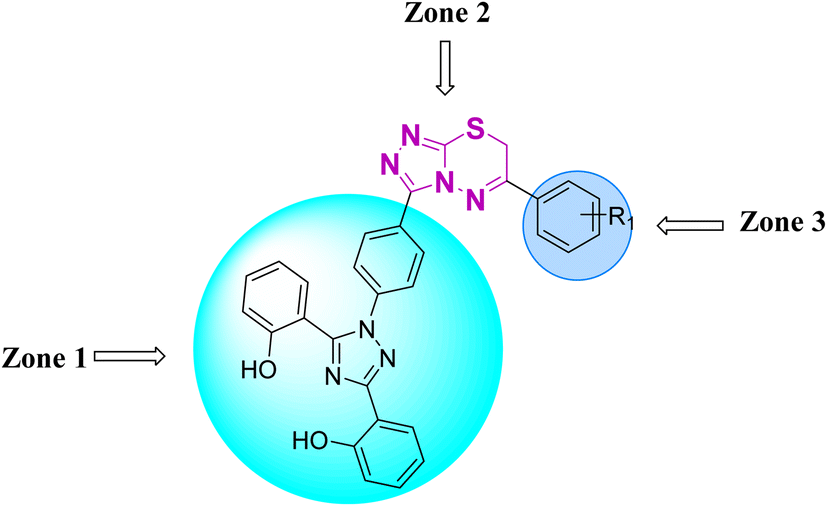
Regarding cholinesterase, all the derivatives were tested against AChE and BChE. In case of AChE, compound 11, a hydrazide derivative of deferasirox, showed greater inhibition potential with IC50 values of 0.31 ± 0.02 μM, which is due to the presence of an electron-donating group (–NH2). After, the activity of compound 13, the 1,3,4-triazole-2-thione amine derivative of deferasirox, was observed, showing an IC50 value of 0.71 ± 0.16 μM. The presence of a free NH2 group and five-membered ring having three nitrogen atoms makes it the most potent derivative of in this series. In contrast, the other compounds showed no activity against AChE. In the case of BChE, the compounds exhibited poor to excellent inhibition potential and this variation in activity is due to the presence of electron-withdrawing/donating groups at different positions in zone 3. Among them, the most potent activities were observed by compound 9, deferasirox, with an IC50 value of 0.11 ± 0.005 μM, 11, the hydrazide derivative, with an IC50 value of 0.094 ± 0.001 μM, 13, the triazoleamine derivative of deferasirox, with an IC50 value of 0.18 ± 0.02 μM, triazolo-thiadiazine derivative 19 having a methoxy group at the para position of zone 3 of the phenyl group with an IC50 value of 0.13 ± 0.02 μM and compound 24 having a hydroxy group at the para position of the phenyl ring in zone 3 with an IC50 value of 0.17 ± 0.02 μM. Low activity was shown by compound 16 with an IC50 value of 6.88 ± 0.39, compound 20 with an IC50 value of 0.46 ± 0.05 μM and compound 23 with an IC50 value of 13.99 ± 1.11 μM having electron-donating groups (–NO2, –OH, and –Br) on ortho and meta position of the phenyl ring in zone 3, respectively. Compound 19 showed good inhibition against BChE due to the presence of an electron-withdrawing group at the para position. Thus, according to the activity chart, it was observed that the presence of substitution at the para position of the phenyl ring in zone 3 makes these compounds more potent compared to substitution at the ortho and meta positions.24
In the case of both monoamine oxidases A and B, the presence of a carboxylic group in compound 9 made it the most potent inhibitor. The next efficient activities were observed by compound 11, a hydrazide derivative of deferasirox, where the presence of free NH2 and amide groups was the reason for its enhanced activity against both isozymes MAO A and B. Activity against the MAO A enzyme was observed by deferasirox-substituted triazolothiadiazole derivative 19 having methoxy group at the para position of the phenyl ring in zone 3, 22 having a coumarinyl group in zone 3 and 25 having two chloro groups at the 2 and 6 positions of the phenyl ring in zone 3. The presence of a methoxy group, fused phenyl rings and chloro groups in zone 3 enhanced the activity of compounds 19, 22, and 25.25 The other compounds showed moderate activity against MAO-A, while no activity was observed against MAO-B for the rest of compounds.
In the case of cyclooxygenase-2 activity, compound 13, the triazole amine derivative of deferasirox, showed good inhibition potential, while the other compounds showed moderate activity or no activity. In the case of 5-Lox, the most potent results were observed by compound 13, the triazole amine derivative of deferasirox, and compound 11, the hydrazide derivative of deferasirox, while the other compounds showed no activity against 5-LOX.26
2.4. Molecular docking investigations
To assess the binding affinities and inhibition profiles of the compounds within the active pocket of the targeted enzymes such as AChE, BuChE, MAO-A, MAO-B, COX-2, and LOX-5, a molecular docking analysis was conducted using the Molecular Operating Environment (MOE) software.27 Initially the co-crystal ligands were re-docked within the active sites of the targets to validate the docking protocol. After validation, the synthesized hybrids were allowed to dock within the active pockets of the targeted proteins. The docking poses of the compounds exhibited a similar conformation as that of the respective co-crystal ligands, validating the docking protocol, and in all cases, suitable RMSD values were observed. The crystallographic structure of the targets was obtained from the Protein Data Bank in the PDB format, specifically using the PDB accession code 1EVE for AChE,28 4BDS for BuChE,29 2Z5X for MAO-A,30 2V5Z for MAO-B,31 1CX2 for COX-2 (ref. 32) and 6N2W for 5-LOX.33 The binding score of each compound within the active pocket of targeted enzyme is given in the Tables S4–S6.†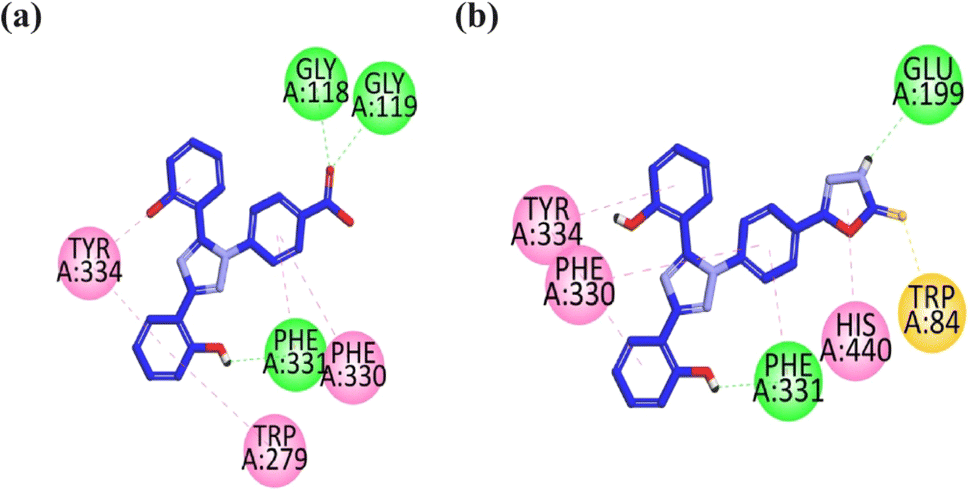 | ||
| Fig. 2 2D interaction plot of compounds 9 (a) and 13 (b) in the binding site of AChE (1EVE), modeled using the Discovery Studio Visualizer. | ||
The visual inspection of compound 9 within the active site of BChE revealed that the carbonyl group forms hydrogen bond interactions with the TRP82 and TRP430 amino acid residues. The two aromatic rings containing hydroxyl groups form hydrophobic interaction (pi–pi) with the GLY115, GLY116, TRP231, and PHE329 amino acid residues. The triazole ring forms hydrophobic interaction (pi–pi) with the HIS428 amino acid residue. The hydrophilic interaction with GLY117 is formed by the nitrogen atom of the triazole ring (Fig. 3a).
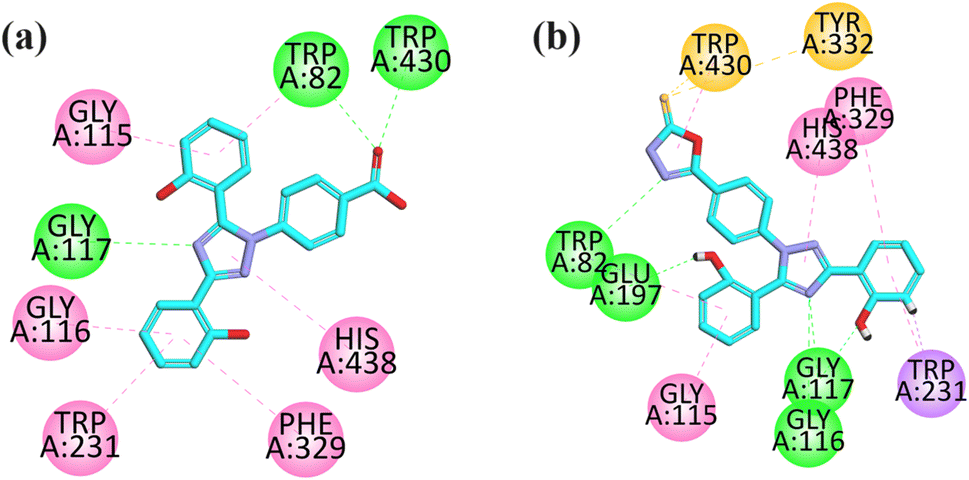 | ||
| Fig. 3 2D interaction plot of compounds 9 (a) and 12 (b) in the binding site of BuChE (4BDS), modeled by using the Discovery Studio Visualizer. | ||
The visual inspection of the binding mode of compound 12 in the binding site of BChE revealed that five hydrogen bond interactions are formed by this compound with the TRP82, GLU197, GLU117, and GLU116 amino acid residues. Three hydrophobic interactions (pi–pi) were formed by compound 12, where two are formed between HIS438 and PHE329 with a triazole ring and one is formed between the phenyl ring and GLY115 amino acid residue. In addition, a pi–sigma interaction is also formed by an aromatic ring with TRP231 (Fig. 3b).
Visual inspection of compound 18 in the active site of MAO-A revealed that the compound exhibited three hydrogen bond interactions with TYR69, ASN181, and ILE207 due to two –OH groups attached to the aromatic ring. The aromatic rings formed hydrophobic interaction (pi–pi) with TYR40 and PHE352 amino acid residues. One of the pi–lone pair interactions was observed by the triazole ring with GLN215. Two pi–sulfur interactions were displayed by the sulfur atom present in the ring with Cys406 and Trp397 (Fig. 4a).
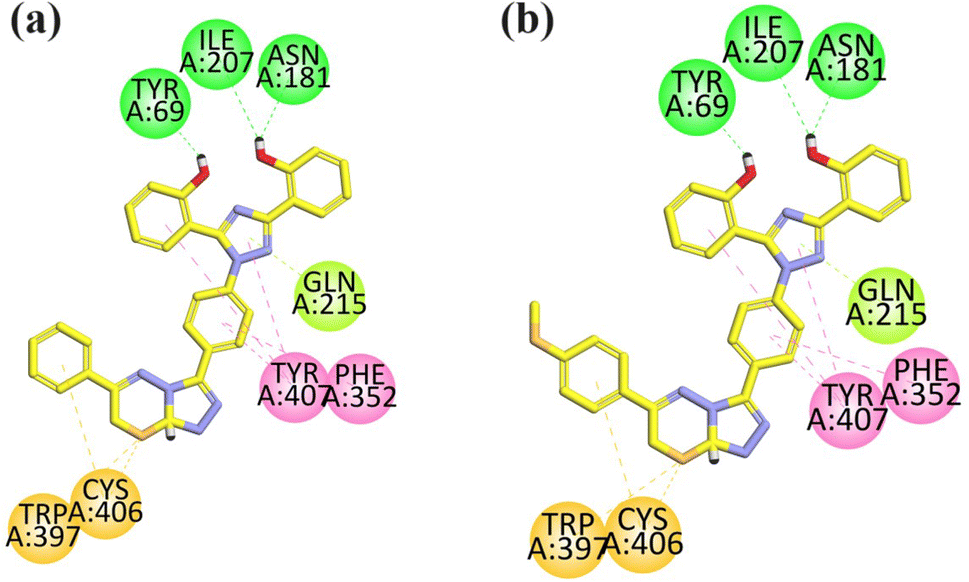 | ||
| Fig. 4 2D interaction plot of compounds 18 (a) and 21 (b) in the binding site of MAO-A (2Z5X), modeled using Discovery Studio Visualizer. | ||
The compound 21 was analyzed for its interactions within the active site of MAO-A. Structural analysis revealed that the compound showed three key hydrogen bond interactions involving TYR69, ASN181, ILE207, facilitated by two hydroxyl groups (–OH) attached to its aromatic ring, which helped in the stability of the complex. Additionally, the aromatic rings of the compound participated in hydrophobic (pi–pi) interactions with TYR40 and PHE352. The triazole ring of the compound formed a π–lone pair interaction with GLN215. Furthermore, the sulfur atom within the ring structure engaged in two π–sulfur interactions with CYS406 and TRP397 (Fig. 4b).
Visual inspection of the binding mode of compound 9 in the binding site of MAO-B revealed that one hydrogen bond interaction is formed between the carbonyl group and TRP388, and another hydrogen bond interaction is formed by the hydroxyl group of the aromatic ring with TYR60 toward the C-terminal (substrate binding site). The aromatic rings display pi–sulfur interactions with MET436, CYS397, and CYS172 (substrate binding site). TYR435 and TYR398 exhibit hydrogen bond interaction with the aromatic region in the substrate binding site, and also show hydrophobic interaction (pi–pi) with the aromatic ring of GLY57. The important interactions displayed by this compound are with the TYR435, TYR398, CYS172, and TYR60 amino acid residues (substrate binding region) (Fig. 5a).
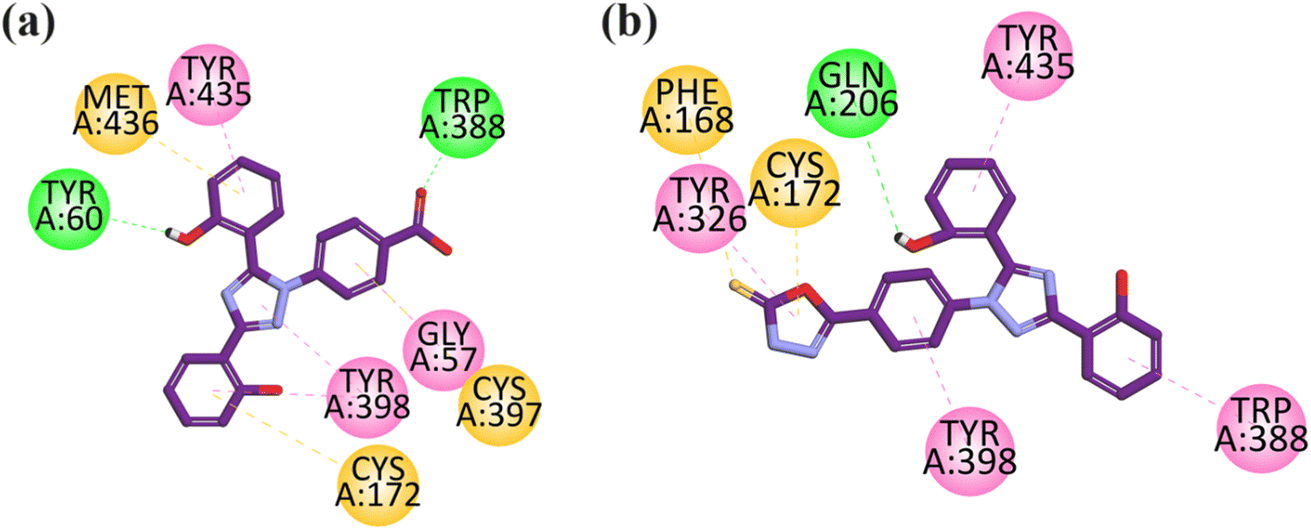 | ||
| Fig. 5 2D interaction plot of compounds 9 (a) and 12 (b) in the binding site of MAO-B (2V5Z), modeled using Discovery Studio Visualizer. | ||
Visual inspection of compound 12 in the binding site of MAO-B revealed that TYR435, TYR398, TYR326, and TRP588 lie in the aromatic region and exhibit hydrophobic interaction (pi–pi) towards the C-terminal (substrate binding site). The –OH group of the aromatic rings display hydrogen bond interaction with Gln 206 toward the entrance cavity (Fig. 5b).
The visual inspection of the binding mode of compound 13 in the binding site of COX-2 revealed that 4-amino-2,4-dihydro-3H-1,2,4-triazole-3-thione has strong binding interactions within the selective pocket region. It forms pi–sulfur interaction with HIS90, hydrophilic interaction with ARG513, and hydrophobic interaction with GLN192 and LEU352. The aromatic ring attached to triazole displays pi–sigma interaction in the selective pocket region with SER353. The triazole ring displays hydrophilic interaction with TYR355. The hydroxyl group containing an aromatic ring displays pi–pi interaction with PHE518. This compound orients toward the selective pocket region of COX-2 via HIS90, ARG513, SER353 and PHE518 (Fig. 6a).
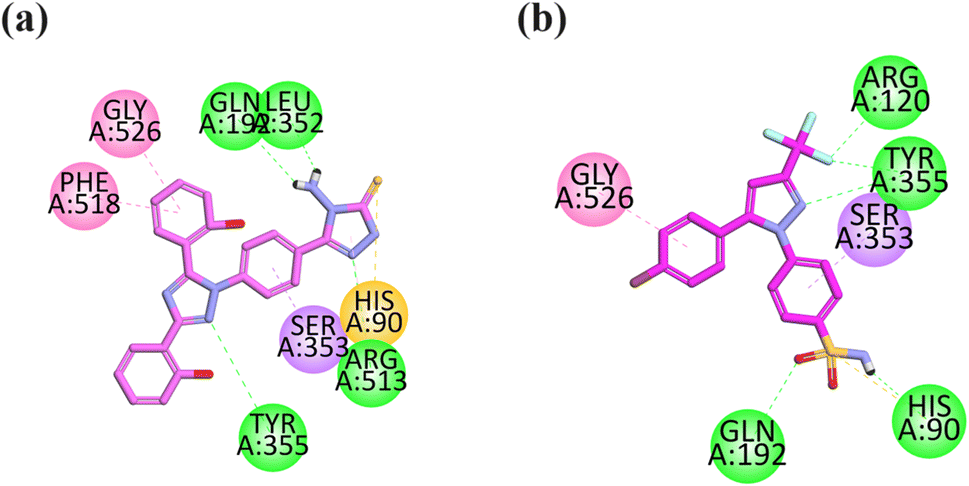 | ||
| Fig. 6 2D interaction plot of compounds 13 (a) and celecoxib (b) in the binding site of COX-2 (1CX2), modeled using Discovery Studio Visualizer. | ||
The co-crystalized selective inhibitor (native ligand) of COX-2 forms binding affinities with important residues including hydrophilic binding affinities with the COX-2 residue (ARG120, TYR355, GLN192, and HIS90), hydrophobic interactions with GLY526 and pi–sigma interaction with SER353 (Fig. 6b).
Visual inspection revealed that compound 13 orients itself in the pocket of 5-LOX with two –OH groups attached to the aromatic ring forming H-bonds (hydrophilic binding affinity) with important residues including ILE673 and ASN407, which is an important interaction explaining the stability of the drug–ligand complex. The hydrophobic interaction (pi–pi) with ALA410 is displayed by an aromatic ring attached with a hydroxyl ring at one end and a triazole ring at another end. The carbonyl group of 4-amino-2,4-dihydro-3H-1,2,4-triazole-3-thione displays three pi–sulfur interactions with PHE359 and HIS600 (Fig. 7a).
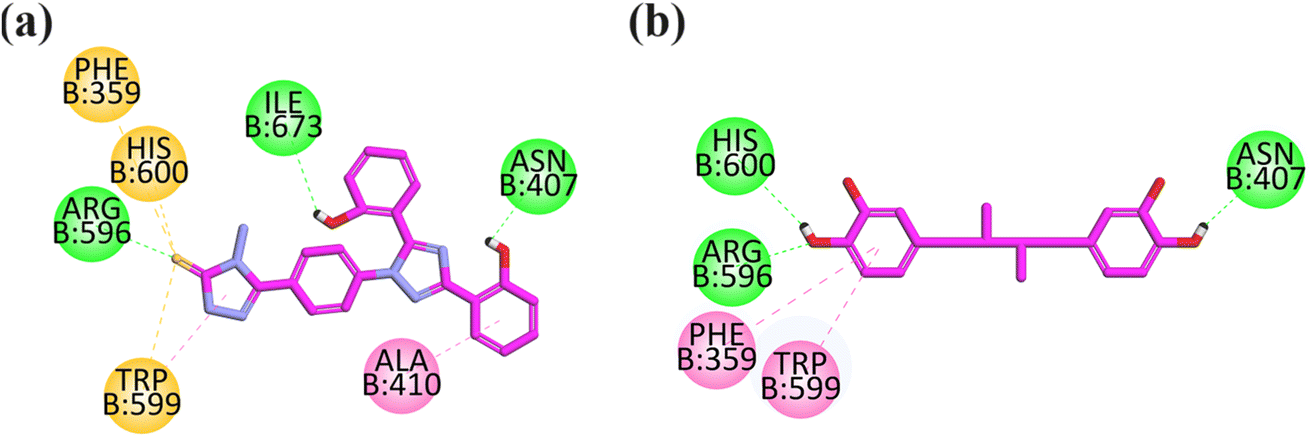 | ||
| Fig. 7 2D interaction plot of compounds 13 (a) and nordihydroguaiaretic acid (b) in the binding site of 5-LOX (6N2W), modeled using the Discovery Studio Visualizer. | ||
The co-crystallized structure of 5-lipoxygenase (6N2W) with nordihydroguaiaretic acid (NDGA) was selected for the docking analysis. NDGA showed hydrogen binding affinities with important residues including HIS600, ARG596, and ASN407, and hydrophobic interaction with PHE359 and TRP599 (Fig. 7b).
According to the molecular docking results, the results were found to be consistent with the in vitro investigations, suggesting that compound 11 is the most potent compound as a multi-targeting agent.
2.5 Molecular dynamics simulation
The molecular dynamics simulation was carried out to evaluate the strength, movement of atoms, and stiffness of the highest ranked docked pose of protein complexes 11 (AChE and MAO-B) and 13 (COX2). This was accomplished by utilizing the online web server iMOD (https://imods.iqf.csic.es/, which was accessed on August 23, 2024) and the outcomes of the docked complexes are shown in Fig. 7. The graphs describing the deformability of the protein complexes are shown in Fig. 8. The placement of various hinges, both high and low, illustrates the area of residues that are distorted in the docked complexes. Through the use of the experimental B-factor, the value of root mean square (RMS) was established, hence revealing the unpredictability of atoms inside the protein complexes. According to the supplied data, the eigenvalues of compounds 11 and 13 were found to be 2.732398 × 10−33, 8.716516 × 10−44, and 9.838933 × 10−44 for AChE, MAO-B, and COX-2, respectively. The eigenvalue is linked to the normal mode and demonstrates motion inflexibility, and it is proportional to the binding energy that is associated with the process of deforming the globular structure of the protein. The variance graph of amino acid residues is shown in Fig. 9, illustrating how the eigenvalue is inversely proportional to the variance. Alternatively, the covariance matrix, as shown in Fig. 7–9, stipulates that the pairing between amino acid residues experiences movements in distinct colors. These colors include red (correlated), white (uncorrelated), and blue (anti-correlated). The coupling pair of atoms that are considered to be related with springs is characterized by the elastic model. As can be seen in Fig. 8–10, the various dots, which are grey in color, that are present in the graph represent one spring between the interrelating atoms that constitute the inelastic springs.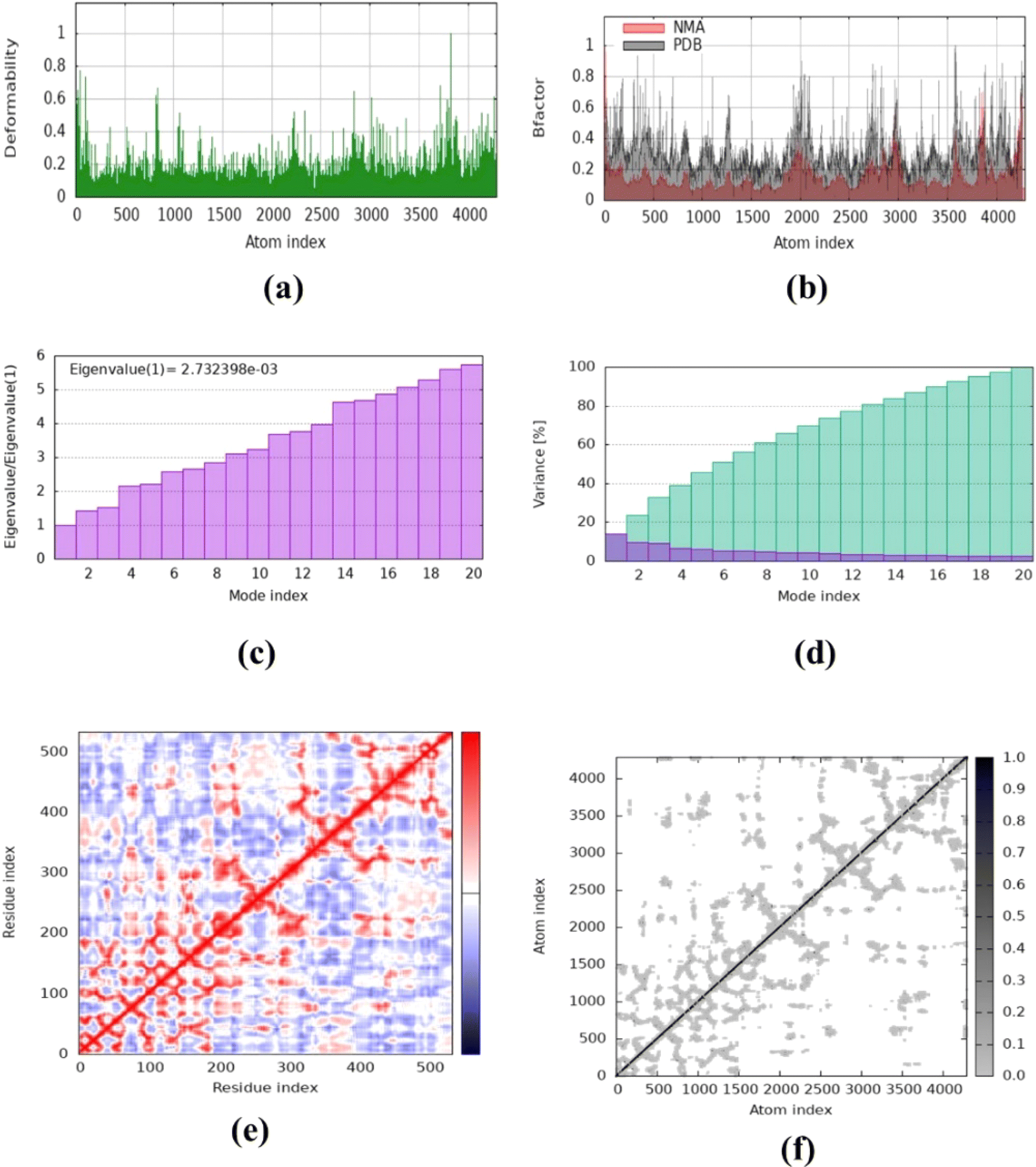 | ||
| Fig. 8 Molecular dynamics simulation of complex 11 (AChE) using iMODS. (a) Deformability, (b) B-factor values, (c) eigenvalue, (d) variance, (e) co-variance and (f) elastic network model. | ||
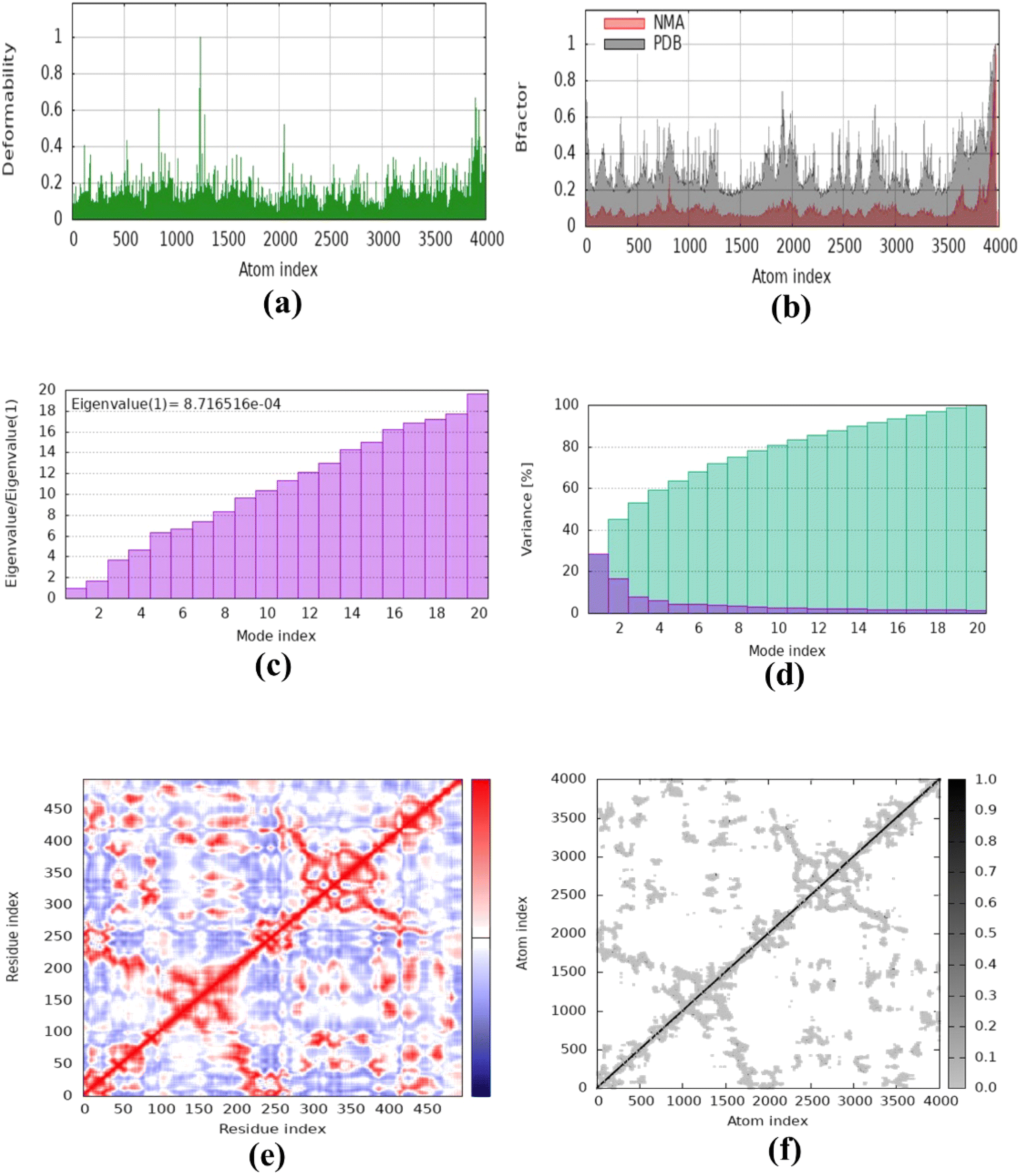 | ||
| Fig. 9 Molecular dynamics simulation of complex 11 (MAO-B) using iMODS. (a) Deformability, (b) B-factor values, (c) eigenvalue, (d) variance, (e) co-variance and (f) elastic network model. | ||
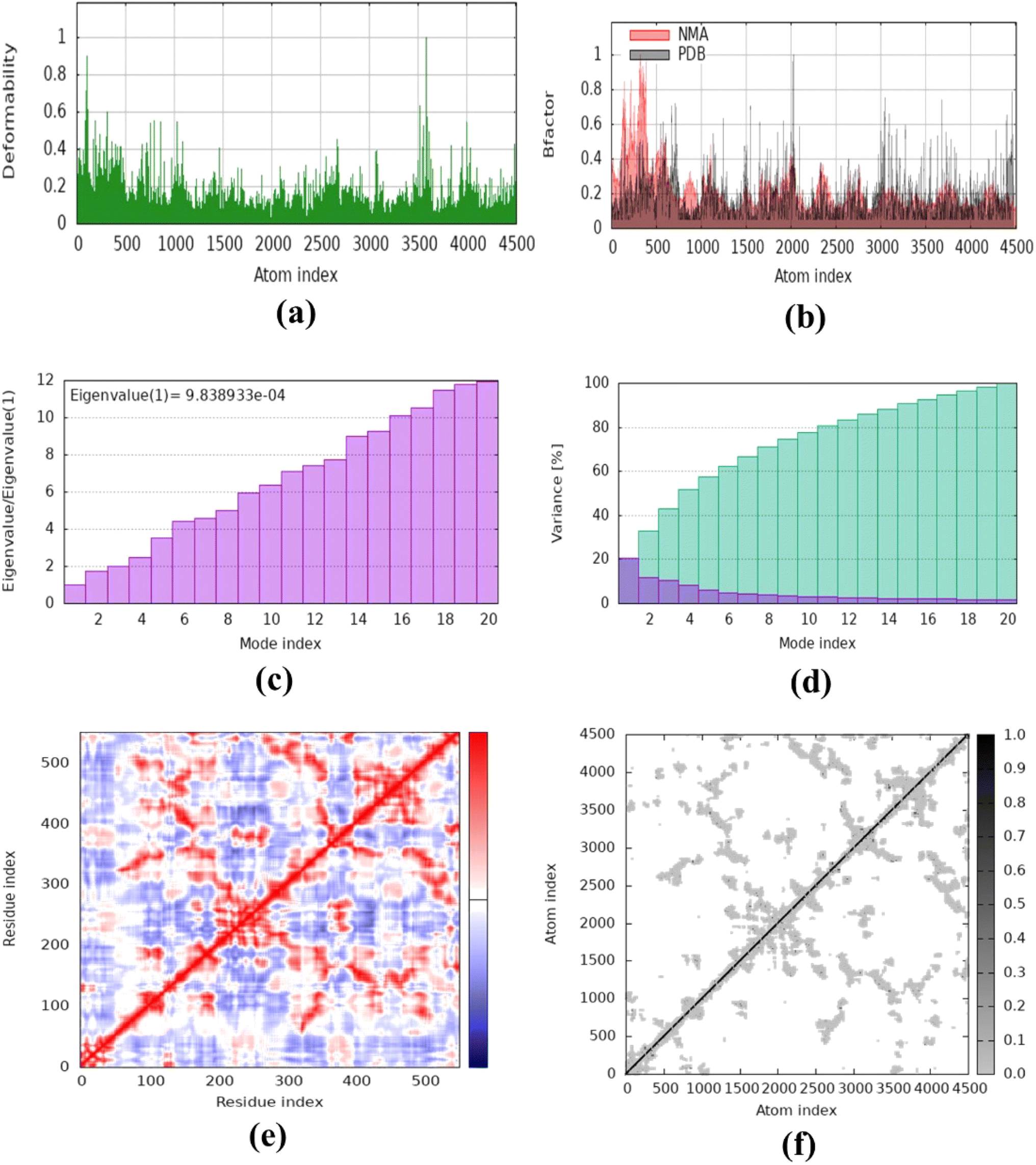 | ||
| Fig. 10 Molecular dynamics simulation of complex 13 (COX-2) using iMODS. (a) Deformability, (b) B-factor values, (c) eigenvalue, (d) variance, (e) co-variance and (f) elastic network model. | ||
2.6. ADMET analysis
The investigation focused on assessing the physicochemical characteristics of the synthesized hybrid compounds 9–11, 13, 15 and 16–25 utilizing the SwissADME tool.34 The detailed description is provided in the ESI File.†3 Conclusions
This study described the synthesis and evaluation of new compounds aimed at addressing Alzheimer's disease (AD), a major cause of dementia worldwide. Given the complex nature of AD, characterized by both neuroinflammation and disruptions in lipid metabolism, there is a significant need for drugs that can target multiple pathways simultaneously. This study involved the synthesis of triazolo-thiadiazine derivatives of deferasirox, aiming to create multi-target compounds that can inhibit the above-mentioned enzymes. In particular, compound 11 shows potential as a lead molecule due to its strong inhibitory effects on AChE, MAO-B, and COX-2. Other compounds, such as 9 and 13, also showed dual inhibition, with their performance surpassing that of the standard drugs. The simulations and physicochemical properties of the lead compounds support their potential as promising candidates for treating multifactorial diseases such as AD, which involve neuro-inflammation. Conclusively, this research paves the way for the further development of these hybrid compounds as potential treatments for AD, aiming to provide more effective disease-modifying therapies.4 Experimental
4.1. Materials and methods
All chemicals and reagents: deferasirox, hydrazine monohydrate, carbon disulfide (CS2), potassium hydroxide (KOH), substituted acetophenones, acetic acid, sulphuric acid (H2SO4), n-hexane, ethyl acetate, diethyl ether, acetone, methanol and ethanol were purchased from Sigma Aldrich.Transparent oily liquid. Yield: 55%, Rf = 0.7, (n-hexane/ethyl acetate = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), M.P: 194–196 °C, FT-IR (KBr disc) γmax (cm−1): 3280 (O–H), 2920 (C–H), 1721 (C
:3), M.P: 194–196 °C, FT-IR (KBr disc) γmax (cm−1): 3280 (O–H), 2920 (C–H), 1721 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1587 (CC), 1491 (CN), 1465 (C–N), 1277 (C–O). 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H, OH), 8.15–8.10 (m, 2H), 8.03 (dd, J = 13.0, 7.9 Hz, 4H), 7.57 (dd, J = 12.6, 7.8 Hz, 3H), 7.38 (q, J = 7.5 Hz, 3H), 7.00 (m, 5H), 6.86 (d, J = 8.2 Hz, 1H), 3.86 (s, 3H).21
O), 1587 (CC), 1491 (CN), 1465 (C–N), 1277 (C–O). 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H, OH), 8.15–8.10 (m, 2H), 8.03 (dd, J = 13.0, 7.9 Hz, 4H), 7.57 (dd, J = 12.6, 7.8 Hz, 3H), 7.38 (q, J = 7.5 Hz, 3H), 7.00 (m, 5H), 6.86 (d, J = 8.2 Hz, 1H), 3.86 (s, 3H).21
White solid. Yield: 90%, Rf = 0.4, (n-hexane/ethyl acetate = 3:2), M.P: 259–261 °C, FT-IR (KBr disc) γmax (cm−1): 3297 (O–H),3185 (N–H), 3058 (C–H), 1702 (CO), 1608 (CC), 1492 (CN), 1460 (C–N), 1289 (C–O). 1H NMR (400 MHz, DMSO-d6) δ: 10.83 (s, 1H, OH-24), 10.08 (s, 1H, NH-28), 9.87 (s, 1H, OH-25), 8.04 (d, 1H, Ar-H-9), 7.87 (d, J = 8.3 Hz, 2H, Ar-H-20 & Ar-H-22), 7.56–7.48 (m, 3H, Ar-H-18, Ar- 34 H-19 & Ar-H-23), 7.42–7.33 (m, 2H, Ar-H-11 & Ar-H-16), 7.06–6.94 (m, 3H, Ar-H-10, ArH-12 & Ar-H-17), 6.86 (d, J = 8.2 Hz, 1H, Ar-H-15), 4.55 (s, 2H, NH2-29), 13C NMR (150 MHz, DMSO-d6) δ 165.1, 160.2, 156.8, 155.7, 152.4, 140.2, 132.9, 132.5, 131.8, 131.5, 129.0, 128.4, 127.2, 123.8, 123.7, 120.1, 119.8, 117.5, 116.6, 114.9, 114.1.
Light-yellowish solid, Yield: 70%, Rf = 0.6, (n-hexane/ethyl acetate = 2:3), M.P: 294–296 °C, FT-IR (KBr disc) γmax (cm−1): 3308 (O–H), 3209 (N–H), 3075 (C–H), 1615 (CC), 1584 (CN), 1514 (C–N), 1294 (C–O),1155 (CS), 1H NMR (400 MHz, DMSO-d6) δ: 14.02 (s, 1H, NH-29), 10.82 (s, 1H, OH-24), 10.15 (s, 1H, OH-25), 8.10 (d, J = 8.5 Hz, 2H, Ar-H-20 & Ar-H-22), 8.05 (dd, J = 7.8, 1.7 Hz, 1H, Ar-H-9), 7.60 (d, 2H, Ar-H-19 & Ar-H-23), 7.56 (dd, J = 7.7, 1.7 Hz, 1H, Ar-H-18), 7.43–7.33 (m, 2H, Ar-H-11 & Ar-H-16), 7.06–6.95 (m, 3H, Ar-H-10, Ar-H-12 & Ar-H-17), 6.88 (d, J = 8.2 Hz, 1H, Ar-H-15), 5.80 (s, 2H, NH2-30). 13C NMR (100 MHz, DMSO-d6) δ: 167.7, 160.3, 156.8, 155.7, 152.4, 149.1, 139.7, 133.0, 131.9, 131.6, 129.3, 127.2, 126.1, 123.9, 120.2, 119.9, 117.5, 116.6, 114.9, 114.1.21
:3), M.P: 250–252 °C, FT-IR (KBr disc) γmax (cm−1): 3061 (O–H), 2850 (C–H), 1612 (CC), 1587 (CN), 1453 (C–N), 1249 (C–O), 848 (C–Cl), 760 (C–S–C), 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.12 (s, 1H), 8.16 (d, J = 8.2 Hz, 2H), 8.12–8.02 (m, 5H), 7.65 (d, J = 8.6 Hz, 2H), 7.57 (dd, J = 7.7, 1.7 Hz, 1H), 7.45–7.33 (m, 2H), 7.06–6.95 (m, 3H), 6.88 (d, J = 8.2 Hz, 1H), 4.47 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 159.8, 156.3, 155.2, 154.9, 151.9, 150.9, 142.8, 139.1, 137.6, 133.0, 132.5, 131.4, 131.1, 128.7, 128.4, 126.7, 125.6, 123.6, 119.7, 119.4, 118.2, 117.0, 116.1, 114.4, 114.0, 113.7, 56.0, 23.2, HRMS (ESI) m/z, Calcd. for (C30H20ClN7O2S + H+) 578.05, Found 569.2614.:2), M.P: 233–235 °C, FT-IR (KBr disc) γmax (cm−1): 3252 (O–H), 3088 (C–H), 1621 (CC), 1587 (CN), 1530 (ONO), 1494 (C–N), 1253 (C–O), 760 (C–S–C); 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.13 (s, 1H), 8.76 (t, J = 2.0 Hz, 1H), 8.43 (ddd, J = 13.1, 7.8, 2.0 Hz, 2H), 8.12–8.00 (m, 3H), 7.87 (t, J = 8.0, 1.3 Hz, 1H), 7.69–7.61 (m, 2H), 7.57 (dd, J = 7.7, 1.7 Hz, 1H), 7.38 (m, 2H), 7.06–6.94 (m, 3H), 6.88 (d, J = 8.2 Hz, 1H), 4.52 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.3, 156.8, 155.7, 155.0, 152.4, 151.3, 148.7, 143.4, 139.5, 135.5, 134.1, 132.9, 131.9, 131.5, 131.2, 129.2, 127.2, 126.8, 126.0, 124.0, 122.8, 120.1, 119.9, 117.5, 116.5, 114.9, 114.1, 23.4, HRMS (ESI) m/z, Calcd. for (C30H20N8O4S + H+) 588.6, Found 589.6000.:2), M.P: 280–282 °C. FT-IR (KBr disc) γmax (cm−1): 3319 (O–H), 3059 (C–H), 2226 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1612 (CC), 1587 (CN), 1455 (C–N), 1299 (C–O), 759 (C–S–C)1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.12 (s, 1H), 8.16 (d, J = 8.2 Hz, 2H), 8.06 (dd, J = 8.5, 3.1 Hz, 5H), 7.65 (d, J = 8.4 Hz, 2H), 7.57 (dd, J = 7.6, 1.7 Hz, 1H), 7.45–7.33 (m, 2H), 7.06–6.95 (m, 3H), 6.88 (d, J = 8.2 Hz, 1H), 4.47 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.3, 156.8, 155.7, 155.3, 152.4, 151.3, 143.3, 139.5, 138.0, 133.4, 133.0, 131.9, 131.5, 129.2, 128.8, 127.2, 126.0, 124.1, 120.1, 119.9, 118.7, 117.5, 116.5, 114.9, 114.5, 114.1, 23.2 HRMS (ESI) m/z, Calcd. for (C31H20N8O2S + H+) 568.6, Found 569.6130.:3), M.P: 282–284 °C. FT-IR (KBr disc) γmax (cm−1): 3632 (O–H), 3058 (C–H), 1613 (CC), 1582 (CN), 1457 (C–N), 1299 (C–O), 757 (C–S–C) 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.11 (s, 1H), 8.12–7.97 (m, 5H), 7.69–7.53 (m, 6H), 7.45–7.33 (m, 2H), 7.07–6.95 (m, 3H), 6.89 (d, J = 8.3 Hz, 1H), 4.45 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.3, 156.8, 156.7, 155.7, 152.4, 151.2, 143.4, 139.4, 133.8, 132.96, 132.5, 131.9, 131.5, 129.6, 129.1, 128.1, 127.2, 126.2, 124.1, 120.1, 119.9, 117.5, 116.5, 114.9, 114.1, 23.2; HRMS (ESI) m/z, Calcd. for (C30H21N7O2S + H+) 543.63, Found 544.6383.:1), M.P: 258–260 °C. FT-IR (KBr disc) γmax (cm−1): 3193 (O–H), 3055 (C–H), 1588 (CC), 1513 (CN), 1459 (C–N), 1299 (C–O), 753 (C–S–C).1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.12 (s, 1H), 8.08 (dd, J = 11.0, 8.3 Hz, 3H), 8.02–7.95 (m, 2H), 7.64 (d, J = 8.5 Hz, 1H), 7.57 (dd, J = 7.6, 1.7 Hz, 2H), 7.39 (d, J = 8.3 Hz, 2H), 7.12 (d, J = 8.7 Hz, 1H), 7.07–6.93 (m, 4H), 6.88 (d, J = 8.3 Hz, 1H), 4.40 (s, 1H), 3.86 (s, 4H); 13C NMR (100 MHz, DMSO-d6, DEPT-135) δ: 132.9, 131.9, 131.5, 130.2, 129.0, 127.2, 124.0, 120.1, 117.5, 116.5, 115.0, 23.4, HRMS (ESI) m/z, Calcd. for (C31H23N7O3S + H+) 573.63, Found 574.6562.:3), M.P: 273–275 °C. FT-IR (KBr disc) γmax (cm−1): 3260 (O–H), 3047 (C–H), 1622 (CC), 1588 (CN), 1462 (C–N), 1285 (C–O), 751 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 10.81 (s, 1H), 10.10 (s, 1H), 8.09–8.01 (m, 3H), 7.67–7.62 (m, 2H), 7.65–7.50 (m, 3H), 7.42–7.32 (m, 2H), 6.99 (ddd, J = 16.7, 7.9, 3.5 Hz, 5H), 6.87 (d, J = 8.2 Hz, 1H), 4.28 (s, 2H); 13C NMR (100 MHz, DMSO-d6, DEPT-135) δ:135.6, 132.9, 132.3, 131.8, 131.5, 129.0, 127.2, 124.0, 120.1, 119.8, 119.2, 117.5, 116.5, 23.4.:3), M.P: 230–232 °C. FT-IR (KBr disc) γmax (cm−1): 3066 (O–H), 2981 (C–H), 1620 (CC), 1583 (CN), 1460 (C–N), 1353 (C–O), 750 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 10.14 (s, 1H), 8.11–8.04 (m, 3H), 7.86 (d, J = 8.4, 1H), 7.60–7.56 (m, 3H), 7.41–7.31 (m, 4H), 7.02–6.95 (m, 4H), 6.87 (d, J = 8.4, 1H), 5.80 (s, 2H), 1.39 (s, 3H, SCH3), HRMS (ESI) m/z, Calcd. for (C31H23N7O2S2 + H+) 589.60, Found 590.6937.O), 1620 (CC), 1587 (CN), 1461 (C–N), 1353 (C–O), 752 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.12 (s, 1H), 8.16 (d, J = 8.2 Hz, 2H), 8.06 (m, 3H), 7.65 (d, J = 8.4 Hz, 2H), 7.57 (dd, J = 7.6, 1.7 Hz, 1H), 7.45–7.33 (m, 2H), 7.30–7.10 (m, 3H), 7.06–6.95 (m, 3H), 6.88 (d, J = 8.2 Hz, 1H), 4.47 (s, 2H).O), 1620 (CC), 1587 (CN), 1461 (C–N), 1353 (C–O), 828 (C–Br), 752 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.13 (s, 1H), 8.76 (t, J = 2.0 Hz, 1H), 8.43 (m, 2H), 8.12–8.00 (m, 3H), 7.87 (t, J = 8.0 Hz, 1H), 7.69–7.61 (m, 2H), 7.57 (dd, J = 7.7, 1.7 Hz, 1H), 7.38 (m, 2H), 7.06–6.94 (m, 3H), 6.88 (d, J = 8.2 Hz, 1H), 4.52 (s, 2H).C), 1514 (CN), 1457 (C–N), 1228 (C–O), 752 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.12 (s, 1H), 8.16 (d, J = 8.2 Hz, 2H), 8.12–8.02 (m, 4H), 7.65 (d, J = 8.6 Hz, 2H), 7.57 (dd, J = 7.7, 1.7 Hz, 1H), 7.45–7.33 (m, 2H), 7.06–6.95 (m, 3H), 6.72 (s, 1H), 6.88 (d, J = 8.2 Hz, 1H), 4.47 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 161.8, 160.2, 156.8, 156.3, 155.8, 152.4, 150.9, 143.3, 139.3, 132.9, 131.9, 131.5, 130.1, 129.0, 127.2, 126.4, 124.0, 120.1, 119.8, 117.5, 116.5, 116.4, 114.9, 114.1, 22.9.C), 1580 (CN), 1491 (C–N), 1242 (C–O), 982 (C–Cl), 751 (C–S–C), 1H NMR (400 MHz, DMSO-d6) δ: 10.81 (s, 1H), 10.09 (s, 1H), 8.05 (d, 1H, J = 8.6), 7.91 (d, 1H, J = 8.3), 7.88–7.85 (m, 3H), 7.79–7.74 (m, 4H), 7.37 (m, 3H), 6.08–6.87 (m, 3H), 4.32 (s, 2H), 13C NMR (101 MHz, DMSO-d6) δ 160.2, 156.8, 156.5, 155.6, 152.3, 139.6, 136.8, 133.5, 133.2, 132.9, 131.9, 131.5, 131.1, 131.1, 130.9, 130.6, 130.3, 129.1, 128.0, 127.9, 127.2, 124.1, 120.1, 119.9, 117.5, 116.6, 114.8, 114.1, 67.9,23.4, HRMS (ESI) m/z, Calcd. for (C30H19Cl2N7O2S) 612.49, Found 612.4974.
N), 1612 (CC), 1587 (CN), 1455 (C–N), 1299 (C–O), 759 (C–S–C)1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.12 (s, 1H), 8.16 (d, J = 8.2 Hz, 2H), 8.06 (dd, J = 8.5, 3.1 Hz, 5H), 7.65 (d, J = 8.4 Hz, 2H), 7.57 (dd, J = 7.6, 1.7 Hz, 1H), 7.45–7.33 (m, 2H), 7.06–6.95 (m, 3H), 6.88 (d, J = 8.2 Hz, 1H), 4.47 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.3, 156.8, 155.7, 155.3, 152.4, 151.3, 143.3, 139.5, 138.0, 133.4, 133.0, 131.9, 131.5, 129.2, 128.8, 127.2, 126.0, 124.1, 120.1, 119.9, 118.7, 117.5, 116.5, 114.9, 114.5, 114.1, 23.2 HRMS (ESI) m/z, Calcd. for (C31H20N8O2S + H+) 568.6, Found 569.6130.:3), M.P: 282–284 °C. FT-IR (KBr disc) γmax (cm−1): 3632 (O–H), 3058 (C–H), 1613 (CC), 1582 (CN), 1457 (C–N), 1299 (C–O), 757 (C–S–C) 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.11 (s, 1H), 8.12–7.97 (m, 5H), 7.69–7.53 (m, 6H), 7.45–7.33 (m, 2H), 7.07–6.95 (m, 3H), 6.89 (d, J = 8.3 Hz, 1H), 4.45 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.3, 156.8, 156.7, 155.7, 152.4, 151.2, 143.4, 139.4, 133.8, 132.96, 132.5, 131.9, 131.5, 129.6, 129.1, 128.1, 127.2, 126.2, 124.1, 120.1, 119.9, 117.5, 116.5, 114.9, 114.1, 23.2; HRMS (ESI) m/z, Calcd. for (C30H21N7O2S + H+) 543.63, Found 544.6383.:1), M.P: 258–260 °C. FT-IR (KBr disc) γmax (cm−1): 3193 (O–H), 3055 (C–H), 1588 (CC), 1513 (CN), 1459 (C–N), 1299 (C–O), 753 (C–S–C).1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.12 (s, 1H), 8.08 (dd, J = 11.0, 8.3 Hz, 3H), 8.02–7.95 (m, 2H), 7.64 (d, J = 8.5 Hz, 1H), 7.57 (dd, J = 7.6, 1.7 Hz, 2H), 7.39 (d, J = 8.3 Hz, 2H), 7.12 (d, J = 8.7 Hz, 1H), 7.07–6.93 (m, 4H), 6.88 (d, J = 8.3 Hz, 1H), 4.40 (s, 1H), 3.86 (s, 4H); 13C NMR (100 MHz, DMSO-d6, DEPT-135) δ: 132.9, 131.9, 131.5, 130.2, 129.0, 127.2, 124.0, 120.1, 117.5, 116.5, 115.0, 23.4, HRMS (ESI) m/z, Calcd. for (C31H23N7O3S + H+) 573.63, Found 574.6562.:3), M.P: 273–275 °C. FT-IR (KBr disc) γmax (cm−1): 3260 (O–H), 3047 (C–H), 1622 (CC), 1588 (CN), 1462 (C–N), 1285 (C–O), 751 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 10.81 (s, 1H), 10.10 (s, 1H), 8.09–8.01 (m, 3H), 7.67–7.62 (m, 2H), 7.65–7.50 (m, 3H), 7.42–7.32 (m, 2H), 6.99 (ddd, J = 16.7, 7.9, 3.5 Hz, 5H), 6.87 (d, J = 8.2 Hz, 1H), 4.28 (s, 2H); 13C NMR (100 MHz, DMSO-d6, DEPT-135) δ:135.6, 132.9, 132.3, 131.8, 131.5, 129.0, 127.2, 124.0, 120.1, 119.8, 119.2, 117.5, 116.5, 23.4.:3), M.P: 230–232 °C. FT-IR (KBr disc) γmax (cm−1): 3066 (O–H), 2981 (C–H), 1620 (CC), 1583 (CN), 1460 (C–N), 1353 (C–O), 750 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 10.14 (s, 1H), 8.11–8.04 (m, 3H), 7.86 (d, J = 8.4, 1H), 7.60–7.56 (m, 3H), 7.41–7.31 (m, 4H), 7.02–6.95 (m, 4H), 6.87 (d, J = 8.4, 1H), 5.80 (s, 2H), 1.39 (s, 3H, SCH3), HRMS (ESI) m/z, Calcd. for (C31H23N7O2S2 + H+) 589.60, Found 590.6937.O), 1620 (CC), 1587 (CN), 1461 (C–N), 1353 (C–O), 752 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.12 (s, 1H), 8.16 (d, J = 8.2 Hz, 2H), 8.06 (m, 3H), 7.65 (d, J = 8.4 Hz, 2H), 7.57 (dd, J = 7.6, 1.7 Hz, 1H), 7.45–7.33 (m, 2H), 7.30–7.10 (m, 3H), 7.06–6.95 (m, 3H), 6.88 (d, J = 8.2 Hz, 1H), 4.47 (s, 2H).O), 1620 (CC), 1587 (CN), 1461 (C–N), 1353 (C–O), 828 (C–Br), 752 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.13 (s, 1H), 8.76 (t, J = 2.0 Hz, 1H), 8.43 (m, 2H), 8.12–8.00 (m, 3H), 7.87 (t, J = 8.0 Hz, 1H), 7.69–7.61 (m, 2H), 7.57 (dd, J = 7.7, 1.7 Hz, 1H), 7.38 (m, 2H), 7.06–6.94 (m, 3H), 6.88 (d, J = 8.2 Hz, 1H), 4.52 (s, 2H).C), 1514 (CN), 1457 (C–N), 1228 (C–O), 752 (C–S–C). 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 10.12 (s, 1H), 8.16 (d, J = 8.2 Hz, 2H), 8.12–8.02 (m, 4H), 7.65 (d, J = 8.6 Hz, 2H), 7.57 (dd, J = 7.7, 1.7 Hz, 1H), 7.45–7.33 (m, 2H), 7.06–6.95 (m, 3H), 6.72 (s, 1H), 6.88 (d, J = 8.2 Hz, 1H), 4.47 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 161.8, 160.2, 156.8, 156.3, 155.8, 152.4, 150.9, 143.3, 139.3, 132.9, 131.9, 131.5, 130.1, 129.0, 127.2, 126.4, 124.0, 120.1, 119.8, 117.5, 116.5, 116.4, 114.9, 114.1, 22.9.C), 1580 (CN), 1491 (C–N), 1242 (C–O), 982 (C–Cl), 751 (C–S–C), 1H NMR (400 MHz, DMSO-d6) δ: 10.81 (s, 1H), 10.09 (s, 1H), 8.05 (d, 1H, J = 8.6), 7.91 (d, 1H, J = 8.3), 7.88–7.85 (m, 3H), 7.79–7.74 (m, 4H), 7.37 (m, 3H), 6.08–6.87 (m, 3H), 4.32 (s, 2H), 13C NMR (101 MHz, DMSO-d6) δ 160.2, 156.8, 156.5, 155.6, 152.3, 139.6, 136.8, 133.5, 133.2, 132.9, 131.9, 131.5, 131.1, 131.1, 130.9, 130.6, 130.3, 129.1, 128.0, 127.9, 127.2, 124.1, 120.1, 119.9, 117.5, 116.6, 114.8, 114.1, 67.9,23.4, HRMS (ESI) m/z, Calcd. for (C30H19Cl2N7O2S) 612.49, Found 612.4974.4.2. Biological assay
COX-2 inhibition experiments were conducted using the Glassman and White methodology with minor modifications. The COX-2 enzyme (300 units per mL) was activated on ice for 5 min prior to the assay, followed by the introduction of a co-factor solution (50 μL) comprised of N,N,N,N-tetramethyl-p-phenylenediamine dihydrochloride (TMPD) (0.24 mM), glutathione (0.9 mM), and hematin (0.1 mM) in 0.1 M Tris HCl buffer at pH 8.0. Solutions of tested substances (20 μL) with varying concentrations from 31.25 to 1000 μg mL−1 and enzyme solutions (60 μL) were kept at room temperature for 5 min, after which arachidonic acid (30 mM, 20 μL) was added to initiate the reaction. The reaction mixture was incubated for 5 min, following which the absorbance was measured at 570 nm using a UV-visible spectrophotometer. The percentage of COX-2 inhibition was calculated based on the absorbance value per unit of time. Celecoxib served as a positive control for COX-2. The IC50 values (in μM) were determined by plotting percentage inhibition versus concentration of the sample solution.37
000 U mL−1) were used for the assay. Linoleic acid (80 mM) was used as the substrate, while phosphate (50 mM) was used as the buffer with pH 6.3. Mixing of the buffers and the samples and lipoxidase enzyme solution (0.25 mL) was carried out and the mixture was incubated for 5 min. After, linoleic acid (1 mL, 0.6 mM) was added to the mixture. Using a spectrophotometer, the absorbance was measured at a wavelength of 234 nm. The experiment was repeated in triplicate. Zileuton was used as the standard drug. The percent inhibition was calculated using the following equation: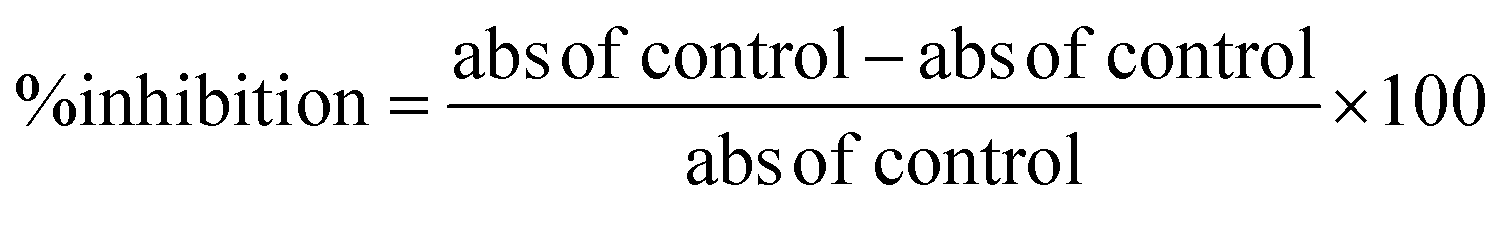
The IC50 values (μM) were determined by plotting inhibition against sample solution concentration.37
4.3. Methodology for molecular docking
The docking studies were conducted using Molecular Operating Environment (MOE 2016.08). The Protein Data Bank (PDB code 1CX2) was utilized to retrieve the enzyme structures, which were validated through re-docking with their native ligands. Previously reported methods were employed for the preparation of the ligands, downloading the enzymes, energy minimization, 3D protonation, and determination of the binding site.1,29–33 The Ligand.mdb database of compounds were drawn using the Builder option in MOE. The energy minimization up to 0.01 Gradient was done by using the MMFF94X force field. The MOE window was used to open the enzyme structures. Removal of water molecules (if present) was carried out. The 3D protonation was done for all atoms in the environment at a temperature of 300 K, pH of 7, and salt concentration of 0.1 M. By using the MMFF94X force field, the energy of the complete structure was minimized, followed by docking the compounds in the binding sites of the enzyme. The docking parameters were established for each compound, and ten distinct conformations were generated. The analysis of the lowest binding energy ligand–enzyme complexes was conducted using the MOE ligand interaction module. For the three-dimensional interaction plot, the Discovery Studio Visualizer was employed.34,384.4. Molecular dynamics simulations
A molecular dynamics (MD) simulation was conducted to examine the extensive mobility, stiffness, and sub-cellular functions of proteins. The molecular dynamics simulations were conducted following the docking analysis. The MD simulations were conducted using the online software iMODS.1 iMODS provides a suitable framework for the analysis of magnified normal modes within their coordinate system. This web server demonstrates flexibility and responsiveness across all major browsers and contemporary devices. It provides information on various parameters, including B-factor values, eigenvalue computation, variance, protein structure deformability, elastic network models, and covariance maps.2,3 The results were obtained within a few minutes by uploading the PDB file of the docked complex to the online server as the input file, while maintaining all parameters at their default settings.364.5. Physicochemical and pharmacokinetic characteristics
SwissADME (https://www.swissadme.ch/) was used to ascertain the physicochemical and pharmacokinetic characteristics of the synthesized derivatives. The thorough evaluation of many pharmacokinetic characteristics, such as absorption, distribution, metabolism, and excretion (ADME), was made easier using this computational method.7 The bioavailability radar chart algorithms is based on sophisticated statistical and machine learning techniques that have been tested on big molecular datasets with distinct characteristics.8 Important ADME characteristics were clearly defined within the parameters of the BOILED-Egg model, including passive gastrointestinal absorption (HIA), permeability glycoprotein classification as either substrate or non-substrate, and blood–brain barrier (BBB) penetration.9Data availability
Data are available from the authors upon request.Conflicts of interest
There are no conflicts to declare.References
- R. J. Perry and J. R. Hodges, Attention and executive deficits in Alzheimer's disease: A critical review, Brain, 1999, 122(3), 383–404 CrossRef.
- K. R. Bales, Interaction of the β-amyloid Protein in Alzheimer's Disease and the Choline Transporter, Doctoral Dissertation, Indiana University, 2007 Search PubMed.
- Acetylcholinesterase (AChE) terminates the ACh-mediated neurotransmission and is mostly found in neurons. The therapeutic efficacies of AChE inhibitors in AD have been shown to be due to augment synaptic ACh levels in the cerebral cortex and improve cholinergic transmissions.
- T. H. Ferreira-Vieira, I. M Guimaraes, F. R Silva and F. M Ribeiro, Alzheimer's disease: targeting the cholinergic system, Curr. Neuropharmacol., 2016, 14(1), 101–115 CrossRef CAS.
- G. T. Grossberg, Cholinesterase inhibitors for the treatment of Alzheimer's disease:: getting on and staying on, Curr. Ther. Res., 2003, 64(4), 216–235 CrossRef CAS.
- C. G. Ballard, N. H. Greig, A. L. Guillozet-Bongaarts, A. Enz and S. Darvesh, Cholinesterases: roles in the brain during health and disease, Curr. Alzheimer Res., 2005, 2(3), 307–318 CrossRef CAS PubMed.
- L. Pezzementi, F. Nachon and A. Chatonnet, Evolution of acetylcholinesterase and butyrylcholinesterase in the vertebrates: an atypical butyrylcholinesterase from the Medaka Oryzias latipes, PLoS One, 2011, 6(2), e17396 CrossRef CAS PubMed.
- A. S. Kalgutkar, N. Castagnoli Jr and B. Testa, Selective inhibitors of monoamine oxidase (MAO-A and MAO-B) as probes of its catalytic site and mechanism, Med. Res. Rev., 1995, 15(4), 325–388 CrossRef CAS.
- M. G. Erkkinen, M. O. Kim and M. D. Geschwind, Clinical neurology and epidemiology of the major neurodegenerative diseases, Cold Spring Harbor Perspect. Biol., 2018, 10(4), a033118 CrossRef.
- L. Minghetti, Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases, J. Neuropathol. Exp. Neurol., 2004, 63(9), 901–910 CrossRef CAS PubMed.
- P. S. Khansari and L. Coyne, NSAIDs in the treatment and/or prevention of neurological disorders, Inflammopharmacology, 2012, 20(3), 159–167 CrossRef CAS PubMed.
- M. Yan, S. Zhang, C. Li, Y. Liu, J. Zhao, Y. Wang and L. Zhang, 5-Lipoxygenase as an emerging target against age-related brain disorders, Ageing Res. Rev., 2021, 69, 101359 CrossRef CAS PubMed.
- T. A. E. R. Farghaly, M. A. Abdallah and H. K. MMAHMOUD, Synthesis of novel 1, 2, 4-triazoles and triazolo-thiadiazines as anticancer agents, Turk. J. Chem., 2015, 39(5), 955–969 CrossRef CAS.
- Z. A. Kaplancıklı, G. Turan-Zitouni, A. Özdemir and G. Revial, New triazole and triazolothiadiazine derivatives as possible antimicrobial agents, Eur. J. Med. Chem., 2008, 43(1), 155–159 CrossRef PubMed.
- I. Khan, A. Ibrar, S. Zaib, S. Ahmad, N. Furtmann, S. Hameed and J. Iqbal, Active compounds from a diverse library of triazolothiadiazole and triazolothiadiazine scaffolds: Synthesis, crystal structure determination, cytotoxicity, cholinesterase inhibitory activity, and binding mode analysis, Bioorg. Med. Chem., 2014, 22(21), 6163–6173 CrossRef CAS PubMed.
- I. Khan, A. Ibrar and N. Abbas, Triazolothiadiazoles and triazolothiadiazines–Biologically attractive scaffolds, Eur. J. Med. Chem., 2013, 63, 854–868 CrossRef CAS PubMed.
- I. Khan, et al., Exploration of a library of triazolothiadiazole and triazolothiadiazine compounds as a highly potent and selective family of cholinesterase and monoamine oxidase inhibitors: design, synthesis, X-ray diffraction analysis and molecular docking studies, RSC Adv., 2015, 5(27), 21249–21267 RSC.
- S. M. A. Abid, S. Aslam, S. Zaib, S. M. Bakht, M. Ahmad, M. M. Athar, J. M. Gardiner and J. Iqbal, Pyrazolobenzothiazine-based carbothioamides as new structural leads for the inhibition of monoamine oxidases: design, synthesis, in vitro bioevaluation and molecular docking studies, Medchemcomm, 2017, 8(2), 452–464 RSC.
- I. Khan, et al., Active compounds from a diverse library of triazolothiadiazole and triazolothiadiazine scaffolds: Synthesis, crystal structure determination, cytotoxicity, cholinesterase inhibitory activity, and binding mode analysis, Bioorg. Med. Chem., 2014, 22(21), 6163–6173 CrossRef CAS.
- R. K. P. Tripathi and S. R. Ayyannan, Monoamine oxidase-B inhibitors as potential neurotherapeutic agents: An overview and update, Med. Res. Rev., 2019, 39(5), 1603–1706 CrossRef CAS.
- U. M. Zahid, A. S. Sohail, A. Mohammed, I. Hasher, R. Sanwa, S. A. Shakir and A. Mumtaz, An extensive experimental and DFT studies on highly selective detection of nitrobenzene through deferasirox based new fluorescent sensor, Spectrochim. Acta, Part A, 2024, 306, 123607 CrossRef.
- S. Alcaro, M. L. Bolognesi, A. T. García-Sosa and S. Rapposelli, Multi-target-directed ligands (MTDL) as challenging research tools in drug discovery: From design to pharmacological evaluation, Front. Chem., 2019, 7, 71 CrossRef CAS PubMed.
- (a) M. Mahmood, et al., Evaluation of 3,4-diethoxy substituted thioureas and their thiazole derivatives as potent anti-Alzheimer's agents: Synthesis, DFT, biological activity and molecular modeling investigations, J. Mol. Struct., 2024, 1314, 138760 CrossRef CAS; (b) M. Rudrapal, et al., Dual synergistic inhibition of COX and LOX by potential chemicals from Indian daily spices investigated through detailed computational studies, Sci. Rep., 2023, 13(1), 8656 CrossRef CAS.
- F. A. Larik, et al., New cholinesterase inhibitors for Alzheimer's disease: Structure activity relationship, kinetics and molecular docking studies of 1–butanoyl–3–arylthiourea derivatives, Int. J. Biol. Macromol., 2018, 116, 144–150 CrossRef CAS PubMed.
- S. Rauhamäki, et al., Structure-Activity Relationship Analysis of 3-Phenylcoumarin-Based Monoamine Oxidase B Inhibitors, Front. Chem., 2018, 6 DOI:10.3389/fchem.2018.00041.
- C. Almansa, et al., Synthesis and Structure−Activity Relationship of a New Series of COX-2 Selective Inhibitors: 1,5-Diarylimidazoles, J. Med. Chem., 2003, 46(16), 3463–3475 CrossRef CAS PubMed.
- MOE (Molecular Operating Environment) Version 2016.01, Chemical Computing Group, (CCG), https://www.chemcomp.com/MOE/Molecular_Operating_Environment.htm Search PubMed.
- J. R. López-Blanco, J. I. Aliaga, E. S. Quintana-Ortí and P. Chacón, iMODS: Internal coordinates normal mode analysis server, Nucleic Acids Res., 2014, 42, W271–W276 CrossRef PubMed.
- J. R. López-Blanco, J. I. Garzón and P. Chacón, iMod: multipurpose normal mode analysis in internal coordinates, Bioinformatics, 2011, 27(20), 2843–2850 CrossRef.
- J. Kovacs, P. Chacón and R. Abagyan, Predictions of Protein Flexibility: First Order Measures, Proteins:Struct., Funct., Bioinf., 2004, 56(4), 661–668 CrossRef CAS PubMed.
- A. Daina and V. Zoete, Application of the SwissDrugDesign online resources in virtual screening, Int. J. Mol. Sci., 2019, 20(18), 4612 CrossRef CAS PubMed.
- C.-Y. Jia, et al., A drug-likeness toolbox facilitates ADMET study in drug discovery, Drug discovery today, 2020, 25(1), 248–258 CrossRef CAS PubMed.
- C. A. Lipinski, et al., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7(1), 42717 CrossRef.
- G. L. Ellman, et al., A new and rapid colorimetric determination of acetylcholinesterase activity, Biochem. Pharmacol., 1961, 7(2), 88–95 CrossRef CAS.
- S. U. Qazi, et al., Semicarbazones, thiosemicarbazone, thiazole and oxazole analogues as monoamine oxidase inhibitors: Synthesis, characterization, biological evaluation, molecular docking, and kinetic studies, Bioorg. Chem., 2021, 115, 105209 CrossRef CAS PubMed.
- M. Mahmood, Z. Hussain, M. Z. Ullah, S. A. Ejaz, A. Fayyaz, U. Rashid, J. Iqbal and A. Mumtaz, Evaluation of 3,4-diethoxy substituted Thioureas and Their Thiazole Derivatives as Potent anti-Alzheimer’s agents: Synthesis, DFT, Biological Activity and Molecular Modeling Investigations, J. Mol. Struct., 2024, 1314, 138760 CrossRef CAS.
- A. Munir, A. Khushal, K. Saeed, A. Sadiq, R. Ullah, G. Ali, Z. Ashraf, E. U. Mughal, M. Saeed Jan, U. Rashid and I. Hussain, Amara Mumtaz Synthesis, in-vitro, in-vivo anti-inflammatory activities and molecular docking studies of acyl and salicylic acid hydrazide derivatives, Bioorg. Chem., 2020, 104, 104168 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06916a |
| This journal is © The Royal Society of Chemistry 2025 |